

Abstract
Yao-Feng Tsao, Wen-Wen Lin, Chia-Hao Chang, Takayoshi Ueda, Nian-Hong Jang-Liaw, Ya-Hui Zhao, and Hsiao-Wei Kao (2016) Rose bitterling, Rhodeus ocellatus, is a small cyprinid fish distributed in East Asia. To infer its phylogeography and genetic structure, specimens from Taiwan, China, and Japan were collected, and complete mitochondrial cytochrome b (cyt b) DNA sequences were amplified and sequenced. Phylogenetic analyses identified seven mitochondrial lineages (A-G). Among them, three lineages (A, B, and C) distributed in mainland China. Lineages D, E, and F distributed in Japan, Korea, and Taiwan, respectively. Lineage G distributed in both China and Japan. The results of the Bayesian Binary MCMC analysis (BBM) suggested that the most recent common ancestor of R. ocellatus was from Lower Yangtze region. Divergence times among lineages inferred by molecular clock ranged from 7.55 to 1.44 million years ago. We propose that topography and climate changes by uplift of the Tibetan Plateau in the Late Miocene-Pliocene and the glacial-interglacial cycles in the Pleistocene might account for population expansion and genetic differentiation. Divergence times among lineages A, B, and C in Yangtze River basin ranged from 7.55 to 2.27 million years ago that might result from changes of flow directions of rivers from westward to eastward driven by the uplift of the Tibetan Plateau. The glacial-interglacial cycles in the Pleistocene might further cause population expansion to the northward of lineage G at about 0.19 million years ago. Lineage D in Japan was dispersed from the mainland China before the opening of the Sea of Japan, and lineage F in Taiwan was dispersed from the mainland China through the land bridge in the Pleistocene. Because of the genetic differentiation is statistically significant among populations, protection of genetic diversity and distinctness of R. ocellatus should be considered in the future conservation management.
Keywords: Cytochrome b, Molecular clock, Population expansion
BACKGROUND
Historical events, such as topography and climate changes, can shape the distribution, phylogenetic pattern, and genetic structure of a species (Hewitt 2000, 2004; Yap 2002). In East Asia, the uplift of the Tibetan Plateau since the Miocene has profoundly changed the environment (Clift et al. 2004; Jia et al. 2003; Zhang et al. 2000; Zheng et al. 2013). The topographic trend in China was reversed from right tilt to left tilt, with the flow direction of the main river systems turned eastward to the sea (Clark et al. 2004; Zheng et al. 2013). In addition, the uplift also affected the formation of the East Asia monsoon. The southeast monsoon started to develop in the east of China, while the arid deserts in the inland expanded. When the plateau is getting higher, the monsoon is getting stronger, and the influence area is getting wider (An et al. 2001). Moreover, there are several islands at the margin of East Asia that were connected to the continent during the glacial periods (Maruyama et al. 1997; Voris 2000), providing opportunities for faunal exchange between the continent and those islands. The combination of these factors could have influenced the demographic and distributional patterns of species in East Asia, which can be inferred by phylogenetic analyses (Avise 2009).
Because the dispersal abilities of freshwater fish are limited, the distribution patterns and genetic structures of freshwater fish are expected to reflect historical changes, and that makes them an ideal model in phylogeographic research. The bitterling is a small cyprinid fish of the family Acheilognathidae, which has the unusual reproductive behavior of using freshwater mussels as spawning sites (Smith et al. 2004). In this family, the rose bitterling, Rhodeus ocellatus (Kner, 1866), is a widespread species with high genetic diversity in East Asia (Chang et al. 2014; Kawamura et al. 2014). It consists of two subspecies: (1) R. o. ocellatus (Kner, 1866), which distributed in China, Korea, and Taiwan. (2) R. o. kurumeus Jordan and Thompson, 1914, which distributed only in western Japan. R. o. ocellatus was accidentally introduced from the Yangtze River basin of China into Japan and dispersed rapidly (Nakamura 1955). Hybridization between the two subspecies was detected in several populations in Japan that caused genetic introgression by R. o. ocellatus into R. o. kurumeus (Nagata et al. 1996; Kawamura et al. 2001b). R. o. kurumeus is now an endangered cyprinid species, however, its genetic distinctness has been spoiled by hybridization (Kawamura et al. 2001b).
The freshwater fish in East Asia are facing threats from human activities, especially for the bitterling species which suffer from population decline and loss of genetic diversity, caused mainly by habitat degradation and invasions of exotic species (or population) (Kawamura et al. 2001a; Kawamura 2005; Kubota et al. 2010; Miyake et al. 2010; Kitanishi et al. 2013; Kubota and Watanabe 2013; Kitazima et al. 2015). The conservation status of R. ocellatus is categorized as “data deficient” in the IUCN Red List of Threatened Species and requires more information in its native range (Huckstorf 2013). For the future conservation management, it is important to understand the phylogeographic pattern and genetic structure of the species (Moritz 1994; Winter et al. 2013). Previous studies were focused on the populations of R. ocellatus in Japan (Nagata et al. 1996; Kawamura et al. 2001a; Kawamura et al. 2001b; Miyake et al. 2001; Kawamura 2005; Onikura et al. 2013). In this study, specimens of R. ocellatus were collected from China, Japan, and Taiwan. The mitochondrial cytochrome b (cyt b) DNA sequences were used as genetic markers to infer phylogeny, divergence time, and demography of R. ocellatus in East Asia. The specific aims of this study were as follows: (1) to investigate genetic diversity and population differentiation of R. ocellatus in East Asia (2) to infer whether historical events were accounted for genetic divergence of R. ocellatus. This is the first study to investigate the phylogeographic pattern and genetic structure of R. ocellatus in East Asia. The results are important for understanding the history and the future conservation management of R. ocellatus.
MATERIALS AND METHODS
Sample collection
A total of 133 specimens of R. ocellatus from 31 localities across China, Korea, Japan, and Taiwan were used for analysis (Fig. 1). Among them, 20 cyt b DNA sequences of the specimens were downloaded from GenBank (including 9 sequences of R. o. ocellatus, 4 sequences of R. o. kurumeus, and 7 sequences of R. ocellatus without subspecies designation) and 113 specimens were sequenced in this study (Additional file 1) (Saitoh et al. 2006; Yang et al. 2011; Kawamura et al. 2014; Watanabe unpublished data). The 16 sampling localities in China were categorized into five regions based on the biogeographic regions of freshwater fishes in China (Kang et al. 2014). These five Chinese regions are Northeastern (I), Yellow River (II), Lower Yangtze (III), Upper- Middle Yangtze (IV), and Southern (V) region. The six sampling localities in Japan were categorized into the Japan region (VI) and the eight sampling localities in Taiwan were categorized into Taiwan region (VIII) because Japan and Taiwan are islands that are geographically separated from the Asian continent by the Sea of Japan and Taiwan Strait, respectively. One sampling locality from the Korean Peninsula was assigned as Korea region (VII) because of the topographic isolation. All specimens were stored in 95% ethanol before DNA extraction.
Fig. 1.
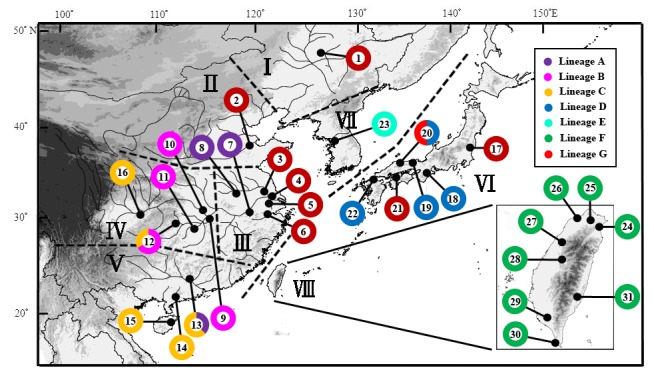
Fig. 1. Map of the sampling localities and the population structures of Rhodeus ocellatus in East Asia. Black circles refer to sampling localities and Pie charts refer to lineage compositions in each locality with the numbers correspond to the locality numbers of Additional file 1. Roman numerals represent the biogeographic regions: Northeastern (I), Yellow River (II), Lower Yangtze (III), Upper-Middle Yangtze (IV), Southern (V), Japan (VI), Korea (VII), and Taiwan (VIII) region. The colors used in the pie charts correspond to the lineages in figure 2.
DNA extraction, polymerase chain reaction (PCR), DNA sequencing
Total DNA was extracted from ethanol- preserved muscle tissue by a Genomic DNA Purification Kit (Gentra Systems, Minneapolis City, MN, USA). To amplify the cyt b gene, primers were designed based on conserved regions adjacent to and within the cyt b DNA sequences of R. o. kurumeus (AB070205) (Saitoh et al. 2006), Labeo batesii (AB238967) (Saitoh et al. 2006), Acheilognathus typus (AB239602) (Saitoh et al. 2006), R. sinensis (NC_007885) (Kim et al. 2006), and R. ocellatus (NC_011211) (He et al. 2008). Two pairs of primers were used in PCR amplification and DNA sequencing. The first pair was Glu-14355(+) (5’-CGTTGTARYTCAACTACAAGAAC-3’, Y representing C or T according to IUB codes) and Cytb-15029(-) (5’-TCGGCGTCRGARTTTAGG CCG-3’, R representing A or G according to IUB codes), located in tRNAGlu and cyt b, respectively. The second pair was Cytb-14905(+) (5’-CTAACCCGATTTTTTCGCCTT-3’) and Thr- 15569(-) (5’-TCTTCGGATTACAAGACCGAT GC-3’), located in cyt b and tRNAThr, respectively. PCR reactions were performed in 100 μl of reaction solution containing 50-200 ng of DNA template, 2 μl of each 10 pM primer, 8 μl of 2.5 mM dNTP, 10 μl of 10 × reaction buffer, 1 μl ProTaq TM DNA polymerase (Protech Technology Enterprise, Taipei City, Taiwan), and distilled water to bring the sample up to volume. PCR reactions were performed with a GeneAmp PCR System 2700 (Applied Biosystems, Foster City, CA, USA). The thermal cycle began with an initial denaturation at 94°C for 4 minutes; then 40 cycles of denaturation at 94°C for 1 minute, annealing at 45-55°C for 1 minute, and extension at 72°C for 1.5 minutes. There was also a final extension step at 72°C for 10 min. PCR products were checked by electrophoresis on 1% agarose gel in 1 × TBE buffer. DNA products were purified with a Micro- Elute DNA Clean/Extraction Kit (GeneMark, Tainan City, Taiwan) following manufacturer protocols. Sequencing reactions followed the protocols of the BigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Forster City, CA, USA). Different segments of the cyt b gene were concatenated by BioEdit version 7.0.9.0 (Hall 1999).
Sequence analyses
The DNA sequences of cyt b were checked by BLAST on the website of the National Center for Biotechnology information Entrez (NCBI) website (http://www.ncbi.nlm.nih.gov/). Sequence alignment was conducted using ClustaW implemented in BioEdit version 7.2.5. Nucleotide diversity (π) and haplotype diversity (h) were calculated using DnaSP version 5.10.01 (Librado and Rozas 2009).
Phylogenetic trees and phylogenetic network
The phylogenetic trees were constructed from cyt b sequences using Bayesian inference (BI) and maximum likelihood (ML) under the codon partition model conducted in MrBayes 3.2.2 (Huelsenbeck and Ronquist 2001) and GARLI (Zwickl 2006), respectively. Three cyt b DNA sequences download from GenBank were used as the outgroup: A. macropterus (AB366454), R. sericeus (AB366518), and Tanakia tanago (AB366539) (Kawamura et al. 2014) (Additional file 1). The best-fit models for first, second, and third codons of cyt b were K80 + I (I = 0.778), F81, and TIM2 + I + G (I = 0.084 and G = 2.107), selected under the Corrected Akaike information criterion (cAIC) using jModelTest 2.1.3 (Guindon and Gascuel 2003; Darriba et al. 2012). MrBayes was run with 2 × 106 generations of the MCMC chain. Trees were saved every 100 generations and the first 5000 trees (25%) were discarded as burn-in. Bootstrap values for ML were estimated from 100 pseudoreplicates using GARLI. Relationships among haplotypes within different mitochondrial lineages were further examined by a minimum spanning network using Arlequin version 3.11 (Excoffier et al. 2005). Genetic distances (p-distances) between and within lineages were calculated using MEGA 5 (Tamura et al. 2011).
Divergence time estimation
The divergence time of each lineage was estimated using BEAST v1.8.0 (Drummond and Rambaut 2007). Five cyt b DNA sequences download from GenBank were used as the outgroup, including A. macropterus (AB366454), A. meridianus (AB366464), T. lanceolate (AB366534), T. tanago (AB366539), R. sericeus (AB366518) (Kawamura et al. 2014). We used the strict clock method with three different fixed evolutionary rates for the cyt b: (1) 0.52% per site per million year in genus Capoeta (Levin et al. 2012), (2) 0.76% per site per million year in European cyprinids (Zardoya and Doadrio 1999), (3) 1.05% per site per million year in North American plagopterins (Dowling et al. 2002). The GTR + I + G model was used according to the result of jModelTest 2.1.3. Three independent runs with 5 × 107 generations of MCMC chains were performed in all analyses. The data were sampled every 1000 generations and the first 5000 samples (10%) were discarded as burn-in. The results of three independent runs were combined using Tracer v1.5 and the effective sample size (ESS) of all parameters was > 200. Three independent trees were combined using LogCombiner v1.8.0 (part of the BEAST package) and assessed using TreeAnnotator v1.8.0 (part of the BEAST package). Annotated times and the phylogenetic tree were visualized using FigTree v1.4.0 (Rambaut 2009). To determine which estimation is most plausible, we compared these results with two fossil records: (1) the fossil pharyngeal teeth of Acheilognathidae in the early Miocene (Yasuno 1984), (2) the fossil of T. lanceolate in the late Miocene (Yoshio et al. 1977). The result which is most consistent with the fossil records will be chosen for further discussion.
Population demography and genetic structure
Population demography was estimated by mismatch distribution using Arlequin. Tajima’s D test and Fu’s Fs test were performed to confirm population size change. The Bayesian skyline plot (BSP) was used for inferring changes in the historical demography using BEAST v1.8.0. We used the strict clock method with the same fixed evolutionary rates for the cyt b that used in divergence time estimation. The TN93 + G model was used according to the results of jModelTest 2.1.3. Three independent runs with 5 × 107 generations of MCMC chains were performed. The data were sampled every 1000 generations and the first 5000 samples (10%) were discarded as burn-in. The results of three independent runs were combined using Tracer v1.5 and the effective sample size (ESS) of all parameters was >200. The parameters of three independent trees were combined using LogCombiner v1.8.0 (part of the BEAST package).
For the hierarchical genetic structure of R. ocellatus in East Asia, an analysis of molecular variance (AMOVA) using the Tamura and Nei model was performed. Three levels of genetic structure were analyzed with 1000 permutations: among regions, among sample localities within regions, and within sample localities. The pairwise Φ-statistic with the Tamura and Nei model and 1000 permutations was calculated for population differentiation. The Korean specimen was excluded from the regional demographic analyses because only one cyt b sequence was available for this region.
Ancestral range reconstruction
To determine the ancestral ranges of R. ocellatus in East Asia, we conducted the Bayesian Binary MCMC analysis (BBM) using RASP 3.2 (Yu et al. 2015). The post-burnin trees and condensed tree from the BEAST analysis were used as the input data. The distributions of haplotypes were categorized into eight biogeograpical regions (I-VIII) as mentioned before. The JC + G model was chosen, and the number of maximum areas was set from 4-8 in the different analyses, respectively. The MCMC chains were run for 5 × 106 generations in all analyses. The data were sampled every 1000 generations, and the temperature for heating the chains was 0.1.
RESULTS
Sequence variation
The coding region of cyt b of R. ocellatus was 1140 bp long. A total of 70 haplotypes was observed from all 133 specimens (Additional file 1 and Table 1, Genbank accession numbers: JQ622686-JQ622802). Nucleotide and haplotype diversities of cyt b were 0.05069 ± 0.00238 and 0.978 ± 0.005, respectively. The greatest nucleotide diversity (0.03536 ± 0.01259) was observed in Southern region and the greatest haplotype diversity (0.968 ± 0.025) was observed in the Lower Yangtze region.
Table 1. Genetic diversity of cyt b DNA sequences of Rhodeus ocellatus in each region.
| Region | Sample size | Parsimony informative site | Nucleotide diversity ± SD (10-2) | Number of haplotype | Haplotype diversity ± SD |
| Northeastern region (I) | 9 | 2 | 0.122 ± 0.028 | 5 | 0.806 ± 0.120 |
| Yellow River region (II) | 9 | 4 | 0.283 ± 0.063 | 6 | 0.889 ± 0.091 |
| Lower Yangtze region (III) | 20 | 74 | 2.286 ± 0.973 | 15 | 0.968 ± 0.025 |
| Upper-Middle Yangtze region (IV) | 16 | 40 | 1.544 ± 0.430 | 11 | 0.908 ± 0.063 |
| Southern region (V) | 13 | 109 | 3.536 ± 1.259 | 5 | 0.538 ± 0.161 |
| Japan region (VI) | 26 | 69 | 2.795 ± 0.166 | 18 | 0.957 ± 0.027 |
| Korea region (VII) | 1 | - | - | 1 | - |
| Taiwan region (VIII) | 39 | 11 | 0.280 ± 0.033 | 12 | 0.862 ± 0.042 |
| Total | 133 | 222 | 5.069 ± 0.238 | 70 | 0.978 ± 0.005 |
Phylogeny
Phylogenetic trees of BI and ML were similar and consisted of seven lineage (lineages A-G) (Fig. 2). The genetic distances between lineages ranged from 0.028 to 0.100, and the distances within lineages ranged from 0.001 (lineage C) to 0.054 (lineage A) (Additional file 2). The haplotypes of subspecies R. o. kurumeus were clustered in the lineage D with other haplotypes of Japan. Previous phylogenetic studies using mitochondrial DNA sequences showed that R. o. kurumeus and R. o. ocellatus in Japan constituted a monophyletic group, respectively (Miyake et al. 2001). Because mitochondrial DNA is inherited maternally in most vertebrates (Birky 1995), all the haplotypes of the lineage D of Japan belong to the maternal lineage of R. o. kurumeus. Subspecies R. o. ocellatus was a paraphyletic group because lineage D was not included in it. The haplotypes of the lineages A, C, and G distributed in at least two biogeographic regions. Among them, the haplotypes of lineage G had the widest distribution, which distributed in Northeastern, Yellow River, Lower Yangtze, and Japan regions. All haplotypes of the Taiwan region were clustered in the lineage F only.
Fig. 2.
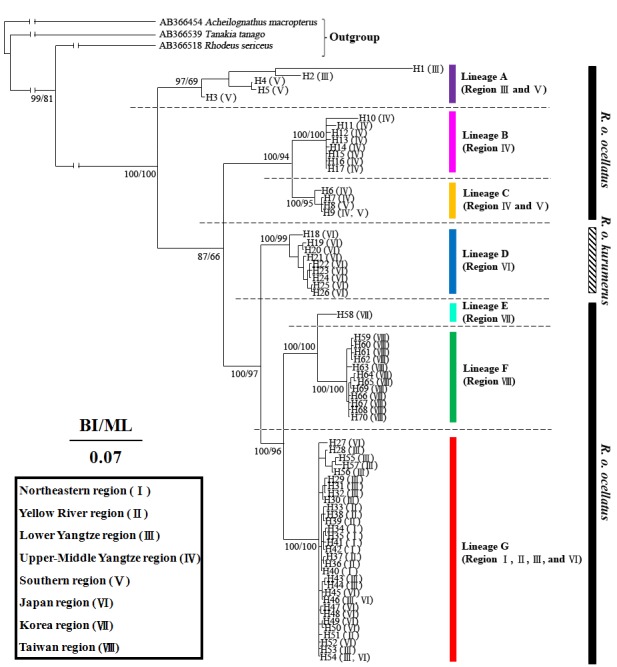
Fig. 2. Phylogenetic tree of Rhodeus ocellatus in East Asia, based on the cyt b gene by Bayesian inference (BI). Posterior probability for BI and bootstrap value for maximum likelihood (ML) are represented at the nodes. The Roman numerals of biogeographic regions are parenthesized behind haplotypes. The spatial distributions of the lineages are represented in figure 1 with the same color usage.
Divergence time
The divergence of R. ocellatus started at 7.55-15.25 million years ago (mya) that separated lineage A from others. The following divergence times among lineages were 5.67-11.44 (between lineagesB+CandD+E+F+G),3.61-7.30 (between lineages D and E + F + G), 2.86- 5.78 (between lineages E + F and G), 2.27- 4.59 (between lineages B and C), and 1.44-2.91 (between lineages E and F) mya (Fig. 3, Additional file 3).
Fig. 3.

Fig. 3. The divergence times (million years ago) of Rhodeus ocellatus in East Asia. The results estimated by 1.05% per site per million years of the evolutionary rate are shown at the main nodes with the 95% highest posterior densities (HPD) intervals in the parenthesis. The ranges of 95% HPD intervals are represented by the gray bars. Numbers in reverse color at main nodes correspond to Additional file 3. The spatial distributions of the lineages are represented in figure 1 with the same color usage.
Genetic structure
Seven major clusters were observed in the minimum spanning network that was congruent with the seven mitochondrial lineages in the phylogenetic tree (Fig. 4). Haplotypes from the same biogeographic region were not always connected to each other, except for the haplotypes from Taiwan region. Shared haplotypes in different biogeographic regions were observed, including H9 in lineage C which distributed in Upper-Middle Yangtze and Southern regions. H46 and H54 in lineage G distributed in Lower Yangtze and Japan regions. In lineage G, all the haplotypes distributed in Japan region were connected to H46 and H54. Moreover, H54 connected to 9 haplotypes from Northeastern, Yellow River, Lower Yangtze, and Japan regions. This part of the network was star shape.
Fig. 4.
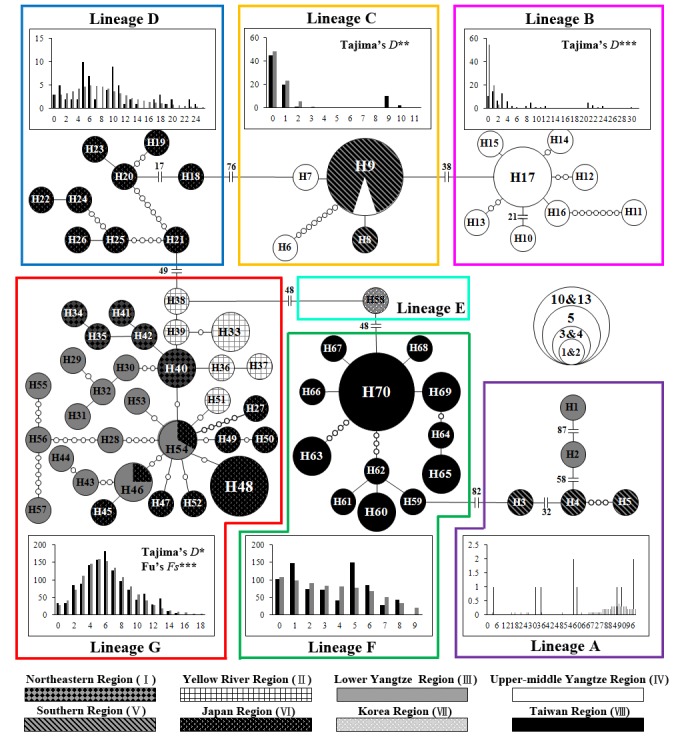
Fig. 4. Minimum spanning network and mismatch distribution from cyt b DNA sequences of Rhodeus ocellatus in each lineage. The colors correspond to the lineage in figure 2. The size of each circle represents the number of specimens of each haplotype. The small, open circle between different haplotypes represents the number of mutations. The number of mutations > 8 is shown as the mutation number beside the traverse lines. The results of mismatch distribution are shown in the histograms. The abscissa and ordinate of the histograms indicate the number of pairwise difference between specimens and the frequency of each value, respectively. The black and gray bars represent the frequency distribution of the observed and expected pairwise difference respectively under the sudden expansion model.
The highest proportion of the genetic variance was among regions (69.59%; Table 2). Genetic differentiations among regions, among sample localities within regions, and within sample localities were significant (ΦCT = 0.676, ΦSC = 0.522, ΦST = 0.845, All P < 0.001). The pairwise ΦST values between regions were all significant, except the values between Northeastern and Lower Yangtze regions (ΦST = 0.087, P = 0.087) and between Yellow River and Lower Yangtze regions (ΦST = 0.084, P = 0.801; Table 3). The highest ΦST value was between populations of the Northeastern and Taiwan regions (ΦST = 0.946, P < 0.001).
Table 2. The analysis of molecular variance (AMOVA) of Rhodeus ocellatus in East Asia.
| Source of variation | d.f. | Sum of squares | Variance components | Percentage of variation (%) | Φ statistics |
| Among regions | 7 | 4464.912 | 37.08484 | 69.59 | ΦCT = 0.676*** |
| Among localities within regions | 23 | 900.836 | 8.64607 | 16.23 | ΦSC = 0.522*** |
| Within localities | 102 | 770.722 | 7.5561 | 14.18 | ΦST = 0.845*** |
*** P < 0.001. ΦCT, ΦSC, and ΦST represented the genetic differentiation among regions, among sample localities within regions, and within sample localities, respectively. Significant values are in bold.
Table 3. Pairwise ΦST between different regions of Rhodeus ocellatus of cyt b DNA sequences.
| Region I | Region II | Region III | Region IV | Region V | Region VI | |
| Northeastern region (I) | - | |||||
| Yellow River region (II) | 0.284*** | - | ||||
| Lower Yangtze region (III) | 0.087 (P = 0.087) | 0.084 (P = 0.801) | - | |||
| Upper-Middle Yangtze region (IV) | 0.874*** | 0.868*** | 0.771*** | - | ||
| Southern region (V) | 0.751*** | 0.744*** | 0.673*** | 0.444*** | - | |
| Japan regoin (VI) | 0.306** | 0.284** | 0.213** | 0.706*** | 0.621*** | - |
| Taiwan region (VIII) | 0.946*** | 0.940*** | 0.813*** | 0.917*** | 0.859*** | 0.736*** |
** P < 0.01, *** P < 0.001.
Population demography
The mismatch distribution of R. ocellatus in East Asia was multimodal (Fig. 5A), and the sum of squared deviation (SSD) and Harpending’s raggedness index (R) were both significantly (SSD = 0.018, P < 0.05; R = 0.005, P < 0.01), suggesting the rejection of the sudden expansion model (Table 4). For the results in each lineage, all the SSD and R were not significant (Table 4). However, only the mismatch distribution of lineage G was clearly unimodal (Fig. 4) with significant negative Tajima’s D and Fu’s Fs values (Tajima’s D = -1.484, P < 0.05; Fu’s Fs = -16.716, P < 0.001). Tajima’s D test also gave significant negative value to lineage B (Tajima’s D = -2.238, P < 0.001) and C (Tajima’s D = -2.112, P < 0.01), but no significant Fu’s Fs values were detected in these lineages.
Fig. 5.
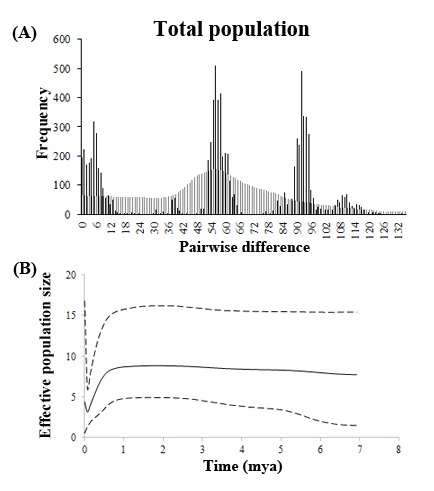
Fig. 5. Demographic inference from cyt b sequences of Rhodeus ocellatus in East Asia. (A) Mismatch distribution of total population. The abscissa and ordinate of the histograms indicate the number of pairwise difference between specimens and the frequency of each value, respectively. The black and gray bars represent the frequency distribution of the observed and expected pairwise difference respectively under the sudden expansion model. (B) Bayesian skyline plot (BSP) of total population estimated by 1.05% per site per million year of evolutionary rate. The black line indicates the mean curve of BSP. The dotted line indicates the 95% highest posterior density intervals of the BSP. The x-axis indicates the time (million years ago, mya). The y-axis is the estimated effective population size.
Table 4. Demographic parameters of cyt b DNA sequences of Rhodeus ocellatus in East Asia.
| Sum of Squared deviation (SSD) | Harpending’s Raggedness index (R) | Tajima’s D | Fu’s Fs | |
| Lineage A | 0.126 | 0.280 | 0.007 | 1.770 |
| Lineage B | 0.389 | 0.042 | -2.238*** | -0.121 |
| Lineage C | 0.024 | 0.190 | -2.112** | 0.660 |
| Lineage D | 0.029 | 0.048 | -0.643 | -0.640 |
| Lineage E | - | - | - | - |
| Lineage F | 0.020 | 0.054 | -0.116 | -1.846 |
| Lineage G | 0.002 | 0.010 | -1.484* | -16.716*** |
| Total populations | 0.018* | 0.005** | 0.407 | -0.002 |
* P < 0.05, ** P < 0.01, *** P < 0.001. Significant values are in bold.
Because the results of the mismatch distribution and the statistics of Tajima’s D and Fu’s Fs values suggested the population expansion of lineage G, we further estimated the time since population expansion (t) with the equation t = τ/2u, where τ is calculated from the mismatch distribution, and u is evolutionary rate × sequence length × generation time (Rogers and Harpending 1992). The value of τ was 4.57 for lineage G. The estimated evolutionary rate was by 1.05% per site per million years. Generation time of R. o. ocellatus is one year (Smith et al. 2004). The estimated time since the population expansion was about 0.19 mya during the Pleistocene. The result of BSP suggested the population size of R. ocellatus in East Asia was constant until about 0.5- 1.0 mya. In the middle Pleistocene, R. ocellatus experienced a population bottleneck resulting from a severe decline in population size that did not recover until about 0.2 mya (Additional file 4 and Fig. 5B).
Ancestral range
The results BBM analysis using RASP showed that the most recent common ancestor (MRCA) of R. ocellatus distributed in the Lower Yangtze region (III) with relative probability 95.25%. The inferred MRCA of lineages A, B, C, D, F, and G distributed in the Lower Yangtze (III), Upper-middle Yangtze (IV), Upper-Middle Yangtze (III), Japan (VI), Taiwan (VIII), and Lower Yangtze (III) regions, respectively with relative probabilities more than 95% (Fig. 6, Additional file 5). The ancestral ranges of the lineage E (Korea) was unable to be inferred by BBM analysis because it contained only one specimen.
Fig. 6.
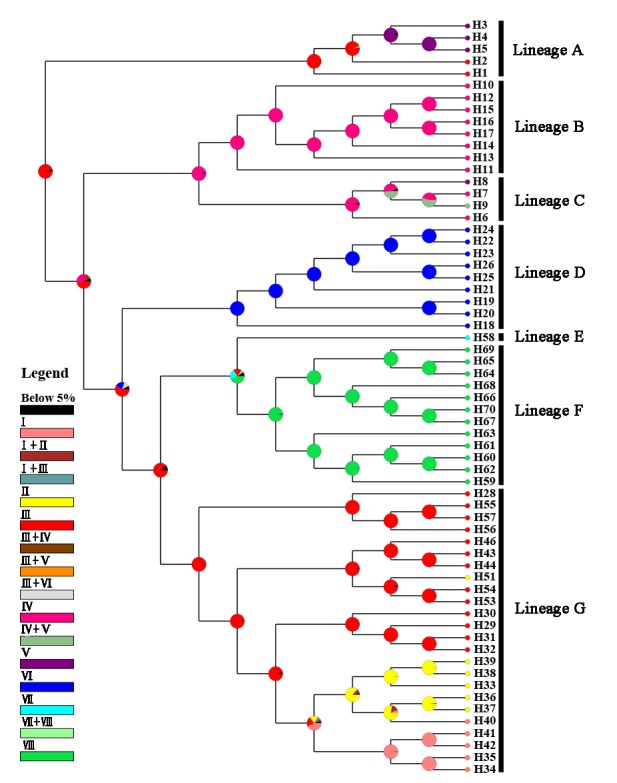
Fig. 6. Ancestral distributions inferred by RASP. The probabilities of alternative ancestral ranges at each node were shown by different colors in the pie charts. Black in the pie charts is the alternative ancestral range with the relative probabilities below 50%
DISCUSSION
Previous studies of R. ocellatus in Japan using mitochondrial DNA sequences showed that R. o. kurumeus is phylogenetically distinct from R. o. ocellatus (Kawamura et al. 2001b; Miyake et al. 2001). However, hybridization between the two subspecies has occurred due to artificial introduction of R. o. ocellatus from mainland China to Japan (Nagata et al. 1996; Kawamura et al. 2001b). In this study, we showed that R. ocellatus differentiated into seven mitochondrial lineages in East Asia (Fig. 2). Divergence times among lineages ranged from 7.55 mya to 1.44 mya (Fig. 3), coincident with the environmental changes due to the uplift of the Tibetan Plateau since the Miocene and climatic fluctuation during Quaternary. The phylogeographic pattern and genetic structure shaped by these historical events have also been reported in the freshwater fishes of Hemibarbus labeo (Lin et al. 2010), Opsariichthys bidens (Li et al. 2009), Rhynchocypris oxycephalus (Yu et al. 2014), and Schizothorax species (He and Chen 2006; Yang et al. 2012) in East Asia.
The Lower Yangtze and Upper-middle Yangtze regions had the haplotypes from four different lineages that might result from the history of the Yangtze River basin. The paleo-Yangtze River basin was confined to the Lower Yangtze region. The river flowed eastward to the East China Sea. The present Upper-middle Yangtze River flows southward to the South China Sea (Clark et al. 2004; Yang et al. 2006; Zheng et al. 2013). About eight million years ago, the altitude of the Tibetan Plateau increased significantly. The subsequent uplift occurred from 3.6-1.7 mya (Harrison et al. 1992; Molnar et al. 1993; Shi et al. 1999; Zheng et al. 2000). During the uplift of the Tibetan Plateau, river captures and flow reversals caused regional extension of the Yangtze River basin to the eastern Tibetan Plateau (Clark et al. 2004; Fan et al. 2005). Furthermore, the uplift also intensified the summer monsoon (An et al. 2001; Shi et al. 1999). The enhanced rainfall in East Asia also accelerated the expansion of the basin (Clift et al. 2008; Fan et al. 2005; Yang et al. 2006; Zheng et al. 2013). The divergence time of R. ocellatus in the Yangtze River basin was coincident to the time of Tibetan Plateau uplift. The results of the Bayesian Binary MCMC analysis showed that the most recent common ancestor of R. ocellatus distributed in the Lower Yangtze region (III). Range expansion of R. ocellatus to Upper-middle Yangtze region (lineages B and C) occurred from 7.55 to 2.27 mya (Fig. 3 and Fig. 6).
Among the lineages that distributed in the Yangtze River basin, lineages A and C contained the haplotypes distributed in Southern region (Fig. 1 and 2). Our BBM analyses showed that the most recent common ancestor of the lineage A distributed in the Lower Yangtze region (Fig. 6). The population further dispersed to the Southern region during the Pleistocene that might be driven by the environmental change due to the glacial- interglacial cycles (Fig. 3 and Fig. 6).
The haplotype 9 distributed in both Upper- middle Yangtze and Southern regions (Fig. 4), and this haplotype widely distributed in Southern region, including localities 13a, 14, and 15. The results of BBM analysis supported range expansion from Upper-Middle Yangtze to Southern regions in the lineage C (Fig. 6). Lingqu Canal was constructed about 2300 years ago which connected the Yangtze and Pearl Rivers. R. ocellatus in Upper-Middle Yangtz region might disperse to the Perl River of Southern region through the canal. Similar dispersal event has also been proposed in Opsariichthys bidens (Li et al. 2009).
The haplotypes of lineage G had wide distribution, including the Northeastern, Yellow River, Lower Yangtze, and Japan regions (Fig. 1). Artificial introduction of R. o. ocellatus from the Yangtze River basin to Japan has been reported in previous studies (Kawamura 2005; Kawamura et al. 2001b). The result of the minimum spanning network showed that two haplotypes (H46 and H54) in lineage G distributed in both the Lower Yangtze and Japan regions (Fig. 4). In addition, haplotypes (H27, H45, H47-50, and H52) from Japan in lineage G connected to H46 and H54 (Fig. 4). The results support the introduction of R. o. ocellatus from the Yangtze River basin to Japan.
The haplotypes of lineage G were widely distributed in the Asian continent that might result from population expansion due to climate oscillation in Pleistocene. The glacial-interglacial cycles of the Pleistocene had great effects on environment. In the glacial period, the rainfall decreased, and the arid region expanded. On the contrary, the climate during the interglacial period was warm and humid (Wang et al. 1999; Zhang 1981; Zhang et al. 2000). These climate changes can influence the species distribution (Zhang et al. 2000). In the minimum spanning network, H40 and H54 were in the central which connected to more than 5 different haplotypes. Most haplotypes from Northeastern and Yellow River regions connected to H40 with a few nucleotide differences. The star- like structure is a pattern of population expansion. Mismatch distribution also supported population expansion in lineage G, and the time since the population expansion was about 0.19 mya. In addition, the MRCA of lineage G distributed in the Lower Yangtze region. We proposed that lineage G experienced northward population expansion during the Pleistocene. When the climate became cold and dry in the glacial period, the population distribution of R. ocellatus in the Asian continent might be restricted to the south. The southern population might expand northward in the interglacial period. In addition, the development of the Yellow River basin which flowed into the Yellow Sea shelf during the Pleistocene might facilitate the dispersal events (Li et al. 1998; Yang et al. 2001). The unimodal mismatch distribution and the expansion time of lineage G suggested that the population expansion during the warmer periods of the Luchan-Dali Interglacial Period (0.2-0.1 mya) (Duan et al. 1980). The result of BSP showed that the population of R. ocellatus in East Asia declined during the mid-Pleistocene and recovered at about 0.2 mya (Fig. 5B).
Although the land bridge formation in the Pleistocene can provide the opportunity for fauna exchange between continent and islands, our result showed the divergence of the Japanese lineage (lineage D, R. o. kurumeus) occurred before the Pleistocene (at about 3.61 mya). The Japanese Archipelago was formed by separating from the edge of the Asian continent in the Late Miocene (Maruyama et al. 1997). The western part of the Japanese Archipelago had been connected to the continent until the opening of the southern channel of the Sea of Japan at about 3 mya in the Late Pliocene (Kitamura and Kimoto 2006; Tada 1994). The Japanese lineage might disperse from the Asian continent before the opening of the southern channel in the Late Pliocene.
Taiwan has been rising from the sea floor since 4 mya (Hsu 1990), and later Pleistocene glaciation lowered the sea level to expose a land bridge between the Asian continent and Taiwan (Boggs et al. 1979). The land bridge might provide the opportunity for R. ocellatus to disperse from Asian continent to Taiwan at about 1.44 mya.
CONCLUSIONS
Phylogeographic pattern and genetic structure of R. ocellatus in East Asia were inferred using cyt b gene sequences. The results suggest the phylogeographic pattern of R. ocellatus were mainly shaped by the topography and climate changes associated with the uplift of the Tibetan Plateau in the Late Miocene-Pliocene and the glacial-interglacial cycles in the Pleistocene. Multiple mitochondrial lineages distributed in the Yangtze River basin that might reflect the developmental history of the Yangtze River. Population expansion during the Pleistocene was detected in the populations distributed in lineage G due to the glacial-interglacial cycles. Genetic differentiation of R. ocellatus is statistically significant among populations. Our results and inference may benefit to the conservation management of R. occelatus because the status of R. ocellatus is categorized as data deficient in the IUCN (Huckstorf 2013).
Acknowledgments
Acknowledgments: This study is founded in parts by Department of Life Science, National Chung Hsing University, Taiwan.
References
- An Z, Kutzbach J E, Prell W L, Porter S C. Evolution of Asian monsoons and phased uplift of the HimalayaTibetan plateau since Late Miocene times. Nature. 411:62–66. doi: 10.1038/35075035. [DOI] [PubMed] [Google Scholar]
- Avise J C. Phylogeography: retrospect and prospect. J Biogeogr. 36:3–15. [Google Scholar]
- Birky CW Jr. Uniparental inheritance of mitochondrial and chloroplast genes: mechanisms and evolution. Proc. Natl Acad Sci. 1995;92:11331–11338. doi: 10.1073/pnas.92.25.11331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boggs S J, Wang W, Lweis F S, Chen J. Sediment properties and water characteristics of Taiwan shelf and slope. Acta Oceanogr Taiwan. 10:10–49. [Google Scholar]
- Chang C H, Li F, Shao K T, Lin Y S, Morosawa T, Kim S, Koo H, Kim W, Lee J S, He S, Smith C, Richard M, Miya M, Sado T, Uehara K, Lavoué S, Chen W J, Mayden R L. Phylogenetic relationships of Acheilognathidae (Cypriniformes: Cyprinoidea) as revealed from evidence of both nuclear and mitochondrial gene sequence variation: Evidence for necessary taxonomic revision in the family and the identification of cryptic species. Mol Phyl Evol. 81:182–194. doi: 10.1016/j.ympev.2014.08.026. [DOI] [PubMed] [Google Scholar]
- Clark M K, Schoenbohm L M, Royden L H, Whipple K X, Burchfiel B C, Zhang X, Tang W, Wang E, Chen L. Surface uplift, tectonics, and erosion of eastern Tibet from largescale drainage patterns. Tectonics; 2004. 23 [Google Scholar]
- Clift P D, Layne G D, Blusztajn J. Marine sedimentary evidence for monsoon strengthening, Tibetan uplift and drainage evolution in East Asia. Geophysical Monograph Series. 149:255–258. [Google Scholar]
- Clift P D, Long H V, Hinton R, Ellam R M, Hannigan R, Tan M T, Blusztajn J, Duc N A. Evolving east Asian river systems reconstructed by trace element and Pb and Nd isotope variations in modern and ancient Red River-Song Hong sediments. 2008.
- Darriba D, Taboada G L, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nature Methods; 2012. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling T E, Tibbets C A, Minckley W L, Smith G R. Evolutionary relationships of the Plagopterins (Teleostei: Cyprinidae) from cytochrome b sequences. Copeia. pp. 665–678.
- Drummond A J, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol; 2007. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan W, Pu Q, Wu X. Climatic variations in China during the quaternary. GeoJournal. 4:515–524. [Google Scholar]
- Excoffier L, Laval G, Schneider S. Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform. 1:45–50. [PMC free article] [PubMed] [Google Scholar]
- Fan D, Li C, Yokoyama K, Zhou B, Li B, Wang Q, Yang S, Deng B, Wu G. Monazite age spectra in the Late Cenozoic strata of the Changjiang delta and its implication on the Changjiang run-through time. Sci China Ser D. 48:1718–1727. [Google Scholar]
- Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- Hall T A. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/ NT. Nucleic Acids Symp Ser. 41:95–98. [Google Scholar]
- Harrison T M, Copeland P, Kidd Wsf, Yin A. Raising Tibet. Science. 255:1663–1670. doi: 10.1126/science.255.5052.1663. [DOI] [PubMed] [Google Scholar]
- He D, Chan Y. Biogeography and molecular phylogeny of the genus Schizothorax (Teleostei: Cyprinidae) in China inferred from cytochrome b sequences. J Biogeogr. 33:1448–1460. [Google Scholar]
- He S, Gu X, Mayden R L, Chen W J, Conway K W, Chen Y. Phylogenetic position of the enigmatic genus Psilorhynchus (Ostariophysi: Cypriniformes): evidence from the mitochondrial genome. Mol Phylogenet Evol. 47:419–425. doi: 10.1016/j.ympev.2007.10.012. [DOI] [PubMed] [Google Scholar]
- Hewitt G M. The genetic legacy of the Quaternary ice ages. Nature. 405:907–913. doi: 10.1038/35016000. [DOI] [PubMed] [Google Scholar]
- Hewitt G M. Genetic consequences of climatic oscillations in the Quaternary. Philos Trans R Soc Lond B Biol Sci. 359:183–195. doi: 10.1098/rstb.2003.1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huckstorf V. Rhodeus ocellatus. The IUCN Red List of Threatened Species.
- Hsu V. Seismicity and tectonics of a continent-island arc collision zone at the island of Taiwan. J Geophys Res. 95:4725–4734. [Google Scholar]
- Huelsenbeck J P, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 17:754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- Jia G, Peng P, Zhao Q, Jian Z. Changes in terrestrial ecosystem since 30 Ma in East Asia: Stable isotope evidence from black carbon in the South China Sea. Geology. 31:1093–1096. [Google Scholar]
- Kang B, Deng J, Wu Y, Chen L, Zhang J, Qiu H, Lu Y, He D. Mapping China's freshwater fishes: diversity and biogeography. Fish Fish. 15:209–230. [Google Scholar]
- Kawamura K. Low genetic variation and inbreeding depression in small isolated populations of the Japanese rosy bitterling, Rhodeus ocellatus kurumeus. Zool Sci. 22:517–524. doi: 10.2108/zsj.22.517. [DOI] [PubMed] [Google Scholar]
- Kawamura K, Nagata Y, Ohtaka H, Kanoh Y, Kitamura J. Genetic diversity in the Japanese rosy bitterling, Rhodeus ocellatus kurumeus (Cyprinidae) Ichthyol Res. 48:369–379. [Google Scholar]
- Kawamura K, Ueda T, Arai R, Nagata Y, Saitoh K, Ohtaka H, Kanoh Y. Genetic Introgression by the Rose Bitterling, Rhodeus ocellatus ocellatus, into the Japanese Rose Bitterling, R. o. kurumeus (Teleostei: Cyprinidae). Zool Sci. 2001;18:1027–1039. [Google Scholar]
- Kawamura K, Ueda T, Arai R, Smith C. Phylogenetic relationships of bitterling fishes (Teleostei: Cypriniformes: Acheilognathinae), inferred from mitochondrial cytochrome b sequences. Zool Sci. 31:321–329. doi: 10.2108/zs130233. [DOI] [PubMed] [Google Scholar]
- Kim B C, Kang T W, Kim M S, Kim C B. The complete mitogenome of Rhodeus uyekii (Cypriniformes, Cyprinidae) DNA Seq. 17:181–186. doi: 10.1080/10425170600806928. [DOI] [PubMed] [Google Scholar]
- Kitamura A, Kimoto K. History of the inflow of the warm Tsushima Current into the Sea of Japan between 3.5 and 0.8 Ma. Palaeogeogr Palaeoclimatol Palaeoecol. 236:355–366. [Google Scholar]
- Kitanishi S, Nishio M, Sagawa S, Uehara K, Ogawa R, Yokoyama T, Ikeya K, Edo K. Strong population genetic structure and its implications for the conservation and management of the endangered Itasenpara bitterling. Conserv Genet. 2013;14:901–906. [Google Scholar]
- Kitazima J, Matsuda M, Mori S, Kokita T, Watanabe K. Population structure and cryptic replacement of local populations in the endangered bitterling Acheilognathus cyanostigma. Ichthyol Res. 62:122–130. [Google Scholar]
- Kubota H, Watanabe K. Loss of Genetic Diversity at an MHC Locus in the Endangered Tokyo Bitterling Tanakia tanago (Teleostei: Cyprinidae) Zool Sci. 30:1092–1101. doi: 10.2108/zsj.30.1092. [DOI] [PubMed] [Google Scholar]
- Kubota H, Watanabe K, Suguro N, Tabe M, Umezawa K, Watanabe S. Genetic population structure and management units of the endangered Tokyo bitterling, Tanakia tanago (Cyprinidae) Conserv Genet. 11:2343–2355. [Google Scholar]
- Levin B A, Freyhof J, Lajbner Z, Perea S, Abdoli A, Gaffaroğlu M, Özuluğ M, Rubenyan H R, Salnikov V B, Doadrio I. Phylogenetic relationships of the algae scraping cyprinid genus Capoeta (Teleostei: Cyprinidae) Mol Phylogenet Evol. 62:542–549. doi: 10.1016/j.ympev.2011.09.004. [DOI] [PubMed] [Google Scholar]
- Li F, Zhang X, Li Y. Buried Paleo-delta in the south Yellow Sea. Acta Geogr Sin. 53:238–244. [Google Scholar]
- Li G Y, Wang X Z, Zhao Y H, Zhang J, Zhang C G, He S P. Speciation and Phylogeography of Opsariichthys bidens (Pisces: Cypriniformes: Cyprinidae) in China: analysis of the cytochrome b gene of mtDNA from diverse populations. Zool Stud. 48:569–583. [Google Scholar]
- Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- Lin C, Lin H, Wang J, Chao S, Chiang T. Phylogeography of Hemibarbus labeo (Cyprinidae): secondary contact of ancient lineages of mtDNA. Zool Scripta. 39:23–35. [Google Scholar]
- Maruyama S, Isozaki Y, Kimura G, Terabayashi M. Paleogeographic maps of the Japanese Islands: Plate tectonic synthesis from 750 Ma to the present. Isl Arc. 6:121–142. [Google Scholar]
- Miyake T, Nakajima J, Onikura N, Ikemoto S, Iguchi K, Komaru A, Kawamura K. The genetic status of two subspecies of Rhodeus atremius, an endangered bitterling in Japan. Conserv Genet. 12:383–400. [Google Scholar]
- Miyake K, Tachida H, Oshima Y, Arai R, Kimura S, Imada N, Honjo T. Genetic variation of the cytochrome b gene in the rosy bitterling, Rhodeus ocellatus (Cyprinidae) in Japan. Ichthyol Res. 48:105–110. [Google Scholar]
- Molnar P, England P, Martinod J. Mantle dynamics, uplift of the Tibetan Plateau, and the Indian Monsoon. Rev Geophys. 31:357–396. [Google Scholar]
- Moritz C. Defining 'Evolutionarily Significant Units' for conservation. Trends Ecol Evol. 9:373–375. doi: 10.1016/0169-5347(94)90057-4. [DOI] [PubMed] [Google Scholar]
- Nagata Y, Tetsukawa T, Kobayashi T, Numachi K. Genetic markers distinguishing between the two subspecies of the rosy bitterling, Rhodeus ocellatus (Cyprinidae) Ichthyol Res. 43:117–124. [Google Scholar]
- Nakamura M. On the freshwater fishes introduced and propagated in Kanto Plains. Bull Biogeogr Soc Japan. 16:333–337. [Google Scholar]
- Onikura N, Miyake T, Nakajima J, Fukuda S, Kawamato T, Kawamura K. Predicting potential hybridization between native and non-native Rhodeus ocellatus subspecies: the implications for conservation of a pure native population in northern Kyushu. Japan. Aquat Invasions. 8:219–229. [Google Scholar]
- Rambaut A. FigTree v1.3.1. Available at: http://tree.Bio. Ed.Ac.Uk/software/figtree.
- Rogers A R, Harpending H. Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol. 9:552–569. doi: 10.1093/oxfordjournals.molbev.a040727. [DOI] [PubMed] [Google Scholar]
- Saitoh K, Sado T, Mayden R L, Hanzawa N, Nakamura K, Nishida M, Miya M. Mitogenomic evolution and interrelationships of the Cypriniformes (Actinopterygii: Ostariophysi): the first evidence toward resolution of higher-level relationships of the world's largest freshwater fish clade based on 59 whole mitogenome sequences. J Mol Evol. 63:826–841. doi: 10.1007/s00239-005-0293-y. [DOI] [PubMed] [Google Scholar]
- Shi Y, Li J, Li B, Yao T, Wang S, Li S, Cui Z, Wang F, Pan B, Fang X, Zhang Q. Uplift of the Qinghai-Xizang (Tibetan) Plateau and East Asia environmental change during late Cenozoic. Acta Geogr Sin. 66:10–20. [Google Scholar]
- Smith C, Reichard M, Jurajda P, Przybylski M. The reproductive ecology of the European bitterling (Rhodeus sericeus) J Zool Lond. 262:107–124. [Google Scholar]
- Tada R. Paleoceanographic evolution of the Japan Sea. Palaeogeogr Palaeoclimatol Palaeoecol. 108:487–508. [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voris H K. Map of Pleistocene sea levels in Southeast Asia: shorelines, river systems and time durations. J Biogeogr. 27:1153–1167. [Google Scholar]
- Wang L, Sarnthein M, Erlenkeuser H, Grimalt J, Grootes P, Heilig S, Ivanova E, Kienast M, Pelejero C, Pflaumann U. East-Asian monsoon climate during the Late Pleistocene: High-resolution sediment records from the South China Sea. Mar Geol. 156:245–284. [Google Scholar]
- Winter M, Devictor V, Schweiger O. Phylogenetic diversity and nature conservation: where are we? Trends Ecol Evol. 28:199–204. doi: 10.1016/j.tree.2012.10.015. [DOI] [PubMed] [Google Scholar]
- Yang J, Yang J X, Chen X Y. A re-examination of the molecular phylogeny and biogeography of the genus Schizothorax (Teleostei: Cyprinidae) through enhanced sampling. China. J Zoolog Syst Evol Res. 50:184–191. [Google Scholar]
- Yang Q, Zhu Y, Xiong B, Liu H. Acheilognathus changtingensis sp. nov., a new species of the cyprinid genus Acheilognathus (Teleostei: Cyprinidae) from Southeastern China based on morphological and molecular evidence. Zool Sci. 28:158–167. doi: 10.2108/zsj.28.158. [DOI] [PubMed] [Google Scholar]
- Yang S, Cai J, Li C, Deng B. New discussion about the run-through time of the Yellow River. Mar Geol Quat Geol (Beijing) 21:15–20. [Google Scholar]
- Yang S, Li C, Yokoyama K. Elemental compositions and monazite age patterns of core sediments in the Changjiang Delta: Implications for sediment provenance and development history of the Changjiang River. E Earth Planet Sci Lett. 245:762–776. [Google Scholar]
- Yap S Y. On the distributional patterns of SoutheastEast Asian freshwater fish and their history. J Biogeogr. 29:1187–1199. [Google Scholar]
- Yasuno K. Fossil pharyngeal teeth of the Rhodeinae fish from the Miocene Katabira Formation of the Kani Group. Bull Mizunami Fossil Mus. 11:101–105. [Google Scholar]
- Yoshio T, Haruto K, Tsuneo N, Toshikatsu Y. Fossil freshwater fishes from Japan. Mem Geol Soc Japan. 14:221–243. [Google Scholar]
- Yu D, Chen M, Tang Q, Li X, Liu H. Geological events and Pliocene climate fluctuations explain the phylogeographical pattern of the cold water fish Rhynchocypris oxycephalus (Cypriniformes: Cyprinidae) in China. BMC Evol Biol; 2014. 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Harris A J, Blair C, He X J. RASP (Reconstruct Ancestral State in Phylogenies): a tool for historical biogeography. Mol Phylogenet Evol. 87:46–49. doi: 10.1016/j.ympev.2015.03.008. [DOI] [PubMed] [Google Scholar]
- Zardoya R, Doadrio I. Molecular evidence on the evolutionary and biogeographical patterns of European cyprinids. J Mol Evol. 49:227–237. doi: 10.1007/pl00006545. [DOI] [PubMed] [Google Scholar]
- Zhang L. The influence of the uplift of Qinghai-Xizang Plateau on the Quaternary environmental evolution in China. J Lanzhou Univ Nat Sci. 3:142–155. [Google Scholar]
- Zhang D, Liu F. Eco-environmental effects of the Qinghai-Tibet Plateau uplift during the Quaternary in China. Environ Geol. 39:1352–1358. [Google Scholar]
- Zheng H, Clift P D, Wang P, Tada R, Jia J, He M. Pre-Miocene birth of the Yangtze River. Proc Natl Acad Sci U S A. 110:7556–7561. doi: 10.1073/pnas.1216241110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Cmca Powell, An Z, Zhou J, Dong G. Pliocene uplift of the northern Tibetan Plateau. Geology. 28:715–718. [Google Scholar]
- Zwickl D J. Genetic algorithm approaches for the phylogenetic analysis of large biological sequence datasets under the maximum likelihood criterion. Dissertation. 2006.