


Abstract
The development of synthetic riboswitches has always been a challenge. Although a number of interesting proof-of-concept studies have been published, almost all of these were performed with the theophylline aptamer. There is no shortage of small molecule-binding aptamers; however, only a small fraction of them are suitable for RNA engineering since a classical SELEX protocol selects only for high-affinity binding but not for conformational switching. We now implemented RNA Capture-SELEX in our riboswitch developmental pipeline to integrate the required selection for high-affinity binding with the equally necessary RNA conformational switching. Thus, we successfully developed a new paromomycin-binding synthetic riboswitch. It binds paromomycin with a KD of 20 nM and can discriminate between closely related molecules both in vitro and in vivo. A detailed structure–function analysis confirmed the predicted secondary structure and identified nucleotides involved in ligand binding. The riboswitch was further engineered in combination with the neomycin riboswitch for the assembly of an orthogonal Boolean NOR logic gate. In sum, our work not only broadens the spectrum of existing RNA regulators, but also signifies a breakthrough in riboswitch development, as the effort required for the design of sensor domains for RNA-based devices will in many cases be much reduced.
INTRODUCTION
The traditional role of RNA in cells was thought to be restricted to carrying genetic information. However, the full importance of RNA was realized in recent decades, in particular when its role in gene control and catalysis became evident (1,2). Various classes of non-coding RNAs are an important part of the regulation of cellular processes, and RNA-based synthetic devices have shown distinct advantages over protein-based systems (3–5). Indeed, fast regulatory response, genetic modularity and portability, shared with the possibility of combining different platforms to realize a wide range of regulatory outputs, are the hallmarks of such devices. Amongst these tools, riboswitches represent their own class. They have the ability to sense metabolites inside of the cell and directly modulate gene expression depending on the concentration of the detected target molecule (6). Due to these special properties, synthetic riboswitches are of great interest as tools not only for conditional gene expression control or for the design of synthetic genetic circuits, but also for several biotechnological applications, e.g. for the optimization of producer strains that enable non-invasive real-time detection of the produced molecule within the cell (7).
Synthetic riboswitches as control elements have several advantages. They have a simple structure, their sensory and regulatory parts are connected, and consist of RNA, so no further protein factors are required for regulation. In addition, the sensory part, i.e. the so-called aptamer domain, recognizes the ligand with high affinity and specificity (7). However, the greatest advantage is that such an aptamer domain can in principle be selected against any desired target molecule using a method called SELEX (8,9). This is an iterative process where specific binding RNA molecules can be selected from a very extensive library. A wealth of adroit proof-of-concept studies demonstrating the application of synthetic riboswitches are available nowadays (10,11). However, all studies to date are exclusively limited to a handful of aptamer domains, mostly the theophylline and the tetracycline aptamer (7). Only the ciprofloxacin riboswitch was added within the last 10 years (12). This small number effectively prevents a wider application of synthetic riboswitches. To remedy this lack of applications and to stimulate and enliven the field of synthetic riboswitch development, our ultimate goal is to expand the repertoire of aptamers suitable and available for riboswitch design. The main reason for the limited suitability of most aptamers is that both excellent binding properties and conformational switching are essential (13–15), yet the latter is a feature not addressed during the process of in vitro selection (12). To find aptamers that combine superior binding properties and the ability to undergo conformational switching, cellular screening after in vitro selection is required. Such screening systems have been established (16–19) and their functionality has been successfully demonstrated e.g. for the neomycin and the ciprofloxacin aptamer (12,20). However, the probability of finding an aptamer with riboswitching properties in an enriched library by screening is extremely low. Deep sequencing analyses have shown that it is below 0.01%, and consequently massive screening efforts have to be made (12). In fact, we screened several enriched pools against other ligands and could not find any aptamer with regulatory properties.
Therefore, we considered that screening alone may not be sufficient to reliably identify aptamers that undergo a conformational change upon ligand binding. We propose to change the SELEX strategy in such a way that not only aptamers with superior binding properties, but also those that switch their conformation are selected. To us, the use of a so-called Capture-SELEX appeared to be a promising solution (14,21–24). This strategy involves the insertion of a small defined motif inside of the random region of the library. This so-called ‘docking sequence’ is hybridized through Watson–Crick interactions to a small complementary oligonucleotide containing biotin via a linker molecule at its end so that it can be used to fix the complex onto streptavidin magnetic beads. Elution with the free ligand frees the aptamers while undergoing a rearrangement. Thus, only structure-switching aptamers should be eluted from the beads. Since a major characteristic of riboswitches is a structural change upon ligand binding (25,26), we assume that a Capture-SELEX approach will allow substantial acceleration of the process of riboswitch development, while simultaneously reducing screening efforts. In addition, with the use of Capture-SELEX, the need to immobilize the ligand, i.e. a requirement often not trivial for small molecule targets, becomes obsolete.
In this study, we demonstrate the application of Capture-SELEX resulting in a significantly faster development of a new synthetic riboswitch. We chose paromomycin (PARO), an aminoglycoside antibiotic. Applying Capture-SELEX, we were able to enrich a PARO-specific RNA library that was also unable to recognize the very close compound neomycin due to the implementation of a counter-selection step. In vivo screening led to the identification of an aptamer with riboswitching properties that we further engineered into a PARO-responsive riboswitch. We characterized the secondary structure and the binding properties of the new riboswitch. In addition, the exclusive binding of PARO allowed the new riboswitch to be combined with other riboswitches to generate orthogonal Boolean gates that allow to fine-tune downstream gene expression.
In sum, with the implementation of the Capture-SELEX protocol, we have succeeded in significantly improving the speed and efficiency of the development of synthetic riboswitches. We now select not only aptamers that bind well, but also those that have the potential to change their conformation so that the subsequent screening step is significantly shorter and more successful. We expect that this protocol has the potential to lead to an ultimate breakthrough and unprecedented acceleration of the field of synthetic riboswitches.
MATERIALS AND METHODS
Pool preparation
The initial library was designed so that the RNA contained a 13 nt fixed docking sequence (DS), responsible for binding the capture oligonucleotide (CO) within a 50 nucleotide (nt) randomized region, along with 40 random nt on the 5′ side of the DS and 10 random nt on the 3′side. This library was flanked by a 27 nt long primer-binding sequence on the 5′side and a 33 nt long primer-binding sequence on the 3′ side. This resulted in the following library composition: 5′ GGGCACUCCA AGCUAGAUCUACCGGU N40CUACUGGCUUCUAN10 AAAAUGGCUAGCAAAGGAGAAG AACUUUUCACU 3′ with the DS underlined (see also Figure 1). The pool was amplified from the template oligonucleotide Pool_template using the following polymerase chain reaction (PCR) conditions: 10 mM Tris–HCl, pH 9.0, 50 mM KCl, 1.5 mM MgCl2, 0.1% Triton X-100, 0.2 mM dNTPs (each), 200 nM template oligonucleotide Pool_template, 2 μM primer Pool_fwd, 2 μM primer Pool_rev, 50 U/ml Taq DNA Polymerase (NEB). The sequences of the oligonucleotides Pool_template, Pool_fwd and Pool_rev are given in Supplementary Table S1. A total of 1015 molecules of the pool were amplified in a 15 ml PCR reaction equally aliquoted into a 96-well plate for only five cycles of amplification (95°C 2 min, 5 cycles of {95°C 1 min, 54°C 1 min, 72°C 1 min}, 72°C for 3 min) to reduce PCR-induced bias. The PCR products were ethanol precipitated and dissolved in MQ-H2O. The deionized water was purified using ion exchange resin and filtered through a Biofilter (ELGA) to remove possible RNase contamination. One tenth of the purified DNA pool (∼5 × 1015 sequences) was in vitro transcribed using T7 RNA polymerase with the following conditions: 200 mM Tris–HCl, pH 8.0, 20 mM Mg-acetate, 2 mM Spermidine, 4 mM NTP (each) and 20 mM DTT, 50 U/ml T7 Polymerase (homemade) and 40 U/ml ribonuclease inhibitor (moloX). After overnight transcription, the RNA pool was ethanol precipitated, dissolved in MQ-H2O, mixed with an equal volume of RNA loading dye (formamide containing 25 mM ethylenediaminetetraacetic acid (EDTA)) and loaded on a 6% denaturing polyacrylamide gel (8 M urea). The RNA was visualized by UV shadowing, sliced out and eluted overnight in 300 mM Na-acetate (pH 6.5). Finally, eluted RNA was ethanol precipitated and the pellet was dissolved in a suitable amount of MQ-H2O.
Figure 1.

Pool design for the Capture-SELEX. The 5′ and 3′ primer binding sites (boxed) are complementary to the vector sequence up- and downstream the two restriction sites AgeI and NheI (italic and underlined) to allow homologous recombination. The docking sequence (boxed) is 13 nt long and complementary to the capture oligonucleotide. The capture oligonucleotide is extended at its 3′ end by a linker and a biotin tag (Biotin TEG). The randomized regions are marked in bold.
In vitro selection
A total of 1 × 1015 RNA molecules from the purified RNA library were mixed with 300 kCPM of 5′ 32P-labelled RNA. The RNA mixture was afterwards mixed with 2 nmol of the biotinylated CO and 40 μl of 5× SELEX buffer (final concentrations: 40 mM HEPES, 250 mM KCl, 20 mM NaCl, 5 mM MgCl2) and filled up to 200 μl with MQ-H2O. The mixture was then heated at 65°C for 5 min and cooled down to 21°C using a thermoblock for 30 min. After this incubation step, the RNA-CO complex was mixed with 1 ml of Dynabeads® M-270 (ThermoFischer) at room temperature with gentle agitation for 1 h. The beads were washed in advance three times with 500 μl B&W buffer (5 mM Tris–HCl, pH 7.5, 1 M NaCl, 0.5 M EDTA) and then equilibrated in 300 μl 1× SELEX buffer. After the immobilization step, the beads now bound to the RNA were transferred on a magnetic rack for 1 min and the supernatant was removed. The beads were resuspended in 500 μl of 1× SELEX buffer and incubated 5 min under mild agitation. This washing step was repeated three times. Afterwards, RNA was specifically eluted with 1 mM of PARO for 5 min (round 1–7) or 100 μM PARO (round 8–11) in 1× SELEX buffer, respectively, in the same conditions as above. The eluted RNA in the supernatant was recovered and ethanol-precipitated in the presence of 0.3 M Na-acetate (pH 6.5) and 15 μg GlycolBlue as co-precipitant (Ambion). After two steps of washing with 70% ethanol, the pellet was air-dried and dissolved in 50 μl MQ-H20, reverse transcribed and amplified (RT-PCR). RT-PCR was carried out as previously described (12). Afterwards, the DNA was analysed via agarose gel electrophoresis. For the following rounds, 10 μl of the RT-PCR were mixed with 40 mM Tris–HCl, pH 8.0, 5 mM DTT, 2.5 mM NTPs (each), 15 mM MgCl2, 100 U T7 RNA Polymerase (homemade), 40 U ribonuclease inhibitor (moloX) and 33 nM 32P-α-UTP (Hartmann analytics) in a total volume of 100 μl. Transcription was carried out overnight at 37°C. Afterwards, the transcribed RNA was NH4-acetate/ethanol-precipitated and washed twice with 70% EtOH. The pellet was then dissolved in 50 μl MQ-H2O. About 100 kCPM of radiolabelled RNA were used for the following selection rounds. As the diversity of the library is already reduced due to the first round of selection, smaller volumes of consumables were used. All subsequent rounds were performed with 150 μl magnetic beads, 0.2 nmol of CO, 100 kCPM of RNA diluted in 50 μl of 1× SELEX buffer and a volume of washing and elution of 200 μl.
Plasmid cloning and doped pool generation
All oligonucleotides used in this study are listed in Supplementary Table S1. All plasmids are described in Supplementary Table S2 along with their respective plasmid maps (Supplementary Figure S1) and sequences (Supplementary Sequence Information S1–3). The plasmid pCBB05 contains the gfp+ gene driven by the ADH1 promoter based on the design of the pWHE601* (16). An AgeI restriction site is located immediately before the start codon of the gfp+ gene with a 5′ UTR length of 39 nt. The restriction site NheI is immediately behind the start codon (16). Like in pWHE601, pCBB05 contains an ampicillin resistance gene and an origin of replication to allow its passage through E. coli. However, the 2μ replication sequence was exchanged to a CEN6/ARS4 sequence resulting in a centromeric plasmid. In addition, the plasmid contains the mCherry gene under control of the constitutive TEF promoter used later on for normalization of GFP expression. This plasmid served as a positive control (GFP expression on, GFP positive). In pCBB06, the Kozak sequence and the start codon of the GFP gene in pCBB05 were replaced by a CTCTTC sequence to ensure GFP expression only after successful integration of the insert carrying the aptamer sequence along with the start codon. Consequently, pCBB06 served as a negative control (GFP negative). This vector was used for the in vivo screening. pCBB07 served as an additional positive control with the neomycin riboswitch (20) integrated in front of the GFP gene (switching control).
For cloning, 40 bp overlapping oligonucleotides were designed and amplified using Q5® High-Fidelity DNA polymerase (NEB) according to the supplier’s instructions. The resulting PCR product was purified (QIAquick PCR Purification Kit, Qiagen), mixed in a 5 molar excess with the double digested (AgeI-HF and NheI-HF (NEB)) plasmid pCBB06 and used for yeast homologous recombination (Frozen-EZ Yeast Transformation II Kit (Zymo Research)). The doped pool was generated using the oligonucleotides Doped_pool_fwd and Doped_pool_3.0 rev (Supplementary Table S1) (Microsynth AG), based on the construct P11.2H2 as template and amplified using Q5® High-Fidelity DNA polymerase (NEB) according to the supplier’s instructions. Again, the library was cloned in pCBB06 using yeast homologous recombination and plated on SCD-ura plates with an efficiency guarantying a yield of more than 10 000 plasmids.
Cultivation of yeast and GFP measurements
Saccharomyces cerevisiae strain RS453α (MATα ade2- 1 trp1–1 can1–100 leu2–3 his3–1 ura3–52) was transformed using Frozen-EZ Yeast Transformation II Kit (Zymo Research). Transformed cells were plated on SCD-ura plates [0.2% YNB w/o AA (Difco), 0.55% ammonium sulphate (Roth), 2% glucose (Roth), 12 μg/ml adenine (SIGMA), 1 × MEM amino acids (SIGMA), 2% Agar (Oxoid)] and incubated at 30°C for 2 days in a humidified incubator. Single colonies were picked and cultured in 1.5 ml SCD-ura for 24 h (450 rpm, 30°C, 24 well plates) before they were diluted 1:1000 in fresh media with and without 250 μM PARO. After 48 h incubation, 20 μl of cells were transferred into 180 μl 1× PBS and both GFP and mCherry fluorescence were measured by flow cytometry for 20 000 cells per candidate. All cytometry measurements were performed on a CytoFlex S (Beckman Coulter) equipped with a 488 nm laser and a 561 nm laser for excitation of GFP and mCherry, respectively. Emission light was bandpass-filtered at 510/20 nm or 610/20 nm. pCBB05, pCBB06 and pCBB07 were analysed in parallel as positive, negative and switching control, respectively. The negative control (pCBB06) was used as a blank and its value was subtracted from all data. GFP values were normalized to the positive control without PARO. GFP measurement was performed simultaneously for four independent colonies (biological replicates) per candidate. This measurement was repeated on three independent days.
In vivo screening
Library preparation and in vivo screening were performed according to the established protocol (20) with modifications described in (16). The aptamer inserts for the plasmid library were prepared from the RT-PCR products from round 8 and 11 of the PARO SELEX with the primers HR_fwd and HR_rev to create a 40 nt overlap identical to the pCBB06 backbone to facilitate homologous recombination. The target vector was digested with the AgeI and NheI, gel purified with Zymoclean Gel DNA Recovery Kit (Zymo Research) and transformed into RS463α with a 10× molar excess of insert using Frozen-EZ Yeast Transformation II Kit according the supplier’s instructions (ZymoResearch). The transformed cells were transferred into 125 ml SCD-ura medium and 1:10 diluted after 48 h into 50 ml of SCD-ura, 1:30 after 72 h and 100 times after 96 h. This serial dilution was done to force plasmid segregation of multiple vector transformants (27). Twenty-four hours after the last dilution, the cells were sorted by FACS to remove cells not expressing GFP (i.e. cells with plasmids without an insert or with an insert that had a highly stable aptamer structure). Recovered cells were spread on several SCD-ura agar plates to achieve a density of about 200 colonies per plate. After 2 days of incubation at 30°C, colonies were picked and transferred into 96-well plates containing 200 μl of SCD-ura media. After sealing the plate with BREATHseal (Greiner Bio-One), the cells were incubated 24 h at 30°C with high speed shaking (1200 rpm) on a Titramax 1000 shaker (Heidolph). Then, the pre-culture was 1:10 diluted into either a 96-well plate containing only 200 μl SCD-ura or into a 96-well plate containing the SCD-ura supplemented with 100 μM of PARO. For the final measurement, experiments were performed in 24-well plates in 1.5 ml media, the pre-culture was diluted 1:1000 in media without or with 250 μM ligand and incubated for 48 h.
For in vivo screening, cells were 1:10 diluted in 1× PBS after 24-h incubation and both GFP and mCherry fluorescence were measured by flow cytometry. The plasmids pCBB05, pCBB06 and pCBB07 were always measured simultaneously as positive, negative and switching control. Fluorescence was afterwards normalized to the controls and to the mCherry signal. About 20 000 cells were measured per sample, the GFP fluorescence median value was taken and the ratio was calculated based on the median value of GFP fluorescence of the same clone with ligand and normalized to the mCherry measurement. Positive hits were isolated and streaked out on SCD-ura plates. Four different colonies were re-analysed under the same condition as before. Confirmed hits were then grown in 4 ml SCD-ura cultures and their plasmid was recovered and sequenced.
RNA synthesis for in vitro analysis
For in vitro analysis (in-line probing and ITC measurements), RNA was transcribed from PCR-amplified templates, all containing three guanosyl residues at the 5′ end of the desired RNA sequence to facilitate transcription using T7 RNA polymerase. For this, two oligonucleotides were designed with an overlap of 30 nt and amplified using Q5® High-Fidelity DNA polymerase (NEB) according to the supplier’s instructions. After ethanol precipitation, the DNA template was used for in vitro transcription with T7 RNA polymerase (homemade) as reported previously (28). The RNA was gel-purified and molarity was determined by spectrophotometric measurement using NanoDrop 1000 Spectrophotometer (Thermo Scientific).
In-line probing experiments
For in-line probing, RNA was dephosphorylated and 32P-labelled at the 5′ end as previously described (29). After gel purification, 35 kCPM of 32P-labelled RNA were incubated for 68 h at 22°C in in-line reaction buffer (10 mM Tris–HCl, pH 8.3, 10 mM MgCl2, 100 mM KCl). To generate a size marker, the 32P-labelled RNAs were subjected to alkaline hydroxylation by incubation for 3 min at 96°C in 50 mM Na2CO3, pH 9.0 or incubated for 3 min at 55°C with 20 U RNase T1 at denaturing conditions to identify guanine residues (30). After in-line reaction, alkaline hydroxylation or RNase T1 treatment, reactions were ethanol-precipitated and the pellet was dissolved in 5 M urea. All reactions were separated by denaturing polyacrylamide gel electrophoresis. Afterwards, gels were dried and analysed using phosphoimaging (GE Healthcare).
Isothermal titration calorimetry (ITC)
RNA folding and buffer compositions were chosen according to the SELEX experiments. About 100 μM PARO solution was prepared in the same buffer. ITC experiments were carried out with a MicroCal PEAQ-ITC (Malvern Instruments) with the sample cell (200 μl) containing 10 μM RNA and 100 μM PARO solution in the injector syringe (40 μl). After thermal equilibration at 25°C, an initial 150 s delay and one initial 0.4 μl injection was carried out. Then, 19 serial injections of 2.0 μl at intervals of 150 s and at a stirring speed of 750 rpm were performed. Raw data were recorded as power (μcal s−1) over time (min). The heat associated with each titration peak was integrated and plotted against the corresponding molar ratio of PARO and RNA. The dissociation constant (KD) was extracted from a curve fit of the corrected data by use of the one-site binding model provided by MicroCal PEAQ-ITC Analysis Software (Ver, 1.1.0.1262). Measurements were repeated at least twice.
Bioinformatic evaluation of sequenced libraries
Libraries for high-throughput sequencing were prepared from double-stranded DNA as described previously (12). The libraries were sequenced on an Illumina MiSeq System (Microsynth) and afterwards demultiplexed into the different pools according to their attached internal barcode. Within each library, same sequences were summed up and for every sequence, the reads per million (RPM) were calculated by dividing the read count by total number of reads per round for inter-library comparison. Furthermore, Gini indices were calculated for each library to assess the enrichment status as measure of imbalance of the respective library (31). Here, the cumulative distribution function (CDF) of the RPM were plotted against all sequences according to their rank in the library (sorted from most to least abundant). Both parameters, CDF of RPM and rank in library, were scaled to the boundaries zero and one. Based on this scaling, first the area under the curve (AUC) and subsequently the Gini index (2*AUC-1) were calculated. The Gini index gives a measure of the imbalance of the pool. Given an unenriched pool, the Gini index equals zero and a fully enriched pool (only one sequence) equals 1. This also allows for graphical visualization of the enrichment process.
Shannon’s informational entropy 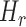 was evaluated after calculation of the frequency
was evaluated after calculation of the frequency 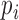 of each RNA species i for each SELEX round r with following formula:
of each RNA species i for each SELEX round r with following formula:
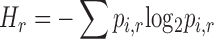 |
Shannon’s entropy is a measure for the expected value of the information content in a random variable such as an RNA sequence. The information content, or surprisal, of an event is defined as the negative logarithm of its probability. Consequently, the information decreases for enriched species within the overall sequence space; eventually vanishing for a pure population of only one specific sequence. Hence, the value of information entropy maximizes for an equally distributed sample in which each species is equally likely to appear. As the distribution shifts towards a few dominating species, we expect the entropy to decrease. Therefore, the information entropy is a good measure for sample/population complexity.
To explore the effect of Capture-SELEX in comparison to traditional or conventional SELEX approaches, we calculated the secondary structures of each RNA sequence in libraries 1, 4, 8 and 11 using mfold 3.6 (32,33). Folding was conducted under the assumption of a temperature of 37°C, and a window parameter of 1. Suboptimal energy structures were outputted if their folding free energy did not exceed a 5% limit above the lowest free folding energy. We evaluated the number of folds complying to these rules and recorded their free folding energies, giving a distribution of folding energies for each RNA sequence. We calculated the span of the folding free energies defined as the minimum folding free energy subtracted from the maximum folding free energy. Densities were calculated using the ggplot2 package (34) for the statistical programming language R.
RESULTS
Pool design for the RNA Capture-SELEX
The pool design was inspired by a previously described pool used for a DNA Capture-SELEX (21). A 50 nt long randomized region was asymmetrically split in N40 and N10 by a 13 nt long docking sequence (DS). The DS was designed to be unstructured and partially complementary to the 5′ primer binding site. Thus, we attempted to enhance the switching capability of the generated aptamers by giving a refolding possibility to the docking sequence when released from the capture oligonucleotide (CO) upon ligand binding. In addition, the shorter randomized part (N10) was positioned on the 3′ side. With this strategy, the folding of the aptamer should happen close to the start codon that was located in the 3′ primer binding site. Such a position would lead to higher probability of finding riboswitches. The constant regions (primer binding sites) were chosen based on the local genetic context in which the aptamers were to be screened in vivo in a subsequent step. They therefore corresponded to the sequence of the screening vector pCBB06 (see Figure 1). The 5′ primer binding site corresponded to the 3′ end of the ADH1 promotor followed by the AgeI restriction site and the 18 nt long 5′ UTR of the GFP gene located upstream of the restriction site on the vector. The 3′ primer binding site started with an optimized Kozak sequence for yeast (35) and the start codon (AAAATG) followed by the NheI restriction site and the first 21 nt of the gfp+ gene. The 5′ primer used for the SELEX experiment corresponded to the 5′ primer binding site extended by the sequence for the T7 promoter and three G residues to allow efficient transcription in vitro. With this strategy, we created a ready-to-clone library in which the sequence of the primer binding sites is unlikely to be associated with off-target effects on gene expression later, as it has already been taken from the vector that will be used for in vivo screening.
Selection of paromomycin-specific aptamers using Capture-SELEX
A library containing 1015 different RNA sequences was used as a starting pool for the first round of selection. A radiolabelled tracer was incorporated to determine the amount of RNA in each step of the selection. In the first round, magnetic beads with a binding capacity of 2 nmol of single-stranded DNA (1 ml) were used to be able to immobilize 1015 RNA molecules. About 150 μl of magnetic beads were used for all the other cycles. In the first seven rounds, the RNA was eluted with low stringency (1 mM PARO) to ensure that most of the binders were eluted. After a confirmed enrichment of the pool towards PARO in round 6 (Figure 2A), the concentration of PARO used for specific elution was decreased to 100 μM (rounds 8–11). With this strategy, only aptamers with a low KD can detach from the CO, bind to the target and remain bound there. Increasing the stringency in this way will force the selection of aptamers with the best binding affinities. After two rounds, the amount of specifically eluted RNA increased again so that stringency could be further increased in the following round. A counter elution step with 100 μM neomycin (NEO) was performed in the two last rounds (rounds 10 and 11) to specifically enrich for PARO binding aptamers that do not recognize the closely related molecule NEO. After two rounds of counter elution, the amount of RNA eluted with PARO was twice as high as that eluted in the counter elution (Supplementary Figure S2). Figure 2 illustrates the selection; a detailed summary can be found in the Supplementary Table S3.
Figure 2.

Capture-SELEX against PARO. (A) Results of the SELEX experiment. The black bars represent the amount of RNA specifically eluted by addition of 1 mM (rounds 1–7) and 100 μM (rounds 8–11) PARO. The grey bars represent the amount of RNA eluted at the last wash step. To create specificity towards PARO, a counter selection with 100 μM NEO was performed at rounds 10 and 11 (CE NEO). The dashed lines represent an increase in stringency (e.g. elution with 100 μM instead of 1 mM PARO or counter elution). Rounds 8 and 11 that were selected for in vivo screening are marked. (B) Chemical structure of PARO. The hydroxyl group on the carbon 6 of the first hexose that is an amino group in NEO is highlighted.
In vivo screening for a paromomycin riboswitch
Enriched libraries from rounds 8 and 11 were cloned into the 5′ UTR of a GFP expressing gene in S. cerevisiae via homologous recombination. Individual cells were analysed for fluorescence changes upon addition of PARO. Candidates that showed a decrease in fluorescence upon ligand addition were selected for further characterization under the assumption that ligand binding stabilizes the aptamer in the 5′ UTR. This in turn prevents scanning of the ribosome in a road block-like manner as already shown for the tetracycline aptamer (36). As a first sorting step, fluorescence-based cell sorting of 2 million cells was performed to eliminate cells without GFP expression (i.e. cells with an inserted RNA structure that was too stable or cells with only the empty vector). For round 8, only 20% of the cells were removed but for round 11 this value increased to 50%, indicating that structured RNA elements were significantly enriched after 11 rounds of selection (data not shown). In total, fluorescence of more than 1200 candidates from both rounds (512 from round 8 and 682 from round 11) was then measured both in the absence and presence of 100 μM PARO. All GFP values were normalized to mCherry expression. The regulatory activity calculated as the ratio of GFP fluorescence in the absence divided by the fluorescence value in the presence of PARO is displayed in the box blot in Figure 3A. The candidate P11.2H2 (indicated by an arrow) showed the best regulatory activity with a value of 1.8-fold.
Figure 3.

In vivo screening for PARO riboswitches and development of the paromomycin riboswitch. (A) The boxplot displays regulation factors of the candidates identified by in vivo screening, the total number of candidates analysed in brackets. The analysis was done with the SELEX pool of rounds 8 and 11 and a doped pool of candidate P11.2H2 (DP 3%). The candidate P11.2H2 we found in the pool of round 11 is highlighted. Note that the other two candidates with ∼1.7-fold regulation proved to be false positives. (B) A doped pool of candidate (3% mutation rate) was screened for mutants with a gain- and loss-of-function phenotype (GOF and LOF, respectively). The heat map represents the frequency of GOF and LOF mutations found in every position amongst the 100 strongest GOF and 200 LOF candidates. Most frequent mutations are highlighted. (C) Comparison of the PARO riboswitch (PARO-RS, black bars) with the originally identified candidate P11.2H2 (grey bars). Displayed is the GFP expression in the absence and presence of 250 μM PARO and NEO, respectively. The results were normalized to GFP expression without ligand set as 100%. The error bars represent standard deviations amongst triplicates; the measurement was repeated at least twice. (D) Summary of the 10 most frequent GOF mutations. The GFP expression level was measured in the absence (black bars) and presence of 250 μM PARO (white bars), the mean regulation factor and the standard deviation are displayed above the bars. The measurements were done in triplicates and repeated twice.
Improving the paromomycin riboswitch by doped mutagenesis
To further improve regulation, we partially randomized the candidate P11.2H2 on each position between the first nucleotide of the 5′ untranslated region and the start codon of the GFP coding region. Based on our previous work, we chose 3.0% randomization of the pool to generate a library containing between one and three point mutations (12). This library was cloned into the plasmid pCBB06 in front of the GFP start codon and 1520 clones were specifically screened for gain- and loss-of-function (GOF and LOF, respectively). Hundred clones with improved regulation could be identified (GOF mutants, Figure 3A shaded). The sequencing results of these and of 200 LOF mutants were plotted on a heat map (Figure 3B). The heat map revealed that three regions contained almost no mutations in the GOF group but concentrated most of the LOF mutants (nt 38–42, 50–63 and 73–90), thus indicating that these regions may be important for ligand binding. Two other regions (nt 33–37 and 64–72) included most of the GOF mutations. Ten most frequently found GOF mutants were selected (C34U, C36G, A45C, A49U, U64C, U66A, C68U, C71U, U72G, U76G) and their impact on the switching factor was tested independently (Figure 3D). All mutations showed a slight increase of regulation. To further improve regulation, we cloned different combinations of three up to five mutations (Supplementary Figure S3). The construct that showed the best regulation was the combination of A45C, A49U and C71U with an 8.5-fold regulation (Figure 3C) that also gives a clear switching signal at lower ligand concentrations (Supplementary Figure S4). In the following, this improved construct was called PARO riboswitch.
In sum, initial screening, subsequent partial randomization and the combination of beneficial mutations led to a new synthetic PARO riboswitch. With 8.5-fold regulatory activity, the dynamic window is comparable to other synthetic riboswitches, e.g. the tetracycline, neomycin or ciprofloxacin riboswitches (12,20,36) and other RNA-based devices that control gene expression in eukaryotes.
Characterization of the binding specificities of the PARO riboswitch
To investigate the ligand binding specificity of the newly developed PARO riboswitch, we tested the switching behaviour with a set of closely related aminoglycoside antibiotics. As shown in Supplementary Figure S5, only PARO was able to trigger the riboswitch indicating a high specificity. These data fit well with in vitro binding studies we performed for P11.2H2. We could show binding only for PARO, but not for neomycin, ribostamycin, kanamycin, tobramycin, apramycin, streptomycin or G418 (Supplementary Figure S5). These results underline the distinct specificity of the riboswitch towards PARO both in vitro and in vivo. It is remarkable that the riboswitch does not even recognize neomycin, although a substitution of one hydroxyl group into an amino group is the only difference here. Failure of neomycin recognition proves that the counter elution step with neomycin was very efficient. We also demonstrated portability of the new riboswitch. P11.2H2 could regulate not only GFP but also mCherry expression (Supplementary Figure S6).
Next, we determined the dissociation constants for both P11.2H2 and the PARO riboswitch by isothermal titration calorimetry (ITC, Figure 4A and Supplementary Figure S7). The analysis resulted in a KD of 21 nM for the P11.2H2 and 10 nM for the optimized riboswitch, values that can be considered in the same range. Interestingly, the same observation has been made for the ciprofloxacin-binding riboswitch, where all the mutations that led to the gain-of-function did not change the ligand binding affinity (12). The KD of the PARO riboswitch for NEO was determined to be above 10 μM, indicating that neomycin does not specifically interact with the riboswitch. This result confirms that RNA Capture-SELEX can indeed generate highly specific aptamers that can discriminate closely related molecules.
Figure 4.

Biochemical characterization of the PARO riboswitch. (A) Determination of the binding constant of P11.2H2 with ITC against PARO (left) and NEO (right). Top panels: power required to maintain the temperature of the RNA solution recorded over the time until saturation was reached (baseline-corrected). Bottom panel: integrated heats of interaction plotted against the molar ratio of ligand over RNA and fitted to a single binding site model (MicroCal PEAQ-ITC Analysis Software 1.1.0). (B) In-line probing experiment for PARO-RS. Shown is the cleavage pattern in the absence (−) and presence (+) of 10 μM PARO under alkaline conditions. As references and for nucleotide position assignment, hydroxyl reaction (OH) and nuclease T1 digestion (T1) were loaded onto the gel. G nucleotides are highlighted. Colour coding for identified stem and loop regions follows the coding for the proposed secondary structure. The docking sequence is represented by the underlined nucleotides (68–80). 5′ and 3′ primer binding site regions are framed with dotted lines. Proposed secondary structure of the PARO-RS riboswitch including two stems (P1 and P2), two loop regions (L1 and L2), an asymmetric internal bulge (B2) and an unstructured spacer region (46–50). The mutated nucleotides compared to P11.2H2 are encircled. (C) Comparison of the GFP gene regulation between the P11.2H2 candidate and the constructs Delta S1 and Shuffle S1. As observed, none of the constructions delete the switching properties, but slightly reduce the switching factor. This indicates that the S1 plays probably a minor role in the switching properties of the riboswitch.
Structure–function analysis of the PARO riboswitch
The secondary structure prediction for the PARO riboswitch using mfold (37) proposed two consecutive stem–loop structures (Figure 4). We performed in-line probing to confirm this prediction. The cleavage pattern is shown in Figure 4B. It supports the existence of a first 8 bp long stem P1 closed by a pentaloop L1, preceded by an unstructured ‘spacer’ region. The probing also confirmed the proposed second stem P2 closed by a heptaloop L2. Stem P2 is disturbed by two bulged nucleotides (C61 and A80) and an asymmetric internal loop at the bottom. This internal loop is composed of two nucleotides on the 5′ side opposed by a 5 nt bulge B2 at the 3′ side. The probing pattern proposes that B2 undergoes a structural change upon the addition of PARO because the nucleotides A84-A87 are protected by PARO (Figure 4B, inset). This suggests that this region may represent the PARO-binding pocket. Moreover, we observed that the majority of the LOF mutants are located in this region, which indicates its importance. Based on the secondary structure, we can also explain some GOF mutations. Many are found because they cause a higher stability of the structure and therefore a decrease in expression level, e.g. U64C leads to a change of a GU wobble to a more stable GC bp. No obvious explanation can be given for A49U and C71U.
Interestingly, in-line probing appears to indicate an increase in flexibility for the nts 30–32 and A43 located in P1 after addition of the ligand. In addition, the GOF mutation A45C lead to an improvement of the switching behaviour by stabilizing P1 through the formation of a GC bp. In contrast, P1 could be deleted or shuffled without loss of regulation (Figure 4B). This information suggests that the structural flexibility of this riboswitch may be more complex than expected, perhaps due to the conditions imposed by Capture-SELEX. Finally, we suggest that P2 is the actual riboswitch and P1 may not be necessary for riboswitching. However, it may be able to modulate the activity of P2, perhaps by slowing down the scanning ribosome.
Impact of Capture-SELEX on finding synthetic riboswitches—an NGS point of view
This is the first time we introduced Capture-SELEX into our workflow to generate synthetic riboswitches. To assess the usefulness and power of the novel adaption of Capture-SELEX, we performed next-generation sequencing (NGS) of all selection rounds (libraries).
Sequencing all 11 rounds by Illumina sequencing resulted in 30.6 million reads that were equally distributed over each sequenced library. First, we performed a statistical analysis of the PARO SELEX and determined the enrichment of the hundred most enriched sequences (Top100, Figure 5A). An exponential enrichment could only be observed from round four to six. Before and after, the enrichment was rather linear. Up to round four, no significant changes could be detected and the proportion of the Top100 remained almost unchanged. The enrichment even levelled out towards the end, which could be a specific feature of this type of SELEX. Moreover, the calculated median of the RPM of the Top100 slightly decreased in the last rounds, meaning that an increase in stringency by counter selection or a reduced ligand concentration had a substantial impact on sequence distribution. Interestingly, our riboswitch candidate P11.2H2 continued to enrich in the latter rounds. To assess the total enrichment and to obtain a visual representation of the changes from round to round, we calculated the CDF of each round for each sequence based on their RPM. Then, we calculated the respective area under the curve (AUC) and transformed it into the Gini index. The Gini index is a measure of the imbalance from a certain starting point (i.e. with the starting library assuming equal distribution of nucleotide composition and sequence composition). Thus, its output represents the state of enrichment (Figure 5B). In our selection, the Gini index remained at 4% up to round three, and then exponentially increased from round four to six to then level out (see also Supplementary Table S4). The visual representation shows that an optimum of enrichment can be found in round seven and eight while maintaining a certain degree of enrichment. Additionally, we can observe a maximum of enrichment in round 11.
Figure 5.

Evaluation of the PARO SELEX by next-generation sequencing. (A) Plot of the RPMs of the Top100. Shown is the minimum and the maximum RPM found in each round (dotted lines) and the median for the Top100 (solid line). The Q1 and Q3 quartile are shown in light grey. The PARO riboswitch precursor P11.2H2 is displayed as a dotted green line. (B) Calculation of the cumulative distribution function (CDF) over all calculated reads per million (RPM) of each sequence within the respective library (for details see ‘Materials and methods’ section and the main text). Increase in AUC and also Gini-index over the course of the experiment can be seen (colour code as inlay). (C) Plot of the information entropy over all sequence frequencies of each selection round for PARO and CFX SELEX. A significantly larger decrease of pool complexity in Capture-SELEX (green line) compared to conventional SELEX (blue line) can be observed from round five on. (D) Density plot of the differences between maximum and minimum folding free energies of each accessible alternative secondary structure for each sequence in the Capture-SELEX and conventional SELEX approach. Comparison of the rounds before enrichment (both times in grey) and the last selection rounds (green for Capture-SELEX, blue for conventional SELEX) for both SELEX strategies. The folding energy span of the PARO riboswitch is displayed as dotted vertical line at 1.8 kcal/mol. (E) Comparison of the PARO Capture-SELEX approach (green line) versus previously published conventional CFX SELEX (blue line). The first 10 000 sequences of the respective last rounds are shown on the x-axis with their corresponding occurrences in reads per million on the y-axis. The precursors of the final riboswitches, namely P11.2H2 (for PARO) and 10A (for CFX) are highlighted. The slopes of the lines show that we obtain a higher degree of enrichment ( = better partitioning during the experiment) with the PARO Capture-SELEX approach. The figure also demonstrates that PARO Capture-SELEX facilitates the finding of the riboswitch precursor (compare P11.2H2 in Top100 to 10A in Top10 000).
Despite the fact that we found a steady increase of enrichment between rounds 4 and 7, the Shannon entropy still decreased until round 11 (Figure 5C). Early on, the entropy remained constant (rounds 1–4). However, as soon an enrichment was observed during the selection process (see Figure 2A), entropy decreased significantly. Even when the selection process seems to come to a halt (see Gini index), we observe a drop in entropy until the very end of the selection process.
To further assess the potential of the Capture-SELEX approach in the process of riboswitch development, we computed the most probable secondary structures for each RNA sequence and plotted the distance between the minimum and maximum folding free energies (span) for each structure as density plot for the Capture-SELEX (Figure 5D). Shown is the round immediately before enrichment (round 4) and the last selection round 11 (all other investigated rounds can be found in Supplementary Figure S8). The energy distribution span in round four displayed a sharp peak at 0 kcal/mol, revealing that most sequences have either only one accessible secondary structure or all structures have the same folding free energy. This peak was almost completely absent in round 11, which shows that all sequences with either only one structure or only structures with similar folding free energies have been evaluated. A peak centred at about 1.5 kcal/mol could be seen in round 11, which indicates the most common folding free energy span. This peak was present only as a part of a broader and significantly smaller band between 0.5 and 2 kcal/mol in round 4 but is already fully present in round 8 (Supplementary Figure S8).
We did a similar analysis for the ciprofloxacin (CFX) riboswitch that was found with a conventional SELEX instead of a Capture-SELEX approach (12). Although we are aware of the limitations and ambiguities of a comparison of two individual selection processes carried out with different ligands, we still consider a cautious and measured comparative assessment valid. Proceeding with care, we were able to observe several distinct effects and differences. Compared to the Capture-SELEX, there was no shift in the distribution of the folding energy spans from round 4 to the last round of the CFX SELEX (R10) (Figure 5D, blue curves and Supplementary Figure S8). This finding suggests that sequences with multiple low folding energy structures are enriched in the Capture-SELEX process. This interpretation is further substantiated considering the folding energy span of the selected riboswitch, which falls directly into the increasing peak (Supplementary Figure S8, dotted line). Moreover, when looking at the Shannon entropy, the decrease of the traditional SELEX was rather moderate compared to the PARO SELEX. This could be interpreted as Capture-SELEX showing a greater speed of enrichment of specific secondary structures than traditional SELEX. However, to substantiate these hypotheses, analysis of many more selections, preferably with the same ligand, would be required.
Comparing the enrichment of the riboswitch candidates found in the PARO and the CFX SELEX, respectively, also led to interesting results. We found P11.2H2 to be amongst the Top100 on rank 65, whereas the candidate for the CFX riboswitch (10A) ranked 3476 (Figure 5D). For P11.2H2, the RPM was calculated to 1049.9, whereas we only obtained 43.5 for 10A. Consequently, about 30 000 clones had to be screened in theory to find 10A, whereas the likelihood to find P11.2H2 was 1 in 952. In other words, P11.2H2 was about 25 times easier to find than 10A. Whether this is due to the Capture-SELEX method remains to be seen.
Additionally, the slope of the PARO SELEX (green line in Figure 5D) is steeper compared to the one of the conventional CFX SELEX (blue line), meaning that we also have a greater partitioning effect with the Capture-SELEX approach over the course of the experiment. Consequently, not only the enrichment of the binding RNA species (aka aptamers) is higher, but also the ones that show regulation are more enriched in terms of their rank in the library.
In sum, our data suggest that the new approach considerably streamlines the process of synthetic riboswitch identification. Thus, finding a riboswitch is no longer a matter of chance associated with enormous screening efforts, but rather a directed search with a much improved rate of success.
Construction of an NOR Boolean logic gate
In a final step, we aimed to apply the novel PARO riboswitch in combination with another synthetic riboswitch (20) for the assembly of an orthogonal Boolean NOR logic gate. As the PARO riboswitch is not triggered by NEO and the NEO riboswitch is not triggered by PARO (15), we decided for the NEO riboswitch (NEO-PARO). Since we found P1 to be irrelevant for regulation, we replaced it with another riboswitch to build a logic gate. We also designed a construct in which the NEO riboswitch was cloned downstream of P2 (PARO-NEO) and one with the NEO riboswitch in front of the complete PARO riboswitch (NEO-fullPARO). The GFP expression was measured without ligand, with 250 μM neomycin, with 250 μM PARO and with 250 μM of both (Figure 6B). For all constructs, we observed a response with one ligand input that further increased with both.
Figure 6.
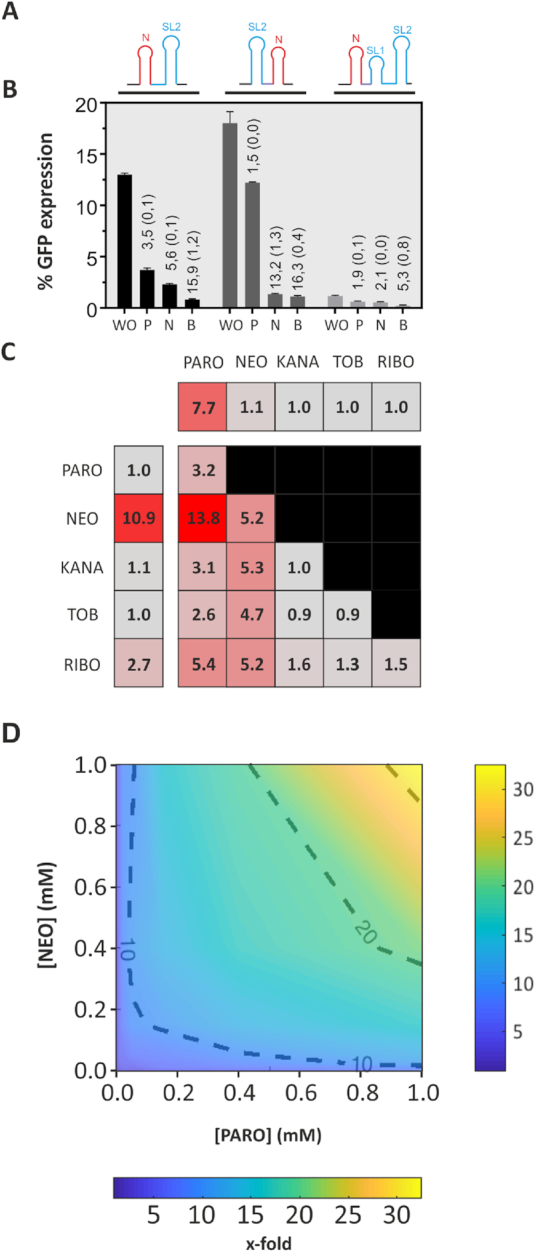
Engineering of an NOR Boolean Logic gate. (A) Representation of the construct NEO-PARO (I), PARO-NEO (II) and NEO-full PARO (III). The neomycin riboswitch (N) is represented in red. The paromomycin riboswitch containing the first stem loop (SL1) and second stem loop (SL2) in blue. A CAA spacer region was sometimes introduced and is represented in purple. All these construct were inserted in front of the GFP gene. (B) Comparison of the GFP expression of three tandem constructs NEO-PARO (black), PARO-NEO (dark grey) and NEO-full PARO (light grey): Experiment was made either without ligand (WO), with 250 μM PARO (P), with 250 μM NEO (N) or with 250 μM of both antibiotic (B). (C) Heat map of the switching factor of the PARO-RS (first line), the NEO-RS (first column) and the logic gate NEO-PARO. Both riboswitches were tested against 250 μM of each of these antibiotics. For the PARO switch, only PARO triggered the switch. However, for NEO, RIBO is also able to slightly trigger the switch. The logic gate NEO-PARO was tested against each five antibiotics previously tested, but as well on all the combination of two different antibiotics at 250 μM each. The lower the switching factor is, the more the colour of the square will turn grey. As expected, the NEO-PARO shows the best switching factor when the combination of both NEO and PARO are added to the media. (D) Dose–response experiment of the NEO-PARO construct in function of the PARO and NEO concentration. The darker blue the colour is, the lower the switching factor is. The redder, the higher is the regulatory activity. Results between each measured concentrations were interpolated.
The analysis of the gates led to some interesting observations. The neomycin riboswitch was actually able to replace P1 of the PARO riboswitch, which leads to even better regulation in the presence of both ligands. The combination of the neomycin with the complete PARO riboswitch, however, strongly inhibited expression already in the absence of any ligand. This indicates that the introduced secondary structures may be too stable. Regulation can be observed, but almost at background level (Figure 6A). When comparing the constructs without P1, the PARO-NEO construct showed a weak response to PARO alone but a strong response to NEO alone. On the other hand, the NEO-PARO construct showed an input in the same range for both antibiotics alone and a strong downregulation upon the addition of both antibiotics combined. The data show that riboswitches can be combined to logical gates that have a better performance than a single riboswitch. However, the genetic context still plays an important role and influences the switching behaviour.
Finally, the NEO-PARO construct was tested with different combinations of antibiotics at 250 μM for each target and the switching factor was plotted on a heat map (Figure 6C). The NEO-PARO construct showed expected behaviour depending of the ligand added to the media. As the NEO switch was also slightly triggered by RIBO, there was a minor effect on the NEO-PARO construct in the presence of RIBO. The combination of both NEO and PARO in the media resulted in the best switching factor of up to 13.8-fold. The behaviour of this logic gate was studied in a dose–response manner with a varying input of both antibiotics. Yeast cells were incubated in 64 different conditions with 8 different concentrations (0, 2, 10, 20, 50, 150, 400 and 1000 μM) of both antibiotics supplemented to the media. The fluorescence of each condition was measured and the regulatory activity was plotted (Figure 6D). As expected, the regulatory activity was higher when the concentration of one or the other antibiotic was higher. However, the results showed that a small input of both antibiotics leads to a higher regulatory activity than a high concentration of only one antibiotic. This proves the high reactivity of riboswitches towards their ligand. In fact, a small input of both target molecules was able to trigger the regulatory activity of the riboswitch better than a high input of only one.
DISCUSSION
In the present study, we succeeded in finding a new synthetic riboswitch. It is a 44 nt long RNA aptamer that, when inserted into the 5′ untranslated region of a reporter gene, leads to the repression of gene expression when the ligand PARO is bound. The dynamic range of regulation of the new riboswitch is comparable to other synthetic riboswitches identified so far (12,13,31). The ligand binds with high affinity (KD = 20 nM for PARO) and specificity (>10 μM for NEO). The combination of several point mutations identified by screening for gain-of-function of the partially mutagenized candidate P11.2H2 considerably improved switching efficiency. A look at the identified secondary structure reveals that the nucleotides that lead to improved regulation are exclusively located in the peripheral regions and not in the proposed ligand binding pocket. A similar observation was made with the CFX riboswitch (12). Also in this case, screening of the partially randomizsed initial riboswitch candidate did not change ligand binding affinity but improved regulation, with gain-of-function mutations also located in peripheral regions but not within the postulated binding pocket.
The most notable feature of the novel riboswitch is the selective discrimination between NEO and PARO. NEO differs from PARO only in one amino group, yet its binding is reduced by a factor of 10 000. Consequently, the PARO riboswitch is triggered only by PARO, not by NEO, whereas a NEO riboswitch (20) is triggered exactly the opposite way. Despite the fact that the two riboswitches bind almost identical ligands, they do not share any sequence similarities. For the NEO riboswitch, both the NEO- and the PARO-bound structure have been resolved by NMR spectroscopy (15,38). The two structures are nearly identical but only NEO and not PARO can flip the switch. A detailed view into the structure revealed that the local loss of only a few key interactions, namely, the hydrogen bonds between the amino group and the 4-CO group of U10 and also to the negatively charged backbone phosphate group of G9 is translated into global changes in RNA conformational dynamics (15). It will be interesting to investigate whether there is a similar mechanism for the PARO riboswitch. The combination of different riboswitches, e.g. the PARO and the NEO switch can be used to build logical gates, which leads to even better switching behaviour. It allows different input signals to be integrated in one regulatory device. Interestingly, the concentration dependence in the 2D matrix shows that the overall amount of inducer required is lower when two riboswitches are combined. This enhancing effect has already been observed in other studies (39,40).
The number of small molecule-binding aptamers that can be used for RNA engineering to date is very limited and only one novel regulatory aptamer, the CFX riboswitch, was discovered within the last 10 years (12). This riboswitch, however, was found by conventional SELEX and not by Capture-SELEX. We were therefore interested to ask whether we could improve the workflow of finding riboswitches by introducing an RNA-adapted Capture-SELEX approach. We obtained a significant enrichment from first to last round but also a high occurrence in the rank compared to the CFX riboswitch from the conventional SELEX (rank 65 versus 3476). So, the chance to find a riboswitch in a Capture-SELEX was considerably high (25× higher when comparing the PARO to CFX selection). Another interesting observation for the Capture-SELEX was that despite the fact that increasing stringency was applied during selection (by counter-selection and decreasing the ligand concentration for elution), the median of all sequences decreased, yet the riboswitch continued to rise. Moreover, this may emerge as a specific property of Capture-SELEX, we observed an enrichment of sequences that not only have one predicted secondary structure (i.e. rigid aptamers), but can also fold into different, but energetically similar structures. There appears to be a small band of sequences with folding energy spans around 1.5 kcal/mol that is enriched over the Capture-SELEX in which the folding energy span of the selected riboswitch is located. This further hints to the finding that sequences with several low folding energy structures and hence more structural flexibility are predominantly selected by Capture-SELEX. Such flexibility may be an important feature for riboswitch functionality. Furthermore, this enrichment process was finished at around round 8 thar hints to a faster convergence speed of Capture-SELEX when compared to traditional SELEX. These preliminary findings will be the subject of further investigations.
In sum, we have not only simplified and optimized the laboratory work (faster, no need for ligand immobilization) but also significantly improved the success rate when it comes down to find new synthetic riboswitches (because we selected not only for binding but also for functionality). Thus, our implementation of the Capture-SELEX has streamlined the process of riboswitch development even further, an improvement that will inspire and accelerate the field of synthetic riboswitches. Many exciting applications are now conceivable not only for the conditional control of gene expression, but also as intracellular biosensors in biotechnology for strain optimization or for the real-time monitoring of metabolic flows in living cells. We look forward to the future with excitement and anticipation as to what new developments our new technology platform for riboswitch development may bring.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank their colleague François-Xavier Lehr for his help in designing of the logic gate 2D surface graphic.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Marie Skłodowska-Curie grant agreement No. [642738] (H2020 of the European Union ITN MetaRNA); Deutsche Forschungsgemeinschaft [SFB902/A2]; LOEWE CompuGene. Funding for open access charge: Deutsche Forschungsgemeinschaft.
Conflict of interest statement. None declared.
REFERENCES
- 1. Doudna J.A., Cech T.R.. The chemical repertoire of natural ribozymes. Nature. 2002; 418:222–228. [DOI] [PubMed] [Google Scholar]
- 2. Evans D., Marquez S.M., Pace N.R.. RNase P: interface of the RNA and protein worlds. Trends Biochem. Sci. 2006; 31:333–341. [DOI] [PubMed] [Google Scholar]
- 3. Green A.A., Silver P.A., Collins J.J., Yin P.. Toehold switches: de-novo-designed regulators of gene expression. Cell. 2014; 159:925–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chappell J., Westbrook A., Verosloff M., Lucks J.B.. Computational design of small transcription activating RNAs for versatile and dynamic gene regulation. Nat. Commun. 2017; 8:1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nelson J.W., Breaker R.R.. The lost language of the RNA World. Sci. Signal. 2017; 10:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Serganov A., Nudler E.. A decade of riboswitches. Cell. 2013; 152:17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Berens C., Groher F., Suess B.. RNA aptamers as genetic control devices: The potential of riboswitches as synthetic elements for regulating gene expression. Biotechnol. J. 10:246–257. [DOI] [PubMed] [Google Scholar]
- 8. Tuerk C., Gold L.. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990; 249:505–510. [DOI] [PubMed] [Google Scholar]
- 9. Ellington A.D., Szostak J.W.. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990; 346:818–822. [DOI] [PubMed] [Google Scholar]
- 10. Groher F., Suess B.. Synthetic riboswitches - a tool comes of age. Biochim. Biophys. Acta. 2014; 1839:964–973. [DOI] [PubMed] [Google Scholar]
- 11. Chang A.L., Wolf J.J., Smolke C.D.. Synthetic RNA switches as a tool for temporal and spatial control over gene expression. Curr. Opin. Biotechnol. 2012; 23:679–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Groher F., Bofill-Bosch C., Schneider C., Braun J., Jager S., Geißler K., Hamacher K., Suess B.. Riboswitching with ciprofloxacin-development and characterization of a novel RNA regulator. Nucleic Acids Res. 2018; 46:2121–2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Weigand J.E., Schmidtke S.R., Will T.J., Duchardt-Ferner E., Hammann C., Wöhnert J., Suess B.. Mechanistic insights into an engineered riboswitch: A switching element which confers riboswitch activity. Nucleic Acids Res. 2011; 39:3363–3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nutiu R., Li Y.. In vitro selection of structure-switching signaling aptamers. Angew. Chem. Int. Ed. 2005; 44:1061–1065. [DOI] [PubMed] [Google Scholar]
- 15. Duchardt-Ferner E., Gottstein-Schmidtke S.R., Weigand J.E., Ohlenschläger O., Wurm J.P., Hammann C., Suess B., Wöhnert J.. What a difference an OH Makes: Conformational dynamics as the basis for the ligand specificity of the Neomycin-Sensing riboswitch. Angew. Chem. Int. Ed. 2016; 55:1527–1530. [DOI] [PubMed] [Google Scholar]
- 16. Schneider C., Suess B.. Identification of RNA aptamers with riboswitching properties. Methods. 2016; 97:44–50. [DOI] [PubMed] [Google Scholar]
- 17. Townshend B., Kennedy A.B., Xiang J.S., Smolke C.D.. High-throughput cellular RNA device engineering. Nat. Methods. 2015; 12:989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lynch S.A., Gallivan J.P.. A flow cytometry-based screen for synthetic riboswitches. Nucleic Acids Res. 2009; 37:184–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Eckdahl T.T., Campbell A.M., Heyer L.J., Poet J.L., Blauch N., Snyder N.L., Atchley D.T., Baker E.J., Brown M., Brunner C. et al.. Programmed Evolution for optimization of orthogonal metabolic output in bacteria. PLoS One. 2015; 10:e0118322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Weigand J.E., Sanchez M., Gunnesch E.-B., Zeiher S., Schroeder R., Suess B.. Screening for engineered neomycin riboswitches that control translation initiation. RNA. 2008; 14:89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stoltenburg R., Nikolaus N., Strehlitz B.. Capture-SELEX: Selection of DNA aptamers for aminoglycoside antibiotics. J Anal Methods Chem. 2012; 2012:415697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lauridsen L.H., Doessing H.B., Long K.S., Nielsen A.T.. Jensen MK, Keasling JD. A Capture-SELEX Strategy for Multiplexed Selection of RNA Aptamers Against Small Molecules. Synthetic Metabolic Pathways: Methods and Protocols. 2018; NY: Springer; 291–306. [DOI] [PubMed] [Google Scholar]
- 23. Rajendran M., Ellington A.D.. In vitro selection of molecular beacons. Nucleic Acids Res. 2003; 31:5700–5713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Feagin T.A., Soh H.T.. Strategies for creating structure-switching aptamers. ACS Sens. 2018; 3:1611–1615. [DOI] [PubMed] [Google Scholar]
- 25. Suess B., Fink B., Berens C., Stentz R., Hillen W.. A theophylline responsive riboswitch based on helix slipping controls gene expression in vivo. Nucleic Acids Res. 2004; 32:1610–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Batey R.T. 2013; 2:299–311.NIH Public Access. [Google Scholar]
- 27. Scanlon T.C., Gray E.C., Griswold K.E.. Quantifying and resolving multiple vector transformants in S. cerevisiae plasmid libraries. BMC Biotechnol. 2009; 9:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Groher F., Suess B.. In vitro selection of antibiotic-binding aptamers. Methods. 2016; 106:42–50. [DOI] [PubMed] [Google Scholar]
- 29. Seetharaman S., Zivarts M., Sudarsan N., Breaker R.R.. Immobilized RNA switches for the analysis of complex chemical and biological mixtures. Nat. Biotechnol. 2001; 19:336–341. [DOI] [PubMed] [Google Scholar]
- 30. Regulski E.E., Breaker R.R.. Wilusz J. In-Line Probing Analysis of Riboswitches. Post-Transcriptional Gene Regulation. 2008; Totowa, NJ: Humana Press; 53–67. [DOI] [PubMed] [Google Scholar]
- 31. Atkinson A.B. On the measurement of inequality. J. Econ. Theory. 1970; 2:244–263. [Google Scholar]
- 32. Rouillard J.M., Zuker M., Gulari E.. OligoArray 2.0: Design of oligonucleotide probes for DNA microarrays using a thermodynamic approach. Nucleic Acids Res. 2003; 31:3057–3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mathews D.H., Sabina J., Zuker M., Turner D.H.. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure11Edited by I. Tinoco. J. Mol. Biol. 1999; 288:911–940. [DOI] [PubMed] [Google Scholar]
- 34. Wickham H. Ggplot2: Elegant Graphics For Data Analysis. 2016; Switzerland: Springer. [Google Scholar]
- 35. Hamilton R., Watanabe C.K., de Boer H.A.. Compilation and comparison of the sequence context around the AUG startcodons in Saccharomyces cerevisiae mRNAs. Nucleic Acids Res. 1987; 15:3581–3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Suess B., Hanson S., Berens C., Fink B., Schroeder R., Hillen W.. Conditional gene expression by controlling translation with tetracycline-binding aptamers. Nucleic Acids Res. 2003; 31:1853–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003; 31:3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Duchardt-Ferner E., Weigand J.E., Ohlenschläger O., Schmidtke S.R., Suess B., Wöhnert J.. Highly modular structure and ligand binding by conformational capture in a minimalistic riboswitch. Angew. Chem. Int. Ed. 2010; 49:6216–6219. [DOI] [PubMed] [Google Scholar]
- 39. Schneider C., Bronstein L., Diemer J., Koeppl H., Suess B.. ROC’n’Ribo: Characterizing a riboswitching expression system by modeling Single-Cell data. ACS Synth. Biol. 2017; 6:1211–1224. [DOI] [PubMed] [Google Scholar]
- 40. Beilstein K., Wittmann A., Grez M., Suess B.. Conditional control of mammalian gene expression by Tetracycline-Dependent hammerhead ribozymes. ACS Synth. Biol. 2015; 4:526–534. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.