

Abstract
The threat of a deliberate release of chemical nerve agents has underscored the need to continually improve field effective treatments for these types of poisonings. The oxime containing HLö-7 is a potential second-generation therapeutic reactivator. A synthetic process for HLö-7 is detailed with improvements to the DIBAL reduction and ion exchange steps. HLö-7 was visualized for the first time within the active site of human acetylcholinesterase and its relative ex vivo potency confirmed against various nerve agents using a phrenic nerve hemidiaphragm assay.
Keywords: Acetylcholinesterase, VX, HLö, -7, nerve agent, organophosphate, reactivator
The recent nerve agent attack against the Russian defector Skripal in Salisbury, England, along with a string of other attacks in Macau and Syria stressed the need for potent therapeutics to counter the threat from chemical nerve agents.1−3 Chemical nerve agents and many pesticides share an organophosphate (OP) core and inhibit human acetylcholinesterase (hAChE) activity. G-type nerve agents such as soman (GD; O-pinacolyl methylphosphonofluoridate), sarin (GB; O-isopropyl methylphosphonofluoridate), and cyclosarin (GF; (cyclohexyl methylphosphonofluoridate) have the same core as diisopropylfluorophosphate (DFP), a commercial pesticide. All incorporate a fluoride leaving group and a phosphonate methyl group except for DFP, which differs from the G-type agents replacing the methyl with an O-isopropyl group. Tabun (GA) has a cyanide leaving group. In VX (ethyl N-2-diisopropylaminoethyl methylphosphonothiolate) and other V-agents, the fluoride leaving group of the G-agents is replaced with a thiolate.4 Toxicity from nerve agents poisoning occurs by nucleophilic attack by active site serine (S203) of hAChE on these agent’s electrophilic phosphorus moiety. This covalent phosphorylation inhibits hAChE. Dealkylation of the phosphate moiety “ages” the covalently modified enzyme5 and increases resistance to enzyme reactivation.
A current OP poisoning treatment includes a seizure controlling benzodiazepine, muscarinic antagonizing atropine, and hAChE reactivating 2-PAM.6 2-PAM belongs to an oxime containing class of nerve agent therapeutics known as reactivators that nucleophilically dephosphorylate the active site serine returning hAChEs from an inhibited state to its native active form. Over the last 50 years, substantially more effective oximes relative to 2-PAM have been discovered, such as TMB-4, Obidoxime, HI-6, and HLö-7 (Figure 1).7 HLö-7 has been reported to have several fold to orders of magnitude better efficacy than 2-PAM, depending on the nerve agent.8 HLö-7 contains two pyridinium ring systems as in TMB-4, Obidoxime, and HI-6 but differs by having two oxime groups on one ring. The ring containing the nonreactivating acetamide group in HLö-7 has been shown to interact with a peripheral anionic site (PAS) of mouse AChE (mAChE).9
Figure 1.
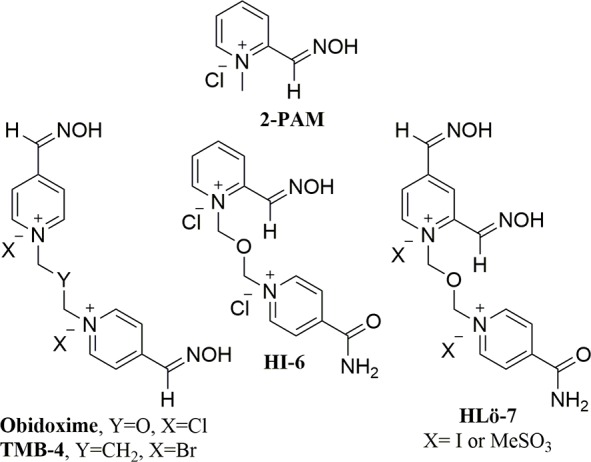
Oxime-based reactivators.
The synthesis of HLö-7 diiodide and HLö-7 dimethanesulfonate (DMS) along with their pharmacological properties have been reported.10−12 Similar to HI-6, HLö-7 diiodide has a 12 h half-life at physiological pH and temperature. This fact necessitates, for possible therapeutic use, a wet/dry autoinjection system for any reactivator with such aqueous stability. Thus, the potential reactivator must exhibit rapid aqueous dissolution. HLö-7 diiodide has limited water solubility (approximately 20 mg/mL) and releases elemental iodine in solution even in the absence of light. These properties led to the synthesis of the more readily water-soluble HLö-7 DMS. Herein, an improved gram-scale synthesis of HLö-7 DMS is presented. Leveraging these improvements, HLö-7 DMS was visualized for the first time in the human hAChE active site. Also, the potency of HLö-7 DMS was evaluated using an ex vivo mouse model system.
The synthesis of HLö-7 DMS is shown in Scheme 1. The chemistry does not differ in synthetic design from the literature, but the methodology has been improved through modification of the reaction conditions, workups, and purifications.
Scheme 1. Synthesis of HLö-7 DMS.
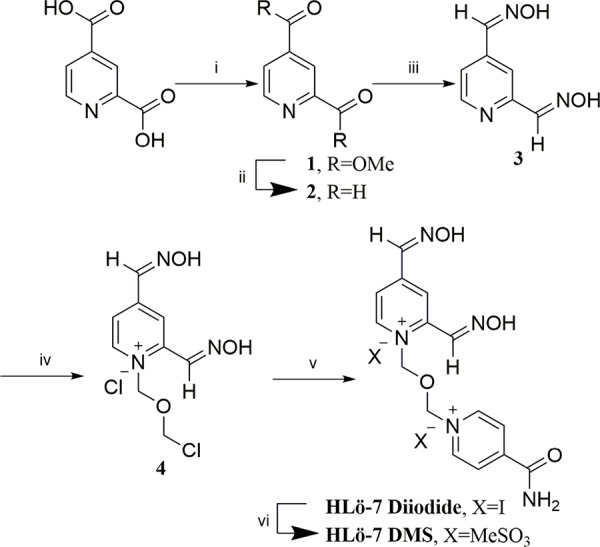
The reported synthesis proceeded through the intermediate bis-aldehyde 2, whose synthesis has been described.13 The report contains a temperature controlled bismethyl-ester 1 to bis-aldehyde 2 reduction employing neat diisobutylaluminumhydride (DIBAL) as the reducing agent in toluene at −65 °C. Attempts at reproducing this procedure revealed that the starting bis-ester 1 was not completely soluble in toluene below −35 °C at the indicated concentration. Furthermore, neat DIBAL addition decreased the reaction yield through over-reduction presumably due to difficulties with temperature control. Finally, the reported solid extraction process was inefficient. In our hands, the reduction process was improved by replacing toluene with dichloromethane (DCM) and in addition of 1 M DIBAL in DCM to the bis-ester 1. These changes alleviated the solubility and temperature control problems. Quenching the reaction with glacial acetic acid below −65 °C was followed by the addition of hydrated Celite to the mixture at 0 °C. This improved efficiency by allowing for a single filtration of the aluminum solids requiring no subsequent salt extraction. (DIBAL quenching methodologies employing tartaric acid, sodium sulfate decahydrate, or monobasic potassium citrate all resulted in inefficient recovery of the bis-aldehyde 2 from the workup mixtures.) The bis-aldehyde 2 was purified via column chromatography. The conversion of compound 2 to bis-oxime 3 was followed by monoalkylation with bischloromethyl ether (BCME)14 to provide intermediate 4.
Addition of isonicotinamide to compound 4 was accomplished by heating in the presence of excess sodium iodide to provide HLö-7 diiodide after recrystallization. The literature-based ion exchange procedure to HLö-7 DMS called for the use of silver methanesulfonate in a methanol/water/acetonitrile solvent system.12 In our hands, this methodology was compromised by unacceptable yields and incomplete ion exchange as indicated by melting point and 1H NMR analyses. An alternative ion exchange process was adapted from the literature15 using methanesulfonic acid treated Dowex monosphere 550A resin. The HLö-7 DMS was obtained in a consistent quality and yield.
A one-pot BCME bis-alkylation of bis-oxime 3 and isonicotinamide failed. Additionally, the utilization of bismethanesulfonatemethyl ether (BMSME) as the alkylating agent was performed as reported.11 The moisture sensitive reagent proved to be impractical in its synthesis, storage, and utility.
BCME is a known carcinogen.16 The use of BCME in the production of HLö-7 DMS might hinge upon its presence in the final product. Trace BCME analysis was conducted by GC–MS. Using synthesized BCME, the instrument method detection limit (MDL) was determined to be 27.7 ppb (ng/mL) with a signal-to-noise ratio ≥5 using single ion mode (SIM) detection. Intermediate 4 was stirred in DCM (100 mg/mL) for 24 h. The organic solution was analyzed, and an 8.68 μg/g ratio of BCME to intermediate 4 was determined. The same DCM extraction procedure was applied to HLö-7 DMS, and no quantifiable amount of BCME was detected in the DCM extract. Thus, the processes employed in Scheme 1 eliminated the BCME carcinogen to an undetectable level. If BCME is present in HLö-7 DMS, it is below 0.277 μg/g.
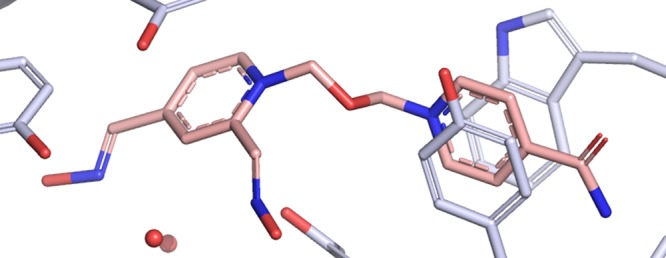
Molecular insight into how HLö-7 interacts with the active site of hAChE was obtained through X-ray crystallography (Table S1). Crystals of hAChE were soaked in cryogenic freezing solution containing HLö-7 DMS prior to being frozen. These crystals yielded a 2.4 Å data set. Upon solving by molecular replacement using 4EY4(17) as a model, Fo – Fc simulated annealing omit map density matching the entirety of HLö-7 was observed within the hAChE active site (Figures 2a and S1). The acetamide containing pyridinium ring of HLö-7 is found to form π–π stacking with the PAS site by inserting itself between Tyr72 and Trp286. The oxime containing pyridinium ring is positioned deeper in the active site and stabilized by intermolecular forces with the backbone amine of Asp74, waters, and the induced dipole interaction with Tyr124.
Figure 2.

X-ray crystal structure of HLö-7 bound to hAChE and mAChE. (a) The active site of hAChE (light blue) with HLö-7 (pink). Waters are shown as red balls. Hydrogen bonds represented by black dashed lines. Measurements are in angstroms and labeled in red. (b,c) hAChE-HLö-7 in (a) overlaid with the active site of mAChE (green) bound to HLö-7 (tan).
Previously, HLö-7 had only been observed within the active site of mAChE.9 Comparing the hAChE-HLö-7 to the mAChE-HLö-7, HLö-7 is observed to be in a noticeably different pose. Specifically, the HLö-7 in the mAChE is located deeper in the active site relative to HLö-7 in hAChE. In this position, HLö-7 is stabilized by multiple intermolecular interactions. This includes a hydrogen bond network with the catalytically important residues Ser203, Gly121, and Gly122 suggesting this pose of HLö-7 is unlikely to be accommodated by a mAChE that is inhibited by an OP (Figure 2b). The oxime containing ring of HLö-7 in hAChE does not adopt this positioned because HLö-7 in hAChE makes use of π–π stacking with Tyr34, which appears to prevent HLö-7 moving deeper into the active site (Figure 2b). To accommodate this orientation within the hAChE active site, Asp74 is swung away from the position found in mAChE. Overall, HLö-7 appears to be stabilized in hAChE at a more standoff distance from the catalytic Ser203 than found in the mAChE-HLö-7 structure.
Both hAChE and mAChE demonstrate π–π stacking with HLö-7 at their respective PAS’s; however, the orientation of the acetamide containing pyridinium ring differs radically. In hAChE, the acetamide containing pyridinium ring forms π–π stacking with Trp286 and Tyr72, which allows the acetamide to produce stabilizing hydrogen bond interactions with the main chain of Glu285 and the side chain of Val282 (Figure 2c). In mAChE, the acetamide containing ring π–π stacks with Trp286 and Tyr124, while the acetamide is oriented toward Ser298 leaving the possibility for hydrogen bonding. Overall, the position of Trp286 and Tyr124 in the HLö-7-bound hAChE structure resembles that found in mAChE in complex with obidoxime.18 Whereas the one found in mAChE is more indicative of HI-6 in complex with mAChE and of HI-6 in complex with mAChE inhibited by sarin.19,20
The divergent orientation of HLö-7 in the PAS of hAChE and mAChE may be the cause of the differing position of oxime containing pyridinium ring in these structures. However, comparing these uninhibited AChE structures bound to HLö-7 with the structure of mAChE inhibited by tabun with HLö-7 appears to refute this assertion (Figure S2). Specifically, the PAS interaction with HLö-7 found in a tabun-inhibited mAChE structure appears to resemble that of the uninhibited counterpart (Figure S2). Although the HLö-7 in the mAChE tabun-inhibited structure does not take advantage of π–π stacking with Tyr341, it does more closely resemble the stacking found in the hAChE-HLö-7 structure. This suggests that the placement of the oxime containing pyridinium ring within the active site of AChE has to do more with its stabilization than that of the acetamide pyridinium ring interaction with the PAS. This raises the issue of why HLö-7 is found in two different poses between mAChE and hAChE given that the active site residues between these AChEs are well-conserved.21 When aligning the active sites of AChEs from these two species, residues outside of the PAS do not fully line up such as Tyr337, Try341, and Phe295. Some of these residues have been implicated in species to species differences observed for inhibition and reactivation of AChEs by VX and another dual ring reactivator.21 As a result, the hAChE bound HLö-7 structure appears to further support the idea that the AChE active site is dynamic, and the degree of loop flexibility may differ between species.
To further probe the efficacy of HLö-7 DMS against nerve agents in an ex vivo setting, a mouse phrenic nerve hemidiaphragm (MPNH) assay was utilized. The ex vivo mouse phrenic nerve hemidiaphragm tissue bath prep is a well-established assay21−24 and serves as an intermediate step between an in vitro assay and the in vivo evaluation of reactivators in mice and guinea pigs. The assay provides a physiological measure of the reactivation potential of compounds as well as having the ability to measure AChE activity in the homogenized tissue at the conclusion of the experiment (Figure 3). The 200 μM dose of reactivator that was used is considered the highest blood plasma concentration generally achieved during in vivo studies of oxime therapy.21 The lower concentrations are similar to those achieved in plasma after therapeutic administration in vivo.21 The data clearly shows that HLö-7 DMS is effective at reactivating VX-, GF-, and GB-inhibited diaphragm muscle AChE activity. These results are similar to AChE reactivation by HLö-7 DMS in studies using paraoxon (diethyl-4-nitrophenylphosphate)-inhibited MPNH assays.22,23
Figure 3.

HLö 7 restoration of mouse diaphragm AChE activity. mAChE activity inhibited by VX (a), GB (b), and GF (c) is reversed with treatment by HLö 7 at various concentrations.
Reactivation is also seen through restoring muscle function in the MPNH prep (Figure S3). Reactivation of at least 60% of muscle force was seen against VX, GF, and GB using 50–200 μM HLö-7 DMS concentrations. Additionally, HLö-7 DMS reactivation of GF and GB inhibited muscle was compared to 2-PAM, HI-6, and MMB-4 (Figure S4). HLö-7 DMS reactivation was marginally better than HI-6 and MMB-4 and significantly better than 2-PAM. This data is consistent with a previous report of HLö-7 diiodide muscle force restoration of GA-, GB-, and GD-inhibited rat diaphragms.24
In summary, an improved gram-scale synthetic methodology for the generation of HLö-7 DMS was accomplished. The process reported here utilized an improved DIBAL reduction methodology. With a resin-based ion exchange, HLö-7 DMS was generated in a 25% yield from intermediate 3. This represents an improvement over the BMSME methodology (17% yield) and the two step BCME alkylation with silver methanesulfonate mediated salt exchange (20% yield) from the same intermediate.11 The HLö-7 DMS synthesized was able to provide the structural insights into hAChe reactivation and ex vivo efficacy via a MPNH assay.
Experimental Procedures
General Methods
Starting materials were purchased from Aldrich Chemical Co. (Milwaukee, WI). GC–MS data was obtained on an Agilent 6850 Series II GC with an Agilent 5975 Inert XL Mass Selective Detection. Flash chromatography was performed on a Grace Reveleris X2 semiautomatic purification system. NMR spectra were obtained on a JEOL 400 MHz spectrometer, and chemical shifts (δ) are reported in parts per million (ppm) downfield from tetramethylsilane. Elemental analysis was run on a PerkinElmer Series II 2400 CHNS/O Elemental Analyzer with cysteine as the standard. HRMS data was obtained on a Waters Acquity UPLC Synapt G2-S equipped with an electrospray ionization (ESI) interface. Data was acquired in positive ion scan mode over a mass range of 50–1200 Da with leucine enkephalin as the reference standard.
Chemical Synthesis
Dimethyl Pyridine-2,4-dicarboxylate 1
A suspension of pyridine-2,4-dicarboxylic acid hydrate (6.00 g, 35.8 mmol) and 60 mL of MeOH was stirred in an ice water bath. Thionyl chloride (11.54 g, 97.0 mmol) was added dropwise with an addition funnel. The ice bath was removed, and the mixture was allowed to come to room temperature. The mixture was heated at reflux temperature for 20 h. The reaction mixture was allowed to cool to room temperature, and the volatiles were evaporated. The residue was taken up with saturated aqueous sodium bicarbonate and chloroform. The organic layer was separated. The aqueous layer was washed two additional times with chloroform. The combined organic extracts were dried with sodium sulfate, filtered, and the volatiles were evaporated providing 5.48 g of a white solid in 78% yield. mp 58–61 °C; 1H NMR (CDCl3) δ 8.89 (d, 1H, J = 4.58 Hz), 8.64 (s, 1H), 8.02 (dd, 1H, J = 5.03, 1.37 Hz), 4.02 (s, 3H), 3.98 (s, 3H); 13C NMR (CDCl3) δ 165.07, 164.83, 150.80, 149.07, 138.85, 126.21, 124.48, 53.25, 53.10.
Pyridine-2,4-dicarbaldehyde 2
Dimethyl pyridine-2,4-dicarboxylate 1 (2.00 g, 10.3 mmol) was dissolved in 30 mL of dry DCM at room temperature under a nitrogen atmosphere in a three-necked flask with an internal thermometer. The solution was cooled to −78 °C with a dry ice/acetone bath. Diisobutylaluminum hydride (DIBAL) (27 mL of a 1 M solution in DCM, 27.0 mmol) was slowly added with a syringe at a rate that did not allow the solution temperature to rise above −65 °C. The solution was kept at −65 °C or below for 4 h. The reaction was quenched by the dropwise addition of 2.40 mL acetic acid in 2.40 mL of DCM. The quench was added at such a rate as to maintain the solution temperature below −65 °C. The solution was allowed to come to 0 °C and a slurry of 2.40 g of Celite wetted with 2.40 mL of water was slowly added to control bubbling and was stirred at room temperature overnight. The mixture was filtered through Celite. The filtrate was concentrated under reduced pressure. The residue was flash chromatographed using 30% to 50% ethyl acetate in hexanes gradient elution. This provided 0.92 g of a tan solid in a 67% yield. 1H NMR (CDCl3) δ 10.16 (s, 2H), 9.04 (d, 1H, J = 5.04 Hz), 8.34 (br s, 1H), 7.95 (dd, 1H, J = 5.03, 1.37 Hz); 13C NMR (CDCl3) δ 192.44, 190.40, 154.42, 151.70, 142.68, 125.45, 120.66.
Pyridine-2,4-dialdoxime 3
Pyridine-2,4-dicarbaldehyde 2 (1.90 g, 14.1 mmol) was dissolved in 40 mL of MeOH at room temperature. K2CO3 (4.10 g, 29.7 mmol) was added, and the mixture was cooled in an ice bath under a nitrogen atmosphere. Hydroxylamine hydrochloride (2.05 g, 29.5 mmol) in 8 mL of water was added dropwise with an addition funnel. The ice bath was removed, and the resulting suspension was stirred at room temperature for 16 h. The mixture was filtered, and the collected solids were washed with methanol. The filtrate was concentrated to dryness under reduced pressure. The residue was taken up in MeOH and filtered. The collected solids were washed with hot MeOH. The filtrate was again concentrated under reduced pressure. The residue was taken up in water, stirred at room temperature for 30 min, and cooled in a refrigerator overnight. The off-white solid was collected by filtration to provide 1.66 g of compound 3 in a 74% yield. mp = 204–206 °C; For NMR spectra, major isomer data is reported: 1H NMR (CD3OD) δ 8.48 (d, 1H, J = 5.04 Hz), 8.09 (br s, 2H), 7.99 (br s, 1H), 7. 54 (dd, 1H, J = 5.03, 1.37 Hz); 13C NMR (CD3OD) δ 152.84, 149.11, 147.91, 146.09, 142.31, 120.67, 117.51.
Intermediate salt 4
Pyridine-2,4-dialdoxime 3 (1.20 g, 6.96 mmol) was suspended in 39 mL of THF and under a nitrogen atmosphere. Bis-chloromethyl ether (2.84 g, 24.7 mmol) was added dropwise with an addition funnel. The suspension was heated to 47 °C internal temperature and was stirred for 4 days. The solids were collected by filtration and washed with THF then diethyl ether, which provided 1.06 g of a yellow solid in a 52% yield. 1H NMR (CD3OD) δ 9.00 (d, 1H, J = 6.87 Hz), 8.64–8.63 (m, 2H), 8.34 (s, 1H), 8.19 (dd, 1H, J = 6.04, 2.06 Hz), 6.26 (s, 2H0, 5.73 (s, 2H). The compound was not stable enough for 13C NMR analysis in CD3OD.
HLö-7 Diiodide
Intermediate salt 4 (0.80 g, 2.87 mmol) was suspended in 57 mL of acetone under a nitrogen atmosphere. Isonicotinamide (0.35 g, 2.87 mmol) and sodium iodide (1.72 g, 11.48 mmol) were sequentially added. The mixture was stirred at room temperature for 16 h. The solid material was filtered and washed with acetone then diethyl ether. The collected solid was recrystallized from water to provide 0.91 g of a brown solid in a 54% yield. 1H NMR (D2O) δ 9.13 (d, 2H, J = 6.41 Hz), 8.91 (d, 1H, J = 6.87 Hz), 8.57 (s, 1H), 8.50 (d, 1H, J = 1.83 Hz), 8.38 (d, 2H, J = 6.87 Hz), 8.29 (s, 1H), 8.14 (dd, 1H, J = 6.41, 1.83 Hz), 6.28 (s, 2H), 6.21 (s, 2H); 13C NMR (D2O) δ 166.37, 151.10, 150.90, 147.33, 146.00, 145.38, 144.61, 141.88, 126.81, 124.39, 123.96, 86.94, 85.28.
HLö-7 DMS
Dowex monosphere 550A(OH) resin (13 g, 25–35 mesh) was suspended in 140 mL of 1 M methanesulfonic acid and was stirred for 1 h. The mixture was added to a column, and the liquid was eluted. The resin was washed with deionized water until it measured pH = 7. HLö7 diiodide (0.88 g, 1.50 mmol) in 15 mL of deionized water was added and eluted with deionized water at a rate of 1 drop/s. The yellow eluent was collected and concentrated under reduced pressure. The liquid residue was treated with warm MeOH and refrigerated overnight. The mixture was filtered, and the collected solid was recrystallized from water/MeOH to provide 0.71 g of a tan solid in an 89% yield. mp 194–196 °C (dec). 1H NMR (D2O) δ 9.08 (d, 2H, J = 7.33 Hz), 8.84 (d, 1H, J = 6.87 Hz), 8.53 (s, 1H), 8.47 (d, 1H, J = 1.83 Hz), 8.34 (d, 2H, J = 6.87 Hz), 8.25 (s, 1H), 8.10 (dd, 1H, J = 6.64, 2.06 Hz), 6.25 (s, 2H), 6.16 (s, 2H), 2.65 (s, 6H); 13C NMR (D2O) δ 166.33, 151.09, 150.91, 147.37, 145.94, 145.30, 144.56, 141.80, 126.74, 124.25, 123.79, 86.92, 85.22, 38.44. Elemental Analysis calculated for C17H23N5O10S2: C, 39.15; H, 4.45; N, 13.43; S, 12.30. Found: C, 39.24; H, 4.54; N, 13.50; S, 12.66. HRMS (ESI-TOF) m/z calculated for C15H17N5O4 [M]2+ 165.5640, found 165.5638.
Acknowledgments
X-ray data were collected at Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline at the Advanced Photon Source, Argonne National Laboratory. Supporting institutions may be found at https://www.ser.aps.anl.gov/www.ser-cat.org/images/SER-CAT18_Member_list.jpg. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38. For the ex vivo mouse diaphragm assay, the experimental protocol was approved by the Animal Care and Use Committee at the United States Army Medical Research Institute of Chemical Defense, and all procedures were conducted in accordance with the principles stated in the Guide for the Care and Use of Laboratory Animals (National Research Council, 2011) and the Animal Welfare Act of 1966 (P.L. 89-544), as amended. The authors thank Mr. Kenneth Sumpter for running the elemental analysis.
Glossary
ABBREVIATIONS
- PAS
peripheral anionic site
- AChE
acetylcholinesterase
- hAChE
human AChE
- mAChE
mouse AChE
- HLö-7
[(Z)-[1-[(4-carbamoylpyridin-1-ium-1-yl)methoxymethyl]-2-[(Z)-hydroxyiminomethyl]pyridin-4-ylidene]methyl]-oxoazanium
- GD
O-pinacolyl methylphosphonofluoridate
- GA
ethyl dimethylphosphoramidocyanidate
- GB
O-isopropyl methylphosphonofluoridate
- GF
fluoro-methyl-phosphoryloxycyclohexane
- VX
ethyl N-2-diisopropylaminoethyl methylphosphonothiolate
- DDFP
N,N′-diisopropyldiamidofluorophosphate
- 2-PAM
[(E)-(1-methylpyridin-2-ylidene)methyl]-oxoazanium
- OP
organophosphate
- TMB
4,oxo-[[1-[3-[4-(oxoazaniumylmethylidene)pyridin-1-yl]propyl]pyridin-4-ylidene]methyl]azanium
- DIBAL
diisobutylaluminumhydride
- DCM
dichloromethane
- BCME
bischloromethyl ether
- BMSME
bismethanesulfonatemethyl
- PEG
polyethylene glycol
- DMS
dimethanesulfonate
- MPNH
mouse phrenic nerve hemidiaphragm
- SIM
single ion mode
- MDL
method detection limit
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00021.
X-ray figures with experimental details, structural data, PDB information; MPNH assay figures with experimental details; HLö-7 DMS NMR spectra (PDF)
Author Contributions
All authors contributed to this manuscript.
This work was funded by the Defense Threat Reduction Agency projects: CB#3889 “Elucidation of the mechanisms and physical properties of the molecular targets of chemical nerve agents” (to J.J.H. and S.D.P.), CB#3943 “Improved Nerve Agent Treatment System (INATS)” (to D.R.A.), and CB#10243 “Medicinal Chemistry Synthesis” (to F.L.H. and A.J.W.).
The authors declare no competing financial interest.
Supplementary Material
References
- Kingsley P.; Barnard A.. Banned Nerve Agent Sarin Used in Syria Chemical Attack, Turkey Says. In The New York Times, 2017. [Google Scholar]
- Paddock R. C.; Sang-Hun C.; Wade N.. In Kim Jong-nam’s Death, North Korea Lets Loose a Weapon of Mass Destruction. In The New York Times, 2017. [Google Scholar]
- Booth W.British lab confirms Novichok was used in spy attack but can’t determine source. In The Washington Post, 2018. [Google Scholar]
- Coulter P. B.; Callahan J. J.; Link R. S.. Physical constants of thirteen V agents (U). U.S. Army Chemical Warfare Laboratories Technical Report 1959, CWLR 2346. [Google Scholar]
- Worek F.; Thiermann H.; Szinicz L.; Eyer P. Reactivation of organophosphate-inhibited human acetylcholinesterase by isonitrosoacetone (MINA): a kinetic analysis. Chem.-Biol. Interact. 2011, 194 (2–3), 91–96. 10.1016/j.cbi.2011.09.001. [DOI] [PubMed] [Google Scholar]
- Buckley N. A.; Roberts D.; Eddleston M. Overcoming apathy in research on organophosphate poisoning. BMJ. 2004, 329 (7476), 1231–1233. 10.1136/bmj.329.7476.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korabecny J.; Soukup O.; Dolezal R.; Spilovska K.; Nepovimova E.; Andrs M.; Nguyen T. D.; Jun D.; Musilek K.; Kucerova-Chlupacova M.; Kuca K. From Pyridinium-based to Centrally Active Acetylcholinesterase Reactivators. Mini-Rev. Med. Chem. 2014, 14, 215–221. 10.2174/1389557514666140219103138. [DOI] [PubMed] [Google Scholar]
- Worek F.; Thiermann H.; Szinicz L.; Eyer P. Kinetic analysis of interactions between human acetylcholinesterase, structurally different organophosphorus compounds and oximes. Biochem. Pharmacol. 2004, 68 (11), 2237–2248. 10.1016/j.bcp.2004.07.038. [DOI] [PubMed] [Google Scholar]
- Ekstrom F. J.; Astot C.; Pang Y. P. Novel nerve-agent antidote design based on crystallographic and mass spectrometric analyses of tabun-conjugated acetylcholinesterase in complex with antidotes. Clin. Pharmacol. Ther. 2007, 82, 282–293. 10.1038/sj.clpt.6100151. [DOI] [PubMed] [Google Scholar]
- Löffler M.Quartäre Salze von Pyridin-2, 4-dialdoxim als Gegenmittel für Organophosphat Vergiftungen. University of Freiburg: Freiburg (Breisgau), 1986. [Google Scholar]
- Eyer P.; Ladstetter B.; Schafer W.; Sonnenbichler J. Studies on the stability and decomposition of the Hagedorn-oxime HLo 7 in aqueous solution. Arch. Toxicol. 1989, 63, 59–67. 10.1007/BF00334636. [DOI] [PubMed] [Google Scholar]
- Eyer P.; Hagedorn I.; Klimmek R.; Lippstreu P.; Loffler M.; Oldiges H.; Spohrer U.; Steidl I.; Szinicz L.; Worek F. HLo 7 dimethanesulfonate, a potent bispyridinium-dioxime against anticholinesterases. Arch. Toxicol. 1992, 66, 603–621. 10.1007/BF01981499. [DOI] [PubMed] [Google Scholar]
- Queguiner G.Study of the reduction of ethyl pyridinedicarboxylates. In Synthesis in the Pyridine Series; Bull. Soc. Chim. Fr., 1969; pp 3678–3683. [Google Scholar]
- Buc S. R.Bischloromethyl ether. In Organic Syntheses Collections; Organic Syntheses, Inc.: New York, 1956; p 1. [Google Scholar]
- Kuca K.; Stodulka P.; Hrabinova M.; Jun D.; Dolezal B. Convenient preparation of the oxime HI-6 (dichloride and DMS) – antidote against nerve agents. Def. Sci. J. 2008, 58, 399–404. 10.14429/dsj.58.1660. [DOI] [Google Scholar]
- Zajdela F.; Croisy A.; Barbin A.; Malaveille C.; Tomatis L.; Bartsch H. Carcinogenicity of chloroethylene oxide, an ultimate reactive metabolite of vinyl chloride, and bis(chloromethyl)ether after subcutaneous administration and in initiation-promotion experiments in mice. Cancer Res. 1980, 40, 352–356. [PubMed] [Google Scholar]
- Cheung J.; Rudolph M. J.; Burshteyn F.; Cassidy M. S.; Gary E. N.; Love J.; Franklin M. C.; Height J. J. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J. Med. Chem. 2012, 55, 10282–10286. 10.1021/jm300871x. [DOI] [PubMed] [Google Scholar]
- Ekstrom F.; Pang Y. P.; Boman M.; Artursson E.; Akfur C.; Borjegren S. Crystal structures of acetylcholinesterase in complex with HI-6, Ortho-7 and obidoxime: structural basis for differences in the ability to reactivate tabun conjugates. Biochem. Pharmacol. 2006, 72 (5), 597–607. 10.1016/j.bcp.2006.05.027. [DOI] [PubMed] [Google Scholar]
- Ekstrom F.; Hornberg A.; Artursson E.; Hammarstrom L. G.; Schneider G.; Pang Y. P. Structure of HI-6*sarin-acetylcholinesterase determined by X-ray crystallography and molecular dynamics simulation: reactivator mechanism and design. PLoS One 2009, 4 (6), e5957 10.1371/journal.pone.0005957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bester S. M.; Guelta M. A.; Cheung J.; Winemiller M. D.; Bae S. Y.; Myslinski J.; Pegan S. D.; Height J. J. Structural Insights of Stereospecific Inhibition of Human Acetylcholinesterase by VX and Subsequent Reactivation by HI 6. Chem. Res. Toxicol. 2018, 31 (12), 1405–1417. 10.1021/acs.chemrestox.8b00294. [DOI] [PubMed] [Google Scholar]
- Tattersall J. E. Ion channel blockade by oximes and recovery of diaphragm muscle from soman poisoning in vitro. Br. J. Pharmacol. 1993, 108, 1006–1015. 10.1111/j.1476-5381.1993.tb13498.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiermann H.; Eyer P.; Worek F. Muscle force and acetylcholinesterase activity in mouse hemidiaphragms exposed to paraoxon and treated by oximes in vitro. Toxicology 2010, 272, 46–51. 10.1016/j.tox.2010.04.002. [DOI] [PubMed] [Google Scholar]
- Thiermann H.; Eyer P.; Worek F.; Szinicz L. Effects of oximes on muscle force and acetycholinesterase activity in isolated mouse hemidiaphragms exposed to paraoxon. Toxicology 2005, 214, 190–197. 10.1016/j.tox.2005.06.013. [DOI] [PubMed] [Google Scholar]
- Alberts P. A new H-oxime restores rat diaphragm contractility after esterase inhibition in vitro. Eur. J. Pharmacol. 1990, 184 (1), 191–194. 10.1016/0014-2999(90)90682-V. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.