

Abstract
Osteoarthritis (OA) presents a major global health burden and is projected to become even more prevalent in coming decades. Therefore, it is of utmost importance to uncover novel therapies for the treatment and prevention of this disease. In the present study, we investigated the effects of exenatide, a specific glucagon-like peptide (GLP) agonist, on degradation of type II collagen and aggrecan, the two main components of the articular extracellular matrix, in human primary chondrocytes. Our results reveal that exenatide could ameliorate degradation of type II collagen and aggrecan by inhibiting expression of metalloproteinases (MMPs) and a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS) induced by advanced glycation end-products. We also found that exenatide reduces oxidative stress and inhibits activation of nuclear factor-κB through the p38 cellular signaling pathway. Taken together, the findings of this study indicate that exenatide may have potential as a novel treatment for osteoarthritis.
Keywords: Osteoarthritis, NF-κB, exenatide, ROS
Introduction
Osteoarthritis (OA) is a major global disease burden that primarily affects the elderly population. The prevalence of OA is forecasted to greatly increase in coming decades as the average age of the general populace increases [1]. Advanced glycation end-products (AGEs) accumulate in tissues over time and are very resilient to degradation. It has been shown that the presence of AGEs in tissues leads to the release of various pro-inflammatory factors, such as tumor necrosis factor α (TNF-α), nuclear factor κB (NF-κB), interleukin 1β (IL-1β), and production of reactive oxygen species (ROS) resulting from oxidative stress [2,3]. AGEs have also been shown to enhance expression of metalloproteinases (MMPs) and a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS) in chondrocytes [4]. Of these, MMP-3, MMP-13, ADAMTS-4, and ADAMTS-5 are most relevant in OA. These factors trigger and sustain the pathological development of OA, which is primarily characterized by excessive degradation of the articular extracellular matrix (ECM) [5].
MMPs and ADAMTS are well-recognized as critical factors in the progression of OA. Termed as collagenases and aggrecanases, MMPs and ADAMTS respectively degrade type II collagen and aggrecan, which are the main components of the articular ECM [5,6]. Under normal physiological conditions, MMPs and ADAMTS play an important role in maintaining homeostasis by triggering regular cell turnover in the articular ECM as well as other joint components, such as articular cartilage, the synovial membrane, and subchondral bone [7]. While aggrecan, which gives cartilage its shock-absorptive property, has a relatively high rate of turnover, type II collagen, the proverbial backbone of the ECM, has a very low rate of turnover. Therefore, excessive degradation of type II collagen is largely considered to be irreversible [8,9]. Accelerated degradation or reduced regeneration of these components eventually leads to joint failure, which is extremely painful and debilitating for the patient. Presently, arthroplasty is the first-line therapy for advanced OA. While this approach has a high success rate of roughly 85%, it is costly, and recovery can be quite difficult in elderly patients [10]. Therefore, novel drug therapies against the development and progression of OA are of great value.
Exenatide is a glucagon-like peptide 1 (GLP-1) agonist belonging to the family of synthetic exendin-based incretin mimetics and was approved by the FDA for the treatment of type II diabetes mellitus in 2005 [11,12]. GLP-1 is an incretin hormone present in the gut that plays a role in triggering glucose-induced insulin secretion, inhibiting the release of glucagon, delaying gastric emptying, and reducing appetite. The resulting weight-loss effect has brought GLP-1 receptor agonists to be widely used in the treatment of type II diabetes [13]. Interestingly, exenatide has displayed anti-inflammatory and antioxidant capacities in various tissues and cells. For example, exenatide significantly attenuated AGEs-induced IL-6 and TNF-α production, the receptor of advanced glycation endproducts expression, and cell death in rat mesangial cells (RMC) [14]. Another study reported that exenatide treatment inhibits the calcification of human calcifying vascular smooth muscle cell (CVSMC) via suppressing the receptor activator of nuclear kappa B (NF-κB)/nuclear factor-κB ligand (NF-κB/RANKL) signaling pathway [15]. Administration of exendin-4 is able to inhibit lipopolysaccharide (LPS)-induced osteoclast formation and bone resorption by suppressing LPS-induced TNF-α production in macrophages [16]. It has recently been demonstrated that GLP-1-knockout mice exhibited reduced bone strength and poor bone quality, and that gut incretin hormones may play a role in regulating bone formation and bone remodeling [17]. Therefore, we were prompted to investigate whether the GLP-1 agonist exenatide has an inhibitory effect on degradation of the articular ECM induced by accumulation of AGEs.
Materials and methods
Chondrocyte isolation, culture, and treatment
Experiments with human subjects were designed in accordance with the World Medical Association Declaration of Helsinki Ethical Principles for Medical Research Involving Human Subjects. Human subject experiments were approved by the ethics committee of our institute (NO. 20160041). Signed written informed consent was submitted by all participants.
Healthy human knee joint cartilage specimens were obtained from 18 adults undergoing hip replacement. Briefly, cartilage slices were collected and cut into small pieces, followed by treatment with 0.5 mg/ml trypsin for 20 min at 37°C. Samples were then incubated overnight with 2 mg/ml clostridial collagenase at 37°C and centrifuged at 500× g at RT for 10 min. Cells were suspended in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S) in a humid incubator with 5% CO2 at 37°C. Cells were stimulated with 100 μg/ml AGEs in the presence or absence of exendin-4 (a form of exenatide) at concentrations of 10 and 20 nM [18] for 24 h.
Elisa
Secreted levels of TNF-α, IL-1β, MMP-3, and MMP-13 in the supernatants of cultured chondrocytes were assessed using a commercial ELISA kit (R&D Systems, USA). After the indicated treatment, plates were coated with primary antibodies against TNF-α, IL-1β, MMP-3, and MMP-13 and incubated overnight at 4°C. After washing 3 times with PBS, plates were blocked with goat serum at room temperature (RT) for 1 h. Then, 50 μl of samples were then loaded and incubated at 4°C overnight. After washing with PBS for 3 times, biotinylated sheep polyclonal antibodies were loaded and incubated at RT for 1 h, followed by incubation with 50 μl of avidin-HRP (diluted 1:5000). H2SO4 was added to stop the reaction. OD values recorded at 490 nm were used to index secreted levels of target proteins.
Determination of reactive oxygen species (ROS)
Intracellular ROS in cultured cells was measured by staining using the 2,7-dichlorofuorescin diacetate (DCFH-DA) dye method. Briefly, cells were gently washed 3 times with HBSS. Cells were then loaded with 1 μM DCFH-DA (Sigma-Aldrich, USA) in culture medium without FBS and cultured for 15 min in darkness at 37°C. Cells were then washed with PBS and photographed with a DM500 fluorescence microscope (Leica Microsystems, Germany). The fluorescent images of ROS staining were quantified by fluorescence density using the software Image J. Briefly, regions of interest (ROI) was defined in the fluorescent image and the average number of cells present in the defined ROI was counted. The integrated density value (IDV) in ROI was calculated and divided by the average number of cells. Results were used to represent average level of intracellular ROS.
RNA extraction and quantitative real-time PCR
Total RNA was isolated from cultured cells using a Qiazol reagent according to the manufacturer’s instructions. RNA concentration and quality were determined by NanoDrop analysis. cDNA was synthesized by reverse transcription using a cDNA synthesis kit (Bio-rad, USA). Real-time PCR experiments were performed with the synthesized cDNA products and SYBR Green Supermix (Bio-Rad Laboratories) on an ABI 7500 real-time PCR machine. Expression of target genes was calculated by the 2-ΔΔct method and normalized to GAPDH.
Immunoblot analysis
Cell lysates were prepared from cultured cells using RIPA buffer. Protein concentrations were determined using a BCA commercial kit. Samples were subjected to 8%-12% SDS-PAGE to separate proteins. Samples were then transferred to PVDF membranes. PVDF membranes were blocked with 5% non-fat milk, followed by sequential incubation with primary antibodies at 4°C overnight and horse radish peroxidase (HRP)-conjugated secondary antibody for 2 h at RT. Immunoblots were visualized using the chemiluminescence technique (Santa Cruz Biotechnology, USA). The bands of blot were carefully scanned and total optical density of target bands was quantified by Kodak Digital Science 1D software (Eastman Kodak Company, USA). Target bands were selected and the background was subtracted to quantify signal intensities. Results were exported for statistical analysis. Expression of target proteins was normalized to β-actin.
Luciferase reporter gene assay
NF-κB promoter-luciferase activity assay was used to determine the transcriptional activity of NF-κB. NF-κB promoter-luciferase and β-galactosidase plasmids were obtained from Clontech and transfected into human chondrocytes. At 12 h post transfection, cells were stimulated with 100 μg/ml AGEs in the presence or absence of exendin-4 at the concentrations of 10 and 20 nM for 24 h. Luciferase and β-galactosidase activities were measured using a Secrete-PairTM Dual luminescence assay kit. Luciferase activity was normalized to β-galactosidase activity.
Reduced glutathione (GSH) assay
Intracellular levels of reduced glutathione (GSH) were determined by a fluorometric assay. After the indicated treatment, cells were collected and sonicated, followed by centrifugation at 14000× g for 5 min at RT. Supernatants were mixed with OPAME (Sigma-Aldrich, USA) in methanol and borate buffer and incubated for 15 min at RT. Fluorescent signals were recorded (350 nm excitation and 420 nm emission).
Statistical analysis
Experimental data are presented as means ± S.E.M from at least three separate experiments. Statistical significance of differences among treatment groups were evaluated using the one-way analysis of variance (ANOVA) method. P < 0.05 was considered statistically significant.
Results
Exenatide ameliorates AGEs-induced oxidative stress
Exendin-4, a form of exenatide, was used in the following experiments. As shown in Figure 1, HPCs were exposed to 100 µg/ml AGEs in the presence or absence of 10 and 20 nM exendin-4 for 24 h to determine the effects on ROS ge-neration and expression of glutathione (GSH) induced by AGEs. The results of DCFH-DA staining reveal that treatment with 10 and 20 nM exendin4 significantly reduced AGEs-induced generation of ROS in a dose-dependent manner (Figure 1A). Additionally, 10 and 20 nM exendin-4 rescued AGEs-induced reduction in GSH levels in a dose-dependent manner, as shown in Figure 1B.
Figure 1.
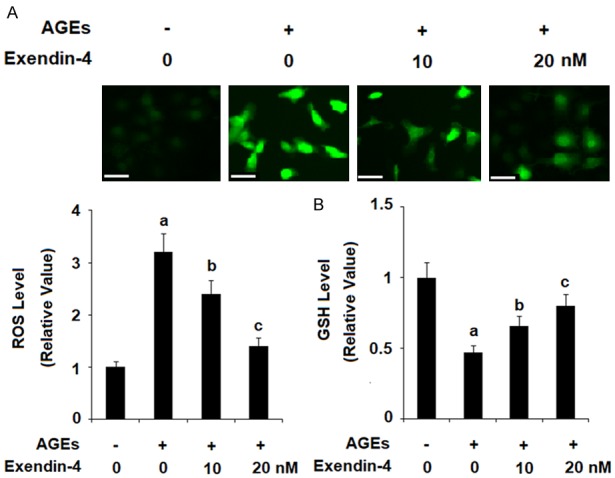
Exenatide ameliorated advanced glycation end products (AGEs)-induced oxidative stress in human primary chondrocytes. Human primary chondrocytes were stimulated with 100 μg/ml AGEs in the presence or absence of exendin-4 (a form of exenatide) at the concentrations of 10 and 20 nM for 24 h. A. Intracellular ROS was determined by DCFH-DA staining; Scale bar, 100 μm; B. Intracellular GSH levels were determined by a fluorometric assay (a, b, c, P < 0.01 vs. previous column group).
Exenatide ameliorates AGEs-induced cytokine production
TNF-α and IL-1β have been shown to play a major role in the progression of various diseases including OA. As shown in Figure 2, exposure to 100 µg/ml AGEs for 24 h significantly increased production of the pro-inflammatory cytokines TNF-α and IL-1β in HPCs, while the presence of 10 and 20 nM exendin-4 significantly ameliorated AGEs-induced production of TNF-α and IL-1β in a dose-dependent manner at both the mRNA and protein levels (Figure 2A and 2B, respectively).
Figure 2.
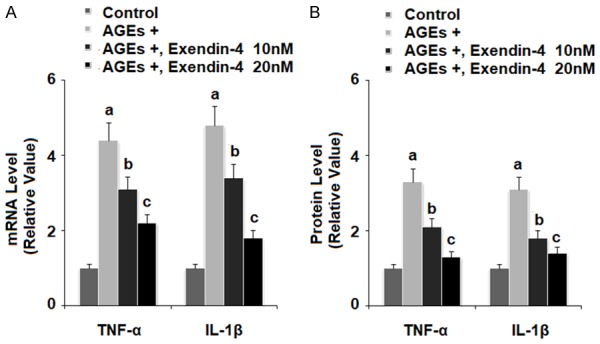
Exenatide ameliorated advanced glycation end products (AGEs)-induced production of inflammatory cytokines in human primary chondrocytes. Human primary chondrocytes were stimulated with 100 μg/ml AGEs in the presence or absence of exendin-4 (a form of exenatide) at the concentrations of 10 and 20 nM for 24 h. A. mRNA levels of TNF-α and IL-1β determined by real-time PCR; B. Protein levels of TNF-α and IL-1β determined by ELISA (a, b, c, P < 0.01 vs. previous column group).
Exenatide ameliorates AGEs-induced expression of MMP-3 and MMP-13
MMP-3 and MMP-13 are recognized as the main collagenases responsible for degradation of type II collagen in the articular ECM. The results in Figure 3 reveal that exposure of HPCs to 100 µg/ml AGEs for 24 h led to a significant increase in expression of both MMP-3 and MMP-13, while the presence of 10 and 20 nM exendin-4 ameliorated AGEs-induced expression of MMP-3 and MMP-13 in a dose-dependent manner at both the mRNA and protein levels.
Figure 3.
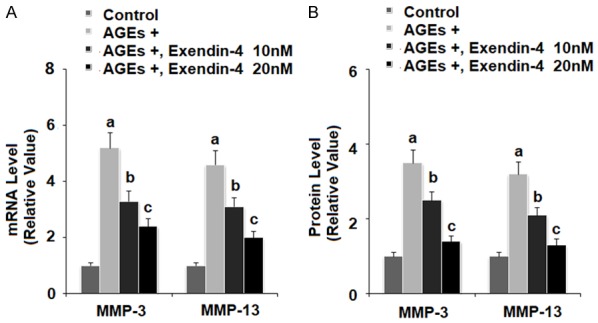
Exenatide ameliorated advanced glycation end products (AGEs)-induced expression of MMP-3 and MMP-13 in human primary chondrocytes. Human primary chondrocytes were stimulated with 100 μg/ml AGEs in the presence or absence of exendin-4 (a form of exenatide) at the concentrations of 10 and 20 nM. A. At 24 h post treatment, expressions of MMP-3 and MMP-13 at the mRNA levels were measured by real-time PCR; B. At 48 h post treatment, expressions of MMP-3 and MMP-13 at the protein levels were measured by ELISA (a, b, c, P < 0.01 vs. previous column group).
Exenatide ameliorates AGEs-induced degradation of type II collagen
We confirmed the effects of exendin-4 on AGEs-induced expression of MMP-3 and MMP-13 by measuring degradation of type II collagen in HPCs. As shown in Figure 4, HPCs were exposed to 100 µg/ml AGEs in the presence or absence of 10 and 20 nM exendin-4 for 48 h. The results of western blot analysis reveal that 100 µg/ml AGEs significantly increased degradation of type II collagen, while 10 and 20 nM exendin-4 ameliorated this effect in a dose-dependent manner.
Figure 4.
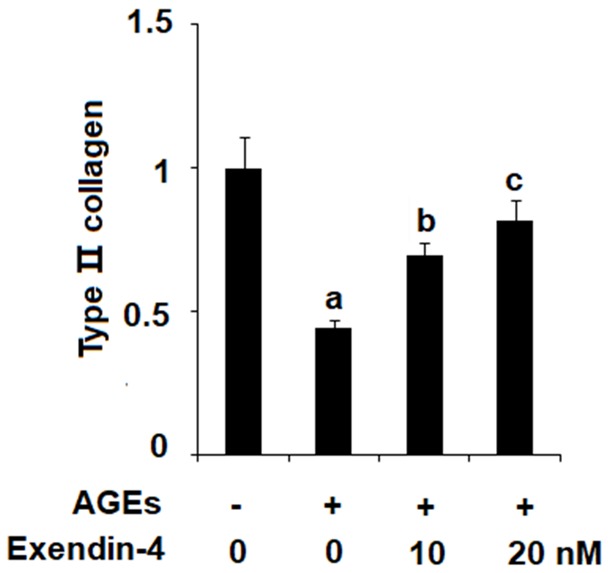
Exenatide ameliorated advanced glycation end products (AGEs)-induced degradation of type II collagen in human primary chondrocytes. Human primary chondrocytes were stimulated with 100 μg/ml AGEs in the presence or absence of exendin-4 (a form of exenatide) at the concentration of 10 and 20 nM for 48 h. Expression of type II collagen was determined (a, b, c, P < 0.01 vs. previous column group).
Exenatide ameliorates AGEs-induced expression of ADAMTS-4 and ADAMTS-5
The aggrecanases ADAMTS-4 and ADAMTS-5 degrade aggrecan in the articular ECM. To determine the effects of exendin-4 on AGEs-induced expression of ADAMTS-4 and ADAMTS-5, we exposed HPCs to 100 µg/ml AGEs in the presence or absence of 10 and 20 nM exendin-4 for 24 and 48 h. As shown in Figure 5A and 5B, 10 and 20 nM exendin-4 significantly reduced AGEs-induced expression of AD-AMTS-4 and ADAMTS-5 at both the mRNA and protein levels in a dose-dependent manner.
Figure 5.
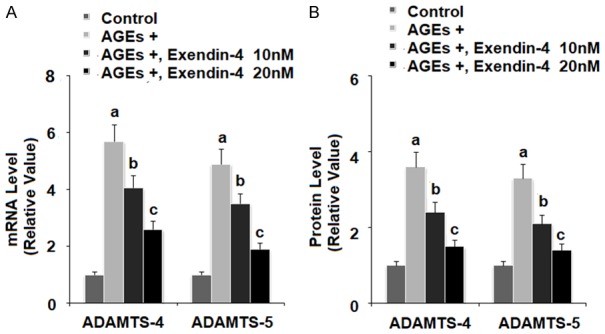
Exenatide ameliorated advanced glycation end products (AGEs)-induced expression of ADAMTS-4 and ADAMTS-5 in human primary chondrocytes. Human primary chondrocytes were stimulated with 100 μg/ml AGEs in the presence or absence of exendin-4 (a form of exenatide) at the concentrations of 10 and 20 nM. A. At 24 h post treatment, expressions of ADAMTS-4 and ADAMTS-5 at the mRNA levels were measured by real-time PCR; B. At 48 h post treatment, expressions of ADAMTS-4 and ADAMTS-5 at the protein levels were measured by ELISA (a, b, c, P < 0.01 vs. previous column group).
Exenatide ameliorates AGEs-induced degradation of aggrecan
To confirm our findings regarding the effects of exendin-4 on AGEs-induced expression of AD-AMTS-4 and ADAMTS-5, we measured the extent of degradation of aggrecan upon exposure to 100 µg/ml AGEs in the presence or absence of 10 and 20 nM exendin-4 for 48 h. As the results in Figure 6 reveal that exendin-4 rescued AGEs-induced degradation of aggrecan in a dose-dependent manner, with 20 nM exendin-4 nearly restoring aggrecan to baseline levels.
Figure 6.
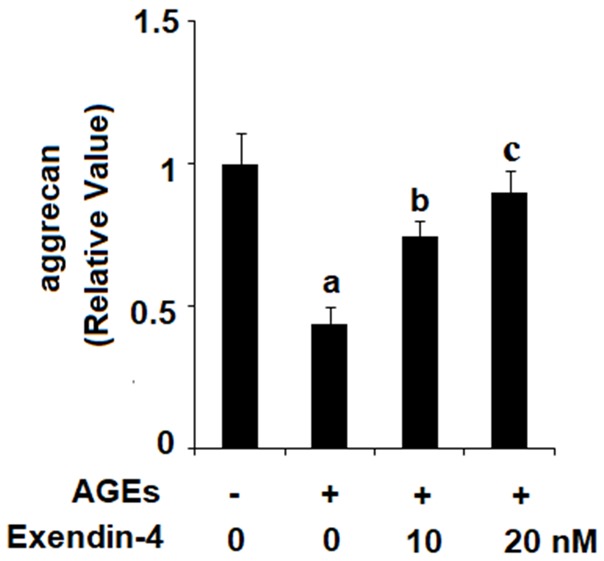
Exenatide ameliorated advanced glycation end products (AGEs)-induced degradation of aggrecan in human primary chondrocytes. Human primary chondrocytes were stimulated with 100 μg/ml AGEs in the presence or absence of exendin-4 (a form of exenatide) at the concentrations of 10 and 20 nM for 48 h. Expression of aggrecan was determined (a, b, c, P < 0.01 vs. previous column group).
Exenatide ameliorates AGEs-induced activation of p38
Activation of the p38 pathway via phosphorylation of p38 protein is recognized as an important event in the initiation of the inflammatory response. To determine the effects of exendin-4 on activation of the p38 pathway, HPCs were exposed to 100 µg/ml AGEs in the presence or absence of 10 and 20 nM exendin-4 for 2 h. As shown in Figure 7, exendin-4 reduced AGEs-induced phosphorylation of p38 in a dose-dependent manner, while the level of total p38 remained constant.
Figure 7.
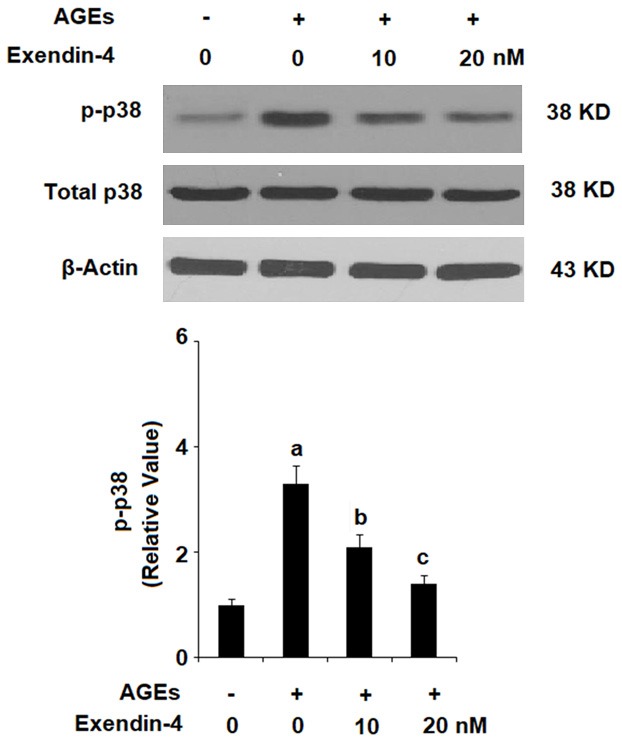
Exenatide ameliorated advanced glycation end products (AGEs)-induced activation of p38 in human primary chondrocytes. Human primary chondrocytes were stimulated with 100 μg/ml AGEs in the presence or absence of exendin-4 (a form of exenatide) at the concentrations of 10 and 20 nM for 2 h. Phosphorylated and total levels of p38 was determined by western blot analysis (a, b, c, P < 0.01 vs. previous column group).
Exenatide ameliorates AGEs-induced activation of NF-κB
Activation of NF-κB is a major contributor to inflammation and has been shown to drive the pathological development of numerous diseases including OA. To determine the effects of exendin-4 treatment on AGEs-induced activation of NF-κB, we measured nuclear translocation of p65 and luciferase activity of NF-κB by exposing HPCs to 100 µg/ml AGEs in the presence or absence of 20 nM exendin-4 for 24 h. The immunostaining results in Figure 8A reveal that exendin-4 significantly ameliorated AGEs-induced nuclear translocation of p65. The results in Figure 8B demonstrate that exendin-4 significantly inhibited AGEs-induced increase in luciferase activity of NF-κB in a dose-dependent manner.
Figure 8.
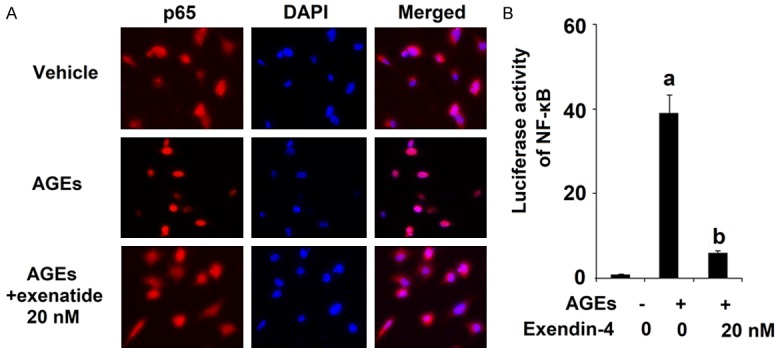
Exenatide ameliorated advanced glycation end products (AGEs)-induced activation of NF-κB in human primary chondrocytes. Human primary chondrocytes were stimulated with 100 μg/ml AGEs in the presence or absence of exendin-4 (a form of exenatide) at the concentrations of 10 and 20 nM for 24 h. A. Nuclear translocation of p65; B. Luciferase activity of NF-κB (a, b, c, P < 0.01 vs. previous column group).
Discussion
The factors contributing to the development and progression of OA are complicated. Among them, excessive degradation of the ECM due to dysregulation of expression of enzymes and proinflammatory cytokines is recognized as a hallmark of the pathological progression of the disease [17]. The risk factors for OA include excessive mechanical loading, obesity, and injury, but the primary risk factor is ageing. In 2018, statistics from the Centers for Disease Control (CDC) reported that over 30 million people in the United States suffer from OA, and that this number is expected to increase by more than double by 2030 [19]. AGEs result from non-enzymatic protein glycation and have been shown to accumulate in tissues over time. The receptor for AGEs (RAGE) has been shown to be expressed on the surface of chondrocytes [20]. In cartilage, accumulation of AGEs inhibits regeneration of type II collagen and accelerates its degradation via activation of the Jak/Stat pathway through the interaction between AGEs and RAGE, which leads to increased expression of MMP-13 and ADAMTS-4/5 [21]. Accumulation of AGEs has also been shown to elevate production of ROS, which further increases the formation of AGEs and leads to the sustained presence of a harmful oxidative stress environment. Under normal physiological conditions, expression of MMPs and ADAMTS, and the presence of ROS serve to maintain homeostasis by facilitating cell turnover. However, under pathological conditions, the delicate balance between these processes is shifted toward a pathological state [22].
Another major factor in the progression of OA is the activation of inflammatory signaling pathways. The NF-κB transcription factor is an ubiquitously expressed heterodimer that has been extensively studied for its key role in the inflammatory response. Activation of the p38/mitogen activated protein kinase (MAPK) signaling pathway inhibits the activity of protein kinase Cζ, resulting in nuclear translocation of p65, one of the heterodimers of NF-κB, and activation of NF-κB. The NF-κB family plays important roles in a wide range of physiological processes including inflammatory response, cell proliferation, and cell differentiation in human chondrocytes. Activation of the NF-κB pathway is essential to induce a variety of inflammation-related mediators, including inducible nitric oxide synthase (iNOS), IL-1β, and TNF-α, and these induced cytokines further activate the signaling cascade [23]. These results in increased expression of MMPs, ADAMTS, and generation of ROS, thereby driving degradation of type II collagen and aggrecan [24-27]. Therefore, NF-κB signaling has been widely associated with OA pathophysiology through a diversity of effects and is activated in OA chondrocytes during aging and inflammation. Although numerous studies have explored the mechanisms behind the role of inflammatory signaling in OA, the subject remains to be fully elucidated.
In the present study, we found that treatment of HPCs with 100 µg/ml AGEs drastically increased production of ROS, expression of MMPs and ADAMTS, degradation of type II collagen and aggrecan, and activation of p38 and NF-κB. Administration of 10 and 20 nM exenatide (in the form of exendin-4) successfully ameliorated these negative effects of AGEs in HPCs. This indicates that exenatide may have potential as a safe and effective therapy for the treatment of OA. Various studies have demonstrated the anti-degradative effects of exendin-4 in OA [28-30]. In regard to its mechanism of action, exenatide acts as an incretin hormone mimetic by binding to the GLP-1 receptor in a fashion similar to naturally occurring GLP-1 in humans [31]. The role of incretin hormones in bone formation and regeneration has been studied in patients with type II diabetes mellitus and obesity. It was found that GLP-1 has an anabolic effect on bone regeneration in patients with glucose intolerance [32]. Furthermore, the authors of a study performed in 2015 concluded that treatment with a GLP-1 receptor agonist increased bone formation by 16% in obese women after weight loss. Taken together, the findings of this study and others indicate that exenatide, a GLP-1 agonist, may have potential as a therapeutic agent for the prevention and treatment of age-related OA resulting from excessive accumulation of AGEs. Additional research using in vivo animal models is recommended to better understand the benefits of this treatment demonstrated in the present study.
Disclosure of conflict of interest
None.
References
- 1.Cross M, Smith E, Hoy D, Nolte S, Ackerman I, Fransen M, Laslett LL. The global burden of hip and knee osteoarthritis: estimates from the global burden of disease 2010 study. Ann Rheum Dis. 2014;73:1323–1330. doi: 10.1136/annrheumdis-2013-204763. [DOI] [PubMed] [Google Scholar]
- 2.Nowotny K, Jung T, Höhn A, Weber D, Grune T. Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules. 2015;5:194–222. doi: 10.3390/biom5010194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ott C, Jacobs K, Haucke E, Santos AN, Grune T, Simm A. Role of advanced glycation end products in cellular signaling. Redox Biol. 2015;2:411–429. doi: 10.1016/j.redox.2013.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang CY, Lai KY, Hung LF, Wu WL, Liu FC, Ho LJ. Advanced glycation end products cause collagen II reduction by activating Janus kinase/signal transducer and activator of transcription 3 pathway in porcine chondrocytes. Rheumatology (Oxford) 2011;50:1379–1389. doi: 10.1093/rheumatology/ker134. [DOI] [PubMed] [Google Scholar]
- 5.Kapoor M, Martel-Pelletier J, Lajeunesse D, Pelletier JP, Fahmi H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat Rev Rheumatol. 2011;7:33–42. doi: 10.1038/nrrheum.2010.196. [DOI] [PubMed] [Google Scholar]
- 6.Echtermeyer F, Bertrand J, Dreier R, Meinecke I, Neugebauer K, Fuerst M, Lee YJ, Song YW, Herzog C, Theilmeier G, Pap T. Syndecan-4 regulates ADAMTS-5 activation and cartilage breakdown in osteoarthritis. Nat Med. 2009;15:1072–1076. doi: 10.1038/nm.1998. [DOI] [PubMed] [Google Scholar]
- 7.Martel-Pelletier J, Boileau C, Pelletier JP, Roughley PJ. Cartilage in normal and osteoarthritis conditions. Best Pract Res Clin Rheumatol. 2008;22:351–384. doi: 10.1016/j.berh.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 8.Ismail HM, Miotla-Zarebska J, Troeberg L, Tang X, Stott B, Yamamoto K, Nagase H, Fosang AJ, Vincent TL, Saklatvala J. Brief report: JNK-2 controls aggrecan degradation in murine articular cartilage and the development of experimental osteoarthritis. Arthritis Rheumatol. 2016;68:1165–1171. doi: 10.1002/art.39547. [DOI] [PubMed] [Google Scholar]
- 9.Goldring MB, Otero M. Inflammation in osteoarthritis. Curr Opin Rheumatol. 2011;23:471–478. doi: 10.1097/BOR.0b013e328349c2b1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosshirt N, Engbarth T, Gotterbarm T, Hagmann S, Moradi B. M1/M2 macrophages induce chondral MMP/ADAMTS enzyme secretion in a direct co-culture experiment. Osteoarthritis Cartilage. 2018;26:S129. [Google Scholar]
- 11.Neumiller JJ. Differential chemistry (structure), mechanism of action, and pharmacology of GLP-1 receptor agonists and DPP-4 inhibitors. J Am Pharm Assoc. 2009;49:S16–29. doi: 10.1331/JAPhA.2009.09078. [DOI] [PubMed] [Google Scholar]
- 12.Mafong DD, Henry RR. Exenatide as a treatment for diabetes and obesity: implications for cardiovascular risk reduction. Curr Atheroscler Rep. 2008;10:55–60. doi: 10.1007/s11883-008-0009-z. [DOI] [PubMed] [Google Scholar]
- 13.Meier JJ, Nauck MA. Glucagon-like peptide 1 (GLP-1) in biology and pathology. Diabetes Metab Res Rev. 2005;21:91–117. doi: 10.1002/dmrr.538. [DOI] [PubMed] [Google Scholar]
- 14.Chang JT, Liang YJ, Hsu CY, Chen CY, Chen PJ, Yang YF, Chen YL, Pei D, Chang JB, Leu JG. Glucagon-like peptide receptor agonists attenuate advanced glycation end products-induced inflammation in rat mesangial cells. BMC Pharmacol Toxicol. 2017;18:67. doi: 10.1186/s40360-017-0172-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhan JK, Tan P, Wang YJ, Wang Y, He JY, Tang ZY, Huang W, Liu YS. Exenatide can inhibit calcification of human VSMCs through the NF-kappaB/RANKL signaling pathway. Cardiovasc Diabetol. 2014;13:153. doi: 10.1186/s12933-014-0153-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shen WR, Kimura K, Ishida M, Sugisawa H, Kishikawa A, Shima K, Ogawa S, Qi J, Kitaura H. The glucagon-like peptide-1 receptor agonist exendin-4 inhibits lipopolysaccharide-induced osteoclast formation and bone resorption via inhibition of TNF-α expression in macrophages. J Immunol Res. 2018;2018:5783639. doi: 10.1155/2018/5783639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fernandes JC, Martel-Pelletier J, Pelletier JP. The role of cytokines in osteoarthritis pathophysiology. Biorheology. 2002;39:237–246. [PubMed] [Google Scholar]
- 18.Lee YS, Jun HS. Glucagon-like peptide-1 receptor agonist and glucagon increase glucose-stimulated insulin secretion in beta cells via distinct adenylyl cyclases. Int J Med Sci. 2018;15:603–609. doi: 10.7150/ijms.24492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Corsi M, Alvarez C, Callahan LF, Cleveland RJ, Golightly YM, Jordan JM, Nelson AE, Renner J, Tsai A, Allen KD. Contributions of symptomatic osteoarthritis and physical function to incident cardiovascular disease. BMC Musculoskelet Disord. 2018;19:393. doi: 10.1186/s12891-018-2311-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qu H, Li J, Wu LD, Chen WP. Trichostatin A increases the TIMP-1/MMP ratio to protect against osteoarthritis in an animal model of the disease. Mol Med Rep. 2016;14:2423–2430. doi: 10.3892/mmr.2016.5523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang CY, Lai KY, Hung LF, Wu WL, Liu FC, Ho LJ. Advanced glycation end products cause collagen II reduction by activating Janus kinase/signal transducer and activator of transcription 3 pathway in porcine chondrocytes. Rheumatology. 2011;50:1379–1389. doi: 10.1093/rheumatology/ker134. [DOI] [PubMed] [Google Scholar]
- 22.Ott C, Jacobs K, Haucke E, Santos AN, Grune T, Simm A. Role of advanced glycation end products in cellular signaling. Redox Biol. 2015;2:411–429. doi: 10.1016/j.redox.2013.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marcu KB, Otero M, Olivotto E, Borzi RM, Goldring MB. NF-kappaB signaling: multiple angles to target OA. Curr Drug Targets. 2010;11:599–613. doi: 10.2174/138945010791011938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roman-Blas JA, Jimenez SA. NF-κB as a potential therapeutic target in osteoarthritis and rheumatoid arthritis. Osteoarthritis Cartilage. 2006;14:839–848. doi: 10.1016/j.joca.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 25.Mohetaer M, Li G, Wang Y, Cao L. Protective effects of gemigliptin against type II collagen degradation in human chondrocytes. Biomed Pharmacother. 2018;104:590–594. doi: 10.1016/j.biopha.2018.04.018. [DOI] [PubMed] [Google Scholar]
- 26.Mengshol JA, Vincenti MP, Coon CI, Barchowsky A, Brinckerhoff CE. Interleukin-1 induction of collagenase 3 (matrix metalloproteinase 13) gene expression in chondrocytes requires p38, c-Jun N-terminal kinase, and nuclear factor kappaB: differential regulation of collagenase 1 and collagenase 3. Arthritis Rheum. 2000;43:801–811. doi: 10.1002/1529-0131(200004)43:4<801::AID-ANR10>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 27.Zandi E, Rothwarf DM, Delhase M, Hayakawa M, Karin M. The IκB kinase complex (IKK) contains two kinase subunits, IKKα and IKKβ, necessary for IκB phosphorylation and NF-κB activation. Cell. 1997;91:243–252. doi: 10.1016/s0092-8674(00)80406-7. [DOI] [PubMed] [Google Scholar]
- 28.Berenbaum F, Bougault C, Attali C, inventors. No. 9,592,272. Washington, DC: U.S. Patent and Trademark Office; U. S. Patent. 2017
- 29.Chen J, Xie JJ, Shi KS, Gu YT, Wu CC, Xuan J, Ren Y, Chen L, Wu YS, Zhang XL, Xiao J, Wang DZ, Wang XY. Glucagon-like peptide-1 receptor regulates endoplasmic reticulum stress-induced apoptosis and the associated inflammatory response in chondrocytes and the progression of osteoarthritis in rat. Cell Death Dis. 2018;9:212. doi: 10.1038/s41419-017-0217-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haack T, Wagner M, Henkel B, Stengelin S, Evers A, Bossart M, Kadereit D, inventors. Application No. 14/569,326. U. S. Patent. 2017
- 31.Koole C, Reynolds CA, Mobarec JC, Hick C, Sexton PM, Sakmar TP. Genetically encoded photocross-linkers determine the biological binding site of exendin-4 peptide in the N-terminal domain of the intact human glucagon-like peptide-1 receptor (GLP-1R) J Biol Chem. 2017;292:7131–7144. doi: 10.1074/jbc.M117.779496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nuche-Berenguer B, Moreno P, Esbrit P, Dapía S, Caeiro JR, Cancelas J, Haro-Mora JJ, Villanueva-Peñacarrillo ML. Effect of GLP-1 treatment on bone turnover in normal, type 2 diabetic, and insulin-resistant states. Calcif Tissue Int. 2009;84:453–461. doi: 10.1007/s00223-009-9220-3. [DOI] [PubMed] [Google Scholar]