

Abstract
Endoplasmic reticulum aminopeptidase 1 (ERAP1) is an intracellular enzyme that helps generate peptides presented by Major Histocompatibility Complex Class I (MHC class I) molecules and is an emerging target for immunotherapy applications. Despite almost two decades of research on ERAP1, lack of high-resolution crystal structures has hampered drug-development efforts. By optimizing the protein construct, we obtained a high-resolution (1.60 Å) crystal structure of the closed-conformation of ERAP1 with a potent phosphinic pseudopeptide inhibitor bound in its active site. The structure provides key insight on the mechanism of inhibition as well as selectivity toward homologous enzymes and allows detailed mapping of the internal cavity of the enzyme that accommodates peptide-substrates. Bis-tris propane and malic acid molecules, found bound in pockets in the internal cavity, reveal potential druggable secondary binding sites. The ability to obtain high-resolution crystal structures of ERAP1 removes a major bottleneck in the development of compounds that regulate its activity and will greatly accelerate drug-discovery efforts.
Keywords: Aminopeptidase, enzyme, inhibitor, structure, X-ray, mechanism, antigen, peptide, immune system
Endoplasmic reticulum aminopeptidase 1 (ERAP1) is an intracellular enzyme that helps generate antigenic peptides that are presented to the immune system by Major Histocompatibility Complex Class I molecules (MHC-I).1 It affects qualitative and quantitative aspects of the cellular immunopeptidome and influences cytotoxic responses and has thus been termed a “quintessential editor” of antigenic peptides.2−5 ERAP1 is polymorphic and several coding missense single nucleotide polymorphisms in its gene have been associated with predisposition to human diseases, most notably human leukocyte antigen (HLA)-associated inflammatory autoimmunity but extending to viral infections and cancer.6−8 The importance of ERAP1 in regulating immune responses has attracted interest in drug-discovery, aiming to modulate its activity for applications in cancer immunotherapy or the control of autoimmunity.9 First generation active-site inhibitors for ERAP1 have been reported and shown to have activity in regulating immune response in model systems opening opportunities for clinical applications.10,11
Previous crystallographic analysis of ERAP1 has been limited to medium-to-low resolution structures (2.7–3.0 Å) that however have provided much insight on function.12 It has been crystallized in two distinct conformations, an open and a closed one, and it has been hypothesized that cycling between these two conformations is necessary during catalytic turnover.13−15 Four distinct domains (I–IV) have been identified, with domain II being the catalytic one, domain IV shielding the catalytic site upon its closure, domain III acting as a hinge that allows for the open-close-open transition, and domain I stabilizing the closed conformation through its interactions with both domains II and IV. Despite the growing interest in the development of small MW compounds that regulate ERAP1 activity as part of drug-discovery efforts, little structural information exists on complexes of ERAP1 with known inhibitors. The first two structures of ERAP1 contain bestatin, a broad-range aminopeptidase inhibitor that is a very weak inhibitor of ERAP1, in the active site, determined however with relatively weak electron density.14,15 No other ERAP1-inhibitor structures have been reported during the last 7 years, highlighting crystallographic analysis as an important bottleneck on the preclinical development of compounds that modulate ERAP1 activity. In this study, we present the first high-resolution crystal structure of ERAP1 determined at 1.60 Å. ERAP1 was cocrystallized with a potent, nanomolar affinity, phosphinic pseudotripeptidic inhibitor (IC50 of 86 nM, Figure S1, compound 14 in ref (16), henceforth named DG046) that was found bound in the active site and stabilized by an unusual extensive network of π-stacking interactions. This high-resolution structure allows detailed mapping of protein–inhibitor interactions that can guide optimization efforts as well as mapping of the internal peptide binding site of ERAP1. Two buffer components, bis-tris propane and malic acid molecules, were found bound in pockets of the internal cavity that might constitute opportunities for modulating peptide selectivity.
ERAP1 was found in the closed conformation, adopting an overall architecture considerably similar to the previously solved structure of ERAP1 bound with bestatin15 and to a lesser extent similar to the closed structures of ERAP2 and IRAP (50% sequence identity), the other members of the oxytocinase subfamily of M1 aminopeptidases, when cocrystallized with other phosphinic pseudotripeptides.10,17 However, the present structure being in remarkably high resolution for a protein molecule of that size, allows an unprecedentedly detailed structural analysis of an enzyme of the family. This high resolution was achieved by using an optimized ERAP1 construct that is described in detail in the Supporting Information. This construct features a removable C-terminal purification tag and has a 28-amino acid sequence (amino acids 486−513), encoded by exon 10 and part of exon 11 of the ERAP1 gene, substituted by a short loop consisting of a GSG sequence. While this short sequence has been reported to be important for interactions with an ER chaperone ERp44 via mixed disulfide formation,18 it was not visible in previous ERAP1 structures and presumed to be unstructured.14,15 Specific activity of this ERAP1 construct was measured to be 0.34 ± 0.02 mol/s mol for the L-AMC assay, which is similar and possibly slightly higher than reported value for previous ERAP1 constructs, suggesting that the substitution did not have any deleterious effects on ERAP1 activity.19,20
DG046 is coordinated to the zinc ion via its phosphinic group with a geometry that imitates a transition state analogue (Figure 1). The phosphinic group preserves its active site position compared to the previously described phosphinic pseudotripeptide ligands bound on ERAP2 or IRAP10,17 and the amine group forms hydrogen bonds with the carboxyl oxygens of Glu183, Glu320, and Glu376. However, the homophenylalanine side chain (A), which is buried deep into the S1 cavity, adopts a different orientation forming a T-shaped π–π interaction with Phe433 from domain II. The propargyl group which points toward the S1′ site adopts a spatial conformation that optimizes its π–π interactions with the aromatic cloud of the zinc coordinating histidine His353, while the main chain carbonyl group forms a hydrogen bond with the glycine amide of the juxtaposed GAMEN motif (Figure 1). Despite the clear ligand binding in the present study, compared to the previously reported closed structure of ERAP1,15 the enzyme does not present ligand-induced rearrangements, unlike in the homologous IRAP.17 The phenyl moiety (B) of the phenylalanine residue of DG046 interacts via T-shaped aromatic interactions both with Tyr438 of ERAP1 and intramolecularly with the phenyl group (A) of the homophenylalanine. The later interaction is probably causing the altered conformation of homophenylalanine when bound into the S1 cavity. It appears that the lack of an aromatic residue at S2′ of ERAP1, contrary to ERAP2 and IRAP (Ser869 in ERAP1 vs Tyr892 in ERAP2 and Tyr961 in IRAP), releases the P2′ site of the ligand from being stacked in a parallel mode with any aromatic group from domain IV, adopting a totally different rotamer (Figure 2). This altered conformation results in a more distant placement of its C-terminal carbonyl group, which in DG046 is located by ∼3 Å farther from the metal ion, compared to the corresponding group of DG013A when bound on ERAP2. This network of π–π interactions greatly limits the conformational freedom of the ligand and appears optimized for the active site of ERAP1 (see Figure S2 for a depiction of the B-factors of the atoms of the inhibitor). This probably contributes to the potency of this inhibitor, which is comparable to the larger compound DG013A.16
Figure 1.
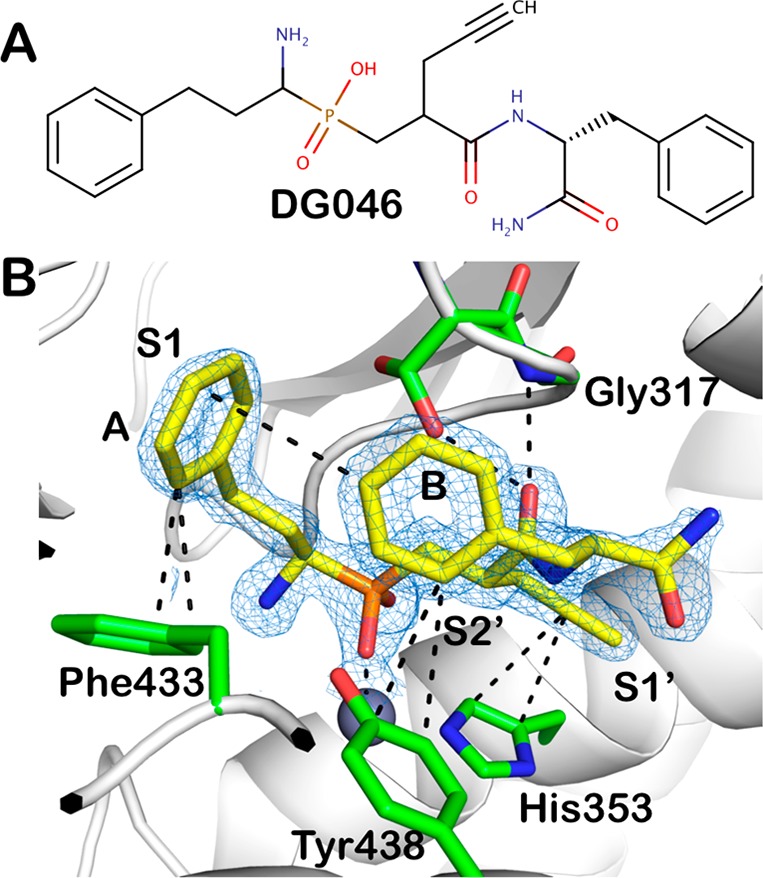
(A) Chemical structure of the phosphinic pseudopeptide DG046. (B) DG046 bound in the ERAP1 active site. The inhibitor is shown in yellow sticks and the main residues in ERAP1 that make interactions are shown in green. Oxygen atoms are in red, nitrogen in blue, phosphorus in orange. The active site zinc(II) is shown as a gray sphere. 2Fo-Fc electron density map is shown in blue mesh (contour level 2.0 sigma). The approximate location of the active site specificity pockets S1, S1′, and S2′ are indicated.
Figure 2.
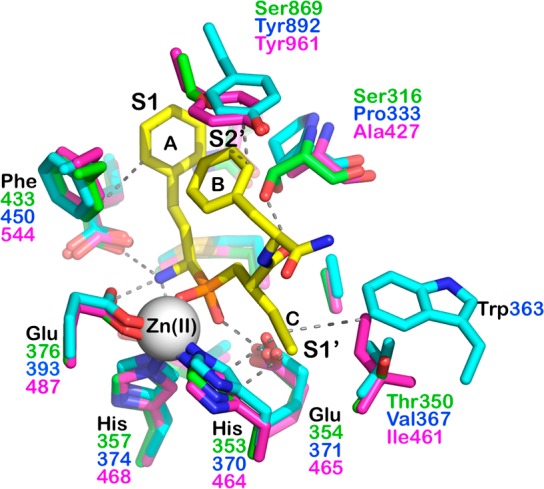
Superposition of active-site residues of ERAP1 (in green) to homologous ERAP2 (PDB code 5AB0, in cyan) and IRAP (PDB code 5MJ6, in magenta). Catalytic zinc(II) atom is shown as a gray sphere. Numbering of each residue is indicated in different colors depending on the enzyme. Phosphinic inhibitor carbon atoms are shown in yellow, oxygen atoms are in red, nitrogen in blue, phosphorus in orange. Only residues that come within 4 Å of the inhibitor are shown. The active site zinc(II) is shown as a gray sphere. The approximate location of the active site specificity pockets S1, S1′, and S2′ are indicated.
To address selectivity of the inhibitor for the three enzymes of the oxytocinase subfamily, we superimposed the structures of ERAP2 and IRAP on the structure of ERAP1 and examined the locations of active-site residues that could impact inhibitor binding (Figure 2). DG046 has been characterized to be a potent (nanomolar) inhibitor of all three enzymes, with similar potency for ERAP1 and ERAP2 and a greater potency for IRAP (43 ± 4 nM for ERAP1, 37 ± 4 nM for ERAP2, and 2 ± 1 nM for IRAP).16 In ERAP1, the inhibitor’s phenyl group B comes in close proximity with Ser869. In both ERAP2 and IRAP, the equivalent position has a Tyr residue (892 and 961, respectively), which could provide favorable stacking interactions. Given the network of π-stacking interactions between Phe433, phenyl A, and phenyl B, such interactions can be highly stabilizing in ERAP2 and IRAP. However, both ERAP2 and IRAP lack the favorable H-bonding interaction of Ser316, present in ERAP1, which may in part ameliorate the increase in potency due to Tyr892 and Tyr961. ERAP2 furthermore has a bulky Trp373 that is proximal to the C-terminal end of the inhibitor. The interplay between additional positive interactions and negative ones may explain the similar levels of potency for ERAP2. IRAP however has a hydrophobic Ile461 that could make van der Waals interactions with the propargyl group in the inhibitor, helping in properly orienting it to stack with His357, leading to increased potency. Regardless of small differences in potency, however, most of the inhibitor–protein interactions are with residues that are conserved in all of the three enzyme’s active sites, leading to the limited selectivity of this compound.
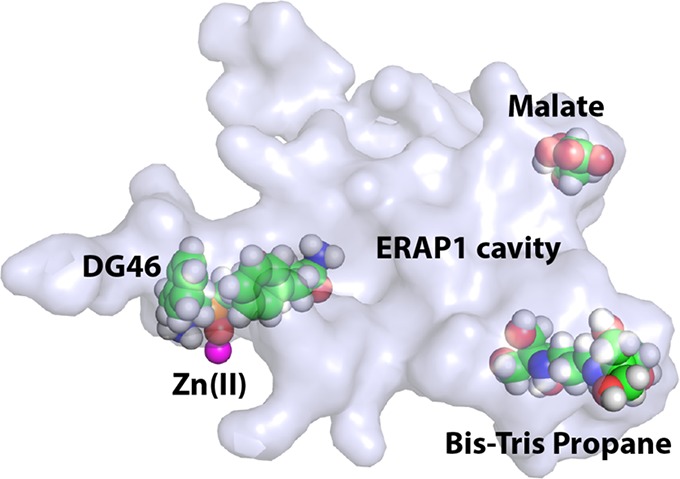
Additional electron density within the internal cavity of the enzyme but away from the catalytic site was interpreted to belong to other ligands present during the crystallization process (Figures S3 and S4). Based on the constituents of the crystallization conditions and the high quality of the electron density maps we identified bis-tris propane and malic acid bound on distinct clefts inside the enzyme. The average B-factors of the bis-tris propane and malic acid atoms were 23.2 Å2 and 36.2 Å2, respectively, which are similar to the values of the main chain atoms of their immediate protein environment. Figure 3 illustrates how bis-tris propane is wedged between domains II and IV. Trp921 of the C-terminal domain α-helix shields the side walls of this novel binding cleft, which we term “side pocket” and interacts extensively via van der Waals interactions with the hydroxymethyl groups of bis-tris propane. The buffer molecule is being stabilized further by a number of H-bonds with residues Asp406, Glu409, Asn641, and Gln675 from the three neighboring α-helices (two from domain IV and one from domain II) and also via an H-bond with the domain III lysine 551, which protrudes toward the side pocket. The side pocket is also present in the previously reported structure of the closed ERAP1, albeit empty from any ligand. Herein, the high resolution of the structure and the presence of a bound ligand allowed us to trace the side chains of residues that previously were not detected. Comparing the current ERAP1 structure with the previous “closed” ERAP1 (PDB ID 2YD0) shows that the binding of bis-tris propane to the side pocket is not accompanied by conformational changes in any of the stabilizing α-helices, indicating probably that bis-tris propane is uninvolved in the closure of domain IV. However, as the side pocket is located between the functionally important domains II and IV and close to the hinge domain III, its possible role as an allosterically modulating site cannot be excluded. This pocket is framed by four α-helices and is widely accessible for ligands from the direction of the catalytic site. The total volume of the side pocket, which apart from the space occupied by bis-tris propane extends beyond toward the limiting α-helices of domains II and IV, was calculated at 2216 Å3.
Figure 3.
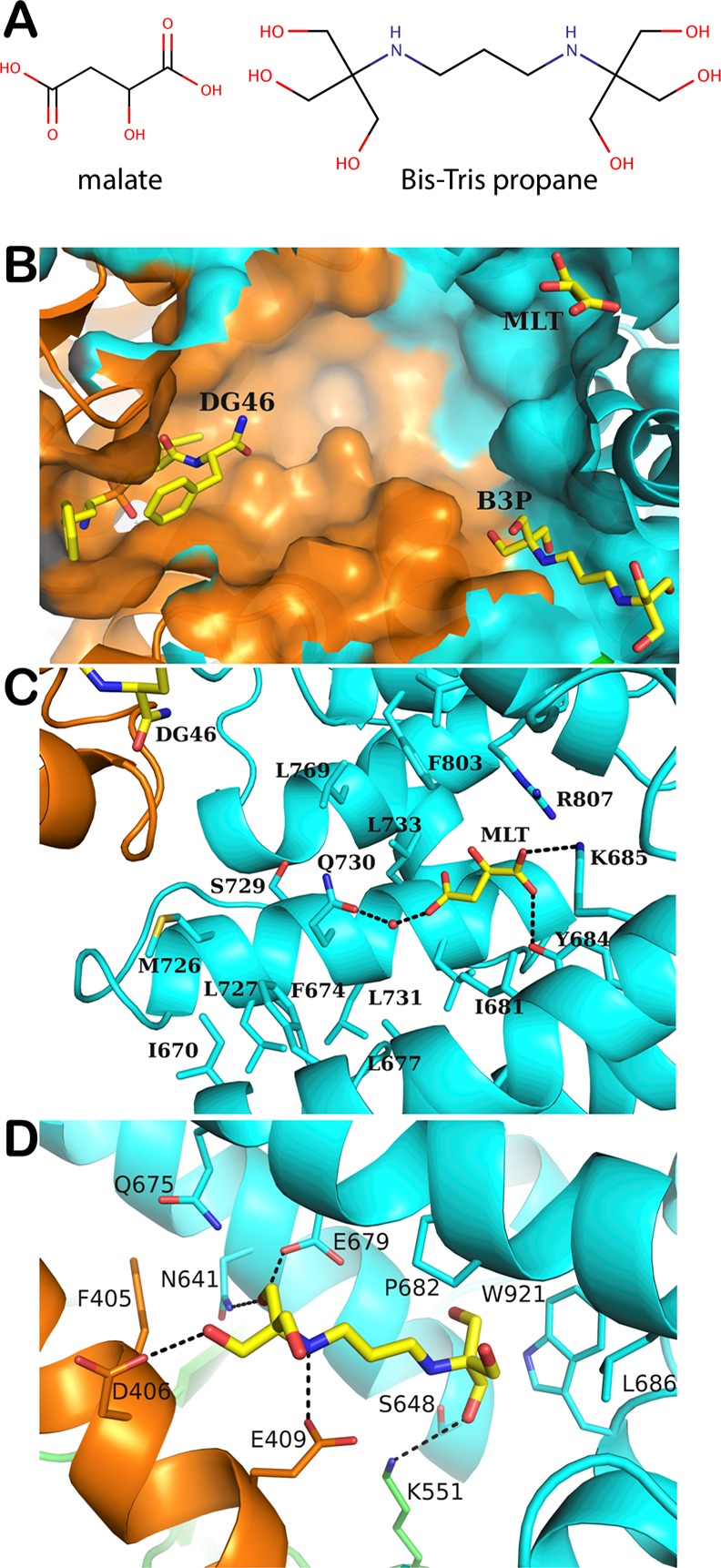
(A) Chemical structures of malic acid and bis-tris propane. (B) Bird’s eye view of the internal cavity of ERAP1 showing the binding of the inhibitor DG046, malic acid (MLT), and bis-tris propane (B3P) molecules. (C) Interactions between MLT and ERAP1 residues. (D) Interactions between B3P and ERAP1 residues. Domain II of ERAP1 is in orange and domain IV is in cyan. Carbon atoms are shown in yellow, oxygen atoms are in red, nitrogen in blue, phosphorus in orange.
Malic acid was found bound close to the nozzle of the domain IV channel sealing the internal cavity from the outer environment (Figure 3). Its binding occurs via strong H-bonds of one carboxyl group with the side chains of the domain IV residues Lys685 and Tyr684 as well as with water-mediated H-bonds of its other carboxyl group with Gln730. This binding pocket is occupied on the one side by polar or charged residues (mostly basic), which adopt an extended conformation and converge toward the spatial position of one of the malic acid carboxylates. Gln730, which stabilizes the second carboxylate of malic acid via the water-mediated interaction, is one of ERAP1’s polymorphic residues (Q730E) that has been consistently shown to associate with disease predisposition and antigen presentation.20 Our crystal structure shows that Gln730 lies deep in the internal cavity of ERAP1 and conceptually can interact with residues of a bound peptide. Thus, the other common variant for this position, glutamic acid, would alter such interactions.
This local difference of the surface electrostatic potential of the internal cavity could contribute to changes in substrate specificity and/or to the conformational dynamics of the enzyme, especially since this residue lies among extended hydrophobic patches (Figure 3C). Recently, the crystal structure of a single chain bimodular protein comprising the C-terminal domain of ERAP1 (residues 529–944) fused with part of a well-known ovalbumin-derived epitope (IINFEKL) was published, showing strong interactions of the IINFEKL peptide with the same pocket of domain IV of a symmetry related ERAP1 molecule.21 Interestingly, malic acid in the present structure adopts a binding pose that partially overlaps with the carboxyl group of the IINFEKL peptide and forms a similar pattern of interactions. Enzymatic studies have shown ERAP1’s allosteric activation for the hydrolysis of both peptides and small pseudosubstrates, by small peptides such as IINFEKL, and is believed that part of the elaborate activation mechanism comprises binding of the C-terminus of the modulating peptides at the site where malic acid was bound.14,21 Although malic acid was part of the crystallization condition and is not a known binder or regulator of ERAP1’s enzymatic activity, its spatial coincidence with the C-terminus carboxyl group of the IINFEKL peptide indicates a noticeable affinity of that pocket for carboxylate-containing species. Intriguingly, in the previously reported closed structure of ERAP1 this region is occupied by strong electron density that was not attributed to any compound.15 Our attempt to fit the cacodylate ion, which was a constituent of the crystallization buffer, was successful in eliminating the residual electron density from the particular site and improving the refinement statistics (Figure S5).
Examination of the internal cavity of the enzyme provides insight on function and substrate recognition. Compared to the open conformation of ERAP1, in the closed conformation, domain IV moves toward domain II forming a large internal cavity of ∼10 974 Å3 that fully occludes the catalytic site from the external environment (Figure 4). The volume and dimensions of the internal cavity are sufficient for the accommodation of peptides even as long as 16-residues long, a hallmark of ERAP1 catalytic properties.22 Using the 3 V Web server23 and an oxygen atom as probe, three narrow channels connecting the internal cavity with the outer environment were detected. Two are framed by domains I, II, and IV, while the third one threads among α-helices of domain IV (H14, H15, H16, and H20). Notably, one of the former channels is located above the S1 pocket and in our crystal structure is occupied by several water molecules and one ethylene glycol, which was a component of the cryoprotecting solution (Figure 4). The position of this channel is such that could facilitate postcleavage amino acid release to allow processive peptide trimming. Although its width in the crystal structure is small and is thus unlikely, it can accommodate larger amino acids, its size may be larger when the enzyme is in solution, and crystal packing restraints are not present.24
Figure 4.
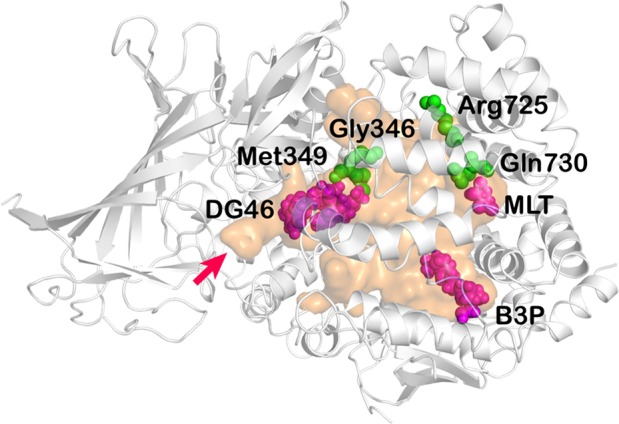
Substrate cavity of ERAP1 is shown in orange surface representation. Crystallized ligands are shown in magenta, zinc(II) in white. Common SNPs in ERAP1 that line the cavity are indicated in green spheres. The S1 pocket narrow channel is indicated by a red arrow.
Overall, the three ligands detected in our crystal structure occupy three distal edges of the cavity (Figure 4). Tracing from the catalytic site where DG046 is located toward MLT or B3P suggests possible trajectories for long peptide substrate binding. Thus, it is very likely that the MLT and B3P ligands reveal secondary binding pockets for long peptides. Interestingly, along that path lie four ERAP1 SNPs that have been associated with disease predisposition and have been shown to affect peptide processing,25,26 suggesting that direct interactions between these SNPs and peptides trapped in the cavity may underlie their functional effects. Furthermore, optimization of compounds that bind into one of those secondary sites may affect binding of some peptidic substrates, altering the specificity of ERAP1, something that may translate to changes in the cellular immunopeptidome, an exciting prospect for immunotherapy approaches that rely on modulating antigen presentation.27
In summary, we present the first high-resolution crystal structure of ERAP1, in complex with a potent inhibitor. The inhibitor is found in the active site making key interactions that explain both potency and selectivity. We also found additional ligands bound within the enzyme’s large internal cavity that suggest possible trajectories for the binding of large peptides and potential druggable pockets that can regulate the specificity of the enzyme.
Acknowledgments
We thank the beamline scientists of EMBL-Hamburg for their assistance during data collection at the P13 beamline of PETRA III, Hamburg, Germany. This research was financed by the Harry J. Lloyd Charitable Trust through a grant to E.S. and by the project “NCSRD–INRASTES research activities in the framework of the national RIS3” (Grant MIS 5002559), which is implemented under the “Action for the Strategic Development on the Research and Technological Sector”, funded by the Operational Programme “Competitiveness, Entrepreneurship and Innovation” (Grant NSRF 2014–2020) and cofinanced by Greece and the European Union (European Regional Development Fund). We also acknowledge support of this work by the project “INSPIRED-The National Research Infrastructures on Integrated Structural Biology, Drug Screening Efforts and Drug Target Functional Characterization” (Grant MIS 5002550), which is implemented under the Action “Reinforcement of the Research and Innovation Infrastructure”, funded by the Operational Programme “Competitiveness, Entrepreneurship and Innovation” (Grant NSRF 2014–2020) and cofinanced by Greece and the European Union (European Regional Development Fund). Synchrotron access was funded by iNEXT (Proposal ID 5589) and by EMBL-Hamburg (Proposal MX-619).
Glossary
Abbreviations
- ERAP1
endoplasmic reticulum aminopeptidase 1
- MHC-I
Major Histocompatibility Complex Class I
- ER
endoplasmic reticulum
- HLA
human leukocyte antigen
- MW
molecular weight
- ERAP2
endoplasmic reticulum aminopeptidase 2
- IRAP
insulin-regulated aminopeptidase
- SNPs
single nucleotide polymorphisms
- L-AMC
leucine-7-amino-4-methylcoumarin.
Biography
Efstratios Stratikos studied chemistry at the University of Athens in Greece and then moved on to receive a Ph.D. in biochemistry from the University of Illinois at Chicago in 1999. After postdoctoral training in structural and molecular biology at Harvard University, he returned to Greece in 2005 to establish the Protein Chemistry Laboratory at the National Centre for Scientific Research “Demokritos” in Athens, where he is still employed at the rank of research director. During the last decade his research has focused on the pathway of intracellular antigen processing and its role in regulating adaptive immune responses in human disease.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00002.
Detailed description of the protein construct, purification, crystallization conditions, and structure determination; table showing data collection and refinement statistics; supporting figures showing the inhibition curve of the crystallized ERAP1 construct from DG046, the B-factors of DG046 atoms, the models of malic acid and bis-tris propane built into 2Fo-Fc electron density; and the cacodylate ion built in residual electron density of the ERAP1 crystal structure with PDB code 2YD0 (PDF)
Author Contributions
P.G. crystallized the complex, performed diffraction experiments, and solved and interpreted the structure. M.N. and P.R. designed the construct and screened crystallization conditions. E.S. supervised the project, helped design the protein construct, interpreted the data, and wrote the paper in collaboration with P.G. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Notes
Atomic coordinates and structure factors have been deposited in the Protein Data Bank (www.pdb.org), with PDB code 6Q4R.
Supplementary Material
References
- Evnouchidou I.; Papakyriakou A.; Stratikos E. A new role for Zn(II) aminopeptidases: antigenic peptide generation and destruction. Curr. Pharm. Des. 2009, 15, 3656. 10.2174/138161209789271816. [DOI] [PubMed] [Google Scholar]
- Hammer G. E.; Gonzalez F.; James E.; Nolla H.; Shastri N. In the absence of aminopeptidase ERAAP, MHC class I molecules present many unstable and highly immunogenic peptides. Nat. Immunol. 2007, 8, 101. 10.1038/ni1409. [DOI] [PubMed] [Google Scholar]
- James E.; Bailey I.; Sugiyarto G.; Elliott T. Induction of Protective Antitumor Immunity through Attenuation of ERAAP Function. J. Immunol. 2013, 190, 5839. 10.4049/jimmunol.1300220. [DOI] [PubMed] [Google Scholar]
- Hammer G. E.; Gonzalez F.; Champsaur M.; Cado D.; Shastri N. The aminopeptidase ERAAP shapes the peptide repertoire displayed by major histocompatibility complex class I molecules. Nat. Immunol. 2006, 7, 103. 10.1038/ni1286. [DOI] [PubMed] [Google Scholar]
- York I. A.; Brehm M. A.; Zendzian S.; Towne C. F.; Rock K. L. Endoplasmic reticulum aminopeptidase 1 (ERAP1) trims MHC class I-presented peptides in vivo and plays an important role in immunodominance. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 9202. 10.1073/pnas.0603095103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez de Castro J. A.; Alvarez-Navarro C.; Brito A.; Guasp P.; Martin-Esteban A.; Sanz-Bravo A. Molecular and pathogenic effects of endoplasmic reticulum aminopeptidases ERAP1 and ERAP2 in MHC-I-associated inflammatory disorders: Towards a unifying view. Mol. Immunol. 2016, 77, 193. 10.1016/j.molimm.2016.08.005. [DOI] [PubMed] [Google Scholar]
- Stratikos E.; Stamogiannos A.; Zervoudi E.; Fruci D. A role for naturally occurring alleles of endoplasmic reticulum aminopeptidases in tumor immunity and cancer pre-disposition. Front. Oncol. 2014, 4, 363. 10.3389/fonc.2014.00363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ombrello M. J.; Kastner D. L.; Remmers E. F. Endoplasmic reticulum-associated amino-peptidase 1 and rheumatic disease: genetics. Curr. Opin. Rheumatol. 2015, 27, 349. 10.1097/BOR.0000000000000189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratikos E. Modulating antigen processing for cancer immunotherapy. Oncoimmunology 2014, 3, e27568. 10.4161/onci.27568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zervoudi E.; Saridakis E.; Birtley J. R.; Seregin S. S.; Reeves E.; Kokkala P.; Aldhamen Y. A.; Amalfitano A.; Mavridis I. M.; James E.; Georgiadis D.; Stratikos E. Rationally designed inhibitor targeting antigen-trimming aminopeptidases enhances antigen presentation and cytotoxic T-cell responses. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 19890. 10.1073/pnas.1309781110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiadis D.; Mpakali A.; Koumantou D.; Stratikos E. Inhibitors of ER aminopeptidase 1 and 2: from design to clinical application. Curr. Med. Chem. 2018, 10.2174/0929867325666180214111849. [DOI] [PubMed] [Google Scholar]
- Stratikos E.; Stern L. J. Antigenic peptide trimming by ER aminopeptidases--insights from structural studies. Mol. Immunol. 2013, 55, 212. 10.1016/j.molimm.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papakyriakou A.; Stratikos E. The Role of Conformational Dynamics in Antigen Trimming by Intracellular Aminopeptidases. Front. Immunol. 2017, 8, 946. 10.3389/fimmu.2017.00946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T. T.; Chang S. C.; Evnouchidou I.; York I. A.; Zikos C.; Rock K. L.; Goldberg A. L.; Stratikos E.; Stern L. J. Structural basis for antigenic peptide precursor processing by the endoplasmic reticulum aminopeptidase ERAP1. Nat. Struct. Mol. Biol. 2011, 18, 604. 10.1038/nsmb.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochan G.; Krojer T.; Harvey D.; Fischer R.; Chen L.; Vollmar M.; von Delft F.; Kavanagh K. L.; Brown M. A.; Bowness P.; Wordsworth P.; Kessler B. M.; Oppermann U. Crystal structures of the endoplasmic reticulum aminopeptidase-1 (ERAP1) reveal the molecular basis for N-terminal peptide trimming. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 7745. 10.1073/pnas.1101262108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokkala P.; Mpakali A.; Mauvais F. X.; Papakyriakou A.; Daskalaki I.; Petropoulou I.; Kavvalou S.; Papathanasopoulou M.; Agrotis S.; Fonsou T. M.; van Endert P.; Stratikos E.; Georgiadis D. Optimization and Structure-Activity Relationships of Phosphinic Pseudotripeptide Inhibitors of Aminopeptidases That Generate Antigenic Peptides. J. Med. Chem. 2016, 59, 9107. 10.1021/acs.jmedchem.6b01031. [DOI] [PubMed] [Google Scholar]
- Mpakali A.; Saridakis E.; Harlos K.; Zhao Y.; Kokkala P.; Georgiadis D.; Giastas P.; Papakyriakou A.; Stratikos E. Ligand-induced conformational change of Insulin-regulated aminopeptidase: insights on catalytic mechanism and active site plasticity. J. Med. Chem. 2017, 60, 2963. 10.1021/acs.jmedchem.6b01890. [DOI] [PubMed] [Google Scholar]
- Hisatsune C.; Ebisui E.; Usui M.; Ogawa N.; Suzuki A.; Mataga N.; Takahashi-Iwanaga H.; Mikoshiba K. ERp44 Exerts Redox-Dependent Control of Blood Pressure at the ER. Mol. Cell 2015, 58, 1015. 10.1016/j.molcel.2015.04.008. [DOI] [PubMed] [Google Scholar]
- Stamogiannos A.; Maben Z.; Papakyriakou A.; Mpakali A.; Kokkala P.; Georgiadis D.; Stern L. J.; Stratikos E. Critical Role of Interdomain Interactions in the Conformational Change and Catalytic Mechanism of Endoplasmic Reticulum Aminopeptidase 1. Biochemistry 2017, 56, 1546. 10.1021/acs.biochem.6b01170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamogiannos A.; Koumantou D.; Papakyriakou A.; Stratikos E. Effects of polymorphic variation on the mechanism of Endoplasmic Reticulum Aminopeptidase 1. Mol. Immunol. 2015, 67, 426. 10.1016/j.molimm.2015.07.010. [DOI] [PubMed] [Google Scholar]
- Sui L.; Gandhi A.; Guo H. C. Crystal structure of a polypeptide’s C-terminus in complex with the regulatory domain of ER aminopeptidase 1. Mol. Immunol. 2016, 80, 41. 10.1016/j.molimm.2016.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S. C.; Momburg F.; Bhutani N.; Goldberg A. L. The ER aminopeptidase, ERAP1, trims precursors to lengths of MHC class I peptides by a ″molecular ruler″ mechanism. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 17107. 10.1073/pnas.0500721102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss N. R.; Gerstein M. 3V: cavity, channel and cleft volume calculator and extractor. Nucleic Acids Res. 2010, 38, W555. 10.1093/nar/gkq395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein M.; Chothia C. Packing at the protein-water interface. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 10167. 10.1073/pnas.93.19.10167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evnouchidou I.; Kamal R. P.; Seregin S. S.; Goto Y.; Tsujimoto M.; Hattori A.; Voulgari P. V.; Drosos A. A.; Amalfitano A.; York I. A.; Stratikos E. Coding single nucleotide polymorphisms of endoplasmic reticulum aminopeptidase 1 can affect antigenic peptide generation in vitro by influencing basic enzymatic properties of the enzyme. J. Immunol. 2011, 186, 1909. 10.4049/jimmunol.1003337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves E.; Edwards C. J.; Elliott T.; James E. Naturally occurring ERAP1 haplotypes encode functionally distinct alleles with fine substrate specificity. J. Immunol. 2013, 191, 35. 10.4049/jimmunol.1300598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratikos E. Regulating adaptive immune responses using small molecule modulators of aminopeptidases that process antigenic peptides. Curr. Opin. Chem. Biol. 2014, 23, 1. 10.1016/j.cbpa.2014.08.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.