


Abstract
Next-generation sequencing (NGS) technologies have been employed in several phage display platforms for analyzing natural and synthetic antibody sequences and for identifying and reconstructing single-chain variable fragments (scFv) and antigen-binding fragments (Fab) not found by conventional ELISA screens. In this work, we developed an NGS-assisted antibody discovery platform by integrating phage-displayed, single-framework, synthetic Fab libraries. Due to limitations in attainable read and amplicon lengths, NGS analysis of Fab libraries and selection outputs is usually restricted to either VH or VL. Since this information alone is not sufficient for high-throughput reconstruction of Fabs, we developed a rapid and simple method for linking and sequencing all diversified CDRs in phage Fab pools. Our method resulted in a reliable and straightforward platform for converting NGS information into Fab clones. We used our NGS-assisted Fab reconstruction method to recover low-frequency rare clones from phage selection outputs. While previous studies chose rare clones for rescue based on their relative frequencies in sequencing outputs, we chose rare clones for reconstruction from less-frequent CDRH3 lengths. In some cases, reconstructed rare clones (frequency ∼0.1%) showed higher affinity and better specificity than high-frequency top clones identified by Sanger sequencing, highlighting the significance of NGS-based approaches in synthetic antibody discovery.
INTRODUCTION
Phage display is a widely used technology for generating, identifying and engineering fully human antibodies, and it has delivered numerous antibodies for research and clinical applications (1). A typical antibody phage display experiment consists of three major steps: (i) generation and display of an antibody library, such as antigen-binding fragment (Fab) library or single-chain variable fragment (scFv) library; (ii) enrichment of antigen-binding antibodies by multiple rounds of binding selection and amplification and (iii) recovery of antigen-specific antibodies from phage selection pools (2). Sanger sequencing is commonly used at each of these steps to decode sequences of antibody fragments recovered from phage pools. It requires the isolation of individual phagemids from phage pools and is practically limited to sequencing a few hundred clones routinely. Since each selection round in phage display gives an output of up to ∼106 sequences, the use of next-generation sequencing (NGS) becomes necessary if one must capture and interrogate the entire sequence diversity present in phage selection outputs. With NGS, entire phage pools can be subjected to sequencing (isolation of individual phagemids is not required) and ∼106–109 sequences can be readily obtained (3,4). Up to ten NGS platforms exist, each with its own advantages and preferred applications, with primary variables being read length, data quality and quantity, time and cost (5,6). Three commonly used NGS platforms in the antibody field include 454 (Roche), MiSeq (Illumina) and Ion Torrent (Life Technologies).
The Fischer lab was the first to use NGS in antibody phage display (2). They used the Illumina platform to sequence the complementarity-determining region H3 (CDRH3) from a multiple-framework synthetic scFv library. NGS analysis was used to assess the quality of the library and to follow changes in heavy chain germline usage and CDRH3 length distribution over three rounds of selection. They also developed a strategy to rescue rare scFv clones from phage pools using fragment assembly (2). The Fischer lab used the same approach to sequence the CDRH3 region of semi-synthetic scFv libraries and to rescue rare scFv clones from phage selection outputs (7,8). The Lerner lab used Roche’s 454 to sequence the VH region of a phage selection output from a natural scFv library (9). Three strategies were tested for recovering scFv clones based on CDRH3 sequences: fragment assembly, rolling circle amplification and hybridization using biotin probes (9). A few labs have used Roche’s 454 to sequence the VH and VL regions of synthetic Fab/scFv libraries, and the NGS information was used to assess the quality of libraries (10–12). The Bradbury lab used the Ion Torrent platform to sequence the CDRH3 region of two different selection outputs from a natural scFv library (13). A rescue strategy was developed to isolate scFv clones from selection outputs using inverse polymerase chain reaction (PCR) and ligation (13,14). Recently, MiSeq (Illumina) was used to sequence the CDRH2–CDRH3 region of a selection output from a single-framework synthetic Fab library (15), and the VH region of selection outputs from immune scFv libraries (16). Both groups used CDRH3 information for analyzing the amino acid composition, for monitoring the enrichment process and for retrieving rare clones by fragment assembly (15,16).
For antibody engineering, a key aspect is to obtain the entire sequence of highly diverse VH and VL chains. PacBio offers the longest read length but is more expensive and less commonly used. Among the commonly used NGS platforms, Roche’s 454 offers the longest read length (700 bp), which is only sufficient to cover one of the variable chains (17). Due to this read-length limitation in short-read DNA sequencing platforms, NGS analysis of phage-displayed Fab/scFv libraries or selection outputs is usually restricted to VH or VL. CDRH3 is typically the most diversified CDR in Fab/scFv libraries due to its dominant role in antigen recognition. CDRH3 sequencing has been used to assess the quality of libraries, to monitor the evolution of Fabs during selections and to characterize changes in CDR length or amino acid distribution during selections (2,13). Reconstruction of rare clones, however, is not the same as sequence identification. To reconstruct full-length Fab or scFv clones from NGS information, CDRH3 information alone is not sufficient, as antibody libraries typically contain two, four or all six diversified CDRs. To circumvent this, a few strategies have been tested to rescue Fab/scFv clones from selection outputs based only on CDRH3 information. These strategies use both hybridization- and PCR-based cloning techniques to rescue the entire sequences of Fab/scFv clones (2,9,13,15,16). Gene synthesis has also been used to reconstruct scFv clones from NGS information (18).
The objective of this work was to develop an NGS-assisted antibody discovery platform. First, we developed a rapid and simple method for linking and sequencing all diversified CDRs in phage Fab pools without losing the CDR pairing information. Our method resulted in a reliable and straight-forward platform for converting NGS information into Fab clones. Next, we used the NGS-assisted antibody discovery platform to reconstruct and test low-frequency rare Fab clones from phage selection outputs. While previous studies chose rare clones for rescue based on their relative frequencies in sequencing outputs, we chose rare clones for reconstruction from less-frequent CDRH3 lengths. In some cases, reconstructed rare clones (frequency ∼0.1%) showed higher affinity and better specificity than high-frequency top clones isolated by Sanger sequencing, highlighting the significance of NGS-based approaches in synthetic antibody discovery.
MATERIALS AND METHODS
Phage display selections
Recombinant Fc-extracellular domain fusions of human Jagged-2 and Notch-3 were purchased from R&D Systems and used as selection targets. Solid-phase panning of Library-S (19) and Library-F (20) was conducted according to previously described protocols (21,22). Briefly, phages from the frozen master library were precipitated, deselected for binding to the Fc protein, cycled through rounds of binding selection with the target protein immobilized on 96-well Maxisorp plates and amplified in XL1-Blue Escherichia coli cells. After four rounds of selections, phage clones were plated as individual colonies for isolation, sequencing and manipulation of phagemid DNA.
Ion Torrent sequencing and data analysis
Ion Torrent sequencing of diversified CDRs was accomplished in three major steps: PCR amplification of CDRs, emulsion PCR on Ion sphere particles (ISPs) and sequencing enriched ISPs on an Ion semiconductor chip (23). To PCR amplify CDRs from phage pools, we designed primers that hybridize to the fixed framework regions of the phagemid that flank the CDR region. Primers contained barcodes for multiplexing purposes and adapter sequences to facilitate emulsion PCR. We PCR amplified the CDR of interest from phage samples, checked the purity, concentration and length of PCR products using a 2100 bio-analyzer (Agilent Technologies), prepared the template for emulsion PCR by pooling multiple PCR products, performed emulsion amplification of the amplicon library on the Ion OneTouch 2 instrument (Life Technologies), loaded the enriched ISPs into an Ion 314 Semiconductor chip, and sequenced the loaded ISPs on the V2 Ion Personal Genome Machine (Thermo-Scientific), according to manufacturer’s instructions.
Ion Torrent sequencing of one diversified CDR was accomplished in three steps: (i) The CDR of interest was PCR amplified from phage selection pools using barcoded forward and reverse primers (Supplementary Table S1). The PCR reaction mix (50 μl) contained 32.5 μl of nuclease-free H2O, 10 μl of 5× Phusion High-Fidelity buffer (New England BioLabs), 1 μl of dNTP mix (10 mM of each nucleotide), 1 μl of phage solution (1012 PFU/ml), 2.5 μl of 10 μM Forward primer, 2.5 μl of 10 μM Reverse primer and 0.5 μl of Phusion Hot-Start Flex DNA Polymerase (New England BioLabs). The reaction mix was subjected to PCR using the following conditions: initial denaturation at 98°C for 30 s, 25 amplification cycles each consisting of a denaturing step at 98°C for 10 s, an annealing step at 56°C for 10 s and an extension step at 72°C for 5 s, and a final extension at 72°C for 15 sec. (ii) PCR amplicons were purified, quantified, multiplexed and subjected to emulsion PCR using the Ion OneTouch template kit. (iii) Enriched ISPs were loaded on an Ion 314 chip and sequenced using the Ion PGM kit.
Ion Torrent sequencing of the L3-H3 CDR strip was accomplished in six steps: (i) ssDNA was extracted from amplified phage selection outputs (1013 PFU) using the Spin M13 kit. (ii) About 500 ng of ssDNA was subjected to Kunkel mutagenesis (24) to delete framework regions between diversified CDRs. In the mutagenesis reaction, one oligonucleotide, L3-H3 Seq (Supplementary Table S1) was used to link the L3-H3 regions together. Phosphorylation of L3-H3 Seq, annealing of L3-H3 Seq to the ssDNA template and in vitro synthesis of CCC-dsDNA were carried out as described previously (25,26). (iii) DNA from the mutagenesis reaction was run on an agarose gel and the right-sized product (CCC-dsDNA) was excised and purified using a gel-extraction kit. (iv) The L3-H3 CDR strip was PCR amplified from the purified CCC-dsDNA template using barcoded L3-Fwd and H3-Rev primers (Supplementary Table S1). The PCR reaction mix (50 μl) contained 28.5 μl of nuclease-free H2O, 10 μl of 5X Phusion High-Fidelity buffer, 1 μl of dNTP mix, 5 μl of CCC-dsDNA (50 ng), 2.5 μl of 10 μM L3-Fwd, 2.5 μl of 10 μM H3-Rev and 0.5 μl of Phusion Hot-Start Flex DNA Polymerase. The reaction mix was subjected to PCR using the following conditions: initial denaturation at 98°C for 30 s, 25 amplification cycles each consisting of a denaturing step at 98°C for 10 s, an annealing step at 56°C for 10 s and an extension step at 72°C for 5 s, and a final extension at 72°C for 15 s. (v) PCR amplicons were purified, quantified, multiplexed and subjected to emulsion PCR using the Ion PGM Template OT2 200 kit. (vi) Enriched ISPs were loaded on an Ion 314 Chip and sequenced using the Ion PGM Sequencing 200 V2 kit.
Ion Torrent sequencing of the L3-H1-H2-H3 CDR strip was accomplished in six steps: (i) ssDNA was extracted from amplified phage selection outputs (1013 PFU) using the Spin M13 kit. (ii) About 500 ng of ssDNA was subjected to Kunkel mutagenesis (24) to delete framework regions between four diversified CDRs. In the mutagenesis reaction, three oligonucleotides L3-H1 Seq, H1-H2 Seq and H2-H3 Seq were used to link the L3-H1-H2-H3 regions together (oligonucleotide sequences in Supplementary Table S1). Phosphorylation of oligonucleotides, annealing of oligonucleotides to the ssDNA template and in vitro synthesis of CCC-dsDNA were carried out as described previously (25,26). (iii) DNA from the mutagenesis reaction was run on an agarose gel and the right-sized product (CCC-dsDNA) was excised and purified using a gel-extraction kit. (iv) The L3-H1-H2-H3 CDR strip was PCR amplified from 50 ng of purified CCC-dsDNA using barcoded L3-Fwd and H3-Rev primers (see reaction setup and conditions above). (v) PCR amplicons were purified, quantified, multiplexed and subjected to emulsion PCR using the Ion PGM Template OT2 400 kit. (vi) Enriched ISPs were loaded on an Ion 314 Chip and sequenced using the Ion PGM Sequencing 400 kit.
We built a custom workflow for NGS data processing and analysis (Supplementary Figure S1). Sequences were base called and separated by barcode on the Ion PGM Torrent Server and exported in FASTQ format. Sequences were imported into the Galaxy server (27,28), where they were trimmed based on quality score (>17), converted to FASTA and then run on a custom R script (29) to parse the CDR, and to translate and count CDR sequences (Supplementary Figure S2). CDR sequences were processed using the Biostrings package (30) and length distribution plots were generated using the ggplot2 package (31).
Reconstruction of rare Fab clones
To reconstruct rare Fab clones from the CDR strip sequencing information, we cloned desired CDR combinations into a Hu4D5-Fab-encoding phagemid by Kunkel mutagenesis (24). A Hu4D5-Fab phagemid whose CDRs have been replaced with NotI sites was used as a template to reconstruct Fab clones. Oligonucleotides were designed to encode for a desired CDR sequence and to hybridize to either side of the CDR. Two or four oligonucleotides were used in the mutagenesis reaction for reconstructing rare Fabs from Library-S (19) or Library-F (20) selections, respectively. Phosphorylation of oligonucleotides, annealing of oligonucleotides to the uracil-inserted ssDNA template and in vitro synthesis of CCC-dsDNA were carried out as described previously (25,26). Following mutagenesis, the reaction product was transformed into dut+/ung+E. coli to eliminate the wild-type template strand. Positive Fab clones were screened by NotI restriction digestion analysis.
Fab expression and purification
Fab sequences from the phagemid vector were sub-cloned into a modified pCW-LIC Fab expression vector using standard molecular biology procedures. Briefly, Fab sequences were amplified from phagemids by PCR and ligated into the SacI/XhoI-digested pCW-LIC vector using Gibson assembly (32). Gibson assembly reactions were electroporated into BL21 E. coli cells and three colonies from each reaction were screened for Fab expression using bio-layer interferometry. Briefly, single colonies were transferred to 1 ml of Overnight Express Terrific Broth (TB) auto-induction medium (EMD Millipore) supplemented with 100 μg/ml carbenicillin in 96-well deep-well plates and incubated for 18 h at 25°C and 200 rpm. Cells were pelleted by centrifugation and lysed with 200 μl of B-PER bacterial protein extraction reagent (Pierce). Cells were centrifuged again, and 50 μl of the clarified supernatant was transferred to 384-well plates. Fab expression was detected using anti-Fab CH1 biosensors and anti-HIS biosensors in the ForteBio Octet RED384 system (Pall Corporation), according to manufacturer’s instructions. Positive clones were transferred into 30 ml of TB auto-induction medium supplemented with 100 μg/ml carbenicillin and incubated for 18 h at 25°C with shaking at 200 rpm. Cells were pelleted by centrifugation, suspended in protein-L binding buffer (20 mM Na2HPO4, 0.15 M NaCl, pH 8), containing 1:100 dilution of protease inhibitor cocktail (Sigma) and lysed with a cell disruptor (Constant Systems). Clarified supernatant was incubated with 200 μl of Protein-L resin (GenScript) for 1 h at 4°C. The Protein-L resin was collected by centrifugation and washed 5X with Protein-L binding buffer. Fabs were eluted with IgG elution buffer (Thermo-Scientific) and neutralized with 1 M Tris–HCl (pH 9). Eluted Fabs were dialyzed against PBS and stored at −20°C. Fab purity was verified using 2100 bio-analyzer, and Fab concentration was determined by UV–visible spectrometry.
Enzyme-linked immunosorbent assays
Phage-ELISA was performed to check the binding of phage-displayed Fabs to immobilized target proteins. The Fab-encoding phagemid was electroporated into M13KO7-infected electro-competent SR320 E. coli cells for phage production. Cells were rescued with pre-warmed SOC media and incubated for 30 min at 37°C. The culture was transferred to 30 ml of 2YT media supplemented with 100 μg/ml carbenicillin and 25 μg/ml kanamycin and incubated overnight at 37°C with shaking at 200 rpm. Phages were precipitated from the culture supernatant using 6 ml of ice-cold PEG/NaCl solution, resuspended in PBT buffer (PBS containing 0.5% BSA and 0.05% Tween) and quantified using UV spectrometry.
To conduct phage-ELISA, target and control proteins were immobilized at 5 μg/ml on Maxisorp plates (Nunc) by overnight incubation at 4°C. Wells were subsequently blocked with PB buffer (PBS containing 0.5% BSA) for 90 min at RT before washing four times with PT buffer PBS containing 0.05% Tween. Wells were exposed to PBT-diluted phage solution (1012 PFU/ml) for 30 min, washed 8× with PT buffer and incubated with a 1:3000 dilution of HRP-conjugated anti-M13 antibody (GE healthcare) for 30 min at RT. Plates were washed again 6× with PT buffer and 2× with PBS. Wells were developed with 3,3′,5,5′-tetramethylbenzidine (TMB) substrate for 5 min and quenched with equal volume of 1 M H3PO4. Plates were read at 450 nm using a SpectraMax 340PC plate reader (Molecular devices).
Fab-ELISA was performed to check the binding of purified Fabs to immobilized target proteins. Proteins were immobilized at 5 μg/ml on Maxisorp plates by overnight incubation at 4°C. Wells were subsequently blocked with PB buffer for 90 min at RT before washing 4× with PT buffer. Wells were exposed to 100 μl of Fab diluted in PT for 30 min, washed 10× with PT buffer and incubated with a 1:3000 dilution of HRP-conjugated anti-HIS antibody (Rockland Biosciences) for 30 min at RT. Plates were washed again 6× with PT buffer and 2× with PBS. Wells were developed with TMB substrate for 5 min, and quenched with equal volume of 1 M H3PO4. Plates were read at 450 nm using a SpectraMax 340PC plate reader (Molecular devices). Single-point Fab-ELISA was used to assess Fab specificity, and multi-point Fab-ELISA was used to calculate the EC50 for Fab binding to the immobilized target. In multi-point Fab ELISA, ABS450 values were obtained for a range of Fab concentrations and EC50 was calculated by fitting the data to the one-site specific-binding equation in Prism (Graphpad).
Bio-layer interferometry
The ForteBio Octet RED384 system (Pall Corporation) was used to measure binding kinetics between purified Fabs and target proteins. Fabs were immobilized on amine-reactive generation-2 biosensors (for KD < 5 nM) or anti-Fab CH1 biosensors (for KD > 5 nM), according to manufacturer’s instructions. Immobilized Fabs were exposed to increasing concentrations of target proteins, and association and dissociation rates were measured by the shift in wavelength (nm). All reactions were performed at 25°C in PBS. For each sensor-immobilized Fab, at least four different target protein concentrations were used, and KD (equilibrium dissociation constant) was obtained by fitting the data to 1:1 binding model. Data were collected with Octet Data Acquisition version 7.1.0.87 (ForteBio), and analyzed using Octet Data Analysis version 7.1 (ForteBio).
RESULTS
Integrating Ion Torrent sequencing with antibody phage display
Ion Torrent sequencing consists of three basic steps: (i) PCR amplification of a short region of interest, (ii) emulsion PCR on proprietary ion sphere particles (ISPs) and (iii) sequencing enriched ISPs on an Ion semiconductor chip (23). Ion semiconductor chips offer three different read lengths (100, 200 or 400 bp). Our phage-displayed synthetic Fab libraries contained up to four diversified CDRs within a fixed Hu4D5-8 Fab framework (33). The combined length of the four diversified CDRs was only 200 bp, which was shorter than the read length offered by the 400 bp Ion semiconductor chip. Therefore, we sought to link the diversified CDRs in phage pools next to each other by deleting the intervening framework regions to produce a ‘CDR strip’, containing DNA that encodes mainly the CDRs of interest. Since the template for the framework deletion was phage-derived, single-stranded phagemid DNA (ssDNA), we used Kunkel mutagenesis; a method that introduces site-directed mutations when ssDNA is converted into double-stranded DNA in vitro (24) to produce the CDR strip. The overall strategy for CDR strip generation and sequencing is illustrated in Figure 1. We generated two types of CDR strips using this method, one containing L3-H3 CDRs and the other containing L3-H1-H2-H3 CDRs. Originally, we developed the method to generate L3-H3 CDR strips from a library containing only two diversified CDRs (Library-S) (19) and to sequence them using a 200 bp chip. As the Ion Torrent sequencing technology improved, we extended our method to generate L3-H1-H2-H3 strips from a library containing four diversified CDRs (Library-F) (20) and to sequence them using a 400 bp chip. Oligonucleotides used in the Kunkel reaction are 30 bases long and anneal to the 3′ region of one CDR and the 5′ region of adjacent CDR within the same ssDNA. For example, out of 30 bases of the L3-H1 primer, 15 bases anneal to the 3′ region of CDRL3 and 15 bases anneal to the 5′ region of CDRH1. One oligonucleotide was used to bring the L3-H3 regions together, and three oligonucleotides were used to bring the L3-H1-H2-H3 regions together. The mutagenesis reaction was run on an agarose gel and the correct size product was excised. The gel-purified product was used as a template to generate a PCR amplicon of the CDR strip. Quantified, multiplexed amplicons were subjected to emulsion PCR and Ion Torrent sequencing.
Figure 1.

Strategy for CDR strip generation and sequencing. ssDNA rescued from round-3 phage pools is subjected to Kunkel mutagenesis for deleting the intervening framework regions between diversified CDRs. This step links the diversified CDRs next to each other (L3-H3 from Library-S selections and L3-H1-H2-H3 from Library- F selections) without losing the CDR pairing information. The product from the mutagenesis reaction is used as a template to generate a PCR amplicon of the CDR strip. Quantified, multiplexed amplicons are subjected to emulsion PCR and Ion Torrent sequencing. L3-H3 and L3-H1-H2-H3 strips are sequenced on 200 and 400 bp chips, respectively.
To reconstruct Fab clones from the CDR strip sequencing information, we cloned desired CDR combinations into the Fab-4D5 encoding phagemid by Kunkel mutagenesis (Figure 2). A Hu4D5-8 Fab-encoding phagemid whose CDRs have been replaced with NotI sites was used as a template to reconstruct Fab clones. Primers were designed to encode for a desired CDR sequence and to hybridize to either side of the CDR. Two or four primers were used to reconstruct Fab clones from Library-S (19) or Library-F (20) selections, respectively. Following mutagenesis, the reaction was transformed into E. coli and positive clones were screened by NotI digestion. Kunkel reactions outlined in Figures 1 and 2 serve different purposes. In Figure 1, the Kunkel reaction was used to link diversified CDRs together with fixed primer sequences (ssDNA template was variable). In Figure 2, the Kunkel reaction was used to reconstruct Fab phagemids using variable primers (ssDNA template was constant for all Fabs). The second Kunkel reaction contained a uracil-containing ssDNA template and the mutagenesis reaction was transformed into E. coli to eliminate the undesired wild-type strand.
Figure 2.

Strategy for reconstructing Fab clones from NGS information. CDR strips (L3-H3 or L3-H1-H2-H3) generated from round-3 phage pools are subjected to Ion Torrent Sequencing. Following NGS analysis, desired CDR combinations are reconstructed by cloning CDR-encoding oligonucleotides into the Hu4D5 Fab-encoding template phagemid by Kunkel mutagenesis.
Validation of the NGS-assisted Fab reconstruction platform
To demonstrate the feasibility of our Fab reconstruction approach, we chose Jagged-2 as a target. We conducted four rounds of solid-phase selections against the full-length extracellular domain of human Jagged-2. Briefly, we immobilized the recombinant Jagged-2-Fc fusion protein on Maxisorp plates and incubated target-coated wells with Library-S deselected for binding to the Fc protein. Upon elimination of non-specific phages by washing, we eluted bound phages and amplified them in E. coli overnight for subsequent rounds of panning. Random clone picking and Sanger sequencing resulted in 13 sequences from round-3 and round-4 phage pools. Seven were clones with unique sequences for Jagged-2. Phage-ELISA showed that all Jagged-2 Fab clones bound to the target but not to BSA or the Fc protein (Figure 3A). Next, we subcloned four Jagged-2 Fab clones into a Fab expression vector and expressed and purified Fabs from E. coli using Protein-L affinity chromatography. We determined EC50 values for Fabs binding to Jagged-2 using Fab-ELISA. J2/S/1, J2/S/2 and J2/S/4 bound to Jagged-2 with an EC50 value of ∼1 nM, and J2/S/5 had an EC50 value of ∼ 50 nM (Figure 3B and Supplementary Figure S3). Next, we measured kinetics of Fab binding using bio-layer interferometry (BLI). Briefly, Fabs were immobilized on amine-reactive sensors and sensor tips were exposed to increasing concentrations of Jagged-2. Association and dissociation rates were assessed by a wavelength shift and kinetic data sets were globally fit using a 1:1 binding model. Fabs J2/S/1 and J2/S/2 possessed sub-nM KD,apparent values, 0.17 and 0.69 nM, respectively. Fab J2/S/4 was the highest-affinity binder (KD,apparent = 0.14 nM). Fab J2/S/5 bound to Jagged-2 with a KD,apparent of 3.8 nM (Figure 3B and Supplementary Figure S3). The KD,apparent values were lower than the EC50 values, which was most likely due to the avidity in the BLI measurement. In the BLI experiment, the monovalent Fabs were immobilized on biosensors and exposed to bivalent Fc-Jagged-2 in solution. In Fab-ELISA, Fc-Jagged-2 was immobilized on Maxisorp plates and exposed to monovalent Fabs. Next, to assess Fab specificity, we tested the binding of Fabs to Notch and Jagged receptor ectodomains using Fab-ELISA. At 100 nM Fab concentration, Jagged-2 Fabs did not cross-react with Jagged-1 and exhibited target-specific binding (Figure 3B). Together, these results indicated that top clones isolated from Library-S bound with high affinity and selectively to Jagged-2.
Figure 3.
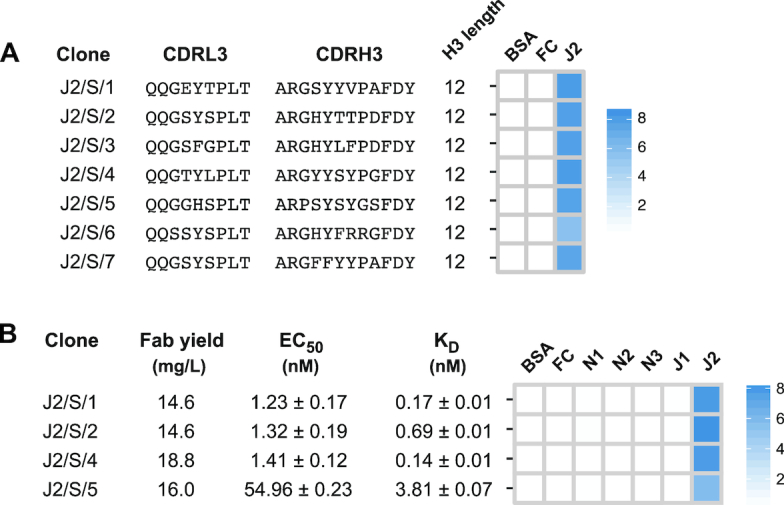
Jagged-2 Fabs from Library-S. (A) Diversified CDR sequences and phage-displayed Fab binding characteristics for seven Jagged-2 Fabs isolated by Sanger sequencing. (B) Fab yield, affinity and specificity of purified Jagged-2 Fabs. Fab yield was estimated for 1 L bacterial culture. EC50 values and KD values for Fabs binding to Jagged-2 were determined by multi-point Fab-ELISA and bio-layer interferometry, respectively. Error values represent the standard error of regression. Fab specificity was determined using single- point Fab-ELISA at 100 nM Fab concentration. Phage-ELISA and specificity-ELISA Abs450 values are shown as heat maps. In ELISA heat maps, Notch-1/2/3 and Jagged-1/2 target proteins are indicated as N1, N2, N3, J1 and J2.
Like previous phage display selections (2,7), we observed a significant enrichment in target-specific CDRH3 lengths and sequences after the third round of selection; therefore, we chose to use the round-3 phage pool for L3-H3 strip sequencing. We generated and sequenced the L3-H3 strip and ranked L3-H3 sequences based on their relative frequencies. A list of enriched CDR-combinations (with frequencies >1%) is shown in Figure 4A. The seven CDRL3-CDRH3 sequences identified by random clone picking and Sanger sequencing identically matched to seven CDR-combinations in the list. Next, we obtained the CDRL3 and CDRH3 regions and analyzed them in terms of length distribution (Figure 4B). In agreement with Sanger sequencing results, a significant enrichment was observed for CDRL3 length of 9 residues and CDRH3 length of 12 residues. Also, a minor enrichment was noted with two CDRH3 lengths (10 and 14 residues). We extracted sequences containing a CDRH3 length of 12 residues and generated sequence logos for CDRL3 and CDRH3 regions (Figure 4C). Enrichment of specific amino acids was observed in the diversified region of both CDRs. Notably, Gly91 in CDRL3 and Gly95, Tyr97 and Pro100 in CDRH3. J2/S/5 had mismatches in two of these positions (Pro95 and Gly100) and bound weaker than other three Fab clones.
Figure 4.

NGS-assisted reconstruction of rare Fab clones from the Jagged-2 selection. (A) Enriched sequences (>1%) from the round-3 CDRL3-CDRH3 strip sequencing output. Seven Fabs isolated by Sanger sequencing are indicated with arrows. (B) CDRL3 and CDRH3 length distribution of the Jagged-2 selection output (round-3). (C) Sequence logos showing the positional amino acid composition of CDRL3 and CDRH3 sequences from the Jagged-2 selection output. The Kabat scheme was used for numbering amino acids. (D) Diversified CDR sequences and phage-displayed Fab binding characteristics for two Jagged-2 rare clones reconstructed from NGS information. (E) Fab yield, affinity and specificity of purified rare Fabs J2/S/R1 and J2/S/R2. EC50 values were determined by Fab-ELISA. Error values represent the standard error of regression. Fab specificity was determined at 100 nM Fab concentration. Phage-ELISA and specificity-ELISA Abs450 values are shown as heat maps.
Next, to assess binding properties of clones corresponding to less frequent CDRH3 lengths, we reconstructed two clones with CDRH3 lengths of 10 or 14 residues. J2/S/R1 had a CDRH3 length of 10 residues and was present at a frequency of 0.03%. J2/S/R2 had a CDRH3 length of 14 residues and had the highest frequency (2.8%) among reconstructed clones. J2/S/R1 and J2/S/R2 showed moderate binding in phage-ELISA (Figure 4D). Purified Fab J2/S/R1 bound to Jagged-2 with an EC50 value of 200 nM. J2/S/R1 also had a strict specificity for Jagged-2. The EC50 of the purified Fab J2/S/R2 (2.23 nM) was similar to the EC50 values of clones identified in Sanger sequencing. J2/S/R2 did not cross-react with other targets in Fab-ELISA (Figure 4E and Supplementary Figure S4). Together, these results indicated that less frequent clones with low-nM affinity and high specificity could be reconstructed from phage selection outputs.
Reconstruction of Fab clones with improved binding properties
In selections against Notch-3, clones isolated by Sanger sequencing had low affinity and specificity; therefore, we used our Fab reconstruction approach to check whether clones with better binding properties could be reconstructed from the Notch-3 selection output. We subjected the round-3 phage pool to L3-H3 strip sequencing and analyzed sequences in terms of relative abundance and length distribution. Sequence analyses and characterization of phage-displayed Fabs and purified Fabs are summarized in Figure 5. Ten Sanger sequences gave rise to five unique clones with CDRH3 lengths of eight residues. In phage-ELISA, two clones (N3/S/1 and N3/S/2) showed strong binding to Notch-3, whereas three clones (N3/S/3, N3/S/4 and N3/S/5) showed moderate to weak binding (Figure 5B). When phage-displayed Fabs were converted into soluble Fabs, N3/S/1 and N3/S/2 showed weak binding to Notch-3 in Fab-ELISA (Figure 5C and Supplementary Figure S5). CDRH3 length analysis confirmed the enrichment of CDRH3 sequences with eight residues in the Notch-3 selection and in addition showed a minor enrichment for sequences with 16 residues (Figure 5A). We reconstructed the most-frequent Fab sequence (0.16%) from CDRH3 length of 16 residues and assessed its affinity and specificity for Notch-3. Phage-displayed Fab N3/S/R1 showed moderate binding in phage-ELISA (Figure 5B). Purified Fab N3/S/R1 had EC50 and KD,apparent values of 16.04 nM (Figure 5C) and 2.65 nM (Supplementary Figure S5), respectively. In Fab-ELISA, N3/S/R1 cross-reacted with Notch-1 and Jagged-2 (Figure 5C). Characterization of Fab N3/S/R1 indicated that clones reconstructed from NGS information can possess higher affinity than clones isolated by Sanger sequencing.
Figure 5.

Notch-3 Fabs from Library-S. (A) CDRL3 and CDRH3 length distributions of the Notch-3 selection output (round-3). (B) Diversified CDR sequences and phage-displayed Fab binding characteristics for Notch-3 Fabs. (C) Fab yield, affinity and specificity of Notch-3 Fabs. EC50 values were determined by Fab-ELISA. Error values represent the standard error of regression. Fab specificity was determined at 1 μM Fab concentration. Phage-ELISA and specificity-ELISA Abs450 values are shown as heat maps. In panels (B) and (C), reconstructed rare clones are highlighted in gray.
Reconstruction of Fab clones from Library-F selections
To demonstrate the feasibility of reconstructing Fabs with four diversified CDRs, we chose Jagged-2 as a target. We panned Library-F against the Jagged-2 extracellular domain and subjected the round-3 phage pool to L3-H1-H2-H3 strip sequencing. We obtained CDRL3 and CDRH3 regions and analyzed them in terms of length distribution (Figure 6A). A significant enrichment was observed for CDRL3 sequences with 8 residues (70.4%) and CDRH3 sequences with 10 residues (56.8%). CDRH3 sequences with 7, 8 and 14 residues were present between 5 and 10%, and CDRH3 sequences with 11, 12, 15, 16, 17, 18 and 23 residues were present between 1 and 5%. Random clone picking resulted in 18 Sanger sequences from round-3 and round-4 phage pools containing 9 Fab clones with unique sequences. These clones had CDRH3 lengths of 7, 8, 10, 14, 17 and 18 residues. We reconstructed seven clones from six less-frequent CDRH3 lengths (11, 12, 15, 16, 18 and 23 residues). The frequencies of reconstructed rare clones ranged from 0.7% to 0.1%. Phage-ELISA indicated that 15 out of 16 clones bound to Jagged-2 but not to control proteins (Figure 6B). One reconstructed clone (J2/F/R1) bound to BSA and the Fc protein in addition to Jagged-2. We converted 13 phage-displayed Fabs into soluble Fabs encompassing different CDRL3 and CDRH3 lengths and assessed their affinity and specificity using Fab-ELISA (Figure 6C and Supplementary Figure S6). Among the seven Sanger clones, five Fabs possessed low-nM EC50 values for Jagged-2. However, all five Fabs cross-reacted with Jagged-1. J2/F/5 and J2/F/8 had low affinities for Jagged-2, nonetheless had high Fab expression rates in E. coli. Interestingly, J2/F/5 had the highest frequency in the round-3 phage pool (45%). Among the six reconstructed clones, five Fabs possessed low-nM EC50 values for Jagged-2. The reconstructed clone J2/F/R7 with the lowest frequency (0.1%) had an EC50 of 24.62 nM. Strikingly, three out of six reconstructed clones (J2/F/R4, J2/F/R6 and J2/F/R7) possessed strict specificity for Jagged-2. Together, these results indicated that reconstructed clones could not only bind tighter than clones identified by Sanger sequencing, but also be more specific.
Figure 6.

Jagged-2 Fabs from Library-F. (A) CDRL3 and CDRH3 length distribution of the Jagged-2 selection output (round-3). (B) Diversified CDR sequences and phage-displayed Fab binding characteristics of 9 top Fabs isolated by Sanger sequencing and 7 rare Fabs reconstructed from L3-H1-H2-H3 NGS information. (C) Fab yield, affinity and specificity of purified Fabs. EC50 values were determined by Fab-ELISA. Error values represent the standard error of regression. Fab specificity was determined at 1 μM Fab concentration. Phage-ELISA and specificity-ELISA Abs450 values are shown as heat maps. In panels (B) and (C), reconstructed rare clones are highlighted in gray.
Comparison of NGS sequences from CDR strip generation with other methods
To evaluate the bias introduced by CDR strip generation, we looked at the frequency of clones identified by Sanger sequencing versus frequency of clones after CDR strip generation using NGS data. Even with the small number of clones sequenced using the Sanger method, 13 for J2/S, 10 for N3/S and 18 for J2/F, there was a statistically significant correlation between the NGS and Sanger sequencing results using either a Pearson (r = 0.8793, P value < 0.0001) or Spearman correlation (r = 0.6685; P value = 0.0013). To further evaluate the reproducibility of the method, we compared CDRH3 frequency in pre- and post-CDR strip generation (Supplementary Figure S7). The two methods were highly correlated (Pearson = 0.9175) for all targets combined, showing the reproducibility of the method.
DISCUSSION
In recent years, NGS technologies have revolutionized both fundamental and translational aspects of biological research. Sequencing of functional antibody repertoires and subsequent bioinformatics analyses has provided unprecedented insights into the mechanisms of B-cell development in humans and other species and has transformed our understanding of immune responses to autoimmunity, vaccination, infection and cancer (34–36). Sequencing of antibody repertoires is also used for identifying critical biomarkers and epitopes targeted by functional antibody responses for developing vaccines, immunomodulatory drugs and diagnostic tools, and for discovering antibodies (34–36). NGS technologies have also been employed in several phage display platforms for analyzing natural and synthetic antibody fragment sequences and for identifying and reconstructing scFv and Fab binders not found by conventional ELISA screens (4,37).
In this work, we developed an NGS-assisted antibody discovery platform. We demonstrated seamless integration of antibody phage display with short-read DNA sequencing using single-framework synthetic Fab libraries rationally engineered to include length and amino acid diversities within CDRs (19,20). Due to limitations in attainable read lengths, NGS of Fab libraries and selection outputs is usually restricted to VH or VL (2,13). Since CDRH3 sequencing alone is not sufficient for high-throughput reconstruction of Fabs, we developed a method for linking and sequencing all diversified CDRs in phage Fab pools without losing the CDR pairing. Our method determined sequences of paired CDRs in selection outputs and allowed the straightforward conversion of NGS information into Fab clones. Using a defined single-framework, sequencing efforts were directed only toward the diversified CDR regions. NGS primers were designed to anneal the framework regions. During sequence analysis, the use of a single-framework eliminated complications associated with the occurrence of multiple frameworks and framework mutations present in natural antibody repertoires enabling fast and accurate interpretation of NGS data sets. The method is rapid and simple and can be adapted to any synthetic scaffold-based phage library including the HuCal and Ylanthia libraries from MorphoSys (11,38–40) by incorporating additional oligonucleotides that target the synthetic frameworks for these libraries during amplification. The method can also be applied to naïve and immunized Fab libraries to remove the constant light (CL) and intervening region of ∼540 bp between the framework light 4 (FRL4) and framework heavy 1 (FRH1) region, using a single oligonucleotide complementary to the start of CL and preceding FRH1 (Figure 7). This reduces the overall length of the library amplicon from ∼1200 to ∼630 bp, which is necessary on HiSeq and MiSeq instruments where clustering libraries larger than ∼1000 bp requires higher loading concentrations and libraries ∼1500 bp may fail to cluster altogether. NextSeq and MiniSeq instruments require libraries of <700 bp for efficient clustering.
Figure 7.
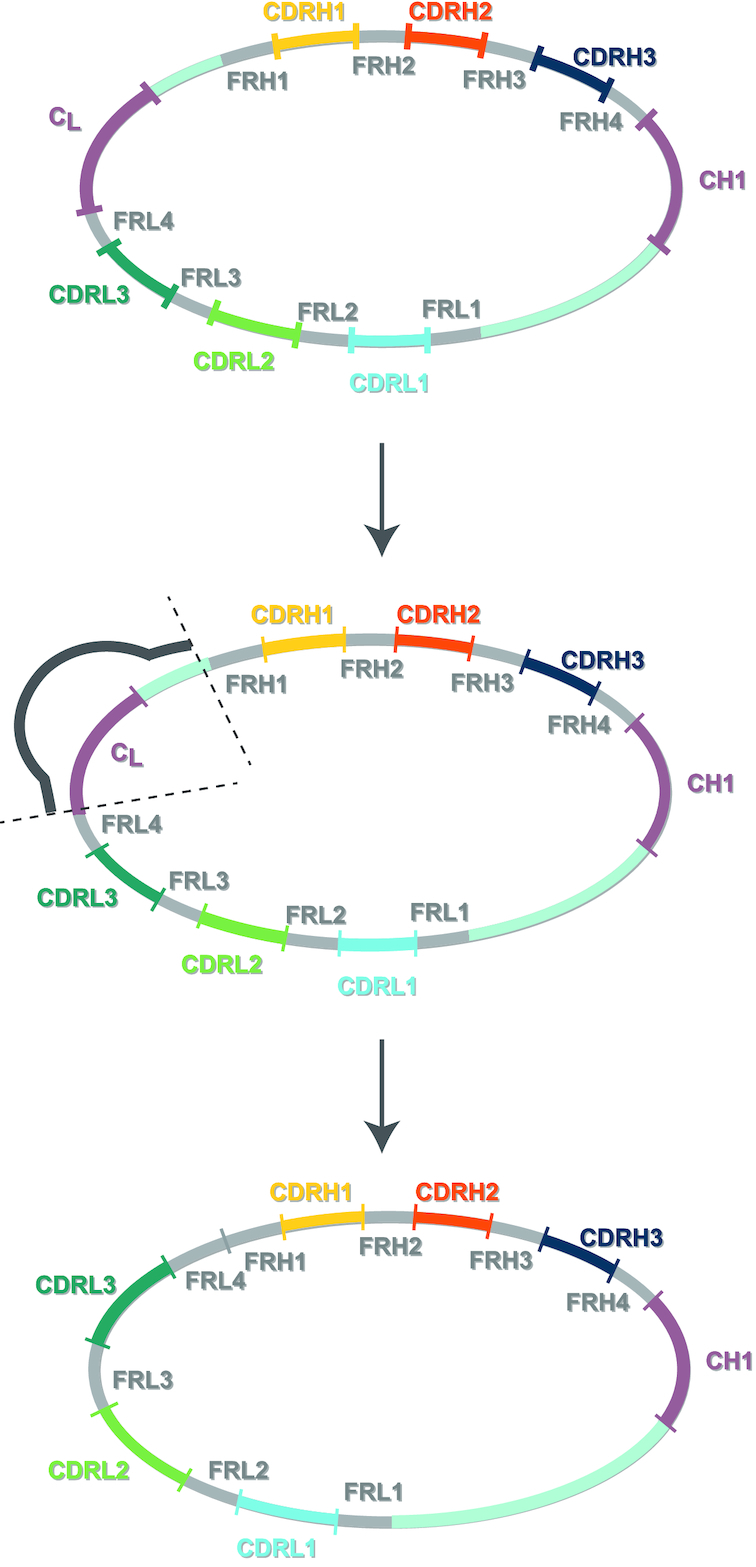
General strategy for strip generation with naïve libraries. Strip generation can be used to reduce the amplicon size for any NGS platform and library. Kunkel mutagenesis using a primer with regions complementary to the constant light (CL) and upstream of the framework heavy chain 1 (FRH1) region will result in a template with reduced size for subsequent PCR amplification and NGS sequencing.
We used Ion Torrent sequencing, which can deliver up to 5.5 × 106 non-paired reads with 100–400 bp read lengths in a short span of time (5 h) and at low cost. 454 and PacBio sequencers have longer read lengths; however, 454 sequencers have been discontinued and PacBio instruments are primarily aimed at read lengths greater than 10 kb, far in excess of what is required here. The two remaining commonly used short-read DNA sequencing platforms are Illumina and Ion Torrent. Both these instruments can benefit from generating CDR strip amplicons. Strip amplicon generation is required on the Ion Torrent as it cannot perform paired-end sequencing reads and has a maximum read length of 400 bp. MiSeq Reagent Kit v3 from Illumina provides up to 25 × 106 reads with 2 × 300 bp read lengths (paired-end reads); however, libraries of <1000 bp are preferred for efficient clustering and the CDR strip amplicon strategy can be used to create libraries of smaller lengths. All other instruments from Illumina that produce >108 reads (HiSeq2500, HiSeq3000, HiSeq4000, HiSeqX and NovaSeq) have read lengths <300 bp, and CDR strip amplicon can aid in deep sequencing samples using these instruments.
Previous studies have showed that single-framework synthetic Fab libraries allow facile reformatting of Fabs between different vector systems for affinity maturation, bacterial expression and IgG conversion (41,42). In this work, we demonstrate an additional advantage of rapid and reliable reconstruction of Fabs from NGS information.
Typically, to recover antigen-specific Fabs from phage selection pools, hundreds to thousands of colonies from selection outcomes are first interrogated by binding assays (phage/Fab ELISA) in 96/384 well plates and then identities of ELISA-positive clones are determined by Sanger sequencing (2). Due to phage propagation biases in E. coli, many binders present in earlier selection rounds do not get enriched and remain at very low frequencies in later selection rounds. Therefore, the conventional clone recovery method leads to the repeated identification of the same enriched clones from phage selection pools (43,44). NGS-based reconstruction of antibody clones can avoid the repeated identification of growth-advantaged, high frequency binders, and can identify and recover many unique clones that are present in low frequencies in phage pools (8,14,15,37). Antibody clone identification following phage display analysis using NGS has been primarily based on clone frequency in the last round of selection. Reconstruction of rare Fab clones using length as part of the analysis allowed us to recover Fabs with new and potentially better binding solutions than looking at frequency alone. As evidence of this, some low-frequency rare Fabs (∼0.1% frequency) reconstructed in this work showed higher affinity and specificity than high-frequency Fabs isolated by Sanger sequencing. Another strategy to pick clones from phage display selections is to look at enrichment over different rounds of selections. The problem with this method is that enrichment does not always correlate with enhanced affinity. In our selections, some of the reconstructed clones peaked in round 3 and decreased in round 4 and round 5, yet had similar affinities to clones that did not decrease. Full CDR sequencing allows new variables and more sophisticated analyses to be developed to select rare clones. Future studies will be based on selecting clones on their amino acid properties. Rare clones could be selected based on charge frequency in different CDRs or hydrophobicity in specific CDRs combined with charge in other CDRs to form coordinated binding surfaces. Performing analysis beyond the single dimension of frequency and looking at length or amino acid properties offers a new way to select sequences from phage display selections.
Synthetic antibody technology is well known for high-throughput generation of antibodies. In this work, the following factors contributed to the high-throughput reconstruction and Fabs from selection outputs: (i) multiplexing of up to 20 selection outputs in one sequencing run; (ii) semi-automated NGS data processing and analysis; (iii) synthesis of CDR-encoding oligonucleotides in 96-well format; (iv) optimized procedures for cloning CDR-encoding oligonucleotides into the Fab-encoding template phagemid; (v) replacement of Sanger sequencing with restriction digestion analysis for screening positive phagemid clones; (vi) optimized procedures for sub-cloning ELISA-positive phage-Fab clones into Fab-expression vector in 96-well format, and (vii) confirmation of Fab sub-cloning and expression by bio-layer interferometry in 96-well format. We isolated top Fab clones by Sanger sequencing and compared them with rare Fab clones for emphasizing the need for NGS-based reconstruction approaches in antibody phage display. This step is now excluded in routine phage display selections. Following three rounds of selection, the selection output is subjected to L3-H3 or L3-H1-H2-H3 sequencing, promising clones are identified by NGS analysis, reconstructed, and tested for binding. This approach eliminated the use of Sanger sequencing from antibody phage display (from phage selections to Fab KD determination) and resulted in a reliable and high-throughput antibody discovery platform.
In this work, we reconstructed rare Fab clones in the phagemid vector and sub-cloned the ELISA-positive clones into the Fab expression vector. In future, desired CDR combinations can be reconstructed directly in suitable Fab/scFv/IgG expression vectors and tested for binding to relevant antigens using bio-layer interferometry in a high-throughput format. The NGS approach described in this work can be used for identifying and reconstructing Fabs from biopanning experiments where isolating target-specific Fabs is more difficult than from solid-phase selections (45). NGS analysis can be used for comparing CDR sequences from negative and positive selections and enriched CDR combinations in target phage pools can be used for reconstruction. In this work, we obtained only ∼10 000 sequences per selection round due to multiplexing of numerous selection outputs in one chip. This number was sufficient to interrogate the selection output and isolate a few low-frequency rare clones from the selection output. Advances in depth and length of NGS reads will help to obtain more diverse Fab clones and to reconstruct ultra-low frequency Fabs from earlier rounds of selection. This NGS-assisted approach can also be used for screening Fabs against an array of antigens pooled in different configurations, and subsequently deconvoluting the resulting selection outputs to deduce Fab sequences specific for each pooled antigen (46). NGS information can also be used for mapping binding energy landscapes, designing affinity maturation libraries and improving biophysical properties of antibodies, especially when crystal structures of antigen–antibody complexes are also available for analysis (47–49).
Supplementary Material
ACKNOWLEDGEMENTS
BVM likes to thank the Department of Biochemistry, University of Saskatchewan and the Government of Saskatchewan for Graduate Scholarships and Awards.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Western Economic Diversification Canada [#12939]. Funding for open access charge: University of Saskatchewan.
Conflict of interest statement. None declared.
REFERENCES
- 1. Geyer C.R., McCafferty J., Dubel S., Bradbury A.R., Sidhu S.S.. Recombinant antibodies and in vitro selection technologies. Methods Mol. Biol. 2012; 901:11–32. [DOI] [PubMed] [Google Scholar]
- 2. Ravn U., Gueneau F., Baerlocher L., Osteras M., Desmurs M., Malinge P., Magistrelli G., Farinelli L., Kosco-Vilbois M.H., Fischer N.. By-passing in vitro screening- next generation sequencing technologies applied to antibody display and in silico candidate selection. Nucleic Acids Res. 2010; 38:e193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fischer N. Sequencing antibody repertoires: the next generation. mAbs. 2011; 3:17–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Glanville J., D’Angelo S., Khan T.A., Reddy S.T., Naranjo L., Ferrara F., Bradbury A.R.. Deep sequencing in library selection projects: what insight does it bring. Curr. Opin. Struct. Biol. 2015; 33:146–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Metzker M.L. Sequencing technologies- the next generation. Nat. Rev. Genet. 2010; 11:31–46. [DOI] [PubMed] [Google Scholar]
- 6. Loman N.J., Misra R.V., Dallman T.J., Constantinidou C., Gharbia S.E., Wain J., Pallen M.J.. Performance comparison of benchtop high-throughput sequencing platforms. Nat. Biotechnol. 2012; 30:434–439. [DOI] [PubMed] [Google Scholar]
- 7. Venet S., Ravn U., Buatois V., Gueneau F., Calloud S., Kosco-Vilbois M., Fischer N.. Transferring the characteristics of naturally occurring and biased antibody repertoires to human antibody libraries by trapping CDRH3 sequences. PLoS One. 2012; 7:e43471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ravn U., Didelot G., Venet S., Ng K.T., Gueneau F., Rousseau F., Calloud S., Kosco- Vilbois M., Fischer N.. Deep sequencing of phage display libraries to support antibody discovery. Methods. 2013; 60:99–110. [DOI] [PubMed] [Google Scholar]
- 9. Zhang H., Torkamani A., Jones T.M., Ruiz D.I., Pons J., Lerner R.A.. Phenotype-information-phenotype cycle for deconvolution of combinatorial antibody libraries selected against complex systems. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:13456–13461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhai W., Glanville J., Fuhrmann M., Mei L., Ni I., Sundar P.D., Van Blarcom T., Abdiche Y., Lindquist K., Strohner R. et al.. Synthetic antibodies designed on natural sequence landscapes. J. Mol. Biol. 2011; 412:55–71. [DOI] [PubMed] [Google Scholar]
- 11. Tiller T., Schuster I., Deppe D., Siegers K., Strohner R., Herrmann T., Berenguer M., Poujol D., Stehle J., Stark Y. et al.. A fully synthetic human Fab antibody library based on fixed VH/VL framework pairings with favorable biophysical properties. mAbs. 2013; 5:445–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mahon C.M., Lambert M.A., Glanville J., Wade J.M., Fennell B.J., Krebs M.R., Armellino D., Yang S., Liu X., O'Sullivan C.M. et al.. Comprehensive interrogation of a minimalist synthetic CDR-H3 library and its ability to generate antibodies with therapeutic potential. J. Mol. Biol. 2013; 425:1712–1730. [DOI] [PubMed] [Google Scholar]
- 13. D’Angelo S., Kumar S., Naranjo L., Ferrara F., Kiss C., Bradbury A.R.. From deep sequencing to actual clones. Protein Eng. Des. Sel. 2014; 27:301–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Spiliotopoulos A., Owen J.P., Maddison B.C., Dreveny I., Rees H.C., Gough K.C.. Sensitive recovery of recombinant antibody clones after their in silico identification within NGS datasets. J. Immunol. Methods. 2015; 420:50–55. [DOI] [PubMed] [Google Scholar]
- 15. Lopez T., Nam D.H., Kaihara E., Mustafa Z., Ge X.. Identification of highly selective MMP-14 inhibitory Fabs by deep sequencing. Biotechnol. Bioeng. 2017; 114:1140–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yang W., Yoon A., Lee S., Kim S., Han J., Chung J.. Next-generation sequencing enables the discovery of more diverse positive clones from a phage-displayed antibody library. Exp. Mol. Med. 2017; 49:e308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. DeKosky B.J., Kojima T., Rodin A., Charab W., Ippolito G.C., Ellington A.D., Georgiou G.. In-depth determination and analysis of the human paired heavy- and light- chain antibody repertoire. Nat. Med. 2015; 21:86–91. [DOI] [PubMed] [Google Scholar]
- 18. Lövgren J., Pursiheimo J.P., Pyykko M., Salmi J., Lamminmaki U.. Next generation sequencing of all variable loops of synthetic single framework scFv-Application in anti-HDL antibody selections. N. Biotechnol. 2016; 33:790–796. [DOI] [PubMed] [Google Scholar]
- 19. Maruthachalam B.V., El-Sayed A., Liu J., Sutherland A.R., Hill W., Alam M.K., Pastushok L., Fonge H., Barreto K., Geyer C.R.. A Single-Framework synthetic antibody library containing a combination of canonical and variable complementarity determining regions. ChemBioChem. 2017; 18:2247–2259. [DOI] [PubMed] [Google Scholar]
- 20. Persson H., Ye W., Wernimont A., Adams J.J., Koide A., Koide S., Lam R., Sidhu S.S.. CDR-H3 diversity is not required for antigen recognition by synthetic antibodies. J. Mol. Biol. 2013; 425:803–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fellouse F.A., Sidhu S.S.. Howard GC, Kaser MR. Making antibodies in bacteria. Making and Using Antibodies: A Practical Handbook. 2006; San Francisco: Taylor and Francis; 157–180. [Google Scholar]
- 22. Rajan S., Sidhu S.S.. Simplified synthetic antibody libraries. Methods Enzymol. 2012; 502:3–23. [DOI] [PubMed] [Google Scholar]
- 23. Rothberg J.M., Hinz W., Rearick T.M., Schultz J., Mileski W., Davey M., Leamon J.H., Johnson K., Milgrew M.J., Edwards M. et al.. An integrated semiconductor device enabling non-optical genome sequencing. Nature. 2011; 475:348–352. [DOI] [PubMed] [Google Scholar]
- 24. Kunkel T.A., Roberts J.D., Zakour R.A.. Rapid and efficient site-specific mutagenesis without phenotypic selection. Methods Enzymol. 1987; 154:367–382. [DOI] [PubMed] [Google Scholar]
- 25. Tonikian R., Zhang Y., Boone C., Sidhu S.S.. Identifying specificity profiles for peptide recognition modules from phage-displayed peptide libraries. Nat. Protoc. 2007; 2:1368–1386. [DOI] [PubMed] [Google Scholar]
- 26. Nelson B., Sidhu S.S.. Synthetic antibody libraries. Methods Mol. Biol. 2012; 899:27–41. [DOI] [PubMed] [Google Scholar]
- 27. Goecks J., Nekrutenko A., Taylor J.. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010; 11:R86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Blankenberg D., Von Kuster G., Coraor N., Ananda G., Lazarus R., Mangan M., Nekrutenko A., Taylor J.. Galaxy: a web-based genome analysis tool for experimentalists. Curr. Protoc. Mol. Biol. 2010; doi:10.1002/0471142727.mb1910s89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. R Core Team R: A Language and Environment for Statistical Computing. 2013; Vienna: R Foundation for Satistical Computing. [Google Scholar]
- 30. Pagès H., Aboyoun P., Gentleman R., DebRoy S.. Biostrings: String objects representing biological sequences, and matching algorithms. 2016; R package version 2.42.1. [Google Scholar]
- 31. Wickham H. ggplot2: Elegant Graphics for Data Analysis. 2009; NY: Springer-Verlag. [Google Scholar]
- 32. Gibson D.G., Young L., Chuang R.Y., Venter J.C., Hutchison C.A., Smith H.O.. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 2009; 6:343–345. [DOI] [PubMed] [Google Scholar]
- 33. Fellouse F.A., Esaki K., Birtalan S., Raptis D., Cancasci V.J., Koide A., Jhurani P., Vasser M., Wiesmann C., Kossiakoff A.A. et al.. High-throughput generation of synthetic antibodies from highly functional minimalist phage-displayed libraries. J. Mol. Biol. 2007; 373:924–940. [DOI] [PubMed] [Google Scholar]
- 34. Georgiou G., Ippolito G.C., Beausang J., Busse C.E., Wardemann H., Quake S.R.. The promise and challenge of high-throughput sequencing of the antibody repertoire. Nat. Biotechnol. 2014; 32:158–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Robinson W.H. Sequencing the functional antibody repertoire–diagnostic and therapeutic discovery. Nat. Rev. Rheumatol. 2015; 11:171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lavinder J.J., Horton A.P., Georgiou G., Ippolito G.C.. Next-generation sequencing and protein mass spectrometry for the comprehensive analysis of human cellular and serum antibody repertoires. Curr. Opin. Chem. Biol. 2015; 24:112–120. [DOI] [PubMed] [Google Scholar]
- 37. Naso M.F., Lu J., Panavas T.. Deep sequencing approaches to antibody discovery. Curr. Drug Discov. Technol. 2014; 11:85–95. [DOI] [PubMed] [Google Scholar]
- 38. Prassler J., Thiel S., Pracht C., Polzer A., Peters S., Bauer M., Nörenberg S., Stark Y., Kölln J., Popp A. et al.. HuCAL PLATINUM, a synthetic fab library optimized for sequence diversity and superior performance in mammalian expression systems. J. Mol. Biol. 2011; 413:261–278. [DOI] [PubMed] [Google Scholar]
- 39. Rothe C., Urlinger S., Löhning C., Prassler J., Stark Y., Jäger U., Hubner B., Bardroff M., Pradel I., Boss M. et al.. The human combinatorial antibody library HuCAL GOLD combines diversification of all six CDRs according to the natural immune system with a novel display method for efficient selection of High-Affinity antibodies. J. Mol. Biol. 2008; 376:1182–1200. [DOI] [PubMed] [Google Scholar]
- 40. Knappik A., Ge L., Honegger A., Pack P., Fischer M., Wellnhofer G., Hoess A., Wölle J., Plückthun A., Virnekäs B.. Fully synthetic human combinatorial antibody libraries (HuCAL) based on modular consensus frameworks and CDRs randomized with trinucleotides11Edited by I. A. Wilson. J. Mol. Biol. 2000; 296:57–86. [DOI] [PubMed] [Google Scholar]
- 41. Miersch S., Sidhu S.S.. Synthetic antibodies: concepts, potential and practical considerations. Methods. 2012; 57:486–498. [DOI] [PubMed] [Google Scholar]
- 42. Adams J.J., Sidhu S.S.. Synthetic antibody technologies. Curr. Opin. Struct. Biol. 2014; 24:1–9. [DOI] [PubMed] [Google Scholar]
- 43. Derda R., Tang S.K., Li S.C., Ng S., Matochko W., Jafari M.R.. Diversity of phage-displayed libraries of peptides during panning and amplification. Molecules. 2011; 16:1776–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Saggy I., Wine Y., Shefet-Carasso L., Nahary L., Georgiou G., Benhar I.. Antibody isolation from immunized animals: comparison of phage display and antibody discovery via V gene repertoire mining. Protein Eng. Des. Sel. 2012; 25:539–549. [DOI] [PubMed] [Google Scholar]
- 45. Tomic J., McLaughlin M., Hart T., Sidhu S.S., Moffat J.. Sidhu SS, Geyer CR. Leveraging synthetic Phage-Antibody libraries for panning on the mammalian cell surface. Phage Display in Biotechnology and Drug Discovery. 2015; Boca Raton: Taylor and Francis; 113–122. [Google Scholar]
- 46. Larman H.B., Xu G.J., Pavlova N.N., Elledge S.J.. Construction of a rationally designed antibody platform for sequencing-assisted selection. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:18523–18528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Whitehead T.A., Chevalier A., Song Y., Dreyfus C., Fleishman S.J., De Mattos C., Myers C.A., Kamisetty H., Blair P., Wilson I.A. et al.. Optimization of affinity, specificity and function of designed influenza inhibitors using deep sequencing. Nat. Biotechnol. 2012; 30:543–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Koenig P., Lee C.V., Sanowar S., Wu P., Stinson J., Harris S.F., Fuh G.. Deep Sequencing-guided design of a high affinity dual specificity antibody to target two angiogenic factors in neovascular Age-related macular degeneration. J. Biol. Chem. 2015; 290:21773–21786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Koenig P., Lee C.V., Walters B.T., Janakiraman V., Stinson J., Patapoff T.W., Fuh G.. Mutational landscape of antibody variable domains reveals a switch modulating the interdomain conformational dynamics and antigen binding. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:E486–E495. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.