

Abstract

To obtain a new anticancer drug, we focused on FER tyrosine kinase. Starting with high-throughput screening with our in-house chemical library, compound 1, which has a pyridine moiety, was found. Referring to their X-ray crystal structure with FES proto-oncogene tyrosine kinase, as a surrogate of FER followed by chemical modification including scaffold hopping of the pyridine template, we discovered pyrido-pyridazinone derivatives with potent FER kinase inhibitory activity. Here, we disclose the structure–activity relationship on the scaffold and representative compound 21 (DS21360717), which showed in vivo antitumor efficacy in a subcutaneous tumor model.
Keywords: FER, inhibitor, tyrosine kinase, FES, pyrido-pyridazinone, antitumor
FER is a nontransmembrane receptor tyrosine kinase in the FES family. It plays a pivotal role in cell migration through the tyrosine phosphorylation of cortactin (CTTN) and direct interaction with phosphatidic acid.1,2 In breast cancer cell lines, FER kinase controls migration and metastasis by regulating integrin-dependent adhesion. High FER expression in human invasive breast cancer is an independent prognostic factor that correlates with high-grade basal/triple-negative tumors and worse overall survival.3 Recently, a fusion gene of mannosidase alpha class 2A member 1-FER tyrosine kinase gene (MAN2A1-FER) was discovered in liver tumors, esophageal adenocarcinoma, glioblastoma multiforme, prostate tumors, nonsmall cell lung tumors, and ovarian tumors. The expression of MAN2A1-FER led to increases of proliferation and invasiveness of cancer cell lines, and conferred liver oncogenic activity in mice.4 These reports suggest that FER has the potential to be a target for anticancer therapy. However, potent FER kinase inhibitors have not been investigated intensively and few reports on an FER inhibitor have been published.5 Here, we disclose and discuss highly potent FER kinase inhibitors discovered by high-throughput screening (HTS) and subsequent scaffold hopping and chemical modification based on the structural information obtained from X-ray crystallography.
Through an HTS strategy with an in-house chemical library, a pyridine derivative 1 with moderate FER inhibitory activity was identified. In a preliminary study, we investigated the basic structure–activity relationship (SAR) of compound 1 by incorporating additional groups and modifying its side chains.
First, incorporation of a substituent at the 5-position of the pyridine scaffold was investigated, and we found that hydrogen can be replaced with Cl, phenyl, or nitrile. Interestingly, the inhibitory activity was enhanced by Cl or nitrile. As shown in Table 1, compound 4 having nitrile at this position showed potent activity. We then progressed to the preliminary derivatization of the side chains on C-2, C-3, and C-6 of compound 4.
Table 1. Preliminary SAR for C-5 Position in the Pyridine Scaffolda.

| compound | R | FER IC50 (nM) |
|---|---|---|
| 1 (HTS hit) | H | 480 (400–590) |
| 2 | Cl | 25 (23–26) |
| 3 | Ph | 510 (440–600) |
| 4 | CN | 2.6 (2.1–3.2) |
IC50 values were calculated from duplicate experiments by the least-squares method along with 95% confidence intervals (95% Cl) in parentheses. The assay details are shown in the Supporting Information.
The results are shown in Table 2. In almost all of the compounds produced by these modifications, maintenance of the activities of compound 4 was difficult. Only compound 10 possessed moderate activity, with IC50 of 57 nM. Removal of the phenyl group from the 2-position (compound 5), replacement of the aniline with a benzyl moiety (compound 6), removal of the carbamoyl group at the 3-position (compound 7), or methylation to the carbamoyl amine (compound 8) resulted in large loss of activity. As for the 6-position, the results clearly showed that cis-cyclohexyl diamine was the optimal structure (compounds 10 and 11). Terminal amine nitrogen and the hydrogens on it in the cyclohexyl diamine moiety are necessary for the activity (compounds 9 and 12). We confirmed that the side chains in compound 4 as a target for modification already had good effects on FER inhibitory activities within the primary derivatization.
Table 2. Side Chain SAR for C-2, C-3, and C-6 Positions in the Pyridine Scaffolda.
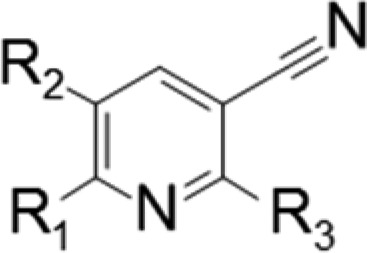

IC50 values were calculated from duplicate experiments by the least-squares method along with 95% confidence intervals (95% Cl) in parentheses. The assay details are shown in the Supporting Information.
Next, we determined the profile of compound 4 in a cellular assay, as shown in Figure 2. Compound 4 possessed potent growth inhibition activity against constructed FER-driven Ba/F3 cells and Ba/F3-Mock. At this stage, the compound showed a preferable profile, but we were concerned about it from a structural perspective, due to a hydrophilic carbamoyl group.6 To address this issue, we next focused on the design of new compounds to reduce the number of hydrogen bond donors. In particular, we considered performing scaffold hopping to modify the carbamoyl group.
Figure 2.
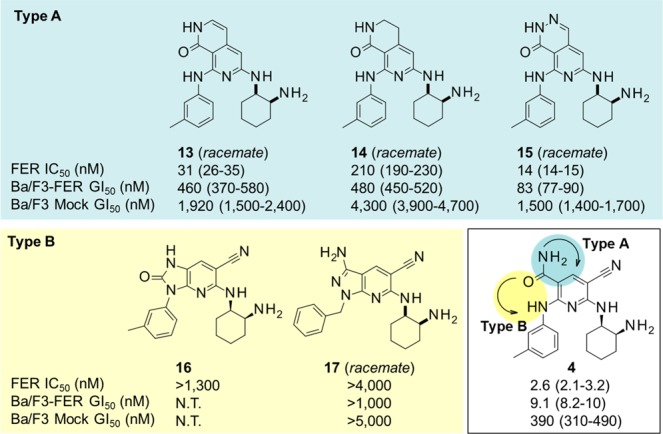
New scaffolds identified by cyclization. IC50 values were calculated from duplicate experiments by the least-squares method, and GI50 values were calculated from quadruplicate experiments along with 95% confidence intervals (95% CI) in parentheses. The assay details are shown in the Supporting Information.
To obtain further insight into the mode of binding of compound 4 to the target protein, we used FES as a surrogate protein for FER because FES has high homology to FER. As shown in Figure 1, we obtained the X-ray crystal structure of FES in complex with compound 4.
Figure 1.
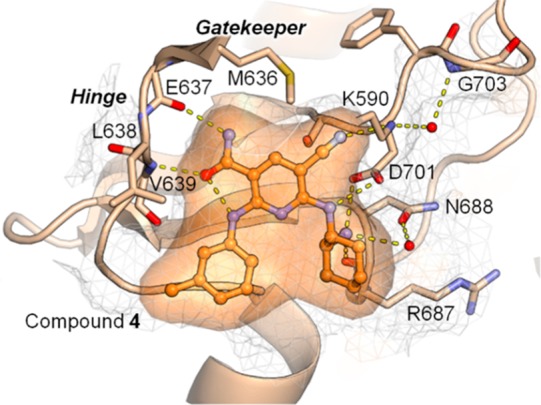
Crystal structure of compound 4 in complex with FES (PDB code: 6JMF). FES is depicted as a cartoon, sticks, and a grid surface in gray. N atoms are in blue, O atoms are in red, and an S atom is in yellow. Modeled FES is in light orange. Compound 4 is shown as a ball-and-stick model in orange. The hydrogen bond network around the compound is shown as dashed yellow lines. Two water molecules participating in the network are shown as spheres in red. The final model and structure factors were deposited in PDB with accession code 6JMF. The image was prepared using PyMOL (The PyMOL Molecular Graphics System, Version 1.8; Schrödinger, LLC).7
In this complex, compound 4 occupies the ATP-binding site of FES. The 3-, 5-, and 6-position substituent groups of the pyridine scaffold of compound 4 undergo hydrogen bonding with FES: (a) The amine of the 3-carbamoyl group forms a hydrogen bond with the backbone carbonyl oxygen atom of E637. The oxygen atom of the 3-carbamoyl group forms inter- and intramolecular hydrogen bonds with the backbone nitrogen atom of V639 and the nitrogen atom of the 2-aniline moiety, respectively. The criticality of these interactions between the 3-carbamoyl group and the FES hinge region for the binding of compound 4 to FES was experimentally supported by the lack of FER inhibitory activity for compound 7. (b) The nitrogen atom of the 5-cyano group forms a hydrogen bond with the side-chain amino group of catalytic lysine (K590). (c) The linker amine of the 6-cyclohexyldiamino group forms a hydrogen bond with the side-chain oxygen atom of D701. The terminal amine of the 6-cyclohexyldiamino group forms three hydrogen bonds with the backbone carbonyl oxygen atom of R687, the side-chain oxygen atom of N688 via a bridged water molecule, and the side-chain oxygen atom of D701. These interactions between the terminal amine of the 6-cyclohexyldiamino group and FES explain the more than 100-fold decrease in FER inhibitory activity observed in the comparison between compounds 4 and 9. In addition to the hydrogen bonds, numerous hydrophobic interactions are also observed as a result of high shape complementarity between compound 4 and the ATP-binding site of FES (see Supporting Information). In this way, the analysis of the crystal structure provided us with a sufficient explanation of the high potency, where 2-anilino, 3-carbamoyl, 5-cyano, and 6-cyclohexyldiamino groups play important roles in the binding to the ATP-binding site of the protein. These observations are consistent with the SARs shown in Tables 1 and 2.
We then focused our attention on enhancing the membrane permeability by modifying the carbamoyl moiety. Our strategy for this conversion consists of the cyclization depicted in Figure 2 to generate a new bicyclic scaffold. Two types of cyclization, Type A and Type B, were investigated: Type A cyclization of 3-carbamoyl to the 4-position of the pyridine template and Type B cyclization of 3-carbamoyl to the 2-position of the pyridine template.
Although these two types appeared to fit in the binding site according to the model, the results that we obtained differed markedly from what we anticipated. Type B compounds did not maintain the activities, while Type A compounds showed moderate to strong activities. For Type B, which includes compounds 16 and 17, their ring structures might prevent those side chains from being arranged at a suitable angle or direction to maintain their activities. However, compound 15 possessing a pyrido-pyridazino scaffold had remarkable activity, even though it did not have the substituents necessary for strong activity in the case of the pyridine scaffold, that is, a halogen or cyano group at the 5-position. In fact, compound 15 was 30 times more potent than parent compound 1. By conversion to a bicyclic structure like Type A, it could make better use of occupying the ATP-binding site. As for inhibitory activities for Ba/F3-Mock cells, which could be thought as an indicator showing the selectivity for FER, compounds 13, 14, and 15 show not so strong inhibitory activities compared with FER IC50 value and Ba/F3-FER IC50 value. Therefore, it could be considered that these bicyclic structure were not nonselective and worthy for derivatization.
Next, the optimization of pyrido-pyridazinone derivatives was performed, focusing on the cyclohexane diamine moiety. The results are displayed in Table 3. We investigated the best configuration of the two amines in the cyclohexane portion. The difference between compounds 15 and 18 clearly showed that (1R,2S)-2-aminocyclohexyl isomer had higher activity. Next, we incorporated a halogen or cyano group into the 8-position of the pyrido-pyridazino scaffold to obtain more potent compounds. The results showed that all compounds exhibited higher biochemical activity, while halogeno variants did not possess very potent cellular activity. Among the derivatives thus far synthesized, compound 21 showed the highest inhibitory activity in biochemical and cellular growth assays, which encouraged us to evaluate its in vivo antitumor activity.
Table 3. Modification of Pyrido-pyridazino Scaffolda.

IC50 values were calculated from duplicate experiments by the least-squares method, and Gl50 values were calculated from at least triplicate quadruplicate experiments along with 95% confidence intervals (95% Cl) in parentheses. The assay details are shown in the Supporting Information.
We characterized the pharmacokinetic profiles of compound 4 and DS21360717, which are displayed in Table 4. Although there were no significant differences in the CLtot between the two, BA of DS21360717 was improved in comparison with that of compound 4, which was presumably attributable to an improved membrane permeability coefficient (Pe). The two compounds showed almost the same moderate total body clearance (CLtot) metabolic stability and protein binding (% bound) in mouse microsomes (% remaining), even though the solubility of compound 21 is poor. However, the bioavailability (BA) of DS21360717 was better than that of compound 4. These results implied that the improvement of Pe could confer better BA, via reduction in the number of hydrogen bond donors as the result of scaffold hopping.
Table 4. Pharmacokinetic Properties of 4 and 21 (DS21360717) in Mouse.

| compound | metabolic stabilitya (% remaining) | solubility (pH 6.8/FaSSIF)b (μg/mL) | protein bindingc (% bound) | Pe (pH 7.4) (10–6 cm/s) | CLtotd (mL/min/kg) | AUCe (μg·h/mL) | Cmaxe (μg/mL) | BA (%) |
|---|---|---|---|---|---|---|---|---|
| 4 | 100 | 35/49 | 99 | 16 | 12 | 2.4 | 0.42 | 17 |
| 21(DS21360717) | 96 | 0.9/2.1 | 99 | >50 | 12 | 4.8 | 0.36 | 34 |
Remaining rate of test compounds (1.0 μM) after incubation for 30 min in mouse liver microsomes (1.0 mg/mL).
Solubility: μg/mL in pH 6.8 buffer and FaSSIF.
Protein binding was tested by BALB/c mice serum samples.
Compounds were dosed iv at 1 mg/kg.
Compounds were dosed po at 10 mg/kg. The statistics are shown in the Supporting Information.
We thought that it was worthwhile subjecting DS21360717 to an in vivo test and thus carried out antitumor study using a Ba/F3-FER subcutaneous tumor model, the results of which are shown in Figure 3. As envisioned, DS21360717 exhibited tumor growth inhibitory activity in a dose-dependent manner without significant body weight loss. Taking into consideration the fact that mean unbound plasma concentration upon oral dosing at 10 mg/kg was 3.1 nM, exceeding GI50 for Ba/F3-FER, the antitumor efficacy observed at doses of more than 12.5 mg/kg was considered reasonable.
Figure 3.
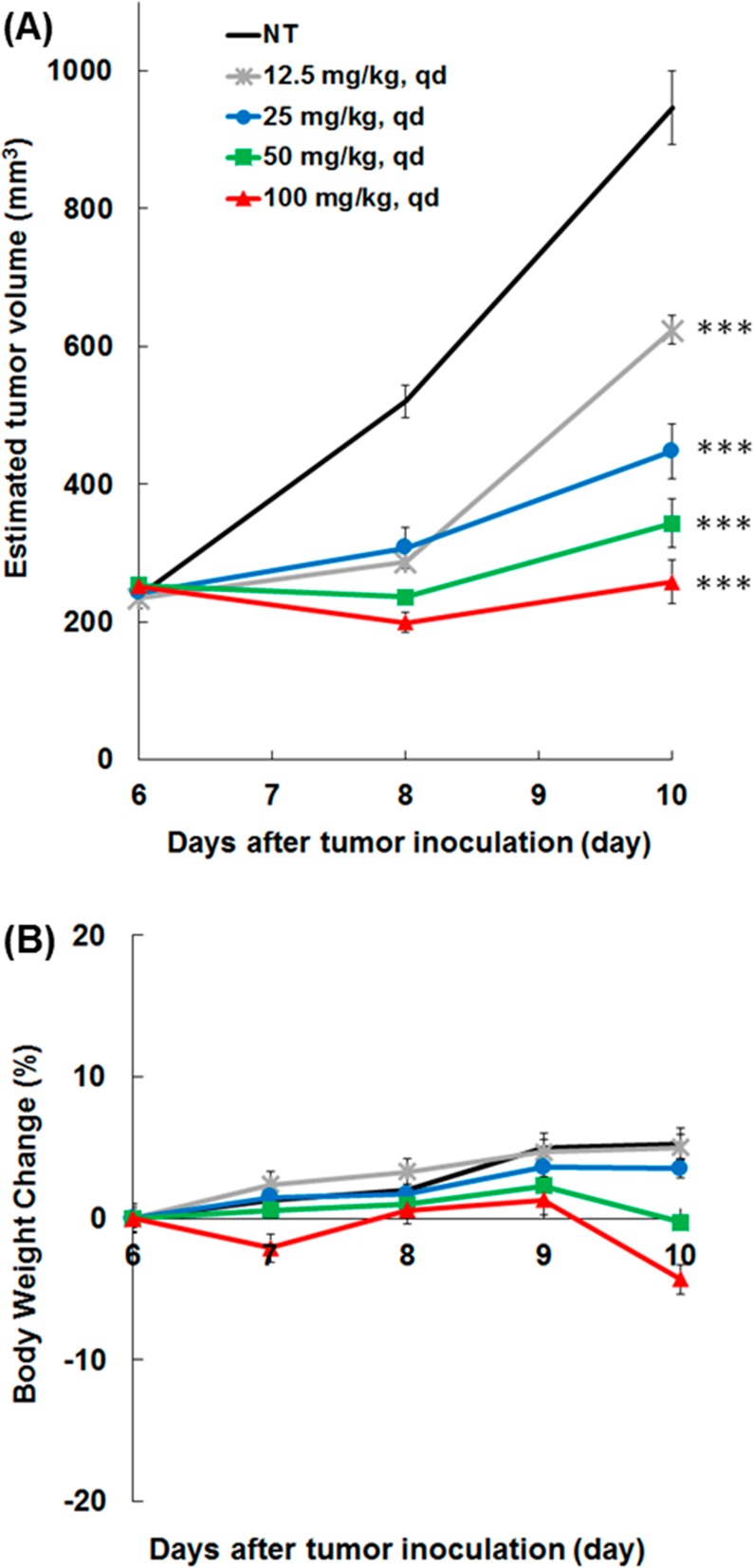
Antitumor efficacy of DS21360717 in a Ba/F3-FER subcutaneous tumor model. (A) Tumor volume of each group (n = 5), **p < 0.01 and ***p < 0.001 vs control. (B) Body weight change from the start of treatment in mice treated with DS21360717 (see Supporting Information).
The docking model of compound 21 with FES is shown in Figure 4. It suggests that, while Type A cyclization retains the hydrogen bonds between the inhibitors and FES, the shape complementarity around the gatekeeper residue (M636) is clearly improved compared with that of compound 4. Further, the additional interactions between the pyridazinone ring and FES were observed by focusing on the binding mode of compound 21 and FES; CH/π interactions with A588 Cβ or L690 Cδ1, aliphatic-CH···aromatic-CH interactions with M636 Cβ or Cε, and divalent-S···aromatic-CH interactions with M636 Sδ. As shown in this figure, their typical distances were considered to be suitable for the preferred affinity for FES.8 The above might be the reason why compound 21 shows high FER inhibitory activity (see Supporting Information).
Figure 4.

Superposition of modeled binding mode of compound 21. (A) Compound 21 and the crystal structure of compound 4 in complex with FES. The drawing style of the crystal structure (FES/compound 4) and its colors are the same as in Figure 1. Compound 21 is shown as a ball-and-stick model in green. Its molecular surface is also shown. (B) Additional interactions observed between FES and cyclized atoms of compound 21 (docked model; green): cyan dashed lines are the CH/π interaction between A588 Cβ or L690 Cδ1 and the center of the cyclized ring (pale cyan sphere). Violet dashed lines are interactions between M636 Cβ, Sδ, or Cε and aromatic CHs. Distances of the additional interactions are in Ångstrom units.
To further evaluate DS21360717, screening was conducted against a panel of 68 kinases (screened at a concentration of 200 nM). In addition to strong inhibition of FER and FES, 14 kinases conferring greater than 90% inhibition were identified, which were ALK, FLT3, FMS, KIT, PYK2, ROS, SYK, TRKA, CDK2/CycA2, CHK2, HGK, IRAK4, MLK1, and TSSK1. The details are shown in the Supporting Information.
Syntheses of pyridine derivative 4 are shown in Scheme 1. Compound 4 was prepared from commercially available dichloropyridine 22.9,10 SNAr reaction of 22 afforded aniline 23. Then, another SNAr reaction of compound 23 furnished N-Boc amine 24. Carboxylic acid 25 was obtained by hydrolysis of the corresponding ethyl ester 24, and the following condensation reaction afforded carbamoyl variant 26. N-Boc deprotection of compound 26 gave pyridine derivative 4.
Scheme 1. Synthesis of Pyridine Derivative 4.

Reagents: (a) 3-methylaniline, DIPEA, MeCN, rt, 96%; (b) tert-butyl [(1S,2R)-2-aminocyclohexyl]carbamate, DIPEA, MeCN, 70 °C, 83%; (c) LiOH, THF, H2O, rt–50 °C, quant.; (d) NH3 aq., WSCI·HCl, HOSu, DMF, rt, 84%; (e) 2 N HCl-EtOH, rt, quant.
Syntheses of pyrido-pyridazinone derivative 21 are shown in Scheme 2. Compound 21 was prepared from commercially available 4-methyl ester 27. Converting the ester part to aldehyde via alcohol gave 29. Then, protection of the aldehyde group with trimethyl orthoformate furnished ketal 30. Ester incorporation into the 3-position of pyridine with lithium diisopropylamide (LDA) and methyl chloroformate afforded methyl ester 31. Hydrolysis of the ketal part gave aldehyde 32, and the following hydrazidation afforded N-Boc hydrazide 33. Ring closure was induced by deprotection of the Boc group to provide pyrido-pyridazinone 34.11 Subsequently, consecutive SNAr reactions of compound 34 with 3-methylaniline and then tert-butyl [(1S,2R)-2-aminocyclohexyl]carbamate furnished compound 36. 8-Bromo derivative 37 was synthesized by bromination of compound 36 with N-bromosuccinimide (NBS).12,13 Finally, incorporation of the cyano group was carried out by zinc cyanide and palladium(0) catalyst on compound 37,14 followed by deprotection of the Boc group to furnish compound 21 (DS21360717).
Scheme 2. Synthesis of Pyrido-pyridazinone Derivative 21 (DS21360717).

Reagents: (a) NaBH4, EtOH, reflux, quant.; (b) Dess–Martin periodinane, CH2Cl2, rt, quant.; (c) trimethyl orthoformate, p-TsOH-H2O, MeOH, toluene, reflux, quant.; (d) LDA, chloroformic acid methyl ester, THF, −78 °C, 27%; (e) TFA, CH2Cl2, rt, quant.; (f) carbazic acid tert-butyl ester, 1,4-dioxane, rt, quant.; (g) TFA, CH2Cl2, rt, 99%. (h) 3-methylaniline, NMP, microwave, 160 °C, quant.; (i) tert-butyl [(1S,2R)-2-aminocyclohexyl]carbamate, NMP, microwave 170 °C, 45%; (j) NBS, DMF, rt, 87%; (k) zinc cyanide, tetrakis(triphenylphosphine) palladium(0), DMF, microwave, 120–130 °C, 55%. (l) TFA, CH2Cl2, rt, 87%.
As for other pyridine derivatives 2–12 and bicyclic derivatives 13–20, their syntheses and characterizations are described in the Supporting Information.
In summary, initial SAR study on HTS hits, followed by scaffold hopping and optimization, furnished a potent, drug-like, and in vivo-active compound, DS21360717. Further optimization and characterization of the lead compound will be reported in due course.
Glossary
ABBREVIATIONS
- HTS
high-throughput screening
- Pe
permeability coefficient
- CLtot
total body clearance
- BA
bioavailability
- DIPEA
N-ethyl-N,N-diisopropylamine
- MeCN
acetonitrile
- THF
tetrahydrofuran
- WSCI·HCl
1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide
- HOSu
N-hydroxysuccinimide
- DMF
N,N-dimethylformamide
- EtOH
ethyl alcohol
- LDA
lithium diisopropylamide
- NBS
N-bromosuccinimide
- p-TsOH
p-toluenesulfonic acid
- MeOH
methyl alcohol
- TFA
trifluoroacetic acid
- NMP
N-methylpyrrolidone
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00631.
Synthetic procedures and characterization data of all compounds, and information on additional studies (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Sangrar W.; Gao Y.; Scott M.; Truesdell P.; Greer P. A. Fer-Mediated Cortactin Phosphorylation Is Associated with Efficient Fibroblast Migration and Is Dependent on Reactive Oxygen Species Generation during Integrin-Mediated Cell Adhesion. Mol. Cell. Biol. 2007, 32, 6140–6152. 10.1128/MCB.01744-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh T.; Hasegawa J.; Tsujita K.; Kanaho Y.; Takenawa T. The Tyrosine Kinase Fer Is a Downstream Target of the PLD-PA Pathway that Regulates Cell Migration. Sci. Signaling 2009, 2 (87), ra52. 10.1126/scisignal.2000393. [DOI] [PubMed] [Google Scholar]
- Ivanova I. A.; Vermeulen J. F.; Ercan C.; Houthuijzen J. M.; Saig F. A.; Vlug E. J.; van der Wall E.; van Diest P. J.; Vooijs M.; Derksen P. W. B. FER Kinase Promotes Breast Cancer Metastasis by Regulating α6- and β1-Integrin-Dependent Cell Adhesion and Anoikis Resistance. Oncogene 2013, 32, 5582–5592. 10.1038/onc.2013.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z.; Yu Y. P.; Tao J.; Liu S.; Tseng G.; Nalesnik M.; Hamilton R.; Bhargava R.; Nelson J. B.; Pennathur A.; Monga S. P.; Luketich J. D.; Michalopoulos G. K.; Luo J. MAN2A1-FER Fusion Gene Is Expressed by Human Liver and Other Tumor Types and Has Oncogenic Activity in Mice. Gastroenterology 2017, 153, 1120–1132. 10.1053/j.gastro.2016.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkis Y.; Cohen M.; Yaffe E.; Satmary-Tusk S.; Feldman T.; Hikri E.; Nyska A.; Feiglin A.; Ofran Y.; Shpungin S.; Nir U. A Novel Fer/FerT Targeting Compound Selectively Evokes Metabolic Stress and Necrotic Death in Malignant Cells. Nat. Commun. 2017, 8, 1–17. 10.1038/s41467-017-00832-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu H.; Yamasaki T.; Yoneda Y.; Muro F.; Hamada T.; Yasukochi T.; Tanaka S.; Toki T.; Yokoyama M.; Morishita K.; Iimura S. Discovery of Imidazo[1,2-b]pyridazines as IKKβ Inhibitors. Part 3: Exploration of Effective Compounds in Arthritis Models. Bioorg. Med. Chem. Lett. 2011, 21, 4550–4555. 10.1016/j.bmcl.2011.05.115. [DOI] [PubMed] [Google Scholar]
- The PyMOL Molecular Graphics System, Version 1.8.6.2; Schrödinger, LLC: New York, 2018. [Google Scholar]
- Bissantz C.; Kuhn B.; Stahl M. J. Med. Chem. 2010, 53, 5061–5084. 10.1021/jm100112j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko S.; Arita T.; Nagasawa Y.; Kato M.. Jpn. Kokai Tokkyo Koho JP 2008/013499, 2008.
- Zhaozhong J.; Song Y.; Xu Q.; Kane B.; Bauer S. M.; Pandey A.. WO2012/61418, 2012.
- Vasudevan A.; Penning T. D.; Chen H.; Liang B.; Wang S.; Yang L.; Gao Y.. WO2012/97682, 2012.
- Senanayake C. H.; Fredenburgh L. E.; Reamer R. A.; Liu J.; Roberts F. E.; Humphrey G.; Thompson A. S.; Reider P. J.; Shinkai I. New Approach to the Imidazolutidine Moiety of MK-996. Heterocycles 1996, 42, 821–830. 10.3987/COM-95-S91. [DOI] [Google Scholar]
- Reiser U.; Madden J.. WO2015/25019, 2015.
- Alterman M.; Hallberg A. Fast Microwave-Assisted Preparation of Aryl and Vinyl Nitriles and the Corresponding Tetrazoles from Organo-halides. J. Org. Chem. 2000, 65, 7984–7989. 10.1021/jo0009954. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.