

Abstract
Acute lung injury (ALI) is a major pathological issue characterized by serious inflammatory response, and a major clinically critical illness with high morbidity and mortality. Glycyrrhizic acid (GA) is a major bioactive constituent isolated from traditional Chinese herb licorice, which has been reported to have positive effects on inflammation. Nevertheless, the effects of GA on lipopolysaccharide (LPS)-treated ALI in mice have not been reported. The purpose of our study is to investigate the inhibitory effects of GA on ALI treated by LPS and to elucidate its possible mechanisms. We found that GA significantly attenuated lung injury and decreased the production of inflammatory factors TNF-α, IL-1β, and HMGB1 with LPS treatment. GA induced autophagy which was showed by enhanced number of autophagosomes through upregulating the protein levels of LC3-II/I and Beclin-1 and downregulating SQSTM1/P62. Moreover, pre-treatment of 3-Methyladenine (3-MA), an autophagy inhibitor, reversed the inhibiting effects of GA on the secretion of inflammatory factors in ALI. The PI3K/AKT/mTOR pathway was associated with GA-induced autophagy under ALI induced by LPS. In conclusion, this study indicated that GA inhibited the production of inflammatory factors in LPS-induced ALI by regulating the PI3K/AKT/mTOR pathway related autophagy, which may provide a novel therapeutic perspective of GA in ameliorating ALI.
Keywords: Glycyrrhizic acid (GA), ALI, autophagy, inflammation, LPS
Introduction
Acute lung injury (ALI) is a clinical syndrome characterized by pulmonary inflammation and increased microvascular permeability, which caused by a variety of pathogenic factors, including acute pneumonia, sepsis, severe trauma, and acute pancreatitis, and eventually develops into acute respiratory distress syndrome [1,2], with a high mortality rate of 35-55% [3,4]. Currently, there are few effective therapies for ALI [5,6]. Among all pathogenic factors, bacterial infection is one of the most common causes. Lipopolysaccharide (LPS), the main component of the cell wall of gram-negative bacteria can induce ALI [7,8]. Once stimulated by LPS, production of inflammatory cytokines, such as tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β), is triggered and facilitates in the progression of ALI [7,8]. Therefore, it is essential to treat inflammation in ALI.
Autophagy is a highly conserved biological process that exists in eukaryotes and maintains cell homeostasis and viability by recycling and reusing energy [9,10]. The process of autophagy mainly includes five stages: initiation, phagocytic vacuoles, autophagosomes, autophagic lysosome formation and phagosome degradation [9,11]. The major proteins involved in autophagy process are called autophagy-related proteins, such as LC3-II/Atg8, Beclin-1/Atg6, SQSTM1/P62, and et al., and the key signaling pathways regulating autophagy include PI3K/AKT/mTORC1 and Bcl-2 protein family [12,13]. 3-Methyladenine (3-MA) is an inhibitor of the phosphoinositide 3-kinase (PI3K) that blocks the formation of autophagosomes [14,15]. Autophagy is generally considered as the protective mechanism of cells, while excessive autophagy leads to autophagic cell death [16,17]. Studies have shown that autophagy promotes the survival or death of cells depending on factors, such as cell types, environmental conditions, and specific stimuli [17,18]. Autophagy plays a crucial role in LPS-induced ALI [19], but its specific mechanism remains unknown.
Glycyrrhizic acid (GA), known as glycyrrhizin or qianglining, is one of the most important bioactive ingredients of Glycyrrhiza uralensis. GA has many beneficial effects, such as immune regulation, anti-viral, anti-cancer, and anti-inflammatory effects [20,21]. Studies have found that GA inhibits the inflammatory response in ALI, but the mechanism involved is unknown [22,23]. In this study, our result indicated that GA could induce autophagy to restrain the production of inflammatory factors, such as TNF-α, IL-1β, and HMGB1, in LPS-induced ALI and the PI3K/AKT/mTOR pathway could be involved in GA induced autophagy.
Materials and methods
Antibodies and reagents
Antibodies against LC3 (18725-1-AP, Proteintech), Beclin-1 (11306-1-AP, Proteintech), P62/SQSTM1 (18420-1-AP, Proteintech), PI3K (20584-1-AP, Proteintech), AKT (10176-2-AP, Proteintech), mTOR (20657-1-AP, Proteintech), and HMGB1 (10829-1-AP, Proteintech), TNF-α (11948, Cell Signaling), phospho-PI3K (p-PI3K) (4228, Cell Signaling), phospho-AKT (p-AKT) (4060, Cell Signaling), phospho-mTOR (p-mTOR) (5536, Cell Signaling), IL-1β (bs-0812R, Bioss) were employed. GA (G0150) was obtained from Shanghai TCI Chemical Industry Co.. Dojindo Inc (Japan) provided Cell Counting Kit-8 (CCK-8, CK04), and 3-Methyladenine (3-MA) (S2767) was obtained from Selleck (USA) and LPS (L2880) from Sigma.
Cell culture and treatments
Mouse macrophage RAW264.7 cells were obtained from the Cell Resources Center of the Chinese Academy of Science (Shanghai, China). Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Hyclone, USA) supplemented with 10% fetal bovine serum (FBS) (Gibco, USA), 100 units/ml penicillin and 100 μg/ml streptomycin, and placed in a 37°C 5% CO2 humidified incubator. The logarithmic growth phase cells were selected for the further experiment. The cells were pretreated with 3-MA (1 mM) or GA (100 μg/ml) for 1.5 h or 1 h, respectively. Then the cells were incubated with LPS (1 μg/ml) for 24 h and then harvested for further investigation.
CCK8 assay
RAW264.7 cells were treated as described above and seeded at a density of 1×104 cells/well in 96-well plates and kept overnight for 24 h at 37°C with 5% CO2. Afterwards, 10 μl of CCK-8 reagents were added to each well, and the mixture was continuously incubated at 37°C for 2 h. The absorbance values at 450 nm were detected using an EXL-800 Multiscan Spectrum (BioTek, USA).
Confocal laser scanning microscope (CLSM) analysis
The cells were fixed with 4% paraformaldehyde, washed with PBS, 0.1% Triton X-100 was permeabilized into the cells and then blocked with 5% bovine serum albumin (BSA), and incubated with antibodies of LC3 (1:50) or HMGB1 (1:50) overnight at 4°C. After washing several times with PBS, the cells were incubated with secondary antibodies for 1 h. Then, the cell nucleus was stained with 4’6’-diamino-2-phenylindole (DAPI) and fluorescence images were captured on CLSM (Leica, Germany).
Transmission electron microscopy (TEM)
RAW264.7 cells were washed 3 times with 0.1 M PBS, and fixed in 2.5% glutaraldehyde at 4°C overnight which was followed by dehydration using an acetone gradient, embedded, sectioned and routinely stained. Visualized autophagosomes were observed under a HT7700 Transmission Electron Microscope from HITACHI.
GA and 3-MA exposure in animal models of ALI
Male Balb/c mice (7-8 weeks old) were obtained from Experimental Animals Co. Ltd of SJA (Changsha, China), and randomly divided into four groups (12 mice per group), control group, LPS (10 mg/kg) group, GA (200 mg/kg) + LPS group, and 3-MA (15 mg/kg) + GA + LPS group. 3-MA was administered 0.5 h before GA intervention, and GA was administered 1 h before LPS treatment. Control group was given equal amounts of PBS at the same time. Each group of mice were sacrificed after 24 h of LPS treatment. All drugs were administered through intraperitoneal injection. All experiments were approved by the Experimental Animal Management Ethics Committee of the Hunan Normal University School of Medicine. Animal treatment and experimental operation procedures were conducted in line with the ethical requirements of experimental animals.
Lung weight coefficient
Lung weight coefficient is an important indicator of pulmonary edema. Lung tissues were removed and wiped dry of surface blood and immediately weighed. The lung weight coefficient was evaluated by dividing the lung weight of each mouse by the body weight of the mouse multiplied by 100%.
Pulmonary histopathology
The lung tissues were 4% formaldehyde-fixed, paraffin-embedded, and cut into 5 μm thick sections. Next, the sections were performed hematoxylin-eosin (HE) staining. The changes of pulmonary histopathology were visualized under the microscope and pathological scores were obtained. According to the degree of lung injury, bleeding, edema, exudation, necrosis, congestion, neutrophil infiltration, and atelectasis, they were evaluated on a scale of 0-4 points [24]: no injury = score of 0, lesion field < 25% = score of 1, lesion field 25-50% = score of 2, lesion field 50-70% = score of 3, full field of vision = score of 4. The scores were calculated for statistical analysis.
Western blot analysis
Proteins were extracted from lung tissue and RAW264.7 cells using a lysis buffer with PMSF, and electrophoresed on 10-12% SDS-PAGE gels and then transferred into PVDF membranes (Immobilon, IPVH00010). After blocked with 5% skim milk for 1 h, the membranes were incubated with primary antibodies for 12 h at 4°C. The membranes were washed with TBST and incubated with horseradish peroxidase-linked secondary antibodies for 2 h at room temperature. The immunoreactive bands were analyzed using image analysis software with an ECL system. All values were normalized to the loading control, β-tubulin.
Immunohistochemistry analysis
The protein level of HMGB1 in lung tissue was evaluated using immunohistochemistry. After conventional dewaxing, lung tissues were repaired by thermal repair, and endogenous peroxidase activity was eliminated using a 3% H2O2 solution. The sections were blocked for 2 h and incubated with the primary antibody for 12 h at 4°C. Subsequently, the secondary antibody and ABC solution were added, and incubated for 2 h at room temperature. Then, the samples were dyed with DAB and haematoxylin, and sealed with a neutral gum. Finally, each sample was observed under a microscope.
qRT-PCR
The total RNA was extracted from lung tissues following the instructions of Trizol reagent (Servicebio, China). 2 μg of RNA was used for reverse transcription synthesis of cDNA, as a quantitative real-time PCR template. The mRNA levels of TNF-α and IL-1β were measured according to the instructions of SYBR Green Master Mix kit (Roche), and standardized with β-tubulin, as loading control. The 2-ΔΔCt method was used to calculate the expression level of mRNA. The primers for qRT-PCR were shown in Table 1.
Table 1.
The primers for qRT-PCR
| Genes | Forward (5’-3’) | Reverse (5’-3’) |
|---|---|---|
| TNF-α | CCCTCACACTCACAAACCACC | CTTTGAGATCCATGCCGTTG |
| IL-1β | TCCATCTTCTTCTTTGGGTATTGC | GTGACGTTGACATCCGTAAAGA |
| β-tubulin | GTGACGTTGACATCCGTAAAGA | GTGACGTTGACATCCGTAAAGA |
Statistical analysis
Data were shown as mean ± SEM and obtained from at least four independent samples (n=4). The experimental data was processed through one-way ANOVA analysis using GraphPad 5.0 software (USA). P < 0.05 was considered statistically significant difference.
Results
GA protects cell viability of RAW264.7 cells with LPS treatment
To estimate the effect of GA on cell viability in ALI cell model, we first employed LPS to establish RAW264.7 cells model of ALI, and found that the viability of RAW264.7 cells was significantly decreased with LPS treatment (1, 5 μg/ml) (P < 0.05) (Figure 1A). Then, an autophagy inhibitor, 3-MA, was used to detect whether inhibition of autophagy could decrease viability of RAW264.7 cells, and it was showed that 3 mM of 3-MA obvious reduced viability of the cells (P < 0.01) (Figure 1B), but seriously leading to cell death. From the above mention, we employed 1 μg/ml of LPS and 1 mM of 3-MA as the dose of the reagents for further experiment. Moreover, to detect whether GA could increase the viability of RAW264.7 cells and 3-MA reverse the protective effect of GA, the cells were incubated with 1 μg/ml of LPS, 100 μg/ml GA, and/or 1 mM of 3-MA. It was shown that the cell viability was significantly increased with pre-treatment of GA and reversed by 3-MA and GA treatment simultaneously in LPS-induced RAW264.7 cells (P < 0.05) (Figure 1C). The above-mentioned results indicated that GA has an inhibitory effect on LPS-induced cell injury.
Figure 1.

GA protects cell viability of RAW264.7 cells with LPS treatment. A. After exposure to LPS (0.1, 0.5, 1, 5 μg/ml) for 24 h, the viability of RAW264.7 cells were evaluated by CCK8 assay. B. RAW264.7 cells were treated with 3-MA (0.1, 0.3, 1, 3 mM) for 30 minutes and the cell viability was determined using CCK8 assay. C. The viability was assessed for RAW264.7 cells treated with LPS, and with or without GA and/or 3-MA treatment for 24 h. The experiments were performed four independent times (n=4) and bars represented as mean ± SEM. * P < 0.05, ** P < 0.01 in comparison to control group. # P < 0.05 in comparison to LPS group. && P < 0.01 in comparison to GA + LPS group.
GA inhibits cytokine secretion in RAW264.7 cells with LPS treatment
To investigate the effect of GA on inflammatory cytokines in ALI, 100 μg/ml of GA, 1 μg/ml of LPS, and/or 1 mM of 3-MA were incubated with RAW264.7 cells. The results indicated that GA suppressed the levels of TNF-α, IL-1β, and HMGB1 proteins, but the inhibiting effects of GA were reversed by 3-MA, in RAW264.7 cells treated with LPS (P < 0.05) (Figure 2A-D). Subsequently, the HMGB1 expression was analyzed using immunofluorescence staining. The expression of HMGB1 induced by LPS was significantly reduced with pre-treatment of GA, and reversed by 3-MA in RAW264.7 cells (P < 0.05) (Figure 2E). These data demonstrated that GA decreased LPS-induced inflammatory responses in vitro.
Figure 2.

GA inhibits the production of cytokines in RAW264.7 cells with LPS treatment. A. The levels of TNF-α, IL-1β, and HMGB1 in RAW264.7 cells detected using Western blotting. B-D. Quantitative analysis of the protein production levels of TNF-α, IL-1β, and HMGB1. E. The remarkable images of HMGB1 observed by CLSM in RAW264.7 cells (magnification ×63). The experiments were performed four independent times (n=4) and bars represented as mean ± SEM. ** P < 0.01, *** P < 0.001 compared with control group. # P < 0.05, ## P < 0.01, ### P < 0.001 versus LPS group. & P < 0.05 in comparison to GA + LPS group.
GA induces autophagy through the PI3K/AKT/mTOR pathway in RAW264.7 cells with LPS treatment
To explore whether the protective effect of GA on RAW264.7 cells was associated with autophagy activation, we detected the level of several key autophagy-related proteins using Western blotting. As shown in Figure 3A-D, treatment of LPS significantly upregulated the ratio of LC3-II/LC3-I and the level of Beclin-1, while P62 decreased, suggesting autophagy activation (P < 0.01). GA could further activate autophagy and 3-MA reversed it, in the LPS-induced RAW264.7 cells (P < 0.05). Subsequently, pre-treatment of GA obvious upregulated the fluorescence intensity of LC3 observed under CLSM (Figure 3E), and increased the number of autophagosomes by TEM, which indicating autophagy activation, in the cells with LPS treatment (Figure 3F, 3G). In addition, the fluorescence intensity of LC3 and the number of autophagosomes was significantly decreased in the 3-MA group (P < 0.05). The above results indicated that GA could induce autophagy under ALI in vitro.
Figure 3.
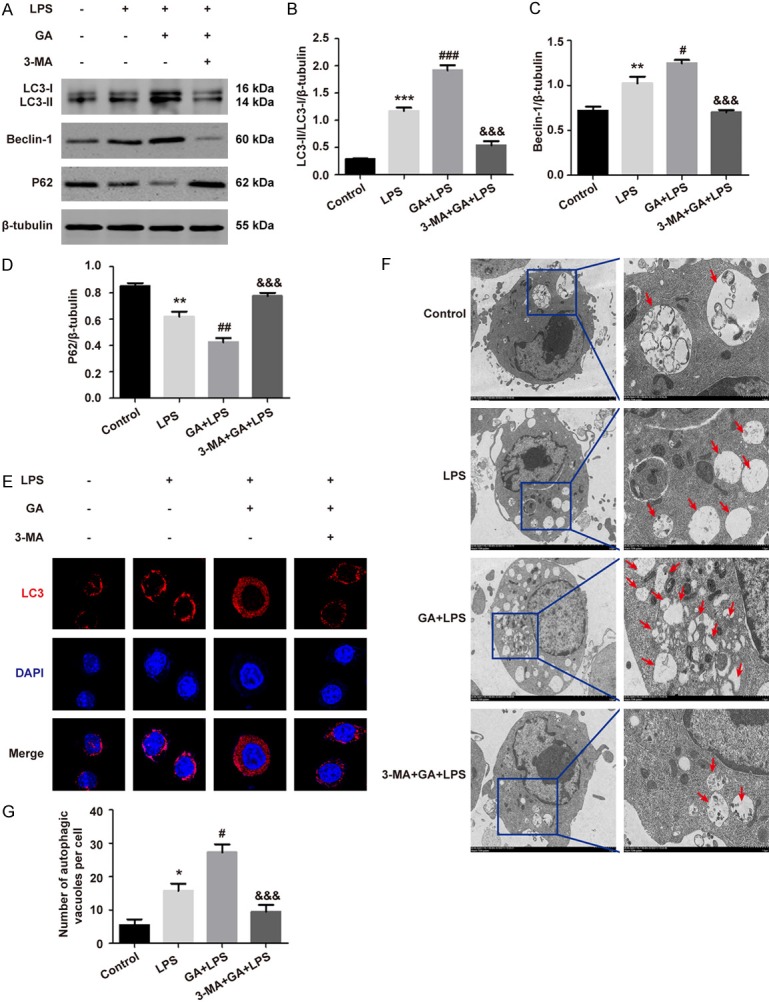
GA induces autophagy in LPS-induced RAW264.7 cells. A-D. The levels of LC3-II/I, Beclin-1, and SQSTM1/P62 were detected by Western blotting in RAW264.7 cells, and the bar graphs showed the quantitative analysis results of the corresponding protein in each group, respectively. E. The representative images of the fluorescence of LC3 in RAW264.7 cells (magnification ×63). F. The remarkable images of autophagosomes ultrastructure by TEM in RAW264.7 cells (magnification ×1500, ×6000, respectively). G. Quantitative analysis of autophagosomes numbers in each group, and autophagosomes were indicated shown by the red arrows. The experiments were performed four independent times (n=4) and bars represented as mean ± SEM. * P < 0.05, ** P < 0.01, *** P < 0.001 in comparison to control group. # P < 0.05, ## P < 0.01, ### P < 0.001 in comparison to LPS group. &&& P < 0.001 versus the GA + LPS group.
The possible mechanisms of autophagy activated by GA in LPS-induced RAW264.7 cells were being further explored. We investigated the PI3K/AKT/mTOR pathway, the classical autophagy-related pathway. It was found that the levels of p-PI3K, p-AKT, and p-mTOR proteins were decreased with LPS treatment, and further suppressed with the treatment of GA (P < 0.05). However, compared with GA group, the levels of p-PI3K, p-AKT, and p-mTOR were downregulated with treatment of 3-MA (P < 0.05) (Figure 4). These results suggested that GA induced autophagy at least partly through regulation of the PI3K/AKT/mTOR pathway in RAW264.7 cells with LPS treatment.
Figure 4.
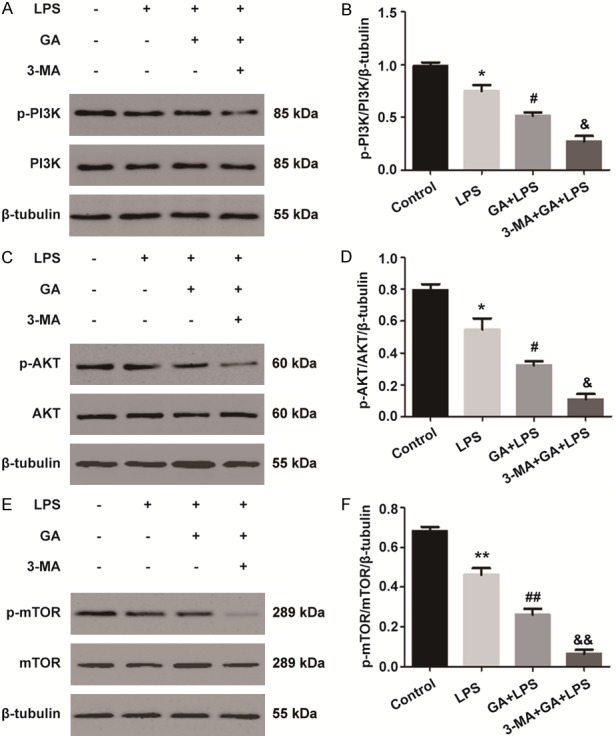
GA enhances autophagy through the PI3K/AKT/mTOR pathway in vitro. Western blotting detected the levels of p-PI3K, PI3K (A, B), p-AKT, AKT (C, D), p-mTOR, and mTOR (E, F). The experiments were performed four independent times (n=4) and bars represented as mean ± SEM. * P < 0.05, ** P < 0.01 in comparison to control group. # P < 0.05, ## P < 0.01 in comparison to LPS group. & P < 0.05, && P < 0.01 versus the GA + LPS group.
GA inhibits lung inflammatory injury through induction of autophagy in LPS-induced ALI mouse model
Autophagy plays crucial role in regulating inflammatory response by eliminating inflammasome, cytokines, and cellular components, which provides an important method for regulation of inflammation. To explain whether GA decreased inflammation of ALI induced by LPS through autophagy activation in vivo, we established the ALI mouse model induced by LPS administration, then the lung weight coefficient and HE staining were to evaluate changes of the pulmonary histopathological features. It was indicated that the lung weight coefficient was significantly increased and the lung tissues accompanied with inflammatory cell infiltration, vascular congestion and bronchial wall thickening in ALI model induced by LPS, compared with control group. Importantly, the histological changes and lung weight coefficients dramatically reduced after GA administration, and these phenomena were reversed with 3-MA treatment (P < 0.05) (Figure 5A, 5B). As shown in Figure 5C, the results of the lung injury score according to the changes of pulmonary histopathology of each group by HE staining which was consistent with the lung weight coefficient results.
Figure 5.

GA inhibits lung inflammatory injury through autophagy activation in vivo. (A) The pulmonary edema was evaluated by the lung weight coefficient. (B) The histopathological changes of lung tissues were examined using H&E staining (magnification ×400), and (C) morphological damage score for the lung tissues. (D, E) The mRNA levels of TNF-α and IL-1β detected using qRT-PCR. (F) The levels of TNF-α, IL-1β, and HMGB1 were assessed by Western blotting. (G-I) Quantitative analysis of TNF-α, IL-1β, and HMGB1 were shown in bar graphs. (J) The representative pictures of immunohistochemical analysis of HMGB1 protein expression. The experiments were performed four independent times (n=4) and bars represented as mean ± SEM. * P < 0.05, ** P < 0.01, *** P < 0.001 compared with control group. # P < 0.05, ## P < 0.01, ### P < 0.001 in comparison to LPS group. &P < 0.05, &&P < 0.01 versus the GA + LPS group.
TNF-α, IL-1β, and HMGB1 are representative proinflammatory factors in many infectious diseases. To further explore the effect of GA on inflammation of ALI in vivo, we first detected the mRNA levels of TNF-α and IL-1β by qRT-PCR, and the data indicated that LPS increased the production of TNF-α and IL-1β (P < 0.01). Nevertheless, pre-treatment of GA significantly restrained the production of TNF-α and IL-1β (P < 0.05), and the inhibitory effects of GA on inflammatory factors were reversed by 3-MA in LPS-induced ALI mouse model (P < 0.05) (Figure 5D, 5E). Moreover, we further evaluated the levels of TNF-α, IL-1β, and HMGB1 using Western blotting and HMGB1 by immunohistochemical analysis. It was consistent with the mRNA expression of the mentioned inflammatory factors. In briefly, GA obvious downregulated the protein production of TNF-α, IL-1β, and HMGB1, while the inhibiting effects of GA on inflammation were reversed by 3-MA administration under ALI in vivo (P < 0.05) (Figure 5F-J). These above results declared that GA decreased lung inflammation of ALI through activation of autophagy in vivo.
GA enhances autophagy through the PI3K/AKT/mTOR pathway in LPS-induced ALI mouse model
To elucidate the effect of GA on autophagy in lung injury induced by LPS, we first evaluated the protein levels of LC3-II/I, Beclin-1, and P62 by Western blotting. As shown in Figure 6, it was shown that the expression of LC3-II/I and Beclin-1 were significantly increased and P62 decreased in the LPS group (P < 0.05). Moreover, pre-treatment of GA could further enhance the levels of LC3-II/I and Beclin-1, reduce P62 in LPS-induced ALI mouse model (P < 0.05). Importantly, the autophagy activation induced by GA was reversed by 3-MA in vivo (P < 0.05). Our results indicated that GA enhanced autophagy in LPS-induced ALI mouse model.
Figure 6.
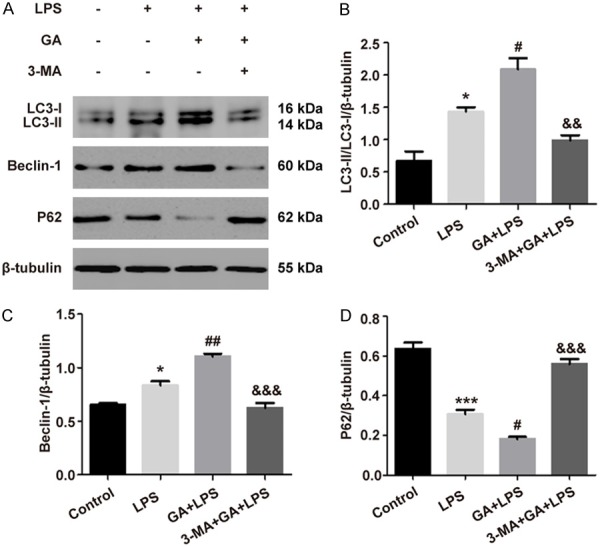
GA induces autophagy in vivo. A. Western blotting detected the levels of LC3-II/I, Beclin-1, and SQSTM1/P62 in lung tissues. B-D. Quantitative analysis of LC3-II/I, Beclin-1, and SQSTM1/p62 were shown in bar graphs, respectively. The experiments were performed four independent times (n=4) and bars represented as mean ± SEM. * P < 0.05, *** P < 0.001 versus control group. # P < 0.05, ## P < 0.01 in comparison to LPS group. && P < 0.01, &&& P < 0.001 versus sGA + LPS group.
In order to further identify the mechanisms of autophagy activated by GA in LPS-induced ALI in vivo, we also investigated the PI3K/AKT/mTOR pathway. It was indicated that the levels of p-PI3K, p-AKT, and p-mTOR proteins were sharply decrease with GA treatment (200 mg/kg) (P < 0.05), and further decreased after addition of 3-MA (P < 0.05), but the total PI3K, AKT, and mTOR did not change significantly (P > 0.05), in the ALI mouse model induced by LPS (Figure 7). These results suggested that the PI3K/AKT/mTOR pathway governed GA enhancement of autophagy under ALI in vivo.
Figure 7.
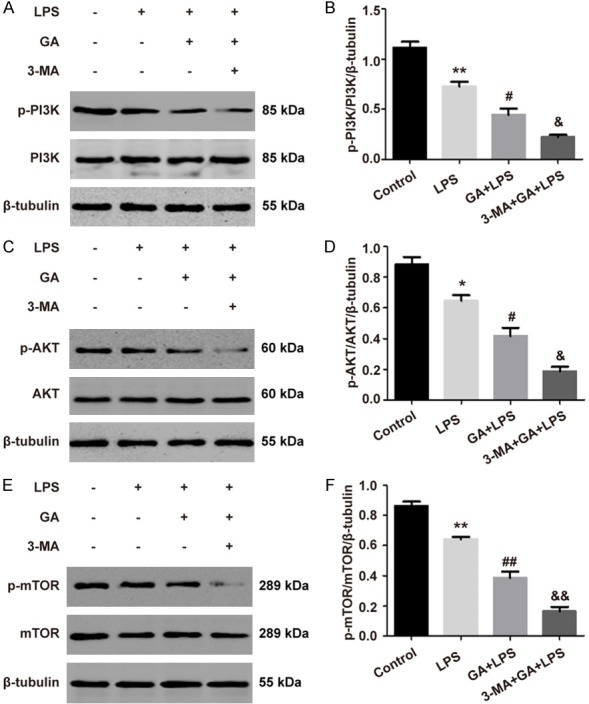
GA induces autophagy through regulating the PI3K/AKT/mTOR pathway in vivo. (A, B) The levels of p-PI3K and PI3K, and (C, D) p-AKT and AKT, and (E, F) p-mTOR and mTOR were evaluated by Western blotting. The experiments were performed four independent times (n=4) and bars represented as mean ± SEM. * P < 0.05, ** P < 0.01 in comparison to control group. # P < 0.05, ## P < 0.01 in comparison to LPS group. & P < 0.05, && P < 0.01 in comparison to GA + LPS group.
Discussion
Currently, there is no effective treatment for ALI, therefore, it is urgent to develop the strategy for ALI treatment [5,25]. The main mechanism of ALI induction by LPS is where multiple intracellular inflammatory signaling pathways cause the excessive release of various inflammatory factors [7,8,26]. The imbalance between proinflammatory and anti-inflammatory responses leads the accumulation of large numbers of inflammatory cells and inflammatory cytokines which infiltrating the lung [26,27]. Nevertheless, the specific mechanism by which inflammation develops into ALI is unknown. Alveolar macrophages are the main resident phagocytes in lung, which participate in the host’s initial defense response through producing inflammatory mediators and chemokines, and regulate the initiation and development of pulmonary inflammation [7,28]. Meanwhile, excessive activation of macrophages is also the key mechanism of inflammatory damage. TNF-α and IL-1β are the key components of cytokine network and the important inflammatory mediators of ALI initiation, which mainly secreted by mononuclear macrophages [8,29]. High-mobility group box 1 (HMGB1), a highly conserved protein, is related to the pathogenesis of inflammation and as a proinflammatory mediator when stimulated by LPS [30,31]. In our study, we examined the levels of the above inflammatory factors and found that LPS induced the production of TNF-α, IL-1β, and HMGB1 in vitro and in vivo.
GA has been reported to be widely used in the protection efficacy of liver injuries [32,33], while the protective effect of GA on lung injury has rarely been reported. Studies have shown that GA has anti-inflammatory and inhibitory effects on HMGB1 [33-35]. In mouse RAW264.7 macrophages induced by LPS, we found that GA increased the cell viability and decreased the production of TNF-α, IL-1β, and HMGB1. Moreover, we also showed that GA suppressed the secretion of inflammatory cytokines in ALI mouse model. On this basis, we further investigated the impact of GA on ALI and tried to reveal its potential mechanism.
Autophagy has attracted extensive attention in recent years due to the effects on various physiological and pathological processes. The process of autophagy involves many autophagy related proteins, which can engulf and kill pathogens to protect cells from pathogens, and inhibits inflammasome and the secretion of inflammation factors [19,36]. It is reported that GA significantly enhances autophagy in hepatocellular carcinoma in vivo and in vitro [37]. Lin et al. shows the anti-cancer effect of GA, which is related to the induction of autophagy in breast cancer cells [38]. In this study, we studied GA could activate autophagy in ALI models induced by LPS, and it was indicated that GA significantly increased the protein ratio of LC3-II/LC3-I and the protein levels of Beclin-1 and decreased P62, upregulated the fluorescence intensity of LC3, and enhanced the number of autophagosomes. During the process of autophagy flux, LC3-I converts to LC3-II, and the ratio of LC3-II/LC3-I is used as a quantitative index of autophagy activity [36,39]. The degradation of P62 protein contributes to the formation of autophagosome, while upregulation of P62 indicates autophagy inhibition [39,40]. Thus, we suggested that GA activated autophagy in ALI induced by LPS, and importantly, the activation of autophagy by GA reversed with 3-MA treatment.
Many cytokines are involved in controlling autophagy. Helper T cell cytokines, such as IL-2, TNF-α, and IFN-γ, are generally considered as autophagy inducers, while Th2 cell-associated cytokines, such as IL-4, IL-10, and IL-13, can restrain autophagy [41,42]. In addition, it has shown that autophagy regulates inflammation factors, such as TNF-α and IL-1β [43-46]. Autophagy reduces the secretion of IL-1β by inhibiting the activation of inflammasome [45,47]. Activation of autophagy inhibits TNF-α secretion [15,19]. As a classical damage-related model molecule, HMGB1 regulates autophagy mainly by binding to Beclin-1 [48,49]. Based on these reports and our previous results, we hypothesized that GA regulated the expression of inflammatory cytokines through regulating autophagy in ALI. The cytokines TNF-α, IL-1β, and HMGB1 were suppressed after inhibition of autophagy in vitro and in vivo. In conclusion, the anti-inflammatory effects of GA might be related to the induction of autophagy.
Zhang et al. shows that isoliquiritigenin is a component extracted from licorice, which promotes autophagy and inhibits gastric cancer cells through the PI3K/AKT/mTOR pathway [50]. PI3K regulates autophagy through different mechanisms, as type I PI3K-mTOR is an inhibitory pathway and type III PI3K is an activated pathway [51,52]. Autophagy plays a protective role in hyperoxia-induced lung injury [53]. Our results showed that GA reduced the levels of p-PI3K, p-AKT, and p-mTOR proteins, while the total PI3K, AKT, and mTOR had no evident changes, under ALI in vitro and in vivo, indicating that GA induced autophagy at least party through the PI3K/AKT/mTOR pathway. Then, 3-MA reduced the levels of p-PI3K, p-AKT, and p-mTOR proteins. Hence, it was indicated that GA ameliorated LPS-induced ALI through autophagy activation by regulating the PI3K/AKT/mTOR pathway.
This study suggested that GA induced autophagy through the PI3K/AKT/mTOR pathway to reduce the secretion of inflammatory factors and ameliorate ALI induced by LPS, as shown in Figure 8. The study first discovered that GA activated autophagy through regulating the PI3K/AKT/mTOR pathway and attenuated LPS-induced ALI, which providing a novel prospection of GA alleviated ALI through inducing autophagy. Nevertheless, there still had some specific molecular mechanisms needed to be explored, such as, how autophagy regulated the changes of cytokines under ALI. Anyhow, this study provided an experimental basis for the application of GA for the clinical treatment of lung injury.
Figure 8.
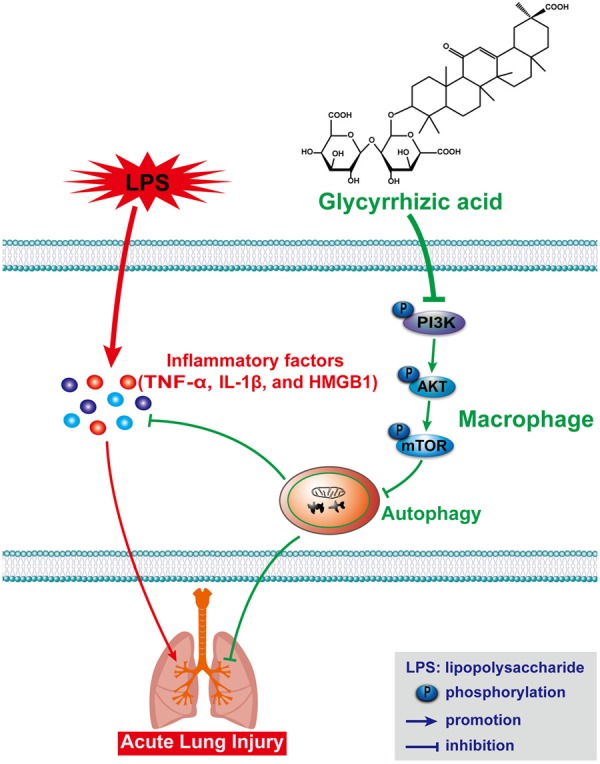
A diagram depicting the mechanism of GA ameliorates LPS-induced ALI by activating autophagy through regulating of the PI3K/AKT/mTOR pathway.
Acknowledgements
The work thanks the National Natural Science Foundation of China (No. 81100054), Hunan Natural Science Foundation (No. 2018JJ2255), and Hunan Natural Science Foundation Joint Project of Science and Health (No. 2018JJ6052) for financial support.
Disclosure of conflict of interest
None.
Abbreviations
- ALI
acute lung injury
- CLSM
confocal laser scanning microscope
- GA
glycyrrhizic acid
- HMG-B1
high-mobility group box 1
- IL-1β
interleukin-1β
- LPS
lipopolysaccharide
- PI3K
phosphoinositide 3-kinase
- TNF-α
tumor necrosis factor-α
- TEM
transmission electron microscopy
- 3-MA
3-Methyladenine
References
- 1.Taylor WRJ, Hanson J, Turner GDH, White NJ, Dondorp AM. Respiratory manifestations of malaria. Chest. 2012;142:492–505. doi: 10.1378/chest.11-2655. [DOI] [PubMed] [Google Scholar]
- 2.Sapoznikov A, Gal Y, Falach R, Sagi I, Ehrlich S, Lerer E, Makovitzki A, Aloshin A, Kronman C, Sabo T. Early disruption of the alveolar-capillary barrier in a ricin-induced ARDS mouse model: neutrophil-dependent and -independent impairment of junction proteins. Am J Physiol Lung Cell Mol Physiol. 2019;316:L255–L268. doi: 10.1152/ajplung.00300.2018. [DOI] [PubMed] [Google Scholar]
- 3.Hayes M, Curley G, Ansari B, Laffey JG. Clinical review: stem cell therapies for acute lung injury/acute respiratory distress syndrome - hope or hype? Crit Care. 2012;16:205. doi: 10.1186/cc10570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levitt JE, Calfee CS, Goldstein BA, Vojnik R, Matthay MA. Early acute lung injury: criteria for identifying lung injury prior to the need for positive pressure ventilation. Crit Care Med. 2013;41:1929–1937. doi: 10.1097/CCM.0b013e31828a3d99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fragoso IT, Ribeiro EL, Gomes FO, Donato MA, Silva AK, Oliveira AC, Araujo SM, Barbosa KP, Santos LA, Peixoto CA. Diethylcarbamazine attenuates LPS-induced acute lung injury in mice by apoptosis of inflammatory cells. Pharmacol Rep. 2017;69:81–89. doi: 10.1016/j.pharep.2016.09.021. [DOI] [PubMed] [Google Scholar]
- 6.Elicker BM, Jones KT, Naeger DM, Frank JA. Imaging of acute lung injury. Radiol Clin North Am. 2016;54:1119–1132. doi: 10.1016/j.rcl.2016.05.006. [DOI] [PubMed] [Google Scholar]
- 7.Zou B, Jiang W, Han H, Li J, Mao W, Tang Z, Yang Q, Qian G, Qian J, Zeng W, Gu J, Chu T, Zhu N, Zhang W, Yan D, He R, Chu Y, Lu M. Acyloxyacyl hydrolase promotes the resolution of lipopolysaccharide-induced acute lung injury. PLoS Pathog. 2017;13:e1006436. doi: 10.1371/journal.ppat.1006436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee H, Zhang D, Laskin DL, Jin Y. Functional evidence of pulmonary extracellular vesicles in infectious and noninfectious lung inflammation. J Immunol. 2018;201:1500–1509. doi: 10.4049/jimmunol.1800264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19:349–364. doi: 10.1038/s41580-018-0003-4. [DOI] [PubMed] [Google Scholar]
- 10.Du J, Zhu X, Guo R, Xu Z, Cheng FF, Liu Q, Yang F, Guan L, Liu Y, Lin J. Autophagy induces G0/G1 arrest and apoptosis in menstrual blood-derived endometrial stem cells via GSK3-beta/beta-catenin pathway. Stem Cell Res Ther. 2018;9:330. doi: 10.1186/s13287-018-1073-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Subramani S, Malhotra V. Non-autophagic roles of autophagy-related proteins. EMBO Rep. 2013;14:143–151. doi: 10.1038/embor.2012.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu L, Chen Y, Tooze SA. Autophagy pathway: cellular and molecular mechanisms. Autophagy. 2018;14:207–215. doi: 10.1080/15548627.2017.1378838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22:124–131. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moon EK, Kim SH, Hong Y, Chung DI, Goo YK, Kong HH. Autophagy inhibitors as a potential antiamoebic treatment for Acanthamoeba keratitis. Antimicrob Agents Chemother. 2015;59:4020–4025. doi: 10.1128/AAC.05165-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu C, Wang W, Zhong J, Lei F, Xu N, Zhang Y, Xie W. Canagliflozin exerts anti-inflammatory effects by inhibiting intracellular glucose metabolism and promoting autophagy in immune cells. Biochem Pharmacol. 2018;152:45–59. doi: 10.1016/j.bcp.2018.03.013. [DOI] [PubMed] [Google Scholar]
- 16.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zeng M, Sang W, Chen S, Chen R, Zhang H, Xue F, Li Z, Liu Y, Gong Y, Zhang H, Kong X. 4-PBA inhibits LPS-induced inflammation through regulating ER stress and autophagy in acute lung injury models. Toxicol Lett. 2017;271:26–37. doi: 10.1016/j.toxlet.2017.02.023. [DOI] [PubMed] [Google Scholar]
- 18.Jin Y, Tanaka A, Choi AM, Ryter SW. Autophagic proteins: new facets of the oxygen paradox. Autophagy. 2012;8:426–428. doi: 10.4161/auto.19258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang D, Zhou J, Ye LC, Li J, Wu Z, Li Y, Li C. Autophagy maintains the integrity of endothelial barrier in LPS-induced lung injury. J Cell Physiol. 2018;233:688–698. doi: 10.1002/jcp.25928. [DOI] [PubMed] [Google Scholar]
- 20.Su X, Wu L, Hu M, Dong W, Xu M, Zhang P. Glycyrrhizic acid: a promising carrier material for anticancer therapy. Biomed Pharmacother. 2017;95:670–678. doi: 10.1016/j.biopha.2017.08.123. [DOI] [PubMed] [Google Scholar]
- 21.Fouladi S, Masjedi M, Ghasemi R, G Hakemi M, Eskandari N. The in vitro impact of glycyrrhizic acid on CD4+ T lymphocytes through OX40 receptor in the patients with allergic rhinitis. Inflammation. 2018;41:1690–1701. doi: 10.1007/s10753-018-0813-8. [DOI] [PubMed] [Google Scholar]
- 22.Zhao H, Zhao M, Wang Y, Li F, Zhang Z. Glycyrrhizic acid prevents sepsis-induced acute lung injury and mortality in rats. J Histochem Cytochem. 2016;64:125–137. doi: 10.1369/0022155415610168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Menegazzi M, Di Paola R, Mazzon E, Genovese T, Crisafulli C, Dal Bosco M, Zou Z, Suzuki H, Cuzzocrea S. Glycyrrhizin attenuates the development of carrageenan-induced lung injury in mice. Pharmacol Res. 2008;58:22–31. doi: 10.1016/j.phrs.2008.05.012. [DOI] [PubMed] [Google Scholar]
- 24.Smith KM, Mrozek JD, Simonton SC, Bing DR, Meyers PA, Connett JE, Mammel MC. Prolonged partial liquid ventilation using conventional and high-frequency ventilatory techniques: gas exchange and lung pathology in an animal model of respiratory distress syndrome. Crit Care Med. 1997;25:1888–1897. doi: 10.1097/00003246-199711000-00030. [DOI] [PubMed] [Google Scholar]
- 25.Graham S, Fairhall S, Rutter S, Auton P, Rendell R, Smith A, Perrott R, Roberts TN, Jugg B. Continuous positive airway pressure: an early intervention to prevent phosgene-induced acute lung injury. Toxicol Lett. 2018;293:120–126. doi: 10.1016/j.toxlet.2017.11.001. [DOI] [PubMed] [Google Scholar]
- 26.Ding X, Tong Y, Jin S, Chen Z, Li T, Billiar TR, Pitt BR, Li Q, Zhang LM. Mechanical ventilation enhances extrapulmonary sepsis-induced lung injury: role of WISP1-alphavbeta5 integrin pathway in TLR4-mediated inflammation and injury. Crit Care. 2018;22:302. doi: 10.1186/s13054-018-2237-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ezegbunam W, Foronjy R. Posttranscriptional control of airway inflammation. Wiley Interdiscip Rev RNA. 2018;9 doi: 10.1002/wrna.1455. [DOI] [PubMed] [Google Scholar]
- 28.Atabai K, Matthay MA. The pulmonary physician in critical care. 5: acute lung injury and the acute respiratory distress syndrome: definitions and epidemiology. Thorax. 2002;57:452–458. doi: 10.1136/thorax.57.5.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cross LJ, Matthay MA. Biomarkers in acute lung injury: insights into the pathogenesis of acute lung injury. Crit Care Clin. 2011;27:355–377. doi: 10.1016/j.ccc.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang Q, Liu X, Yao Z, Mao S, Wei Q, Chang Y. Penehyclidine hydrochloride inhibits the release of high-mobility group box 1 in lipopolysaccharide-activated RAW264.7 cells and cecal ligation and puncture-induced septic mice. J Surg Res. 2014;186:310–317. doi: 10.1016/j.jss.2013.08.015. [DOI] [PubMed] [Google Scholar]
- 31.Musumeci D, Roviello GN, Montesarchio D. An overview on HMGB1 inhibitors as potential therapeutic agents in HMGB1-related pathologies. Pharmacol Ther. 2014;141:347–357. doi: 10.1016/j.pharmthera.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 32.Chen HJ, Kang SP, Lee IJ, Lin YL. Glycyrrhetinic acid suppressed NF-kappaB activation in TNF-alpha-induced hepatocytes. J Agric Food Chem. 2014;62:618–625. doi: 10.1021/jf405352g. [DOI] [PubMed] [Google Scholar]
- 33.Yu JY, Ha JY, Kim KM, Jung YS, Jung JC, Oh S. Anti-Inflammatory activities of licorice extract and its active compounds, glycyrrhizic acid, liquiritin and liquiritigenin, in BV2 cells and mice liver. Molecules. 2015;20:13041–13054. doi: 10.3390/molecules200713041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meng L, Li L, Lu S, Li K, Su Z, Wang Y, Fan X, Li X, Zhao G. The protective effect of dexmedetomidine on LPS-induced acute lung injury through the HMGB1-mediated TLR4/NF-kappaB and PI3K/Akt/mTOR pathways. Mol Immunol. 2018;94:7–17. doi: 10.1016/j.molimm.2017.12.008. [DOI] [PubMed] [Google Scholar]
- 35.Cui Z, Li S, Liu Z, Zhang Y, Zhang H. Interferon regulatory factor 1 activates autophagy to aggravate hepatic ischemia-reperfusion injury by increasing high mobility group box 1 release. Cell Physiol Biochem. 2018;48:328–338. doi: 10.1159/000491732. [DOI] [PubMed] [Google Scholar]
- 36.Shibutani ST, Saitoh T, Nowag H, Munz C, Yoshimori T. Autophagy and autophagy-related proteins in the immune system. Nat Immunol. 2015;16:1014–1024. doi: 10.1038/ni.3273. [DOI] [PubMed] [Google Scholar]
- 37.Zhang X, Yang H, Yue S, He G, Qu S, Zhang Z, Ma B, Ding R, Peng W, Zhang H, Yang Z, Dou K, Tao K, Li X. The mTOR inhibition in concurrence with ERK1/2 activation is involved in excessive autophagy induced by glycyrrhizin in hepatocellular carcinoma. Cancer Med. 2017;6:1941–1951. doi: 10.1002/cam4.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin SC, Chu PY, Liao WT, Wu MY, Tsui KH, Lin LT, Huang CH, Chen LL, Li CJ. Glycyrrhizic acid induces human MDA-MB-231 breast cancer cell death and autophagy via the ROS-mitochondrial pathway. Oncol Rep. 2018;39:703–710. doi: 10.3892/or.2017.6123. [DOI] [PubMed] [Google Scholar]
- 39.Fan X, Wang J, Hou J, Lin C, Bensoussan A, Chang D, Liu J, Wang B. Berberine alleviates ox-LDL induced inflammatory factors by up-regulation of autophagy via AMPK/mTOR signaling pathway. J Transl Med. 2015;13:92. doi: 10.1186/s12967-015-0450-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dong W, He B, Qian H, Liu Q, Wang D, Li J, Wei Z, Wang Z, Xu Z, Wu G, Qian G, Wang G. RAB26-dependent autophagy protects adherens junctional integrity in acute lung injury. Autophagy. 2018;14:1677–1692. doi: 10.1080/15548627.2018.1476811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu TT, Li WM, Yao YM. Interactions between autophagy and inhibitory cytokines. Int J Biol Sci. 2016;12:884–897. doi: 10.7150/ijbs.15194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harris J. Autophagy and cytokines. Cytokine. 2011;56:140–144. doi: 10.1016/j.cyto.2011.08.022. [DOI] [PubMed] [Google Scholar]
- 43.Cho MH, Cho K, Kang HJ, Jeon EY, Kim HS, Kwon HJ, Kim HM, Kim DH, Yoon SY. Autophagy in microglia degrades extracellular beta-amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy. 2014;10:1761–1775. doi: 10.4161/auto.29647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, Fitzgerald KA, Ryter SW, Choi AM. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12:222–230. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 46.Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, Tanaka K, Kawai T, Tsujimura T, Takeuchi O, Yoshimori T, Akira S. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456:264–268. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 47.Harris J, Hartman M, Roche C, Zeng SG, O’Shea A, Sharp FA, Lambe EM, Creagh EM, Golenbock DT, Tschopp J, Kornfeld H, Fitzgerald KA, Lavelle EC. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem. 2011;286:9587–9597. doi: 10.1074/jbc.M110.202911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kang R, Livesey KM, Zeh HJ, Loze MT, Tang D. HMGB1: a novel Beclin 1-binding protein active in autophagy. Autophagy. 2010;6:1209–1211. doi: 10.4161/auto.6.8.13651. [DOI] [PubMed] [Google Scholar]
- 49.Tang D, Loze MT, Zeh HJ, Kang R. The redox protein HMGB1 regulates cell death and survival in cancer treatment. Autophagy. 2010;6:1181–1183. doi: 10.4161/auto.6.8.13367. [DOI] [PubMed] [Google Scholar]
- 50.Zhang XR, Wang SY, Sun W, Wei C. Isoliquiritigenin inhibits proliferation and metastasis of MKN28 gastric cancer cells by suppressing the PI3K/AKT/mTOR signaling pathway. Mol Med Rep. 2018;18:3429–3436. doi: 10.3892/mmr.2018.9318. [DOI] [PubMed] [Google Scholar]
- 51.Scherz-Shouval R, Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci. 2011;36:30–38. doi: 10.1016/j.tibs.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 52.Muller C, Salvayre R, Negre-Salvayre A, Vindis C. Oxidized LDLs trigger endoplasmic reticulum stress and autophagy: prevention by HDLs. Autophagy. 2011;7:541–543. doi: 10.4161/auto.7.5.15003. [DOI] [PubMed] [Google Scholar]
- 53.Tanaka A, Jin Y, Lee SJ, Zhang M, Kim HP, Stolz DB, Ryter SW, Choi AM. Hyperoxia-induced LC3B interacts with the Fas apoptotic pathway in epithelial cell death. Am J Respir Cell Mol Biol. 2012;46:507–514. doi: 10.1165/rcmb.2009-0415OC. [DOI] [PMC free article] [PubMed] [Google Scholar]