


Abstract
Members of the ribonuclease (RNase) III family regulate gene expression by processing dsRNAs. It was previously shown that Escherichia coli (Ec) RNase III recognizes dsRNA with little sequence specificity and the cleavage products are mainly 11 nucleotides (nt) long. It was also shown that the mutation of a glutamate (EcE38) to an alanine promotes generation of siRNA-like products typically 22 nt long. To fully characterize substrate specificity and product size of RNase IIIs, we performed in vitro cleavage of dsRNAs by Ec and Aquifex aeolicus (Aa) enzymes and delineated their products by next-generation sequencing. Surprisingly, we found that both enzymes cleave dsRNA at preferred sites, among which a guanine nucleotide was enriched at a specific position (+3G). Based on sequence and structure analyses, we conclude that RNase IIIs recognize +3G via a conserved glutamine (EcQ165/AaQ161) side chain. Abolishing this interaction by mutating the glutamine to an alanine eliminates the observed +3G preference. Furthermore, we identified a second glutamate (EcE65/AaE64), which, when mutated to alanine, also enhances the production of siRNA-like products. Based on these findings, we created a bacterial Dicer that is ideally suited for producing heterogeneous siRNA cocktails to be used in gene silencing studies.
INTRODUCTION
Found in all kingdoms of life, the ribonuclease III (RNase III) family endoribonucleases regulate gene expression by specifically processing double-stranded (ds) RNAs (1,2). The founding member of the family, Escherichia coli RNase III, was discovered in 1968 (3). Bacterial RNase III plays a major role in ribosomal RNA (rRNA) processing (4). It is also involved in post-transcriptional gene expression control (5) and defense against viral infection (6,7). Knocking out or mutating RNase III in E. coli (Ec) or Bacillus subtilis induces global gene expression changes (8,9). The production of a set of Staphylococcus aureus small RNAs is dependent on RNase III (10,11).
Other representative members of the RNase III family include yeast Rnt1p, human Drosha and human Dicer. Like bacterial RNase III, yeast Rnt1p also functions in the processing of rRNA (12). In addition, it is involved in the processing of small non-coding RNA (13) and the degradation of messenger RNA (mRNA) (14). The two human RNase III enzymes, Drosha and Dicer, are more specialized. Dicer plays a role in the production of small interfering RNA (siRNA). Along with Drosha, it is also involved in the biogenesis of microRNA (miRNA), a master gene regulator in eukaryotic cells (15–20). The RNase III nuclease domain (RIIID) dimerizes, which creates a catalytic valley that accommodates a dsRNA substrate (21,22). Bacterial RNase III and yeast Rnt1p have a single RIIID; these enzymes function as homodimers (23,24). Human Dicer and Drosha have two RIIIDs in the same molecule; these enzymes function as monomers (19,20). Both RNase III and Dicer can digest long dsRNA substrates, for which two distinct mechanisms have been previously described. One mechanism features an end-in strategy, in which the enzyme recognizes the termini of a long dsRNA, cleaves both RNA strands and removes a small RNA duplex upon product release. Both enzymes can adopt this end-in mechanism and successively remove small RNA duplexes from the dsRNA termini (5,20,24). Whereas RNase III typically measures 11 nucleotides (nt) from the termini for cleavage (Figure 1A), Dicer typically measures 22 nt (Figure 1B). A second mechanism features an inside-out scheme, in which multiple RIIID dimers bind consecutively to a long dsRNA and cleave the substrate simultaneously, producing siRNA-like small RNA duplexes, the length of which is the distance between consecutive active centers. This inside-out mechanism has been observed for EcRNase III (Figure 1A) under special conditions (25) and yeast Kluyveromyces polysporus Dcr1 under physiological conditions (Figure 1C) (26).
Figure 1.
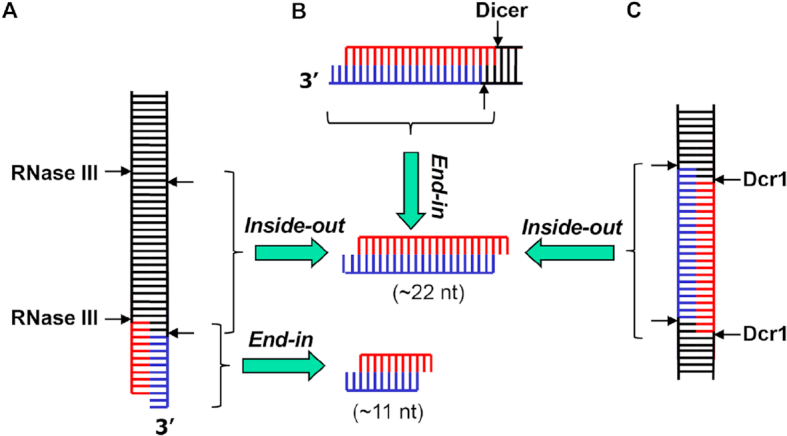
Mechanisms of long dsRNA digestion by RNase III enzymes. (A) A bacterial RNase III dimer recognizes the dsRNA termini, especially those featuring a 2-nt 3′ overhang, cleaves both strands and produces a short RNA duplex of 11 nt in each strand. Under special conditions, two RNase III dimers bind to and cleave dsRNA in a cooperative manner, which produces an RNA duplex of 22 nt in each strand. (B) Human Dicer recognizes the dsRNA termini with a 2-nt 3′ overhang, cleaves both strands and produces an RNA duplex of 22 nt in each strand. (C) Two dimers of yeast Dcr1 cooperatively bind to and cleave a long dsRNA, producing RNA duplexes containing ∼22 nt in each strand.
Given the critical role of RNase III in dsRNA processing, the mechanism of RNase III cleavage is of great interest to the field of RNA biology. Structurally, bacterial RNase III (∼200 amino acid residues) is much simpler than human Dicer (∼2,000 residues). Therefore, bacterial RNase III has served as the principal model system for elucidating the mechanistic details of basic RNase III activity. It consists of an N-terminal RIIID, a flexible linker and a dsRNA-binding domain (dsRBD). Since the discovery of EcRNase III (3), significant progress has been made in studies of the genetics, function, structure and mechanism of bacterial enzymes (1,2). Crystal structures of Aquifex aeolicus (Aa) RNase III play critical roles in describing substrate recognition, scissile bond selection, two-Mg2+-ion catalysis, phosphoryl transfer and product release (2,24,27,28). The AaRNase III structures also provide a high-resolution framework for further basic and translational research. It was found that the E38A mutant of EcRNase III (EcE38A) promotes the inside-out cleavage of a long dsRNA (Figure 1A), leading to the discovery of an economical reagent for the preparation of siRNA cocktails for gene silencing studies (29).
Previously, it was reported that EcRNase III recognizes long dsRNA with little specificity (30). Taking advantage of next-generation sequencing (NGS), we investigated Ec and AaRNase III cleavage events on a large scale using two long dsRNA substrates derived from firefly luciferase (FF-luc) and E. coli maltose-binding protein (MBP), respectively. To our surprise, bacterial RNase IIIs does not cleave long dsRNAs randomly, but mostly at preferred sites. Both Ec and AaRNase III recognize a guanine base near the cleavage site by a highly conserved glutamine side chain (Q165 and Q161 in Ec and AaRNase III, respectively). This interaction contributes to sequence specificity during substrate recognition and cleavage site selection. Using site-directed EcQ165A and AaQ161A mutants, we eliminated this preference among cleavage sites. Guided by the crystal structure (24), we also identified a position where this single mutant of RNase III (E64A and E65A in Aa and EcRNase III, respectively) could also enhance the inside-out cleavage of dsRNA. Based on these findings, we generated a E38A/E65A/Q165A triple mutant of EcRNase III (EcEEQ) that functions as a bacterial Dicer and produces random, heterogeneous 22-nt-siRNA cocktails that are optimal for gene silencing studies.
MATERIALS AND METHODS
Expression vector construction of EcRNase III and mutants
The plasmid expression vector that was used to produce wild-type EcRNase III was constructed by overlap extension PCR (31) and Gateway recombinational cloning. Plasmid pASC, which carries the wild-type EcRNase III gene, was used as the template for PCR. Two oligodeoxyribonucleotides were used as PCR primers: (i) 5′-GGGGACAACTTTGTACAAAAAAGTTGTGGAGAACCTGTACTTCCAGGGTATGAACCCCATCGTAATTA-3′, (ii) 5′-GGGGACAACTTTGTACAAGAAAGTTGCATTATCATTCCAGCTCCAGTTTTTTC-3′. The PCR amplicon was recombined into the entry vector pDONR221 (Invitrogen, Carlsbad, CA), and then the nucleotide sequence of the entire ORF was confirmed. The ORF encoding the mutant was then recombined into pDEST-His-MBP (32) to create the expression vector. All mutants (E38A, E65A, Q165A, E38A/Q165A, E65A/Q165A and E38A/E65A/Q165A) were constructed by QuikChange Lightning site-directed mutagenesis (Agilent Technologies, Santa Clara, CA).
Expression and purification of EcRNase III proteins
The His6-MBP tagged wild-type EcRNase III and its mutants were expressed in E. coli BL21(DE3) CodonPlus-RIL cells (ThermoFisher Scientific, Waltham, MA). The cells were cultivated in Luria-Bertani (LB) broth containing 100 μg ml−1 ampicillin, 30 μg ml−1 chloramphenicol and 0.2% glucose to mid-log phase at 37°C, induced by the addition of isopropyl β-d-1-thiogalactopyranoside (IPTG) to a final concentration of 1 mM, and shaken overnight at 18°C. Harvested cultures were lysed by sonication and, following removal of the insoluble cell debris by centrifugation, the supernatant was applied to a HisTrap FF column (GE Healthcare Life Sciences, Pittsburgh, PA). The His6-MBP tag was removed by incubating overnight with 0.5 mg ml−1 S219V TEV protease (33) at 4°C and passing through a second HisTrap FF column. Proteins were further purified with a HiLoad (26/60) Superdex 200 gel filtration column (GE Healthcare Life Sciences) and the quality was analyzed by sodium dodecyl sulfate-polyacrylamide electrophoresis (SDS-PAGE) and electrospray mass spectrometry (LC-ESMS). The final products were concentrated to 10 mg ml−1 and stored at −80°C.
Expression vector construction of AaRNase III and mutants
Protein expression vectors were generated by Gateway™ recombinational cloning as previously described (34). Briefly, entry clones carried the wild-type RNase III gene or its mutants in the pDONR201 (Invitrogen, Carlsbad, CA) backbone. All mutations were verified by DNA sequencing using primers PE367 (5′-TCG CGT TAA CGC TAG CAT GGA TCT C-3′) and PE240 (5′-GTA ACA TCA GAG ATT TTG AGA CAC-3′). Entry clones were recombined by Gateway LR reaction with pET-DEST42 (Invitrogen, Carlsbad, CA) to produce expression vectors.
An entry clone encoding the E110Q mutant of AaRNase III was constructed for experiments related to this work by overlap extension PCR (31) using four primers: PE1426, 5′-GGG GAC AAG TTT GTA CAA AAA AGC AGG CTT TAA GAA GGA GAT ATA CAT ATG AAA ATG TTG GAG CAA CTT G-3′; PE1427, 5′-CTG CCC AAA GAG CTT GAA ATA CGT CTC CTA-3′; PE1428, 5′-TAG GAG ACG TAT TTC AAG CTC TTT GGG CAG-3′; PE1429, 5′-GGG GAC CAC TTT GTA CAA GAA AGC TGG GTT ATT ATT CTG ATT CCT CCA GTA ATT T-3′. First, two separate PCRs with primer pairs, PE1426-PE1427 and PE1428-PE1429, introduced an E110Q mutation and removed the C-terminal hexahistidine tag from an antecedent E110K mutant vector. Then, the products from the first two PCRs were used in a third reaction with primers PE1426 and PE1429 to generate full-length product for recombination into pDONR201 via the Gateway BP reaction, which produced entry vector pBA1665. The resultant E110Q entry vector was reverted to the wild-type by site-directed mutagenesis using primers PE2679 (5′-GGA GAC GTA TTT GAA GCT CTT TGG GCA GCG G-3′) and PE2680 (5′-CCG CTG CCC AAA GAG CTT CAA ATA CGT CTC C-3′) to generate entry clone pBA2518, which was subsequently recombined into pET-DEST42 to produce pBA2520, the expression vector for untagged, wild-type AaRNase III.
The E37A mutant was produced by site-directed mutagenesis of the wild-type entry clone pBA2518 with primers PE2677 (5′-CTC AAA AAA AGA ACA CTA CGC AAC TCT TGA GTT CCT CGG C-3′) and PE2678 (5′-GCC GAG GAA CTC AAG AGT TGC GTA GTG TTC TTT TTT TGA G-3′) to produce entry clone pBA2519, which was recombined with pET-DEST42 to generate the expression vector pBA2521.
The wild-type (pBA2518) and E37A mutant (pBA2519) entry clones were used as templates in QuikChange Lightning site-directed mutagenesis reactions with primers A.a161-A-F (5′-ATA CTT CAG GAG ATC ACT GCA AAA CGA TGG AAG GAA AGA-3′) and A.a161-A-R (5′-TCT TTC CTT CCA TCG TTT TGC AGT GAT CTC CTG AAG TAT-3′) to produce an entry clone encoding the Q161A mutation alone (pDN2749) and one encoding both E37A and Q161A (pDN2750). pDN2749 and pDN2750 were recombined into pET-DEST42 to generate the Q161A (pDN2755) and E37A/Q161A (pDN2754) expression vector.
Single, double and triple mutants were created by adding the E64A mutation via QuikChange Lightning to the wild-type (pBA2518), Q161A (pDN2749) and E37A/Q161A (pDN2750) entry vectors using primers PE3025 (5′-AGG GAG ATA AAA AGC CTG CCC TTT TGT TGG GGG AA-3′) and PE3026 (5′-TTC CCC CAA CAA AAG GGC AGG CTT TTT ATC TCC CT-3′). Following sequence verification, entry clones pDN2954 (E64A), pDN2955 (E64A/Q161A) and pDN3061 (E37A/E64A/Q161A) were recombined with pET-DEST42 to generate expression vectors pDN2961, pDN2962 and pDN3063, respectively.
Expression and purification of AaRNase III proteins
The AaRNase III protein was overproduced in E. coli and purified as described (28). All expression vectors were transformed into E. coli strain BL21(DE3)-CodonPlus-RIL (ThermoFisher Scientific). Cells were grown to mid-log phase at 37°C in LB broth containing 100 μg ml−1 ampicillin, 30 μg ml−1 chloramphenicol and 0.2% glucose. Overproduction of the recombinant protein was induced with isopropyl β-D-1-thiogalactopyranoside (IPTG) at a final concentration of 1 mM for 4 h at 30°C. The cells were pelleted by centrifugation and stored at −80°C.
All procedures were performed at 4–8°C unless otherwise stated. E. coli cell paste from 6 L of culture, expressing AaRNase III, was suspended in ice-cold 50 mM sodium phosphate (pH 8), 25 mM NaCl buffer (buffer A) containing 1 mM benzamidine and Complete™ EDTA-free protease inhibitor cocktail (Roche Diagnostics Corporation, Indianapolis, IN), and lysed with an APV-1000 homogenizer (SPX Corporation, Charlotte, NC) at 10,000 psi. The homogenate was centrifuged at 30,000 × g for 30 min and the supernatant was heat-treated at 80°C for 20 min. Following a 30-min incubation on ice, insoluble material was removed by centrifugation and the supernatant was filtered through a 0.2-μm polyethersulphone membrane. The sample was applied to a HiPrep 16/10 SP FF column (GE Healthcare Life Sciences) equilibrated in buffer A. The column was washed to baseline with buffer A and eluted with a linear gradient of NaCl to 1 M. Fractions containing recombinant protein were pooled and concentrated using an Ultracel® 10 kDa ultrafiltration disc (EMD Millipore Corporation, Billerica, MA). The concentrate was fractionated on a HiLoad 26/60 Superdex 75 pg column (GE Healthcare Life Sciences) equilibrated in 25 mM Tris–HCl (pH 7.2), 600 mM NaCl buffer. Peak fractions containing AaRNase III were pooled and diluted with 50 mM Tris–HCl (pH 7.5) buffer to reduce the NaCl concentration to 200 mM. The sample was applied to a 15 ml AGPoly(I)·Poly(C)™ Type 6 column (Amersham Biosciences/GE Healthcare Life Sciences) equilibrated in 50 mM Tris–HCl (pH 7.5), 200 mM NaCl buffer. The column was washed to baseline with equilibration buffer and eluted with a linear gradient of NaCl to 1 M. Fractions containing recombinant protein were pooled, concentrated and subjected to a second round of size exclusion chromatography as described above. The final product was diluted with 25 mM Tris–HCl (pH 7.2) buffer to reduce the NaCl concentration to 300 mM and concentrated using an Ultracel® 10 kDa ultrafiltration disc. Concentrations were determined spectrophotometrically using molar extinction coefficients derived from the ExPASy ProtParam tool (35). Aliquots were flash-frozen in liquid nitrogen and stored at −80°C. Purity was judged to be >95% by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The molecular weights were confirmed by LC-ESMS.
Cleavage assays and electrophoresis
As templates for in vitro transcription, pshicheck2 plasmid for Firefly luciferase and gateway pDest-HisMBP plasmid for MBP were PCRed by (i) 5′-TAATACGACTCACTATAGGGAGAATGGCCGATGCTAAGAACATT-3′, (ii) 5′-AATTAACCCTCACTAAAGGGAGATTACACGGCGATCTTGCCGCC-3′, (iii) 5′-AATTAACCCTCACTAAAGGGAGATTACACGGCGATCTTGCCGCC-3′, (iv) 5′-AATTAACCCTCACTAAAGGGAGATTTTTTGTACAAACTTGTGA-3′. The PCR amplicon from (i) and (ii), (iii) and (iv) was purified, respectively. In vitro transcription was carried out by using Maxi script (ThermoFisher Scientific) as per the manufacturer’s instructions.
RNase III cleavage reactions were done using 4 mg of purified His-tagged protein and 500 ng of either FF-luc or MBP dsRNA as the substrate in New England Biolabs Buffer 2 [10 mM Tris–HCl (pH 7.9), 10 mM MgCl2, 50 mM NaCl and 1 mM dithiothreitol] and incubated for 30 min at 37°C (EcRNase III) or 30 min at 60°C (AaRNase III). For protein titration reactions, 4 mg of purified His-tagged protein was used for the highest amount and serially diluted. Reactions were stopped by the addition of DNA loading dye [20 mM EDTA (pH 8.0), 1.25% SDS and 10% glycerol] and incubated for 10 min at 65°C before loading onto a 20% polyacrylamide/TBE gel (Thermo-Fisher Scientific). After electrophoresis, gels were stained with EtBr and imaged using a ChemiDoc Touch imaging system (Bio-Rad, Hurcules, CA).
Library preparation and next generation sequencing
Prior to library preparation, RNAs from the cleavage assay were extracted and quantified using a Qubit 2.0 Fluorometer (ThermoFisher Scientific). Total RNA was also analyzed with an Agilent Bioanalyzer 2100 (Agilent Technologies) to ensure appropriate RNA quality. Sequencing libraries were prepared using an Illumina® TruSeq® RNA sample preparation kit according to the manufacturer’s instructions. The 3′ and 5′ RNA adapters were ligated to the cleavage products sequentially followed by reverse transcription and PCR amplification. The cDNA constructs were recovered from TBE gel electrophoresis and further purified by ethanol precipitation. The cDNA libraries were pooled and sequenced using an Illumina MiSeq Reagent Kit v3. Sequencing resulted in approximately 500,000 reads per sample. For each library, we used 100 ng of RNA as a starting material.
Programming and analysis of next-generation sequencing data
We removed the 3′ adaptor sequences by customized scripts. Reads without the 3′ adaptor sequences or shorter than 6 nt were discarded. Using Bowtie (version 1.1.1) (36) with the command ‘bowtie -v 0 –best –sam,’ we aligned the resulting reads to the coding sequence of either FF-luc or MBP without allowing mismatch. The resulting SAM files were converted to BAM files, sorted, indexed by SAMtools (37) and eventually visualized with IGV (38). Length distribution analysis of mapped reads and cleavage sites analysis were carried out subsequently by customized scripts. The consensuses motif of preferred cleavage sites or mappable reads was identified by Weblogo (39).
RESULTS
Bacterial RNase III cleaves long dsRNA at preferred sites
To investigate how bacterial RNase III cleaves dsRNAs, we purified Ec and AaRNase III proteins and incubated each with the dsRNA substrate corresponding to the FF-luc coding sequence (Figure 2A). Consistent with a previous report (29), both enzymes cleaved the 1676-base pair (bp) FF-luc dsRNA into small RNA fragments, ranging from 10 to 15 nt in length (Figure 2B).
Figure 2.

Bacterial RNase III cleaves long dsRNA at preferred sites. (A) Schematic representation of the in vitro cleavage experiment. (B) dsRNA of FF-luc sequence was cleaved by either Aquifex aeolicus or Escherichia coli RNase III. The cleavage products were separated on 20% polyacrylamide non-denaturing (native) gels and detected by staining. Synthetic single-stranded RNAs with certain length were used as markers. After next-generation sequencing, reads of cleavage products were mapped back to (C) FF-luc or (D) MBP, respectively. Coverage plots are presented.
To further characterize these cleavage events, we converted the library of digestion products into DNA, cloned the fragments and then subjected them to NGS. Four independent cleavage and subsequent NGS experiments were performed with EcRNase III and three were performed with AaRNase III, resulting in 1 026 111 and 509 780 reads, respectively. About 90% of these reads (869,360 and 486,711 for Ec and AaRNase III, respectively) could be mapped without mismatch to either the sense or antisense sequence of the FF-luc dsRNA substrate. These small RNA reads are in sufficient abundance to provide an extensive coverage over the relatively small (1676-bp) dsRNA substrate (∼300–500 fold), demonstrating the robustness of this approach for analyzing in vitro cleavage products of RNase III.
Although the cleavage products cover the entire substrate sequence, the distribution of their abundance is far from even. Whereas most of the small RNAs were detected fewer than 100 times, certain reads were found over 10,000 times. Mapping all of these reads back to the substrate, we identified several ‘hot spots’ along the FF-luc sequence, each of which consists of one or several highly abundant read(s) (Figure 2C). Of note, this coverage distribution pattern is highly consistent among replicates but slightly different between EcRNase III- and AaRNase III-treated samples, indicating that the observed ‘hot spots’ are not a trivial result of sequencing bias.
Similar results were obtained when we replaced the dsRNA substrate with the coding sequence of MBP (Figure 2D), demonstrating that the bacterial RNase III does not cleave dsRNA randomly but rather at preferred sites. More importantly, these results suggest that the underlying mechanism for the formation of these hot spots might involve features that are intrinsic to the long dsRNA sequence per se.
RNase III recognizes +3G in selecting cleavage sites
Given that the substrates used in our assay are dsRNAs without internal structure, we reason that the preferred cleavage sites might contain conserved sequence motifs that are recognized by bacterial RNase III. To test this directly, we first sought to identify hotspots of AaRNase III cleavage sites, which can be defined from either the 5′ or 3′ end of the cleavage products. Based on a total of 973,422 counts of ends (2 × 486,711 reads), 1,674 cleavage sits were identified along the 1,676-bp substrate, indicating that RNase III cleavage was extensive and happened at nearly every possible position. Nonetheless, the frequency varied dramatically. While many cleavage sites were supported by only few reads, several cleavage sites generated over 10,000 corresponding reads (Figure 3A). Using five times the average read as a threshold, we identified 141 highly preferred cleavage sites (Figure 3A). Overlapping nucleotide sequences around these cleavage sites of FF-luc dsRNA revealed that nucleotide G is enriched in the +3 position (+3G) and C is enriched to a lesser extent in the -6 position (-6C), relative to the position of cleavage (Figure 3B). The same analysis of MBP dsRNA cleavage site hotspots (102 unique cleavage site sequences) also generated the +3G/-6C motif (Figure 3B), demonstrating the intrinsic nature of this identified motif.
Figure 3.
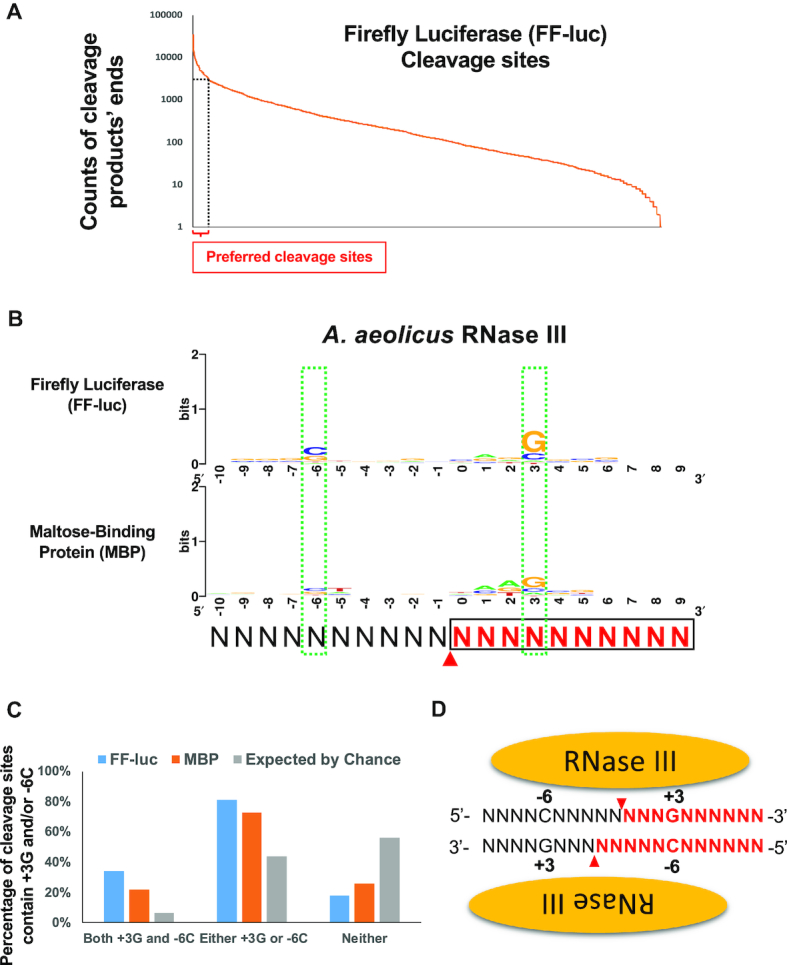
RNase III recognizes +3G in cleavage site selection. (A) Cleavage sites were inferred by the ends of cleavage products. Cleavage sites along FF-luc substrate were sorted by the counts of cleavage products’ ends and plotted. Cleavage sites supported by a count higher than 2,907, which is 5× the average (581.3), were defined as preferred cleavage sites (hotspots) and indicated by the dashed lines. (B) Sequence logos surrounding preferred AaRNase III cleavage sites on FF-luc and MBP are presented. Red arrowheads indicate the cleavage site (between positions 0 and -1), which is inferred from the end of cleavage products. Cleavage product upstream of cleavage site is in black, whereas cleavage products downstream of cleavage site are in red and boxed. Two green dashed-line boxes indicate two consensus nucleotides +3G and -6C. (C) Percentage of cleavage sites that contain +3G and/or -6C is higher than that expected by chance. (D) Schematic illustration of the RNase III:dsRNA complex. The +3G and -6C are symmetric relative to the cleavage sites that are indicated by red arrowheads. Cleavage products on one side of cleavage site are in black, whereas cleavage products on the other side of cleavage site is in red.
Consistent with the identified +3G/-6C motif, further analysis revealed that around 30% of all preferred cleavage sites on FF-luc and MBP contain both the +3G and -6C, which is significantly higher than the percentage expected by chance (1/16 or 6.25%, Figure 3C). However, this still leaves more than half of the preferred cleavage sites unexplained. Since RNase III cleaves dsRNA as a dimer and the cleavage sites on the sense and antisense strands of dsRNA are 2 bp apart, the -6C on the sense strand is equivalent to the +3G on the antisense strand (Figure 3D). This led us to hypothesize that RNase III only recognizes the +3G. The enrichment of -6C is an indirect result of the other RNase III subunit recognizing the +3G on the antisense strand (Figure 3D). In such a scenario, a cleavage site containing either a +3G or a -6C can be recognized by the RNase III dimer. In support of this idea, we found that over 75% of those preferred cleavage sites on FF-luc and MBP have either +3G or -6C and >25% of these have neither (Figure 3C).
Parallel analyses of EcRNase III cleavage products identified 120 and 78 preferred cleavage sites on FF-luc and MBP dsRNA, respectively. Consistent with the results obtained with AaRNase III, a guanine nucleotide is enriched in the +3 position (+3G) (Supplementary Figure S1A) and over 70% of those preferred cleavage sites contain either a +3G or a -6C (Supplementary Figure S1B). These results strongly suggest that the preference of +3G in cleavage site selection is a conserved mechanism for bacterial RNase IIIs. Interestingly, a SS (S stands for G or C) motif is also enriched at the -1/-2 position among the preferred cleavage sites of EcRNase III (Supplementary Figure S1A), indicating that EcRNase III might recognize these two nucleotides in addition to the +3G during target site selection.
Taken together, these results indicate that preferred cleavage sites (hot spots) are a result of sequence-specific enzyme–substrate interaction. The sites that can be recognized by either subunit of the RNase III dimer are cleaved at a higher frequency.
A conserved glutamine side chain in dsRBD recognizes the +3G near the cleavage site
Based on the crystal structure of AaRNase III in complex with dsRNA, the first α-helix of the dsRBD is an important determinant for substrate selection. Residues T154, Q157, E158 and Q161 form hydrogen bonds with 2′-hydroxyl groups for specific recognition of RNA substrates (24). In addition, the Q161 side chain, which is conserved in bacterial RNase IIIs (Supplementary Figure S2A), forms two base-specific hydrogen bonds with the +3G nucleotide, indicating that Q161 is responsible for sequence-specific substrate recognition (Figure 4A). Unlike +3G, the -6C base does not form any hydrogen bonds with the protein (24).
Figure 4.
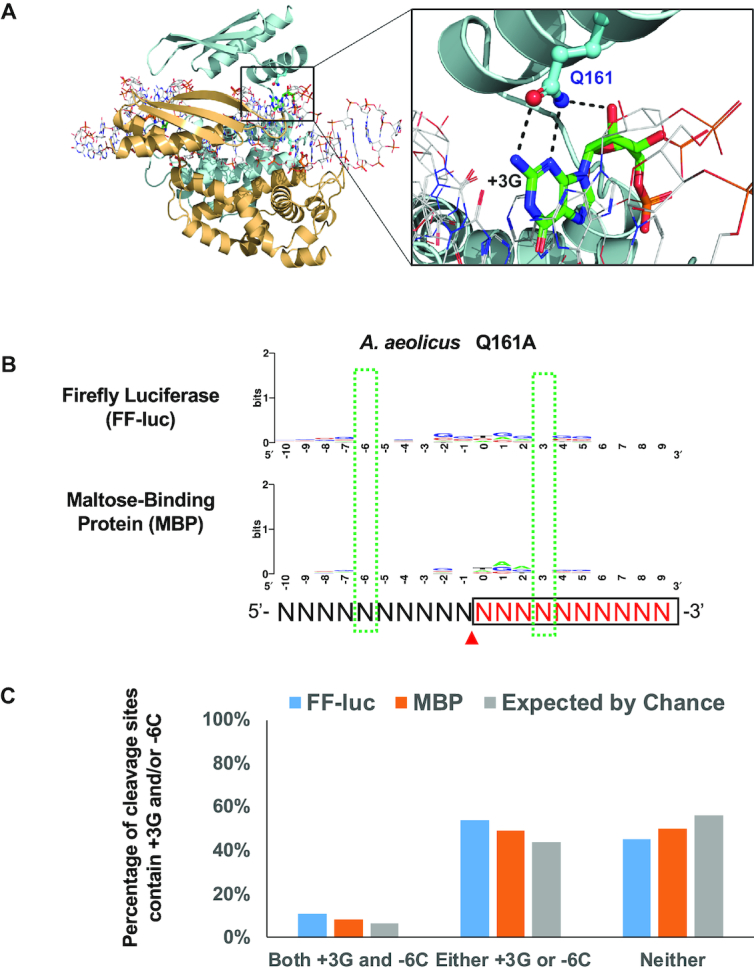
Residue Q161 of AaRNase III recognizes the +3G near the cleavage site. (A) On the left: schematic illustration of the crystal structure of AaRNase III in complex with dsRNA (PDB entry: 2EZ6); On the right: residue Q161 recognizes the +3G by forming two base-specific hydrogen bonds and one hydrogen bond with the 2′-hydroxyl group. (B) The consensus sequence of cleavage site is abolished when residue 161 was mutated from Q to A. Sequence logos were created as illustrated in Figure 3. (C) Percentage of highly preferable cleavage sites of the AaRNase III Q161A mutant, with or without +3G and/or -6C, is similar to that expected by chance.
To test the functional role of Q161 directly, we created the Q161A mutant of AaRNase III (AaQ161A) and performed the cleavage and subsequent NGS analysis. As expected, the consensus sequences surrounding cleavage sites are completely abolished for the sequences of both FF-luc and MBP (Figure 4B). In addition, the percentage of cleavage sites containing +3G and/or -6C dropped to a level close to that expected by chance (Figure 4C). Together, these results indicate that the +3G preference observed in wild-type AaRNase III cleavage products is not due to a bias introduced during cloning or sequencing. Instead, it is caused by the specific interaction between residue Q161 of AaRNase III and dsRNA substrate.
While a wealth of structural information is available for AaRNase III (1,2), 3D structure of EcRNase III remains to be determined. Nonetheless, given that the Q161 side chain is conserved in bacterial RNase IIIs (Supplementary Figure S2A), we hypothesize that the corresponding residue Q165 is responsible for the +3G preference in EcRNase III cleavage site selection. Indeed, mutating Q165 to an alanine (Q165A) completely obliterated the +3G preference among preferred cleavage sites (Supplementary Figure S3). Together, these results demonstrate that the specific interaction between the conserved glutamine side chain and dsRNA substrate contributes to the sequence preference of +3G during cleavage site selection.
Bacterial RNase III cleaves dsRNA into small RNA duplexes with distinct patterns of length distribution
Bacterial RNase III cleaves both natural and synthetic dsRNA into small duplex products ranging from 10 to 18 bp in length (5,40). Consistently, our NGS results show that the Ec and AaRNase IIIs cleave dsRNA into small RNA duplexes within the range of 6–17 and 6–19 bp, respectively (Figure 5A). The dominant length of EcRNase III products is 12 nt in a single strand, followed by 9, 11 and 13 nt, whereas that of AaRNase III are mainly 11 nt. Interestingly, the pattern of product length distribution is species (E. coli or A. aeolicus) dependent but sequence (FF-luc or MBP) independent. Whereas patterns derived from different sequences by the same RNase III are similar, patterns from the same sequence by different RNase IIIs are distinct (Figure 5A), which appears to be determined by certain intrinsic features of RNase III structures. Of note, the cleavage reactions were performed at different temperatures: 37°C for EcRNase III and 60°C for AaRNase III because AaRNase III is inactive at 37°C (Supplementary Figure S4). Although unlikely, we cannot exclude the possibility that the distinctions in cleavage pattern between EcRNase III and AaRNase III are partially due to the difference in cleavage reaction temperature.
Figure 5.

Bacterial RNase III cleaves dsRNA into small RNAs with distinct patterns of length distribution. (A) The length of RNase III cleavage products was measured by the next-generation sequencing analysis. The percentage of reads with certain length to total number of reads was plotted. (B) Cleavage products of FF-luc dsRNA were mapped back to the reference. Coverage plots of reads with certain length are presented. (C) A detailed look of one hotspot. The height of the bar represents the number of cleavage products covering each position. The reference sequence is listed below. Two potential cleavages are indicated: red arrows point to the cleavage sites; +3G (yellow) and -6C (blue) corresponding to each cleavage site are labeled.
The NGS analysis provides detailed insights into the mechanism of cleavage site selection by bacterial RNase III. Mapping cleavage products with various lengths to the substrates yields distinct landscapes of hot spots (Figure 5B), indicating that products with variable lengths were generated by distinctive cleavage events. Details at one ‘hot spot’ of the FF-luc dsRNA are illustrated in Figure 5C. The AaRNase III cleavage of this region created both 11- and 12-nt products. When these two types of dominant products are aligned at their 3′ ends, the 12-nt products exhibit one more nt beyond the 5′ ends of 11-nt products, showing that the enzyme performs either a major cleavage to generate the 11-mer or a minor cleavage to produce the 12-mer. The reference sequence indicates that both cleavage sites are selected according to the +3G/-6C rule, enabling the enzyme to perform cleavages at these two adjacent sites (Figure 5C).
The rate of product release affects the length distribution of RNase III cleavage products
To understand the mechanism that governs the length distribution of RNase III products, we sought insights using site-directed mutagenesis coupled with NGS analysis. The EcE38A mutant has been shown to slow down the release of cleavage product, induce the inside-out cleavage mechanism and promote the production of 23-nt products (29). As shown in Figure 6, the E38 side chain points toward the backbones of dsRNA (24). Therefore, the E38A mutation removes a repulsive protein–RNA interaction and thereby stabilizes the protein:dsRNA complex. Similarly, the E65 side chain also points toward the dsRNA backbone (Figure 6A). We predict that the E65A mutation should also stabilize the protein:dsRNA complex and promote the production of longer RNA products. Unlike E38 and E65, side chain Q165 forms three hydrogen bonds to the +3G (Figure 4A). The Q165A mutation, while eliminating sequence preference (Supplementary Figure S3), should destabilize the protein:dsRNA complex and thereby promote the production of shorter RNA products. Our working hypothesis is that the balance between the stabilization and destabilization effects on the protein:dsRNA complex dictates the product length distribution.
Figure 6.

Length distribution of cleavage products is affected by the rate of product release. (A) On the left: cartoon illustration of the crystal structure of the AaRNase III dimer in complex with dsRNA (PDB entry 2EZ6); on the right: zoom-in window shows relative positioning of side chains E37, E64 and Q161, of which the counterparts in EcRNase III are E38, E65 and Q165, respectively. (B–G) FF-luc and MBP dsRNAs were cleaved by various RNase III mutants. The length of products was compared to that of the wild-type. Fold changes in log scale are plotted against product lengths for EcE38A (B), EcE65A (C), EcQ165A (D), AaE37A (E), AaE64A (F) and AaQ161A (G).
We expressed and purified the EcE38A, EcE65A and EcQ165A mutant proteins. Using the FF-luc and MBP dsRNAs as substrates, the cleavage products were sequenced and analyzed. As expected, the percentage of 22-mer product produced by either EcE38A or EcE65A is significantly increased when compared to the wild-type (Figure 6B and C), whereas the percentage of 22-mer product by EcQ165A is either slightly increased for the MBP or decreased for the FF-luc dsRNA (Figure 6D). Interestingly, the relative abundances of all cleavage products changed, with a turning point around the 12-nt product. Furthermore, the degree of changes is approximately proportional to the product lengths. The corresponding mutants of the AaRNase III produced similar results (Figure 6E, F and G). Taken together, these results support a RNase III cleavage model in which increased protein:substrate interaction (slower product release) promotes the production of longer cleavage products, whereas decreased protein:substrate interaction (faster product release) promotes the production of shorter cleavage products.
The E38A/E65A/Q165A mutant of EcRNase III processes long dsRNA into a heterogeneous mixture of siRNAs of 22 nt in length
The advantage of generating an siRNA cocktail pool instead of designing a specific siRNA is that an siRNA cocktail can potentially target multiple sites within an mRNA target. Bacterial RNase III, compared to human Dicer, is much easier to prepare and therefore much more economical. Consistent with a previous report (29), we found that EcE38A produces a higher percentage of 22-mer products than the corresponding wild-type protein (Figure 7A). In addition, the E65A mutation of EcRNase III also results in the elevation of the 22-mer product although to a lesser extent (Figure 7A). Although similar effects were observed for the E37A mutant of AaRNase III, the absolute percentage of 22-mer products is not as high as that for the EcE38A. Hence, we focused on the E. coli enzyme for further optimization of siRNA production.
Figure 7.
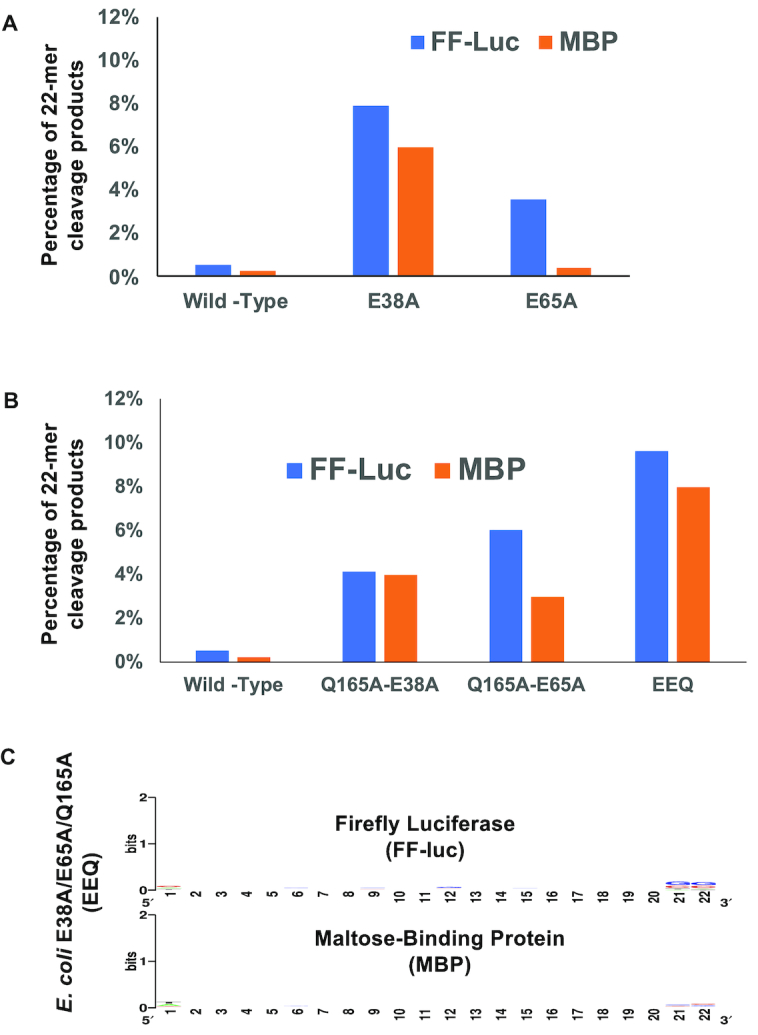
Engineered EcRNase III cleaves in vitro transcribed dsRNA into a heterogeneous mixture of siRNAs with a narrow size distribution centered at 22 nt. (A) The percentage of 22-nt products to the total number of reads was plotted for EcRNase III and its single mutants. (B) The percentage of 22-nt products to the total number of reads was plotted for EcRNase III and its double and triple mutants. (C) No consensus sequence of 22-nt cleavage products were detected in the triple mutant (EEQ) of EcRNase III.
To eliminate the substrate specificity governed by the +3G/-6C rule, we introduced the Q165A mutation into EcE38A and EcE65A, resulting in two double mutants, EcE38A/Q165A and EcE65A/Q165A, respectively. Some decrease in the production of 22-mer may happen because the Q165A mutation weakens the protein:dsRNA interaction. As expected, the two double mutants exhibited elevated production of the 22-mer products but not as much as EcE38A (Figure 7B). To maximize the production of 22-mer products, we generated a triple mutant, EcE38A/E65A/Q165A (EcEEQ). Using the FF-luc and MBP dsRNAs as substrates, we carried out in vitro cleavage and NGS analysis. The results show that the EcEEQ mutant protein produces more 22-nt RNA products than the two single mutants (Figure 7A), the two double mutants and the wild-type enzyme (Figure 7B). In addition, although EcEEQ only abolished the +3G preference during cleavage site selection, there are in general no consensus sequences among the 22-mer products from the triple mutant (Figure 7C).
Furthermore, 87% of 22-mer reads mapping to one strand of FF-Luc dsRNA can pair with a read mapping to the other strand of FF-luc dsRNA, in a manner that both ends have a 2-nt 3′ overhang (Supplementary Figure S5). Similar result (89%) was observed for the 22-mer cleavage products of MBP dsRNA (Supplementary Figure S5B). Of note, these percentages are the upper-bound estimates since the number of reads mapped to each strand of a duplex is often unmatched. Finally, we analyzed the cleavage products of EcEEQ at variable time points. Similar to wild-type EcRNase III, cleavage products were detectable 10 min after reaction initiation, indicating that the activity of EcEEQ is comparable to that of wild-type EcRNase III (Supplementary Figure S6). Consistent with a previous study (29), the 22-mer products generated by the inside-out mechanism were apparently protected by EcEEQ from being further processed and accumulated over time (Supplementary Figure S6).
Together, these results demonstrate that the new reagent EcEEQ cleaves long dsRNAs into a mixture of random, heterogeneous 22-nt products that are ideal for targeting multiple sites on a target mRNA in gene silencing studies.
DISCUSSION
It was previously shown that EcRNase III recognizes dsRNA with little specificity and no specific features are required for cleavage (30), which is in line with the notion that bacterial RNase III recognizes substrate structure rather than sequence (1). Here, we provide compelling evidence demonstrating that bacterial RNase III does recognize a certain sequence feature around the cleavage site through its dsRBD. Interestingly, a recent publication indicates that B. subtilis Mini-III, an RNase III enzyme without dsRBD, can also recognize a specific sequence motif near the cleavage site (41). Together, these results suggest that the mode of RNase III–substrate interaction is more complicated than previously believed. Besides structure, the sequence of RNA substrates also contributes to cleavage specificity and efficacy.
By demonstrating that both AaRNase III and EcRNase III recognize the +3G via the conserved glutamine side chain, we provided one of the first structural insights into the understanding of how bacterial RNase III achieves sequence specificity during cleavage site determination (1,2,42,43). The A-form dsRNA is characterized by a deep and narrow major groove, where access to the bases is hindered, and a wide and shallow minor groove, where the edges of the bases are readily accessible. Both functional and structural information suggest that the dsRBD performs a direct readout of RNA sequence in the minor groove (44). Although the dsRBD of AaRNase III contacts the minor groove extensively, only three base-specific hydrogen bonds are formed between RNA and two highly conserved glutamine side chains, Q157 and Q161, in the first α-helix of its dsRBD, known as RNA-binding motif 1 (RBM1) (24). The side chain Q157 carboxamide group forms one hydrogen bond with either a U base (24) or an A base (27) in the -5 position, indicating that the UA (Supplementary Figure S2B) and AU (Supplementary Figure S2C) bp are functionally equivalent. It has been shown that a CG or GC bp substitution strongly inhibits catalysis and that the Q157A mutation results in a defect in substrate binding (45). In contrast, the Q161 carboxamide group specifically recognizes the +3G base with two hydrogen bonds (Figure 4A), of which the critical role in cleavage site selection is revealed by our in vitro cleavage experiments and the NGS analysis of cleavage products. The cleavage products are analyzed by gel electrophoresis and the cleavage sites are mapped by primer extension. Despite many successes, these approaches are low-throughput in nature. The number of cleavage events that can be investigated in each study is rather limited. Recent advances in sequencing technology are making it possible to monitor many cleavage events in parallel. In this study, using NGS with dsRNAs of FF-Luc and MBP as substrates, we characterized more than 3,000 cleavage events. Each of them was backed up by multiple (up to several thousand) small RNAs detected by NGS. In addition, the results are highly consistent among multiple repeats. Together with two recent reports (41,43,46), our study demonstrates that NGS is a powerful approach in studying RNase III cleavage.
Both EcRNase III and AaRNase III recognize the +3G. Interestingly, the former, but not the latter, has an additional preference of SS (S = G or C) at the -1 and -2 positions (Supplementary Figure S1A). This difference would explain, at least in part, the distinct patterns of preferred cleavage sites (Figure 5B). While EcRNase III’s Q165 is responsible for recognizing the +3G during cleavage, the identity of residues that interact with the -1S/-2S remains elusive. Future high-resolution structures of the complex formed by EcRNase III and its substrate should give additional insights into the underlying mechanisms of the -1S/-2S preference. Finally, our result is highly consistent with a recent study where the transcriptome-wide cleavage sites of EcRNase III were mapped in vivo (43). The enrichments of both +3G and -1S/-2S were observed among sequences surrounding the cleavage sites on endogenous substrates, indicating that our conclusion can apply to EcRNase III cleavages under physiological conditions.
It is intriguing to ask why RNase III has evolved to recognize specific sequences near the cleavage sites. It is likely that just the dsRNA structure is not sufficient to define cleavage sites in cellular targets. The +3G as well as -1S/-2S recognitions may function as additional determinants for the enzyme to perform cleavage only at desired sites. In fact, it has been recently shown that a set of Dicer-like RNase III enzymes in Paramecium cleave dsRNA in a sequence-specific manner to enable precise targeting of transposon-derived IESs, playing important roles in the development of Paramecium (46). RNase III mutants whose sequence preference was abolished will serve as a valuable resource to further interrogate the biological function of RNase III. Given that the interaction between dsRBD and the +3G is observed in both EcRNase III and AaRNase III, we speculate that the same mechanism may be conserved in eukaryotic counterparts as well, in which it might play important functional roles. Future studies should provide more insights into the mechanism of cleavage site selection by eukaryotic RNase III enzymes.
We demonstrate that bacterial RNase III cleaves long dsRNA into a set of small RNAs with distinct patterns of length distribution (Figure 5A). A likely scenario is that RNase III places its cleavage site approximately N nt away from the end of dsRNA substrate. Whereas the ideal value of N is 11 as defined by high-resolution structures (24,27), the range of N is ∼6–19 that is dictated by the recognition of +3G (this study). Therefore, it is the combination of structural requirement and sequence recognition that makes the 11- and 12-mer the dominant species of cleavage products of wild-type RNase III. It was reported that the EcE38A promotes the inside-out cleavage of a long dsRNA by reducing substrate release after cleavage (29). Here, we provide additional evidence supporting this model by demonstrating that other mutants that potentially alter substrate release rate also promote or inhibit the inside-out mode of cleavage.
Finally, our study has important implications for RNA interference (RNAi) technology, specifically for the preparation of heterogeneous siRNA cocktails. To eliminate the sequence specificity and promote the production efficiency, we have developed the EcEEQ mutant protein. In addition to the 22-nt siRNAs, engineered RNase III also generates 21- and 23-nt duplexes but in much smaller amounts. The size distribution of the siRNA cocktail pool generated by EcRNase III mutants (Supplementary Figure S2) is narrow and centered at 22 nt (Supplementary Figure S7), mimicking the typical size distribution of good miRNA libraries previously characterized by large-scale profiling of miRNAs (47). Therefore, bacterial RNase III is an excellent reagent to produce siRNA cocktails and EcEEQ is the most efficient because it not only produces more siRNAs than any other mutant (Supplementary Figure S7), but also produces random, heterogeneous siRNAs that are ideal for gene silencing studies. It has been two decades since the discovery of RNAi (48), which was recognized by the Nobel Prize in Physiology or Medicine in 2006. As a Nobel-winning technique, RNAi got its first-ever drug approval (patisiran) by the Food and Drug Administration in August 2018 (49). This landmark drug shows the promise of RNAi. Our EcEEQ triple mutant serves as an efficient and economical reagent for the discovery of more siRNA drugs that mute disease-causing genes like the Huntington's.
DATA AVAILABILITY
The Small RNA-Seq datasets generated in this study are available on NCBI GEO under the accession number GSE120052 (ylsrecyglzifxwt - reviewer token).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Donald Court and Alexander Wlodawer for reading the manuscript and discussions, Brian Austin for technical assistance, and the Biophysics Resource in the Structural Biophysics Laboratory, Center for Cancer Research, National Cancer Institute for use of the LC/ESMS instrument. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Intramural Research Program of the NIH, Center for Cancer Research, National Cancer Institute. Funding for open access charge: National Institutes of Health, Intramural Research Program.
Conflict of interest statement. None declared.
REFERENCES
- 1. Nicholson A.W. Ribonuclease III mechanisms of double-stranded RNA cleavage. Wiley Interdiscip. Rev. RNA. 2014; 5:31–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Court D.L., Gan J., Liang Y.-H., Shaw G.X., Tropea J.E., Costantino N., Waugh D.S., Ji X.. RNase III: Genetics and Function; Structure and mechanism. Annu. Rev. Genet. 2013; 47:405–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Robertson H.D., Webster R.E., Zinder N.D.. Purification and properties of ribonuclease III from Escherichia coli. J. Biol. Chem. 1968; 243:82–91. [PubMed] [Google Scholar]
- 4. Watson N., Apirion D.. Molecular cloning of the gene for the RNA-processing enzyme RNase III of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 1985; 82:849–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Court D.L. Belasco JG, Brawerman G. Control of Messenger RNA Stability. 1993; NY: Academic Press; 71–116. [Google Scholar]
- 6. Langenberg W.G., Zhang L., Court D.L., Giunchedi L., Mitra A.. Transgenic tobacco plants expressing the bacterial rnc gene resist virus infection. Mol. Breed. 1997; 3:391–399. [Google Scholar]
- 7. Deltcheva E., Chylinski K., Sharma C.M., Gonzales K., Chao Y., Pirzada Z.A., Eckert M.R., Vogel J., Charpentier E.. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011; 471:602–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stead M.B., Marshburn S., Mohanty B.K., Mitra J., Pena Castillo L., Ray D., van Bakel H., Hughes T.R., Kushner S.R.. Analysis of Escherichia coli RNase E and RNase III activity in vivo using tiling microarrays. Nucleic Acids Res. 2011; 39:3188–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Durand S., Gilet L., Bessieres P., Nicolas P., Condon C.. Three essential ribonucleases-RNase Y, J1, and III-control the abundance of a majority of Bacillus subtilis mRNAs. PLoS Genet. 2012; 8:e1002520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lasa I., Toledo-Arana A., Dobin A., Villanueva M., de los Mozos I.R., Vergara-Irigaray M., Segura V., Fagegaltier D., Penades J.R., Valle J. et al.. Genome-wide antisense transcription drives mRNA processing in bacteria. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:20172–20177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lioliou E., Sharma C.M., Caldelari I., Helfer A.C., Fechter P., Vandenesch F., Vogel J., Romby P.. Global regulatory functions of the Staphylococcus aureus endoribonuclease III in gene expression. PLoS Genet. 2012; 8:e1002782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Abou Elela S., Igel H., Ares M. Jr. RNase III cleaves eukaryotic preribosomal RNA at a U3 snoRNP-dependent site. Cell. 1996; 85:115–124. [DOI] [PubMed] [Google Scholar]
- 13. Ghazal G., Ge D., Gervais-Bird J., Gagnon J., Abou Elela S.. Genome-wide prediction and analysis of yeast RNase III-dependent snoRNA processing signals. Mol. Cell Biol. 2005; 25:2981–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gagnon J., Lavoie M., Catala M., Malenfant F., Elela S.A.. Transcriptome wide annotation of eukaryotic RNase III reactivity and degradation signals. PLoS Genet. 2015; 11:e1005000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee R.C., Feinbaum R.L., Ambros V.. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993; 75:843–854. [DOI] [PubMed] [Google Scholar]
- 16. Carthew R.W. Gene silencing by double-stranded RNA. Curr. Opin. Cell Biol. 2001; 13:244–248. [DOI] [PubMed] [Google Scholar]
- 17. Bernstein E., Caudy A.A., Hammond S.M., Hannon G.J.. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature. 2001; 409:363–366. [DOI] [PubMed] [Google Scholar]
- 18. Filippov V., Solovyev V., Filippova M., Gill S.S.. A novel type of RNase III family proteins in eukaryotes. Gene. 2000; 245:213–221. [DOI] [PubMed] [Google Scholar]
- 19. Kwon S.C., Nguyen T.A., Choi Y.G., Jo M.H., Hohng S., Kim V.N., Woo J.S.. Structure of human DROSHA. Cell. 2016; 164:81–90. [DOI] [PubMed] [Google Scholar]
- 20. Liu Z., Wang J., Cheng H., Ke X., Sun L., Zhang Q.C., Wang H.W.. Cryo-EM structure of human dicer and its complexes with a Pre-miRNA substrate. Cell. 2018; 173:1191–1203. [DOI] [PubMed] [Google Scholar]
- 21. MacRae I.J., Li F., Zhou K., Cande W.Z., Doudna J.A.. Structure of Dicer and mechanistic implications for RNAi. Cold Spring Harb. Symp. Quant. Biol. 2006; 71:73–80. [DOI] [PubMed] [Google Scholar]
- 22. Blaszczyk J., Tropea J.E., Bubunenko M., Routzahn K.M., Waugh D.S., Court D.L., Ji X.. Crystallographic and modeling studies of RNase III suggest a mechanism for double-stranded RNA cleavage. Structure. 2001; 9:1225–1236. [DOI] [PubMed] [Google Scholar]
- 23. Liang Y.H., Lavoie M., Comeau M.A., Abou Elela S., Ji X.. Structure of a eukaryotic RNase III postcleavage complex reveals a double-ruler mechanism for substrate selection. Mol. Cell. 2014; 54:431–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gan J., Tropea J.E., Austin B.P., Court D.L., Waugh D.S., Ji X.. Structural insight into the mechanism of double-stranded RNA processing by ribonuclease III. Cell. 2006; 124:355–366. [DOI] [PubMed] [Google Scholar]
- 25. Yang D., Buchholz F., Huang Z., Goga A., Chen C.Y., Brodsky F.M., Bishop J.M.. Short RNA duplexes produced by hydrolysis with Escherichia coli RNase III mediate effective RNA interference in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:9942–9947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Weinberg D.E., Nakanishi K., Patel D.J., Bartel D.P.. The inside-out mechanism of Dicers from budding yeasts. Cell. 2011; 146:262–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gan J., Shaw G., Tropea J.E., Waugh D.S., Court D.L., Ji X.. A stepwise model for double-stranded RNA processing by ribonuclease III. Mol. Microbiol. 2008; 67:143–154. [DOI] [PubMed] [Google Scholar]
- 28. Gan J., Tropea J.E., Austin B.P., Court D.L., Waugh D.S., Ji X.. Intermediate states of ribonuclease III in complex with double-stranded RNA. Structure. 2005; 13:1435–1442. [DOI] [PubMed] [Google Scholar]
- 29. Xiao J., Feehery C.E., Tzertzinis G., Maina C.V.. E. coli RNase III(E38A) generates discrete-sized products from long dsRNA. RNA. 2009; 15:984–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lamontagne B., Elela S.A.. Evaluation of the RNA determinants for bacterial and yeast RNase III binding and cleavage. J. Biol. Chem. 2004; 279:2231–2241. [DOI] [PubMed] [Google Scholar]
- 31. Ho S.N., Hunt H.D., Horton R.M., Pullen J.K., Pease L.R.. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989; 77:51–59. [DOI] [PubMed] [Google Scholar]
- 32. Nallamsetty S., Austin B.P., Penrose K.J., Waugh D.S.. Gateway vectors for the production of combinatorially-tagged His6-MBP fusion proteins in the cytoplasm and periplasm of Escherichia coli. Protein Sci. 2005; 14:2964–2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kapust R.B., Tözsér J., Fox J.D., Anderson D.E., Cherry S., Copeland T.D., Waugh D.S.. Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein. Eng. 2001; 14:993–1000. [DOI] [PubMed] [Google Scholar]
- 34. Blaszczyk J., Gan J., Tropea J.E., Court D.L., Waugh D.S., Ji X.. Noncatalytic assembly of ribonuclease III with double-stranded RNA. Structure. 2004; 12:457–466. [DOI] [PubMed] [Google Scholar]
- 35. Gasteiger E., Gattiker A., Hoogland C., Ivanyi I., Appel R.D., Bairoch A.. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003; 31:3784–3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Langmead B., Trapnell C., Pop M., Salzberg S.L.. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009; 10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. Genome Project Data Processing, S. . The sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Robinson J.T., Thorvaldsdottir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P.. Integrative genomics viewer. Nat. Biotechnol. 2011; 29:24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Crooks G.E., Hon G., Chandonia J.M., Brenner S.E.. WebLogo: a sequence logo generator. Genome Res. 2004; 14:1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Robertson H.D., Dunn J.J.. Ribonucleic acid processing activity of Escherichia coli ribonuclease III. J. Biol. Chem. 1975; 250:3050–3056. [PubMed] [Google Scholar]
- 41. Glow D., Pianka D., Sulej A.A., Kozlowski L.P., Czarnecka J., Chojnowski G., Skowronek K.J., Bujnicki J.M.. Sequence-specific cleavage of dsRNA by Mini-III RNase. Nucleic Acids Res. 2015; 43:2864–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lamontagne B., Abou Elela S.. Evaluation of the RNA determinants for bacterial and yeast RNase III binding and cleavage. J. Biol. Chem. 2004; 279:2231–2241. [DOI] [PubMed] [Google Scholar]
- 43. Altuvia Y., Bar A., Reiss N., Karavani E., Argaman L., Margalit H.. In vivo cleavage rules and target repertoire of RNase III in Escherichia coli. Nucleic Acids Res. 2018; 46:10380–10394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Masliah G., Barraud P., Allain F.H.. RNA recognition by double-stranded RNA binding domains: a matter of shape and sequence. Cell Mol. Life Sci. 2013; 70:1875–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shi Z., Nicholson R.H., Jaggi R., Nicholson A.W.. Characterization of Aquifex aeolicus ribonuclease III and the reactivity epitopes of its pre-ribosomal RNA substrates. Nucleic Acids Res. 2011; 39:2756–2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hoehener C., Hug I., Nowacki M.. Dicer-like enzymes with sequence cleavage preferences. Cell. 2018; 173:234–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chu A., Robertson G., Brooks D., Mungall A.J., Birol I., Coope R., Ma Y., Jones S., Marra M.A.. Large-scale profiling of microRNAs for The Cancer Genome Atlas. Nucleic Acids Res. 2016; 44:e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fire A., Xu S., Montgomery M.K., Kostas S.A., Driver S.E., Mello C.C.. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998; 391:806–811. [DOI] [PubMed] [Google Scholar]
- 49. Adams D., Gonzalez-Duarte A., O’Riordan W.D., Yang C.C., Ueda M., Kristen A.V., Tournev I., Schmidt H.H., Coelho T., Berk J.L. et al.. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med. 2018; 379:11–21. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The Small RNA-Seq datasets generated in this study are available on NCBI GEO under the accession number GSE120052 (ylsrecyglzifxwt - reviewer token).