

Important Compound Classes
Title
Compounds for Tau Protein Degradation
Patent Application Number
WO 2019/014429 A1
Publication Date
January 17, 2019
Priority Application
US 62/531,773
Priority Date
July 12, 2017
Inventors
Gray, N. S.; Haggarty, S. J.; Cai, Q.; Telo Baptista Lima Da Silva, M. C.; Zhang, T.; Ferguson, F. M.
Assignee Company
Dana-Farber Cancer Institute Inc. and The General Hospital Corporation D/B/A Massachusetts General Hospital
Disease Area
Neurological disorders
Biological Target
Tau protein
Summary
Alzheimer’s disease (AD) is characterized by progressive deficits in cognitive, memory, and other mental functions. There are nearly 35 million people that suffer from AD worldwide, which is one of the main causes of mental disability and death among seniors. The exact pathogenesis of AD has remained elusive; however, several hypotheses have been proposed, including pathogenic tau protein, β-amyloid (Aβ) cascade, chronic inflammation, oxidative stress, acetylcholine abnormalities, and so forth. AD studies in the past decades using mouse models and human brain samples have revealed numerous signaling cascades that lead to pathological changes in structure, function, and synapse number. The two main characteristic protein deposits associated with AD are the senile plagues and the neurofibrillary tangles (NFT). The senile plaques involve extracellular accumulation of Aβ in the brain of AD patients, which is the primary driver to the disease progression. The formation of NFT, which contain hyperphosphorylated tau proteins, is the result of an imbalance between the accumulation and clearance of the Aβ. The tau proteins are abundant in neurons and are expressed at very low levels in the central nervous system, and accumulation occurs prior to extensive neuronal damage. Consequently, tau deposits have become a target for determining the progression and treatment of AD. Furthermore, the tau hypothesis states that an excessive or abnormal phosphorylation of tau disassembles microtubules and sequesters normal tau, microtubule associated protein1 (MAP1), MAP2, and ubiquitin into tangles of paired helical filament. Aβ peptides are generated from amyloid precursor protein (APP) and consist of short peptides that are 39–43 amino acids.
The understanding of synaptic failure in AD comes from studies of AD mouse models that overexpress specific AD risk genes, including APP, PSI, PSI, tau, etc. However, none of these approaches have translated into successful clinical applications with disease-modifying outcomes. An early pathological marker in AD patients and animal models shows evidence that suggests intracellular accumulation of Aβ. Electron microscopy studies indicate prior to Aβ plaque formation intracellular Aβ deposition at the axon terminals, mitochondria, lysosomes, and dendrites, which may affect the synaptic plasticity. In addition, intracellular Aβ causes potential long-term memory deficits. These pathological changes have been shown to be caused by also extracellular Aβ. Intracellular Aβ may appear prior to extracellular plaques formation, and a intraneuronal Aβ level decrease does result in extracellular plaque accumulation. The possibility exists that the toxicity and pathological effects caused by extracellular plaques would have resulted from the long-term effect of the intracellular Aβ accumulation.
Several signaling pathways have been shown to participate in the AD process, and their cross functional activities are complicated. Consequently, there exists an urgent need for the understanding of the net effect of signal pathways for AD therapy. A recent therapeutic strategy to reduce or eliminate proteins associated with pathological states such as PROTAC (Proteolysis targeting chimeras) was developed, which involves bifunctional compounds that recruit target proteins to E3 ubiquitin ligases and subsequently induce proteasome-mediated degradation of the target protein. The E3 ubiquitin ligases are proteins that, in combination with an E2 ubiquitin-conjugating enzyme, promote the attachment of ubiquitin to a lysine on a target protein. The ubiquitination of the protein results in degradation of the target protein by the proteasome. This Patent Highlight describes the conjugation of tau binding moieties with an E3 ubiquitin ligase binding moiety such as lenalidomide or thalidomide, which provide compounds that can induce the ubiquitination of tau protein and promote its degradation in cells. Since aggregation of tau protein, in particular hyperphosphorylated tau protein, causes tauopathies in AD, targeting tau protein or any post-translationally modified form and recruiting an E3 ubiquitin ligase such as Cereblon to ubiquitinate the tau protein would mark it for degradation. Consequently, a single bifunctional molecule can promote the degradation of tau protein and would provide new compounds for the treatment of neurological disease such as AD.
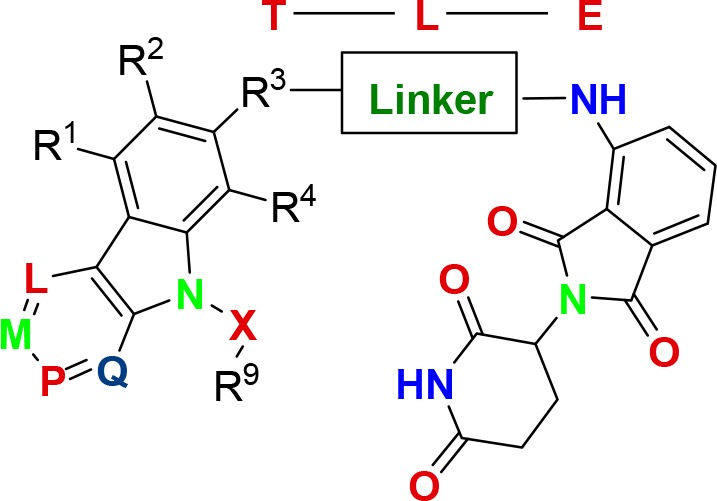
Compounds of this Patent Highlight are pharmaceutical hydrates, solvates, cocrystals, polymorphs, and so forth, where T represents a tau protein binding moiety, E represents E3 ubiquitin ligase binding moiety, and L represents a linker, which is a substituted or unsubstituted alkylene, substituted or unsubstituted alkenylene, arylene, heterocyclylene, heteroalkylene, etc.
Definitions
R1, R2, and R3–R8 are independently H, OH, halogen, NH2, CH3, SO2, NO2, heteroaryl, etc.
R9 = H, −N3, alkynyl, OH, halogen, NH2, N(C1–6 alkyl)2, aryl, heteroaryl, SO2, NH2, etc.
R3 = −(CH2)n–O–, −A–(CH2)n–O–, – (CH2)n–S(O)2NRA, etc.
A = substituted or unsubstituted heterocyclylene, heteroarylene, etc.
n = 0–12;
L = M = P = Q = nitrogen.
Key Structures

Biological Assay
Tau degradation assays demonstrated the ability of the compounds in this Patent Highlight to degrade tau proteins in human cells. Furthermore, compounds were evaluated in an in vivo assay to determine whether they were capable of crossing the blood–brain barrier (BBB).
Biological Data
Most of the compounds in this Patent Highlight degraded
hyperphoshorylated tau and total tau proteins in human tau-A152T neurons
and tau-P301L neurons after treatment of the neurons with requisite
compounds for 24 h. Compound 3 was administered to mice
in a pharmacokinetic study of plasma and brain tissue, and the in vivo results are shown in the following Table.
Recent Review Articles
-
1.
Ma K.-G.; Qian Y.-H.. Neuropeptides. 2019, 73, 96.
-
2.
Zou Y.; Ma D.; Wang Y.; Zou Y.. Cell Biochem. Funct. 2019, 37, 21.
-
3.
Wurz R. P.; Cee V. J.. J. Med. Chem. 2019, 62, 445.
-
4.
Churcher I.J. Med. Chem. 2018, 61, 444.
-
5.
Ottis P.; Crews C. M.. ACS Chem. Biol. 2017, 12, 892.
The author declares no competing financial interest.