

Abstract
Cognitive impairment in Alzheimer’s disease (AD) is usually accompanied by synaptic loss in both the hippocampus and neocortex. In the early stage of AD, amyloid β-induced synapse changes is the main reason, while in the later stage, the accumulation of Tau protein promotes synapse degeneration as the key factor leading to dementia. MicroRNA (miRNA) is closely related to the expression changes of many AD-related genes. One of the most abundant brain-enriched miRNAs is miR-132, which has been shown to regulate both neuron morphogenesis and plasticity. It has been reported that miR-132 is significantly reduced in the brains of Alzheimer’s patients. Genetic deletion of miR-132 in mice promotes Aβ deposition, leading to impaired memory and enhanced Tau pathology, but how the miRNA-mediated gene expression dysregulation contributes to AD pathology remains unclear. Here we found the possible downstream target of miR-132 by in silico analysis, namely C1q. C1q is the primary protein of classical complement cascade, which is highly expressed in the synaptic regions of the central nervous system in Alzheimer’s patients. However, it is not clear whether miR-132 plays a role in AD through regulating C1q. To address this question, the APP/PS1 transgenic mice were transfected with miR-132 and given C1 inhibitors. Behavior tests were conducted to assess memory and cognitive abilities seven days after administration. In addition, we analyzed the expression of PSD95, Synapsin-1 and phosphorylated (p)- Synapsin. We found that the expression levels of the synaptic proteins treated with miR-132 or C1INH were significantly increased compared with the AD group. Further RT-qPCR result suggested that miR-132 might regulate C1q expression in AD.
Key words: miR-132, Alzheimer's disease, synapse, C1q
Introduction
Alzheimer’s disease (AD) is a degenerative disorder in the central nervous system (CNS) characterized by progressive cognitive deficit and behavioral impairment. Pathological changes include amyloid plaque, neurofibrillary tangles, and neuronal reduction, which are closely related to the morphological and functional abnormality of neurons and glial cells. It is generally accepted that the combined presence of amyloid β (Aβ) and tau protein plays a key role in the AD pathogenesis, causing synaptic dysfunction and cognitive decline.1 However, studies have shown that regionalspecific synaptic loss may be more associated with cognitive decline in AD compared with amyloid plaque, neurofibrillary tangles, and neuronal reduction.2,3 Therefore, in the search for understanding the disease mechanism, research is moving increasingly into the role of synaptic proteins at different phases of AD.
Multiple lines of evidence implicate that microRNAs (miRNAs) modulate essential physiological processes, including neuronal differentiation, plasticity and regeneration, and fine-tune post-transcriptional gene expressions.4 miRNAs also control innate immune responses and acquired immune responses in mammalian immune systems.5 They are abundantly expressed in the CNS and play an important role in neural cell development, differentiation and synaptic plasticity.6 Indeed, aberrations in the expression of miRNAs in the CNS result in a variety of neurodegenerative disorders, particularly including Parkinson’s and Alzheimer’s diseases.7,8 Along these lines, it has been reported that miR-132 is a positive regulator of synaptic activity and plasticity, whose expression level is significantly increased in the early AD stage (Braak II phase),9 and significantly decreased in the later stage,10-12 presenting the mode of “increased at first and then decreased”. Interestingly, knockout of miR- 132 gene in AD transgenic mice results in impaired memory, increased Aβ expression, accumulated amyloid plaques, and hyperphosphorylation of tau protein.13 Furthermore, deletion of the mir-132 locus might reduce dendritic spine density and impair the integration of new neurons.14,15 Nevertheless, the miRNA-mediated downstream gene dysregulation in AD-induced aberrant synapse formation remains largely unknown.
In the present study, we found that miR- 132 has a 3’UTR binding site for C1q mRNA through in silico analysis. C1q belongs to the classical complement cascade proteins, components of the innate immune system, which mediate developmental synaptic pruning, a key process in establishing precise synaptic circuits.16 Recent study shows that C1q was increased and involved in synaptic loss before apparent plaque deposition.17 When inhibited, the number of phagocytic microglia reduced and early synaptic loss was observed as well.16,18 In addition, the synaptic expression of AD patients in different areas of the brain, including cerebral cortex, hippocampus and dentate gyrus, decreased significantly, and the expression of C1q increased significantly. However, it is not clear whether miR-132 exerts its effect through regulating C1q expression in this process. To address this, we examined the effect of miR-132 on the expression of synaptic proteins in the APP/PS1 transgenic mice and further assessed whether it involved its possible downstream target C1q. The effect of their changes on learning and memory was also detected by Morris water maze test. Our results showed that the expression levels of the synaptic proteins in the temporal cortex of mice treated with miR-132 or the C1q inhibitor C1INH were significantly increased compared with the AD group.
Materials and Methods
Construction and validation of miR- 132-3p lentivirus
The lentivirus-delivered miR-132-3p overexpression vector and miR-scramble control were designed and synthesized by Wanlei Biotech (Shenyang, China). The ability of the lentivirus vectors to express the gene of interest was evaluated by sequencing. The final titers used for the experiments were 1×108 Tu/mL.
Animals and grouping
Twelve adults male C57BL/6 mice and thirty-six APPswe/PS1dE9 (APP/PS1) transgenic mice were purchased from Liaoning Changsheng biotechnology Co., Ltd (Shenyang, China). The mice were 10 months old (25-30 g) and individually housed under a 12 h light-dark cycle (temperature: 25±1°C; humidity: 50 ± 10%) with ad libitum access to water and food. The AD mice were randomly divided into three groups: AD group (n=12); AD+C1INH group (n=12); AD+miR-132 overexpression group (n=12), and the C57 mice served as the normal control group (n = 12). The experiments were in accordance with the guidelines for the Care and Use of Laboratory Animals from the National Institutes of Health, and were approved by the Animal Experimental Committee of China Medical University.
Stereotaxic intracranial injection
The lentivirus vector and the C1q inhibitor C1INH were microinjected into the right lateral ventricle of AD mice. Briefly, the mice were anesthetized with an intraperitoneal injection of 4% chloral hydrate (0.5 mL/100g body weight) and mounted into a stereotaxic apparatus with a mouse adaptor (RWD Life Science Co., Ltd., Shenzhen, China). The skull was opened using a dental drill, and 2 μL lentivirus vectors were injected into the right lateral ventricle (AP: -2.0 mm, R: -1.5 mm, DV: -3.0 mm, according to bregma) and delivered for 5 min until the entire vector was administered. After another 2-3 min the needle was slowly withdrew. The mice in the control group was subjected to identical surgery. The GFP fluorescence was randomly observed 7 d after administration to verify whether miR-132 was successfully delivered throughout the brain.
Morris water maze behavior test
Seven days after the stereotaxic intracranial injection, Morris water maze (MWM) test was performed as described in our previous study.19 The MWM consisted of a black circular pool (120 cm in diameter, divided into four quadrants) filled 2/3 with water (23 ± 1°C) and additional Titanium dioxide powder. A platform was submerged approximately 2 cm below the water surface during the spatial learning trials. Mice were initially placed into a quadrant (excluding the one with the platform). The time that the mice found the platform was recorded (evacuation latency). If they could not find the escape platform in 60 s, the experimenters would gently guide the mice onto the platform and allowed them to stay there for 20 s. All mice were subjected to an MWM test for 5 consecutive days with 4 acquisition trials per day. On day 6, a probe test was carried out to evaluate spatial memory, where the platform was removed and they were placed at a new random position. The times of mice crossing the platform were calculated. When the MWM test was over, the mice were sacrificed for the following procedure.
Western blot analysis
The mice were anesthetized with an intraperitoneal injection of 4% chloral hydrate (0.5 mL/100g body weight) and immediately perfused transcardially with 0.9% NaCl. The temporal cortex was taken out and homogenized in lysis buffer for 30 min (Beyotime, Shanghai, China), and were homogenized for 20 min and centrifuged at 500 g for 30 min at 4°C. Protein extracts were processed with the following Western blot method. Briefly, Equal amounts of protein (50 mg) were resolved on a 4-12% Bis- Tris gel before transferred onto a polyvinylidene difluoride membrane, and blocked for 1 h at room temperature, in 5% skim milk powder solution. Next, membranes were incubated at 4°C with primary antibodies against PSD95 (1:500, Abcam, Cambridge, MA, USA), Synapsin-1 (1:500, Abcam), and p-Synapsin (1:500, Arigo Biolaboratories Corp., Shanghai, China) and GAPDH (1:5000, Abcam). After washing, the membranes were incubated with goat anti-rabbit IgG (1:5000, Proteintech Group, Inc., Rosemont, IL, USA) secondary antibody for 1 h at 37°C. Blots were imaged with a chemiluminescence imaging system (Bio- Rad, Hercules, CA, USA) and measured with Image J v1.80 software (NIH, Bethesda, MD, USA).
Immunofluorescence analysis
After anesthesia, mice were immediately perfused transcardially with 4% paraformaldehyde (PFA). The brains were fixed in 4% PFA for 24 h after perfusion, and then soaked in 15%, 25% and 30% sucrose for 24 h, 24 h and 48 h. Next, they were immerged in OCT compound and frozen at -80°C. After rinsing with PBST for three times, sections were incubated in blocking solution containing 0.2% Triton X and 5% goat serum for 40 min. Then they were incubated with primary antibody for the detection of PSD95 (1:200, Abcam), Synapsin-1 (1:200, Abcam), and p-Synapsin (1:200, Arigo) at 4°C overnight. Rabbit IgG monoclonal Isotype Control (1:200, Abcam) served as negative control. On the second day, sections were incubated in Goat Anti- Rabbit IgG (H&L) Cy3 secondary antibody (1:500; Abcam) for 2 h, followed by incubation with DAPI (1 μg/mL; Boster Bio, Wuhan, China) for 5 min. Images were captured using a fluorescence microscope (BX51, Olympus, Japan) and the immunopositive cells was analyzed by ImageJ software (version 1.52, NIH, USA) to calculate the calibrated total fluorescence with this formula: Integrated density - (Area × Mean background fluorescence), and then normalized to achieve fold changes (% of control).
Real-time RT-PCR (RT-qPCR) analysis
Total RNA from mice temporal cortex was extracted using Total RNA Isolation Kit (Vazyme Biotech, Nanjing, China) according to the manufacturer’s instructions. Complementary DNA (cDNA) was synthesized by a HiScript II Q RT Super Mix for qPCR (+gDNA wiper) Kit and miRNA 1st Strand cDNA Synthesis Kit (Vazyme Biotech), and RT-qPCR was performed using ChamQ Universal SYBR qPCR Master Mix and miRNA Universal SYBR qPCR Master Mix (Vazyme Biotech). Relative miR-132 levels were calculated following a delta delta Ct method, and Ct represents the cycle threshold. The primer sequence of mir-132: 5’—CGCGTAACAGTCTACAGCCA— 3’ (forward); 5’—AGTGCAGGGTCCGAGGTATT— 3’(reverse); C1ql1: 5’—GCACGGCCACCTATACCAC— 3’ (forward); 5’— CACGTCGTCAAACTTGAGCA—3’ (reverse); GAPDH: 5’—GGTGAAGGTCGGTGAACC— 3’ (forward); 5’— CTCGCTCCTGGAAGATGGTG— 3’(reverse); U6: 5’—CTCGCTTCGGCAGCACA— 3’(forward); 5’— AACGCT TCACGAAT T TGCGT— 3’(reverse).
Statistical analysis
Statistical analyses and calculation of p values were performed using Prism software (version 5.0, GraphPad Software Inc., USA). Student’s t-test was used for pairwise comparisons, and one-way ANOVA followed by Tukey test was used for multiple comparisons. The escape latencies were analyzed by repeated measures ANOVA with post-hoc Holm-Sidak test. A value of P<0.05 was considered to indicate statistical significance.
Results
miR-132 was highly expressed in the temporal cortex following transfection of miRNA-132
To detect the transfection efficiency of miR-132 delivered in the lateral ventricle, we first examined the intensity of GFP fluorescence, for the virus carries a GFP reporter, and all the successfully transfected cells expressed GFP (Figure 1A). The control group received LV-null. Moreover, real-time RT-PCR results showed that the expression of miR-132 in the temporal cortex was significantly increased compared with the AD group (P<0.05) (Figure 1B).
Figure 1.
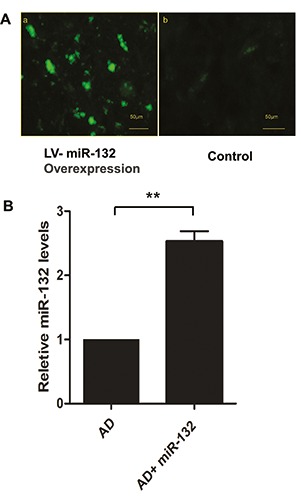
miR-132 was highly expressed in the temporal cortex following transfection of miRNA-132 (A). GFP-miR-132 was observed 14 days after intracerebroventricular injection of lentivirus (a); the control group received LV-null (b). Expression of miR-132 was analyzed by RT-qPCR (B). Data represent the mean ± SD. **P<0.01 compared with AD group.
Intracerebral injection of miR-132 improved the cognitive performance of APP/PS1 mice
To determine whether the injection of miR-132 mimic can ameliorate the memory deficit in APP/PS1 mice, we performed behavioral tests with Morris water maze. During training, spatial learning was assessed by the amount of elapsed time before the animal climbs onto the platform to escape the water (escape latency). The training time was composed of 1-min periods over 5 days. The escape latency and the track distance of the AD+C1INH and AD+Mir- 132 rats were significantly decreased on days 2–5 compared with the AD group (Figure 2 A,B). In the probe trial, we observed the times of mice crossing the platform. Among them, the time of AD+C1INH and AD+ Mir-132 groups crossing the original platform was significantly increased (Figure 2C).
Figure 2.
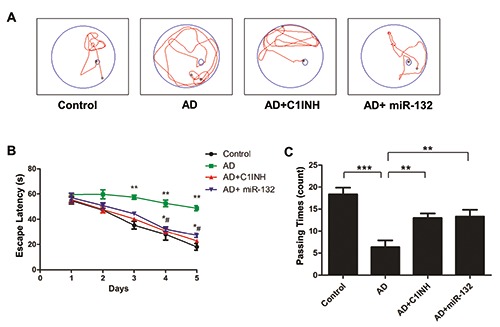
Representative trajectory of each group in place navigation test (A). The escape latency of the four groups in place navigation test (B). **P<0.01 compared with control group; *P<0.05, #P<0.05 compared with AD group. Comparison of the crossing times of the 4 groups in space probe trial across the original platform (C). **P<0.05, ***P<0.001 compared with AD group.
PSD95, Synapsin-1 and p-Synapsin expression levels in the temporal cortex of mice were detected by Western blot
The expression changes in synapserelated proteins were measured in order to investigate whether miR-132 and C1INH expressions affected synaptic plasticity, as AD transgenic models showed reduction of PSD-95 and Synapsin-1 in dendrites of different brain regions. We found that either miR-132 overexpression or C1INH administration resulted in a significant increase in the expression of PSD95, Synapsin-1 and p- Synapsin in the temporal cortex (Figure 3 A-D). The ratio of p-Synapsin over Synapsin-1 in these two groups also increased significantly (Figure 3E). In contrast, the expression of PSD95, Synapsin-1 and p-Synapsin in the AD group revealed decreased.
Figure 3.
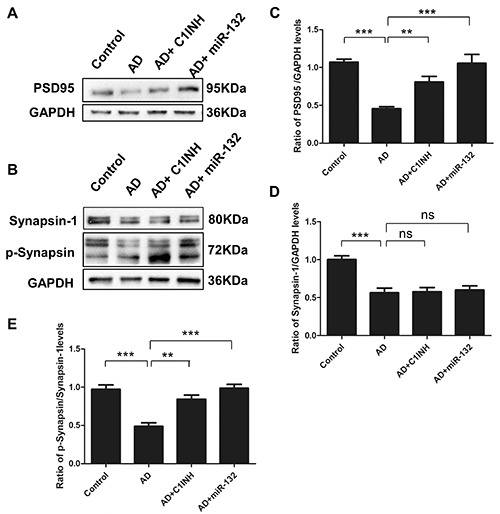
Fourteen days after intracerebroventricular injection of miR-132 lentivirus or C1q inhibitor, the expression of PSD95, Synapsin-1 and p-Synapsin in the temporal cortex was analyzed by western blotting (A and B). Data represent the mean±SD of four independent experiments. Comparison of PSD95 proteins in the four groups (C); Comparison of Synapsin-1 proteins in the four groups (D); The ratio of p-Synapsin over Synapsin-1 (E). **P<0.01, ***P<0.001 compared with AD group.
Immunofluorescent staining results
Representative graphs of immunofluorescent staining are shown in Figures 4A, 5A and 6A. PSD95, Synapsin-1 and p- Synapsin were observed on the cytomembrane. Quantitative analysis showed that the fluorescence intensity of PSD95, Synapsin- 1 and p-Synapsin in the AD group was significantly lower than that of the control group. The fluorescence intensity in the C1INH and miR-132 groups was increased compared with the AD group (Figures 4B, 5B and 6B).
Figure 4.
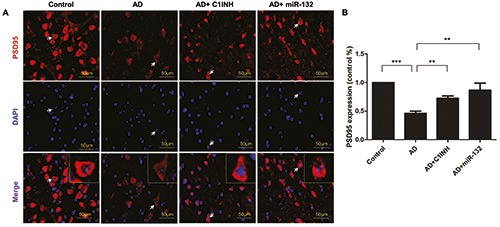
Representative images depicting the immunoreactivity of PSD95 in the temporal cortex (A) and the quantitative analysis of PSD95 expression (B). **P<0.01, ***P<0.001 compared with AD group.
Figure 5.
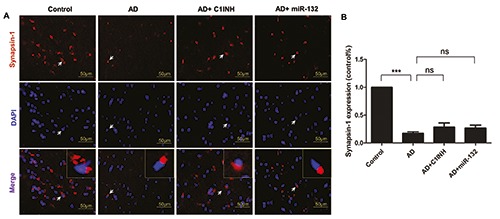
Representative images depicting the immunoreactivity of Synapsin-1 in the temporal cortex (A) and the quantitative analysis of Synapsin-1 expression (B). ***P<0.001 compared with AD group.
Figure 6.
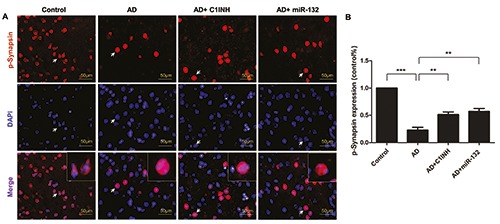
Representative images depicting the immunoreactivity of p-Synapsin in the temporal cortex (A) and the quantitative analysis of p-Synapsin expression (B). **P<0.01, ***P<0.001 compared with AD group.
Expression of C1q after transfection of miRNA-132
Based on the role of miRNAs in translational repression, inhibition of miR-132-3p via miRNA sponge principle could increase the expression of target gene. Bioinformatics analysis identified that the 3’-UTR of the C1q mRNA contained a highly conserved miR-132-3p target sequence (Figure 7A). To validate whether C1q is genuinely regulated by miR-132-3p in vivo, we performed RT-qPCR test. The result confirmed that the mRNA levels of C1q in the temporal cortex were increased in the AD group, and significantly decreased by overexpression of miR-132-3p (Figure 7B).
Figure 7.
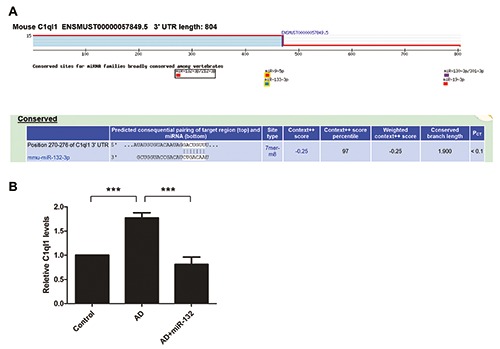
The 3'-UTR of the C1q mRNA was predicted to have a binding site with miR-132-3P through bioinformatics analysis (A). Expression of C1q in the temporal cortex was analyzed by RT-qPCR (B). Data represent the mean±SD of three independent experiments. ***P<0.001 compared with AD group.
Discussion
In the present study, we first verified the transfection effect of lentivirus-delivered overexpression of miR-132-3p injected in the lateral ventricle by immunofluorescence analysis in an independent experiment. Further real-time RT-PCR results confirmed that the expression of miR-132 in the temporal cortex was significantly increased, indicating successful transfection. We also showed that miR-132 mimic could ameliorate the memory deficit in APP/PS1 mice with Morris water maze test, which was partly in agreement with the previous study which indicated that down-regulation of miR-132 exhibited altered learning and probe phases in Barnes maze test.20 Indeed, increasing evidence has shown that aberrant expression of miRNAs is involved in neurodegenerative diseases.9 Therefore, further screening of these miRNAs is inevitably required to determine which miRNAs and downstream molecules are usually involved during disease development.9
MiR-132, one of the most functional miRNAs in the brain, is a highly conserved miRNA transcribed from human chromosome 17 by the transcription factor CREB.21 In mice, the miR-132/212 gene cluster is transcribed from the first intron of a noncoding transcript, AK006051, on Chromosome 11.22 miR-132 has been revealed to play an important role in modulating dendritic morphology and plasticity of synaptic networks. It has been reported that miR-132 was down-regulated in postmortem AD brain occurring even before abnormal aggregation and accumulation of Aβ and tau protein and neuronal loss.23,24 It was induced in cultured neurons by neuronal activity and addition of neurotrophins, and was found to have a specific downstream target p250GAP.25 Besides, several target genes and signaling molecules might be regulated by miR-132 neuroprotective functions. Through online TargetScan analysis, we correlated the down-regulated miR-132 with an up-regulated target in the CNS of the Alzheimer’s disease.26 Indeed, recent study showed that C1q, the key initiating protein of the classical complement cascade, was upregulated before overt plaque deposition and closely related to synapse modulation, and downregulation of C1q rescued early synapse loss.17 It is reported that C1q may have a potential detrimental effect on neuronal integrity via initiating an inflammatory response.27 Zhang et al. found that complement C1 inhibitor was reduced in plasma biomarker analysis in AD patients.28 In addition, C1q mRNA was shown to be upregulated in the neurons of AD patients, and dysregulated in other neurodegenerative diseases as well. 29,30 Thus, it is interesting to know whether miR-132 regulates C1q expression. In this study, we found that inhibition of C1q with its inhibitor C1INH has a positive regulatory effect on cerebral synapsis proteins in AD mice. Then we speculated that C1q might be the possible downstream target of miR-132 by in silico analysis and thus C1q might be involved in this neuroprotection function of miR-132. Next through RT-PCR test, we confirmed that the mRNA levels of C1q in the temporal cortex were significantly decreased by overexpression of miR- 132-3p, suggesting interaction between miR-132-3p and its validated target C1q through regulating synaptogenesis.
In recent years, synaptic plasticity deficits have become increasingly recognized as a cause of memory impairment and central players in the synapse deterioration characteristic of AD pathogenesis.31 The accumulation and deposition of Aβ in the brain is involved in the morphological and functional changes of synapses by disrupting related signaling pathways, thereby diminishing synapses.31 Damage to synaptic structure and plasticity in either hippocampus or cerebral cortex correlates with the severity of the neuropathy and memory deficits present in AD patients.32 Synapserelated proteins are important components in maintaining synaptic morphology and function. Their loss will result in the inevitable deterioration of neurological functions such as cognitive impairment. In AD, synaptic loss is closely related to the severity of dementia, and is mainly caused by mutant proteins in the presynaptic membrane or the postsynaptic densities (PSD), including Synapsin-1, PSD-95, SAP97, Synaptophysin, the NMDA receptor and AMPA receptor. The latter two are essential for glutamatergic synaptic transmission and play key roles in the maintenance of LTP and LTD,33 exhibiting reduced surface levels and total levels in AD brains.34 Although the involvement of various synaptic proteins in AD has been established, their roles are versatile and remain to be further verified. Here we focused on PSD-95, Synapsin-1 and its phosphorylation p- Synapsin, which are closely related to the onset of AD.
PSD-95, which is highly concentrated at the PSD domain, has been characterized as one of the most abundant scaffolding proteins in the excitatory neurons and plays a key role in synaptic plasticity. Changes in PSD-95 levels result in alteration of synapse number, and therefore synaptic PSD-95 reduction in vivo indicates dendritic spine elimination.35 Many consistent results demonstrated that overexpression of PSD-95 significantly increased the amplitude of AMPAR-mediated EPSCs in hippocampal neurons, while AMPAR EPSCs were decreased by knockdown of PSD-95 through RNA interference.36
Synapsin-1 is one of the major phosphoproteins which regulate neurotransmitter release in the nervous system. The function of synapsin-1 is exerted through phosphorylation, though both phosphorylated and non-phosphorylated Synapsin-1 modulates neuronal development. The phosphorylation and de-phosphorylation cycle is important for neurotransmitter release from synaptic vesicle.37 The present study found that the expression of PSD95, Synapsin-1 and p-Synapsin in the AD group was decreased significantly. Meanwhile, either miR-132 overexpression or C1INH administration resulted in a significant increase in the expression of PSD95, Synapsin-1 and p- Synapsin in the temporal cortex, and the ratio of p-Synapsin over Synapsin-1 in these two groups also increased significantly. Similarly, miR-132 loss resulted in increased ERK1/2 activity and elevated TAU phosphorylation in an AD mouse model.38
Our findings, together with those from other groups, suggest a fundamental role of miR-132 in AD, and emphasize the downstream target C1q that might be of potential clinical interest for AD therapy. In particular, this study demonstrates that administration of C1q inhibitor C1INH exerts therapeutic effect in APP/PS1 mice. This effect can largely be attributed to the protective effect of miR-132 on synapses. In the light of this evidence, our study identifies C1INH as a novel synapto-protective candidate drug for repurposing potential in the clinical treatment of AD.
Funding Statement
Funding: This work was supported by grants from the Natural Science Foundation of Liaoning Province, China (No. 2015020459, 201602825 and 20170541011).
References
- 1.Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med 2008;14:837-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang Z, Liu X, Schroeder JP, Chan CB, Song M, Yu SP, et al. 7,8-dihydroxyflavone prevents synaptic loss and memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 2014;39: 638-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scheff SW, Neltner JH, Nelson PT. Is synaptic loss a unique hallmark of Alzheimer’s disease? Biochem Pharmacol 2014;88:517-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Olde Loohuis NF, Kos A, Martens GJ, Van Bokhoven H, Nadif Kasri N, Aschrafi A. MicroRNA networks direct neuronal development and plasticity. Cell Mol Life Sci 2012;69:89-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim J, Yao F, Xiao Z, Sun Y, Ma L. MicroRNAs and metastasis: small RNAs play big roles. Cancer Metastasis Rev 2018;37:5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kosik KS. The neuronal microRNA system. Nat Rev Neurosci 2006;7:911-20. [DOI] [PubMed] [Google Scholar]
- 7.Woldemichael BT, Mansuy IM. Micro- RNAs in cognition and cognitive disorders: Potential for novel biomarkers and therapeutics. Biochem Pharmacol 2016; 104:1-7. [DOI] [PubMed] [Google Scholar]
- 8.Molasy M, Walczak A, Szaflik J, Szaflik JP, Majsterek I. MicroRNAs in glaucoma and neurodegenerative diseases. J Hum Genet 2017;62:105-12. [DOI] [PubMed] [Google Scholar]
- 9.Lau P, Bossers K, Janky R, Salta E, Frigerio CS, Barbash S, et al. Alteration of the microRNA network during the progression of Alzheimer’s disease. EMBO Mol Med 2013;5:1613-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pichler S, Gu W, Hartl D, Gasparoni G, Leidinger P, Keller A, et al. The miRNome of Alzheimer’s disease: consistent downregulation of the miR- 132/212 cluster. Neurobiol Aging 2017;50:167.e1-167.e10. [DOI] [PubMed] [Google Scholar]
- 11.Satoh J-i. MicroRNAs and their therapeutic potential for human diseases: Aberrant microRNA expression in Alzheimer’s Disease brains. J Pharmacol Sci 2010;114:269-75. [DOI] [PubMed] [Google Scholar]
- 12.Salta E, De Strooper B. microRNA- 132: a key noncoding RNA operating in the cellular phase of Alzheimer’s disease. FASEB J 2017;31:424-33. [DOI] [PubMed] [Google Scholar]
- 13.Min SW, Cho SH, Zhou Y, Schroeder S, Haroutunian V, Seeley WW, et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 2010;67:953-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kalia M. Brain development: anatomy, connectivity, adaptive plasticity, and toxicity. Metabolism 2008;57:S2-5. [DOI] [PubMed] [Google Scholar]
- 15.Tau GZ, Peterson BS. Normal development of brain circuits. Neuropsychopharmacology 2010;35:147-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bialas AR, Stevens B. TGF-beta signaling regulates neuronal C1q expression and developmental synaptic refinement. Nat Neurosci 2013;16:1773-82. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 17.Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016;352:712-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, et al. The classical complement cascade mediates CNS synapse elimination. Cell 2007;131:1164-78. [DOI] [PubMed] [Google Scholar]
- 19.Ji LL, Peng JB, Fu CH, Cao D, Li D, Tong L, et al. Activation of Sigma-1 receptor ameliorates anxiety-like behavior and cognitive impairments in a rat model of post-traumatic stress disorder. Behav Brain Res 2016;311:408-15. [DOI] [PubMed] [Google Scholar]
- 20.Hernandez-Rapp J, Smith PY, Filali M, Goupil C, Planel E, Magill ST, et al. Memory formation and retention are affected in adult miR-132/212 knockout mice. Behav Brain Res 2015;287:15-26. [DOI] [PubMed] [Google Scholar]
- 21.Nudelman AS, DiRocco DP, Lambert TJ, Garelick MG, Le J, Nathanson NM, et al. Neuronal activity rapidly induces transcription of the CREB-regulated microRNA-132, in vivo. Hippocampus 2010;20:492-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aten S, Hansen KF, Hoyt KR, Obrietan K. The miR-132/212 locus: a complex regulator of neuronal plasticity, gene expression and cognition. RNA Dis 2016;3:e1375. [PMC free article] [PubMed] [Google Scholar]
- 23.Hernandez-Rapp J, Rainone S, Goupil C, Dorval V, Smith PY, Saint-Pierre M, et al. microRNA-132/212 deficiency enhances Abeta production and senile plaque deposition in Alzheimer’s disease triple transgenic mice. Sci Rep 2016;6:30953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Patrick E, Rajagopal S, Wong HA, McCabe C, Xu J, Tang A, et al. Dissecting the role of non-coding RNAs in the accumulation of amyloid and tau neuropathologies in Alzheimer’s disease. Mol Neurodegener 2017;12:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vo N, Klein ME, Varlamova O, Keller DM, Yamamoto T, Goodman RH, et al. A cAMP-response element binding protein- induced microRNA regulates neuronal morphogenesis. Proc Natl Acad Sci U S A 2005;102:16426-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fonseca MI, Zhou J, Botto M, Tenner AJ. Absence of C1q leads to less neuropathology in transgenic mouse models of Alzheimer’s disease. J Neurosci 2004;24:6457-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crehan H, Hardy J, Pocock J. Microglia, Alzheimer’s disease, and complement. Int J Alzheimers Dis 2012;2012:983640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang R, Barker L, Pinchev D, Marshall J, Rasamoelisolo M, Smith C, et al. Mining biomarkers in human sera using proteomic tools. Proteomics 2004;4:244-56. [DOI] [PubMed] [Google Scholar]
- 29.Johnson SA, Lampert-Etchells M, Pasinetti GM, Rozovsky I, Finch CE. Complement mRNA in the mammalian brain: Responses to Alzheimer’s disease and experimental brain lesioning. Neurobiol Aging 1992;13:641-8. [DOI] [PubMed] [Google Scholar]
- 30.Singhrao SK, Neal JW, Morgan BP, Gasque P. Increased complement biosynthesis by microglia and complement activation on neurons in Huntington’s disease. Exp Neurol 1999; 159:362-76. [DOI] [PubMed] [Google Scholar]
- 31.Chakroborty S, Kim J, Schneider C, West AR, Stutzmann GE. Nitric oxide signaling is recruited as a compensatory mechanism for sustaining synaptic plasticity in Alzheimer’s disease mice. J Neurosci 2015;35:6893-902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klevanski M, Herrmann U, Weyer SW, Fol R, Cartier N, Wolfer DP, et al. The APP intracellular domain is required for normal synaptic morphology, synaptic plasticity, and hippocampus-dependent behavior. J Neurosci 2015;35:16018-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron 2004;44:5-21. [DOI] [PubMed] [Google Scholar]
- 34.Jürgensen S, Antonio LL, Mussi GE, Brito-Moreira J, Bomfim TR, De Felice FG., et al. , Activation of D1/D5 dopamine receptors protects neurons from synapse dysfunction induced by amyloid-beta oligomers. J Biol Chem 2011;286:3270-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cane M, Maco B, Knott G, Holtmaat A. The relationship between PSD-95 clustering and spine stability in vivo. J Neurosci 2014;34:2075-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu W, Schlueter OM, Steiner P, Czervionke BL, Sabatini B, Malenka RC. Molecular dissociation of the role of PSD-95 in regulating synaptic strength and LTD. Neuron 2008;57: 248-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fornasiero EF, Bonanomi D, Benfenati F, Valtorta F. The role of synapsins in neuronal development. Cell Mol Life Sci 2010;67:1383-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Salta E, Sierksma A, Vanden Eynden E, De Strooper B. miR-132 loss de-represses ITPKB and aggravates amyloid and TAU pathology in Alzheimer’s brain. EMBO Mol Med 2016;8:1005-18. [DOI] [PMC free article] [PubMed] [Google Scholar]