

Abstract
Our previous study showed that Cyp1b1 deficiency could protect male mice from high fat diet (HFD) obesity. In this paper, we aim to explore the role of Cyp1b1 in HFD induced learning and memory deficits in female C57BL/6J mice. Methods: Female Cyp1b1 knock-out (KO) and wild type (WT) mice were both randomly divided into normal chew (NC) and HFD groups. All mice were fed research diet after weaning for 24 consecutive weeks. Morris Water Maze was carried out to evaluate the spatial learning and memory. Brain lipoxidation status was evaluated by malondialdehyde (MDA) level and 4-hydroxynonenal (4-HNE)-protein adducts in mice cerebral cortex. Both activity and expression of antioxidant enzymes were determined by commercial kits and realtime RT-PCR. β-amyloid (Aβ) was detected in mice brain by immunohistochemistry. Brain derived neurotrophic factor (BDNF), synaptic plasticity protein Activity-regulated cytoskeleton-associated protein (ARC), neuronal migration and positioning gene Reelin, and Nrf2, a key transcription factor in oxidative stress, and its downstream targets heme oxygenase-1 (HO-1) and NAD(P)H dehydrogenase (quinone) (NQO-1) were measured in cerebral cortex. Results: Cyp1b1 deficiency mice performed better on learning and memory tests compared with WT mice after 24 weeks HFD feeding. HFD elevated brain oxidative stress, lipoxidation in mice cerebral cortex, β-amyloid deposition in hippocampus; suppressed antioxidant genes expression in cerebral cortex, and these effects were ameliorated by Cyp1b1 deletion. BDNF expression increased in Cyp1b1 deficiency mice after HFD feeding compared with WT. HFD activated Nrf2 and its target genes and Cyp1b1 deletion reversed such impact. Conclusion: Cyp1b1 deficiency protects mice from HFD induced cognitive impairment. Sustained BDNF expression, reduced β-amyloid accumulation and brain oxidative stress, and Nrf2 deactivation might be the key events in mice redox system through Cyp1b1-diet interaction.
Keywords: CYP1B1, learning and memory, oxidative stress, antioxidant enzymes
Introduction
Increased brain oxidative stress is considered as an early event in cognitive function impairment and has been implicated in its pathogenesis. Free radical damage from oxidative stress has long been thought to play an important role in age-related neurodegenerative disorders [1]. The feature of the oxidative stress hypothesis for neurodegenerative diseases is that cumulative oxidative damage could account for the slowly progressive nature of these disorders. The direct evidence supporting oxidative stress in AD comes from the increased free radical generation, increased lipid peroxidation, increased 4-HNE, increased DNA oxidation, diminished energy metabolism and aggregated Aβ [2].
Long term consumption of a high fat diet (HFD) results in excess energy intake, increased adiposity and impaired regulation of metabolic processes [3,4]. Long-term exposition to a high fat diet also induced β-amyloid depositions in the brain of C57BL/6J mice [5]. Lin’s lab found that HFD intake significantly promotes the progression of AD like pathology in 5XFAD mice via enhancing oxidative stress and cerebral amyloid angiopathy [6]. The cerebral accumulation of Aβ is a contributor to the initiation and maintenance of neuronal degeneration in AD [7,8]. Some researchers focus on screening natural products which can influence the dynamic equilibrium of Aβ. Supplementation of TQRF nano-emulsion into a high fat-cholesterol diet (HFCD) mitigated memory deficits, lipid peroxidation and soluble Aβ levels in rats caused by HFCD [9]. Long-term consumption of grape powder containing diet lowered Aβ expression and subsequently ameliorated the cognitive decline in aged rats caused by a long-term high-fructose-high-fat diet [10]. Silymarin ameliorated memory deficits in mouse model of HFD induced experimental dementia by its anti-oxidative potential and decreases in Aβ burden [11].
CYP1B1 is a member of the cytochrome P450 family of proteins that metabolize a large number of compounds. This family of enzymes catalyzes many mono-oxygenase reactions targeting both foreign and endogenous lipophilic compounds including fat-soluble vitamins and polyunsaturated fatty acid (PUFA) products [12]. CYPs are abundant in the liver and also expressed in many other tissues including the brain [13]. Although CYPs levels in the brain are much lower, they can potentially contribute to the local metabolism [14]. It has been recognized that P450s can generate mutagenic metabolites and produce reactive oxygen species (ROS) that result in oxidative stress and cell death [15,16]. F. Chen found that inhibition of CYP1B1 reduced both oxidative stress and the activation of JNK signaling, among which oxidative stress is most prominent [17]. But Y.X. Tang demonstrated that CYP1B1 metabolized cell products that modulate intracellular oxidative stress and lack of CYP1B1 leads to increased intracellular oxidative stress in the endothelium [18]. There are also some studies discovered the relationship between CYPs and cognitive function. Wang and Lee found that CYPs metabolized testosterone and estradiol [19], which belongs to neuroactive steroids that have a considerable impact on cognition and memory [20]. Brain-specific CYP46A1 knockout mice produced lower level of brain cholesterol, and CYP46A1 polymorphisms were associated with cognitive impairment in human [21].
Although there have been quite a number of studies investigated the mechanism of oxidative stress and cognitive impairment induced by high fat diet (HFD), the specific role of CYP1B1 in HFD effects on brain aging are poorly understood. To investigate the role of CYP1B1 in oxidative stress and cognitive function, genetic knockout mice have been employed. Our investigation will provide important insight into the mechanisms of CYP1B1 regulated brain aging. Herein, we tested the hypothesis that Cyp1b1 deficiency ameliorates learning and memory deficits caused by HFD through balancing oxidative stress.
Materials and methods
Diets and animals
All animal experiments were approved by the Institutional Animal Care, Use Committee and Wuhan University. Cyp1b1 knock-out (KO) mice were generously sent by Frank Gonzale and C57Bl/6 wild type (WT) mice were obtained from Wuhan University Animal Experimental Center. All animals had free access to food and water, and were housed in cages (22°C temperature and 12-hour daylight cycle). In initial studies, mice were randomly assigned into high fat diet (HFD) or normal chew (NC) groups (8/group): HFD consisting of 60% animal fat (Shanghai Laboratory Animal Center), and their corresponding NC diet consisting of 5% animal fat. Mice were maintained on diet for 24 weeks, with body weight recorded every 2 weeks.
After 24 weeks on their respective diets, all mice were measured for cognitive performance using Morris Water Maze (described below). Afterwards, they were fasted overnight and scarified by either cervical translocation or transcardial perfusion, vena caudal blood was collected for biochemical parameters, brains were collected for molecular measures and immunohistochemistry.
Morris water maze
The Water Maze (Shanghai Mobiledatum Information Technology Co., China) consisted of a circular pool (1.2 m in diameter and 50 cm deep) filled to a depth of 45 cm with water rendered opaque by addition of milk powder. A white platform with 6 cm in diameter was submerged 1 cm below the surface of the water. Water temperature was maintained at 20-22°C. Spatial learning and memory was assessed in the hidden platform version of the Morris task. Mice were placed in the pool facing the wall and allowed to swim until they found the platform or until 120 s had elapsed. If a mouse didn’t find the platform within 120 s, it was placed on it for 15 s. Mice received 8 trails per day during 5 consecutive days with the platform positioned in the southwest quadrant. All trials were recorded with a video camera connected to a computerized system, and escape latency to find the visible platform was used as a parameter for spatial learning. On day 6, the mice performed a probe trial which involved removing the platform from the pool. The trial was performed with the cut-off time of 120 s. The time spent in the target quadrant (previously contained the platform) and the swimming paths were used for spatial memory assessment.
Brain homogenate oxidative stress
Cerebral cortex tissue was homogenized in a saline solution containing 0.9% NaCl. The homogenates were then centrifuged, the supernatants were collected and total protein concentration was determined using the BCA Protein Assay (Beyotime, China). Cerebral cortex homogenate MDA, a common stable marker for oxidative stress induced lipid peroxidation, was determined by a spectrophotometric measurement of thiobarbituric acid-reactive substances (TBARS) according to the instructions (Nanjing jiancheng Biochemistry, China). The absorbance was read at 532 nm on microplate reader for ELISA (Bio-Tek, USA).
Brain homogenate antioxidant enzyme activity
A 10% cerebral cortex homogenate was prepared in 0.1 M phosphate buffer (pH 7.4) containing 0.1 M KCl. Enzyme assays were performed in the supernatant obtained following centrifugation of the homogenate at 9000 × g for 10 min at 4°C. The protein concentration of each sample was determined by the Bradford reagent method with BSA standard.
Cerebral cortex activities of catalase and glutathione level were assayed by the methods of He et al. (2008) [22] and Bi et al. (2008) [23], respectively. These assay kits were purchased from Beyotime Institute of Biotechnology, China. And SOD activity was measured using the SOD Assay Kit-WST (Dojindo Laboratories, Japan), using the protocols provided by the manufacturer.
Immunohistochemistry
After being anesthetized with diethyl ether, mice were transcardially perfused with 25 ml 0.1 M PBS, follow by 100 ml 4% paraformaldehyde. Brains were collected and post-fixed overnight in 4% PFA at 4°C. The brains were transferred to 30% sucrose for overnight, and then were cut into 30-um sections by a cryostat microtome. After several washes in PBS, endogenous peroxidase activity was quenched by 3% hydrogen peroxide in PBS for 30 min, followed by 10 min wash in PBS. The slides were incubated at 4°C with a rabbit polyclonal antibody against Aβ antibody (1:150, Abcam, ab2539) overnight. Slides were washed in PBS before incubated with a HRP conjugated goat anti-rabbit IgG secondary antibody (1:1000, Cell signaling) at room temperature for 1 h. Slides were washed with PBS and visualized by the chromogen 3,3’-diaminobenzidine (Vector Laboratories). Measurement of Aβ density in the CA1, CA3 and DG of the hippocampus were defined as the area covered by Aβ immunoreactivity per mm2 of the total area of the region measured, and quantified in three succeeding slides of the hippocampus for each mouse by the program image J.
Western blot
The brain tissues were homogenized with RIPA buffer containing protease inhibitor cocktail and centrifuged at 10,000 × g for 15 min at 4°C. Equal amounts of total proteins from cortex tissue were resolved on 10% or 15% SDS-PAGE, and then transferred to nitrocellulose membranes. Non-specific binding sites were blocked in TBST containing 5% skim milk at room temperature for 2 hrs. Then membranes were incubated with anti-4-HNE (1:1000; ab46545; Abcam), anti-BDNF (1:500; SC546; Santa Cruz), anti-Nrf2 (1:1000; D1C9; Cell Signaling), anti-eNOS (1:1000; 49G3; Cell Signaling) and anti-actin (1:5000, A5441; Sigma) overnight at 4°C. After three washes with TBST, blots were incubated for 1 h at room temperature with anti-rabbit IgG-HRP-conjugated secondary antibody, followed by three washes with TBST. The membranes were visualized by chemiluminescence reagents (SuperSignal West Pico, Pierce), and the densities of target bands were measured with Quantity One (Bio-Rad) and expressed as relative level with respect to the normal control.
Real-time RT-PCR
Brain cortex was carefully dissected from frozen tissue and total RNA was prepared by Trizol reagent (Invitrogen). The concentration of nucleic acids was determined spectrophotometrically at 260 nm and 280 nm. Total RNA (1 μg) was subjected to reverse transcription with the Omniscript RT in a 20 μL reaction. The RT reaction was diluted ten-fold and 1 μL was amplified by Real-time PCR (quantitative PCR) in 5 μL reaction mixture containing Master Sybr Green (Applied Biosystems). Gene specific primers derived from mice genomic DNA sequences for eNOS, iNOS, nNOS, GPx1, PHGPx, SOD, BDNF, ARC, Reelin, HO-1 and NQO-1 genes (NCBI) will be provided upon request. Relative quantification of gene expression was carried out by comparative Ct method according to manufacture protocol.
Statistical analysis
Statistical analysis was performed using SPSS for Windows 19.0 package and the GraphPad Prism 5.0 software. Each experiment was performed at least three times, and the results were presented as means ± SD. The repeated measures ANOVA was applied for the acquisition phase of the Morris Water Maze (MWM). Differences among groups in biochemical parameters and probe phase of the MWM were tested by multivariate ANOVA, followed by Bonferroni post hoc pairwise comparisons. Statistical outliers were removed from the dataset, and Statistical significance was set at P<0.05.
Results
CYP1B1 deficiency protects mice from excess body weight gained caused by HFD
Right after weaning, mice were put on the NC and HFD. Both WT and KO mice gained significant body weight relative to their NC partners after exposed to HFD for consecutive 24 weeks, but KO mice gained significant less weight than WT ones (Figure 1A). HFD exhibited similar effect on blood glucose in both wild type mice and KO mice (Figure 1B).
Figure 1.
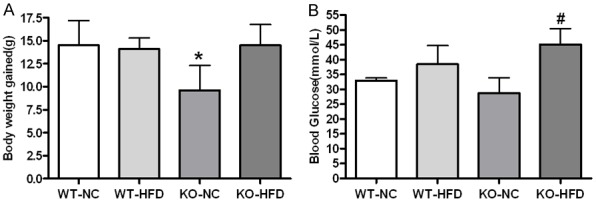
Changes in body weight and blood glucose. Female mice were placed on high fat diets (HFD) or normal chew for 24 weeks and analyzed for changes in body weight (A) and Blood glucose (B). Data are presented as the mean and SD from more than 6 animals per group. *Significantly different from WT-NC, #Significantly different from WT-HFD, P<0.05.
CYP1B1 deficiency ameliorates learning and memory deficits induced by HFD
Previous reports have demonstrated that obesity increased cognitive impairment in experimental animals [24]. To determine effects of CYP1B1 on cognitive function under HFD feeding, WT and KO mice were tested in the Morris Water Maze to assess their spatial learning and memory at the end of the 24 weeks of diet-exposure.
The total swimming distance during all trials was not affected by the increased body weights in the WT and KO mice caused by the HFD, therefore, we analyzed the data without adjusting the body weight.
During acquisition phase, the escape latency to platform significantly decreased in all groups, suggesting that the mice learned the task (Figure 2A). However, HFD fed WT mice displayed a significant increased escape latency compared to their counterparts. KO mice demonstrated a significant decreased escape latency compared to WT mice in HFD feeding (Figure 2A). In the probe phase, KO mice spent significant longer time in platform area compare to the control mice (Figure 2B).
Figure 2.
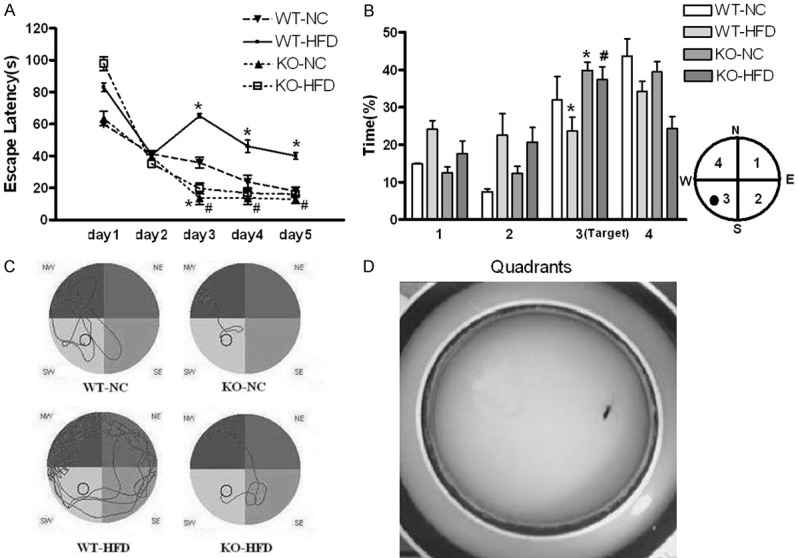
Effects of Cyp1b1 and HFD diet on spatial learning and memory detected by Morris Water Maze. A. The escape latency (mean ± S.E.M) to find the platform was higher both in female WT and KO mice fed HFD. The learning curves indicated that KO mice learned the task faster when comparing with WT mice. B. Probe trial data showed that all groups spent significant longer time in the target quadrant than time spent in quadrants 1 and 2. KO mice performed better in the target area compared to WT controls either fed HFD or NC. C. Typical swimming paths in the probe trial. Compared with WT mice, KO mice found the platform more quickly and efficiently. D. Representative image of forced swimming test. *Significantly different from WT-NC, #Significantly different from WT-HFD, P<0.05.
Furthermore, typical swim paths analysis indicated that WT mice spent more time in the target quadrant than KO mice under normal condition. However, WT mice with HFD swam more randomly showing an obvious impairment to recognize the target, while Cyp1b1 deficiency reversed this impairment (Figure 2C).
CYP1B1 deficiency increases brain BDNF levels in NC group, not HFD groups
BDNF can enhance synaptic plasticity which closely associated with cognitive function [25]. Previous study showed that the diet rich in saturated fat decreases brain-derived neurotrophic factor (BDNF) to the extent that compromises cognitive performance [26]. We found that Cyp1b1 deletion increased both protein and mRNA expression of BDNF compared to WT-NC mice, but did not reverse the inhibition of BDNF by HFD feeding (Figure 3A, 3B). Furthermore, ARC, a marker of synaptic plasticity following memory consolidation in the brain, and Reelin, a marker of neuronal migration and position, were both elevated in Cyp1b1 deficient mice when compared with their counterparts (Figure 3C, 3D).
Figure 3.
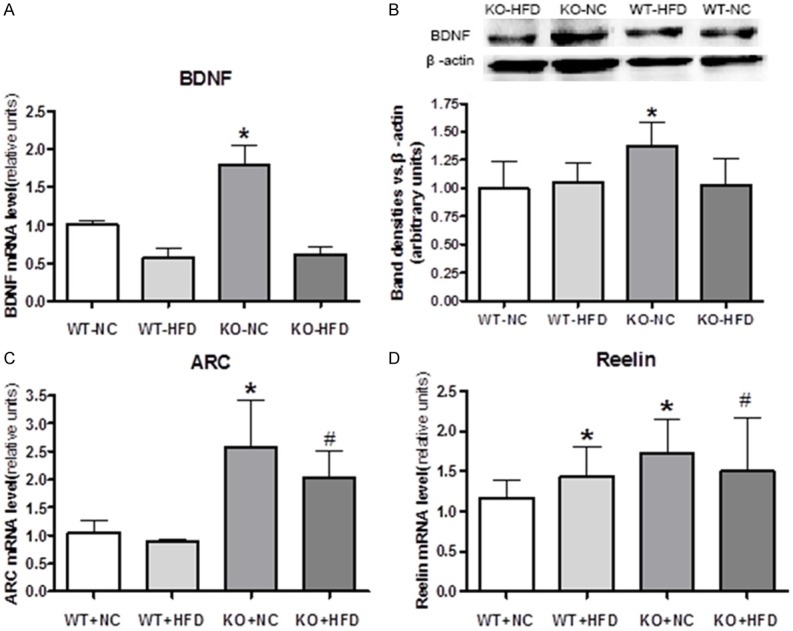
Expression of Brain-derived neurotrophic factor (BDNF) in mice cerebral cortex. A. Brain tissue from Cyp1b1 deficient mice fed with NC had higher BDNF mRNA expression. B. Representative Western blots of BDNF, and quantified data show that BDNF protein levels are higher in KO mice fed with NC than in other groups. C and D. Cyp1b1 deficient mice had higher levels of activity-regulated cytoskeletal protein (ARC) and Reelin when compared with their counterparts. *Significantly different from WT-NC, #Significantly different from WT-HFD, P<0.05.
CYP1B1 deficiency suppresses brain oxidation induced by HFD
Well documented relationship between cognitive impairment and oxidative stress [27] prompted us to investigate the role of oxidative stress in cyp1b1 regulated cognitive function.
Malondialdehyde (MDA), as a lipid peroxidation marker, is widely used as an indicator of free radical-mediated lipid peroxidation injury [28]. We found Cyp1b1 deficiency markedly reduced brain MDA levels in HFD groups, suggesting Cyp1b1 deficiency reduced the brain oxidation (Figure 4A).
Figure 4.
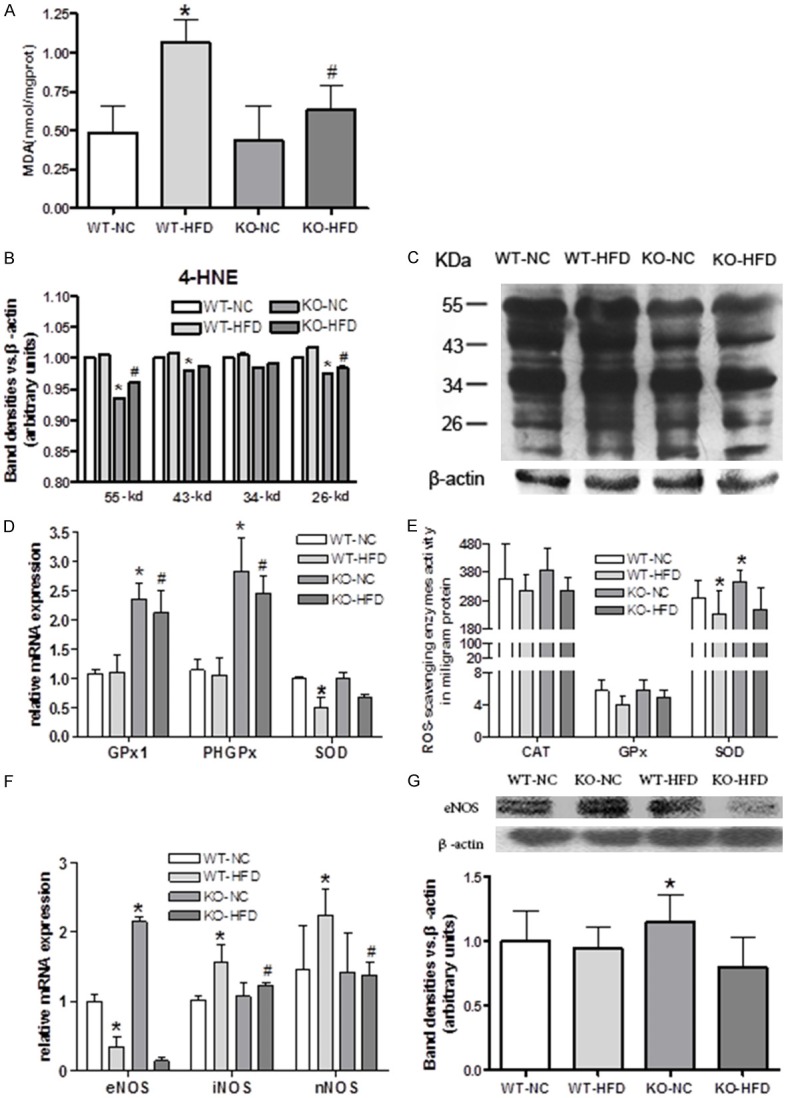
Cyp1b1 deficiency decreased oxidative stress and increased anti-oxidant capacity caused by HFD. A. HFD significantly increased malondialdehyde (MDA) levels and Cyp1b1 deletion suppressed such effect. B. 4-HNE expression decreased in KO mice compared to WT mice. C. Representative Western blots of 4-hydroxynonenal (4-HNE). D. Effect of Cyp1b1 deficiency on antioxidants GPx1, PHGPx and SOD mRNA expression in mice cerebral cortex (β-actin as reference). E. Cyp1b1 deficiency enhanced catalase (CAT), glutathione peroxidase (GPx) and superoxide dismutase (SOD) levels in mice cerebral cortex. F. Cyp1b1 deficiency modulates eNOS, iNOS and eNOS mRNA expression in mice cerebral cortex. G. Representative Western blots of eNOS, and Quantified data. One unit of CAT activity = 1 μmol hydrogen peroxide catalyzed per minute. One unit of GPx activity = 1 nmol NADPH oxidized per minute. One unit of SOD is defined as the amount of the enzyme in 20 μl of sample solution that inhibits the reduction reaction of WST-1 with superoxide anion by 50%. β-actin is loading control for mRNA detection. GAPDH is loading control for protein detection. *Significantly different from WT-NC, #Significantly different from WT-HFD, P<0.05.
4-hydroxynonenal (4-HNE), structurally modified proteins which forming stable adducts termed advanced lipid peroxidation end products [29], may also play a mechanistic role in the pathogenesis of oxidative stress. Previous reports indicated that HFD contributed to oxidative injury in brain by 4-HNE accumulation. We found 4-HNE protein adduct were lower in Cyp1b1 deficiency mice, suggesting Cyp1b1 deficiency might alleviate the effect of oxidation injury induce by HFD (Figure 4B, 4C).
Oxidative stress is essentially an imbalance between reactive oxygen species (ROS) production and the antioxidant defense. Different ROS scavenging enzymes including catalase, GPx, PHGPx and SOD in mice cerebral cortex were measured in both activity and mRNA levels. In general, HFD modestly suppressed these antioxidant enzymes activities compared with NC groups, and Cyp1b1 deficiency displayed obvious higher antioxidant enzyme activities than WT-NC mice. In addition, cyp1b1 deficiency markedly reversed a significant decreased of GPx activity caused by HFD feeding (Figure 4D).
HFD feeding resulted in a significant decrease in GPx1, PHGPx and SOD, mRNA expression in WT mice, while knocking down Cyp1b1 markedly increased GPx1, and PHGPx mRNA levels in both NC and HFD groups when compared to WT mice (Figure 4E).
Oxidative stress is responsible for redox regulation involving not only reactive oxygen species (ROS) but also reactive nitrogen species (RNS)[26]. Nitric oxide (NO), is a reactive free radical produced by three isoforms of NO synthase in brain. Therefore, brain NO synthases (NOS)-endothelial (eNOS), inducible (iNOS) and neuronal (nNOS) were measured. HFD significantly decreased brain eNOS mRNA, whereas increased brain iNOS and nNOS mRNA expressions (Figure 4F). Cyp1b1 deletion reverse the HFD effects on eNOS, iNOS and nNOS mRNA levels. Furthermore, Cyp1b1 deficient mice brain had highest eNOS protein levels; with HFD feeding, the eNOS protein levels became comparable between KO and WT groups (Figure 4G).
CYP1B1 deficiency reduces Aβ accumulation in DG region of hippocampus caused by HFD
Previous study found that HFD stimulates the expressions of APP, BACE1 and Aβ production [30]. Existed findings suggest that oxidative stress is upstream of Aβ in AD and that Aβ might be generated as a compensatory response in neurons attempting to attenuate oxidative stress [31]. Our observation indicated that HFD resulted in significant Aβ deposition in hippocampus of WT mice brains. Cyp1b1 deficiency did not altered Aβ accumulation in NC groups, but did alleviate it in DG region of hippocampus in HFD groups (Figure 5).
Figure 5.
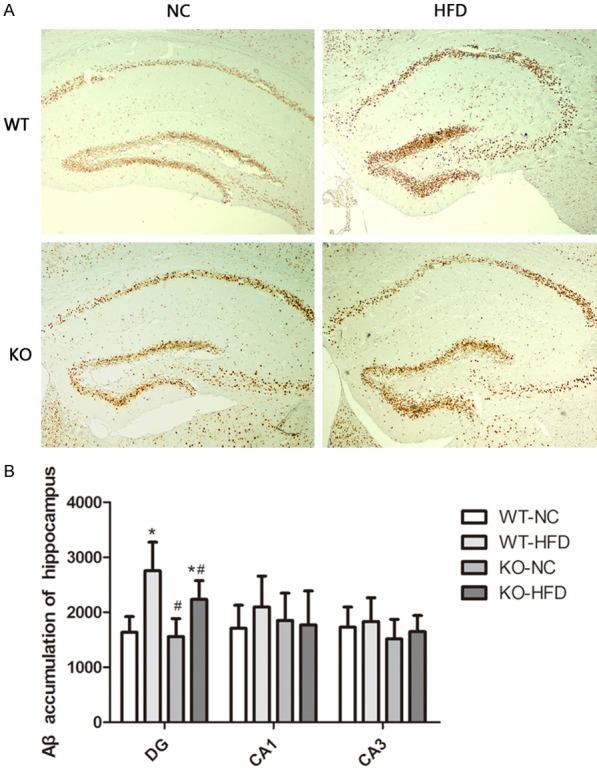
Cyp1b1 deficiency protect HFD induced Aβ deposition. A. Representative low magnification images of Aβ staining in hippocampus. B. Quantified data showed that HFD induced Aβ accumulation and Cyp1b1 deficiency reversed such effect in DG region of hippocampus. *Significantly different from WT-NC, #Significantly different from WT-HFD, P<0.05.
CYP1B1 deficiency rescued brain Nrf2 signaling damage in HFD-fed mice
Nuclear factor-erythroid-2 related factor 2 (Nrf2) is an essential transcription factor maintaining homeostasis in redox systems including antioxidant agents and phase II detoxifying protect against ROS elicited tissue damage. We found that HFD consumption decreased Nrf2 content in cytoplasm (Figure 6A, 6B), and accompanied by markedly increased Nrf2 target genes, including antioxidant heme oxygenase 1 (HO-1) and detoxifying agents NADPH dehydrogenase (quinone 1) 1 (NQO-1), suggesting activation of Nrf2 (Figure 6C). Cyp1b1 deletion partially inhibited Nrf2 activation, and such effects might attribute to its lower oxidative stress and higher antioxidant enzymes (GPx and SOD) levels.
Figure 6.
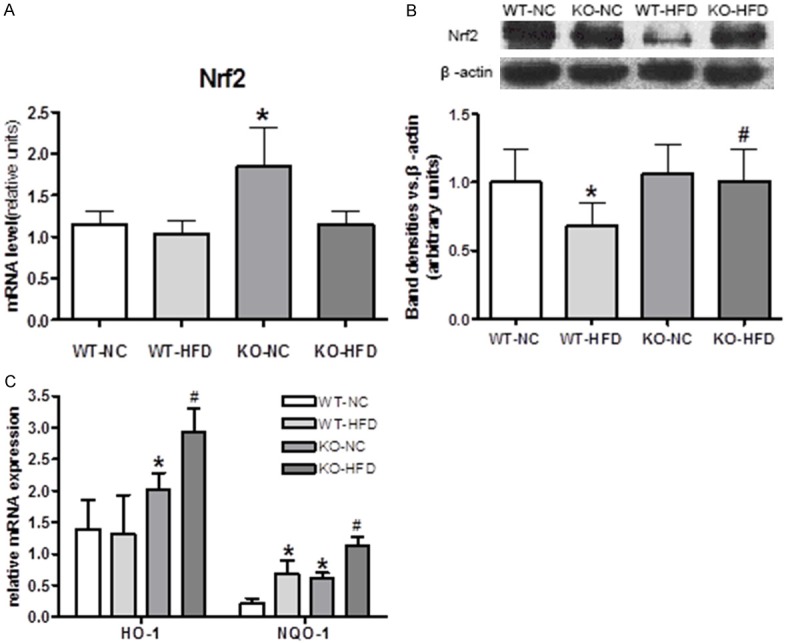
Up-regulation of Nuclear factor-erythroid-2-related factor 2 (Nrf2) protein and its target genes in the face of oxidative stress induced by HFD. A. Cyp1b1 deletion promoted Nrf2 mRNA expression. B. Representative Western blots of Nrf2, and quantified data showed that Cyp1b1 deletion sustained Nrf2 expression suppressed by HFD. C. Nrf2 target genes, heme oxygenase-1 (HO-1), NAD(P)H dehydrogenase (quinone) (NQO-1) were elevated by Cyp1b1 deletion in both NC and HFD groups. *Significantly different from WT-NC, #Significantly different from WT-HFD, P<0.05.
Discussion
We provide evidence that CYP1B1 plays an important role in the HFD-associated cognitive deficits and oxidative damage in animal experiment. In this study, both KO mice and WT mice were given HFD (60% calories from fat), and their cognitive performance and brain oxidative stress were evaluated. Spatial learning and memory assessed by Morris water maze show that HFD did cause the detrimental cognitive performance, which is consistent with the documented relationship between obesity and cognitive impairment [32]. Whereas Cyp1b1 deficient mice perform better than WT mice with shorter escape latency and longer time spent in target quadrant. Compared to WT mice, Cyp1b1 deficient mice achieved greater proficiency in locating the hidden platform and employed spatial search strategy earlier with greater accuracy.
In agreement with previous reports [33], HFD consumption was associated with significant decreases in brain BDNF. However, KO-NC mice had the most abundant BDNF expression in cerebral cortex (Figure 3), which is in accordance with the excellent cognitive performance (Figure 2). However, compare with WT-HFD mice, better spatial learning and memory performance in KO-HFD group could not be explained by BDNF expression. Synaptic plasticity here might play more important role in cognitive function because the upregulation of ARC and Reelin levels by Cyp1b1 deficiency in both NC and HFD groups.
MDA level is not only reflect the extent of lipid peroxidation but also oxidative susceptibility of high- and low-density lipoproteins in serum [34]. Park’s group found that HFD increased the level of MDA and decreased BDNF expression. The high level of MDA reduced the growth of neural progenitor cells (NPCs), BDNF treatment restored NPCs proliferation impaired by HFD [34]. Our MDA findings support previous studies that there is a negative correlation between cognitive function and oxidative damage [35]. We found that 4-HNE level were consistent with the MDA changes, suggesting that CYP1B1 may be a key trigger in HFD-induced oxidation resistance.
SOD functions as one of the primary enzymatic antioxidant defenses against superoxide radicals, and increases in SOD enzyme activity corresponds with enhanced resistance to oxidative stress [36]. GPx is another antioxidant enzyme with a much greater affinity for hydrogen peroxide than catalase. Enhanced expression/activity of the endogenous antioxidant enzymes SOD and GPx has been commonly used as relevant indices of brain oxidative stress [37]. A decrease in the activity of any of these enzymes would lead to cellular death, which is one of the major characteristics of Alzheimer’s disease in the cerebral cortex [38]. We found Cyp1b1 deficiency significantly elevated GPx1, PHGPx mRNA expression and SOD activity in cerebral cortex in both NC and HFD groups when comparing to their counterparts, suggesting that improved antioxidant defense by Cyp1b1 deletion might contribute to their lower oxidative stress and better cognitive performance.
The bioavailability of nitric oxide (NO) can be affected by metabolism including increases in the generation of ROS [39]. Consistent with Tang’s results, Cyp1b1 deletion dramatically decreased brain eNOS expression in HFD-fed mice compared with WT ones [40], but not in NC-fed mice. It’s documented that inactivation of eNOS under oxidative stress results in decreased NO bioavailabilit [41], which is an effective anti-oxidant through the reaction with superoxide to form peroxynitrite [42]. Downregulations of iNOS and nNOS expression were observed in Cyp1b1 deficient mice with HFD. iNOS contributes to lipid peroxidation injuries in the intermittent hypoxia model [43]. NO produced by iNOS and nNOS has been implicated in brain damage, while by eNOS is known to be neuroprotective because of its vasodilative effects [44].
Previous study found that oxidative stress is upstream of Aβ in AD [31]. Sustained HFD over the entire lifespan resulted in impaired perivascular clearance of Aβ from brain [45]. Our observation indicated that Aβ accumulation did not altered by Cyp1b1 deletion in NC groups, but did alleviate it in DG region of hippocampus in HFD feeding groups. BDNF treatment reduced production of toxic Aβ via enhancing a-secretase [46], less Aβ deposition in KO-HFD group might be due to the fact that Cyp1b1 deficiency upregulated BDNF. Furthermore, curcumin, exerting its anti-tumor activity by modulating cytochrome P450 function, was found to reduce Aβ1-42 protein accumulation in brain of mice [47].
Nrf2 is a transcription factor that plays an important role in defending against oxidative stress induced damage. Nrf2 expression was significantly decreased in transgenic AD mice [48]. HFD consumption increased hippocampus oxidative stress and damaged cognitive function, which were correlated with decreased Nrf2 signaling [24]. In the other spectrum of story, the activation of Nrf2/HO-1 pathway attenuated Aβ neurotoxicity [49], and suppressed Aβ induced oxidative stress in a cellular model of AD [50]. Our 24 weeks of feeding study indicated that HFD produced significant oxidative stress, while Cyp1b1 deletion provided a compensatory response including Nrf2 activation and its target genes expression which ameliorates the impact of oxidative stress.
In conclusion, the current data demonstrated that mice with 24 weeks of HFD consumption displayed cognitive impairment, and this effect was associated with an increased oxidative damage within the brain. However, Cyp1b1 deficiency was effective in ameliorating memory deficits and reducing HFD-induced brain oxidative stress, as evidenced by better performance in Morris Water Maze, downregulation of lipid peroxidation and Aβ accumulation, elevation of antioxidant enzymes levels. Nrf2 activation might be the key pathway Cyp1b1 exerts its protection, however, further studies are required to confirm such relationship by applying Nrf2 inhibitor.
Acknowledgements
This work was supported by National Natural Science Foundation of China (Grant No. 30972463, and 81172664). The content of the information does not necessarily reflect the position or the policy of the government, and no official endorsement should be inferred.
Disclosure of conflict of interest
None.
References
- 1.Li RL, Lu ZY, Huang JJ, Qi J, Hu A, Su ZX, Zhang L, Li Y, Shi YQ, Hao CN, Duan JL. SRT1720, a SIRT1 specific activator, protected H2O2-induced senescent endothelium. Am J Transl Res. 2016;8:2876–2888. [PMC free article] [PubMed] [Google Scholar]
- 2.Cheignon C, Tomas M, Bonnefont-Rousselot D, Faller P, Hureau C, Collin F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018;14:450–464. doi: 10.1016/j.redox.2017.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Avalos Y, Kerr B, Maliqueo M, Dorfman M. Cell and molecular mechanisms behind diet-induced hypothalamic inflammation and obesity. J Neuroendocrinol. 2018;30:e12598. doi: 10.1111/jne.12598. [DOI] [PubMed] [Google Scholar]
- 4.Zhang L, Bruce-Keller AJ, Dasuri K, Nguyen AT, Liu Y, Keller JN. Diet-induced metabolic disturbances as modulators of brain homeostasis. Biochim Biophys Acta. 2009;1792:417–422. doi: 10.1016/j.bbadis.2008.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Busquets O, Ettcheto M, Pallas M, Beas-Zarate C, Verdaguer E, Auladell C, Folch J, Camins A. Long-term exposition to a high fat diet favors the appearance of beta-amyloid depositions in the brain of C57BL/6J mice. A potential model of sporadic Alzheimer’s disease. Mech Ageing Dev. 2017;162:38–45. doi: 10.1016/j.mad.2016.11.002. [DOI] [PubMed] [Google Scholar]
- 6.Lin B, Hasegawa Y, Takane K, Koibuchi N, Cao C, Kim-Mitsuyama S. High-fat-diet intake enhances cerebral amyloid angiopathy and cognitive impairment in a mouse model of Alzheimer’s disease, independently of metabolic disorders. J Am Heart Assoc. 2016;5 doi: 10.1161/JAHA.115.003154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 8.Forman MS, Trojanowski JQ, Lee VM. Neurodegenerative diseases: a decade of discoveries paves the way for therapeutic breakthroughs. Nat Med. 2004;10:1055–1063. doi: 10.1038/nm1113. [DOI] [PubMed] [Google Scholar]
- 9.Ismail N, Ismail M, Azmi NH, Bakar M, Yida Z, Stanslas J, Sani D, Basri H, Abdullah MA. Beneficial effects of TQRF and TQ nano- and conventional emulsions on memory deficit, lipid peroxidation, total antioxidant status, antioxidants genes expression and soluble Abeta levels in high fat-cholesterol diet-induced rats. Chem Biol Interact. 2017;275:61–73. doi: 10.1016/j.cbi.2017.07.014. [DOI] [PubMed] [Google Scholar]
- 10.Chou LM, Lin CI, Chen YH, Liao H, Lin SH. A diet containing grape powder ameliorates the cognitive decline in aged rats with a long-term high-fructose-high-fat dietary pattern. J Nutr Biochem. 2016;34:52–60. doi: 10.1016/j.jnutbio.2016.04.006. [DOI] [PubMed] [Google Scholar]
- 11.Neha , Kumar A, Jaggi AS, Sodhi RK, Singh N. Silymarin ameliorates memory deficits and neuropathological changes in mouse model of high-fat-diet-induced experimental dementia. Naunyn Schmiedebergs Arch Pharmacol. 2014;387:777–787. doi: 10.1007/s00210-014-0990-4. [DOI] [PubMed] [Google Scholar]
- 12.Coon MJ. Cytochrome P450: nature’s most versatile biological catalyst. Annu Rev Pharmacol Toxicol. 2005;45:1–25. doi: 10.1146/annurev.pharmtox.45.120403.100030. [DOI] [PubMed] [Google Scholar]
- 13.Warner M, Gustafsson JA. Effect of ethanol on cytochrome P450 in the rat brain. Proc Natl Acad Sci U S A. 1994;91:1019–1023. doi: 10.1073/pnas.91.3.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hedlund E, Gustafsson JA, Warner M. Cytochrome P450 in the brain; a review. Curr Drug Metab. 2001;2:245–263. doi: 10.2174/1389200013338513. [DOI] [PubMed] [Google Scholar]
- 15.Knockaert L, Fromenty B, Robin MA. Mechanisms of mitochondrial targeting of cytochrome P450 2E1: physiopathological role in liver injury and obesity. FEBS J. 2011;278:4252–4260. doi: 10.1111/j.1742-4658.2011.08357.x. [DOI] [PubMed] [Google Scholar]
- 16.Barouki R, Morel Y. Repression of cytochrome P450 1A1 gene expression by oxidative stress: mechanisms and biological implications. Biochem Pharmacol. 2001;61:511–516. doi: 10.1016/s0006-2952(00)00543-8. [DOI] [PubMed] [Google Scholar]
- 17.Chen F, Castranova V, Li Z, Karin M, Shi X. Inhibitor of nuclear factor kappaB kinase deficiency enhances oxidative stress and prolongs c-Jun NH2-terminal kinase activation induced by arsenic. Cancer Res. 2003;63:7689–7693. [PubMed] [Google Scholar]
- 18.Tang Y, Scheef EA, Wang S, Sorenson CM, Marcus CB, Jefcoate CR, Sheibani N. CYP1B1 expression promotes the proangiogenic phenotype of endothelium through decreased intracellular oxidative stress and thrombospondin-2 expression. Blood. 2009;113:744–754. doi: 10.1182/blood-2008-03-145219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang RW, Newton DJ, Scheri TD, Lu AY. Human cytochrome P450 3A4-catalyzed testosterone 6 beta-hydroxylation and erythromycin N-demethylation. Competition during catalysis. Drug Metab Dispos. 1997;25:502–507. [PubMed] [Google Scholar]
- 20.Lee AJ, Cai MX, Thomas PE, Conney AH, Zhu BT. Characterization of the oxidative metabolites of 17beta-estradiol and estrone formed by 15 selectively expressed human cytochrome p450 isoforms. Endocrinology. 2003;144:3382–3398. doi: 10.1210/en.2003-0192. [DOI] [PubMed] [Google Scholar]
- 21.Lorbek G, Lewinska M, Rozman D. Cytochrome P450s in the synthesis of cholesterol and bile acids--from mouse models to human diseases. FEBS J. 2012;279:1516–1533. doi: 10.1111/j.1742-4658.2011.08432.x. [DOI] [PubMed] [Google Scholar]
- 22.He Z, Sun X, Mei G, Yu S, Li N. Nonclassical secretion of human catalase on the surface of CHO cells is more efficient than classical secretion. Cell Biol Int. 2008;32:367–373. doi: 10.1016/j.cellbi.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 23.Bi J, Jiang B, Liu JH, Lei C, Zhang XL, An LJ. Protective effects of catalpol against H2O2-induced oxidative stress in astrocytes primary cultures. Neurosci Lett. 2008;442:224–227. doi: 10.1016/j.neulet.2008.07.029. [DOI] [PubMed] [Google Scholar]
- 24.Morrison CD, Pistell PJ, Ingram DK, Johnson WD, Liu Y, Fernandez-Kim SO, White CL, Purpera MN, Uranga RM, Bruce-Keller AJ, Keller JN. High fat diet increases hippocampal oxidative stress and cognitive impairment in aged mice: implications for decreased Nrf2 signaling. J Neurochem. 2010;114:1581–1589. doi: 10.1111/j.1471-4159.2010.06865.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu R, Liu IY, Bi X, Thompson RF, Doctrow SR, Malfroy B, Baudry M. Reversal of age-related learning deficits and brain oxidative stress in mice with superoxide dismutase/catalase mimetics. Proc Natl Acad Sci U S A. 2003;100:8526–8531. doi: 10.1073/pnas.1332809100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Incalza MA, D’Oria R, Natalicchio A, Perrini S, Laviola L, Giorgino F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul Pharmacol. 2018;100:1–19. doi: 10.1016/j.vph.2017.05.005. [DOI] [PubMed] [Google Scholar]
- 27.Kim GH, Kim JE, Rhie SJ, Yoon S. The role of oxidative stress in neurodegenerative diseases. Exp Neurobiol. 2015;24:325–340. doi: 10.5607/en.2015.24.4.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Butterfield DA, Reed T, Newman SF, Sultana R. Roles of amyloid beta-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer’s disease and mild cognitive impairment. Free Radic Biol Med. 2007;43:658–677. doi: 10.1016/j.freeradbiomed.2007.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luczaj W, Gegotek A, Skrzydlewska E. Antioxidants and HNE in redox homeostasis. Free Radic Biol Med. 2017;111:87–101. doi: 10.1016/j.freeradbiomed.2016.11.033. [DOI] [PubMed] [Google Scholar]
- 30.Kim JY, Lee HJ, Lee SJ, Jung YH, Yoo DY, Hwang IK, Seong JK, Ryu JM, Han HJ. Palmitic Acid-BSA enhances amyloid-beta production through GPR40-mediated dual pathways in neuronal cells: involvement of the Akt/mTOR/HIF-1alpha and Akt/NF-kappaB pathways. Sci Rep. 2017;7:4335. doi: 10.1038/s41598-017-04175-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goldsbury C, Whiteman IT, Jeong EV, Lim YA. Oxidative stress increases levels of endogenous amyloid-beta peptides secreted from primary chick brain neurons. Aging Cell. 2008;7:771–775. doi: 10.1111/j.1474-9726.2008.00423.x. [DOI] [PubMed] [Google Scholar]
- 32.Miller AA, Spencer SJ. Obesity and neuroinflammation: a pathway to cognitive impairment. Brain Behav Immun. 2014;42:10–21. doi: 10.1016/j.bbi.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 33.Yu Y, Wang Q, Huang XF. Energy-restricted pair-feeding normalizes low levels of brain-derived neurotrophic factor/tyrosine kinase B mRNA expression in the hippocampus, but not ventromedial hypothalamic nucleus, in diet-induced obese mice. Neuroscience. 2009;160:295–306. doi: 10.1016/j.neuroscience.2009.01.078. [DOI] [PubMed] [Google Scholar]
- 34.Khalil A, Jay-Gerin JP, Fulop TJ. Age-related increased susceptibility of high-density lipoproteins (HDL) to in vitro oxidation induced by gamma-radiolysis of water. FEBS Lett. 1998;435:153–158. doi: 10.1016/s0014-5793(98)01058-8. [DOI] [PubMed] [Google Scholar]
- 35.Morrison CD, Pistell PJ, Ingram DK, Johnson WD, Liu Y, Fernandez-Kim SO, White CL, Purpera MN, Uranga RM, Bruce-Keller AJ, Keller JN. High fat diet increases hippocampal oxidative stress and cognitive impairment in aged mice: implications for decreased Nrf2 signaling. J Neurochem. 2010;114:1581–1589. doi: 10.1111/j.1471-4159.2010.06865.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsang CK, Liu Y, Thomas J, Zhang Y, Zheng XF. Superoxide dismutase 1 acts as a nuclear transcription factor to regulate oxidative stress resistance. Nat Commun. 2014;5:3446. doi: 10.1038/ncomms4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ilic TV, Jovanovic M, Jovicic A, Tomovic M. Oxidative stress indicators are elevated in de novo Parkinson’s disease patients. Funct Neurol. 1999;14:141–147. [PubMed] [Google Scholar]
- 38.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 39.Kim JA, Montagnani M, Koh KK, Quon MJ. Reciprocal relationships between insulin resistance and endothelial dysfunction: molecular and pathophysiological mechanisms. Circulation. 2006;113:1888–1904. doi: 10.1161/CIRCULATIONAHA.105.563213. [DOI] [PubMed] [Google Scholar]
- 40.Tang Y, Scheef EA, Gurel Z, Sorenson CM, Jefcoate CR, Sheibani N. CYP1B1 and endothelial nitric oxide synthase combine to sustain proangiogenic functions of endothelial cells under hyperoxic stress. Am J Physiol Cell Physiol. 2010;298:C665–C678. doi: 10.1152/ajpcell.00153.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Whitsett J, Picklo MS, Vasquez-Vivar J. 4-Hydroxy-2-nonenal increases superoxide anion radical in endothelial cells via stimulated GTP cyclohydrolase proteasomal degradation. Arterioscler Thromb Vasc Biol. 2007;27:2340–2347. doi: 10.1161/ATVBAHA.107.153742. [DOI] [PubMed] [Google Scholar]
- 42.Gao YT, Roman LJ, Martasek P, Panda SP, Ishimura Y, Masters BS. Oxygen metabolism by endothelial nitric-oxide synthase. J Biol Chem. 2007;282:28557–28565. doi: 10.1074/jbc.M704890200. [DOI] [PubMed] [Google Scholar]
- 43.Zhan G, Fenik P, Pratico D, Veasey SC. Inducible nitric oxide synthase in long-term intermittent hypoxia: hypersomnolence and brain injury. Am J Respir Crit Care Med. 2005;171:1414–1420. doi: 10.1164/rccm.200411-1564OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab. 2001;21:2–14. doi: 10.1097/00004647-200101000-00002. [DOI] [PubMed] [Google Scholar]
- 45.Hawkes CA, Gentleman SM, Nicoll JA, Carare RO. Prenatal high-fat diet alters the cerebrovasculature and clearance of beta-amyloid in adult offspring. J Pathol. 2015;235:619–631. doi: 10.1002/path.4468. [DOI] [PubMed] [Google Scholar]
- 46.Park HR, Park M, Choi J, Park KY, Chung HY, Lee J. A high-fat diet impairs neurogenesis: involvement of lipid peroxidation and brain-derived neurotrophic factor. Neurosci Lett. 2010;482:235–239. doi: 10.1016/j.neulet.2010.07.046. [DOI] [PubMed] [Google Scholar]
- 47.Abou ED, Maher A, Sallam N, El-Brairy A, Kenawy S. Trans-cinnamaldehyde modulates hippocampal Nrf2 factor and inhibits amyloid beta aggregation in lps-induced neuroinflammation mouse model. Neurochem Res. 2018;43:2333–2342. doi: 10.1007/s11064-018-2656-y. [DOI] [PubMed] [Google Scholar]
- 48.Choudhry F, Howlett DR, Richardson JC, Francis PT, Williams RJ. Pro-oxidant diet enhances beta/gamma secretase-mediated APP processing in APP/PS1 transgenic mice. Neurobiol Aging. 2012;33:960–968. doi: 10.1016/j.neurobiolaging.2010.07.008. [DOI] [PubMed] [Google Scholar]
- 49.Wang Y, Miao Y, Mir AZ, Cheng L, Wang L, Zhao L, Cui Q, Zhao W, Wang H. Inhibition of beta-amyloid-induced neurotoxicity by pinocembrin through Nrf2/HO-1 pathway in SH-SY5Y cells. J Neurol Sci. 2016;368:223–230. doi: 10.1016/j.jns.2016.07.010. [DOI] [PubMed] [Google Scholar]
- 50.Eftekharzadeh B, Maghsoudi N, Khodagholi F. Stabilization of transcription factor Nrf2 by tBHQ prevents oxidative stress-induced amyloid beta formation in NT2N neurons. Biochimie. 2010;92:245–253. doi: 10.1016/j.biochi.2009.12.001. [DOI] [PubMed] [Google Scholar]