

Abstract
Background
Hypertension is the major risk factor for cardiovascular disease, the most common cause of death worldwide. Resistance arteries are capable of adapting their diameter independently in response to pressure and flow‐associated shear stress. Ryanodine receptors (RyRs) are major Ca2+‐release channels in the sarcoplasmic reticulum membrane of myocytes that contribute to the regulation of contractility. Vascular smooth muscle cells exhibit 3 different RyR isoforms (RyR1, RyR2, and RyR3), but the impact of individual RyR isoforms on adaptive vascular responses is largely unknown. Herein, we generated tamoxifen‐inducible smooth muscle cell–specific RyR2‐deficient mice and tested the hypothesis that vascular smooth muscle cell RyR2s play a specific role in elementary Ca2+ signaling and adaptive vascular responses to vascular pressure and/or flow.
Methods and Results
Targeted deletion of the Ryr2 gene resulted in a complete loss of sarcoplasmic reticulum–mediated Ca2+‐release events and associated Ca2+‐activated, large‐conductance K+ channel currents in peripheral arteries, leading to increased myogenic tone and systemic blood pressure. In the absence of RyR2, the pulmonary artery pressure response to sustained hypoxia was enhanced, but flow‐dependent effects, including blood flow recovery in ischemic hind limbs, were unaffected.
Conclusions
Our results establish that RyR2‐mediated Ca2+‐release events in VSCMs specifically regulate myogenic tone (systemic circulation) and arterial adaptation in response to changes in pressure (hypoxic lung model), but not flow. They further suggest that vascular smooth muscle cell–expressed RyR2 deserves scrutiny as a therapeutic target for the treatment of vascular responses in hypertension and chronic vascular diseases.
Keywords: BKCa channel, blood pressure, Ca2+ sparks, hypoxia, isoforms, pulmonary hypertension, ryanodine receptors
Subject Categories: Electrophysiology, Calcium Cycling/Excitation-Contraction Coupling, Ion Channels/Membrane Transport, Hypertension, Vascular Biology
Clinical Perspective
What Is New?
Our study demonstrates a specific contribution of ryanodine receptor type 2 in mediating vascular smooth muscle cell Ca2+‐release events relevant for regulating myogenic tone in the systemic circulation.
We also found that ryanodine receptor type 2 provides a negative regulatory role in hypoxic vasoconstriction in the pulmonary circulation but does not contribute to postischemic recovery of blood flow conductance in the hind limb circulation.
What Are the Clinical Implications?
These findings elucidate the role and mechanism of ryanodine receptor type 2 channels in myogenic constriction and in regulating adaptive reductions in arterial diameter in response to pressure, but not flow.
Our study potentially offers new possibilities for therapeutic interventions in systemic and pulmonary hypertension.
Introduction
Arteries adapt to hemodynamic forces through changes in their diameter. These adaptive responses take 2 forms that reflect the operation of 2 independent mechanisms: (1) a decrease in vessel diameter in response to increased transmural pressure, a response termed myogenic constriction (or Bayliss effect) that is intrinsic to smooth muscle (SM) myocytes; and (2) an increase in vessel diameter in response to increased blood flow, reflecting the short‐ and long‐term action of shear stress on the vessel wall, the latter of which reflects outward remodeling of the vascular SM cell (VSMC) layers.1
In arterial SM cells (SMCs), membrane depolarization produces a moderate (100–300 nmol/L) increase in intracellular Ca2+ concentration ([Ca2+]i) because of influx of Ca2+ ions through the voltage‐dependent L‐type Ca2+ channel, CaV1.2.2 This increase in global Ca2+ causes myocyte contraction through activation of Ca2+‐dependent myosin light chain kinase and induction of myosin‐actin cross‐bridge cycling.3 An additional important contributor to the contractility of VSMCs is the ryanodine receptor (RyR), an ion channel in the sarcoplasmic reticulum (SR) that mediates Ca2+ release, detected optically as “Ca2+ sparks.” Ca2+ sparks are elementary Ca2+‐release events generated by a single Ca2+‐release unit composed of a cluster of RyRs in the SR.4, 5 Unlike Ca2+ influx via voltage‐dependent L‐type Ca2+ channels, Ca2+ release from the SR in the form of Ca2+ sparks paradoxically causes vasodilation.6, 7 There are 2 reasons for this counterintuitive effect of Ca2+ sparks. First, a single spark is capable of producing a high (10–100 μmol/L) local (≈1% of the cell volume) increase in [Ca2+]i, 8, 9 but increases global [Ca2+]i by <2 nmol/L.6, 10 Second, Ca2+ sparks occur in close proximity to the cell membrane, where every Ca2+ spark activates numerous large‐conductance Ca2+‐sensitive K+ (BKCa) channels, causing K+ efflux.8, 11, 12, 13 The resultant “spontaneous transient outward currents” (STOCs) cause hyperpolarization of the cell membrane, thereby shutting off tonic Ca2+ entry through Cav1.2 channels through channel deactivation. Therefore, the net result of Ca2+ spark–BKCa channel coupling is decreased global [Ca2+]i and vasorelaxation.6, 11, 12, 13, 14, 15
The ability of RyRs to produce Ca2+ sparks in VSMCs may depend on the RyR isoform. Mammals possess 3 subtypes of RyRs (RyR1, RyR2, and RyR3), each composed of >5000 amino acids with a total molecular mass of ≈565 kDa.16, 17, 18, 19 The 3 RyRs show 66% to 70% overall amino acid identity,20 and all 3 RyR isoforms are expressed in arterial SM.21, 22 Coussin et al23 suggested that both RyR1 and RyR2 contribute to spontaneous Ca2+ sparks in cultured VSMCs from the portal vein, a suggestion supported by the observation that antisense oligonucleotides against RyR3 had no effect on spontaneous Ca2+ spark activity.24 Consistent with this, we previously showed that the peak amplitude, width, and duration of Ca2+ sparks in freshly isolated cerebral VSMCs from RyR3‐deficient (Ryr3 −/−) mice were normal.25 However, the frequency of Ca2+ sparks was higher in RyR3‐deficient VSMCs than in wild‐type cells, suggesting that RyR3 might play a role in modulating RyR1 and/or RyR2 function. A specific role of RyR1 in SR Ca2+ release in pulmonary artery VSMCs was suggested on the basis of the observation that hypoxia‐induced increases in [Ca2+]i and contraction were largely eliminated in Ryr1 +/‐ VSMCs.26 However, results from global gene knockout mouse models are difficult to interpret because of possible confounding effects of compensatory mechanisms. In addition, the contributions of the RyR2 subtype to arterial elementary Ca2+ signaling and adaptive responses in the vasculature remain unclear.
Herein, we established the first tamoxifen‐inducible, SM‐specific RyR2‐knockout (SM‐Ryr2 −/−) mouse model to test the hypothesis that RyR2 contributes to SR Ca2+ release in arterial VSMCs. Unexpectedly, we found that the RyR2 isoform plays a dominant role in local and global VSMC SR Ca2+ release in both systemic and pulmonary arteries. We further found that the expression of VSMC RyR2 is an essential prerequisite for the formation of Ca2+ sparks, which limit arterial myogenic constriction to pressure. Furthermore, we found that RyR2 plays an important role in the pulmonary artery pressure response to sustained hypoxia but does not contribute to flow‐induced increases in pulmonary arterial pressure. Finally, RyR2 does not contribute to postischemic recovery of blood flow in a hind limb occlusion model.
Methods
The data that support the findings of this study are available from the corresponding author on reasonable request.
Mouse Model
Animal care followed American Physiological Society guidelines. All animal protocols were approved by the local animal care committee (LAGeSo, Berlin, Germany) and the animal welfare officers of the Max Delbrück Center for Molecular Medicine. There are no ethical concerns. Mice were maintained in individually ventilated cages (IVC, Techniplast, Germany) under standardized conditions with a 12‐hour dark‐light cycle and free access to standard chow (0.25% sodium; SSNIFF Spezialitäten, Soest, Germany) and drinking water. SM‐Ryr2 −/− conditional knockout mice were created by crossbreeding mice containing an Ryr2 gene containing a loxP‐flanked region (Figure 1A) (crrm1)27 with mice containing tamoxifen‐dependent Cre recombinase28 under the control of the endogenous SM22α gene locus, which is selectively expressed in SM of adult mice.29 The SMAKO mouse,2, 30 carrying loxP‐flanked CaV1.2 channel genes and the SM–tamoxifen‐dependent Cre recombinase knock‐in transgene (SM22α‐CreT2),29 served as a source of the SM22α‐CreT2 in breeding experiments. The floxed variant of the Cav1.2 channel gene was eliminated after 2 generations of breeding, resulting in the Ryr2 flox/flox;SM22α‐CreT2 mice used for experiments. SM‐Ryr2 −/− (Ryr2 flox/flox;SM22α‐CreT2 or Ryr2 flox/flox;SM22α‐CreT2/T2) male mice (20–30 g, 8–14 weeks old) and littermate or age‐matched controls (Ryr2 +/+;SM22α‐CreT2/T2, Ryr2 +/+;SM22α‐CreT2, Ryr2 +/+, Ryr2 flox/+, or Ryr2 flox/flox) were injected IP with tamoxifen (30 μg/mg body weight) on 6 consecutive days. Mice showed no gross anomalies before or after inducing Ryr2 gene deletion in SM by tamoxifen treatment. Isolated arteries were usually obtained after ≈2 to 3 days after tamoxifen treatment. The SM myosin heavy chain–Cre31 line was from Stefan Offermanns (Max‐Planck‐Institut für Herz‐und Lungenforschung, Bad Nauheim, Germany).
Figure 1.
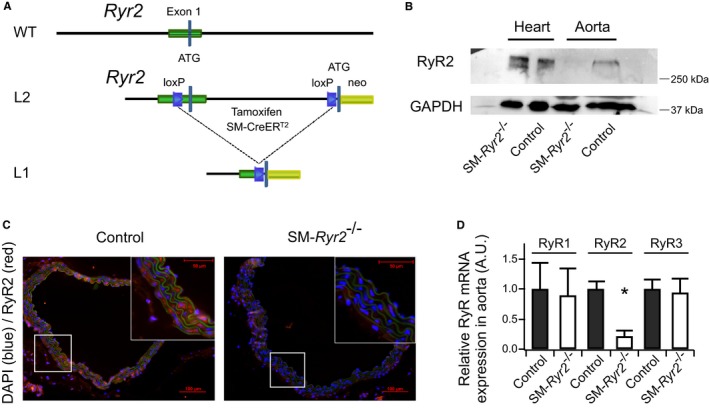
Conditional knockout of ryanodine receptor type 2 (RyR2) in vascular smooth muscle (SM) cells. A, Schematic representation of the RyR2 wild‐type (WT) allele, the allele containing loxP sequences (L2), and the floxed allele after the action of Cre recombinase (L1). B, Western blot analysis of RyR2 protein in aortic and heart tissues from control and SM‐Ryr2 −/− mice; 40 μg aortic tissue and 10 μg heart tissue were loaded per lane. C, Immunohistochemical detection of WT (control) and SM‐Ryr2 −/− aortae. Red indicates RyR2 staining; blue indicates stained nuclei (4’,6‐diamidino‐2‐phenylindole [DAPI]); green represents nonspecific autofluorescence. D, RyR1, RyR2, and RyR3 mRNA expression in aortic tissue. RyR1/2/3 mRNA levels were normalized against 18s mRNA. Mean mRNA expression value was arbitrarily set at 1 for WT control tissue, and relative expression was calculated for SM‐Ryr2 −/− tissue (n=3 mice, RyR1 and RyR3 samples; n=6 mice, RyR2 WT; n=7 mice, RyR2 knockout). A.U. indicates arbitrary unit; SM‐CreERT2, SM–tamoxifen‐dependent Cre recombinase. *P<0.01 vs WT (unpaired t test).
Western Blot Analysis
Thoracic aortae and hearts were isolated from mice and placed into cold physiological saline solution (PSS) previously oxygenated for 30 minutes (95% O2, 5% CO2). The composition of PSS (in mmol/L) was as follows: 119 NaCl, 4.7 KCl, 1.2 KH2PO4, 25 NaHCO3, 1.2 Mg2SO4, 11.1 glucose, and 1.6 CaCl2. Vessels were cleaned of perivascular fat, and all tissues were immediately placed on dry ice and kept at −80°C until use. Samples were homogenized in radioimmunoprecipitation assay buffer (Cell Signaling Technology, Danvers, MA) containing protease inhibitors (Sigma‐Aldrich, Taufkirchen, Germany). Tubes containing homogenates were freeze thawed 3 times at −80°C and 37°C, respectively, and then centrifuged at 9000g for 20 minutes at 4°C. After determining the protein concentration, samples prepared in Laemmli buffer (50 mmol/L Tris, pH 6.8, 10% SDS, 10% glycerol, 5% mercaptoethanol, and 2 mg/mL bromophenol blue) were boiled for 2 minutes, separated by SDS‐PAGE on 7% polyacrylamide gels, and transferred onto polyvinylidene fluoride membranes. Membranes were blocked in 5% nonfat dry milk in PBS containing 0.1% Tween 20, and then incubated overnight at 4°C with primary anti‐RyR2 antibody (catalog No. MA3916, 1:800 final dilution; Pierce Biotechnology). After washing, membranes were incubated with anti‐mouse IgG‐peroxidase–linked secondary antibody (1:5000 final dilution; GE Healthcare, UK) for 1 hour at room temperature. Blots were washed and incubated in enhanced chemiluminescence reagents (ECL Prime; Amersham Bioscience, UK), after which bands were detected using a ChemiDoc XRS+ Imaging System (Bio‐Rad, Hercules, CA). An anti‐GAPDH antibody (catalog No. AM4300, 1:4000 final dilution; Ambion) was used as a loading control, and Precision Plus Protein Prestained Standard (Bio‐Rad, Germany) was used as a molecular weight marker.
Immunohistochemistry
Isolated aortae were fixed in 4% formaldehyde solution for 1 hour on ice, after which they were incubated first in 3 mL of 50 mmol/L NH4Cl for 30 minutes on ice and then in 3 mL of 30% sucrose solution containing 2 mmol/L MgCl2 overnight at 4°C. Fixed aortae were placed in Tissue‐Tek and stored at −80°C. For immunohistochemistry, 5‐μm cryosections of aortae were incubated with rabbit polyclonal anti‐RyR2 primary antibody (AB9080; Merck Millipore, Germany), diluted 1:500. Sections were incubated in 10% normal donkey serum (Jackson ImmunoResearch Laboratories, Inc) for 30 minutes to block nonspecific binding sites, and then incubated with primary antibody for 1 hour at room temperature. All incubations were performed in a humidified chamber. For fluorescence visualization of bound primary antibody, sections were further incubated with anti‐rabbit Cy3‐conjugated secondary antibody (Jackson ImmunoResearch Laboratories, Inc) for 1 hour in a humidified chamber at room temperature. Specimens were examined under a Zeiss Axioplan‐2 imaging microscope and analyzed with AxioVision 4.8 software (Zeiss) by an investigator blinded to group‐assignment information.
RNA Isolation and Quantitative Reverse Transcription–Polymerase Chain Reaction
Total RNA was isolated from snap‐frozen fat‐free aortae using the RNeasy RNA isolation kit (Qiagen). RNA concentration and quality were measured using a NanoDrop‐1000 spectrophotometer (PeqLab Biotechnologie, Germany). cDNA was synthesized by reverse transcription from 2 μg of total RNA (Applied Biosystems). Target mRNA expression levels were analyzed by quantitative reverse transcription–polymerase chain reaction using SYBR green reagents and an Applied Biosystems 7500 Sequence Detector (Applied Biosystems). Expression levels of the target Ryr2 gene, determined using the relative standard curve method, were normalized to those of 18s RNA. The following primer pairs were used: Ryr1, 5′‐ATGACCGTAGGGCTCCTGGCCGTAG‐3′ (forward) and 5′‐GGGTCCTCGATCTCGTCCCCGA‐3′ (reverse); Ryr2, 5′‐CTACCCGAACCTCCAGCGATACT‐3′ (forward) and 5′‐GCAAAAGAAGGAGATGATGGTGTG‐3′ (reverse); Ryr3, 5′‐ATTCTGACCAGCTCTTCCGC‐3′ (forward) and 5′‐TCTTCCGTTCCTGGTCCTGT‐3′ (reverse); and 18s RNA, 5′‐ACATCCAAGGAAGGCAGCAG‐3′ (forward) and 5′‐TTTTCGTCACTACCTCCCCG‐3′ (reverse).
Wire Myography
The thoracic aorta and mesenteric, tibial, femoral, and cerebral arteries were removed immediately after euthanizing the mouse, quickly transferred to cold (4°C), oxygenated (95% O2/5% CO2) PSS (consisting of the following [in mmol/L]: 119 NaCl, 4.7 KCl, 1.2 KH2PO4, 25 NaHCO3, 1.2 Mg2SO4, 11.1 glucose, and 1.6 CaCl2), dissected into 2‐mm rings, and trimmed of perivascular fat and connective tissue. The composition of 60 mmol/L KCl PSS (in mmol/L) was as follows: 63.7 NaCl, 60 KCl, 1.2 KH2PO4, 25 NaHCO3, 1.2 Mg2SO4, 11.1 glucose, and 1.6 CaCl2. Each ring was positioned between 2 stainless steel wires (diameter, 0.0394 or 0.025 mm) in a 5‐mL organ bath of a Mulvany Small Vessel Myograph (DMT 610 M; Danish Myo Technology, Denmark) filled with PSS. The bath solution was continuously oxygenated with a 95% O2/5% CO2 gas mixture and kept at 37°C (pH 7.4). Aortic rings were placed under a tension of 0.3 g,32 whereas mesenteric, tibial, femoral, and cerebral rings were placed under a tension equivalent to that generated at 0.9 times the diameter of the vessel at 100 mm Hg. This normalization procedure was performed to obtain the passive diameter of the vessel at 100 mm Hg.32 Chart5 software (AD Instruments Ltd, Spechbach, Germany) was used for data acquisition and display. Rings were precontracted with 60 mmol/L KCl PSS and equilibrated until a stable resting tension was attained. In some experiments, vessels were treated sequentially with repeated doses of 10 mmol/L caffeine at increasing time intervals (1, 2, 3, 4, and 5 minutes). Drugs were added to the bath solution unless indicated otherwise. Tension is expressed as a percentage of the steady‐state tension (100%) obtained with isotonic external 60 mmol/L KCl.
Video Microscopic Ca2+ and Diameter Imaging
Ca2+ events in mesenteric arteries (third‐ and fourth‐order branches) were studied after loading with the Ca2+‐binding fluorescent dye Fluo‐4‐AM (10 μmol/L) in the presence of pluronic acid (2.5 μg/mL) in a solution consisting of the following (in mmol/L): 134 NaCl, 6 KCl, 10 glucose, 1 MgCl2, 2 CaCl2, and 10 HEPES (pH 7.4). After loading, the tissue was placed in a chamber specialized for Ca2+ imaging and superfused with PSS at a rate of 1 to 2 mL/min at 37°C. Images were collected with a Noran Oz laser‐scanning confocal microscope and Nikon ×60 water‐immersion (numerical aperture, 1.2) fluorescence objectives with the aid of software developed by Prairie Technologies (Middleton, WI). Fluo‐4 was excited with a krypton‐argon laser at 488 nm, and the emitted fluorescence was collected at >500 nm. Each file was recorded for 30 to 60 seconds, and images were collected at a rate of 53 images/s. Imaging fields were 131×131 μm (256×256 pixels).33 The diameter of pressurized arteries was monitored, as previously described.34
Isolation of Arterial VSMCs
VSMCs from tibial and mesenteric arteries were isolated, as described.12, 14 Briefly, arteries were removed and quickly transferred to cold (4°C), oxygenated (95% O2–5% CO2) PSS. The arteries were then cleaned, cut into pieces, and incubated for 50 minutes at 37°C in Ca2+‐free Hank's solution consisting of the following (in mmol/L): 55 NaCl, 80 sodium glutamate, 5.6 KCl, 2 MgCl2, 10 glucose, and 10 HEPES (pH 7.4 with NaOH) containing 1 mg/mL BSA (Sigma, Taufkirchen, Germany), 0.5 mg/mL papain (Sigma), and 1.0 mg/mL dithiothreitol. The segments were then placed in Hank's solution containing 1 mg/mL collagenase (types F and H; ratio, 30% and 70%, respectively; Sigma) and 0.1 mmol/L CaCl2 for 10 minutes at 37°C. After several washes in Ca2+‐free Hank's solution (containing 1 mg/mL BSA), single cells were dispersed from artery segments by gently triturating. Cells were then stored in the same solution at 4°C.
Ca2+ Sparks
VSMCs were seeded onto glass coverslips and incubated with the Ca2+ indicators Fluo‐3‐AM or Fluo‐4‐AM (10 μmol/L) and pluronic acid (0.005%, w/v) for 60 minutes on ice (in the same bath solution used for patch‐clamp measurements).30 After loading, the cells were washed with bath solution for at least 10 minutes at room temperature. Single SMCs were imaged (at 20–59 frames/s) using a Nipkow disc‐based UltraView LCI confocal scanner (Perkin Elmer, Waltham, MA) linked to a fast digital camera. The confocal system was mounted on an inverted Diaphot microscope with a ×40 oil‐immersion objective (numerical aperture, 1.3; Nikon). Images were obtained by illumination with an argon laser at 488 nm and by recording all emitted light >515 nm. Ca2+ spark analyses were performed off‐line using the UltraView Imaging Suite software (Perkin Elmer). Ca2+ sparks were defined as local fractional fluorescence increases that were clearly above the noise level. Ca2+ spark frequency was calculated as the number of detected sparks divided by the total scan time. Line‐scan images were obtained with a Bio‐Rad imaging system (Munich, Germany) attached to a Nikon Diaphot microscope25, 35 by illumination with a krypton‐argon laser at 488 nm and by recording all emitted light >500 nm.
Electrophysiology
K+ currents were measured in the conventional whole‐cell configuration of the patch‐clamp technique at room temperature, as previously described.30, 36 Patch pipettes (resistance, 3–5 MΩ) were filled with a solution containing the following (in mmol/L): 110 K‐Aspartate, 30 KCl, 10 NaCl, 1 MgCl2, and 0.05 EGTA (pH 7.2). For measurement of K+ currents in the whole‐cell perforated‐patch mode, the patch pipette solution was supplemented with 200 μg/mL amphotericin B, dissolved in dimethyl sulfoxide. The external bath solution contained the following (in mmol/L): 134 NaCl, 6 KCl, 1 MgCl2, 2 CaCl2, 10 glucose, and 10 HEPES (pH 7.4). The holding potential was −60 mV. Whole‐cell currents were recorded using an Axopatch 200B (Axon Instruments/Molecular Devices, Sunnyvale, CA) or EPC 7 (List, Darmstadt, Germany) amplifier and digitized at 5 kHz using a Digidata 1440A digitizer (Axon CNS, Molecular Devices) and pClamp software versions 10.1 and 10.2. STOC analysis was performed off‐line using IGOR Pro (WaveMetrics, Lake Oswego, OR) and Excel (Microsoft Corporation, Redmond, WA).
Telemetry
Radiotelemetric blood pressure measurements were performed on adult C57Bl6 SM‐Ryr2 −/− mice, as described elsewhere.37 After implanting blood pressure devices, mice were allowed ≈2 weeks to recover from surgery and exhibit regular diurnal activity rhythms. Targeted deletion of Ryr2 was induced by administration of tamoxifen (see above).
Femoral Artery Occlusion Model and Assessment of Blood Flow by Laser Doppler Imaging
Occlusion of the right femoral artery in 12‐week‐old mice was performed, as described previously.38 For repetitive assessment of hind limb blood flow after occlusion, we used the noninvasive laser Doppler imaging technique.38 The laser Doppler imaging technique depends on the Doppler principle, whereby low‐power light from a monochromatic stable laser (wavelength, 830 nm; laser diode; model LDI2‐HR; Moor Instruments, UK), incident on tissue, is scattered by moving red blood cells, photodetected, and processed to provide a measurement of blood flow. Artery structure of right and left hind limbs was characterized after polyurethane treatment.39
Hypoxic and Caffeine‐Induced Pulmonary Vasoconstriction in Isolated Perfused and Ventilated Mouse Lungs
Lungs were prepared, as described previously.40 Briefly, lungs were perfused with 37°C sterile Krebs‐Henseleit hydroxyethyl amylopectin buffer (1 mL/min; Serag‐Wiesner, Naila, Germany) containing indomethacin (30 μmol/L; Sigma) and Nω‐nitro‐l‐arginine methyl ester hydrochloride (1 mmol/L; Sigma) in a nonrecirculating manner; left atrial pressure was adjusted to 2.2 cm H2O. After isolation, lungs were ventilated with negative pressure in a closed chamber, volume controlled with a tidal volume of ≈8 mL/kg body weight, an end‐expiratory pressure of −2 cm H2O, and a respiratory rate of 90 breaths/min. Hyperinflation (−24 cm H2O) was performed at 4‐minute intervals. Mean pulmonary arterial pressure was continuously monitored, and the difference in mean pulmonary arterial pressure was expressed in cm H2O. After an initial steady‐state period of 10 minutes, lungs were perfused at a flow rate of 0.5, 1, 1.5, and 2 mL/min for 30 seconds each to generate a 4‐point pressure‐flow curve under normoxic conditions. Flow was then set to 1 mL/min and, after another steady‐state period of 4 minutes, mean pulmonary arterial pressure was recorded in response to a change from normoxic to hypoxic (1% O2) ventilation.41 Second and third pressure‐flow curves were generated during hypoxic pulmonary vasoconstriction phase 1 and phase 2, respectively. After 88 minutes of hypoxia, normoxic ventilation was reestablished and, after another steady‐state period of 22 minutes, caffeine (10 mmol/L) was administered to the perfusate for 2 minutes; this latter step was repeated twice at 12‐minute intervals.
Materials
Fluo‐3‐AM and Fluo‐4‐AM were purchased from Molecular Probes (Eugene, OR), and A23187 was from AppliChem (Darmstadt, Germany). The antibody against RyR2 was from Millipore/Chemicon (Schwalbach/Ts, Germany). All salts and other drugs were obtained from Sigma‐Aldrich (Munich, Deisenhofen, or Schnelldorf, Germany) or Merck (Darmstadt, Germany). In cases in which dimethyl sulfoxide was used as a solvent, the maximal dimethyl sulfoxide concentration after application did not exceed 0.5%.
Statistical Analysis
Data are presented as means±SEM. Descriptive statistics were calculated, and variables were examined for meeting assumptions of normal distribution without skewness and kurtosis by using quantile‐quantile plot, Shapiro‐Wilk test, or D'Agostino and Pearson omnibus normality test, as appropriate.42 Statistically significant differences in mean values were determined by Student unpaired t test or one‐way ANOVA. The Mann‐Whitney U test was performed for comparison of dose‐response curves. Statistical tests were performed using GraphPad Prism (San Diego, CA), R (R Foundation for Statistical Computing, Vienna, Austria), or Excel (Microsoft Corporation). In the quantitative polymerase chain reaction experiments, mean mRNA expression value was arbitrarily set at 100% for wild‐type control tissue, and relative expression was calculated for SM‐Ryr2 −/− tissue, as previously described for small amounts of tissues.43 P<0.05 was considered statistically significant.
Results
SM‐Ryr2 −/− mice, generated by SMC‐targeted deletion of the Ryr2 gene by tamoxifen‐dependent Cre recombinase under the control of the endogenous SM22α gene locus (Figure 1A), showed virtually no detectable RyR2 mRNA in aortic, mesenteric, and tibial artery tissues compared with control mice and completely lacked RyR2 protein (Figure 1B through 1D, Figure S1B and S1C).
RyR2s in Global SR Ca2+ Release in Peripheral Arteries
We next studied the contribution of RyR2 to global SR Ca2+ release and arterial contraction. SR Ca2+ release was induced by the pan RyR activator caffeine (10 mmol/L). As shown in Figure 2A and 2B, caffeine induced strong contractions of the aorta and mesenteric, femoral, tibial, and cerebral arteries of control mice. In contrast, caffeine (10 mmol/L) was largely ineffective in inducing contraction in any of these arteries from SM‐Ryr2 −/− mice (Figure 2A and 2B). These data indicate that the RyR2 isoform is the predominant contributor to SR Ca2+‐release–induced contraction of peripheral arteries.
Figure 2.
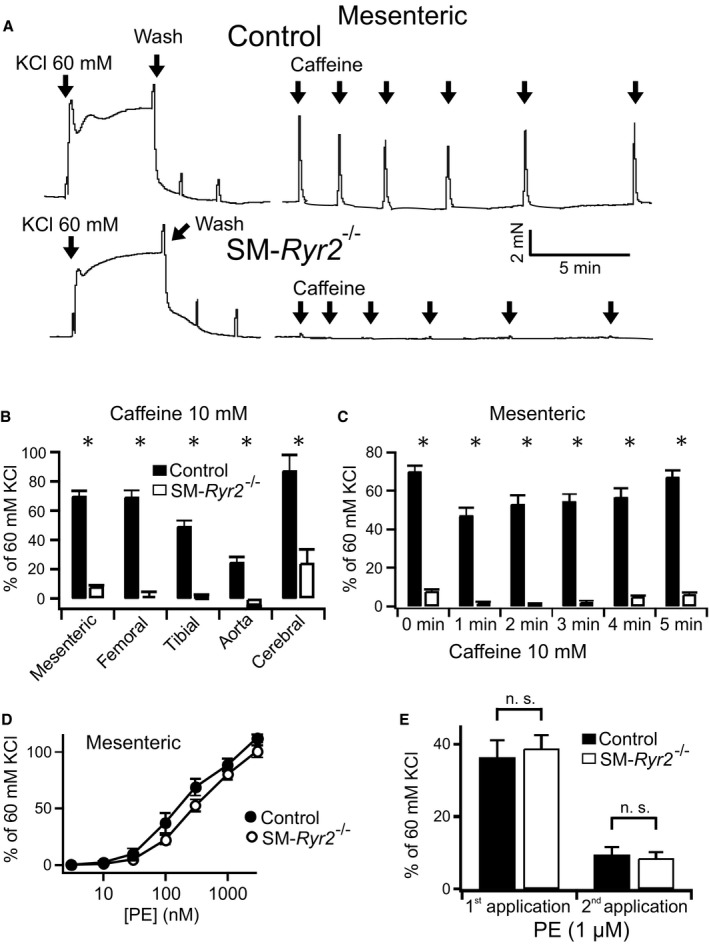
Effects of caffeine on peripheral artery contraction. A, Original recordings of contractile force of mesenteric arteries from wild‐type (control) and smooth muscle (SM)–Ryr2 −/− mice. Addition of caffeine (10 mmol/L) into the bath solution is indicated by arrows. B, Effects of caffeine on contractile force in various arteries from wild‐type mice (n=13 mesenteric arteries of 2 mice; n=4 femoral arteries of 2 mice; n=22 tibial arteries of 3 mice; n=12 aortae of 2 mice; n=10 cerebral arteries of 5 mice) and SM‐Ryr2 −/− mice (n=16 mesenteric arteries of 2 mice; n=6 femoral arteries of 3 mice; n=24 tibial arteries of 4 mice; n=6 aortae and n=8 cerebral arteries of 5 mice). C, Contraction of mesenteric arteries with repeated applications of caffeine (10 mmol/L) at different time intervals (n=13 control rings; n=16 rings). D, Concentration‐response curves for the contractile effects of phenylephrine in mesenteric arteries of control and SM‐Ryr2 −/− mice (n=24 rings). E, Contraction of mesenteric arteries in response to repeated applications of phenylephrine (1 μmol/L) in a Ca2+‐free bath solution (n=24 rings of 3 mice each in C through E). There were no differences between control and SM‐Ryr2 −/− rings. n.s. indicates not significant. *P<0.05.
The role of RyR2 in VSMC Ca2+ release after refilling of SR Ca2+ stores was next investigated using a protocol of repeated caffeine applications at varying time intervals. Mesenteric arteries from control mice showed normal contractions in response to caffeine (10 mmol/L) after an interval of 5 minutes between caffeine applications (Figure 2C), indicating that VSMC RyR‐sensitive SR Ca2+ stores are refilled within 5 minutes after SR Ca2+ depletion by this RyR activator. In contrast, vessels originating from SM‐Ryr2 −/− mice showed weak contractions in response to caffeine at all time intervals (Figure 2A and 2C). These results support the interpretation that RyR2 is the major isoform in VSMCs involved in RyR‐dependent SR Ca2+ release. In keeping with this, caffeine (10 mmol/L) induced a prominent increase in arterial wall [Ca2+]i in control mesenteric arteries (Figure 3, upper), but not in SM‐Ryr2 −/− mesenteric arteries (Figure 3, lower), as measured by video microscopy using Fluo‐4‐AM–loaded vessels. However, vascular contractions in response to the α1‐adrenergic receptor agonist phenylephrine were normal in SM‐Ryr2 −/− arteries in both Ca2+‐containing PSS (Figure 2D) and Ca2+‐free PSS (Figure 2E). These latter results indicate that RyR2 deficiency does not affect phenylephrine‐induced contractions of VSMCs in mesenteric arteries.
Figure 3.
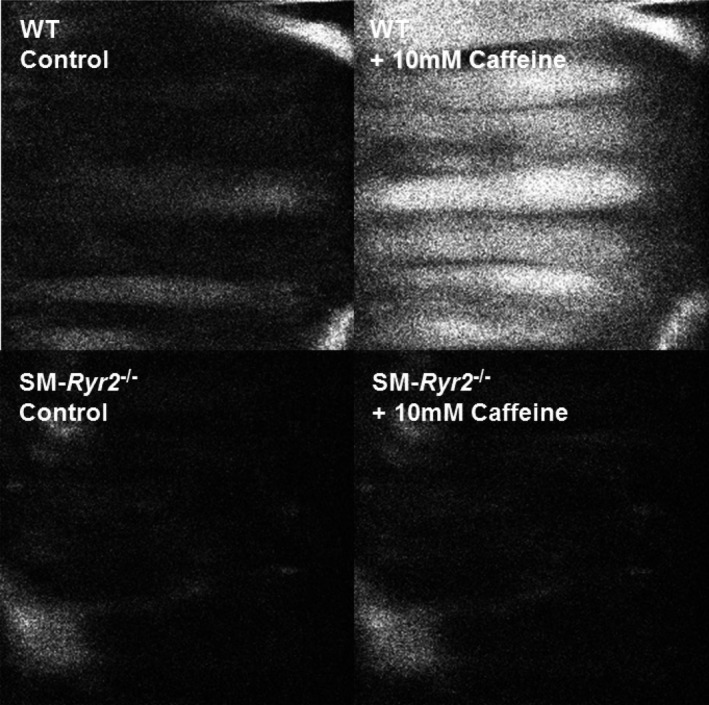
Changes in arterial wall intracellular Ca2+ concentration. Ca2+ fluorescence video microscopy images of mesenteric arteries from wild‐type (WT; upper) and smooth muscle (SM)–Ryr2 −/− (lower) mice before (left) and after (right) application of caffeine.
RyR2s in Local Ca2+ Release in VSMCs of Peripheral Arteries
Ca2+ sparks are local, elementary Ca2+‐release events mediated by RyRs that activate nearby BKCa channels to oppose arterial constriction to elevations in transmural pressure. Using our SM‐Ryr2 −/− mice in conjunction with Nipkow spinning‐disc confocal microscopy, we investigated the role of RyR2 in the generation and properties of Ca2+ sparks.
Ca2+ sparks were readily detected in tibial artery SMCs from wild‐type mice, as shown in Figure 4A through 4C, which depicts fluorescence images of a representative Fluo‐4–loaded cell. Two regions of interest (ROIs), denoted by yellow circles a and b (Figure 4A), are highlighted; these correspond to nonspark and spark sites, respectively, as shown in Figure 4B. Continuous recordings of changes in fluorescence intensity over time revealed recurring Ca2+ sparks in ROI b (Figure 4C, highlighted in detail in the inset), but not in ROI a. Figure 4C also shows that subsequent application of 10 mmol/L caffeine, which causes activation of RyRs and release of SR Ca2+, induced a transient increase in global [Ca2+], detectable in both ROIs. Shortly after the caffeine‐induced increase in global [Ca2+], Ca2+ sparks were no longer detectable in ROI b, consistent with the fact that depletion of SR Ca2+ by caffeine eliminates the Ca2+ gradient that drives RyR‐mediated SR Ca2+ release, causing Ca2+ sparks to disappear. Notably, VSMCs from SM‐Ryr2−/− mice showed little or no global [Ca2+]i transient in response to treatment with 10 mmol/L caffeine (Figure 4D and 4G). Moreover, although the majority (87%) of VSMCs from control mice exhibited Ca2+ sparks (n=15), with an average frequency of 0.59±0.10 Hz, Ca2+ sparks were undetectable in VSMCs from SM‐Ryr2 −/− mice (n=11) (Figure 4E and 4F). We confirmed our results using another transgenic mouse line in which Cre recombinase expression is driven by the SM myosin heavy chain promoter.44 Reverse transcription–polymerase chain reaction showed that RyR2 mRNA was almost undetectable in aortic tissue, as well as mesenteric and tibial arteries, of SM myosin heavy chain–Ryr2 −/− mice compared with control mice (Figure 5A); and tibial artery SMCs from these mice lacked Ca2+ sparks (Figure 5B through 5H). By contrast, Ca2+ sparks were present in 55.3% of VSMCs from control mice (n=123), albeit at a reduced frequency (0.06±0.01 Hz) compared with VSMCs from wild‐type littermates of SM‐Ryr2 −/− mice (Figure 5F and 5G). Additional experiments performed in SMCs from mesenteric arteries showed that 28% of wild‐type (control) cells exhibited Ca2+ sparks (n=127), whereas no Ca2+ sparks were observed in VSMCs from SM‐Ryr2 −/− mice (n=104) (Figure S2). Control VSMCs also showed a strong increase in [Ca2+]i in response to 10 mmol/L caffeine, but VSMCs from SM‐Ryr2 −/− mice were largely unresponsive (Figure S2).
Figure 4.
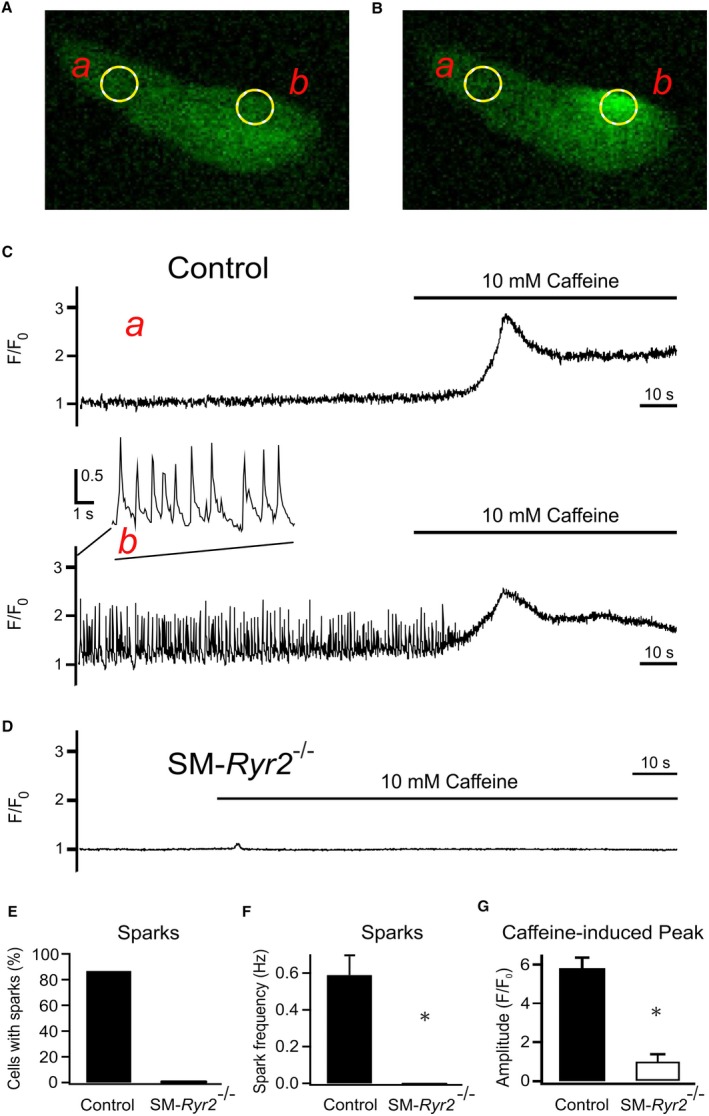
Ca2+‐release events (Ca2+ sparks) in wild‐type and smooth muscle (SM)–Ryr2 −/− vascular SM cells (VSMCs). A, Ca2+ fluorescence image of a Fluo‐4–loaded wild‐type (control) tibial artery SMC. B, Ca2+ fluorescence image of the same cell as in A during the occurrence of a Ca2+ spark. Two‐dimensional images were recorded at a rate of 20/s. C, Time course of Ca2+ fluorescence changes in cellular regions of interest (ROIs) without sparks (ROI a, upper) and with sparks (ROI b, lower). Presence of 10 mmol/L caffeine is indicated by horizontal lines. D, Time course of Ca2+ fluorescence changes in a ROI (similar size as in A) of an SM‐Ryr2 −/− VSMC in the absence and presence of caffeine (10 mmol/L). E, Percentage of VSMCs with Ca2+ sparks. F, Ca2+ spark frequencies in n=15 wild‐type and n=11 SM‐Ryr2 −/− VSMCs. G, Increases in peak Ca2+ fluorescence induced by 10 mmol/L caffeine in wild‐type (control) and SM‐Ryr2 −/− VSMCs (n=14 cells and n=8 cells, respectively; 3 mice). F/F0, flurescence/background fluorescence. *P<0.05.
Figure 5.
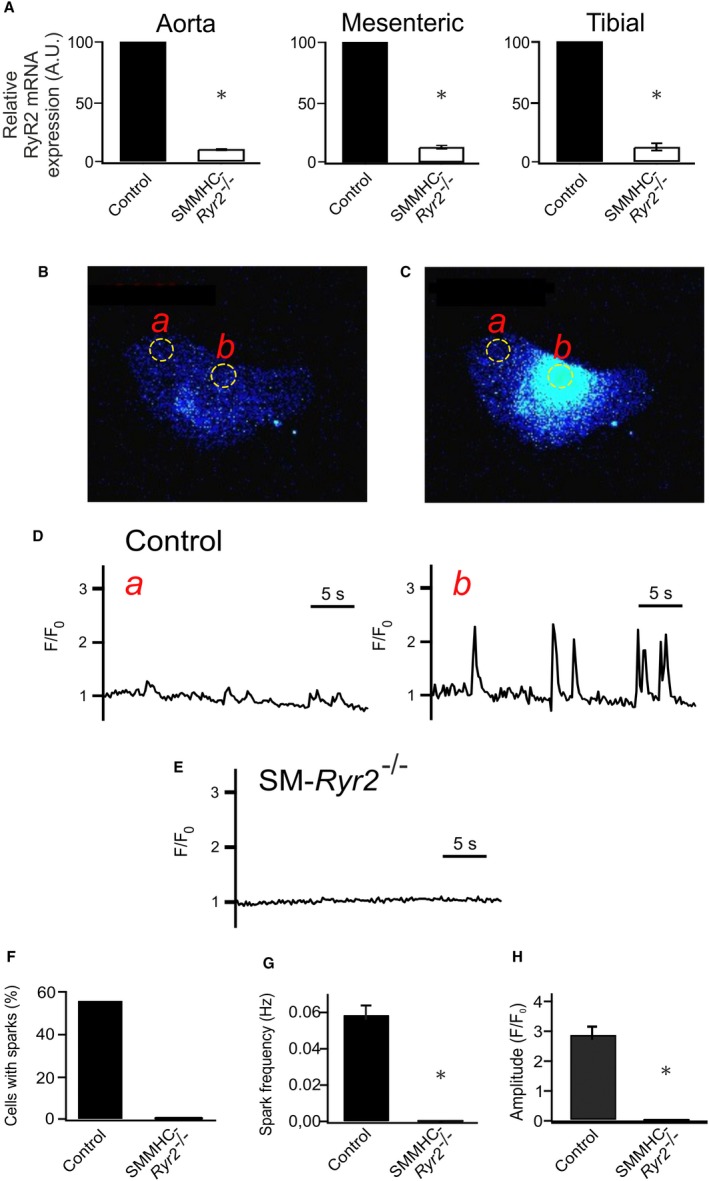
Ca2+‐release events (Ca2+ sparks) in wild‐type and smooth muscle myosin heavy chain (SMMHC)–Ryr2 −/− tibial artery smooth muscle cells (SMCs). A, Ryanodine receptor type 2 (RyR2) mRNA expression in aortic, mesenteric artery, and tibial artery tissues. mRNA levels for RyR2 were normalized against 18s mRNA. The mean mRNA expression value was arbitrarily set at 100 for wild‐type (control) tissue, and relative expression was calculated for SMMHC‐RyR2 −/− tissue (n=4 each). P value is given vs control (1‐sample t test). B, Ca2+ fluorescence image of a Fluo‐4‐AM–loaded control vascular SMC (VSMC). C, Ca2+ fluorescence image of the same cell as in A during the occurrence of a Ca2+ spark. Two‐dimensional images were recorded at a rate of 5/s. D, Time course of Ca2+ fluorescence changes in cellular regions of interest (ROIs) without sparks (ROI a, left) and with sparks (ROI b, right). E, Time course of Ca2+ fluorescence changes in an ROI (similar size as that in A) of an SMMHC‐Ryr2 −/− VSMC. F, Percentage of VSMCs with Ca2+ sparks. G, Ca2+ spark frequencies in VSMCs from control and SMMHC‐Ryr2 −/− mice. H, Amplitude of Ca2+ sparks (n=123 cells of 4 control mice; n=124 cells of 4 SMMHC‐Ryr2 −/− mice). A.U. indicates arbitrary unit. F/F0, flurescence/background fluorescence. *P<0.05.
Although our results indicate an essential role for RyR2 in mediating Ca2+ release in the form of sparks in VSMCs, other reports have suggested that SMCs also express RyR1 and RyR3.21, 22, 45 Therefore, we used high‐resolution, confocal line‐scan imaging of VSMCs to further confirm the role of RyR2 in Ca2+ spark formation, measuring Ca2+ sparks in close proximity to the cell surface.30 Figure 6A shows a confocal line‐scan image of 2 Ca2+ sparks in a representative tibial artery SMC isolated from a control mouse. Ca2+ sparks occurred at a frequency of 0.46±0.10 Hz (n=52) in tibial SMCs from these mice (Figure 6B), but were essentially absent (frequency, 0.02±0.01 Hz; n=42) in VSMCs from SM‐Ryr2 −/− mice. Together, these data demonstrate that RyR2s are indispensable for Ca2+ spark generation in SMCs of tibial and mesenteric arteries.
Figure 6.
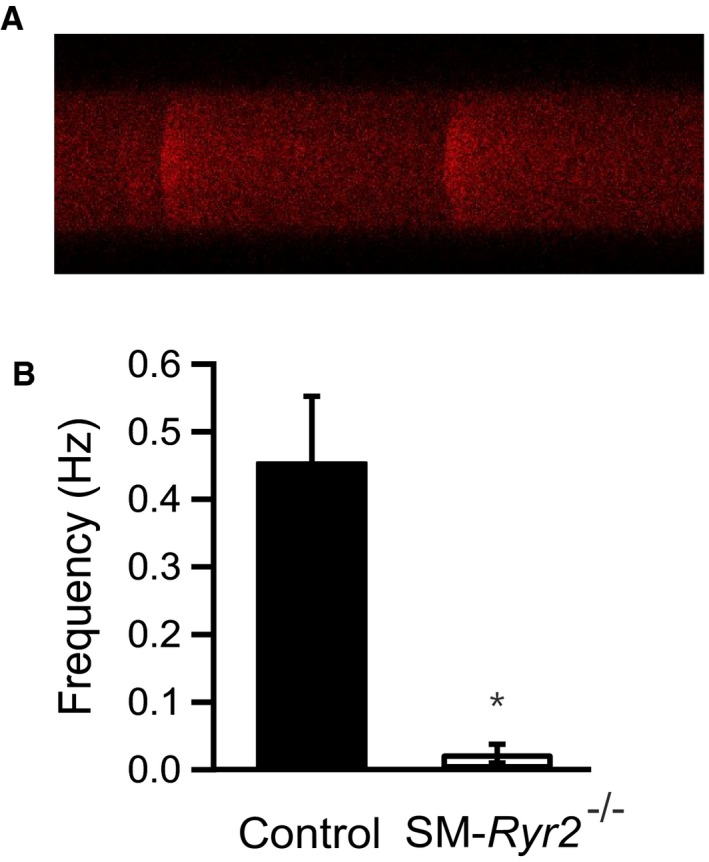
Line‐scan imaging of Ca2+‐release events (Ca2+ sparks) in wild‐type and smooth muscle (SM)–Ryr2 −/− vascular SM cells (SMCs). A, Confocal line‐scan image of a Fluo‐3‐AM–loaded tibial artery wild‐type (control) cell showing the time course of Ca2+ sparks. The line‐scan image duration was 5 s, and each line was 4 ms. B, Ca2+ spark frequency in tibial artery SMCs from control (n=52) and SM‐Ryr2 −/− (n=42) mice. *P<0.05.
RyR2‐Dependent Ca2+ Sparks Induce STOCs
In isolated myocytes under voltage‐clamp conditions, Ca2+ sparks activate a small number of proximate BKCa channels to cause a transient outward current, traditionally referred to as STOCs. STOCs were recorded in tibial artery SMCs from control and SM‐Ryr2 −/− mice using the patch‐clamp technique (Figure 7). A peak amplitude >28 pA above baseline, the criterion for a STOC at a holding potential of ‐20 mV, is 3 times the BKCa single‐channel current based on a conductance of 150 pS.46 Figure 7A shows recordings from a control VSMC at −40, −20, and 0 mV, demonstrating the presence of STOCs at −20 mV and more positive membrane potentials. In contrast, STOCs were absent in VSMCs from SM‐Ryr2 −/− mice (Figure 7B). At −20 mV, 48% of control cells (n=31) exhibited STOCs, whereas no SM‐Ryr2 −/− VSMCs (n=25) exhibited STOCs (Figure 7C). STOCs in control cells exhibited a frequency of 0.44±0.14 Hz (Figure 7D) and a mean amplitude of 26.5±6.0 pA at −20 mV (Figure 7E). Caffeine (10 mmol/L) induced a strong outward BKCa current in control VSMCs (Figure S3A) that is attributable to an increase in global [Ca2+]i in the micromolar range,47 followed by suppression of STOCs because of severe SR Ca2+ depletion. In contrast, caffeine failed to induce BKCa currents in SM‐Ryr2 −/− VSMCs (Figure S3B). These findings support the idea that RyR2s generate Ca2+ sparks in VSMCs, which, in turn, activate BKCa channels to produce STOCs. Notably, the Ca2+ ionophore A23187 (5 or 10 μmol/L) induced large outward BKCa currents in both control and SM‐Ryr2 −/− VSMCs, indicating that BKCa channels are present and their Ca2+ sensitivity is preserved after ablation of Ryr2 (Figure 8A and 8B).
Figure 7.
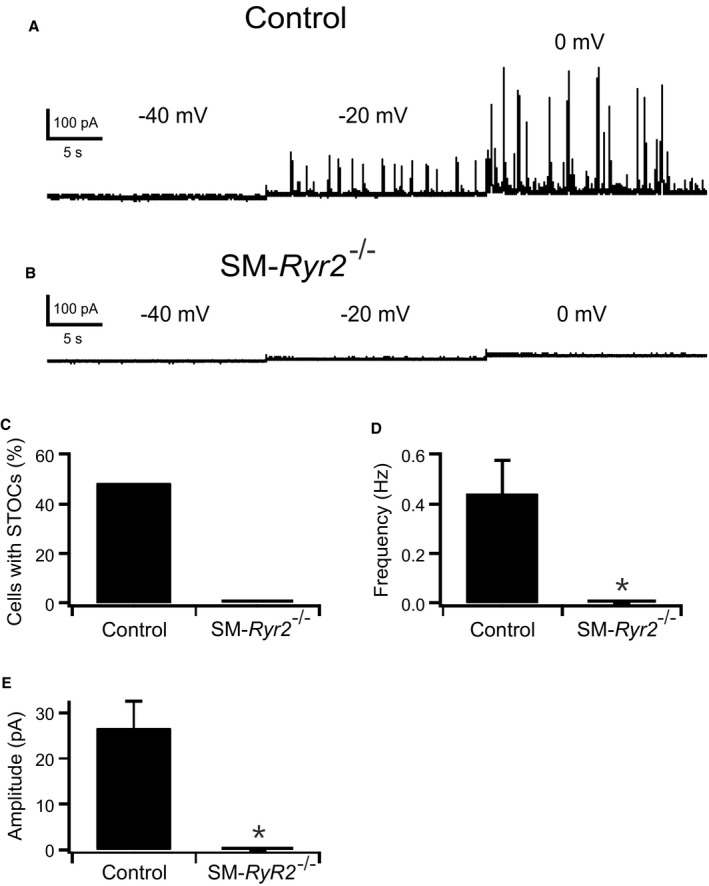
Spontaneous transient outward currents (STOCs) caused by Ca2+‐release events. A and B, Original recordings of STOCs in tibial artery smooth muscle (SM) cells isolated from wild‐type (control) (A) and SM‐Ryr2 −/− (B) mice. The holding potential was stepwise increased in 20‐mV increments from −40 to 0 mV. C through E, Comparison of STOC characteristics at −20 mV. Percentage of cells with STOCs (C), STOC frequencies (D), and mean STOC amplitudes (E). n=31 cells of 11 control mice; n=25 cells of 9 SM‐Ryr2 −/− mice. *P<0.05.
Figure 8.
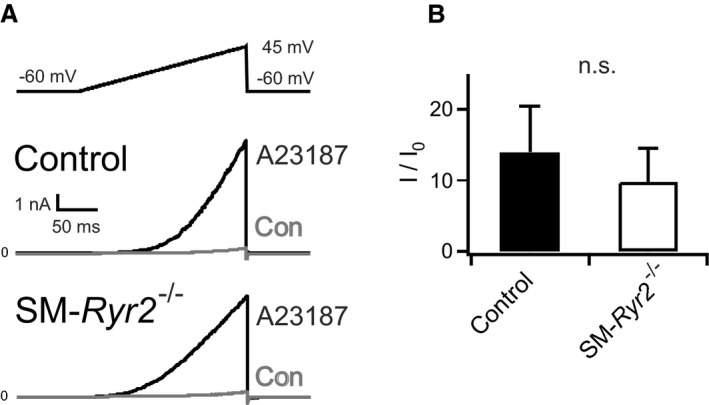
Functional large‐conductance Ca2+‐sensitive K+ channels in wild‐type and smooth muscle (SM)–Ryr2 −/− vascular SM cells (SMCs). A, Original current traces in wild‐type (control) and SM‐Ryr2 −/− tibial artery SMCs before (control) and after application of 5 or 10 μmol/L A23187. B, Summary of the results (n=7 cells of 4 control mice; n=7 cells of 3 SM‐Ryr2 −/− mice). I/I0, A23187‐induced current/basal current. n.s. indicates not significant.
RyR2s in the Myogenic Response of Peripheral Arteries
We next tested the function of the RyR2 Ca2+ spark–BKCa channel pathway in opposing vasoconstriction to elevated transmural pressure (myogenic tone). Isolated mesenteric arteries were pressurized to 80 mm Hg and allowed to develop myogenic tone with a functional Ca2+ spark–BKCa channel pathway34 (Figure 9). The viability of vessels was confirmed by measuring the contractile response to a depolarizing concentration of KCl (60 mmol/L). The degree of myogenic tone in pressurized arteries was expressed relative to maximum diameter, determined by exposure to Ca2+‐free external PSS.34 Inhibition of the Ca2+ spark–BKCa channel pathway with the RyR inhibitor ryanodine (10 μmol/L)48 increased myogenic constriction in wild‐type (control) arteries (Figure 9A), consistent with previous reports.6, 49 In contrast, ryanodine had almost no effect on the diameters of arteries from SM‐Ryr2 −/− mice (Figure 9B and 9C). These results indicate that the Ca2+ spark–BKCa channel pathway in VSMCs limits myogenic tone through RyR2s, without the involvement of other RyR isoforms.
Figure 9.
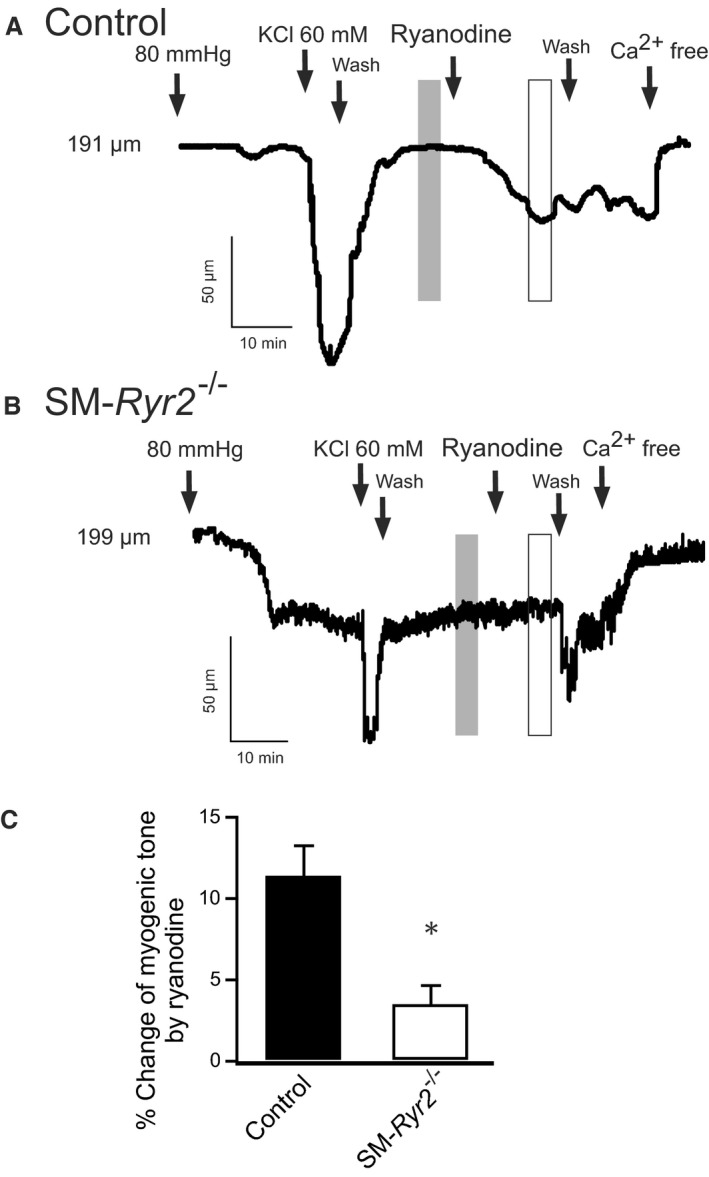
Arterial constriction induced by inhibition of Ca2+‐release events with ryanodine in wild‐type and smooth muscle (SM)–Ryr2 −/− peripheral arteries. A, Application of ryanodine (10 μmol/L) provoked a strong constriction of isolated wild‐type (control) mesenteric arteries. B, Ryanodine‐induced constriction was reduced in SM‐Ryr2 −/− arteries. C, Summary of the results (n=6 cells of 4 control mice; n=6 cells of 5 SM‐Ryr2 −/− mice). The vertical gray and white bars in A and B represent the time points for the analyzed myogenic tone values. *P<0.05.
RyR2s in the Pulmonary Circulation
In the lung, alveolar hypoxia leads to constriction of upstream pulmonary arterioles through retrograde signal transduction via gap junctions.50 Using an ex vivo lung perfusion model, we investigated the role of VSMC RyR2s in the pulmonary artery pressure response to hypoxia. Basal mean pulmonary arterial pressure was not significantly different between control (10.3±0.2 cm H2O) and SM‐Ryr2 −/− (11.0±0.3 cm H2O) lungs. There was also no difference in pulmonary arterial pressure between control and SM‐Ryr2 −/− lungs during the short‐term phase of hypoxia (phase 1) (Figure 10A and 10B). However, under sustained hypoxia (phase 2), the increase in mean pulmonary arterial pressure was more pronounced in SM‐Ryr2 −/− lungs than in control lungs (Figure 10A and 10B). This difference was observed over a wide range of flow rates (0.5–2.0 mL/min) (Figure 10C). Notably, flow‐induced increases in mean pulmonary arterial pressure did not differ between the 2 groups under normoxic conditions or in the short‐term phase of hypoxic pulmonary vasoconstriction (Figure 10C). These results argue for an important role of VSMC RyR2s in the sustained phase of hypoxic pulmonary vasoconstriction, but not in the short‐term phase. It is unlikely that SR Ca2+ release through VSMC RyR1 or RyR3 plays an important role in these effects because caffeine (10 mmol/L) induced a transient pulmonary vasoconstriction in control lungs, but not in SM‐Ryr2 −/− lungs (Figure 10D and 10E).
Figure 10.
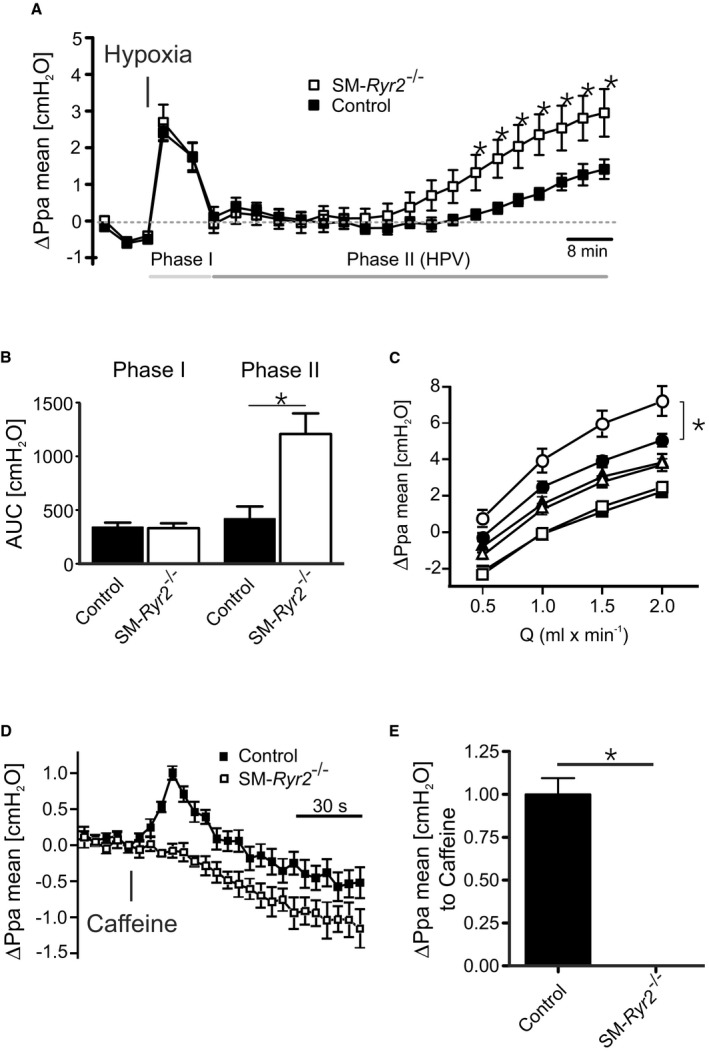
Function of ryanodine receptor type 2 (RyR2) in hypoxic pulmonary vasoconstriction. A, Changes in mean pulmonary arterial pressure (ΔPpa mean) observed in isolated perfused and ventilated lungs from wild‐type (control; closed squares, n=7 mice) and smooth muscle (SM)–Ryr2 −/− (open squares, n=7 mice) mice after induction of hypoxic (1% O2) ventilation. B, Pulmonary pressure response (area under the curve [AUC]) in control and SM‐Ryr2 −/− lungs attributable to short‐term (left, phase 1) and long‐term (right, phase 2) phases of hypoxic pulmonary vasoconstriction. C, Flow‐induced Ppa increase in control (closed symbols) and SM‐Ryr2 −/− (open symbols) lungs under normoxia (squares), acute hypoxia (triangles), and chronic hypoxia (circles). D, Pulmonary pressure response after application of caffeine (10 mmol/L) in control (closed symbols) and SM‐Ryr2 −/− (open symbols) lungs. Similar effects were observed with 3 consecutive applications of caffeine. E, The peak increase in Ppa caused by the first application of caffeine is shown. HPV, hypoxic pulmonary vasoconstriction; Q, flow rate. *P<0.05.
RyR2s in the Ischemic Hind Limb Circulation
RyRs may play a role in postischemic recovery of blood flow in the systemic peripheral circulation.51, 52, 53 In this setting, the magnitude of collateral flow strongly depends on the shear‐stress–induced outward remodeling response of preexisting collateral arteries in the hind limb.54, 55 To investigate the role of RyR2 in hind limb blood flow recovery after ligation of the femoral artery, we monitored perfusion of the right hind limb using the laser Doppler imaging technique and compared it with that in the intact left hind limb. There was no difference in blood flow recovery in the right hind limb in control compared with SM‐Ryr2 −/− mice within 21 days after ligation (Figure 11). These results argue against an important role of RyR2 in postischemic recovery of blood flow. Changes in systemic blood pressure may mask the role of the gene(s) of interest in these effects. However, we found that tamoxifen induced only a 3– to 4–mm Hg increase in mean arterial blood pressure in SM‐Ryr2 −/− mice (Figure 12).
Figure 11.
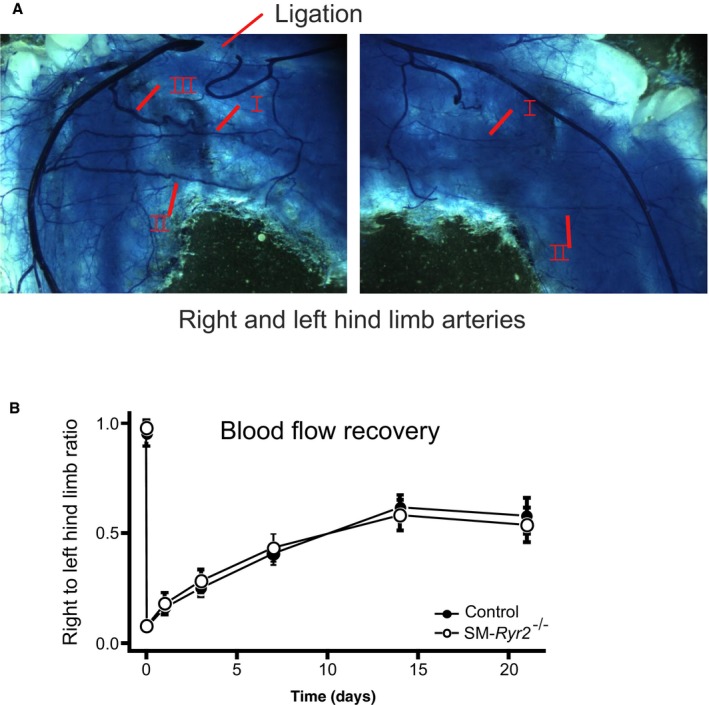
Blood flow recovery after femoral artery ligation. A, Artery structure of right and left hind limbs of a wild‐type (control) mouse after polyurethane treatment. Left: Position of the femoral artery ligation is marked by an arrow. B, Recovery of right hind limb perfusion after femoral artery ligation in control (n=4–10) and smooth muscle (SM)–Ryr2 −/− (n=3–8) mice relative to the left hind limb perfusion. P≥0.05 for all comparisons. Arrows mark native collateral arteries in the ligated and unligated hind limbs, revealing outward remodeling of arteries after femoral artery ligation. Native collateral arteries refer to collaterals I and II. Collateral III is newly developed within 1 to 3 weeks after the ligation.
Figure 12.
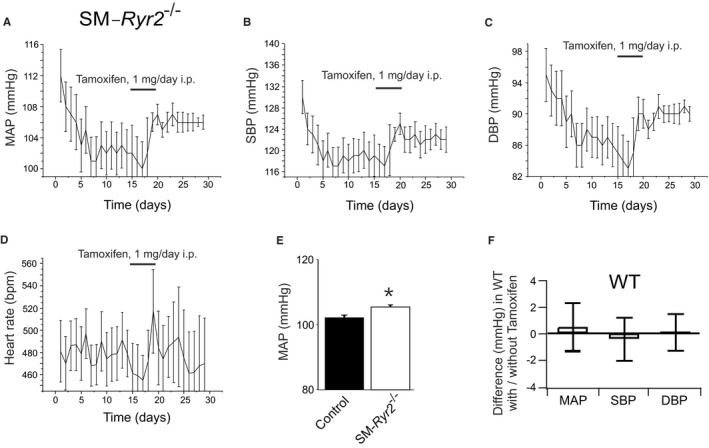
Mean arterial blood pressure (MAP) in smooth muscle (SM)–Ryr2 −/− mice. Time course of MAP (A), systolic blood pressure (SBP; B), diastolic blood pressure (DBP; C), and heart rate (implantation of the telemetric sensor at day 0; D) in SM‐Ryr2 −/− mice. E, Summary of the results. Mean±SEM values were calculated for time intervals before (days 6–14) and after (days 21–29) tamoxifen administration (n=6 mice). Tamoxifen did not change these parameters in wild‐type (WT; control) mice (ΔMAP, −0.8 mm Hg; ΔSBP, −1.2 mm Hg; ΔDBP, 0.0 mm Hg; Δheart rate, 7.7 beats per minute [bpm]; no significant differences). F, MAP, SBP, and DBP did not change in WT mice before and after administration of tamoxifen (n=6 mice each).
Discussion
Resistance arteries exhibit adaptive changes in their diameter in response to pressure and flow that are mediated by distinct mechanisms.1 Increases in transluminal pressure trigger an adaptive reduction in diameter. This myogenic response, which serves to normalize perfusion, is triggered by stretch‐sensitive mechanisms that operate in VSMCs.34 In contrast, increases in blood flow act via an increase in shear stress to trigger an increase in arterial diameter. Short‐term increases in blood flow/shear stress result in arterial dilation through a mechanism that involves the endothelial‐dependent NO pathway, whereas long‐term exposure to elevated shear stress causes a structural increase in the arterial lumen. This adaptive response requires activation of matrix metalloproteases that allow remodeling of the SM layers. In the current study, we tested the contribution of VSMC RyR2s to elementary Ca2+ signals and adaptive vascular responses to pressure and flow.
RyR2s Mediate Elementary SR Ca2+ Events in Arterial SMCs
Our results identify RyR2 as the key SR Ca2+‐release channel involved in generating Ca2+ sparks in VSMCs. In cardiac and skeletal muscle, the intimate association between triggering Cav1.x L‐type channels and target RyRs is a prerequisite for generating Ca2+ sparks from a single Ca2+‐release unit.56, 57, 58, 59, 60, 61 Although RyR2 plays a major role in Cav1.x/RyR coupling in cardiac muscle,62 RyR1 is crucial in skeletal muscle.63, 64 Although the importance of the Cav1.2 Ca2+ channel for proper function of arterial VSMCs is well established, we previously demonstrated that Ca2+ spark generation in these cells does not rely on the intimate association between Cav1.2 L‐type channels and RyRs.30 Instead, Cav1.2 channels contribute to global cytosolic [Ca2+], which, in turn, influences luminal SR Ca2+ and thus Ca2+ sparks.30 In the present study, we investigated the contribution of RyR2s to Ca2+ sparks in arterial VSMCs. We found that RyR2s are essential for the development of Ca2+ sparks/STOCs in VSMCs and subsequent feedback regulation of myogenic tone in response to transmural pressure. We further found that RyR2s contribute to the pulmonary artery pressure response to sustained hypoxia but have no effect on flow‐induced responses. Finally, we found that RyR2s do not contribute to postischemic recovery of blood flow conductance in the peripheral systemic circulation, as shown in a hind limb occlusion model.
VSMC RyR2s in Peripheral Arteries and Vascular Adaptation to Pressure
Our data show that RyR2 is the major RyR isoform responsible for Ca2+ sparks in the VSMC Ca2+‐release unit of systemic arteries. We reach this conclusion for several reasons. First, Ca2+ sparks and STOCs were nearly abolished in SMCs of tibial and mesenteric arteries obtained from SM‐Ryr2 −/− mice, suggesting an essential role for this RyR isoform in this process. This result contrasts with the role of RyR1 in Ca2+ spark generation in cultured VSMCs from portal veins, proposed by Coussin and colleagues,23 who suggested that both RyR1 and RyR2 contribute to spontaneous Ca2+ sparks. However, RyR Ca2+ signaling pathways delineated in cultured cells may reflect changes in protein expression that occur during cell dedifferentiation and may not represent a native feature of VSMCs. The findings reported herein extend our previous studies on Ryr3 −/− mice,25 in which we showed that the frequencies of Ca2+ sparks and STOCs are increased in cerebral VSMCs from Ryr3 −/− mice compared with those from wild‐type mice. To explain these results, we proposed that RyR3 acts as part of the SR Ca2+‐release unit to regulate the generation of Ca2+ sparks by RyR1 and/or RyR2.25 In contrast to the effects of RyR3 ablation on frequency, Ca2+ spark and STOC amplitudes were normal in Ryr3 −/− VSMCs, and caffeine induced normal global Ca2+ release in Ryr3 −/− VSMCs. Second, we found that the pan RyR activator caffeine failed to induce global SR Ca2+ release and subsequent contraction of RyR2‐deficient aortae and femoral, tibial, mesenteric, cerebral, and pulmonary arteries. These results indicate that RyR2 is the only RyR isoform in VSMCs important for overall RyR‐mediated SR Ca2+ release in these vessels. This conclusion is supported by our finding that repeated applications of caffeine failed to induce contractions in mesenteric arteries of SM‐Ryr2 −/− mice. Notably, vascular contractions in response to the α1‐adrenegic receptor agonist phenylephrine were normal in SM‐Ryr2 −/− mesenteric arteries, suggesting a lack of cross talk between RyR2s and IP3‐sensitive Ca2+ pools sensitive to inositol 1,4,5‐trisphosphate (IP3) in the SR of VSMCs, at least in these vessels. Third, an RyR2 deficiency caused a reduction in vascular relaxation through malfunction of the Ca2+ spark–BKCa channel pathway. In these experiments, application of the RyR inhibitor ryanodine constricted control mesenteric arteries but caused almost no constriction of SM‐Ryr2 −/− mesenteric arteries. These findings cannot be explained by a lack of functional BKCa channels in SM‐Ryr2 −/− VSMCs because our patch‐clamp recordings showed normal BKCa channel currents in these cells in response to 5 to 10 μmol/L A23187, which increases global [Ca2+]i to maximally activate BKCa channels.47 Finally, systemic blood pressure was increased in SM‐Ryr2 −/− mice after genetic ablation of Ryr2 in VSMCs. Although this increase was small (3–4 mm Hg), it was statistically significant. Notably, such a small difference is not surprising on the basis of experiments using mice deficient in the BKCa β1 subunit, which exhibit reduced functional coupling of Ca2+ sparks to BKCa channel activation. In these experiments, targeted deletion of the Kcnmb1 gene encoding the BKCa β1 subunit increased systemic blood pressure by ≈10 mm Hg.11, 12, 65, 66 Taken together, these results strongly suggest that elementary SR Ca2+ release in VSMCs is mainly driven by Ca2+ flux through RyR2s. Accordingly, we suggest that RyR2s play a pivotal role in the generation of Ca2+ sparks, which, in turn, activate BKCa channels in VSMCs to limit arterial myogenic constriction in response to transmural pressure. Our results indicate that other RyR isoforms (RyR1 and RyR3) contribute minimally to physiologically relevant Ca2+ sparks in VSMCs of systemic arteries.
VSMC RyR2s in Hypoxic Pulmonary Vasoconstriction and Vascular Adaption to Flow
Using pharmacological tools, Du et al22 suggested that RyRs exert an inhibitory regulatory role in the sustained phase of hypoxic vasoconstriction in the pulmonary circulation. Zheng et al67 attributed part of this hypoxic pulmonary constriction to the action of RyR3, whereas Li et al26 suggested a possible role for RyR1. However, both groups used constitutive (ie, nonconditional) targeting of RyRs, which might be associated with compensatory developmental upregulation of other RyR isoforms. By generating an inducible, SM‐specific RyR2‐knockout mouse model, we were able to investigate RyR2 function in the regulation of pulmonary arterial pressure and hypoxic pulmonary vasoconstriction. In normoxia and in the short‐term phase of hypoxia, we found no differences in the pulmonary arterial pressure response between control and SM‐Ryr2 −/− lungs. However, our experiments revealed an important role of VSMC RyR2s in the sustained phase of hypoxic pulmonary vasoconstriction, although the molecular mechanisms are mostly unclear. Oxidative stress and accumulation of cADP‐ribose and 15‐lipoxygenase metabolites of arachidonic acid (eg, 15‐hydroxyeicosatetraenoic acid) have been implicated in ryanodine‐sensitive SR Ca2+ release in hypoxic pulmonary constriction.68, 69, 70, 71 Our results show that it is unlikely that SR Ca2+ release through RyR1 or RyR3 plays an important role in hypoxic pulmonary vasoconstriction because caffeine (10 mmol/L) was unable to evoke contractile responses in SM‐Ryr2 −/− lungs. Although the BKCa channel itself is sensitive to hypoxia,72 it is possible that this channel is involved in hypoxic pulmonary vasoconstriction, because inhibition of BKCa channels by charybdotoxin (50 ng/mL [≈10 nmol/L]) or iberiotoxin (100 nmol/L) does not decrease hypoxic pulmonary vasoconstriction, but instead enhances vasopressor responses to ventilatory hypoxia in rats73 and rabbits.74 In addition, mice lacking the BKCa β1 subunit show normal pulmonary constriction to acute hypoxia.75 Future studies should explore the mechanisms by which the RyR2 isoform plays a protective role in sustained hypoxic pulmonary vasoconstriction. Plasma membrane stretch‐activated channels,76 TRPC1/6 channels (transient receptor potential canonical channels),77, 78, 79 NADPH oxidase (nicotinamide adenine dinucleotide phosphate oxidase),80 and cytochrome P450–derived epoxyeicosatrienoic acids75 are promising candidate molecules to investigate in this context.
VSMC RyR2s in Hind Limb Ischemia and Vascular Adaptive Responses to Blood Flow
To test if RyR2s affect shear stress–induced responses, we examined the remodeling of native hind limb collateral arteries. It is well established that long‐term increases in shear stress trigger the opening and outward remodeling of these preexisting bypasses.54, 55 An increase in the size of collateral arteries minimizes tissue injury and ischemia caused by vaso‐occlusive diseases, such as atherosclerosis. Using the mouse hind limb femoral artery ligation model38, 81 as an experimental model for monitoring collateral artery recruitment, flow and remodeling characteristics, and tissue ischemia in vivo, we found that SM‐Ryr2 −/− mice and control mice showed comparable blood flow characteristics. We also observed a strong increase in collateral blood flow and collateral diameter in both mouse strains in the short‐term phase as well as long‐term (up to 3 weeks after occlusion). This lack of differences in functional blood flow recovery between the SM‐Ryr2 −/− and wild‐type scenario suggests that, in this time period, RyR2 is not involved in adaptive remodeling of the peripheral vasculature; it neither mediates an acute dilation on changes in flow nor contributes to shear stress–mediated changes in arterial structural properties. Thus, although RyR2s have clear effects on VSMC Ca2+ sparks, shear‐stress–induced remodeling in peripheral hind limb ischemia appears normal in their absence. This is in sharp contrast to mice that are presumed to have altered Cav1.2 channel expression, which affects tonic Ca2+ influx into VSMCs.81 These mice exhibit an almost complete absence of shear‐stress–induced arterial remodeling and collateral blood flow. Taken together, our data point toward a specific contribution of RyR2s in mediating VSMC Ca2+ sparks that is relevant to the regulation of myogenic tone (systemic circulation) and arterial remodeling in response to changes in pressure (lung model), but not flow.
In conclusion, RyR2 is the predominant functional RyR isoform involved in mediating Ca2+ sparks in VSMCs of mice. We found that SM‐specific genetic ablation of Ryr2 substantially reduces Ca2+ sparks, which, in turn, prevents BKCa channel activation, thereby increasing myogenic constriction. We further established that VSMC RyR2s are important in regulating adaptive reductions in arterial diameter in response to pressure, but not flow. We also found that RyR2 provides a negative regulatory role in hypoxic vasoconstriction in the pulmonary circulation but does not contribute to postischemic recovery of blood flow conductance in the hind limb circulation. Our study potentially offers new possibilities for therapeutic interventions in systemic and pulmonary hypertension.
Acknowledgement
We acknowledge support from the Open Access Publication Fund of Charité – Universitätsmedizin Berlin.
Sources of Funding
This study was supported by grants from the Deutsche Akademische Austauschdienst to Gollasch, Szijártó, and Huang; the Deutsche Forschungsgemeinschaft to Witzenrath (SFBTR84 C3 and C6), and Gollasch; the National Institutes of Health (R37 DK053832, R01 HL121706, PO1 HL095488, and R01 HL131181), Fondation Leducq, and the Totman Medical Research Trust to Nelson; and the DZHK (German Centre for Cardiovascular Research) and German Ministry of Education and Research to Gollasch and Kaßmann. García‐Prieto is recipient of Ministerio de Educación Cultura y Deporte, Fundación San Pablo‐CEU, and Banco Santander Fellowships. Fan is recipient of a fellowship from the China Scholarship Council (CSC).
Disclosures
None.
Supporting information
Figure S1. Detection of RyR2 in hearts and different arteries of wild‐type and SM‐Ryr2 −/− mice.
Figure S2. Ca2+ sparks in wild‐type and SM‐Ryr2 −/− mesenteric artery SMCs.
Figure S3. BKCa channel currents in response to caffeine in wild‐type and SM‐Ryr2 −/− VSMCs.
(J Am Heart Assoc. 2019;8:e010090 DOI: 10.1161/JAHA.118.010090.)
References
- 1. Bevan JA, Garcia‐Roldan JL, Joyce EH. Resistance artery tone is influenced independently by pressure and by flow. Blood Vessels. 1990;27:202–207. [DOI] [PubMed] [Google Scholar]
- 2. Moosmang S, Schulla V, Welling A, Feil R, Feil S, Wegener JW, Hofmann F, Klugbauer N. Dominant role of smooth muscle L‐type calcium channel Cav1.2 for blood pressure regulation. EMBO J. 2003;22:6027–6034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nelson MT, Patlak JB, Worley JF, Standen NB. Calcium channels, potassium channels, and voltage dependence of arterial smooth muscle tone. Am J Physiol. 1990;259:C3–C18. [DOI] [PubMed] [Google Scholar]
- 4. Wang SQ, Wei C, Zhao G, Brochet DX, Shen J, Song LS, Wang W, Yang D, Cheng H. Imaging microdomain Ca2+ in muscle cells. Circ Res. 2004;94:1011–1022. [DOI] [PubMed] [Google Scholar]
- 5. Jaggar JH, Wellman GC, Heppner TJ, Porter VA, Perez GJ, Gollasch M, Kleppisch T, Rubart M, Stevenson AS, Lederer WJ, Knot HJ, Bonev AD, Nelson MT. Ca2+ channels, ryanodine receptors and Ca(2+)‐activated K+ channels: a functional unit for regulating arterial tone. Acta Physiol Scand. 1998;164:577–587. [DOI] [PubMed] [Google Scholar]
- 6. Nelson MT, Cheng H, Rubart M, Santana LF, Bonev AD, Knot HJ, Lederer WJ. Relaxation of arterial smooth muscle by calcium sparks. Science. 1995;270:633–637. [DOI] [PubMed] [Google Scholar]
- 7. Knot HJ, Standen NB, Nelson MT. Ryanodine receptors regulate arterial diameter and wall [Ca2+] in cerebral arteries of rat via Ca2+‐dependent K+ channels. J Physiol. 1998;508(pt 1):211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Perez GJ, Bonev AD, Patlak JB, Nelson MT. Functional coupling of ryanodine receptors to KCa channels in smooth muscle cells from rat cerebral arteries. J Gen Physiol. 1999;113:229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Perez GJ, Bonev AD, Nelson MT. Micromolar Ca(2+) from sparks activates Ca(2+)‐sensitive K(+) channels in rat cerebral artery smooth muscle. Am J Physiol Cell Physiol. 2001;281:C1769–C1775. [DOI] [PubMed] [Google Scholar]
- 10. Jaggar JH, Porter VA, Lederer WJ, Nelson MT. Calcium sparks in smooth muscle. Am J Physiol Cell Physiol. 2000;278:C235–C256. [DOI] [PubMed] [Google Scholar]
- 11. Brenner R, Perez GJ, Bonev AD, Eckman DM, Kosek JC, Wiler SW, Patterson AJ, Nelson MT, Aldrich RW. Vasoregulation by the beta1 subunit of the calcium‐activated potassium channel. Nature. 2000;407:870–876. [DOI] [PubMed] [Google Scholar]
- 12. Pluger S, Faulhaber J, Furstenau M, Lohn M, Waldschutz R, Gollasch M, Haller H, Luft FC, Ehmke H, Pongs O. Mice with disrupted BK channel beta1 subunit gene feature abnormal Ca(2+) spark/STOC coupling and elevated blood pressure. Circ Res. 2000;87:E53–E60. [DOI] [PubMed] [Google Scholar]
- 13. Sausbier M, Arntz C, Bucurenciu I, Zhao H, Zhou XB, Sausbier U, Feil S, Kamm S, Essin K, Sailer CA, Abdullah U, Krippeit‐Drews P, Feil R, Hofmann F, Knaus HG, Kenyon C, Shipston MJ, Storm JF, Neuhuber W, Korth M, Schubert R, Gollasch M, Ruth P. Elevated blood pressure linked to primary hyperaldosteronism and impaired vasodilation in BK channel‐deficient mice. Circulation. 2005;112:60–68. [DOI] [PubMed] [Google Scholar]
- 14. Gollasch M, Wellman GC, Knot HJ, Jaggar JH, Damon DH, Bonev AD, Nelson MT. Ontogeny of local sarcoplasmic reticulum Ca2+ signals in cerebral arteries: Ca2+ sparks as elementary physiological events. Circ Res. 1998;83:1104–1114. [DOI] [PubMed] [Google Scholar]
- 15. Filosa JA, Bonev AD, Straub SV, Meredith AL, Wilkerson MK, Aldrich RW, Nelson MT. Local potassium signaling couples neuronal activity to vasodilation in the brain. Nat Neurosci. 2006;9:1397–1403. [DOI] [PubMed] [Google Scholar]
- 16. Marks AR, Tempst P, Hwang KS, Taubman MB, Inui M, Chadwick C, Fleischer S, Nadal‐Ginard B. Molecular cloning and characterization of the ryanodine receptor/junctional channel complex cDNA from skeletal muscle sarcoplasmic reticulum. Proc Natl Acad Sci USA. 1989;86:8683–8687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Takeshima H, Nishimura S, Matsumoto T, Ishida H, Kangawa K, Minamino N, Matsuo H, Ueda M, Hanaoka M, Hirose T, Numa S. Primary structure and expression from complementary DNA of skeletal muscle ryanodine receptor. Nature. 1989;339:439–445. [DOI] [PubMed] [Google Scholar]
- 18. Otsu K, Willard HF, Khanna VK, Zorzato F, Green NM, MacLennan DH. Molecular cloning of cDNA encoding the Ca2+ release channel (ryanodine receptor) of rabbit cardiac muscle sarcoplasmic reticulum. J Biol Chem. 1990;265:13472–13483. [PubMed] [Google Scholar]
- 19. Hakamata Y, Nakai J, Takeshima H, Imoto K. Primary structure and distribution of a novel ryanodine receptor/calcium release channel from rabbit brain. FEBS Lett. 1992;312:229–235. [DOI] [PubMed] [Google Scholar]
- 20. Meissner G. Ryanodine receptor/Ca2+ release channels and their regulation by endogenous effectors. Annu Rev Physiol. 1994;56:485–508. [DOI] [PubMed] [Google Scholar]
- 21. Ledbetter MW, Preiner JK, Louis CF, Mickelson JR. Tissue distribution of ryanodine receptor isoforms and alleles determined by reverse transcription polymerase chain reaction. J Biol Chem. 1994;269:31544–31551. [PubMed] [Google Scholar]
- 22. Du W, Frazier M, McMahon TJ, Eu JP. Redox activation of intracellular calcium release channels (ryanodine receptors) in the sustained phase of hypoxia‐induced pulmonary vasoconstriction. Chest. 2005;128:556S–558S. [DOI] [PubMed] [Google Scholar]
- 23. Coussin F, Macrez N, Morel JL, Mironneau J. Requirement of ryanodine receptor subtypes 1 and 2 for Ca(2+)‐induced Ca(2+) release in vascular myocytes. J Biol Chem. 2000;275:9596–9603. [DOI] [PubMed] [Google Scholar]
- 24. Mironneau J, Coussin F, Jeyakumar LH, Fleischer S, Mironneau C, Macrez N. Contribution of ryanodine receptor subtype 3 to Ca2+ responses in Ca2+‐overloaded cultured rat portal vein myocytes. J Biol Chem. 2001;276:11257–11264. [DOI] [PubMed] [Google Scholar]
- 25. Lohn M, Jessner W, Furstenau M, Wellner M, Sorrentino V, Haller H, Luft FC, Gollasch M. Regulation of calcium sparks and spontaneous transient outward currents by RyR3 in arterial vascular smooth muscle cells. Circ Res. 2001;89:1051–1057. [DOI] [PubMed] [Google Scholar]
- 26. Li XQ, Zheng YM, Rathore R, Ma J, Takeshima H, Wang YX. Genetic evidence for functional role of ryanodine receptor 1 in pulmonary artery smooth muscle cells. Pflugers Arch. 2009;457:771–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Takeshima H, Komazaki S, Hirose K, Nishi M, Noda T, Iino M. Embryonic lethality and abnormal cardiac myocytes in mice lacking ryanodine receptor type 2. EMBO J. 1998;17:3309–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Feil R, Wagner J, Metzger D, Chambon P. Regulation of Cre recombinase activity by mutated estrogen receptor ligand‐binding domains. Biochem Biophys Res Commun. 1997;237:752–757. [DOI] [PubMed] [Google Scholar]
- 29. Kuhbandner S, Brummer S, Metzger D, Chambon P, Hofmann F, Feil R. Temporally controlled somatic mutagenesis in smooth muscle. Genesis. 2000;28:15–22. [DOI] [PubMed] [Google Scholar]
- 30. Essin K, Welling A, Hofmann F, Luft FC, Gollasch M, Moosmang S. Indirect coupling between Cav1.2 channels and ryanodine receptors to generate Ca2+ sparks in murine arterial smooth muscle cells. J Physiol. 2007;584:205–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wirth A, Benyo Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, Orsy P, Horvath B, Maser‐Gluth C, Greiner E, Lemmer B, Schutz G, Gutkind JS, Offermanns S. G12‐G13‐LARG‐mediated signaling in vascular smooth muscle is required for salt‐induced hypertension. Nat Med. 2008;14:64–68. [DOI] [PubMed] [Google Scholar]
- 32. Fesus G, Dubrovska G, Gorzelniak K, Kluge R, Huang Y, Luft FC, Gollasch M. Adiponectin is a novel humoral vasodilator. Cardiovasc Res. 2007;75:719–727. [DOI] [PubMed] [Google Scholar]
- 33. Heppner TJ, Layne JJ, Pearson JM, Sarkissian H, Nelson MT. Unique properties of muscularis mucosae smooth muscle in guinea pig urinary bladder. Am J Physiol Regul Integr Comp Physiol. 2011;301:R351–R362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schleifenbaum J, Kassmann M, Szijarto IA, Hercule HC, Tano JY, Weinert S, Heidenreich M, Pathan AR, Anistan YM, Alenina N, Rusch NJ, Bader M, Jentsch TJ, Gollasch M. Stretch‐activation of angiotensin II type 1a receptors contributes to the myogenic response of mouse mesenteric and renal arteries. Circ Res. 2014;115:263–272. [DOI] [PubMed] [Google Scholar]
- 35. Furstenau M, Lohn M, Ried C, Luft FC, Haller H, Gollasch M. Calcium sparks in human coronary artery smooth muscle cells resolved by confocal imaging. J Hypertens. 2000;18:1215–1222. [DOI] [PubMed] [Google Scholar]
- 36. Gollasch M, Ried C, Bychkov R, Luft FC, Haller H. K+ currents in human coronary artery vascular smooth muscle cells. Circ Res. 1996;78:676–688. [DOI] [PubMed] [Google Scholar]
- 37. Hercule HC, Schunck WH, Gross V, Seringer J, Leung FP, Weldon SM, da Costa Goncalves A, Huang Y, Luft FC, Gollasch M. Interaction between P450 eicosanoids and nitric oxide in the control of arterial tone in mice. Arterioscler Thromb Vasc Biol. 2009;29:54–60. [DOI] [PubMed] [Google Scholar]
- 38. Buschmann I, Pries A, Styp‐Rekowska B, Hillmeister P, Loufrani L, Henrion D, Shi Y, Duelsner A, Hoefer I, Gatzke N, Wang H, Lehmann K, Ulm L, Ritter Z, Hauff P, Hlushchuk R, Djonov V, van Veen T, le Noble F. Pulsatile shear and Gja5 modulate arterial identity and remodeling events during flow‐driven arteriogenesis. Development. 2010;137:2187–2196. [DOI] [PubMed] [Google Scholar]
- 39. Meyer EP, Beer GM, Lang A, Manestar M, Krucker T, Meier S, Mihic‐Probst D, Groscurth P. Polyurethane elastomer: a new material for the visualization of cadaveric blood vessels. Clin Anat. 2007;20:448–454. [DOI] [PubMed] [Google Scholar]
- 40. Tabeling C, Yu H, Wang L, Ranke H, Goldenberg NM, Zabini D, Noe E, Krauszman A, Gutbier B, Yin J, Schaefer M, Arenz C, Hocke AC, Suttorp N, Proia RL, Witzenrath M, Kuebler WM. CFTR and sphingolipids mediate hypoxic pulmonary vasoconstriction. Proc Natl Acad Sci USA. 2015;112:E1614–E1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Spohr F, Busch CJ, Reich C, Motsch J, Gebhard MM, Kuebler WM, Bloch KD, Weimann J. 4‐Aminopyridine restores impaired hypoxic pulmonary vasoconstriction in endotoxemic mice. Anesthesiology. 2007;107:597–604. [DOI] [PubMed] [Google Scholar]
- 42. Lachine NA, Elnekiedy AA, Megallaa MH, Khalil GI, Sadaka MA, Rohoma KH, Kassab HS. Serum chemerin and high‐sensitivity C reactive protein as markers of subclinical atherosclerosis in Egyptian patients with type 2 diabetes. Ther Adv Endocrinol Metab. 2016;7:47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Marko L, Henke N, Park JK, Spallek B, Qadri F, Balogh A, Apel IJ, Oravecz‐Wilson KI, Choi M, Przybyl L, Binger KJ, Haase N, Wilck N, Heuser A, Fokuhl V, Ruland J, Lucas PC, McAllister‐Lucas LM, Luft FC, Dechend R, Muller DN. Bcl10 mediates angiotensin II‐induced cardiac damage and electrical remodeling. Hypertension. 2014;64:1032–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Heinze C, Seniuk A, Sokolov MV, Huebner AK, Klementowicz AE, Szijarto IA, Schleifenbaum J, Vitzthum H, Gollasch M, Ehmke H, Schroeder BC, Hubner CA. Disruption of vascular Ca2+‐activated chloride currents lowers blood pressure. J Clin Invest. 2014;124:675–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Morel JL, Dabertrand F, Porte Y, Prevot A, Macrez N. Up‐regulation of ryanodine receptor expression increases the calcium‐induced calcium release and spontaneous calcium signals in cerebral arteries from hindlimb unloaded rats. Pflugers Arch. 2014;466:1517–1528. [DOI] [PubMed] [Google Scholar]
- 46. Bychkov R, Gollasch M, Ried C, Luft FC, Haller H. Effects of pinacidil on K+ channels in human coronary artery vascular smooth muscle cells. Am J Physiol. 1997;273:C161–C171. [DOI] [PubMed] [Google Scholar]
- 47. Bychkov R, Gollasch M, Ried C, Luft FC, Haller H. Regulation of spontaneous transient outward potassium currents in human coronary arteries. Circulation. 1997;95:503–510. [DOI] [PubMed] [Google Scholar]
- 48. Zucchi R, Ronca‐Testoni S. The sarcoplasmic reticulum Ca2+ channel/ryanodine receptor: modulation by endogenous effectors, drugs and disease states. Pharmacol Rev. 1997;49:1–51. [PubMed] [Google Scholar]
- 49. Watanabe J, Horiguchi S, Karibe A, Keitoku M, Takeuchi M, Satoh S, Takishima T, Shirato K. Effects of ryanodine on development of myogenic response in rat small skeletal muscle arteries. Cardiovasc Res. 1994;28:480–484. [DOI] [PubMed] [Google Scholar]
- 50. Wang L, Yin J, Nickles HT, Ranke H, Tabuchi A, Hoffmann J, Tabeling C, Barbosa‐Sicard E, Chanson M, Kwak BR, Shin HS, Wu S, Isakson BE, Witzenrath M, de Wit C, Fleming I, Kuppe H, Kuebler WM. Hypoxic pulmonary vasoconstriction requires connexin 40‐mediated endothelial signal conduction. J Clin Invest. 2012;122:4218–4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Carrier O Jr, Walker JR, Guyton AC. Role of oxygen in autoregulation of blood flow in isolated vessels. Am J Physiol. 1964;206:951–954. [DOI] [PubMed] [Google Scholar]
- 52. Shimizu S, Bowman PS, Thorne G III, Paul RJ. Effects of hypoxia on isometric force, intracellular Ca(2+), pH, and energetics in porcine coronary artery. Circ Res. 2000;86:862–870. [DOI] [PubMed] [Google Scholar]
- 53. Thorne GD, Paul RJ. Effects of organ culture on arterial gene expression and hypoxic relaxation: role of the ryanodine receptor. Am J Physiol Cell Physiol. 2003;284:C999–C1005. [DOI] [PubMed] [Google Scholar]
- 54. Eitenmuller I, Volger O, Kluge A, Troidl K, Barancik M, Cai WJ, Heil M, Pipp F, Fischer S, Horrevoets AJ, Schmitz‐Rixen T, Schaper W. The range of adaptation by collateral vessels after femoral artery occlusion. Circ Res. 2006;99:656–662. [DOI] [PubMed] [Google Scholar]
- 55. Arras M, Ito WD, Scholz D, Winkler B, Schaper J, Schaper W. Monocyte activation in angiogenesis and collateral growth in the rabbit hindlimb. J Clin Invest. 1998;101:40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cheng H, Lederer WJ, Cannell MB. Calcium sparks: elementary events underlying excitation‐contraction coupling in heart muscle. Science. 1993;262:740–744. [DOI] [PubMed] [Google Scholar]
- 57. Cannell MB, Cheng H, Lederer WJ. The control of calcium release in heart muscle. Science. 1995;268:1045–1049. [DOI] [PubMed] [Google Scholar]
- 58. Lopez‐Lopez JR, Shacklock PS, Balke CW, Wier WG. Local calcium transients triggered by single L‐type calcium channel currents in cardiac cells. Science. 1995;268:1042–1045. [DOI] [PubMed] [Google Scholar]
- 59. Tsugorka A, Rios E, Blatter LA. Imaging elementary events of calcium release in skeletal muscle cells. Science. 1995;269:1723–1726. [DOI] [PubMed] [Google Scholar]
- 60. Klein MG, Lacampagne A, Schneider MF. Voltage dependence of the pattern and frequency of discrete Ca2+ release events after brief repriming in frog skeletal muscle. Proc Natl Acad Sci USA. 1997;94:11061–11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wang SQ, Song LS, Lakatta EG, Cheng H. Ca2+ signalling between single L‐type Ca2+ channels and ryanodine receptors in heart cells. Nature. 2001;410:592–596. [DOI] [PubMed] [Google Scholar]
- 62. Marx SO, Gaburjakova J, Gaburjakova M, Henrikson C, Ondrias K, Marks AR. Coupled gating between cardiac calcium release channels (ryanodine receptors). Circ Res. 2001;88:1151–1158. [DOI] [PubMed] [Google Scholar]
- 63. Takeshima H, Iino M, Takekura H, Nishi M, Kuno J, Minowa O, Takano H, Noda T. Excitation‐contraction uncoupling and muscular degeneration in mice lacking functional skeletal muscle ryanodine‐receptor gene. Nature. 1994;369:556–559. [DOI] [PubMed] [Google Scholar]
- 64. Essin K, Gollasch M. Role of ryanodine receptor subtypes in initiation and formation of calcium sparks in arterial smooth muscle: comparison with striated muscle. J Biomed Biotechnol. 2009;2009:135249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Xu H, Garver H, Galligan JJ, Fink GD. Large‐conductance Ca2+‐activated K+ channel beta1‐subunit knockout mice are not hypertensive. Am J Physiol Heart Circ Physiol. 2011;300:H476–H485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sachse G, Faulhaber J, Seniuk A, Ehmke H, Pongs O. Smooth muscle BK channel activity influences blood pressure independent of vascular tone in mice. J Physiol. 2014;592:2563–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zheng YM, Wang QS, Rathore R, Zhang WH, Mazurkiewicz JE, Sorrentino V, Singer HA, Kotlikoff MI, Wang YX. Type‐3 ryanodine receptors mediate hypoxia‐, but not neurotransmitter‐induced calcium release and contraction in pulmonary artery smooth muscle cells. J Gen Physiol. 2005;125:427–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Connolly MJ, Prieto‐Lloret J, Becker S, Ward JP, Aaronson PI. Hypoxic pulmonary vasoconstriction in the absence of pretone: essential role for intracellular Ca2+ release. J Physiol. 2013;591:4473–4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Dipp M, Evans AM. Cyclic ADP‐ribose is the primary trigger for hypoxic pulmonary vasoconstriction in the rat lung in situ. Circ Res. 2001;89:77–83. [DOI] [PubMed] [Google Scholar]
- 70. Zheng X, Li Q, Tang X, Liang S, Chen L, Zhang S, Wang Z, Guo L, Zhang R, Zhu D. Source of the elevation Ca2+ evoked by 15‐HETE in pulmonary arterial myocytes. Eur J Pharmacol. 2008;601:16–22. [DOI] [PubMed] [Google Scholar]
- 71. Dipp M, Nye PC, Evans AM. Hypoxic release of calcium from the sarcoplasmic reticulum of pulmonary artery smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2001;281:L318–L325. [DOI] [PubMed] [Google Scholar]
- 72. Tano JY, Schleifenbaum J, Gollasch M. Perivascular adipose tissue, potassium channels, and vascular dysfunction. Arterioscler Thromb Vasc Biol. 2014;34:1827–1830. [DOI] [PubMed] [Google Scholar]
- 73. Nossaman BD, Kaye AD, Feng CJ, Kadowitz PJ. Effects of charybdotoxin on responses to nitrosovasodilators and hypoxia in the rat lung. Am J Physiol. 1997;272:L787–L791. [DOI] [PubMed] [Google Scholar]
- 74. Liu R, Ueda M, Okazaki N, Ishibe Y. Role of potassium channels in isoflurane‐ and sevoflurane‐induced attenuation of hypoxic pulmonary vasoconstriction in isolated perfused rabbit lungs. Anesthesiology. 2001;95:939–946. [DOI] [PubMed] [Google Scholar]
- 75. Loot AE, Moneke I, Keseru B, Oelze M, Syzonenko T, Daiber A, Fleming I. 11,12‐EET stimulates the association of BK channel alpha and beta(1) subunits in mitochondria to induce pulmonary vasoconstriction. PLoS One. 2012;7:e46065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Gilbert G, Ducret T, Marthan R, Savineau JP, Quignard JF. Stretch‐induced Ca2+ signalling in vascular smooth muscle cells depends on Ca2+ store segregation. Cardiovasc Res. 2014;103:313–323. [DOI] [PubMed] [Google Scholar]
- 77. Malczyk M, Veith C, Fuchs B, Hofmann K, Storch U, Schermuly RT, Witzenrath M, Ahlbrecht K, Fecher‐Trost C, Flockerzi V, Ghofrani HA, Grimminger F, Seeger W, Gudermann T, Dietrich A, Weissmann N. Classical transient receptor potential channel 1 in hypoxia‐induced pulmonary hypertension. Am J Respir Crit Care Med. 2013;188:1451–1459. [DOI] [PubMed] [Google Scholar]
- 78. Xia Y, Yang XR, Fu Z, Paudel O, Abramowitz J, Birnbaumer L, Sham JS. Classical transient receptor potential 1 and 6 contribute to hypoxic pulmonary hypertension through differential regulation of pulmonary vascular functions. Hypertension. 2014;63:173–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Weissmann N, Sydykov A, Kalwa H, Storch U, Fuchs B, Mederos y Schnitzler M, Brandes RP, Grimminger F, Meissner M, Freichel M, Offermanns S, Veit F, Pak O, Krause KH, Schermuly RT, Brewer AC, Schmidt HH, Seeger W, Shah AM, Gudermann T, Ghofrani HA, Dietrich A. Activation of TRPC6 channels is essential for lung ischaemia‐reperfusion induced oedema in mice. Nat Commun. 2012;3:649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Nagaraj C, Tabeling C, Nagy BM, Jain PP, Marsh LM, Papp R, Pienn M, Witzenrath M, Ghanim B, Klepetko W, Weir EK, Heschl S, Kwapiszewska G, Olschewski A, Olschewski H. Hypoxic vascular response and ventilation/perfusion matching in end‐stage COPD may depend on p22phox. Eur Respir J. 2017;50:1601651. [DOI] [PubMed] [Google Scholar]
- 81. Cristofaro B, Shi Y, Faria M, Suchting S, Leroyer AS, Trindade A, Duarte A, Zovein AC, Iruela‐Arispe ML, Nih LR, Kubis N, Henrion D, Loufrani L, Todiras M, Schleifenbaum J, Gollasch M, Zhuang ZW, Simons M, Eichmann A, le Noble F. Dll4‐Notch signaling determines the formation of native arterial collateral networks and arterial function in mouse ischemia models. Development. 2013;140:1720–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Detection of RyR2 in hearts and different arteries of wild‐type and SM‐Ryr2 −/− mice.
Figure S2. Ca2+ sparks in wild‐type and SM‐Ryr2 −/− mesenteric artery SMCs.
Figure S3. BKCa channel currents in response to caffeine in wild‐type and SM‐Ryr2 −/− VSMCs.