

Abstract
Macrocyclic compounds such as cyclic peptides have emerged as a new and exciting class of drug candidates for inhibition of intracellular protein-protein interactions, which are challenging targets for conventional drug modalities (i.e., small molecules and proteins). Over the past decade, several complementary technologies have been developed to synthesize macrocycle libraries and screen them for binding to therapeutically relevant targets. Two different approaches have also been explored to increase the membrane permeability of cyclic peptides. In this review, we discuss these methods and their applications in the discovery of macrocyclic compounds against protein-protein interactions.
Keywords: Cyclic peptide, Drug discovery, Macrocycle, Protein-protein interaction, Undruggable targets
Summary Statement
Intracellular protein-protein interactions are challenging targets for conventional small molecules or biologics. However, recent development of macrocyle synthesis and delivery technologies has now made it possible to discover cell-permeable and metabolically stable macrocyclic compounds to effectively inhibit intracellular protein-protein interactions.
INTRODUCTION
It is estimated that at any given time, a human cell may contain about 130,000 binary interactions between proteins [1]. Perturbation of any of these protein-protein interactions (PPIs) may lead to altered cellular behaviour and potentially human diseases. It is thus not surprising that PPIs have emerged as an exciting class of targets for inhibitor development, both as research tools for dissecting the complex interaction networks and as therapeutics against a wide variety of human diseases and conditions [2]. There is also significant interest in developing chemical entities to enhance PPIs as a means of disease intervention [3], which will not be the subject of this review.
For convenience, PPIs can be classified into three different classes [4]. The first class of PPIs involves binding of short peptide motifs in their extended conformation to small protein domains, as exemplified by the 120 SH2 domains and 260 PDZ domains in humans [5,6]. The SH2 domains require a phosphotyrosine (pY) for binding, but each recognize a specific sequence immediately C-terminal to the pY residue [7]. PDZ domains, on the other hand, bind to specific partner proteins by recognizing the 3–5 residues at their C-termini [8,9]. The interaction between these protein domains and their peptide ligands resembles in many ways the binding mode of proteases to their peptide substrates, i.e., the peptide ligand is typically in the extended β-strand conformation, with the side chains of key ligand residues plugging into distinct pockets on the protein surface. These PPIs are generally druggable by short peptides, peptidomimetics, or small molecules, and will not be emphasized in this review. The second class of PPIs is mediated primarily by structured epitopes, for example a single α-helix in one of the binding partners. Prominent examples of this class include the interactions between p53 and MDM2 [10], Bcl2 and Bak [11], and estrogen receptor and coactivator [12]. This type of PPIs can be effectively inhibited by stapled peptides which, compared to the linear peptide counterparts, have greatly improved binding affinity and metabolic stability. As demonstrated by the Verdine [13] and Walensky groups [14], stapling can also result in sequence- and staple-dependent improvements in cell permeability. Indeed, a stapled peptide against the p53-MDM2 interaction, ALRN-6924 (Aileron Therapeutics), has already entered phase I clinical trial [15]. Readers interested in stapled peptides are directed to two excellent recent reviews [16,17]. Cyclotides, a family of disulphide-rich cyclic peptides, have been employed as an “ultra-stable” scaffold, onto which known binding motifs were grafted. The engineered cyclotides acted as effective PPI inhibitors against, for example, p53-HdmX [18] and CD2-CD58 interactions [19]. For a comprehensive coverage on the use of cyclotides in drug discovery, readers are referred to a recent review by Craik [20]. Finally, the third class of PPIs involve large, flat interfaces formed by multiple motifs that are distal in the primary structure. These PPI interfaces usually lack any major binding pocket or structurally well-defined binding motif and are challenging to target by the conventional drug modalities. This third class is the focus of the current review.
Because the greater majority of PPIs take place intracellularly, the early efforts of PPI inhibitor development focused on small molecules, which have the ability to passively diffuse across the cell membrane and are orally bioavailable. However, it quickly became clear that PPIs are a challenging class of targets for conventional small molecules [21]. Small molecules excel in binding to large, deep binding pockets, such as those found at the surfaces of enzymes, G protein-coupled receptors, and ion channels. These binding sites can partially or completely envelope the small-molecule ligand, thereby generating many points of contact (and a large binding interface) and high binding affinity. In contrast, the same small-molecule ligand can only interact with a flat PPI interface on one of its sides and the two-dimensional interaction results in much fewer points of contact and greatly reduced binding affinity relative to binding in deep pockets. To compensate for the reduced points of contact associated with a two-dimensional binding mode, one must increase the size of the ligand so that its binding surface area matches that of the PPI interface. One way to generate large binding surfaces is to use another protein such as a monoclonal antibody. In fact, monoclonal antibodies against PPI targets such as tumour necrosis factor-α (TNFα) (e.g., Humira) and programmed cell death protein 1 (e.g. Opdivo) have already become blockbuster drugs on the market. Non-immunoglobulin proteins have also been developed as PPI inhibitors [22]. However, protein drugs generally cannot cross the cell membrane and can only be used to target extracellular PPIs. Additionally, protein drugs are prone to elicit immune responses and have high costs of production.
Over the past decade, macrocycles (or large ring-shaped molecules) including cyclic peptides have emerged as an exciting new modality for inhibition of PPI targets [23,24]. With molecular weights typically in the range of 500–2000 Da, macrocycles are 3–5 times larger than the conventional small-molecule drugs and structural studies of macrocycle-protein complexes revealed that macrocycles possess binding surfaces that are similar in size to those of antibodies and native PPI interfaces [25,26]. The cyclic structures of macrocycles also resemble the constrained peptide loops in the complementarity-determining regions of antibodies. Accordingly, macrocycles have demonstrated antibody-like binding affinity and specificity for challenging targets such as the flat interfaces of PPIs. In the meantime, macrocycles retain many of the drug-like properties of small molecules such as metabolic stability and a lack of immunogenicity. However, macrocycles have historically been underexploited as drugs owing to two formidable technical challenges [27]. First, because of their cyclic nature, macrocycles are difficult to rationally design by fragment-based, computational, or other conventional small-molecule drug discovery approaches. Although the remarkable success of a few macrocyclic natural products (e.g. cyclosporine A) in the clinic has inspired decades of intense search for additional macrocyclic natural products as therapeutics [28], many of the naturally occurring macrocycles (or their analogues) are not suitable as drugs because their structural complexity renders their chemical synthesis commercially non-viable. Second, many macrocycles, including almost all cyclic peptides, are impermeable to the cell membrane and, like proteins, cannot access intracellular targets. Fortunately, several powerful combinatorial library technologies have been developed over the past 10 years to synthesize and screen large libraries of macrocyclic compounds against protein targets. Significant advances have also been made in improving the membrane permeability and oral bioavailability of macrocyclic drugs with analysis of current clinically-relevant macrocycles and natural products [29, 30]. This review provides a summary of the key advances in these two areas.
METHODS FOR DISCOVERY OF MACROCYCLIC PPI INHIBITORS
Phage Display Libraries
Originally pioneered by Smith [31], phage display employs peptide sequences expressed as fusions to virion coat proteins of the M13 phage, an engineered Escherichia coli bacteriophage, and later other phage types including T4 [32], T7 [33], and λ phage [34]. In brief, foreign DNA sequences derived from digests of specific genes or synthetic DNA libraries are inserted into the phage genome to code for peptide fusions at the N- or C-terminus of specific coat proteins. The minor coat protein pIII has been the most popular site for peptide insertion and the resulting recombinant phage maintains infectivity with the addition of up to ~30 residues. The first-generation phage displayed macrocycle libraries involved peptides cyclized through a disulphide bond between two cysteine residues flanking the random peptide sequences (Figure 1A). Screening of such a cyclic peptide library by Wells and co-workers resulted in the discovery of a biological probe to elucidate the binding interface between the antibody Fc fragment and Protein A, a component of the Staphylococcus aureus cell wall [35]. From a naïve library of 4 × 109 cyclic peptides, multiple rounds of affinity-based panning were performed to isolate two consensus 18-mer sequences which inhibited the Fc-protein A interaction with an IC50 value of 5 µM. Subsequent modifications yielded a 13-residue cyclic peptide (Figure 1C, compound 1; IC50 = 25 nM) which was later employed as a probe in competition-based assays to discover small-molecule inhibitors of the interaction.
Figure 1.
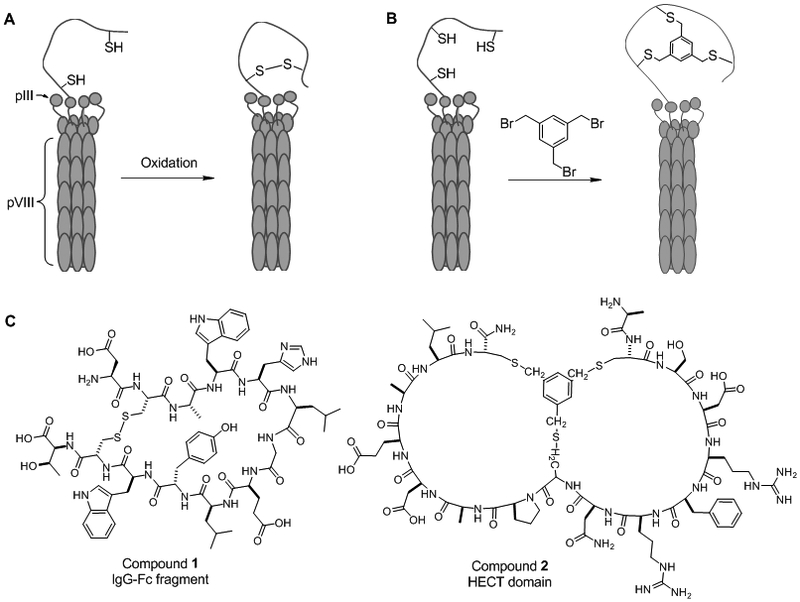
(A,B) Schemes showing examples of phage displayed monocyclic (A) and bicyclic peptide libraries (B). (C) Structures of macrocyclic PPI inhibitors derived from phage display libraries.
A limitation of phage displayed libraries is that generally only proteinogenic amino acids can be used as building blocks. As such, the resulting cyclic peptides, especially conformationally flexible large rings, remain susceptible to proteolytic degradation. High degrees of conformational flexibility also limits the gains in binding affinity and/or specificity provided by macrocyclization. Further structural rigidification of cyclic peptides displayed on phage was recently accomplished through the introduction of a small-molecule scaffold following library expression. By exploiting the unique nucleophilicity of the cysteine side chain, Winter and Heinis [36] treated a phage display library of the general sequence C-X6-C-X6-C (where X is any of the 20 proteinogenic amino acids) with tris(bromomethyl)benzene, resulting in the formation of ~4 × 109 bicyclic peptides (Figure 1B). Three rounds of affinity-based selection resulted in a potent inhibitor against kallikrein, exhibiting an IC50 value of 1.5 nM in an ex vivo intrinsic coagulation assay in human serum. The validity of bicyclic peptides as PPI inhibitors was demonstrated by Mund and co-workers who screened a tris(bromomethyl)benzene-constrained bicyclic peptide library against HECT-type ubiquitin ligases [37]. After multiple rounds of affinity-based selection and amplification, these investigators discovered a lead macrocyclic peptide (compound 2, Figure 1C) which inhibited the ubiquitination of Smurf2 HECT domain (IC50 = 2.5 μM). The excellent specificity of the macrocyclic inhibitor enabled its use as a biological probe in competition assays for the identification of a small-molecule inhibitor against the same interaction.
In an attempt to diversify the building blocks of phage display libraries, Schultz and co-workers introduced unnatural amino acids into phage, by incorporating a nonsense codon (UAA) into the coding sequence and decoding the amber codon with an engineered tRNA charged with the desired unnatural amino acid [38]. This technique can in principle introduce a wide range of unnatural building blocks including pharmacophores into phage displayed macrocycle libraries, but is limited to only one unnatural building block per sequence/library. Alternative small-molecule scaffolds, such as 1,3,5-triacryloyl-1,3,5-triazinane, N,N’,N’’-(benzene-1,3,5-triyl)tris(2-bromoacetamide), and N,N’,N’’-benzene-1,3,5-triyltrisprop-2-enamide have been employed to generate bicyclic peptides with different core structures (i.e., flexibility and size) [39,40]. Phage display libraries containing crown ether-like macrocycles (constructed with N,N’-[1,2-ethanediyl-oxy-2,1-ethanediyl]bis(2-bromoacetamide)) and photo-switchable azobenzene linkers (e.g., 3,3’-bis(sulfonato)-4,4’-bis(chloroacetamido)-azobenzene have also been developed by the Heinis and Derda groups, respectively [41,42].
mRNA Display Libraries
The desire to generate libraries of greater structural diversity than phage display (which is limited to ~109 different molecules by the efficiency of bacterial transformation) led to the development of mRNA display technology by Roberts and Szostak [43]. In an mRNA display library, each peptide is covalently linked to its own encoding mRNA sequence, thus creating a similar genotype-phenotype correlation to that of phage display (Figure 2A). The size of an mRNA display library is limited by the number of encoding DNA molecules that can be chemically synthesized in a research laboratory (~1013 different sequences). The ~10,000-fold increase in library size (relative to phage display) allows mRNA display libraries to exhaustively and redundantly cover the entire sequence space of cyclic peptides of up to 10 residues. The genotype-phenotype connection is accomplished by chemically modifying the 3’-end of each mRNA with the antibiotic puromycin; translation of the modified mRNA results in the formation of an amide bond between puromycin and the nascent polypeptide chain. The resulting RNA-peptide conjugates can be subjected to multiple rounds of affinity-based screening, reverse transcription, amplification, and transcription, allowing the most active compound(s) to be identified. Following isolation of the desired sequence(s), the mRNA construct(s) is converted back into DNA via reverse transcription, amplified using PCR and sequenced. Application of mRNA display libraries for the discovery of macrocyclic ligands was initially achieved through post-translational macrocyclization of the peptide sequence. Typically, a bifunctional crosslinker bearing reactive electrophiles is used to react with the N-terminus and/or the side-chain nucleophiles of cysteine or lysine residues. For example, Schlippe et al. employed dibromoxylene to crosslink cysteine side chains via thioether formation (Figure 2B) [44], whereas Millward et al. used disuccinimidyl glutarate as a general strategy to cyclize between the N-terminus and a lysine side chain near the C-terminus (Figure 2B) [45]. Because of the large library sizes and iterative screening capability, ligands of very high binding affinity and specificity have been identified from mRNA display libraries, often rivalling that of monoclonal antibodies.
Figure 2.
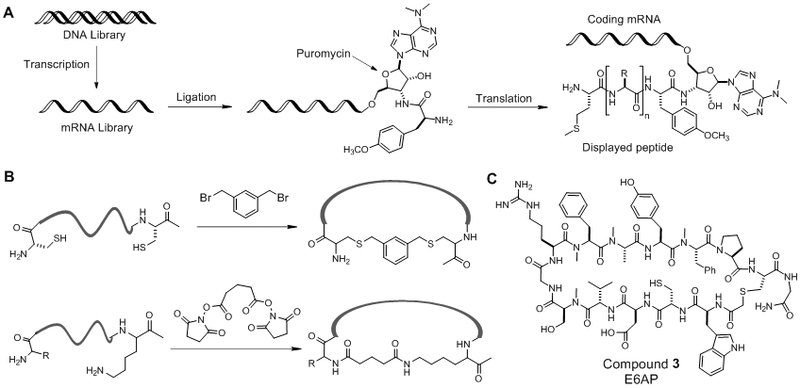
(A) Scheme showing the steps in the synthesis of an mRNA display peptide library. (B) Reactions involved in two different cyclization methods for mRNA display libraries. (C) Structure of a macrocyclic PPI inhibitor against E6AP derived from an mRNA display library.
Since mRNA display libraries are generated in a cell-free system, their chemical space can be expanded beyond the canonical amino acid building blocks through reprogramming the genetic code. Suga and co-workers produced an mRNA display library of natural product-like macrocycles by using the FIT (Flexible In-vitro Translation) system [46]. FIT operates through the use of Flexizyme, an artificial ribozyme which efficiently charges tRNA’s with non-canonical amino acids such as Nα-methylated amino acids and amino acids bearing pharmacophoric side chains [46]. Further work by Iwasaki et al. showed that when the initiator tRNAf-Met was charged with an Nα-chloroacetyl-amino acid, the resulting peptides underwent spontaneous cyclization by forming a thioether bond between the N-terminal chloroacetyl group and a C-terminal cysteine side chain [47]. This macrocyclization method was found to be very general, having few limitations in the primary sequence composition. The Suga group later extended the FIT system into the RaPID (Random non-standard Peptides Integrated Discovery) system by integrating genetic code reprogramming, ribosomal synthesis, and mRNA display [48]. The RaPID system allows the synthesis of very large libraries of natural product-like macrocycles (up to 1012-1014 different compounds), which can be readily screened for binding to challenging protein targets such as PPIs. For example, they generated an mRNA display library of ~1012 macrocycles containing four different Nα-methylated amino acids and subjected the library to multiple rounds of affinity-based screening against ubiquitin ligase E6AP, yielding several inhibitors with sub-to-low nanomolar KD values. The most potent peptide (compound 3, Figure 2C) reduced E6AP-mediated ubiquitination of p53 and Prx1 in a dose-dependent manner [48].
Intein-Based Libraries
Inteins (internal proteins) are protein domains that catalyze post-translational protein splicing, resulting in the ligation of N- and C-terminal flanking sequences (exteins) [49]. Certain inteins, after being split into N- and C-terminal fragments, can reassemble in trans to form a functional domain that catalyzes the ligation of extein sequences fused to the N- and C-terminal intein fragments (i.e., trans-splicing) [50]. Benkovic and co-workers exploited this unique trans-splicing activity to develop the split intein circular ligation of peptides and proteins (SICLOPPS) technology, which permits the synthesis and screening of cyclic peptide libraries inside E. coli cells (Figure 3A) [51]. The diversity of SICLOPPS libraries is limited by the bacterial transformation efficiency, to ~109 members. Whereas other macrocycle libraries can only be screened for binding to protein targets, SICLOPPS libraries are screened phenotypically, e.g., inhibition of intracellular enzyme activities. When integrated with the two-hybrid system, SICLOPPS libraries have been screened for inhibition of intracellular PPIs. For example, Horswill et al. screened ~108 cyclic peptides and identified eight low μM inhibitors that blocked the dimerization of ribonucleotide reductase in an ELISA assay (e.g. compound 4, Figure 3B) [53]. Tavassoli and Benkovic also discovered low μM cyclohexapeptide inhibitors against the homodimerization of aminoimidazole-4-carboamide ribonucleotide transformylase (compound 5, Figure 3B) [54]. Non-canonical amino acids (e.g., 4-benzoylphenylalanine) have been introduced into intein-based libraries through expansion of the genetic code, resulting in the discovery of cyclic peptidyl inhibitors against the HIV protease (compound 6, Figure 3B) [55]. More recently, the intein-based method has been extended to produce and screen macrocycle libraries inside mammalian cells. By adapting the dnaE split inteins previously developed for the bacterial system, Kinsella et al. transfected human B cells with retroviral vectors harbouring ~106 sequences and screened the resulting cyclic peptide library for inhibition of IL-4 signaling. The active hits reduced IL-4 induced transcription of the germ line ε gene [56]. Tavassoli and colleagues reported a cyclic hexapeptide, cyclo(CLLFVY), which inhibits hypoxia inducible factor (HIF) heterodimerisation by binding the PAS‐B domain of HIF‐1α, without affecting HIF‐2α [57]. By screening a cyclononapeptide library, they also discovered cyclic peptides that inhibit the dimerisation of the C‐terminal binding protein (CtBP) transcriptional repressor [58]. One of the cyclic peptides, CP61 (compound 7, Figure 3B), disrupted CtBP homo‐ and heterodimerisation at 20 μM in vitro and inhibited the function of CtBPs in cellulo at 50 μM. Human breast cancer cells treated with the compound showed decreased mitotic fidelity, proliferation, and colony‐forming potential.
Figure 3.
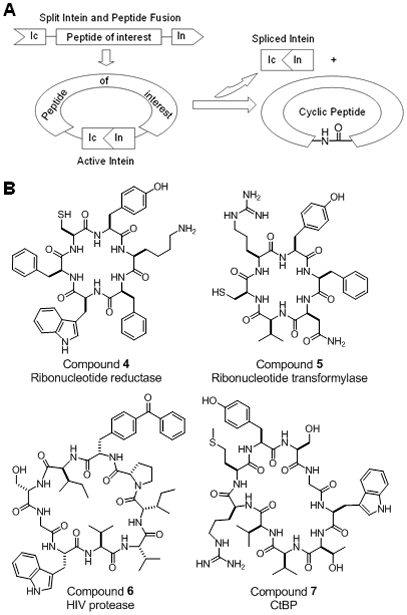
(A) Scheme showing the generation of a SICLOPPS cyclic peptide library. (B) Structures of macrocyclic PPI inhibitors identified from SICLOPPS libraries.
DNA Encoded Libraries
Several investigators independently developed DNA-encoded macrocycle libraries and applied them to ligand discovery. DNA-templated synthesis (DTS), developed by Liu and co-workers [54], has been extensively used for macrocycle-based drug discovery. DTS technology takes advantage of DNA hybridization to enhance the reactivity of individual components. In brief, each building block is covalently attached to a specific encoding DNA sequence, and DNA hybridization brings two reactants into close proximity enhancing their reactivity. At the same time, the DNA tag serves as a record for the reaction steps involved in the construction of the macrocycle. After an active hit is identified from the library, its structure is readily inferred by sequencing the attached DNA sequence. The versatility of DTS in designing and screening macrocyclic PPI inhibitors led to Ensemble Therapeutics leveraging it as its primary drug discovery platform. Seigal et al. [60] screened a focused DTS library against XIAP, an intracellular protein involved in caspase sequestration and apoptosis regulation by binding to the N-termini of caspases with its BIR2/BIR3 domains. Based on a natural tetrapeptide binding motif (AVPI), the researchers designed a library of ~105 unique macrocycles, which were cyclized between azido/alkynyl-containing residues via click chemistry. An initial library hit inhibited the BIR2-SMAC interaction with an IC50 value of 0.36 μM (compound 8, Figure 4A). Optimization by medicinal chemistry led to a dimeric inhibitor which blocked the BIR2/3-SMAC interaction with a nanomolar IC50 value and acted as an effective antitumor agent in a mouse xenograft model (compound 9, Figure 4A) [60]. A different version of DNA-encoded libraries was independently developed by Harbury and co-workers [61,62] and has recently become the core technology for DICE Molecules, a new biopharmaceutical company specializing in macrocyclic drug discovery against challenging targets. Neri and co-workers also pioneered the DNA-encoded self-assembling chemical (ESAC) libraries for lead discovery, which serves as the platform technology behind start-up Philochem AG. [63]. While promising, as of the date of this publication the Harbury and Neri methods have not yet resulted in any published, biologically active macrocyclic PPI inhibitor.
Figure 4.
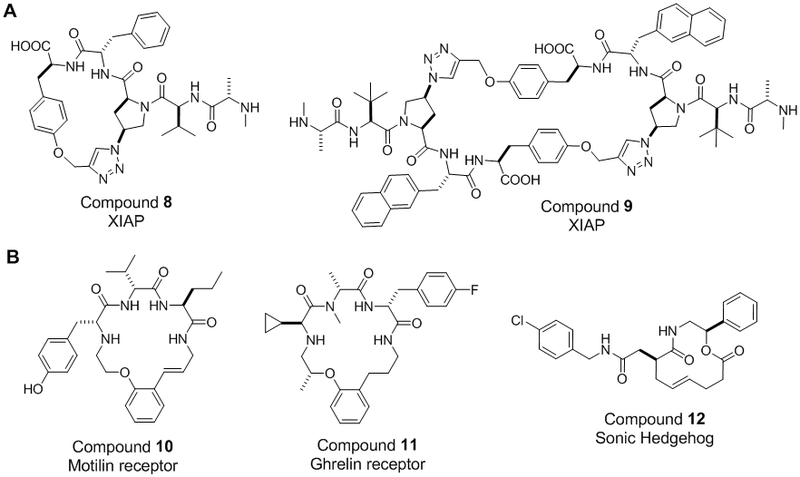
Macrocyclic PPI inhibitors derived from DNA-encoded (A) and DOS libraries (B).
Diversity-Oriented Synthesis Libraries
Diversity-oriented synthesis (DOS) aims to expand the accessible chemical space for drug discovery by generating diverse libraries around natural product-like scaffolds [65]. Starting from a limited number of scaffolds and large sets of diversity elements, DOS can quickly arrive at structurally diverse compound libraries in a limited number of synthetic steps, making it ideal for discovery of initial hit/lead compounds against new targets. For example, researchers at Tranzyme Pharma developed a parallel, solid-phase synthesis of 14- to 18-membered macrocyles by cyclizing a tripeptide sequence around a conformationally rigid scaffold [66]. Fluorescence-based whole-cell screening of the macrocycle library (up to 104 unique members) yielded potent antagonists against the human motilin receptor (e.g. compound 10, Figure 4B). Further optimization through incorporation of unnatural amino acids produced potent, biologically active macrocycles in ex vivo assays [66,67]. Screening of Tranzyme’s proprietary libraries also produced a clinical candidate, TZP-101 (compound 11, Figure 4B), as a ghrelin receptor antagonist [68]. TZP-101 was found to be effective for treating complications of postoperative ileus - a condition that affects the normal mechanical function of the gastrointestinal tract. Some of the macrocyclic hits were found to be orally bioavailable in both rats and monkeys. Meanwhile, researchers at the Broad Institute synthesized a 2070-member aminoalcohol-based macrocycle library in the one macrobead-one stock solution format and used the compounds to generate a small-molecule microarray [69]. Screening of the microarray against Sonic Hedgehog N-terminal peptide (ShhN) produced a moderately potent hit (Figure 4B, compound 12; KD = 9 μM). Other investigators have also applied DOS to prepare various macrocycle libraries [65, 71], although the latter studies have not yet resulted in biologically-active PPI inhibitors. A distinct advantage of DOS is its compatibility with a wide variety of building blocks and chemical reactions. However, DOS library compounds must be screened individually, limiting the number of compounds that can be practically screened in a research laboratory. It is likely that, in order to improve the probability of identifying highly potent PPI ligands, focused DOS libraries will need to be designed for each PPI target.
One-Bead-Two-Compound (OBTC) Libraries
One-bead-one-compound (OBOC) libraries containing up to ~108 different macrocycles, in which each library bead carries multiple copies of a unique macrocycle (e.g., ~100 pmol on a 90-μm TentaGel bead), can be readily prepared by the “split-and-pool” synthesis method of Lam and Houghtern [72,73]. Various strategies were also developed to rapidly screen these libraries against protein targets of interest and isolate the most active hit compound(s) [74]. Structural determination of the macrocyclic hits, however, remained a major challenge until 2006 when Pei and co-workers developed the OBTC libraries [75]. In an OBTC library, each bead is spatially segregated into outer and inner layers with a unique macrocycle synthesized on the bead surface and a corresponding linear peptide synthesized in the inner layer as an encoding sequence (Figure 5A). During library screening against macromolecular targets (e.g., proteins), the target molecules are too large to diffuse into the bead interior and therefore only have access to the macrocycles on the bead surface. Once an active hit is identified, its structure is readily determined by sequencing the linear encoding peptide inside the bead by using the partial Edman degradation-mass spectrometry (PED-MS) technique previously developed in the Kent and Pei laboratories [76,72]. The OBTC method was initially applied to generate large libraries of monocyclic peptides, which were screened for binding to enzymes, cell surface receptors, as well as PPIs. For example, screening a library of cyclohepta- to cyclononapeptides identified an inhibitor against the HIV-1 capsid protein (Figure 5C, compound 13; KD = 0.4 μM), which blocked the capsid protein from binding to lysyl-tRNA synthetase [78]. Wu et al. screened a 3-million member library against the oncogenic K-Ras G12V mutant and identified a modestly potent cyclic peptide inhibitor against K-Ras (Figure 5C, compound 14), which orthosterically blocked the Ras-Raf interaction with an IC50 of 0.7 μM [79].
Figure 5.
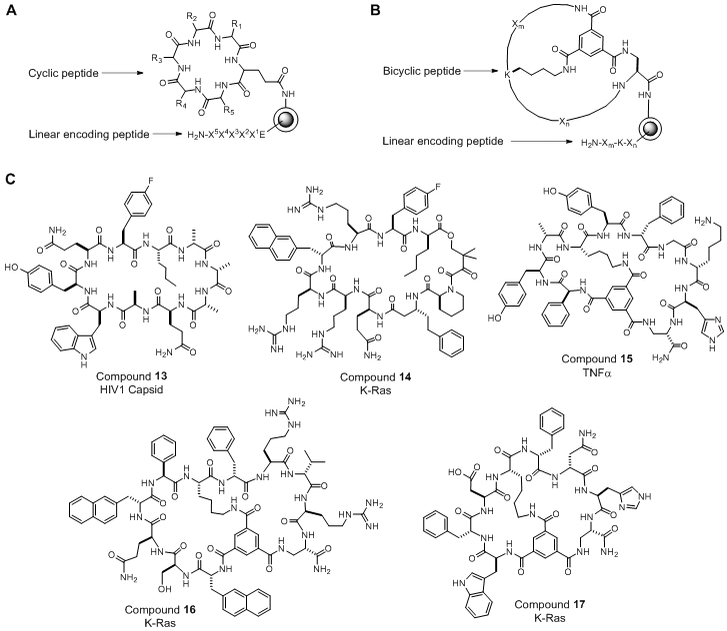
(A) Scheme showing the design of an OBTC monocyclic peptide library. (B) Scheme showing the design of an OBTC bicyclic peptide library. (C) Structures of representative macrocyclic PPI inhibitors obtained from OBTC libraries.
The OBTC methodology was later extended by Lian et al. to synthesize bicyclic peptide libraries [80]. Briefly, after an orthogonally protected linear peptide library is prepared, the linear peptides in the surface layer are selectively deprotected and converted into bicyclic structures by the formation of three amide bonds between a rigid small-molecule scaffold (e.g., trimesic acid) and the N-terminal amine as well as the side chains of a C-terminal (S)-1,3-diaminopropionic acid (Dap) and an internal lysine residue (Figure 5B). With an overall planar geometry, the bicyclic peptides were found to be very effective for binding to flat protein surfaces, such as the PPI interfaces. Screening of a naive library containing 3–5 random residues in each ring against TNFα led to the discovery of a relatively potent low-molecular-weight TNFα antagonist (Figure 5C, compound 15; KD = 0.45 μM), which effectively protected cells from TNFα-induced cell death. Screening of the same library against G12V mutant K-Ras protein resulted in the discovery of two different classes of K-Ras ligands [81]. One class of K-Ras ligands (e.g., compound 16 in Figure 5C) bound apparently to a site at or near the effector protein-binding site and inhibited the Ras-Raf interaction, whereas the second class bound to a yet unidentified site and did not inhibit the Ras-Raf interaction (Figure 5C, compound 17) [82].
METHODS FOR IMPROVING THE CELL PERMEABILITY OF MACROCYCLES
Molecules generally enter mammalian cells through two different mechanisms: passive diffusion - where a molecule spontaneously diffuses across the cell membrane – and active transport – where a molecule binds to a cell-surface receptor and is internalized by an energy-dependent process (e.g. endocytosis). Most of the FDA approved small-molecule drugs enter cells presumably by passive diffusion. In the small-molecule space, Lipinski’s Rule of Five (Ro5 [83], which includes a molecular weight of ≤500, ≤5 hydrogen bond donors, ≤10 hydrogen bond acceptors, and an octanol-water partition coefficient LogP≤5) has provided empirical guidance for achieving passive membrane permeability and desirable pharmacokinetic properties (e.g. oral bioavailability). Macrocycles usually have considerably higher molecular weights (≥500), a higher number of hydrogen bond donors and acceptors, as well as much greater polar surface areas relative to Ro5 molecules. While these attributes make them desirable for targeting the large, flat surface areas at the PPI interfaces, they also greatly impede membrane permeability, as desolvation of a large number of tightly bound water molecules, which is required in order for a molecule to passively diffuse across the hydrophobic region of the lipid bilayer, is energetically very unfavourable. As mentioned earlier, lack of membrane permeability was one of the two key factors that prevented a broader application of macrocycles as therapeutic agents. Since the majority of therapeutically-relevant PPI’s occur inside the cytosol, researchers have been actively exploring both cellular entry mechanisms to improve the membrane permeability and oral bioavailability of macrocycles.
Passive Diffusion
Some naturally-occurring macrocycles, most notably cyclosporine A (CsA, which is a cyclic undecapeptide, Figure 6A), fall well outside the Ro5 space and yet cross the cell membrane by passive diffusion and have respectable oral bioavailability (e.g., F = 29% for CsA [84]). The unique attributes of CsA have inspired medicinal chemists to investigate the molecular basis of these unusual properties and attempt to generalize the mechanism for designing cell-permeable macrocycles. It has been noted that orally bioavailable macrocyclic natural products often contain a high percentage of Nα-methylated peptide bonds. In the case of CsA, 7 out of its 11 peptide bonds are Nα-methylated. Nα-methylation reduces the number of hydrogen bond donors, thus lowering the amount of desolvation energy and increasing the overall hydrophobicity, both of which facilitate passive diffusion through the membrane. However, Lokey and co-workers have shown that global Nα-methylation of all peptide bonds is actually detrimental to membrane permeability and oral bioavailability and, within a given cyclic peptide sequence, the precise number and location of Nα-methylation are critical [85]. Similarly, by generating a combinatorial library of 54 differentially Nα-methylated cyclo(D-Ala-Ala5) peptides, Kessler and colleagues discovered scaffold-specific Nα-methylation patterns that conferred superior intestinal permeability [86]. These and other findings suggest that membrane permeability is likely related to the ability of a macrocycle to adopt alternative conformations in apolar versus aqueous environments. When in an apolar environment (e.g., the hydrophobic region of the lipid bilayer), CsA adopts a closed conformation, forming four intramolecular hydrogen bonds among the backbone amides (Figure 6B), which compensate for the loss of hydrogen bonding interactions with water and effectively lower the desolvation energy [87]. In other words, formation of the intramolecular hydrogen bonds is able to “hide” the hydrogen bond donors and acceptors from the apolar environment. Upon crossing the membrane, CsA relaxes back to the open conformation, in which the backbone amides are available for hydrogen bonding with water and/or protein target. Passive diffusion across the membrane by “beyond the Ro5” macrocycles is certainly not unique to CsA [88]. Ahlbach et al. systematically examined 39 macrocyclic natural products and their derivatives through in silico modeling and identified common structural features amongst the membrane-permeable macrocycles [89]. Most of the membrane-permeable macrocycles formed extensive intramolecular hydrogen bonds and only one of them contained no Nα-methylation. By employing the rules learned from natural products (i.e., Nα-methylation, intramolecular hydrogen bonds, and side-chain hydrophobicity), Hoffman and coworkers [85] as well as investigators at GlaxoSmithKline [86] and Pfizer [87] have designed a number of cyclic peptide model systems with excellent membrane permeability and oral bioavailability. Fairlie and colleagues also dramatically improved the oral bioavailability of a Sanguinamide A derivative through strategic occlusion of backbone amides from solvent by using hydrophobic side-chains (e.g., substitution of tert-leucine for alanine) [92].
Figure 6.
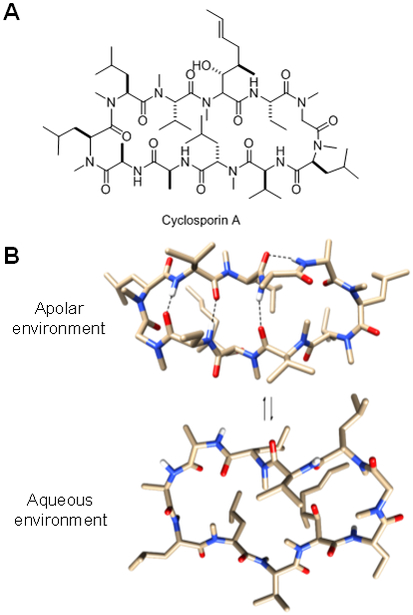
(A) Structure of CsA. (B) CsA exists in a closed conformation when in apolar environment and an open conformation in water.
Given the dramatic effects of Nα-methylation on both bioavailability and proteolytic stability, researchers have developed both chemical and biological methods to introduce Nα-methylated residues into macrocyclic PPI inhibitors. Although less reactive than canonical amino acids, Nα-methylated amino acids can be directly incorporated into macrocycles during solid-phase synthesis by using standard coupling reagents. White et al. also developed a methodology to exhaustively methylate all accessible amide bonds of support-bound peptides [93]. Kodadek and co-workers prepared macrocyclic peptoid libraries containing diverse Nα-alkylated glycine residues while employing traditional solid-phase synthesis and cyclization techniques [95]. Nα-Methylated amino acids can be readily incorporated into ribosomally synthesized macrocycle libraries by applying the RaPID platform [48].
The challenge inherent in developing passively diffusible macrocycles is how to properly balance membrane permeability against aqueous solubility and the ability of the macrocycle to engage a wide variety of protein targets. The very attributes required for membrane permeation (i.e., limited number of hydrogen bond donors/acceptors and lipophilicity) places a major limitation on the aqueous solubility and the types of target sites that they are capable of engaging. Essentially all of the studies described in the preceding section have focused on model systems, with the goals of achieving membrane permeability/oral bioavailability and/or understanding the structural basis of membrane permeability/oral bioavailability. The membrane-permeable cyclic peptides that have been designed so far are predominantly hydrophobic macrocycles, few of which have been evaluated for binding to protein targets. It is likely that these hydrophobic macrocycles will be restricted to targeting hydrophobic PPI interfaces.
Active Transport
A potentially more general approach to imparting membrane permeability is to covalently attach a macrocycle of interest to a cell-penetrating peptide (CPP). CPPs are short peptides (typically 5–30 amino acids) that are capable of crossing the plasma membrane of eukaryotic cells without causing significant damage to the cell membrane [96]. Out of the >1800 different CPPs discovered to date [97], the arginine-rich CPPs such as Tat, octaarginine (R8), and penetratin have been most widely used for delivering cargos including small-molecule drugs, peptides, proteins, nucleic acids, and nanoparticles into mammalian cells. While the detailed mechanism of their cellular entry remains inadequately defined, it is now generally accepted that the CPPs and CPP-cargo conjugates enter cells primarily by endocytosis mechanisms, especially at low concentrations (≤10 μM) [98]. By attaching an R8 sequence to a Grb2 SH2 domain inhibitor, cyclo(AApYVNFFE), Zhang et al. obtained a cell-permeable cyclic peptide inhibitor that disrupted the actin filaments and inhibited the proliferation of breast cancer cells at 20 μM concentration [99]. Desimmie et al. conjugated Tat to a disulfide cyclized nonapeptide inhibitor against the LEDGF/p75 interaction identified from a phage display library (CVMGHPLWC) and the peptide conjugate inhibited HIV replication with an IC50 of 19 μM in cell culture [100]. A major limitation of the CPPs, however, has been their poor cytosolic delivery efficiencies (defined as the ratio of cytosolic over extracellular cargo concentration), which are generally below 5% [101]. The vast majority of the internalized CPPs (or CPP-cargo conjugates) is usually entrapped inside the endosomes. As a result, very high doses are typically required to achieve therapeutic effects, leaving little therapeutic window.
Recently, several research groups independently reported that cyclization of Arg-rich CPPs improved their cellular uptake efficiencies relative to the linear counterparts [102,103,104]. In particular, Pei and co-workers discovered a family of small cyclic peptides as exceptionally active CPPs [e.g., cyclo(FΦRRRRQ), where Φ is 2-naphthylalanine; CPP1 in Figure 7], having cytosolic delivery efficiencies up to 60-fold greater than that of Tat [106,107]. These cyclic CPPs not only serve as powerful vehicles for efficient delivery of cargo molecules (including small molecules, peptides, and proteins) into the cytosol and nucleus of mammalian cells, but have also provided the much needed mechanistic probes which have, for the first time, led to a well-defined mechanism of action for the CPPs [107]. The cyclic CPPs (and the CPP-cargo conjugates) enter cells by endocytosis and efficiently escape from the early endosome by inducing CPP-enriched regions of the endosomal membrane to bud off as small, unstable vesicles which subsequently collapse inside the cytosol. As expected, the cyclic CPPs also have greatly improved metabolic stability over the linear CPPs.
Figure 7.
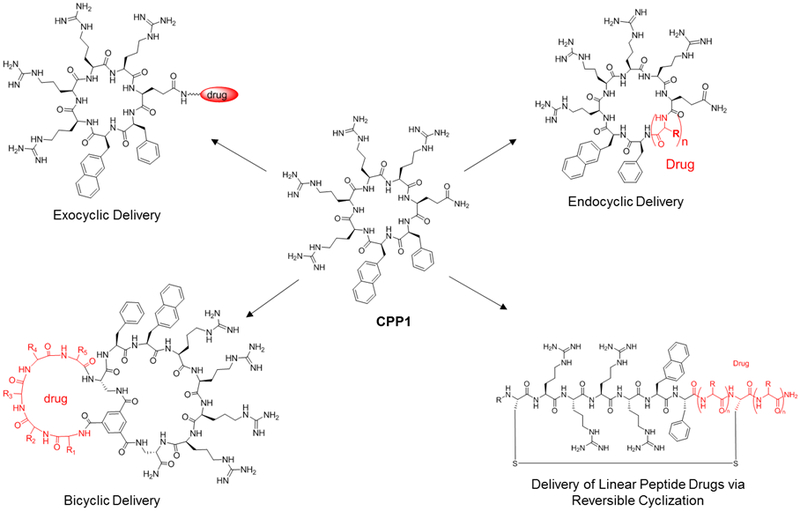
Scheme showing the structure of CPP1 and the four different cargo delivery modes of cyclic CPPs.
The cyclic CPPs provide a flexible platform for delivering cargos in at least four different modes (Figure 7). The most straightforward and cargo tolerant mode is the exocyclic delivery mode, in which the cargo molecule is covalently attached to the side chain of a glutamic acid (Figure 7). A variety of small molecules, linear peptides, and proteins have been efficiently delivered into mammalian cells in vitro using this method [105,106]. Cyclic peptides have been effectively delivered by fusing them with a cyclic CPP to form a bicyclic system (bicyclic delivery, Figure 7). By using this method, Lian et al. generated a potent, highly selective, and cell-permeable bicyclic peptide inhibitor against protein tyrosine phosphatase 1B (PTP1B) (Figure 8, compound 18; KD = 37 nM), which potentiated insulin signaling in HepG2 cells at nanomolar concentrations [103]. Similarly, fusion of a previously discovered, membrane-impermeable cyclic peptide inhibitor of Pin1 with cyclic CPP1 produced a potent bicyclic Pin1 inhibitor that was highly active in cell culture experiments (Figure 8, compound 19; KD = 72 nM). The generality of the bicyclic delivery strategy was further demonstrated by the synthesis of a combinatorial library of 5.7 million cell-permeable bicyclic peptides [109]. Screening of the bicyclic peptide library against the G12V K-Ras protein identified a K-Ras inhibitor that blocked the Ras-Raf interaction with an IC50 value of 3.4 μM, inhibited MEK and AKT phosphorylation, and induced apoptosis of lung cancer cells at low μM concentrations (compound 20, Figure 8,). Short peptidyl cargos have also been directly inserted into the cyclic CPP ring (endocylcic delivery, Figure 7). For example, Upadhyaya et al. synthesized an OBTC library of 1.5 million monocyclic peptides by integrating a CPP-like motif and K-Ras binding sequences into a single ring [81]. Screening of the library identified a cycloundecapeptide which inhibited the Ras-Raf interaction with moderate potency (IC50 = 650 nM) and was marginally cell-permeable. Optimization of the peptide led to an improved compound, cyclorasin 9A5 (compound 21, Figure 8), which has both improved potency against the Ras-Raf interaction (IC50 = 120 nM) and cell-permeability. Cyclorasin 9A5 potently inhibited the activation of MEK, ERK½, and AKT and induced apoptosis of lung cancer cells (LD50 ~3 μM). Finally, some target proteins (e.g., PDZ domains) require the peptide ligand in an extended conformation for binding and cyclization of the peptide ligand reduces or abolishes target binding. Qian et al. reported a reversible cyclization strategy to deliver these linear peptide ligands (Figure 7) [110]. In this case, a peptide of interest is fused with a CPP motif at its N- or C-terminus and the peptide fusion is cyclized with a disulfide bond between the N-terminal (or C-terminal) thiol and the side chain of an internal cysteine. The resulting cyclic peptide has greatly improved cellular uptake efficiency and proteolytic stability. Upon entering the mammalian cytosol (where the target protein is localized), the disulfide is reduced by intracellular glutathione to release the biologically active linear peptide for binding to the intended target. As a proof of principle application, the investigators designed a peptidyl inhibitor against the PDZ domain of CFTR-associated ligand (CAL-PDZ), a protein which binds to the C-terminus of CFTR and chaperones CFTR to the lysosome for degradation (compound 22, Figure 8). Treatment of lung epithelial cells harboring a defective CFTR mutant (ΔF508) with the peptide inhibitor reduced the amount of lysosomal degradation, thereby increasing the amount of membrane-bound CFTR and improving the chloride ion channel activity. This results suggests an improved version of the peptide inhibitor may provide a novel treatment of cystic fibrosis [110]. An improved and more general reversible cyclization method for intracellular delivery of any linear peptidyl drug was recently developed and applied to generate a bicyclic peptide inhibitor against the NEMO-IKK interaction [106].
Figure 8.
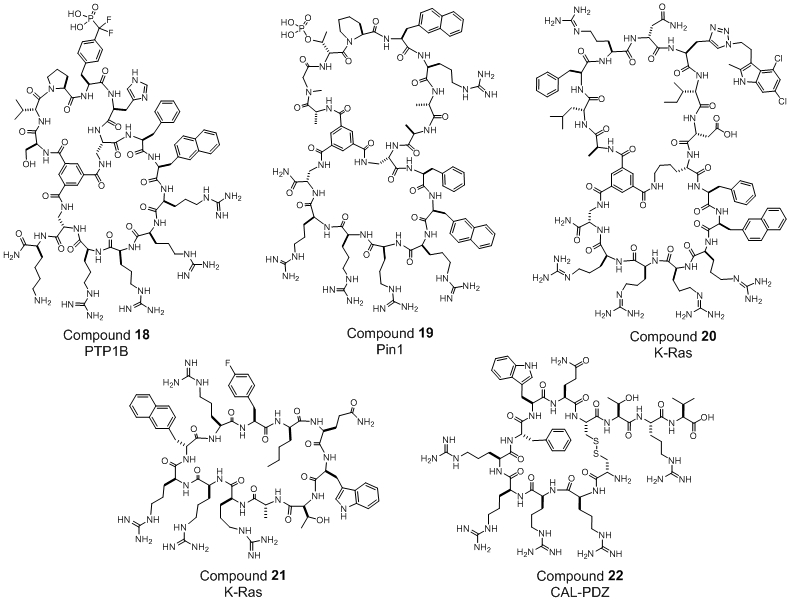
Examples of cell-permeable macrocyclic inhibitors which enter cells by active transport and target intracellular proteins.
CONCLUDING REMARKS AND OUTLOOK
The challenge of targeting intracellular PPIs has fueled an explosive growth in research activities on macrocycles during the past decade. These efforts have now led to several powerful and largely complementary technologies for peptidic macrocycle library synthesis and screening. The availability of these technologies has made the discovery of macrocyclic ligands against protein targets (including those involved in PPIs) a relatively routine exercise, although it still requires considerable effort and resources to obtain a macrocyclic ligand that is capable of binding to the intended protein target with antibody-like affinity and specificity. Significant progress has also been made on understanding the structural features that facilitate membrane permeability and the mechanism by which peptides cross the cell membrane, both by passive diffusion and through active transport. Integration of the technologies from the above two fields has already delivered macrocyclic PPI inhibitors that are highly potent and metabolically stable in cellular assays, with some having demonstrated impressive pharmacodynamic endpoints in animal models. These recent developments suggest that intracellular PPI targets, once considered as “undruggable”, may be druggable after all. Future studies should further assess the generality of macrocycles as PPI inhibitors and advance some of the macrocyclic PPI inhibitors into the clinic.
Acknowledgements
We thank the Pei group members for their contributions to the studies covered in this review.
Funding Information
Work in the Pei laboratory has been supported by grants from the NIH (GM110208 and GM062820).
Abbreviations:
- CPP
cell-penetrating peptide
- CsA
cyclosporine A
- DOS
diversity-oriented synthesis
- DTS
DNA-templated synthesis
- HIF
hypoxia inducible factor
- OBOC
one-bead-one-compound
- OBTC
one-bead-two-compound
- PPI
Protein-protein interaction
- pY
phosphotyrosine
- RaPID
Random non-standard Peptides Integrated Discovery
- Ro5
Rule of Five
- SICLOPPS
split intein circular ligation of peptides and proteins
- TNFα
tumour necrosis factor-α
References
- 1.Venkatesan K, Rual J-F, Vazquez A, Stelzl U, Lemmens I, Hirozane-Kishikawa T, et al. (2009) An empirical framework for binary interactome mapping. Nat. Methods 6, 83–90 doi: 10.1038/nmeth.1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scott DE, Bayly AR, Abell C, and Skidmore J (2016) Small molecules, big targets: drug discovery faces the protein-protein interaction challenge. Nat. Rev. Drug Disc 15∫, 533–550 doi: 10.1038/nrd.2016.29 [DOI] [PubMed] [Google Scholar]
- 3.Giordanetto F, Schäfer A, and Ottmann C (2014) Stabilization of protein-protein interactions by small molecules. Drug Discov. Today: Technologies 19, 1812–1821. doi: 10.1016/j.drudis.2014.08.005 [DOI] [PubMed] [Google Scholar]
- 4.Arkin MR, Tang Y, and Wells JA (2014) Small-molecule inhibitors of protein-protein interactions: progressing toward the reality. Chem. Biol 21, 1102–1114 doi: 10.1016/j.chembiol.2014.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cohen GB, Ren R, and Baltimore D (1995) Modular binding domains in signal transduction proteins. Cell 80, 237–248 doi: 10.1016/0092-8674(95)90406-9 [DOI] [PubMed] [Google Scholar]
- 6.Hung AY, and Sheng M (2002) PDZ domains: Structural modules for protein complex assembly. J. Biol. Chem 277, 5699–5702 doi: 10.1074/jbc.R100065200 [DOI] [PubMed] [Google Scholar]
- 7.Waksman G, Shoelson SE, Pant N, Cowburn D, and Kuriyan J (1993) Binding of a high affinity phosphotyrosyl peptide to the Src SH2 domain: crystal structures of the complexed and peptide-free forms. Cell 72, 779–790 doi: 10.1016/0092-8674(93)90405-F [DOI] [PubMed] [Google Scholar]
- 8.Doyle DA, Lee A, Lewis J, Kim E, Sheng M, and MacKinnon R (1996) Crystal structures of a complexed and peptide-free membrane protein-binding domain: molecular basis of peptide recognition by PDZ. Cell 85, 1067–1076 doi: 10.1016/S0092-8674(00)81307-0 [DOI] [PubMed] [Google Scholar]
- 9.Morais Cabral JH, Petosa C, Sutcliffe MJ, Raza S, Byron O, Poy F, et al. (1996) Crystal structure of a PDZ domain. Nature 382, 649–652 doi: 10.1038/382649a0 [DOI] [PubMed] [Google Scholar]
- 10.Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ et al. (1996) Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 274, 948–953 doi: 10.1126/science.274.5289.948 [DOI] [PubMed] [Google Scholar]
- 11.Sattler M, Liang H, Nettesheim D, Meadows RP, Harlan JE, Eberstadt M et al. (1997) Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science 275, 983–986 doi: 10.1126/science.275.5302.983 [DOI] [PubMed] [Google Scholar]
- 12.Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, Greene GL (1998) The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 95, 927–937 doi: 10.1016/S0092-8674(00)81717-1 [DOI] [PubMed] [Google Scholar]
- 13.Chu Q, Moellering RE, Hilinski GJ, Kim Y-W, Grossman TN, Yeh JT et al. (2015) Towards understanding cell penetration by stapled peptides. Med. Chem. Commun 6, 111–119 doi: 10.1039/C4MD00131A [DOI] [Google Scholar]
- 14.Bird GH, Mazzola E, Opoku-Nsiah K, Lammert MA, Godes M, Neuberg DS et al. (2016) Biophysical determinants for cellular uptake of hydrocarbon-stapled peptide helices. Nat. Chem. Biol. 12, 845–852 doi: 10.1038/nchembio.2153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chang YS, Graves B, Guerlavais V, Tovar C, Packman K, To K-H et al. (2013) Stapled α-helical peptide drug development: A potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc. Natl. Acad. Sci. USA 110, E3445–E3454 doi: 10.1073/pnas.1303002110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walensky LD, and Bird DH (2014) Hydrocarbon-stapled peptides: principles, practice, and progress. J. Med. Chem 57, 6275–6288 doi: 10.1021/jm4011675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cromm PM, Spiegel J, and Grossmann TN (2015) Hydrocarbon stapled peptides as modulators of biological function. ACS Chem. Biol 10, 1362–1375 doi: 10.1021/cb501020r [DOI] [PubMed] [Google Scholar]
- 18.Ji Y, Majumder S, Millard M, Borra R, Bi T, Elnagar AY et al. (2013) In vivo activation of the p53 tumor suppressor pathway by an engineered cyclotide. J. Am. Chem. Soc 135, 11623–11633 doi: 10.1021/ja405108p [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sable R, Durek T, Taneja V, Craik DJ, Pallela S, Gauthier T, et al. (2016) Constrained cyclic peptides as immunomodulatory inhibitors of the CD2:CD58 protein-protein interaction. ACS Chem. Biol 11, 2366–2374 doi: 10.1021/acschembio.6b00486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Northfield SE, Wang CK, Schroeder CI, Durek T, Kan M-W, Swedberg JE, et al. (2014) Disulfide-rich macrocyclic peptides as templates in drug design. Eur. J. Med. Chem 77, 248–257 doi: 10.1016/j.ejmech.2014.03.011 [DOI] [PubMed] [Google Scholar]
- 21.Arkin MR, and Wells JA (2004) Small-molecule inhibitors of protein-protein interactions: progressing toward the dream. Nat. Rev. Drug Discov 3, 301–317 doi: 10.1038/nrd1343 [DOI] [PubMed] [Google Scholar]
- 22.Binz H, Amstutz P, and Pluckthun A (2005) Engineering novel binding proteins from nonimmunoglobulin domains. Nat. Biotech 23, 1257–1268 doi: 10.1038/nbt1127 [DOI] [PubMed] [Google Scholar]
- 23.Marsault E, and Peterson ML (2011) Macrocycles are great cycles: applications, opportunities, and challenges of synthetic macrocycles in drug discovery. J. Med. Chem 54, 1961–2004 doi: 10.1021/jm1012374 [DOI] [PubMed] [Google Scholar]
- 24.Mallinson J, and Collins I (2012) Macrocycles in new drug discovery. Fut. Med. Chem 4, 1409–1438 doi: 10.4155/FMC.12.93 [DOI] [PubMed] [Google Scholar]
- 25.Villar EA, Beglov D, Chennamadhavuni S, Porco JA Jr., Kozakov D, Vajda S, and Whitty A (2014) How proteins bind macrocycles. Nat. Chem. Biol 10 723–731 doi: 10.1038/nchembio.1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doak BC, Zheng J, Dobritzsch D, and Kihlberg J (2016) How Beyond Rule of 5 Drugs and Clinical Candidates Bind to Their Targets J. Med. Chem 59, 2312–2327 doi: 10.1021/acs.jmedchem.5b01286 [DOI] [PubMed] [Google Scholar]
- 27.Driggers EM, Hale SP, Lee J, and Terrett NK (2008) The exploration of macrocycles for drug discovery - an underexploited structural class. Nat. Rev. Drug Discov 7, 608–624 doi: 10.1038/nrd2590 [DOI] [PubMed] [Google Scholar]
- 28.Newman DJ, and Cragg GM (2012) Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod 75, 311–335 doi: 10.1021/np200906s [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Doak BC, Over B, Giordanetto F, and Kihlberg J (2014) Oral Druggable Space beyond the Rule of 5: Insights from Drugs and Clinical Candidates. Chem. Biol 21, 1115–1142 doi: 10.1016/j.chembiol.2014.08.013 [DOI] [PubMed] [Google Scholar]
- 30.Bockus AT, McEwen CM, and Lokey RS (2013) Form and function in cyclic peptide natural products: a pharmacokinetic perspective. Curr. Top. Med. Chem 13, 821–836 doi: 10.2174/1568026611313070005 [DOI] [PubMed] [Google Scholar]
- 31.Smith GP (1985) Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science 288, 1315–1317 doi: 10.1126/science.4001944 [DOI] [PubMed] [Google Scholar]
- 32.Krumpe LR, Atkinson AJ, Smythers GW, Kandel A, Schumacher KM, and McMahon JB (2006) T7 lytic phage displayed peptide libraries exhibit less sequence bias than M13 filamentous phage-displayed peptide libraries. Proteomics 6, 4210–4222, doi: 10.1002/pmic.200500606 [DOI] [PubMed] [Google Scholar]
- 33.Wu J, Tu C, Yu X, Zhang M, Zhang N, and Zhao M (2007) Bacteriophage T4 nanoparticle capsid surface SOC and HOC bipartite display with enhanced classical swine fever virus immunogenicity: a powerful immunological approach. J. Virol. Meth 139, 50–60, doi: 10.1016/j.jviromet.2006.09.017 [DOI] [PubMed] [Google Scholar]
- 34.Cicchini C, Ansuini H, Amicone L, Alonzi T, Nicosia A, Cortese R (2002) Searching for DNA-protein interactions by lambda phage display. J. Mol. Biol 322, 697–706 doi: 10.1016/S0022-2836(02)00851-3. [DOI] [PubMed] [Google Scholar]
- 35.DeLano WL, Ultsch MH, de Vos AM, and Wells JA (2000) Convergent solutions to binding at a protein-protein interface. Science 287, 1279–1283 doi: 10.1126/science.287.5456.1279 [DOI] [PubMed] [Google Scholar]
- 36.Heinis C, Rutherford T, Freund S, and Winter G (2009) Phage-encoded combinatorial libraries based on bicyclic peptides. Nat. Chem. Biol 5, 502–507 doi: 10.1038/nchembio.184 [DOI] [PubMed] [Google Scholar]
- 37.Mund T, Lewis MJ, Maslen S, and Pelham HR (2014) Peptide and small molecule inhibitors of HECT-type ubiquitin ligases. Proc. Nat. Acad. Sci. U.S.A. 111, 16736–16741 doi: 10.1073/pnas.1412152111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tian F, Tsao M, and Schultz PG (2004) A phage display system with unnatural amino acids. J. Am. Chem. Soc 126, 15962–15963 doi: 10.1021/ja045673m [DOI] [PubMed] [Google Scholar]
- 39.Rim C, Lahey LJ, Patel VG, Zhang H, and Son DY (2009) Thiol-ene reactions of 1,3,5-triacryloylhexahydro-1,3,5-triazine (TAT): facile access to functional tripodal thioethers. Tetrahedron Lett. 50, 745–747 doi: 10.1016/j.tetlet.2008.11.094 [DOI] [Google Scholar]
- 40.Chen S, Morales-Sanfrutos J, Angelini A, Cutting B, and Heinis C (2012) Structurally diverse cyclisation linkers impose different backbone conformations in bicyclic peptides. ChemBioChem 13, 1032–1038 doi: 10.1002/cbic.201200049 [DOI] [PubMed] [Google Scholar]
- 41.Fukunaga K,. Hatanaka T, Ito Y,. Minami M, and Taki M (2014) Construction of a crown ether-like supramolecular library by conjugation of genetically-encoded peptide linkers displayed on bacteriophage T7. Chem. Comm 50, 3921–3923 doi: 10.1039/C4CC00811A [DOI] [PubMed] [Google Scholar]
- 42.Jafari MR, Deng L, Kitov PI, Ng S, Matochko WL, Tjhung KF, et al. (2014) Discovery of light-responsive ligands through screening of a light-responsive genetically encoded library. ACS Chem. Biol 9, 443–450 doi: 10.1021/cb4006722 [DOI] [PubMed] [Google Scholar]
- 43.Roberts RW, and Szostak JW (1997) RNA-peptide fusions for the in vitro selection of peptides and proteins. Proc. Natl. Acad. Sci. U.S.A. 94, 12297–12302 doi: 10.1073/pnas.94.23.12297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guillen Schlippe YV, Hartman MCT, Josephson K, and Szostak JW (2012) In vitro selection of highly modified cyclic peptides that act as tight binding inhibitors. J. Am. Chem. Soc 134, 10469–10477 doi: 10.1021/ja301017y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Millward SW, Takahashi TT, and Roberts RW (2005) A general route for post-translational cyclization of mRNA display libraries. J. Am. Chem. Soc 127, 14142–14143 doi: 10.1021/ja054373h [DOI] [PubMed] [Google Scholar]
- 46.Goto Y, Katoh T, and Suga H (2011) Flexizymes for genetic code reprogramming. Nat. Protoc 6, 779–790 doi: 10.1038/nprot.2011.331 [DOI] [PubMed] [Google Scholar]
- 47.Iwasaki K, Goto Y, Katoh T, and Suga H (2012) Selective thioether macrocyclization of peptides having the N-terminal 2-chloroacetyl group and competing two or three cysteine residues in translation. Org. Biomol. Chem 10, 5783–5786 doi: 10.1039/C2OB25306B [DOI] [PubMed] [Google Scholar]
- 48.Yamagishi Y, Shoji I, Miyagawa S, Kawakami T, Katoh T, Goto Y, et al. (2011) Natural product-like macrocyclic N-methyl-peptide inhibitors against a ubiquitin ligase uncovered from a ribosome-expressed de novo library. Chem. Biol 18, 1562–1570 doi: 10.1016/j.chembiol.2011.09.013 [DOI] [PubMed] [Google Scholar]
- 49.Kane PM, Yamashiro C,T, Wolczyk DF, Neff N, Goebl M, and Stevens TH (1990) Protein splicing converts the yeast TFP1 gene product to the 69-kD subunit of the vacuolar H(+)-adenosine triphosphatase. Science 250, 651–657 doi: 10.1126/science.2146742 [DOI] [PubMed] [Google Scholar]
- 50.Wu H, Hu Z, and Liu XQ (1998) Protein trans-splicing by a split intein encoded in a split DnaE gene of Synechocystis sp. PCC6803. Proc. Nat. Acad. Sci. U.S.A. 95, 9226–9231 PMCID: PMC21320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Scott CP, Abel-Santos E, Wall M, Wahnon DC, and Benkovic SJ (1999) Production of cyclic peptides and proteins in vivo. Proc. Nat. Acad. Sci. U.S.A. 96, 13638–13643 doi: 10.1073/pnas.96.24.13638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vidal M, Brachmann RK, Fattaey A, Harlow E, and Boeke JD (1996) Reverse two-hybrid and one-hybrid systems to detect dissociation of protein-protein and DNA-protein interactions. Proc. Nat. Acad. Sci. U.S.A. 93, 10315–10320 doi: 10.1073/pnas.96.24.13638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Horswill AR, Savinov SN, and Benkovic SJ (2004) A systematic method for identifying small-molecule modulators of protein-protein interactions. Proc. Nat. Acad. Sci. U.S.A. 101, 15591–15596 doi: 10.1073/pnas.0406999101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tavassoli A, and Benkovic SJ (2005) Genetically selected cyclic-peptide inhibitors of AICAR transformylase homodimerization. Angew. Chem. Int. Ed 44, 2760–2763 doi: 10.1002/anie.200500417 [DOI] [PubMed] [Google Scholar]
- 55.Young TS, Young DD, Ahmad I, Louis JM, Benkovic SJ, and Schultz PG (2011) Evolution of cyclic peptide protease inhibitors. Proc. Nat. Acad. Sci. U.S.A. 108, 11052–11056 doi: 10.1073/pnas.1108045108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kinsella TM, Ohashi CT, Harder AG, Yam GC, Li W, Peelle B, et al. (2002) Retrovirally delivered random cyclic peptide libraries yield inhibitors of interleukin-4 signaling in human B cells. J. Biol. Chem 277, 37512–37518 doi: 10.1074/jbc.M206162200 [DOI] [PubMed] [Google Scholar]
- 57.Miranda E, Nordgren IK, Male AL, Lawrence CE, Hoakwie F, Cuda F, et al. (2013) A cyclic peptide inhibitor of HIF-1 heterodimerization that inhibits hypoxia signaling in cancer cells. J. Am. Chem. Soc 135, 10418–10425 doi: 10.1021/ja402993u [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Birts CN, Nijjar SK, Mardle CA, Hoakwie F, Duriez PJ, Blaydes JP, et al. (2013) A cyclic peptide inhibitor of C-terminal binding protein dimerization links metabolism with mitotic fidelity in breast cancer cells. Chem. Sci 4, 3046–3057 doi: 10.1039/C3SC50481F [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gartner ZJ, Tse BN, Grubina R, Doyon JB, Snyder TM, and Liu DR (2004) DNA-Templated organic synthesis and selection of a library of macrocycles. Science 305,1601–1605 doi: 10.1126/science.1102629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Seigal BA, Connors WH, Fraley A, Borzelli RM, Carter PH, Emanuel SL, et al. (2015) The discovery of macrocyclic XIAP antagonists from a DNA-programmed chemistry library, and their optimization to give lead compounds with in vivo antitumor activity. J. Med. Chem 58, 2855–2861 doi: 10.1021/jm501892g [DOI] [PubMed] [Google Scholar]
- 61.Halpin DR, and Harbury PB (2004) DNA display I. Sequence-encoded routing of DNA populations. PLoS Biol. 2, E174 doi: 10.1371/journal.pbio.0020173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Halpin DR, and Harbury PB (2004). DNA display II. Genetic manipulation of combinatorial chemistry libraries for small-molecule evolution. PLoS Biol. 2, E174 doi: 10.1371/journal.pbio.0020174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Melkko S, Scheuermann J, Dumelin CE, and Neri D (2004) Encoded self-assembling chemical libraries. Nat. Biotech 22, 568–574 doi: 10.1038/nbt961 [DOI] [PubMed] [Google Scholar]
- 64.Mannocci L, Zhang Y, Scheuermann J, Leimbacher M, De Bellis G, Rizzi E, et al. (2008) High-throughput sequencing allows the identification of binding molecules isolated from DNA-encoded chemical libraries. Proc. Nat. Acad. Sci. U.S.A. 105, 1760–17675 doi: 10.1073/pnas.0805130105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schreiber SL (2000) Target-oriented and diversity-oriented organic synthesis in drug discovery. Science 287, 1964–1969 doi: 10.1126/science.287.5460.1964 [DOI] [PubMed] [Google Scholar]
- 66.Marsault E, Hoveyda HR, Peterson ML, Saint-Louis C, Landry A, Vezina M, et al. (2006) Discovery of a new class of macrocyclic antagonists to the human motilin receptor. J. Med. Chem 49, 7190–7197 doi: 10.1021/jm0606600 [DOI] [PubMed] [Google Scholar]
- 67.Marsault E, Benakli K, Beaubien S, Saint-Louis C, Déziel R, and Fraser G Potent macrocyclic antagonists to the motilin receptor presenting novel unnatural amino acids. Bioorg. Med. Chem. Lett 2007, 17, 4187–4190 doi: 10.1016/j.bmcl.2007.05.043 [DOI] [PubMed] [Google Scholar]
- 68.Hoveyda HR, Marsault E, Gagnon R, Mathieu AP, Vézina M, Landry A, et al. (2011) Optimization of the potency and pharmacokinetic properties of a macrocyclic ghrelin receptor agonist (Part I): development of ulimorelin (TZP-101) from hit to clinic. J. Med. Chem 54, 8305–8320 doi: 10.1021/jm2007062 [DOI] [PubMed] [Google Scholar]
- 69.Peng LF, Stanton BZ, Maloof N, Wang X, and Schreiber SL (2009) Syntheses of aminoalcohol-derived macrocycles leading to a small-molecule binder to and inhibitor of sonic hedgehog. Bioorg. Med. Chem. Lett 19, 6319–6325 doi: 10.1016/j.bmcl.2009.09.089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Beckmann HSG, Nie F, Hagerman CE, Johansson H, Tan YS, Wilcke D, et al. (2013) A strategy for the diversity-oriented synthesis of macrocyclic scaffolds using multidimensional coupling. Nat. Chem 5, 861–867 doi: 10.1038/nchem.1729 [DOI] [PubMed] [Google Scholar]
- 71.Collins S, Barlett S, Nie F, Sore HF, and Spring DR (2016) Diversity-oriented synthesis of macrocycle libraries for drug discovery and chemical biology. Synthesis 48, 1457–1473 doi: 10.1055/s-0035-1561414 [DOI] [Google Scholar]
- 72.Lam KS, Salmon SE, Hersh EM, Hruby V, Kazmierski WM, and Knapp RJ (1991) A new type of synthetic peptide library for identifying ligand-binding activity. Nature 354 82–84 doi: doi: 10.1038/354082a0 [DOI] [PubMed] [Google Scholar]
- 73.Houghten RA, Pinilla C, Blondelle SE, Appel JR, Dooley CT, and Cuervo JH (1991) Generation and use of synthetic peptide combinatorial libraries for basic research and drug discovery. Nature 354, 84–86 doi: 10.1038/354084a0 [DOI] [PubMed] [Google Scholar]
- 74.Mendes K, Ndungu JM, Clark LF, and Kodadek T (2015) Optimization of the magnetic recovery of hits from one-bead-one-compound library screens. ACS Comb. Sci 17, 506–517 doi: 10.1021/acscombsci.5b00090 [DOI] [PubMed] [Google Scholar]
- 75.Joo SH, Xiao Q, Ling Y, Gopishetty B, and Pei D (2006) High-throughput sequence determination of cyclic peptide library members by partial Edman degradation/mass spectrometry. J. Am. Chem. Soc 128, 13000–13009 doi: 10.1021/ja063722k [DOI] [PubMed] [Google Scholar]
- 76.Chait BT, Wang R, Beavis RC, and Kent SB (1993) Protein ladder sequencing. Science 262, 89–92 doi: 10.1126/science.8211132 [DOI] [PubMed] [Google Scholar]
- 77.Wang P, Arabaci G, and Pei D (2001) Rapid sequencing of library-derived peptides by partial Edman degradation and mass spectrometry. J. Comb. Chem 3, 251–254 doi: 10.1021/cc000102l [DOI] [PubMed] [Google Scholar]
- 78.Dewan V, Liu T, Chen K-M, Qian Z, Xiao Y, Kleiman L, et al. (2012) Cyclic peptide inhibitors of HIV-1 capsid-human lysyl-tRNA synthetase interaction. ACS Chem. Biol 7, 761–769 doi: 10.1021/cb200450w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wu X, Upadhyaya P, Villalona-Calero MA, Briesewitz R, and Pei D (2013) Inhibition of Ras-effector interaction by cyclic peptides. Med. Chem. Comm 4, 378–382 doi: 10.1039/C2MD20329D [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lian W, Upadhyaya P, Rhodes CA, Liu Y, and Pei D (2013) Screening bicyclic peptide libraries for protein-protein interaction inhibitors: discovery of a tumor necrosis factor-α antagonist. J. Am. Chem. Soc 135, 11990–11995 doi: 10.1021/ja405106u [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Upadhyaya P, Qian Z, Selner NG, Clippinger SR, Wu Z, Briesewitz R, and Pei D (2015) Inhibition of Ras signaling by blocking Ras-effector interactions with cyclic peptides. Angew. Chem. Int. Ed 54, 7602–7606 doi: 10.1002/anie.201502763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Upadhyaya P, Qian Z, Habir NAA, and Pei D (2014) Direct Ras inhibitors identified from a structurally rigidified bicyclic peptide library. Tetrahedron 70, 7714–7720 doi: 10.1016/j.tet.2014.05.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lipinski CA, Lombardo F, Dominy BW, and Feeney PJ (2001). Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev 46, 3–26. doi: 10.1016/S0169-409X(00)00129-0 [DOI] [PubMed] [Google Scholar]
- 84.Brocks DR, Ala S, and Aliabadi HM (2006) The effect of increased lipoprotein levels on the pharmacokinetics of cyclosporine A in the laboratory rat. Biopharm. Drug Dispos 27, 7–16 doi: 10.1002/bdd.476 [DOI] [PubMed] [Google Scholar]
- 85.Bockus AT, Schwochert JA, Pye CR, Townsend CE, Sok V, Bednarek MA, et al. (2015) Going out on a limb: delineating the effects of β-branching, N-methylation, and side chain size on the passive permeability, solubility, and flexibility of Sanguinamide A analogues. J. Med. Chem 58, 7409–7418 doi: 10.1021/acs.jmedchem.5b00919 [DOI] [PubMed] [Google Scholar]
- 86.Beck JG, Chatterjee J, Laufer B, Kiran MU, Frank AO, Neubauer S et al. (2012) Intestinal permeability of cyclic peptides: common key backbone motifs identified. J. Am. Chem. Soc 134, 12125–12133 doi: 10.1021/ja303200d [DOI] [PubMed] [Google Scholar]
- 87.Witek J, Keller BG, Blatter M, Meissner A, Wagner T, and Riniker S (2016) Kinetic models of cyclosporin A in polar and apolar environments reveal multiple congruent conformational states. J. Chem. Inf. Model. 56, 1547–1562 doi: 10.1021/acs.jcim.6b00251 [DOI] [PubMed] [Google Scholar]
- 88.Matsson P, Doak BC, Over B, and Kihlberg J (2016) Cell permeability beyond the rule of 5. Adv. Drug Deliv. Rev 101, 42–61 doi: 10.1016/j.addr.2016.03.013. [DOI] [PubMed] [Google Scholar]
- 89.Ahlbach CL, Lexa KW, Bockus AT, Chen V, Crews P, Jacobson MP, et al. (2015) Beyond cyclosporine A: conformation-dependent passive membrane permeabilities of cyclic peptide natural products. Fut. Med. Chem 7, doi: 10.4155/fmc.15.78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hess S, Ovadia O, Shalev DE, Senderovich H, Qadri B, Yehezkel T, et al. (2007) Effect of structural and conformation modifications, including backbone cyclization, of hydrophilic hexapeptides on their intestinal permeability and enzymatic stability. J. Med. Chem 50, 6201–6211 doi: 10.1021/jm070836d [DOI] [PubMed] [Google Scholar]
- 91.Thansandote P, Harris RM, Dexter HL, Simpson GL, Pal S, Upton RJ, et al. (2014) Improving the passive permeability of macrocyclic peptides: balancing permeability with other physiochemical properties. Bioorg. Med. Chem 23, 322–327 doi: 10.1016/j.bmc.2014.11.034 [DOI] [PubMed] [Google Scholar]
- 92.Goetz GH, Philippe L, and Shapiro MJ (2014) EPSA: a novel supercritical fluid chromatography technique enabling the design of permable cyclic peptides. ACS Med. Chem. Lett 5, 1167–1172 doi: 10.1021/ml500239m [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nielsen DS, Hoang HN, Lohman RJ, Hill TA, Lucke AJ, Craik DJ, et al. (2014) Improving on nature: making a cyclic heptapeptide orally bioavailable. Angew. Chem. Int. Ed 53, 12059–12063 doi: 10.1002/anie.201405364 [DOI] [PubMed] [Google Scholar]
- 94.White TR, Renzelman CM, Rand AC, Rezai T, McEwen CM, Gelev VM, et al. (2011) On-resin N-methylation of cyclic peptides for discovery of orally bioavailable scaffolds. Nat. Chem. Biol 7, 810–817 doi: 10.1038/nchembio.664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Morimoto J, and Kodadek T (2015) Synthesis of a large library of macrocyclic peptides containing multiple and diverse N-alkylated residues. Mol. BioSys 11, 2770–2779 doi: 10.1039/C5MB00308C [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bechara C, and Sagan S (2013) Cell-penetrating peptides: 20 years later, where do we stand? FEBS Lett. 587, 1693–1702 doi: 10.1016/j.febslet.2013.04.031 [DOI] [PubMed] [Google Scholar]
- 97.Gautam A, Singh H, Tyagi A, Chaudhary K, Kumar R, Kapoor P, et al. (2016) CPPsite: a curated database of cell penetrating peptides database. doi: 10.1093/database/bas015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Richard JP, Melikov K, Vives E, Ramos C, Verbeure B, Gait MJ, et al. (2003) Cell-penetrating peptides: a reevaluation of the mechanisms of cellular uptake. J. Biol. Chem 278, 585–590 doi: 10.1074/jbc.M209548200 [DOI] [PubMed] [Google Scholar]
- 99.Zhang Y, Zhou S, Wavreille A-S, DeWille J, and Pei D (2008) Cyclic peptidyl inhibitors of Grb2 and tensin SH2 domains identified from combinatorial libraries. J. Comb. Chem 10, 247–255 doi: 10.1021/cc700185g [DOI] [PubMed] [Google Scholar]
- 100.Desimmie BA, Humbert M, Lescrinier E, Hendrix J, Vets S, Gijsbers R, et al. (2012) Phage display-directed discovery of LEDGF/p75 binding cyclic peptide inhibitors of HIV replication. Mol. Ther 20, 2064–2075 doi: 10.1038/mt.2012.132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.LaRochelle JR, Cobb GB, Steinauer A, Rhoades E, and Schepartz A (2015) Fluorescence correlation spectroscopy reveals highly efficient cytosolic delivery of certain penta-arg proteins and stapled peptides. J. Am. Chem. Soc 137 2536–2541 doi: 10.1021/ja510391n [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Liu T, Liu Y, Kao H-Y, and Pei D (2010) Membrane permeable cyclic peptidyl inhibitors against human peptidylprolyl isomerase Pin1. J. Med. Chem 53, 2494–2501 doi: 10.1021/jm901778v [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lattig-Tunnermann G, Prinz M, Hoffmann D, Behlke J, Palm-Apergi C, Morano I, et al. (2011) Backbone rigidity and static presentation of guanidinium groups increases cellular uptake of arginine-rich cell-penetrating peptides. Nat. Comm 2, 453 doi: 10.1038/ncomms1459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mandal D, Nasrolahi Shirazi A, and Parang K (2011) Cell-penetrating homochiral cyclic peptides as nuclear-targeting molecular transporters. Angew. Chem. Int. Ed 50, 9633–9637 doi: 10.1002/anie.201102572 [DOI] [PubMed] [Google Scholar]
- 105.Qian Z, LaRochelle JR, Jiang B, Lian W, Hard RL, Selner NG, et al. (2014) Early endosomal escape of a cyclic cell-penetrating peptide allows effective cytosolic cargo delivery. Biochemistry 53, 4034–4046 doi: 10.1021/bi5004102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Qian Z, Liu T, Liu Y, Briesewitz R, Barrios AM, Jhiang SM, and Pei D (2013) Efficient delivery of cyclic peptides into mammalian cells with short sequence motifs. ACS. Chem. Biol 8, 423–431 doi: 10.1021/cb3005275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Qian Z, Martyna A, Hard RL, Wang J, Appiah-Kubi G, Coss C, et al. (2016) Discovery and mechanism of highly efficient cyclic cell-penetrating peptides. Biochemistry 55, 2601–2612 doi: 10.1021/acs.biochem.6b00226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lian W, Jiang B, Qian Z, and Pei D (2014) Cell-permeable bicyclic peptide inhibitors against intracellular proteins. J. Am. Chem. Soc 136, 9830–9833 doi: 10.1021/ja503710n [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Trinh TB, Upadhyaya P, Qian Z, and Pei D (2016) Discovery of a direct Ras inhibitor by screening a combinatorial library of cell-permeable bicyclic peptides. ACS Comb. Sci 18, 75–85 doi: 10.1021/acscombsci.5b00164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Qian Z, Xu X, Amacher JF, Madden DR, Cormet-Boyaka E, and Pei D (2015) Intracellular delivery of peptidyl ligands by reversible cyclization: discovery of a PDZ domain inhibitor that rescues CFTR activity. Angew. Chem. Int. Ed 54, 5874–5878 doi: 10.1002/anie.201411594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Qian Z, Rhodes CA, McCroskey LC, Wen J, Appiah-Kubi G, Wang DJ, Guttridge DC and Pei D (2017) Enhancing the Cell Permeability and Metabolic Stability of Peptidyl Drugs by Reversible Bicyclization. Angew. Chem. Int. Ed 56, in press. doi: 10.1002/anie.201610888 [DOI] [PMC free article] [PubMed] [Google Scholar]