

Abstract
Inhibition of hypoxia/reoxygenation (H/R)-induced insult in cardiac microvascular endothelial cells (CMECs) has been considered as a promising therapeutic strategy for the treatment of ischemic cardiovascular disease. In the present study, we found that H/R significantly increased the expression of dipeptidyl peptidase (DPP)-4 in CMECs. Treatment with the DPP-4 inhibitor sitagliptin, a licensed drug used for the treatment of type 2 diabetes mellitus (T2DM), ameliorated H/R-induced oxidative stress by decreasing the expression of NOX-4 and restoring the intracellular level of GSH in CMECs. Sitagliptin could also improve H/R-induced mitochondrial dysfunction by increasing intracellular MMP and ATP. Additionally, we found that the presence of sitagliptin prevented H/R-induced reduced cell viability and LDH release. Notably, our findings indicate that sitagliptin possesses an anti-inflammatory effect against H/R-induced expression of IL-6, IL-8, and TNF-α as well as secretion of HMGB1. Mechanistically, we found that sitagliptin suppresses activation of p38/NF-κB signaling. These findings suggest that sitagliptin may have potential as a therapeutic agent for the treatment of cardiovascular diseases.
Keywords: Hypoxia/reoxygenation, sitagliptin, oxidative stress, cardiovascular diseases, NF-κB
Introduction
Myocardial infarction is one of the main ischemic heart diseases affecting millions of people worldwide [1]. The mechanisms underlying the pathophysiology of myocardial infarction remain elusive. Endothelial dysfunction has recently been considered as a critical component of the pathological progression of myocardial infarction and heart failure [2]. Hypoxia-reperfusion (H/R) during which the process of myocardial infarction causes damage not only to large vessels but also to the microvasculature of the heart [3]. Cardiac microvascular endothelial cells (CMECs) have been shown to be sensitive to H/R-induced insult. H/R exposure damages a wide range of the physiological activities of endothelial cells, including reduced cell survival, dysregulated energy metabolism, impaired angiogenesis, excessive production of reactive oxygen species (ROS), and mitochondrial dysfunction [4]. Notably, it has been shown that H/R exposure induces an inflammatory response in CMECs. H/R exposure has also been shown to trigger excessive production of pro-inflammatory mediators such as tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), and IL-8 [5]. Several intracellular signaling pathways have been shown to be involved in H/R exposure-induced insult in CMECs. Activation of p38 MAPK plays an important role in mediating oxidative stress and inflammatory response [6,7]. Additionally, H/R exposure has been shown to activate NF-κB, a central regulator of intracellular inflammatory signaling [8]. Cardiovascular derangement has been found in patients with type 2 diabetes mellitus (T2DM) [9]. Dipeptidyl peptidase (DPP)-4 inhibitors have been recently developed for the treatment and management of T2DM [10]. Sitagliptin, a potent and highly selective inhibitor of DPP-4, was developed by Merck & Co. for the treatment of T2DM [11]. In addition to its essential capacity in reducing blood glucose concentration, sitagliptin has displayed a wide spectrum of pharmacological activities in various tissues and cells. For example, an in vivo experiment reported that administration of sitagliptin prevents the pathological progression of hepatic steatosis and insulin resistance in obese mice through suppression of inflammatory responses and activation of autophagy mediated by the AMPK/mTOR signaling pathway [12]. Sitagliptin treatment can also attenuate tubulointerstitial injury, ameliorate inflammatory cell infiltration, and prevent interstitial fibrosis in rodent kidneys with doxorubicin nephropathy by inhibiting NLRP3 inflammasome activation [13]. Sitagliptin significantly ameliorated the development of seizures in a pentylenetetrazol rat model by downregulating the RAGE-JAK2/STAT3 pathway and decreasing the expression of CXCL4/CXCR3 [14]. In the present study, we aimed to investigate whether sitagliptin could protect CMECs against H/R-induced inflammatory response and damage.
Materials and methods
Primary cardiac microvascular endothelial cells (CMECs)
SD rats aged 1 to 4 days old were used to isolate primary cardiac microvascular endothelial cells. Rats were sacrificed, and the cardiac tissues were collected and cut into small pieces, following a brief digestion with collagenase I. CD31 is a marker protein located on the surface of endothelial cells. CMECs were purified with the CD31 antibody-conjugated Dynabeads (Thermo Fisher Scientific, USA) on a magnetic stand. CMECs were cultured using an EGM endothelial cell growth medium bullet kit (Lonza, China) supplemented with 10% FBS. Cells were used for less than 3 passages. Human monocyte cells line THP-1 cells were from ATCC, USA. THP-1 cells were cultured in RPMI 1640 medium (Thermo Fisher Scientific, USA) supplemented with 10% FBS and 1% penicillin/streptomycin (Sigma-Aldrich, USA).
Hypoxia/reoxygenation (H/R) experiments
Isolated CMECs were preincubated with 100, 200 nM sitagliptin (Merk Sharp & Dohme Ltd., UK) or DMSO vehicle for 12 h. H/R experiments were performed by putting cells in an air-tight chamber with pure N2 followed by incubation at 37°C for 6 h. Cells were then cultured with normal medium in a normoxic incubator (95% air plus 5% CO2) for 12 h for reoxygenation.
Real time PCR analysis
Total RNA was isolated from CMECs using Qiazol (Qiagen, USA) in accordance with the manufacturer’s instructions. Total RNA (1 μg) was used to produce complementary DNA (cDNA) with a reverse transcriptase PCR kit (Applied Biosystems, USA) in accordance with the manufacturer’s instructions. Quantitative real time PCR analysis was performed to determine the expressions of target genes with Power SYBR Green PCR Master Mix (Applied Biosystems, USA) on a 7500 Fast Real-time PCR system (Applied Biosystems, USA).
Western blot analysis
Protein extract was prepared in CMECs with cell lysis buffer and complete protease and phosphatase inhibitor mixture. Protein extract was centrifuged at 14,000 × g for 15 min at 4°C. The protein concentration in the cell lysate was measured using a Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, USA). Protein lysates were denatured at 95°C for 5 min. Total protein was resolved in 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene fluoride (PVDF) membranes (Bio-Rad, USA). Membranes were blocked with 5% (w/v) non-fat dry milk for 1 h at room temperature (RT). PVDF membranes were incubated with the indicated antibodies in TBS-T containing 5% (w/v) protease-free BSA overnight at 4°C. After 3 washes, membranes were probed with HRP- conjugated secondary antibodies for 2 h at RT. The results were detected via enhanced chemiluminescence (Thermo Fisher Scientific, USA). The following antibodies were used in this study: DPP-4 (1:1000, #ab28340, Abcam, USA); NOX-4 (1:2000, ab133303, Abcam, USA); p-p38 (1:1000, #9216, Cell Signaling Technology, USA); p38 (1:3000, #8690, Cell Signaling Technology, USA); p65 (1:2000, #9460, Cell signaling technology, USA); Lamin B1 (1:5000, #13435, Cell Signaling Technology, USA); β-actin (1:10000, #3700, Cell Signaling Technology, USA); anti-rabbit IgG, HRP-linked antibody (1:2000, #7074, Cell Signaling Technology, USA); anti-mouse IgG, HRP-linked antibody (1:2000, #7076, Cell Signaling Technology, USA).
Isolation of nuclei
Nuclei were isolated using a Nuclei Isolation Kit (Sigma-Aldrich, USA) in accordance with the manufacturer’s instructions. After the indicated treatment, cells were collected in 500 μl hypotonic buffer with NP40. Homogenates were centrifugated for 10 min at 3000 RPM at 4°C. The pellet is the nuclear fraction. Nuclear fraction was resuspended in 50 μl complete cell extraction buffer and vortexed for 30 min on ice. After centrifugation at 14000 × g for 30 min, supernatant was collected for nuclear protein measurement by western blot analysis.
Enzyme-linked immunosorbent assay (ELISA)
To determine the secretion of various inflammatory cytokines and HMGB1, the cell culture medium was collected to perform the ELISA assay. ELISA kits for IL-6, IL-8, and TNF-α were purchased from R&D Systems, USA and the HMGB-1 ELISA Kit was from Shino Test Corporation, Tokyo, Japan.
Determination of mitochondrial membrane potential (MMP)
The level of MMP in CMECs was determined with tetramethylrhodamine methyl ester (TMRM) (Invitrogen, USA) staining. H/R experiments were performed by putting cells in an air-tight chamber with pure N2 followed by incubation at 37°C for 6 h. Cells were then cultured with normal medium in a normoxic incubator (95% air plus 5% CO2) for 12 h for reoxygenation. Cells were then probed with 20 nmol/L TMRM for 15 min. Fluorescence signals were recorded using a fluorescence microscope (Zeiss, Germany).
Measurement of cell viability
After the necessary treatment, cell viability of CMECs was measured using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Sigma-Aldrich, USA). Upon completion of the indicated treatment, MTT was added to the culture medium at a final concentration of 5 mg/mL and incubated for 4 h at 37°C in darkness. The MTT formazan was dissolved in dimethyl sulfoxide (DMSO). Absorbance was read at 590 nm.
Measurement of adenosine triphosphate (ATP)
The level of ATP in CMECs was assessed using a commercial ATP bioluminescence assay kit (Sigma-Aldrich, USA) in accordance with the manual instructions. Briefly, CMECs were lysed and subjected to a centrifugation (10,000 × g) for 5 min at 4°C. Equal volumes (100 μl) of supernatant and luciferin/luciferase reagent were mixed together to catalyze light production. Light output was immediately read using a microplate luminometer.
Measurement of LDH release
The release of LDH from the cytoplasm into the culture medium was detected using a commercial kit (Thermo Fisher Scientific, USA). Upon completion of the indicated treatment, equal volumes (50 μl) of cell culture medium and substrate were mixed together and incubated for 30 min. Then stop buffer was used to stop the reaction. Absorbance was read at 490 nm.
Reduced glutathione (GSH) assay
The level of reduced GSH in CMECs was measured using a fluorometric assay. Cells were lysed and subjected to centrifugation (500 × g) for 5 min at 4°C. Cells were suspended in 5% meta-phosphoric acid (MPA) on ice, then sonicated and centrifuged at 14000 × g for 5 min. Supernatant was mixed with OPAME (Sigma-Aldrich, USA) in methanol and borate buffer. The fluorescent signal was read at 350 nm excitation and 420 nm emission.
Luciferase reporter gene assay
NF-κB promoter-luciferase and β-galactosidase plasmid (Clontech, USA) were transfected into cells with Lipofectamine 2000 reagent. Cells were pre-incubated with 100 and 200 nM sitagliptin or DMSO vehicle for 12 h, followed by exposure to H/R conditions. Cells were then lysed. Luciferase activity and β-galactosidase activity were measured using a Secrete-Pair/TM Dual luminescence assay kit (Gene Copoeia, MD).
Statistical analysis
Experiments were repeated at least three time. Experimental data are presented as means ± S.D. and analyzed using SPSS 17 software (SPSS Inc., USA). Statistical analysis was performed by one-way analysis of variance (ANOVA). A P-value <0.05 was considered statistically significant.
Results
DPP-4 has been reported to be expressed in endothelial cells. Firstly, we determined whether H/R had an impact on the expression and function of DPP-4. The results of real time PCR analysis in Figure 1A and western blot analysis in Figure 1B indicate that H/R exposure significantly increased the expression of DPP-4 at both the mRNA and protein levels in CMECs. These findings indicate that DPP-4 might play a key role in mediating H/R-induced insults in CMECs.
Figure 1.
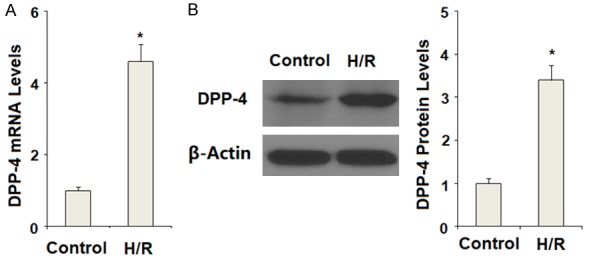
Hypoxia/reoxygenation (H/R) increased the expression of DPP-4 in primary cardiac microvascular endothelial cells (CMECs). Hypoxia/reoxygenation experiments were performed by putting cells in an air-tight chamber with pure N2 followed by incubation at 37°C for 6 h. Cells were then cultured with normal medium in a normoxic incubator (95% air plus 5% CO2) for 12 h for reoxygenation. A. Expression of DPP-4 at the mRNA levels was determined by real time PCR analysis; B. Expression of DPP-4 at the protein levels was determined by western blot analysis (*, P<0.01 vs. the control group, n=6).
To inhibit DPP-4 activity, CMECs were pre-incubated with sitagliptin, a typical DPP-4 inhibitor, at the concentrations of 100 and 200 nM. Patterns of oxidative stress were evaluated by measuring the expression of NOX-4 and the level of reduced intracellular GSH. NOX-4 is a phagocyte-type oxidase, which is responsible for the production of large amounts of intracellular ROS. The results in Figure 2A indicate that H/R significantly increased the expression of NOX-4. Reduced GSH is an important intracellular antioxidant presented in various tissues and cells. Here, our results indicate that H/R significantly decreased the intracellular level of reduced GSH. However, the presence of sitagliptin prevented the decrease in reduced GSH induced by H/R exposure (Figure 2B). These findings suggest that sitagliptin has a protective effect against H/R- induced oxidative stress in CMECs.
Figure 2.
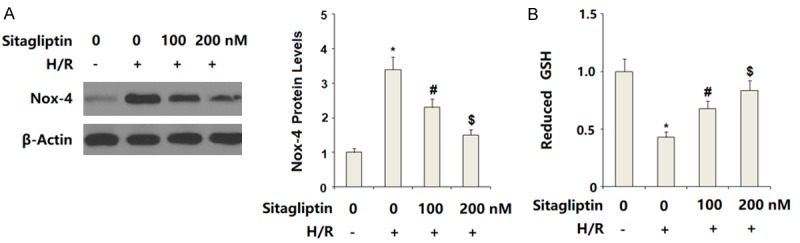
Sitagliptin pretreatment ameliorated hypoxia/reoxygenation (H/R)-induced oxidative stress in primary cardiac microvascular endothelial cells (CMECs). Cells were pre-incubated with 100, 200 nM sitagliptin or DMSO vehicle for 12 h, followed by exposure to hypoxia/reoxygenation (H/R) conditions. A. Western blot analysis of NOX-4; B. Reduced glutathione (GSH) was determined (*, P<0.01 vs. vehicle group; #, P<0.01 vs. H/R group; $, P<0.01 vs. H/R+100 nM sitagliptin, n=5-6).
Impaired mitochondrial function has been associated with brain vascular endothelial dysfunction. Here, mitochondrial function in CMECs was assessed. The intracellular level of MMP was determined by TMRM staining. The results in Figure 3A indicate that H/R-exposure significantly reduced intracellular MMP levels, which could be restored by sitagliptin in a dose-dependent manner. Correspondingly, the presence of sitagliptin ameliorated H/R-induced depletion of intracellular ATP in a dose-dependent manner. These results indicate that sitagliptin improved H/R-induced mitochondrial dysfunction.
Figure 3.
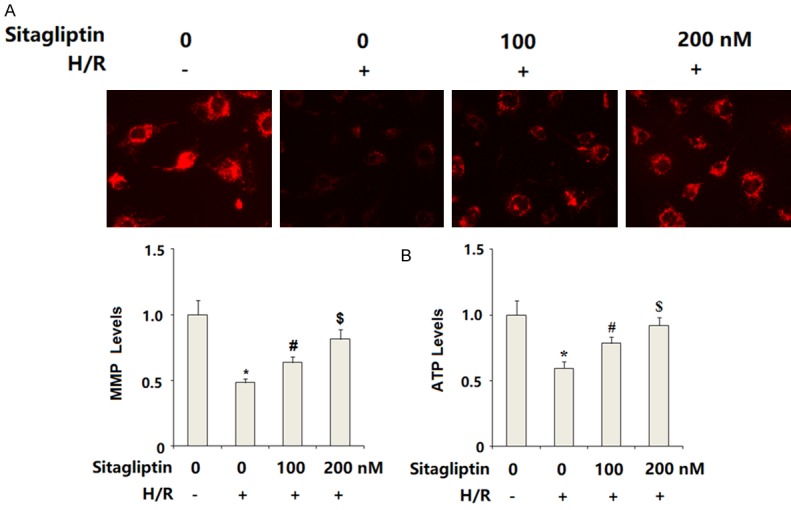
Sitagliptin pretreatment ameliorated hypoxia/reoxygenation (H/R)-induced mitochondrial dysfunction in primary cardiac microvascular endothelial cells (CMECs). Cells were preincubated with 100, 200 nM sitagliptin or DMSO vehicle for 12 h, followed by exposure to hypoxia/reoxygenation (H/R) conditions. A. Mitochondrial membrane potential (MMP) was assayed by the TMRM staining; B. Intracellular levels of ATP were determined (*, P<0.01 vs. vehicle group; #, P<0.01 vs. H/R group; $, P<0.01 vs. H/R+100 nM sitagliptin, n=5-6).
Sitagliptin has displayed a capacity to enhance cell survival. Cell viability was evaluated using the MTT method. The results in Figure 4A indicate that H/R-exposure significantly reduced cell viability of CMECs, which was prevented by the presence of sitagliptin in a dose-dependent manner. Cell death of CMECs was assessed by measuring LDH release. The results shown in Figure 4B indicate that CMECs exposed to H/R exhibited obviously augmented release of LDH into the medium as compared to the control. As expected, sitagliptin treatment significantly ameliorated H/R-exposure-induced LDH release in a dose-dependent manner.
Figure 4.
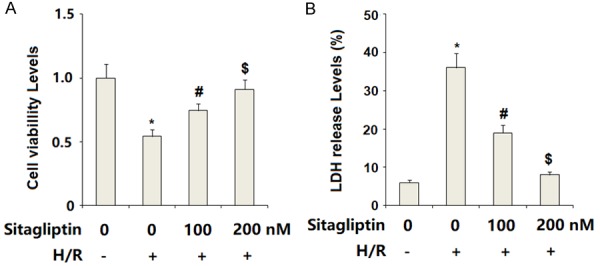
Sitagliptin pretreatment ameliorated hypoxia/reoxygenation (H/R)-induced cell death of primary cardiac microvascular endothelial cells (CMECs). Cells were preincubated with 100, 200 nM sitagliptin or DMSO vehicle for 12 h, followed by exposure to hypoxia/reoxygenation (H/R) conditions. A. Cell viability of CMECs was assessed by MTT assay; B. LDH released from the cytoplasm to the medium was assayed (*, P<0.01 vs. vehicle group; #, P<0.01 vs. H/R group; $, P<0.01 vs. H/R+100 nM sitagliptin, n=5-6).
We then assessed the possible effects of sitagliptin in H/R-exposure-induced inflammatory response. The results of real time PCR analysis in Figure 5A indicate that sitagliptin treatment significantly increased the expression of IL-6, IL-8, and TNF-α, which was prevented by sitagliptin in a dose-dependent manner. Accordingly, the ELISA results in Figure 5B demonstrate that H/R-exposure-induced secretion of IL-6, IL-8, and TNF-α was suppressed by sitagliptin in a dose-dependent manner (Figure 5B). These results suggest that sitagliptin may have an important anti-inflammatory impact against H/R-exposure-induced inflammatory response. Secretion of HMGB1 plays an important role in mediating inflammatory effects. The results of ELISA indicate that H/R-exposure significantly promoted the secretion of HMGB1, which was reduced by the presence of sitagliptin in a dose-dependent manner (Figure 6).
Figure 5.
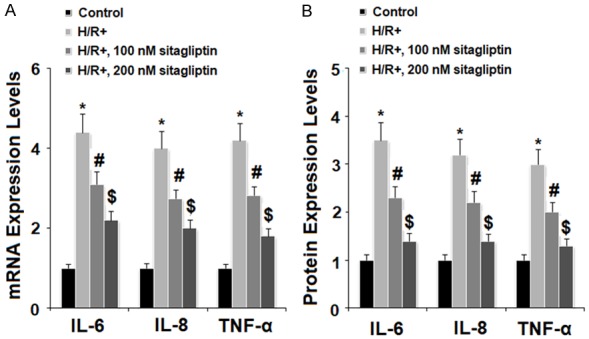
Sitagliptin pretreatment ameliorated hypoxia/reoxygenation (H/R)-induced production of pro-inflammatory cytokines in primary cardiac microvascular endothelial cells (CMECs). Cells were preincubated with 100, 200 nM sitagliptin or DMSO vehicle for 12 h, followed by exposure to hypoxia/reoxygenation (H/R) conditions. A. Expressions of IL-6, IL-8, and TNF-α at the gene levels were measured by real time PCR analysis; B. Secretions of IL-6, IL-8, and TNF-α at the protein levels were determined by ELISA assay (*, P<0.01 vs. vehicle group; #, P<0.01 vs. H/R group; $, P<0.01 vs. H/R+100 nM sitagliptin, n=5-6).
Figure 6.
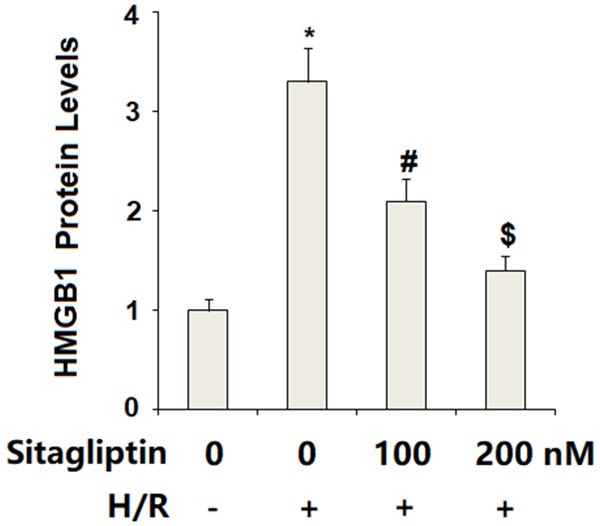
Sitagliptin pretreatment ameliorated hypoxia/reoxygenation (H/R)-induced secretion of high mobility group box 1 (HMGB1) in primary cardiac microvascular endothelial cells (CMECs). Cells were pre-incubated with 100, 200 nM sitagliptin or DMSO vehicle for 12 h, followed by exposure to hypoxia/reoxygenation (H/R) conditions. Secretion of HMGB1 was determined by the ELSA assay (*, P<0.01 vs. vehicle group; #, P<0.01 vs. H/R group; $, P<0.01 vs. H/R+100 nM sitagliptin, n=6).
Finally, we set out to discover the underlying mechanism through which sitagliptin protected CMECs against H/R-exposure-induced insult. Here, we found that H/R-exposure significantly increased phosphorylation of p38, which was inhibited by sitagliptin. However, total levels of p38 remained consistent (Figure 7). We then assessed whether sitagliptin had an influence on activation of NF-κB. Our results indicate that H/R-exposure significantly promoted nuclear translocation of p65, which was prevented by the presence of sitagliptin in a dose-dependent manner (Figure 8A). Importantly, luciferase activity assay results indicate that H/R-exposure caused a significant elevation in NF-κB transcriptional activity, which was successfully suppressed by sitagliptin, thereby verifying the inhibitory effect of sitagliptin on NF-κB activation.
Figure 7.
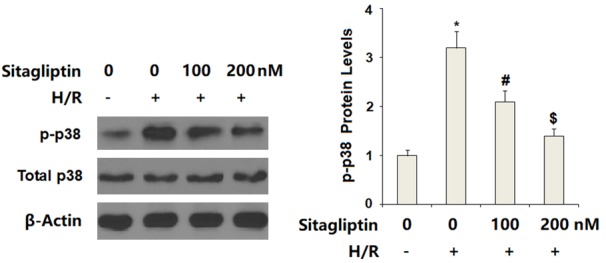
Sitagliptin pretreatment ameliorated hypoxia/reoxygenation (H/R)-induced activation of p38 in primary cardiac microvascular endothelial cells (CMECs). Cells were pre-incubated with 100, 200 nM sitagliptin or DMSO vehicle for 12 h, followed by exposure to hypoxia/reoxygenation (H/R) conditions. Activation of p38 was determined by western blot analysis (*, P<0.01 vs. vehicle group; #, P<0.01 vs. H/R group; $, P<0.01 vs. H/R+100 nM sitagliptin, n=6).
Figure 8.
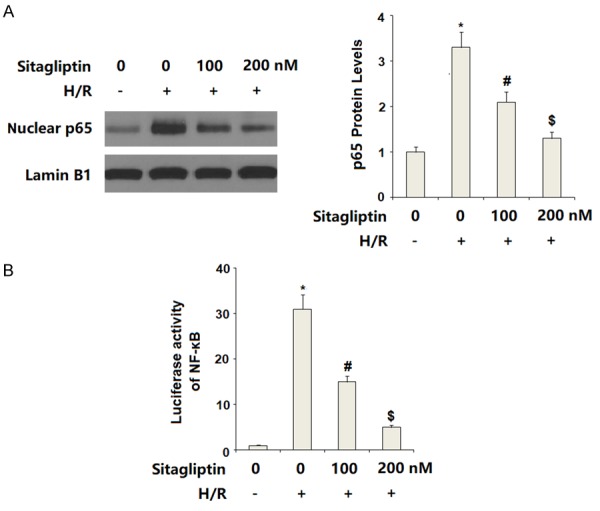
Sitagliptin pretreatment attenuated hypoxia/reoxygenation (H/R)-induced activation of NF-κB signaling in primary cardiac microvascular endothelial cells (CMECs). Cells were pre-incubated with 100, 200 nM sitagliptin or DMSO vehicle for 12 h, followed by exposure to hypoxia/reoxygenation (H/R) conditions. A. Nuclear translocation of p65 was determined by western blot analysis with Lamin B1 as a positive control; B. Luciferase activity of NF-κB (*, P<0.01 vs. vehicle group; #, P<0.01 vs. H/R group; $, P<0.01 vs. H/R+100 nM sitagliptin, n=5-6).
Discussion
Gliptins are a family of medicines developed for the treatment of T2DM. The hypoglycemic effects of gliptins are mediated through inhibition of DPP-4 activity and an increase in glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide 1 (GLP-1) [15]. Increasing evidence has shown that DPP-4 possesses a wide range of capacities to regulate various signaling pathways involved in glucose metabolism, immunity, and inflammatory response [16]. Importantly, DPP-4 has been reported to play a key role in regulating vascular and cardiac function. DPP-4 can be secreted by various cells types including endothelial cells [17]. Long-term and chronic exposure to high glucose concentrations significantly increases the expression of DPP-4 in microvascular endothelial cells [18]. Previous in vitro and in vivo investigations have implied that DPP-4 is a novel link between T2DM and atherosclerosis [19]. DPP-4 inhibitors have displayed their cardiovascular protective capacities in both preclinical studies and clinical trials [20]. In the current study, we aimed to investigate the protective effects and underlying mechanism of the DPP-4 inhibitor sitagliptin in CMECs upon H/R exposure. Firstly, our results indicate that H/R exposure significantly increased the expression of DPP-4. Secondly, the presence of sitagliptin ameliorated H/R exposure-induced oxidative stress and mitochondrial dysfunction. Thirdly, sitagliptin treatment ameliorated H/R-induced cell death. Fourthly, the presence of sitagliptin suppressed the expression of IL-6, IL-8, and TNF-α as well as the secretion of HMGB1. Mechanistically, we found that sitagliptin inhibited activation of p38/ NF-κB signaling.
CMECs, an essential cell type derived from coronary vessels, play an important role in stimulating angiogenesis and carrying nutrients and oxygen to the myocardium. H/R exposure causes dysfunction of CMECs by increasing cellular oxidative stress and subsequent apoptosis [21]. Interestingly, a recent study reported that teneligliptin, another DPP-4 inhibitor, exerts a protective action against H/R-induced CMEC dysfunction via suppression of oxidative stress, improvement of mitochondrial function and amelioration of inflammation and cell death, which is mediated by inhibition of the transcriptional factor Egr-1 [22]. Importantly, activation of MAPK p38 and NF-κB signaling has been shown to play an important role in H/R-induced CMEC damage [23]. In addition to regulating the inflammatory response, NF-κB plays a role in mediating a wide range physiological functions in endothelial cells, including cell proliferation, survival, and differentiation [24]. In the current study, our results show that treatment with sitagliptin inhibited H/R-induced activation of p38 and NF-κB, suggesting that sitagliptin may exert a wide spectrum of protective effects against H/R-induced endothelial dysfunction.
In summary, our results demonstrate that sitagliptin could protect CMECs against H/R-induced injury by mitigating oxidative stress, improving mitochondrial dysfunction, and suppressing inflammation, which is mediated via inactivation of p38/NF-κB signaling. These findings suggest that sitagliptin has the potential to become a promising drug for the treatment of cardiovascular diseases.
Disclosure of conflict of interest
None.
References
- 1.Huang S, Frangogiannis NG. Anti-inflammatory therapies in myocardial infarction: failures, hopes and challenges. Br J Pharmacol. 2018;175:1377–14001. doi: 10.1111/bph.14155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sano M, Minamino T, Toko H, Miyauchi H, Orimo M, Qin Y, Akazawa H, Tateno K, Kayama Y, Harada M, Shimizu I, Asahara T, Hamada H, Tomita S, Molkentin JD, Zou Y, Komuro I. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature. 2007;446:444–448. doi: 10.1038/nature05602. [DOI] [PubMed] [Google Scholar]
- 3.Hausenloy DJ, Yellon DM. Ischemic conditioning and reperfusion injury. Nat Rev Cardiol. 2016;13:193–209. doi: 10.1038/nrcardio.2016.5. [DOI] [PubMed] [Google Scholar]
- 4.Wang Y, Han X, Fu M, Wang J, Song Y, Liu Y, Zhang J, Zhou J, Ge J. Qiliqiangxin attenuates hypoxia-induced injury in primary rat cardiac microvascular endothelial cells via promoting HIF-1α-dependent glycolysis. J Cell Mol Med. 2018;22:2791–2803. doi: 10.1111/jcmm.13572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou Y, Zhang Y, Gao F, Guo F, Wang J, Cai W, Chen Y, Zheng J, Shi G. N-n-butyl haloperidol iodide protects cardiac microvascular endothelial cells from hypoxia/reoxygenation injury by down-regulating Egr-1 expression. Cell Physiol Biochem. 2010;26:839–848. doi: 10.1159/000323993. [DOI] [PubMed] [Google Scholar]
- 6.Barajas-Espinosa A, Basye A, Angelos MG, Chen CA. Modulation of p38 kinase by DUSP4 is important in regulating cardiovascular function under oxidative stress. Free Radic Biol Med. 2015;89:170–81. doi: 10.1016/j.freeradbiomed.2015.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang YQ, Hu SY, Chen YD, Guo MZ, Wang S. Hepatocyte growth factor inhibits hypoxia/reoxygenation-induced activation of xanthine oxidase in endothelial cells through the JAK2 signaling pathway. Int J Mol Med. 2016;38:1055–62. doi: 10.3892/ijmm.2016.2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li D, Wang C, Li N, Zhang L. Propofol selectively inhibits nuclear factor-κB activity by suppressing p38 mitogen-activated protein kinase signaling in human EA. hy926 endothelial cells during intermittent hypoxia/reoxygenation. Mol Med Rep. 2014;9:1460–6. doi: 10.3892/mmr.2014.1946. [DOI] [PubMed] [Google Scholar]
- 9.Campia U, Tesauro M, Di Daniele N, Cardillo C. The vascular endothelin system in obesity and type 2 diabetes: pathophysiology and therapeutic implications. Life Sci. 2014;118:149–55. doi: 10.1016/j.lfs.2014.02.028. [DOI] [PubMed] [Google Scholar]
- 10.Röhrborn D, Wronkowitz N, Eckel J. DPP4 in Diabetes. Front Immunol. 2015;6:386. doi: 10.3389/fimmu.2015.00386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mega C, Teixeira-de-Lemos E, Fernandes R, Reis F. Renoprotective effects of the dipeptidyl peptidase-4 inhibitor sitagliptin: a review in type 2 diabetes. J Diabetes Res. 2017;2017:5164292. doi: 10.1155/2017/5164292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zheng W, Zhou J, Song S, Kong W, Xia W, Chen L, Zeng T. Dipeptidyl-peptidase 4 inhibitor sitagliptin ameliorates hepatic insulin resistance by modulating inflammation and autophagy in ob/ob mice. Int J Endocrinol. 2018;2018:8309723. doi: 10.1155/2018/8309723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jo CH, Kim S, Park JS, Kim GH. Anti-inflammatory action of sitagliptin and linagliptin in doxorubicin nephropathy. Kidney Blood Press Res. 2018;43:987–999. doi: 10.1159/000490688. [DOI] [PubMed] [Google Scholar]
- 14.Liu Y, Hou B, Zhang Y, Fan Y, Peng B, Liu W, Han S, Yin J, He X. Anticonvulsant agent DPP4 inhibitor sitagliptin downregulates CXCR3/RAGE pathway on seizure models. Exp Neurol. 2018;307:90–98. doi: 10.1016/j.expneurol.2018.06.004. [DOI] [PubMed] [Google Scholar]
- 15.Mega C, Teixeira-de-Lemos E, Fernandes R, Reis F. Renoprotective effects of the dipeptidyl peptidase-4 inhibitor sitagliptin: a review in type 2 diabetes. J Diabetes Res. 2017;2017:5164292. doi: 10.1155/2017/5164292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Emson C, Pham TH, Manetz S, Newbold P. Periostin and dipeptidyl peptidase-4: potential biomarkers of interleukin 13 pathway activation in asthma and allergy. Immunol Allergy Clin North Am. 2018;38:611–628. doi: 10.1016/j.iac.2018.06.004. [DOI] [PubMed] [Google Scholar]
- 17.Duan L, Rao X, Xia C, Rajagopalan S, Zhong J. The regulatory role of DPP4 in atherosclerotic disease. Cardiovasc Diabetol. 2017;16:76. doi: 10.1186/s12933-017-0558-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pala L, Mannucci E, Pezzatini A, Ciani S, Sardi J, Raimondi L, Ognibene A, Cappadona A, Vannelli BG, Rotella CM. Dipeptidyl peptidase-IV expression and activity in human glomerular endothelial cells. Biochem Biophys Res Commun. 2003;310:28–31. doi: 10.1016/j.bbrc.2003.08.111. [DOI] [PubMed] [Google Scholar]
- 19.Silva Júnior WS, Godoy-Matos AF, Kraemer-Aguiar LG. Dipeptidyl peptidase 4: a new link between diabetes mellitus and atherosclerosis? Biomed Res Int. 2015;2015:816164. doi: 10.1155/2015/816164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xie W, Song X, Liu Z. Impact of dipeptidyl-peptidase 4 inhibitors on cardiovascular diseases. Vascul Pharmacol. 2018;109:17–26. doi: 10.1016/j.vph.2018.05.010. [DOI] [PubMed] [Google Scholar]
- 21.Xiao X, Xu S, Li L, Mao M, Wang J, Li Y, Wang Z, Ye F, Huang L. The effect of velvet antler proteins on cardiac microvascular endothelial cells challenged with ischemia-hypoxia. Front Pharmacol. 2017;8:601. doi: 10.3389/fphar.2017.00601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Z, Jin X, Yang C, Li Y. Teneligliptin protects against hypoxia/reoxygenation-induced endothelial cell injury. Biomed Pharmacother. 2019;109:468–474. doi: 10.1016/j.biopha.2018.10.016. [DOI] [PubMed] [Google Scholar]
- 23.Guo X, Jiang H, Chen J, Zhang BF, Hu Q, Yang S, Yang J, Zhang J. RP105 ameliorates hypoxia reoxygenation injury in cardiac microvascular endothelial cells by suppressing TLR4/MAPKs/NF-κB signaling. Int J Mol Med. 2018;42:505–513. doi: 10.3892/ijmm.2018.3621. [DOI] [PubMed] [Google Scholar]
- 24.Nakajima H, Mochizuki N. Flow pattern-dependent endothelial cell responses through transcriptional regulation. Cell Cycle. 2017;16:1893–1901. doi: 10.1080/15384101.2017.1364324. [DOI] [PMC free article] [PubMed] [Google Scholar]