


Abstract
HMGN proteins localize to chromatin regulatory sites and modulate the cell-type specific transcription profile; however, the molecular mechanism whereby these ubiquitous nucleosome binding proteins affect gene expression is not fully understood. Here, we show that HMGNs regulate the expression of Rex1, one of the most highly transcribed genes in mouse embryonic stem cells (ESCs), by recruiting the transcription factors NANOG, OCT4 and SOX2 to an ESC-specific super enhancer located in the 5′ region of Rex1. HMGNs facilitate the establishment of an epigenetic landscape characteristic of active chromatin and enhancer promoter interactions, as seen by chromatin conformation capture. Loss of HMGNs alters the local epigenetic profile, increases histone H1 occupancy, decreases transcription factors binding and reduces enhancer promoter interactions, thereby downregulating, but not abolishing Rex1 expression. ChIP-seq analyses show high colocalization of HMGNs and of REX1, a zinc finger protein, at promoters and enhancers. Loss of HMGNs preferentially reduces the specific binding of REX1 to these chromatin regulatory sites. Thus, HMGNs affects both the expression and the chromatin binding specificity of REX1. We suggest that HMGNs affect cell-type specific gene expression by modulating the binding specificity of transcription factors to chromatin.
INTRODUCTION
The dynamic interplay among nuclear components that continuously modify chromatin establishes and maintains the epigenetic landscape necessary for cell-type specific transcription. The key step in establishing cell-type specific genomic landscapes is the binding of tissue specific transcription factors to their specific chromatin binding sites (1–3); however, additional chromatin modifiers, such as the H1 linker histones (4–6) and the high mobility group N (HMGN) (7–9) proteins are known to affect the chromatin organization and transcription levels. The molecular mechanism whereby these chromatin architectural proteins modulate the epigenome and affect gene expression is still not fully understood.
HMGN is a family of structural proteins ubiquitously present in vertebrate cells (10). The proteins bind specifically to nucleosome core particles, the building block of the chromatin fiber, without any known DNA sequence specificity (11). The amount of HMGN protein present in the nuclei is sufficient to bind to ∼1% of the nucleosomes; however, because the interaction of these proteins with chromatin are short lived and very dynamic (12) HMGNs are distributed throughout the nucleus, potentially interacting with the entire genome. The binding of HMGNs to nucleosomes promotes chromatin decompaction most likely by weakening the binding of the linker histone H1 to chromatin (13–15) and by interfering with the interaction of the histone N-terminal tails with the acidic patch of a neighboring nucleosome (16). Genome wide, the major HMGN variants, HMGN1 and HMGN2, colocalize. Furthermore, their chromatin binding is compensatory; loss of one HMGN variant enhances the binding of the remaining variant (17). Significantly, both HMGN1 and HMGN2 bind to chromatin regulatory sites, including to cell type specific super enhancers, regulatory regions known to play a key role in determining cell-type specific gene expression (17,18). In agreement with these observations, experiments with cells derived from double knockout mice lacking both HMGN1 and HMGN2 (DKO), revealed that HMGNs affect gene expression in a tissue-specific manner; they modulate the preexisting, cell-type specific transcription profile (7).
Although the HMGNs bind to numerous regulatory sites their effect on gene expression levels are relatively mild and cannot be predicted; loss of HMGNs leads to both up and down regulation in the expression of numerous genes (7–9,19). For example, transcription analyses of wild type (WT) and DKO embryonic stem cell during the first 8 days of differentiation into embryoid bodies showed that loss of HMGNs leads to significantly altered gene expression levels (20). We now find that most genes were affected only at a specific day of differentiation; the expression of very few genes were consistently different between WT and DKO cells throughout the 8 days of differentiation. One of the consistently down regulated genes in HMGN-DKO cells was Rex1 (Zfp-42), a gene encoding REX1 protein, a pluripotency marker in both human and mouse cells that seems to affect preimplantation development, X-chromosome inactivation and human stem cell pluripotency (21–23). REX1 contains a zinc finger resembling YY1 motif (24) suggesting that it functions as a chromatin binding transcription factor.
As a paradigm for the mechanism whereby HMGNs affect gene expression and chromatin function, we focus on the molecular mechanism whereby HMGN proteins affect both the expression and the binding of REX1 to specific chromatin sites. We demonstrate that HMGNs regulate Rex1 expression by affecting the binding of specific transcription factors to a super-enhancer at the Rex1 locus. Furthermore, we demonstrate that HMGN modulates the binding of REX1 to chromatin. Thus, HMGNs affect not only Rex1 expression but also its specific chromatin interactions. Our studies provide insights into the molecular mechanisms that regulate REX1 expression and in a broader sense demonstrate that by affecting the interaction of cell-type specific transcription factors with chromatin, the ubiquitous HMGNs play a role in epigenetic regulation of gene expression.
MATERIALS AND METHODS
ES cell culture
Mouse ES cell lines derived from 3.5 days blastocysts obtained from Hmgn1+/+n2+/+(WT) and Hmgn1−/− n2−/− (DKO) mice were generated, cultured and differentiated into EBs by withdrawal of LIF/2i as previously described (17,19). Three independent ESC clones for each genotype were used for analysis. The ES cells were co-cultured with mitomycin-c treated mouse primary embryonic fibroblast (MEFs) feeders (Millipore) or cultured under feeder-free conditions in Knock-out Dulbecco's modified Eagle's medium (KO-DMEM, Invitrogen), with 20% serum replacement (SR, Invitrogen), 0.55 mM β-mercaptoethanol (Sigma), 2 mM l-glutamine (Invitrogen), 0.1 mM MEM non-essential amino acid (Invitrogen), 5000 U/ml penicillin/streptomycin (Invitrogen), 1000 U/ml LIF (Millipore) and MEK/GSK3 inhibitors (2i) (Millipore).
ESCs transfection
For siRNA knock-down, 25 nM ON-TARGET SMARTpool siRNA specifically targeting mouse Hmgn1 or Hmgn2 (GE Dharmacon) were transfected into ESCs using Lipofectamine RNAiMAX reagent (Invitrogen). Cells were split and transfected again after first 48 h and harvested after another 48 h. Primers used for qPCR are: Hmgn1F: GAAAAAGGCCGCGGGAAAG, Hmgn1R: ATTTTCTGCAGGCAGCTCTG, Hmgn2F: GCGAATAATCCTGCAGAAAATGG, Hmgn2R: TCAAACTGTACAGTCACCAGAAG, Rex1F: CAATGCCTATGACTCACTTCCAG, Rex1R: TCCTGCACACAGAAGAAAGC
Western blot
Cell lysates were prepared in 1 × SDS PAGE sample buffer (Bio-Rad) supplemented with protease inhibitors (Roche Applied Science). The samples were fractionated on 4–20% pre-cast Criterion gels, transferred to PVDF membranes with iBlot Gel Transfer Stacks (Life Technologies), blocked with non-fat milk in TBST and probed with Rex1 antibody (Thermo Fisher scientific PA5-27567). Chemiluminescent detection using ECL Plus was performed according to manufacturer's (Amersham) recommendations. Western signals were quantified with ImageJ software.
Chromosome conformation capture (3C) assay
1 × 107 WT or DKO ES cells were cross-linked in 1% formaldehyde solution and incubated for 10 min at room temperature, followed by quenching with 125 mM glycine. After washing twice with ice-cold PBS, cell pellets were resuspended in 5 ml of cold lysis buffer and incubated for 10 min on ice. Nuclei were resuspended in 0.5 ml of 1.2× restriction enzyme NEB2.1 buffer (New England Biolabs) containing 0.3% SDS and the mixture was incubated at 37°C for 1 h while shaking (900 rpm). After addition of 50 μl of 20% Triton X-100, the mixtures were incubated for another 1 h at 37°C while shaking. Nuclei were treated with Hind III in the following order: 200 U for 4 h, 200 U for overnight and another 200 U for 4 h. Enzyme was inactivated by incubating for 20 min at 65°C. Ligation was performed with 100 U of T4 DNA ligase for 4 h at 16°C. After de-crosslinking, DNA was purified and dissolved in 150 μl of 10 mM Tris–HCl (pH 7.5). Primers used in 3C assay are: Rex1F: GGTGGCACAGGAATTTGAGT, Rex1R: CCAATAAAACCATTTGAGACGA, GapdhF: CACATTGGGGGTAGGAACAC, GapdhR: AGAACATCATCCCTGCATCC.
Genome-wide DNase I hypersensitivity assay
ES cells were collected and resuspended in buffer A (15 mM Tris–Cl, pH 8.0, 15 mM NaCl, 60 mM KCl, 1 mM EDTA, pH 8.0, 0.5 mM EGTA, pH 8.0) with freshly added 0.5 mM spermidine and 1× protease inhibitor (Roche Applied Science). Cell membranes were disrupted by addition of equal volume of buffer A containing 0.01% IGEPAL, followed by incubation on ice for 10 min. Following centrifugation at 1500 rpm for 5 min, 2 × 107 pelleted nuclei were resuspended in 2.5 ml of DNase I digestion buffer (15 mM Tris–Cl, pH 8.0, 90 mM NaCl, 60 mM KCl, 6 mM CaCl2 1 mM EDTA, pH 8.0, 0.5 mM ethylene glycol tetraacetic acid (EGTA), pH 8.0, 0.5 mM spermidine). DNase I digestion was carried out with 40 U/ml DNase I at 37°C for 3 min followed by termination of reaction with addition of equal volume of stop buffer (50 mM Tris–Cl, pH 8.0, 100 mM NaCl, 0.10% SDS, 100 mM EDTA, pH 8.0, 10 μg/ml RNAse A) and incubated for 4 h at 55°C. After addition of proteinase K (50 μg/ml), samples were further incubated for overnight at 55°C. On the following day, DNA was recovered by phenol/chloroform extraction. DNA fragments with size between 100 and 500 bp were isolated by sucrose gradient centrifugation. DNaseI library construction was performed according to Illumina's protocol and sequenced with HiSeq 2000.
Chromatin immunoprecipitation, Illumina library construction and sequencing
WT or DKO ES cells were cross-linked in culture medium with 1% formaldehyde (v/v) for 10 min at room temperature on a rocking platform, followed by quenching with 125 mM glycine. Crosslinked cells were washed twice with ice-cold phosphate-buffered saline and 1 × 107 cells were incubated in 1 ml Chromatin Prep Buffer containing 1 μl proteinase inhibitor cocktail (PIC) and 1 μl of 100 mM PMSF (Active Motif) for 10 min on ice followed by centrifuging for 3 min at 1250 × g at 4°C. The pellets were re-suspended in 250 μl ChIP Buffer with 2.5 μl PIC and 2.5 μl 100 mM PMSF and sonicated for 10 cycles with Bioruptor (30 s on/30 s off). Aliquots of 25 μl of sonicated chromatin were used to generate the input DNA. 5 μg of specific antibodies were then added to the rest of the chromatin samples and incubated overnight at 4°C with rotation. Following incubation 30 ul of protein G agarose beads (Active Motif) were then added to each reaction and the mixtures were further incubated for 3 h at 4°C. The beads were washed five times with Wash Buffer AM1 (Active Motif). ChIP DNA was eluted in 100 μl Elution Buffer AM4 (Active buffer). Cross-links were reversed at 65°C for overnight in the presence of 3 μl of 10% SDS and 5 μl of proteinase K (20 mg/ml). The DNA samples were eluted in 21 μl of elution buffer using MiniElute kit (Qiagen). ChIP-seq library was prepared following the manufacturer's instructions (Illumina). Briefly, immunoprecipitated and input DNA were blunt ended, ligated to adapters, amplified with PCR and size selected. The ChIP templates were sequenced at 75 bp single read length with Illumina NextSeq 500 system or 101 bp paired end with HiSeq2000 by the NIH CCR-sequencing facility (for details see data submission file). Sequence reads were aligned to the Build 37 assembly of the National Center for Biotechnology Information mouse genome data (NCBI37/mm9). The antibodies used in chromatin immunoprecipitation include: Rabbit polyclonal to Rex1 (Thermo Fisher Scientific PA5-27567), Rabbit polyclonal to Nanog (Active Motif, 61419), Rabbit polyclonal to Oct4 (abcam, ab19857), Rabbit polyclonal to SOX2 (Abcam #ab97959), and Rabbit Polyclonal Anti-CTCF (Millipore Sigma #07-729), Rabbit polyclonal to H3K27ac (abcam, ab4729), Rabbit polyclonal to H3K4me1(abcam, ab8895); Rabbit polyclonal to H3K4me3 (abcam, ab8580); Rabbit polyclonal to H3K9ac (abcam, ab4441), Rabbit monoclonal to H2BK5ac (abcam, ab40886).
Histone H1 ChIP-qPCR
Anti-calf histone H1 antibodies were generated in our laboratory. Primers used in Histone H1 ChIP-qPCR are as follows: Rex_enhancer1_F: TAGGACGGATATGGCTTTGC; Rex_enhancer1_R: TGTCCTTTCACCCAAGCATC; Rex_enhancer2_F: AAAAAGAGGCTA GGGTGTAGGC; Rex_enhancer2_R: AATCAGGGCTCTTAGGAGGTG; Rex_enhancer3_F: GGTTCCGTAGTGAAGCTTTGTG; Rex_enhancer3_R: ATGGAAAGCCCGTTCCTTAC; Rex_Nonreg1_F: GGATTTTGAGAACGCTGGAC; Rex_Nonreg1_R: CAAAGTAGACGG CATCTCTGC; Rex_Nonreg2_F: ATGAAGGCAGGCCCATTATC; Rex_Nonreg2_R: TGGCTGGGTAGAAGCATTTC Rex_Nonreg3_F: CCGGGCAATGGATAATACTG; Rex_Nonreg3_R: CCTCAGAAGGCAAGGAAATC; Rex_Nonreg4_F: GCTTGCCTGCATCTT TTGTC; Rex_Nonreg4_R: TGGCCTACCTGTCCAAAAAG; Rex_Nonreg5_F AAGTGGCGGCCAGTAGTATG; Rex_Nonreg5_R: AAACAGTCCCACCCTGTTTG.
Bioinformatic analyses
Unfiltered sequencing reads were aligned to the mouse reference genome (NCBI build 37, mm9) using BowTie (PMID:19261174). After alignment, the data were subjected to ChIP-seq pipeline from Kundaje lab (https://github.com/kundajelab/chipseq_pipeline), which is based on ENCODE transcription factor and histone ChIP-seq pipeline specifications. The peak regions were controlled by irreproducible discovery rate (IDR) with FDR 0.05. To test for differentially binding sites, we analyzed the peaks with the R/Bioconductor package ‘DiffBind’. For annotation analysis, the significant sites were analyzed with GREAT (http://great.stanford.edu/).
MEME-ChIP (http://meme.suite.org/tools/meme-chip) was used for motif analysis with default setting. ChIP-seq peaks were visualized by IGVTools. EaSeq or ngs.plot was used to create density plot based on the read count per million mapped reads (RPM).
RESULTS
HMGN proteins regulate expression of Rex1 gene in mouse embryonic stem (ES) cells
Mouse ESCs grown in suspension in the presence of LIF/2i maintain their pluripotency; however, upon withdrawal of LIF/2i from the culture they start to form embryoid bodies (EB). Within 8 days of LIF withdrawal, the EB gradually differentiate into cell lineages representing the three germ layers, a process associated with continuous changes in the transcription profile of the differentiating cells (25). By morphological criteria, the differentiation kinetics of WT ESCs into EB is indistinguishable from that of ESCs derived from double knock-out (DKO) mice, lacking both HMGN1 and HMGN2. Yet, transcription analysis of the differentiating cells, taken at 2-day intervals, revealed that loss of HMGNs leads to changes in the transcription level of multiple genes (20). A remaining unanswered question is whether loss HMGNs affects the expression of a specific set of genes throughout the differentiation process, or whether at each differentiation stage a unique set of genes is affected by loss of HMGNs.
We now find that at day 0, 2, 4, 6 following LIF/2i withdrawal, the expression of 176, 239, 447 and 592 genes respectively, was lower in DKO than in WT cells (Figure 1A). Although a relatively low fold of change (1.3) was used to compare affected genes, there was little overlap between the downregulated genes affected at various stages of EB differentiation; only 13 genes were consistently downregulated at all differentiation stages. These results suggest that by and large, at each differentiation stage HMGNs modulate the expression of a distinct set of genes.
Figure 1.

HMGN proteins affect Rex1 expression. (A) Venn diagrams showing the overlap between down-regulated genes in DKO as compared to WT embryonic stem cells, at various days following LIF/2i withdrawal. The total number of genes with down regulated expression in DKO (1.3-fold; P < 0.05) at each EB differentiation stage is shown at the periphery of the diagram. The 13 genes listed on the right of the diagram are downregulated throughout the course of differentiation. (B) Transcription level of Rex1 in WT and DKO ESCs at indicated days following LIF/21 withdrawal. Shown are average value of three biological replicates measured by mRNA-seq. (C) IGV snapshot of 3 biological replicates showing mRNA level of Rex1 in ESCs. (D) IGV snapshot showing mRNA level of Actb as controls. (E) Down regulated expression of REX1 in DKO ESCs. Shown are western blots of three biological replicates. Histone H3 serves as loading control. Right: quantifications of the of REX1 expression relative to H3 using ImageJ. The expression of REX1 in WT ESCs is defined as 1.0. (F) siRNA mediated knock-down of Hmgn1 and Hmgn2 expression in WT ESCs reduces Rex1 expression. Average of two independent experiments.
Among the 13 genes consistently down regulated in DKO cells, throughout all days of early EB differentiation, we focused on Rex1(Zfp-42), a zinc finger protein known to affect the pluripotency (21). Rex1 is highly expressed in mouse embryonic stem cell; quantitative RNA-seq indicates that its transcript levels are in the top 2% of all the transcripts in the cells. The robust Rex1 expression seen in ESCs (day 0 in Figure 1B), is markedly downregulated as soon as 2 days after LIF/2i withdrawal (Figure 1B). In DKO ESCs, the expression of Rex1 was more than 3-fold lower that in WT ESCs, and Rex1 transcription remains lower in DKO cells throughout all 6 days of EB differentiation (Figure 1B and C). In contrast, loss of HMGNs has little effect on the expression of housekeeping genes such as Actb (Figure 1D). Western blots verify that also the REX1protein levels are over 50% lower in DKO than in WT cells (Figure 1E). The downregulation of Rex1 transcripts is indeed due to loss of HMGNs since control experiments with WT ESCs in which the expression of either Hmgn1, Hmgn2 or both Hmgn1 and Hmgn2, is down regulated by specific siRNAs, also shows reduction in Rex1 expression (Figure 1F).
Loss of HMGN proteins alters the epigenetic landscape at Rex1 regulatory sites
Toward understanding the mechanism whereby HMGNs regulate Rex1 expression we first examined whether loss of HMGNs alters chromatin organization at the regulatory regions of this gene. Chromatin immunoprecipitation (ChIP) with antibodies to both HMGN1 and HMGN2 reveals high HMGN occupancy in the 5′ region of the Rex1 gene and at two upstream regions enriched in H3K27ac and H3K4me1, histone modifications that mark enhancer regions (red and brown stars in Figure 2). The distal, more upstream regulatory region, located 14 kb from the Rex1 gene (brown star in Figure 2) shows high relative DNaseI sensitivity and corresponds to a known ESC-specific super enhancer (26). At this super-enhancer, loss of HMGNs leads to a marked reduction in the DNaseI sensitivity and in H3K27ac and H3K4me1 levels (arrows in Figure 2). In addition, loss of HMGNs leads to a reduction in the levels of H3K27ac and H3K4me1 (arrows) and of H3K4me3, H3K9Ac and H2BK5ac in the more proximal 5′ region of the gene (dashed box in Figure 2); however, the downregulation of these marks of transcriptionally active chromatin was less pronounced than those seen in the distal super enhancer region. In summary, our ChIP-seq analyses of WT and DKO ESCs indicate that loss of HMGN proteins reduces the levels of active chromatin marks and the DNase I hypersensitivity at the Rex1 regulatory sites, a finding that is fully consistent with the downregulated Rex1 expression in DKO cells.
Figure 2.
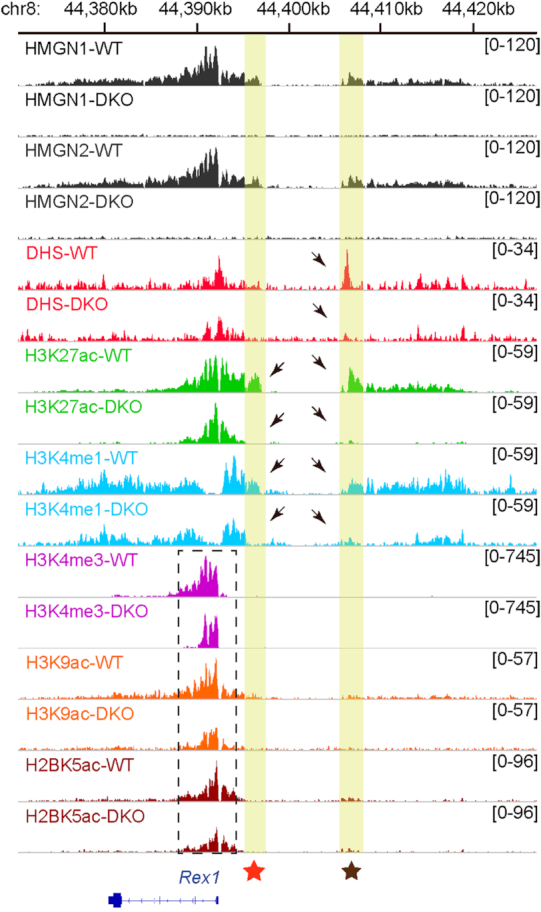
Loss of HMGNs alters the chromatin regulatory landscape at the Rex1 locus. Shown are IGV snapshots depicting HMGN1and HMGN2 occupancy, DNase I hypersensitivity (DHS) and the levels of the indicated histone modifications at the Rex1 locus of ESCs isolated from WT and DKO mice. The yellow columns highlight the position of the proximal (red star) and distal (brown star) enhancer regions. Arrows point to the loss of DNase1, H2K27ac and H3K4me1 in DKO cells. Epigenetic changes in the proximal 5′ region of the Rex1 gene are seen in the box demarcated by dashed lines.
Loss of HMGN proteins reduces the binding of OCT4, NANOG and SOX2 transcription factors to the Rex1 super-enhancer
Rex1 expression in ESCs is known to be regulated by the transcription factors OCT4, NANOG and SOX2 (27,28). Since our mRNA-seq analyses show that loss of HMGN proteins did not affect the transcription levels of either Oct4, Nanog or Sox2 (Figure 3A), we examined the possibility that HMGNs regulates Rex1 expression by modulating the binding of OCT4, NANOG or SOX2 to chromatin. ChIP-seq experiments with antibodies specifically recognizing these transcription factors revealed high specific occupancy of all the three transcription factors at the super-enhancer region located 5′ to the Rex1 gene (Figure 3B, brown star) but not at the proximal enhancer regions (Figure 3B red star), or at the adjacent promoter regions. Significantly, loss of HMGN totally abolished the binding of OCT4, NANOG and reduced the binding of SOX2 to the Rex1 super-enhancer (Figure 3A, arrows). Thus, at the Rex1 locus, loss of HMGNs abolishes the binding of transcription factors to regulatory regions and reduces the levels of histone marks that characterize active chromatin (Figure 2). Taken together, the data indicates that HMGNs facilitate the establishment and maintenance of a chromatin regulatory landscape that optimizes Rex1 expression.
Figure 3.

HMGN-H1 interplay regulates the binding of transcription factors to the Rex1 super-enhancer. (A) Loss of HMGNs does not affect the expression of Oct4, Nanog, or Sox2. Shown are average of three biological replicates of mRNA-seq. (B) IGV snapshots showing loss of OCT4, NANOG and SOX2 binding at the Rex1 super-enhancer in DKO ESCs. Shown are two biological replicates. Red and brown stars indicate location of the proximal enhancer and super-enhancer regions, respectively (as in Figure 2). Arrows point to the location of the transcription factors. Select histone marks at these regulatory regions shown at the bottom. (C) Genomic regions selected for histone H1 ChIP-qPCR experiment. For exact location see primer sequence in Materials and Methods. Brown star denotes the super enhancer region (compare to panel A). (D) Relative H1 occupancy in WT and DKO ESCs at the five non-regulatory and three super-enhancer regions of Rex1 shown on panel C. (E) Elevated H1 occupancy at the Rex1 super-enhancer, but not at adjacent nonregulatory chromatin regions, in DKO cells. Average of values obtained in WT cells are normalized to 1.0. *P < 0.05; **P < 0.01; ****P < 0.0001.
Loss of HMGN proteins enhances the binding of histone H1 to the Rex1 super-enhancer
In considering the mechanism whereby loss of HMGN decreased the binding of transcription factors to the Rex1 super-enhancer, we note that the binding of HMGN to nucleosomes is known to decrease the interaction of the linker histone H1 with chromatin (14,15,29). H1 promotes and stabilizes chromatin condensation, potentially reducing access to chromatin regulatory sites (4,5). Therefore, we tested the possibility that loss of HMGN leads to increase binding of linker histone H1 to the Rex1 super-enhancer. To determine whether HMGNs affects histone H1 chromatin occupancy we used affinity pure antibody to histone H1 to perform ChIP-qPCR on three regions overlapping the Rex1 super-enhancer and five adjacent regions which are not enriched in epigenetic marks characteristic of regulatory chromatin (Figure 3C). In WT cells, the occupancy of H1 at all the three regions overlapping the Rex1 super-enhancer is noticeably lower than in each of the five regions overlapping the adjacent non-regulatory regions (compare blue bars in Figure 3D), a finding that is in full agreement with previous observation that H1 occupancy is decreased at chromatin regulatory sites (30). Importantly, we find that loss of HMGNs leads to a marked upregulation of H1 occupancy at each of the three sites located in the Rex1 super-enhancer but not at any of the five sites located at the adjacent non-regulatory sites (Figure 3D). The average occupancy of H1 at the non-regulatory sites in WT cells was the same as that in DKO cells but at the super-enhancer sites, loss of HMGN leads to a 50% increase in H1 occupancy (Figure 3E). The increased H1 occupancy at the Rex1 super enhancer in cells lacking HMGNs provides a possible explanation for the decreased occupancy of transcription factors and loss of epigenetic marks of active chromatin at this regulatory site.
HMGNs facilitates interaction between Rex1 promoter and enhancer
Transcription factor mediated interaction between gene regulatory elements such as enhancers and promoters are known to regulate gene expression (31,32). Previous ChIA-PET results (33) revealed significant interactions between the Rex1 distal super-enhancer and the Rex1 promoter (Figure 4A), raising the possibility that chromatin looping connecting these regulatory sites controls Rex1 expression levels. To test this possibility, we performed chromatin conformation capture (3C) assay and compared the Rex1 promoter-enhancer interaction in WT and DKO ES cells. We constructed 3C libraries by digestion of nuclei isolated from WT and DKO ES cells with Hind III restriction enzyme, followed by ligation with T4 DNA ligase and PCR amplification with primers targeting defined genomic sites (Figure 4B).
Figure 4.

Loss of HMGNs disrupts chromatin interactions at the Rex1 5′ regulatory region. (A) ChIA-PET analysis of interactions between chromatin regulatory regions at the Rex1 locus (data from (33)). Regulatory region and select histone marks are indicated at the top. Each line in the figure indicates a detected interaction between two genomic loci. Below the lines is a genomic outline showing the approximate location of HindIII restriction sites and of the primers (arrows) used in 3C analysis (panel B). (B) Outline of the 3C procedure used to measure chromatin interactions in the 5′ regulatory region of Rex1. (C) DNA fragment resulting from PCR amplification of the 3C library using the primers located in the 5′ regulator region of Rex1 (see arrows in panel A). The absence of signal in DKO cells indicates loss of chromatin interaction. Gapdh primers are used as loading controls. (D) Sanger sequencing confirms that the PCR products correspond to the junction of the two regions located near the regulatory sites in the 5′ region of Rex1.
PCR amplification of the 3C library prepared from WT ESCs with the primers targeting the 5′ upstream Rex1 regulatory regions (arrows next to red and brown stars, Figure 4A) yielded a clear band indicating high interaction frequency between the regulatory elements in the chromatin of these cells. In contrast, the same set of primers failed to amplify the 3C library generated from DKO cells, although the same amount of DNA was loaded, as shown by the amplified Gapdh control (Figure 4C). Sanger sequencing confirms that the PCR products derived from WT libraries consist of DNA sequence adjacent to the Rex1 enhancer and promoter regions (Figure 4D). In sum, both the ChIA-PET and the chromatin conformation capture experiments indicate that in the chromatin of WT cells, the Rex1 regulatory site located in the 5′ proximal region of the gene interacts with the distal super-enhancer located more than 14 kb upstream of the transcription start site. In DKO cell lacking HMGNs the promoter enhancer reaction is not seen.
The interaction between distal regulatory elements is known to be facilitated by chromatin looping which often is mediated by the zinc finger protein CTCF (32,34). To test whether CTCF plays a role in regulating enhancer-promoter interaction at the Rex1 gene, we performed ChIP-seq experiment using antibody against CTCF. We find relatively high CTCF occupancy at sites flanking the Rex1 gene locus, including its regulatory sites (Figure 5A, upper two tracks). Significantly, loss of HMGN did not alter CTCF occupancy (Figure 5A, lower tracks), suggesting that the HMGN mediated changes in Rex1 chromatin organization and gene expression are not due to changes in CTCF chromatin binding. Likely, HMGNs optimize Rex1 expression by facilitating the transcription factor mediated interactions between Rex1 regulatory sites within a chromatin loop stabilized by CTCF molecules. We suggest that within this loop, HMGNs reduces the binding of H1, but enhances the binding of transcription factors to the Rex1 super enhancer thereby facilitating its interaction with the proximal regulatory sites and facilitating robust Rex1 transcription. A model depicting the effect of HMGN on Rex1 locus chromatin organization and gene expression with a chromatin loop is shown in Figure 5B.
Figure 5.

HMGN mediated chromatin interactions enhance Rex1 transcription by facilitating transcription factor binding at the Rex1 locus. (A) Loss of HMGNs does not affect CTCF binding at Rex1 locus. Shown are two biological replicates of CTCF occupancy at the Rex1 locus, as determined by ChIP-seq assay. (B) Model depicting HMGN effects on chromatin interactions and Rex1 expression. The Rex1 locus is located within a chromatin loop formed by the interaction of CTCF molecules flanking the upstream and downstream regions of the locus. In WT cells (left side of panel) the binding of NANOG, OCT4 and SOX2 at the super-enhancer located 5′ to the Rex1 gene facilitates interaction with proximal regulatory elements thereby optimizing Rex1 expression. Loss of HMGN (right side) does not affect CTCF binding but increases H1 occupancy, abolishes the binding of transcription factors to the super-enhancer, and reduces both the DNase I sensitivity and the levels of histone modification that mark active chromatin. These epigenetic changes prevent contact between the Rex1 super-enhancer and Rex1 proximal regulatory sites, thereby downregulating Rex1 expression.
Altered REX1 Chromatin occupancy in cells lacking HMGNs
REX1 is a transcription factor that has been shown to bind to chromatin (23,24,28,35). Since loss of HMGNs reduces but does not abolish the cellular level of REX1 protein, we used ChIP-seq analysis to map the genome-wide binding sites of REX1 in WT and DKO ESCs and examine whether HMGNs affect the chromatin binding specificity of REX1. In the chromatin of WT ESCs, we identified a total of 4843 REX1 binding sites, while in the chromatin of DKO cells we identified only 2113 sites (Figure 6A). Globally, the top sequence motifs underlying the REX1 binding sites in both WT and DKO cells correspond to the known REX1 binding motif as recorded in the HOCOMOKO database (36) (Figure 6B). Genome wide 73% of REX1 binding sites overlap regulatory regions marked by H3K27ac (Figure 6C). Over 90% of the REX1 sites located at these regulatory sites colocalize with both HMGN1 and HMGN2 binding sites. In contrast, less than 50% of REX1 sites localized at non-regulatory regions colocalize with HMGNs (Figure 6C). Thus, HMGNs and REX1 preferentially colocalize at regulatory regions. Genome wide, the REX1 occupancy at both promoters (Figure 6D and E) and at enhancers (Figure 6F) was significantly lower in DKO cells, suggesting that loss of HMGN reduces REX1 binding to chromatin regulatory sites.
Figure 6.

Loss of HMGNs leads to a global reduction in REX1 binding to chromatin. (A) Venn diagram showing the number of REX1 binding sites detected by ChIP-seq in WT and DKO ESCs. (B) Top DNA sequence motif underlying the REX1 binding sites in WT and DKO cells. (C) Overlap between REX1 and HMGNs occupancy at chromatin regulatory sites. (D, E) Decreased REX1 occupancy at TSS and neighboring 4 kb regions in DKO cells. (F) Box plots showing decreased REX1 binding at enhancers of DKO cells. ****P < 0.0001.
Given the decreased levels of REX1 protein in DKO cells it is not surprising that the overall REX chromatin occupancy is decreased in these cells. Scatter plot of the ChIP analyses show high correlation between two biological replicates of either WT or DKO cells (Figure 7A and B). In contrast, the scatter plot in Figure 7C, which compares REX1 binding in WT to that in DKO cells, reveals two subsets of REX1 binding sites. One subset, located at the diagonal (named ‘unaltered’ in Figure 7C), shows equal binding in both WT and DKO cells suggesting that this subset of REX1 binding was not significantly affected by the loss of HMGNs; the REX1 occupancy at these sites in WT cells was the same as that measured in DKO cells. The second subset (named ‘altered’ in Figure 7C) shows significantly lower REX1 binding in DKO cells suggesting decreased REX1 binding at these sites.
Figure 7.

Altered REX1 Chromatin occupancy in cells lacking HMGNs. (A) Scatter plots comparing intensities of REX1 peaks between two WT biological replicates. (B) Scatter plots comparing intensities of REX1 peaks between two DKO biological replicates. (C) Scatter plots comparing intensities of REX1 peaks between WT and DKO cells. (D) Heat map showing HMGN1 and HMGN2 occupancy at top altered and top unaltered REX1 binding sites. (E) HMGN occupancy at top altered (left) or unaltered (right) REX1 binding sites. (F) IGV snapshot showing colocalization of HMGNs and REX1 in the chromatin of WT cells and loss of REX1 binding in DKO cells (G) Distribution of top unaltered (left panel) and altered (right panel) REX1 binding sites relative to the nearest TSS. (H) Top DNA sequence motifs underlying the most unaltered and most altered REX1 binding sites in DKO cells. Note that the top motif underlying the unaltered REX1 sites (3rd row) contains the REX1 binding motif in the reverse complimentary orientation.
To further examine the effect of HMGNs on the binding of REX1 to chromatin we analyzed the sequence motifs underlying either the unaltered or the altered sites, i.e. the REX1 binding sites that in Figure 7C are located either at, or below the diagonal. In this analysis we examined only the top altered and unaltered sites; for the unaltered sites we included only the sites that showed the least significant change (FDR > 0.1, fold of change < 0.5) while for the altered sites we included only sites that showed the most significant change (FDR < 0.01, fold of change > 2) in the DKO cell. By these criteria, 428 REX1 binding sites were unaltered, i.e. had the same occupancy in WT and DKO cells and 656 REX1 sites were most significantly altered.
The occupancy of HMGN at the altered REX1 sites was significantly larger than at the non-altered sites (Figure 7D–F); almost 90% of altered REX1 sites, but <50% of unaltered sites showed HMGN occupancy in WT cells (Figure 7E). Mapping the altered sites reveals that 422 sites localize within 5 kb of a TSS (Figure 7G, right); the top motif underlying these altered sites corresponds to ATGGC (E-value: 5.7e-402) the ‘canonical’ REX1 binding motif (Figure 7G, compare top to second line). In contrast, mapping the 428 unaltered REX1 binding sites reveals that only 72 sites localize within 5 kb of a TSS (Figure 7G, left). Most of the unaltered binding sites are located at genomic regions positioned >5 kb away from transcription start sites (TSS). Interestingly, the top sequence underlying these unaltered sites (E-value: 3.1e–369) (Figure 7H, third line) is a composite of two binding motifs: the reverse complimentary sequence of the REX1 and the binding motif of MYOD1, raising the possibility that at these sites the presence of MYOD1 helps retain REX1 binding even in the absence of HMGN proteins. The next prevalent binding motif in this group GGRKGCKGCCTMAGC (E-value: 8e–208) contains the binding motif of the transcription factor MAFK but lacks the canonical REX1 binding site. The third most prevalent binding motif in the ‘unaltered site’ GGCGGKYCCCGGMCC (E-value = 2.0e–267) has no match for any known transcription factor binding motif.
In sum, specific REX1 binding sites co-localize with HMGNs. Loss of HMGNs reduces REX1 binding at the ‘canonical’ REX1 binding motif located near a TSS to a larger degree than at sites that are distal to TSS. At the sites that are not near TSS, the canonical REX1 binding motif is either adjacent to the binding motif of the transcription MYOD1, or not detectable. Thus, the presence of HMGNs enhances the specific binding of REX1 to sites containing only the canonical REX1 binding motif to a larger degree than at sites that do not contain only the REX1 binding motif. We conclude that HMGNs facilitates the interaction of REX1 with its specific chromatin binding sites.
DISCUSSION
Our study provides insights into the mechanisms that regulate the expression and chromatin binding of REX1 (ZFP-42) in embryonic stem cells, and in a broader sense, provides new information on the molecular mechanisms whereby the ubiquitous HMGN nucleosome binding proteins impact cell-type specific gene expression.
We find that at the Rex1 locus, the chromatin occupancy of HMGN1 and HMGN2 proteins is elevated at chromatin regions enriched in both H3K27ac and H3K4me1, histone modifications that mark gene regulatory sites (37). At the 5′ Rex1 regulatory sites, and especially at the distal 5′ super enhancer, loss of HMGN reduces the levels of H3K27ac and H3K4me1 and the DNase I hypersensitivity, a hallmark of transcriptionally active chromatin. Significantly, loss of HMGNs leads not only to reduced histone marks but also to a marked reduction in the chromatin occupancy of NANOG, OCT4 and to a lesser degree in that of SOX2, transcription factors that have been shown to affect Rex1 expression (27,28). Importantly, the 5′ distal regulatory region where these epigenetic changes occur corresponds to a known embryonic stem cell super-enhancer. Super-enhancers are known to play a key role in regulating cell-type specific gene expression and stabilize cell identity (2). Significantly, we find that loss of HMGN increases the occupancy of the linker histone H1 at the Rex1 super-enhancer. It is well documented that histone H1 stabilizes chromatin compaction and reduces access to chromatin (4,5). In living cells, both H1 and HMGN bind dynamically to chromatin, and their chromatin binding is interdependent (6,13,15). It has been suggested that the dynamic interplay between these architectural proteins is part of the molecular mechanism that fine tunes gene expression (6). Indeed, we demonstrated that at the Olig1 and Olig2 promoter loss of HMGNs increases the occupancy of H1 thereby enhancing the recruitment of EZH2 leading to the downregulation of Olig1 and Olig2 expression (20). Our present study shows that in embryonic stem cells the HMGN-H1 interplay affects the binding of tissue specific transcription factors to a regulatory site in chromatin.
At the structural level, our 3C studies indicate that loss of HMGNs disrupts the interaction between the Rex1 promoter and enhancer, an interaction that we suggest occurs within a chromatin loop anchored by CTCF proteins bound to sites that bracket the Rex1 locus. We note that Rex1 is the only gene present in this chromatin loop; it remains to be seen whether the location of the loop itself affects the degree to which HMGNs affect the interaction of the transcription factors with their cognate binding sites. Thus, proper Rex1 expression involves the binding of three transcription factors to a regulatory site located in the 5′ region of the Rex1 gene. The binding of the transcription factors leads to changes in histone modification and facilitates enhancer-promoter interactions that upregulate Rex1 transcription. Significantly, the binding of the transcription factors and the subsequent epigenetic changes are contingent on presence of HMGN1 and HMGN2, structural proteins that bind to nucleosomes without DNA specificity.
Given the down regulation of Rex1 expression and reduced REX1 levels in DKO cells it is not surprising that the REX1 protein chromatin occupancy in DKO cells is lower than in WT cells (Figure 6). However, the REX1 chromatin occupancy is not uniformly reduced at all REX1 chromatin binding sites. As elaborated in the results section, the most significant loss occurs at binding sites that contain an isolated ‘canonical’ REX1 binding motifs; at ‘non-canonical’ sites REX1 binding is not significantly affected by loss of HMGNs. The results indicate that HMGNs strengthen the specific interaction of REX1with its specific chromatin binding sites. Thus, HMGNs affect both the expression of REX1 and the specificity of its chromatin interactions.
It is well documented that HMGNs associate with chromatin regulatory sites, reduce chromatin compaction, and impact the cellular transcription profile (9,11,18). Significantly, HMGNs do not regulate the expression of a specific set of genes or a specific biological pathway; they modulate the fidelity of the cellular transcriptional profile in a tissue specific manner (7,8), a finding that raises the possibility that they impact the action of cell-type specific transcription factors. Indeed, we recently reported that HMGNs stabilize cell identity, most likely by facilitating the binding of tissue specific transcription factors to cell-type specific super enhancers that maintain specific cell identity (18). These previous studies suggested, but did not show, that HMGNs modulate the action of transcription factors. This study presents experimental evidence for this possibility and provides a paradigm for the mechanism whereby the ubiquitous HMGN proteins fine tune tissue specific gene expression.
DATA AVAILABILITY
Primary bioinformatic data available at accession number: PRJNA513065.
ACKNOWLEDGEMENTS
Authors Contributions: S.Z., T.D., B.H., T.F. performed experiments and analyzed data, W.T. S.Z. and S.A. performed bioinformatic analyses, S.Z. and M.B. conceived the project, analyzed data and wrote the manuscript. M.B. supervised the project. We thank Dr Yuri V. Postnikov for critical remarks on the manuscript.
FUNDING
Center for Cancer Research; Intramural Research Program of the National Cancer Institute, NIH. Funding for open access charge: National Institutes of Health, Bethesda, MD; Intramural Program.
Conflict of interest statement. None declared.
REFERENCES
- 1. Zhu F., Farnung L., Kaasinen E., Sahu B., Yin Y., Wei B., Dodonova S.O., Nitta K.R., Morgunova E., Taipale M. et al.. The interaction landscape between transcription factors and the nucleosome. Nature. 2018; 562:76–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Whyte W.A., Orlando D.A., Hnisz D., Abraham B.J., Lin C.Y., Kagey M.H., Rahl P.B., Lee T.I., Young R.A.. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013; 153:307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hochedlinger K., Jaenisch R.. Induced pluripotency and epigenetic reprogramming. Cold Spring Harb. Perspect. Biol. 2015; 7:a019448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fyodorov D.V., Zhou B.R., Skoultchi A.I., Bai Y.. Emerging roles of linker histones in regulating chromatin structure and function. Nat. Rev. Mol. Cell Biol. 2018; 19:192–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Izzo A., Schneider R.. The role of linker histone H1 modifications in the regulation of gene expression and chromatin dynamics. Biochim. Biophys. Acta. 2016; 1859:486–495. [DOI] [PubMed] [Google Scholar]
- 6. Bustin M., Catez F., Lim J.H.. The dynamics of histone H1 function in chromatin. Mol. Cell. 2005; 17:617–620. [DOI] [PubMed] [Google Scholar]
- 7. Kugler J.E., Horsch M., Huang D., Furusawa T., Rochman M., Garrett L., Becker L., Bohla A., Holter S.M., Prehn C. et al.. High mobility group N proteins modulate the fidelity of the cellular transcriptional profile in a tissue- and variant-specific manner. J. Biol. Chem. 2013; 288:16690–16703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rochman M., Taher L., Kurahashi T., Cherukuri S., Uversky V.N., Landsman D., Ovcharenko I., Bustin M.. Effects of HMGN variants on the cellular transcription profile. Nucleic Acids Res. 2011; 39:4076–4087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kugler J.E., Deng T., Bustin M.. The HMGN family of chromatin-binding proteins: dynamic modulators of epigenetic processes. Biochim. Biophys. Acta. 2012; 1819:652–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gonzalez-Romero R., Eirin-Lopez J.M., Ausio J.. Evolution of high mobility group nucleosome-binding proteins and its implications for vertebrate chromatin specialization. Mol. Biol. Evol. 2015; 32:121–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Postnikov Y., Bustin M.. Regulation of chromatin structure and function by HMGN proteins. Biochim. Biophys. Acta. 2010; 1799:62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Catez F., Lim J.H., Hock R., Postnikov Y.V., Bustin M.. HMGN dynamics and chromatin function. Biochem. Cell Biol. 2003; 81:113–122. [DOI] [PubMed] [Google Scholar]
- 13. Postnikov Y.V., Bustin M.. Functional interplay between histone H1 and HMG proteins in chromatin. Biochim. Biophys. Acta. 2016; 1859:462–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Murphy K.J., Cutter A.R., Fang H., Postnikov Y.V., Bustin M., Hayes J.J.. HMGN1 and 2 remodel core and linker histone tail domains within chromatin. Nucleic Acids Res. 2017; 45:9917–9930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Catez F., Brown D.T., Misteli T., Bustin M.. Competition between histone H1 and HMGN proteins for chromatin binding sites. EMBO Rep. 2002; 3:760–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kato H., van Ingen H., Zhou B.R., Feng H., Bustin M., Kay L.E., Bai Y.. Architecture of the high mobility group nucleosomal protein 2-nucleosome complex as revealed by methyl-based NMR. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:12283–12288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Deng T., Zhu Z.I., Zhang S., Postnikov Y., Huang D., Horsch M., Furusawa T., Beckers J., Rozman J., Klingenspor M. et al.. Functional compensation among HMGN variants modulates the DNase I hypersensitive sites at enhancers. Genome Res. 2015; 25:1295–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. He B., Deng T., Zhu I., Furusawa T., Zhang S., Tang W., Postnikov Y., Ambs S., Li C.C., Livak F. et al.. Binding of HMGN proteins to cell specific enhancers stabilizes cell identity. Nat. Commun. 2018; 9:5240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang S., Zhu I., Deng T., Furusawa T., Rochman M., Vacchio M.S., Bosselut R., Yamane A., Casellas R., Landsman D. et al.. HMGN proteins modulate chromatin regulatory sites and gene expression during activation of naive B cells. Nucleic Acids Res. 2016; 44:7144–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Deng T., Postnikov Y., Zhang S., Garrett L., Becker L., Racz I., Holter S.M., Wurst W., Fuchs H., Gailus-Durner V. et al.. Interplay between H1 and HMGN epigenetically regulates OLIG1&2 expression and oligodendrocyte differentiation. Nucleic Acids Res. 2017; 45:3031–3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Climent M., Alonso-Martin S., Perez-Palacios R., Guallar D., Benito A.A., Larraga A., Fernandez-Juan M., Sanz M., de Diego A., Seisdedos M.T. et al.. Functional analysis of Rex1 during preimplantation development. Stem Cells Dev. 2013; 22:459–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Son M.Y., Choi H., Han Y.M., Cho Y.S.. Unveiling the critical role of REX1 in the regulation of human stem cell pluripotency. Stem Cells. 2013; 31:2374–2387. [DOI] [PubMed] [Google Scholar]
- 23. Gontan C., Achame E.M., Demmers J., Barakat T.S., Rentmeester E., van I.W., Grootegoed J.A., Gribnau J.. RNF12 initiates X-chromosome inactivation by targeting REX1 for degradation. Nature. 2012; 485:386–390. [DOI] [PubMed] [Google Scholar]
- 24. Kim J.D., Faulk C., Kim J.. Retroposition and evolution of the DNA-binding motifs of YY1, YY2 and REX1. Nucleic Acids Res. 2007; 35:3442–3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Murry C.E., Keller G.. Differentiation of embryonic stem cells to clinically relevant populations: lessons from embryonic development. Cell. 2008; 132:661–680. [DOI] [PubMed] [Google Scholar]
- 26. Hnisz D., Abraham B.J., Lee T.I., Lau A., Saint-Andre V., Sigova A.A., Hoke H.A., Young R.A.. Super-enhancers in the control of cell identity and disease. Cell. 2013; 155:934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shi W., Wang H., Pan G., Geng Y., Guo Y., Pei D.. Regulation of the pluripotency marker Rex-1 by Nanog and Sox2. J. Biol. Chem. 2006; 281:23319–23325. [DOI] [PubMed] [Google Scholar]
- 28. Ben-Shushan E., Thompson J.R., Gudas L.J., Bergman Y.. Rex-1, a gene encoding a transcription factor expressed in the early embryo, is regulated via Oct-3/4 and Oct-6 binding to an octamer site and a novel protein, Rox-1, binding to an adjacent site. Mol. Cell Biol. 1998; 18:1866–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Catez F., Ueda T., Bustin M.. Determinants of histone H1 mobility and chromatin binding in living cells. Nat. Struct. Mol. Biol. 2006; 13:305–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cao K., Lailler N., Zhang Y., Kumar A., Uppal K., Liu Z., Lee E.K., Wu H., Medrzycki M., Pan C. et al.. High-resolution mapping of h1 linker histone variants in embryonic stem cells. PLoS Genet. 2013; 9:e1003417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li W., Notani D., Rosenfeld M.G.. Enhancers as non-coding RNA transcription units: recent insights and future perspectives. Nat. Rev. Genet. 2016; 17:207–223. [DOI] [PubMed] [Google Scholar]
- 32. Sun F., Chronis C., Kronenberg M., Chen X.F., Su T., Lay F.D., Plath K., Kurdistani S.K., Carey M.F.. Promoter-enhancer communication occurs primarily within insulated neighborhoods. Mol. Cell. 2019; 73:250–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kieffer-Kwon K.R., Tang Z., Mathe E., Qian J., Sung M.H., Li G., Resch W., Baek S., Pruett N., Grontved L. et al.. Interactome maps of mouse gene regulatory domains reveal basic principles of transcriptional regulation. Cell. 2013; 155:1507–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ron G., Globerson Y., Moran D., Kaplan T.. Promoter-enhancer interactions identified from Hi-C data using probabilistic models and hierarchical topological domains. Nat. Commun. 2017; 8:2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim J., Chu J., Shen X., Wang J., Orkin S.H.. An extended transcriptional network for pluripotency of embryonic stem cells. Cell. 2008; 132:1049–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kulakovskiy I.V., Vorontsov I.E., Yevshin I.S., Sharipov R.N., Fedorova A.D., Rumynskiy E.I., Medvedeva Y.A., Magana-Mora A., Bajic V.B., Papatsenko D.A. et al.. HOCOMOCO: towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-Seq analysis. Nucleic Acids Res. 2018; 46:D252–D259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Strahl B.D., Allis C.D.. The language of covalent histone modifications. Nature. 2000; 403:41–45. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Primary bioinformatic data available at accession number: PRJNA513065.