

Abstract
Background
The mechanistic target of rapamycin complex 1 (mTORC1) is an important intracellular energy sensor that regulates gene expression and protein synthesis through its downstream signaling components, the S6‐kinase and the ribosomal S6 protein. Recently, signaling arising from mTORC1 has been implicated in regulation of the cardiovascular system with implications for disease. Here, we examined the contribution of mTORC1 signaling to the regulation of vascular function.
Methods and Results
Activation of mTORC1 pathway in aortic rings with leucine or an adenoviral vector expressing a constitutively active S6‐kinase reduces endothelial‐dependent vasorelaxation in an mTORC1‐dependent manner without affecting smooth muscle relaxation responses. Moreover, activation of mTORC1 signaling in endothelial cells increases reactive oxygen species (ROS) generation and ROS gene expression resulting in a pro‐oxidant gene environment. Blockade of ROS signaling with Tempol restores endothelial function in vascular rings with increased mTORC1 activity indicating a crucial interaction between mTORC1 and ROS signaling. We then tested the role of nuclear factor‐κB transcriptional complex in connecting mTORC1 and ROS signaling in endothelial cells. Blockade of inhibitor of nuclear factor κ‐B kinase subunit β activity with BMS‐345541 prevented the increased ROS generation associated with increased mTORC1 activity in endothelial cells but did not improve vascular endothelial function in aortic rings with increased mTORC1 and ROS signaling.
Conclusions
These results implicate mTORC1 as a critical molecular signaling hub in the vascular endothelium in mediating vascular endothelial function through modulation of ROS signaling.
Keywords: endothelial cell, endothelial function, mechanistic target of rapamycin complex 1, NFκB, oxidative stress, vascular biology
Subject Categories: Basic Science Research, Endothelium/Vascular Type/Nitric Oxide, Oxidant Stress, Physiology, Vascular Biology
Clinical Perspective
What Is New?
Stimulation of mechanistic target of rapamycin complex 1 signaling results in impaired endothelial‐mediated vasorelaxation responses.
The vascular endothelial dysfunction evoked by mechanistic target of rapamycin complex 1 activation involves a pro‐oxidant gene program with enhanced reactive oxygen species generation.
What Are the Clinical Implications?
Mechanistic target of rapamycin complex 1 signaling is a direct and crucial mediator of vascular endothelial function and dysregulation of this pathway may contribute to endothelial dysfunction.
Mechanistic target of rapamycin complex 1 signaling is a potential targeting point for a therapeutic approach to mitigate vascular endothelial dysfunction, a major cardiovascular risk.
Introduction
Cellular energy sensing pathways are fundamental to the regulation of intracellular signaling, redox sensing, and responses to alterations in homeostasis by coordinating energy metabolism within cells. One of the major pathways contributing to cellular energy sensing is the mechanistic target of rapamycin complex 1 (mTORC1). This complex serves as a key energy sensor that controls several important cellular processes such as mitochondrial biogenesis and function, cellular proliferation, and autophagy. The mTORC1 consists of a highly conserved catalytic subunit (mTOR), the unique regulatory subunit Raptor (regulatory associated protein of mTOR) and several accessory proteins that integrate a variety of energy signals such as growth factors, hormones, amino acids, and glucose/insulin signaling. The mTORC1 integrates these signals through modulation of cellular transcription and translation processes including the p70 ribosomal S6 kinase (S6K) and its downstream target the S6 ribosomal protein conferring gene and protein programming within cells.
Vascular endothelial dysfunction is widely recognized as a hallmark of a variety of cardiovascular‐related diseases such as hypertension, atherosclerosis, and metabolic disorders.1, 2 A myriad of signaling pathways have been established that underlie these deficits in endothelial‐mediated relaxation including oxidative stress, inflammation, glucose/insulin metabolism, and the renin‐angiotensin system with considerable complexity arising in the crosstalk between these pathways and the resulting integration of cardiovascular responses. A key integrating molecular hub for these signaling pathways in the vascular system is the mTORC1 pathway. Previous studies have demonstrated that mTORC1 activity in vascular endothelial cells is elevated in response to angiotensin II,3 glucose,4 insulin,3 and oxidative stress5 suggesting a potential role in mediating the vascular functional effects of these signaling pathways. However, the overall contribution of mTORC1‐dependent activity in the regulation of these vascular endothelial signaling pathways and ultimately vascular function remains unclear.
In this study, we tested the hypothesis that vascular mTORC1 signaling is directly involved in the regulation of vascular endothelial function. We developed 2 ex vivo models of mTORC1 signaling activation in vascular rings and assessed the effects of mTORC1 signaling on vascular relaxation pathways. Additionally, we investigated mTORC1 signaling in a cultured mouse endothelial cell line and described a mechanism by which mTORC1 signaling may regulate vascular endothelial function that involve reactive oxygen species signaling.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Animals
A total number of 114 male wild‐type C57BL/6J mice (aged 12–36 weeks) from our own colonies were used in the current studies (ex vivo vascular culture studies and molecular analyses). Mice were housed with standard mouse chow and provided water ad libitum. All animal protocols were reviewed and approved by the Institutional Animal Care and Use Committee at the University of Iowa and adhere to animal guidelines of the National Institutes of Health.
Ex Vivo Vascular Ring Culture
Thoracic aortas and mesenteric arteries were isolated, cleaned of debris and cut into 2‐ to 3‐mm length rings. Rings were rinsed in PBS (1x) and placed in a 24‐well tissue culture plate with 1‐mL DMEM/F12 (1:1) and 1% penicillin‐streptomycin (Gibco; #11320033) for 24 hours in an incubator. Leucine stimulation studies were performed as follows: DMEM/F12 media supplemented with leucine (l‐leucine, Sigma; #L8000) for a final concentration of 10 mmol/L leucine in the media. Some rings were exposed to the same concentration of valine (l‐valine, RPI; #1005498) which does not activate mTORC1 signaling.6 Control DMEM/F12 media had a final concentration of 0.45 mmol/L leucine or valine. Adenoviral infection studies were performed as follows: DMEM/F12 media with the addition of equal titer (2×108 pfu/mL) of adenoviral vector expressing a green fluorescent protein (Ad‐GFP), adenoviral S6‐kinase dominant negative (Ad‐S6KDN), or adenoviral S6‐kinase constitutively active (Ad‐S6KCA) for 24 hours in an incubator.6, 7 After 24 hours, rings were rinsed with PBS (1×) and either snap frozen for molecular analysis or mounted on a wire myograph for vascular function assessments.
Wire Myography
The rings were mounted on a wire myograph (DMT, Model 610M) for measurements of isometric tension. Resting tension was set at ≈0.5 g and rings were equilibrated for 45 minutes in heated (37°C) Krebs solution containing the following: 118.3 mmol/L NaCl, 4.7 mmol/L KCl, 1.2 mmol/L MgSO4, 1.2 mmol/L KH2PO4, 25.0 mmol/L NaHCO3, 2.5 mmol/L CaCl2, 11.0 mmol/L glucose at pH 7.4. Rings were submaximally contracted with prostaglandin F2α (PGF2α; ≈30%–50%) (Lutalyse; Zoetis Inc; supplied by University of Iowa Pharmacy) and cumulative response curves to acetylcholine (1 nmol/L–100 μmol/L) and sodium nitroprusside (SNP, 1 nmol/L–100 μmol/L) were performed. A subset of aortic rings was preincubated with Tempol (100 μmol/L; 30 minutes) or NG‐nitro‐L‐arginine methyl ester (L‐NAME) (100 μmol/L; 30 minutes) before performing the cumulative concentration response curves. Additionally, a subset of aortic rings were infected with Ad‐GFP or Ad‐S6KCA for 24 hours and incubated for the final 16 hours with BMS‐345541 (300 nmol/L) before performing the cumulative concentration response curves. Dose response to PGF2α (1 μmol/L–10 mmol/L) and KCl depolarization (100 mmol/L) were also assessed. Data are expressed as percentage of relaxation or fold change of maximal contraction to 100 mmol/L KCl.
Cell Culture
Mouse lung endothelial cells (MLECs)8 were cultured in MCDB‐131 media and used for studies between passages 12 to 18. Leucine‐stimulated cell culture experiments were performed as follows: cells were stimulated with 10 mmol/L leucine under serum free conditions overnight (16 hours) and then cells were processed for protein or mRNA analysis. Adenoviral cell culture experiments were performed as follows: cells were infected (2×108 pfu/mL titer) with Ad‐GFP (control) or Ad‐S6KCA for 48 hours. This adenoviral titer was chosen as Ad‐GFP infection demonstrated nearly 100% GFP expression in MLECs. Cells were then processed for immunocytochemistry, protein, or mRNA analysis as described below.
Immunochemistry
Aortic and mesenteric arterial rings were incubated in 4% paraformaldehyde overnight at 4oC and were cryopreserved in 30% sucrose. Fixed rings were washed 3 times with PBS and mounted in optimal cutting temperature solution, then cryosectioned at 10‐μm thickness. MLECs were fixed in 4% paraformaldehyde for 20 minutes at room temperature. Immunohistochemistry was performed on ring sections or cell slides to detect phosphorylated S6 ribosomal protein by using a 1:250 dilution of a rabbit polyclonal phospho‐S6 antibody (Cell Signaling, #5364). Processed arterial ring sections or cell slides were mounted using VectaShield mounting medium with 4′,6‐diamidino‐2‐phenylindole (DAPI). Images were visualized using confocal microscopy (Zeiss LSM710) and analyzed using ImageJ software. An independent experiment consists of 4 photographs per experimental condition taken on an individual day. Each photo consists of 16 separate areas of analysis which are then averaged for that individual independent experiment. This was then repeated 3 to 6 separate times for the overall data analysis. Data are presented as percentage corrected total cell fluorescence with representative images ‐presented.
Quantitative Real Time Polymerase Chain Reaction
RNAs were isolated from MLECs using a column‐based purification RNA kit (Qiagen). mRNAs were reverse transcribed using an iScript cDNA synthesis kit (Bio‐Rad). Quantitative polymerase chain reaction was performed using an iQ5 real‐time polymerase chain reaction machine (Bio‐Rad) and iScript SYBR green master mix (Bio‐Rad). Primer sequences used in this study are provided in Table S1. Data were normalized to β‐actin (internal control) and expressed as fold change normalized to control. An independent experiment unit consists of RNA isolated from 2 separate dishes per experimental condition run in duplicate for the genes presented. This was then repeated 6 separate times for the overall data analysis.
Western Blotting
Proteins were isolated from aortic rings and MLECs through homogenization in a lysis buffer containing the following: 50 mmol/L Tris‐Cl pH7.5, 0.1 mmol/L ethylenediaminetetraacetic acid (EDTA), 0.1 mmol/L ethylene glycol‐bis(β‐aminoethyl ether (EGTA), 1 mmol/L phenylmethylsulfonyl fluoride (PMSF), 1 mmol/L NaVO4, 10 mmol/L NaF, 1 mmol/L sodium pyrophosphate, 0.001% sodium deoxycholic acid (wt/vol), 1% Tergitol‐type (NP)‐40 (vol/vol), 0.1% SDS (wt/vol), and a protease inhibitor cocktail tablet (Roche). Proteins were then subjected to SDS‐PAGE and transferred to polyvinylidene fluoride membranes. Membranes were blocked with 5% non‐fat dry milk followed by incubation with primary antibodies for phospho‐S6 (1:5000; Cell Signaling #5364), total S6 (1:1000; Cell Signaling #2217), total S6 kinase (1:1000; Cell Signaling #2708) phospho‐eNOS (1:1000; Cell Signaling #9571; BD Biosciences #612393), total eNOS (1:1000; BD Biosciences #610297), gp91phox (1:1000; BD Biosciences #611415), superoxide dismutase‐2 (1:1000, Cell Signaling #13141), and β‐actin (1:50 000; Proteintech #60008). Proteins were detected using anti‐rabbit (1:10 000; Cell Signaling #7074) or anti‐mouse horseradish peroxidase (1:10 000; Cell Signaling #7076) conjugated secondary antibodies. Protein expression was visualized by an ECL Prime chemiluminescent detection kit (GE Healthcare) on film, and densitometry (arbitrary units) was calculated using ImageJ. An independent experiment unit consists of protein isolated from a 60‐mm dish per experimental condition and probed for proteins of interest. This was then repeated 6 separate times for the overall data analysis.
Dihydroethidium Staining
Reactive oxygen species (ROS) production in adenoviral‐infected MLECs (48 hours) or aortic rings (24 hours) was evaluated using dihydroethidium staining (Sigma). A subset of MLECs (48 hours) or aortic rings (24 hours) was infected with Ad‐S6KCA or Ad‐GFP (control). 16 hours before the end of the infection period cells or aortic rings were treated with vehicle (PBS) or BMS‐345541 (300 nmol/L; Tocris; #4806) to block IKKβ activity. MLECs and aortic rings were fixed with 4% paraformaldehyde for 20 minutes at room temperature, then incubated for 30 minutes at 37oC in PBS (1×) containing 8 μmol/L dihydroethidium. The cells or aortic rings were washed 3 times with 1× PBS and mounted with VectaShield mounting medium with 4′,6‐diamidino‐2‐phenylindole. Dihydroethidium images were visualized with confocal microscopy (Zeiss LSM710) at an excitation/emission of 488/568 nm, and analyzed using ImageJ software for its relative intensity and expressed as a percentage of corrected total cell fluorescence/cell for MLECs and aortic rings. For MLECs, an independent experiment consists of 4 photographs per experimental condition taken on an individual day. Each photo consists of 16 separate areas of analysis which are then averaged for that particular independent experiment. This was then repeated 6 separate times for overall data. For aortic rings, an independent experiment consists of 4 Z‐stack photographs combined to form a representative three‐dimensional image using ImageJ. We then used 10 separate Z‐stack three‐dimensional images for quantification (fluorescence) on an individual day per experimental condition. This was repeated 3 separate times for overall data.
Immunoprecipitation
To test mTOR interaction with the IKKβ and p65 subunit of the nuclear factor‐κB (NFκB) transcriptional complex in cultured MLEC, cellular protein samples (800 μg) were subjected to immunoprecipitation assay with 5 μg anti‐mTOR (Cell Signaling, #2972), anti‐IKKβ (Proteintech, #15649), or anti‐p65 (Cell Signaling, #3033) antibodies. Immunocomplex was separated on a 9% SDS‐PAGE gel, transferred to polyvinylidene fluoride membrane, then probed with anti‐IKKβ (1:1000) or anti‐p65 antibody (Cell Signaling, #6956, 1:1000), followed by either a secondary anti‐rabbit horseradish peroxidase goat antibody (Cell Signaling, #7074, 1:10 000) or a goat anti‐mouse horseradish peroxidase (Cell Signaling, #7076, 1:10 000). Protein expression was visualized with ECL detection kit (GE Healthcare) and exposed to film.
Statistical Analysis
All data are expressed as means±SEM, analyzed and graphed with GraphPad Prism 7 software. Sample sizes were determined a priori by power analysis calculation based on pilot studies in our laboratory. Group comparisons were made via Student t test, a 1‐way ANOVA with a Tukey multiple comparison test, or multiple t test analysis with corrections for multiple comparisons using the Holm‐Sidak method. Vascular function assays were analyzed using a 2‐way ANOVA with or without repeated measures and a Tukey (≥3 groups) or Bonferroni (2 groups) post hoc test when appropriate. Significance was accepted with P<0.05.
Results
Leucine‐Induced Activation of mTORC1 Impairs Endothelial‐Mediated Relaxation
To assess the importance of mTORC1 signaling for vascular function, we took advantage of the unique ability of the branched chained amino acid leucine to activate mTORC1 signaling. Indeed, leucine (10 mmol/L) robustly activated mTORC1 signaling in cultured mouse vascular rings as indicated by increases in the phosphorylated ribosomal S6 protein in both aortic and mesenteric arterial rings via immunohistochemistry (Figure 1A) and confirmed by Western blot in aortic rings (Figure 1B). Co‐localization of pS6 and Von Willebrand Factor staining was observed in leucine‐stimulated aortic rings but not control rings (denoted by arrow). As expected, equal concentration of valine (10 mmol/L) did not activate mTORC1 signaling in cultured aortic rings (Figure S1A). To demonstrate that the effect of leucine stimulation is mediated by mTORC1 signaling, we infected the vascular rings with an adenoviral S6‐kinase dominant negative construct (Ad‐S6KDN) to inhibit mTORC1 signaling before stimulation with leucine.6, 7 Blockade of mTORC1 signaling with the Ad‐S6KDN construct prevented leucine‐induced S6 activation similar to control levels (Figure 1B). Efficacy of the Ad‐S6KDN construct to infect aortic rings in culture was evident given the increased expression of total S6 kinase in the transfected aortic rings (Figure 1B).
Figure 1.
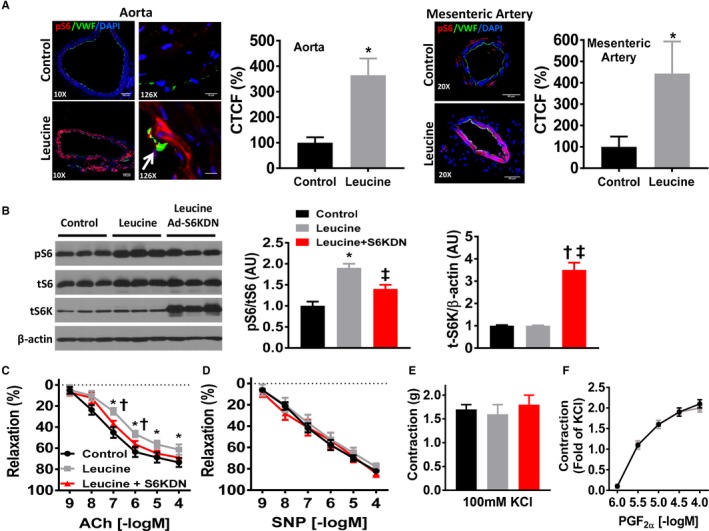
Leucine‐induced activation of mTORC1 impairs endothelial‐mediated relaxation. A, Representative images of aortic and mesenteric arterial rings cultured for 24 hours in leucine‐supplemented (10 mmol/L) media compared with control (0.45 mmol/L) media. Phospho‐S6 (pS6; red) denotes mTORC1 signaling and Von Willebrand Factor (VWF, green) staining denotes the endothelium. White arrow denotes co‐localization of pS6 and Von Willebrand Factor staining. Images taken from 3 to 4 independent experiments and quantification data of aortic and mesenteric arterial rings expressed as percentage corrected total cell fluorescence (CTCF%) are shown. B, Representative Western blot images of aortic rings cultured in control, leucine‐supplemented and leucine‐supplemented+Ad‐S6KDN (2×108 pfu/mL) media for phospho‐S6, total S6, total S6 kinase and β‐actin. Quantification data are expressed as arbitrary units (n=6–9/group). Vascular reactivity responses of cultured aortic rings to (C) endothelial‐dependent acetylcholine, (D) endothelial‐independent sodium nitroprusside (SNP), and contractile responses to (E) 100 mmol/L potassium chloride (KCl) and (F) prostaglandin F2α (PGF2α) (n=10/group). ACh indicates acetylcholine; CTCF %, percentage corrected total cell fluorescence; mTORC1, mechanistic target of rapamycin complex 1; SNP, sodium nitroprusside; *P<0.05 leucine vs control; † P<0.05 leucine vs leucine+S6KDN, ‡P<0.05 control vs leucine+S6KDN.
Next, we sought to determine the consequences of leucine‐induced activation of mTORC1 signaling on vascular endothelial and smooth muscle relaxation responses. Aortic rings stimulated with leucine exhibited impaired endothelial‐dependent relaxation evoked by acetylcholine (Figure 1C; Pinteraction<0.05). Furthermore, aortic rings infected with Ad‐S6KDN restored endothelial function toward control levels although the maximal relaxation responses were not statistically different from leucine‐stimulated aortic rings (P=0.11). Leucine stimulation significantly altered the EC50 response to acetylcholine (P<0.05) but did not alter the maximal response (Emax, Table S2). These data indicate that the leucine‐induced impairment in endothelial‐dependent vasorelaxation responses is mTORC1 signaling‐dependent. No changes were found in endothelial‐independent vasorelaxation responses to SNP in the aortic ring culture models (Figure 1D) suggesting that mTORC1 signaling activation evoked by leucine affects endothelial but not smooth muscle vasorelaxation responses. Similarly, no differences were found in the contractile responses evoked by 100 mmol/L KCl (Figure 1E) or PGF2α (Figure 1F). Stimulation of aortic rings with valine (10 mmol/L) did not alter maximal relaxation responses to acetylcholine (Figure S1B), SNP (Figure S1C), or maximal contraction to PGF2α (Figure S1D).
Leucine‐Induced Activation of mTORC1 Promote Pro‐Oxidant Environment in Endothelial Cells
To elucidate potential signaling pathways underlying the impaired endothelial‐mediated relaxation responses evoked by leucine, we used MLEC.8 MLECs were exposed, overnight (16 hours) under serum‐free conditions, to leucine (10 mmol/L) which robustly activated mTORC1 signaling as indicated by the increased phosphorylated S6 protein compared with control (Figure 2A). Because nitric oxide (NO) is the predominant vasodilator in aortic rings, we investigated the effect of leucine on eNOS expression in MLECs. However, phospho‐eNOS (Ser1177) and total eNOS expression were not altered by leucine stimulation in MLECs (Figure 2A). We next sought to determine expression of markers of oxidative stress and inflammation as oxidants and cytokines are integral regulators of vascular endothelial function. Leucine stimulation did not alter the protein expression of superoxide dismutase 2 (SOD2) in MLECs (Figure 2A) but decreased SOD2 mRNA expression (Figure 2B). No changes were noted for the mRNA levels of superoxide dismutase 1 (SOD1) or catalase. Notably, leucine stimulation increased mRNA expression of NADPH oxidase 2 (NOX2) without altering mRNA expression of NOX1 or NOX4 (Figure 2C). Additionally, no changes in mRNA levels were found for the proinflammatory interleukin‐1β (IL‐1β) or the anti‐inflammatory interleukin‐10 (IL‐10) (Figure 2D). Taken together, these data suggest that leucine‐induced activation of mTORC1 signaling results in a pro‐oxidant gene environment in cultured endothelial cells.
Figure 2.
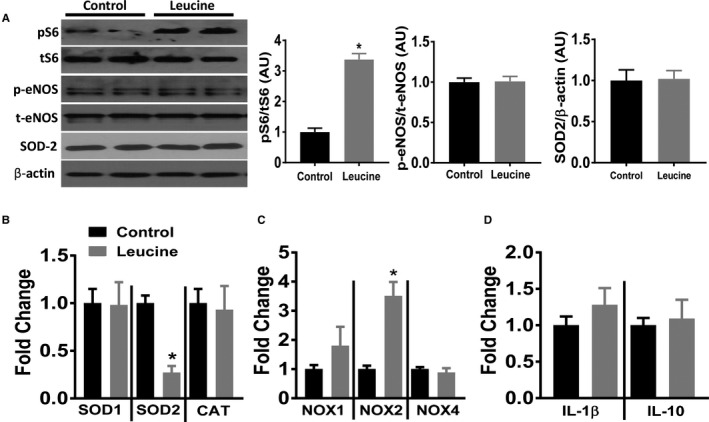
Leucine‐induced activation of mTORC1 promote pro‐oxidant environment in endothelial cells. Mouse lung endothelial cells were stimulated with 10 mmol/L leucine for 16 hours under serum‐free conditions. A, Representative Western blot images and quantification data (expressed as arbitrary units) of mouse lung endothelial cells for indicated proteins (n=6 independent experiments). mRNA levels of indicated (B) Reactive oxygen species degrading enzymes, (C) NADPH oxidases, and (D) inflammatory markers (n=6 independent experiments). CAT indicates catalase; IL, interleukin; mTORC1, mechanistic target of rapamycin complex 1; NOX, NADPH oxidase; SOD, superoxide dismutase. *P<0.05 leucine vs control.
Genetic Activation of mTORC1 Signaling Increases ROS in Endothelial Cells
To further demonstrate that increased mTORC1 signaling promotes a pro‐oxidant gene environment, we used an adenoviral vector expressing a constitutively active S6‐kinase (Ad‐S6KCA).6, 7 Infection of MLECs with Ad‐S6KCA for 48 hours increased expression of pS6 compared with Ad‐GFP infected control cells and this increased mTORC1 signaling resulted in increased ROS generation via dihydroethidium staining (Figure 3A). Similar to leucine stimulated MLECs, activation of mTORC1 signaling with Ad‐S6KCA did not result in changes in phospho‐ or total eNOS expression (Figure 3B). Additionally, no changes in SOD‐2 protein expression were detected (Figure 3B). However, similar to leucine stimulation in MLECs, Ad‐S6KCA infection reduced mRNA levels of SOD2 without changing SOD1 or catalase expression (Figure 3C). Ad‐S6KCA infection robustly increased mRNA levels for NOX1 and NOX2 expression without altering NOX4 levels (Figure 3D). No differences were found in IL‐1β or IL‐10 mRNA levels in MLECs infected with the Ad‐S6KCA (Figure 3E).
Figure 3.
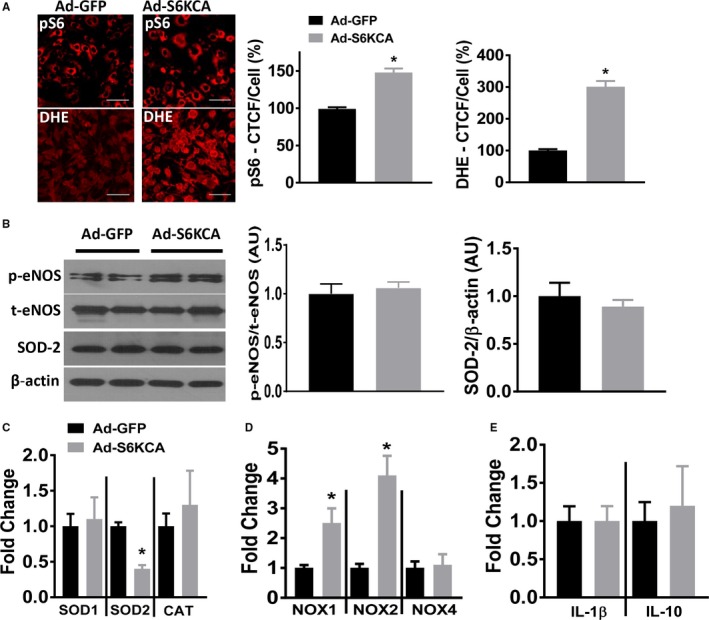
Genetic activation of mTORC1 signaling increases reactive oxygen species in endothelial cells. A, Mouse lung endothelial cells were infected for 48 hours with Ad‐GFP or Ad‐S6KCA and subjected to phospho‐S6 (red) immunocytochemistry (top; n=5 independent experiments) or dihydroethidium reactive oxygen species (red) fluorescence (bottom; n=6 independent experiments). Quantification data expressed as percentage corrected total cell fluorescence. B, Representative Western blot images and quantification data (expressed as arbitrary units) of adenoviral infected mouse lung endothelial cells for indicated proteins (n=6 independent experiments). mRNA levels of indicated (C) Reactive oxygen species degrading enzymes, (D) NADPH oxidases, and (E) inflammatory markers (n=6 independent experiments). Ad‐S6KCA indicates adenoviral S6‐kinase constitutively active; CAT, catalase; DHE, dihydroethidium; GFP, green fluorescent protein; IL, interleukin; mTORC1, mechanistic target of rapamycin complex 1; NOX, NADPH oxidase; SOD, superoxide dismutalse. *P<0.05 vs Ad‐GFP.
Impairment of Endothelial‐Mediated Relaxation Elicited by mTORC1 Activation Involves Oxidative Stress
Next, we examined the relevance of the molecular changes we observed in MLECs to the impaired endothelial‐mediated relaxation evoked by activation of mTORC1 signaling using Ad‐S6KCA. Efficacy of the infection of Ad‐S6KCA is demonstrated by the increased expression of total S6 kinase compared with Ad‐GFP‐infected aortic rings (Figure 4A). Moreover, infection of mouse aortic rings with Ad‐S6KCA resulted in a significant induction of phospho‐S6 compared with control Ad‐GFP‐infected aortic rings (Figure 4A). Because mTORC1 activation evoked by leucine resulted in increased NOX2 mRNA levels in mouse endothelial cells, we measured gp91phox, the catalytic subunit of NOX2, in Ad‐S6KCA–infected aortic rings. We found increased gp91phox expression compared with Ad‐GFP–infected control (P<0.05; Figure 4A). Oxidative stress has also been demonstrated to regulate the bioavailability of NO. Thus, we investigated the effect of Ad‐S6KCA infection on aortic eNOS expression. Surprisingly, aortic rings infected with Ad‐S6KCA demonstrated increased expression of total eNOS (Figure 4A). However, the ratio of phosphorylated eNOS (Ser1177) to total eNOS expression was unaltered.
Figure 4.
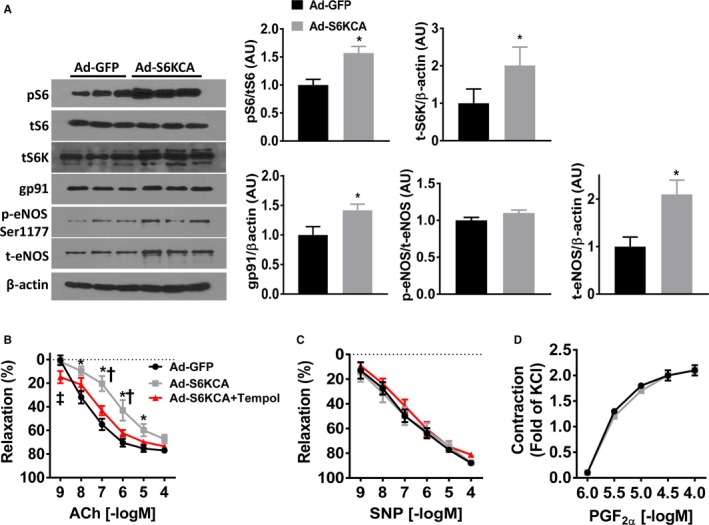
Impairment of endothelial‐mediated relaxation elicited by mTORC1 activation involves oxidative stress. A, Representative Western blot images and quantification data (expressed as arbitrary units) of aortic rings infected (24 hours) with Ad‐GFP or Ad‐S6KCA for indicated proteins (n=6–9/group). Vascular reactivity responses of adenoviral infected aortic rings to (B) endothelial‐dependent acetylcholine, (C) endothelial‐independent sodium nitroprusside, and contractile responses to (D) prostaglandin F2α (n=8/group). A subset of aortic rings was pre‐incubated with Tempol (100 μmol/L; 30 minutes) before cumulative response curves. ACh indicates acetylcholine; Ad‐S6KCA, adenoviral S6‐kinase constitutively active; GFP, green fluorescent protein; mTORC1, mechanistic target of rapamycin complex 1; PGF2α, prostaglandin F2α; SNP, sodium nitroprusside. *P<0.05 vs Ad‐GFP; †P<0.05 vs Ad‐S6KCA+Tempol; ‡P<0.05 Ad‐GFP vs Ad‐S6KCA+Tempol.
We then assessed the relaxation responses of Ad‐S6KCA–infected aortic rings and found significant impairment in endothelial‐mediated relaxation compared with Ad‐GFP–infected control rings (P interaction<0.001), although the maximal relaxation responses were not different (P=0.14; Figure 4B). Similar to leucine‐stimulation, Ad‐S6KCA infection shifted the EC50 compared with Ad‐GFP control without altering the Emax (Table S2). The increased expression of NOX2 in Ad‐S6KCA–infected endothelial cells and aortic rings led us to test the effect of preincubating the infected aortic rings with the free radical scavenger Tempol. Tempol treatment restored endothelial‐mediated relaxation responses back toward control levels (Figure 4B). These data highlight the importance of ROS signaling in mediating mTORC1 activation‐induced endothelial dysfunction. Furthermore, we assessed the effect of NOS blockade with L‐NAME on acetylcholine‐induced vasorelaxation responses in a separate cohort of mice (Figure 5A). L‐NAME significantly inhibited vasorelaxation response to acetylcholine (Figure 5A) in aortic rings infected with Ad‐GFP and Ad‐S6KCA. However, the inhibition of acetylcholine‐induced vasorelaxation was similar between Ad‐GFP and Ad‐S6KCA infected aortic rings demonstrating no compensatory NO‐independent vasorelaxation responses in Ad‐S6KCA–infected aortic rings with increased mTORC1 signaling. No differences were noted in SNP relaxation (Figures 4C and 5B) or PGF2α contractile (Figure 4D) responses further indicating that mTORC1 signaling affects endothelial as opposed to smooth muscle vasorelaxation responses.
Figure 5.
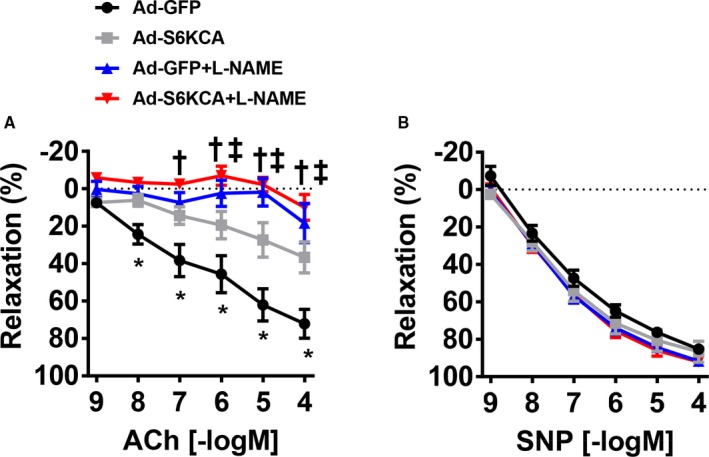
L‐NAME incubation reduces endothelial function in Ad‐S6KCA infected aortic rings. Vascular reactivity responses of adenoviral infected aortic rings with or without L‐NAME preincubation for (A) endothelial‐dependent acetylcholine and (B) endothelial‐independent sodium nitroprusside. ACh indicates acetylcholine; Ad‐S6KCA, adenoviral S6‐kinase constitutively active; GFP, green fluorescent protein; SNP, sodium nitroprusside. N=7/group *P<0.05 Ad‐GFP vs Ad‐S6KCA, † P<0.05 Ad‐GFP vs Ad‐GFP+LNAME, ‡Ad‐S6KCA vs Ad‐S6KCA+LNAME.
mTOR Physically Interacts With Subunits of the NFκB Transcription Complex
Next, we sought to determine a potential transcriptional mediator that may connect increased mTORC1 signaling with the generation of oxidative stress that affects endothelial‐mediated vasorelaxation. The NFκB transcriptional complex is a master regulator of oxidant and inflammatory signaling and can contribute to oxidative stress and inflammation in the endothelium.9 To test if NFκB signaling contributes to ROS generation in response to increased mTORC1 signaling, we subjected MLECs to immunoprecipitation using antibodies against mTOR and the IKKβ and p65 subunits of the NFκB transcriptional complex. We were able to pull down the mTOR catalytic subunit with either the IKKβ subunit (Figure 6A) or the p65 subunit (Figure S2A) indicating the existence of a physical interaction between mTORC1 and the NFκB transcriptional complex. To determine if this physical interaction between mTORC1 and NFκB is involved in the ROS generation in response to increased mTORC1 signaling, Ad‐S6KCA–infected MLECs were treated with BMS‐345541 (IKKβ inhibitor) and subjected to dihydroethidium fluorescence staining. Blockade of IKKβ with BMS‐345541 (300 nmol/L) prevented the increased ROS signaling in response to Ad‐S6KCA infection (Figure 6B) highlighting the significance of the interaction between mTORC1 and NFκB for ROS signaling in endothelial cells. We then assessed the effect of blockade of IKKβ signaling on endothelial function. However, endothelial dysfunction evoked by mTORC1 signaling activation with Ad‐S6KCA infection was unaffected by BMS‐345541 incubation (Figure 6C) and was not associated with changes in endothelial‐independent SNP relaxation (Figure S2B). We confirmed that ROS production is elevated in Ad‐S6KCA–infected aortic rings (Figure 6D). BMS‐344541 (300 nmol/L) tended to attenuate the increase in dihydroethidium fluorescence in Ad‐S6KCA‐infected aortic rings, but this was not statistically significant (P=0.31). Of note, in a pilot study we found that increasing BMS‐345541 concentrations (up to 10 μmol/L) failed to improve endothelial function to acetylcholine in Ad‐S6KCA–infected aortic rings (data not shown). Together, these data suggest that activation of mTORC1 signaling in aortic rings results in increased vascular ROS production and that blockade of NFκB signaling arising from this complex is not sufficient to reduce ROS signaling and improve endothelial function in Ad‐S6KCA–infected aortic rings.
Figure 6.
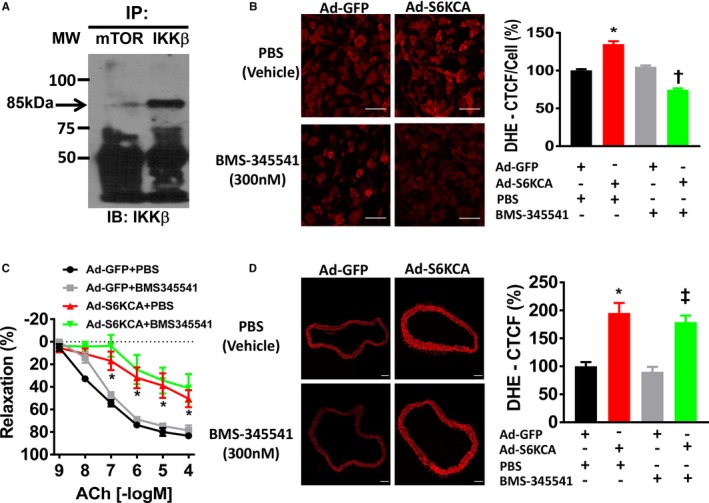
mTOR physically interacts with subunits of the nuclear factor‐κB transcription complex. A, Representative Western blot image of MLECs immunoprecipitated with antibodies for mTOR and IKKβ subunit of the nuclear factor‐κB transcriptional complex. B, Representative images of MLECs infected with Ad‐GFP or Ad‐S6KCA for 48 hours and treated for the final 16 hours with PBS (vehicle) or BMS‐345541 (300 nmol/L) for dihydroethidium fluorescence. Quantification data are expressed as percentage corrected total cell fluorescence. Data shown are from 6 independent experiments. C, Vascular reactivity responses of adenoviral infected aortic rings treated with BMS‐345541 (300 nmol/L) or vehicle (PBS) for 16 hours to endothelial‐dependent acetylcholine (n=5/group). D, Representative images of aortic rings infected with Ad‐GFP or adenoviral S6‐kinase constitutively active for 24 hours and treated for the final 16 hours with PBS (vehicle) or BMS‐345541 (300 nmol/L) for dihydroethidium. Quantification data expressed as percentage corrected total cell fluorescence. Data shown are from 3 independent experiments. ACh indicates acetylcholine; Ad‐S6KCA, adenoviral S6‐kinase constitutively active; CTCF %, percentage corrected total cell fluorescence; DHE, dihydroethidium; GFP, green fluorescent protein; IKK, inhibitor of nuclear factor κ‐β kinase; mTOR mechanistic target of rapamycin. *P<0.05 vs Ad‐GFP; † P<0.05 vs Ad‐S6KCA; ‡ P<0.05 vs Ad‐GFP+BMS‐345541.
Discussion
In this study, we report that vascular mTORC1 signaling is an important regulatory pathway contributing to the maintenance of vascular endothelial function and homeostasis. Activation of vascular mTORC1 signaling, either upstream (via the branched‐chain amino‐acid leucine) or downstream (via constitutively active S6 kinase infection), results in impaired endothelial‐mediated vasorelaxation responses. Mechanistically, we demonstrate that elevated mTORC1 signaling promotes a pro‐oxidant gene program with enhanced ROS generation in mouse endothelial cells and vascular rings leading to vascular endothelial dysfunction. Our study also shows that blockade of the IKKβ subunit activity of the NFκB transcriptional complex can prevent the ROS generation in response to mTORC1 signaling in endothelial cells. However, this does not improve endothelial function where ROS production is elevated highlighting a complex and integrated physiological connection between mTORC1, NFκB, and oxidative stress signaling in mediating vascular endothelial homeostasis and function. Taken together, these results implicate that vascular mTORC1 signaling is a critical regulator of vascular endothelial function and provide a potential targeting point for a therapeutic approach to mitigate vascular endothelial dysfunction, a major cardiovascular risk.
Recently, the mTORC1 signaling pathway has been implicated in the regulation of the cardiovascular system and dysregulation of this pathway may be involved in pathophysiological complications associated with altered energy balance, metabolic disorders, and cardiovascular disease.10 Indeed, our laboratory has previously demonstrated that leucine‐induced activation of the hypothalamic mTORC1 signaling pathway affects sympathetic nerve activity and arterial blood pressure.6 In the context of the vascular system, angiotensin II has been shown to increase mTORC1 activity in endothelial and smooth muscle cells.3, 11 This increased mTORC1 signaling in angiotensin II‐mediated endothelial dysfunction can be ameliorated by treatment with the rapalog rapamycin (inhibitor of mTORC1)3 highlighting the importance of mTORC1 signaling for vascular endothelial dysfunction evoked by angiotensin II. In other models where mTORC1 activity is chronically activated such as vascular aging, rapamycin‐induced inhibition of mTORC1 signaling can improve endothelial dependent vasorelaxation through inhibition of superoxide production and enhanced NO bioavailability.12 On the other hand, infusion of wild‐type mice with rapamycin increases systolic blood pressure through a mechanism involving vascular endothelial dysfunction and altered eNOS phosphorylation13 highlighting the need for balanced mTORC1 activity to maintain vascular endothelial function. Indeed, long‐term immunosuppression via rapamycin in kidney transplant patients has been demonstrated to reduce insulin sensitivity and increase peripheral insulin resistance14 which can directly affect vascular endothelial function. Similarly, in a study on the chronic effects of rapamycin in HEK293 or PC12 cells, it was shown that rapamycin causes partial‐complete loss of Rictor function of the mTORC2 complex in addition to blockade of mTORC1.15 These data suggest that prolonged use of rapamycin affects both mTORC1 and mTORC2 and may contribute to hypertension in organ transplant patients on rapamycin.16
Our current study demonstrates that upstream or downstream activation of mTORC1 signaling results in impairment of endothelial‐mediated relaxation responses in aortic rings to acetylcholine while leaving endothelial‐independent relaxation responses to sodium nitroprusside unaffected. These findings of mTORC1 signaling activation and vascular dysfunction are supported by both human and animal studies. In humans with associated endothelial dysfunction such as hypertension17, 18, 19 and type 2 diabetes mellitus,20 circulating levels of leucine are elevated and strongly correlate with the occurrence of cardiovascular events. In animal models of endothelial dysfunction such as diet‐induced obesity, circulating leucine levels are increased21 and mTORC1 signaling is elevated in the aorta22 and perivascular adipose tissue.23 Collectively, our data implicate and support that mTORC1 signaling is a direct and crucial mediator of vascular endothelial function and that dysregulation of this pathway may contribute to endothelial dysfunction where mTORC1 signaling is elevated.
To investigate potential mechanisms involved in mediating mTORC1 signaling on vascular endothelial function, we utilized both cultured vascular rings and a mouse endothelial cell line to study signaling pathways activated in response to elevated mTORC1 activity. It is well established that vascular endothelial function is regulated by the activity and bioavailability of NO with oxidative stress, via increased production of ROS, and proinflammatory cytokines as mechanisms by which NO and endothelial function can be impaired.1 Specifically in the vasculature, NOX2, a key reactive oxygen species producing enzyme, has been implicated as a critical regulator of oxidative stress and endothelial function in the context of various cardiovascular diseases.24, 25 In this study, we found that activation of mTORC1 signaling in vascular rings resulted in increased gp91phox expression, the catalytic subunit of NOX2, and implicates oxidative stress in the impaired endothelial function in response to elevated mTORC1 activity. This finding is further supported by our studies in cultured mouse endothelial cells where activation of mTORC1 signaling elevated mRNA levels of NOX2 and ROS generation. Furthermore, incubation of Ad‐S6KCA infected aortic rings with Tempol, a free ROS scavenger, ameliorated the impaired endothelial‐mediated relaxation responses evoked by mTORC1 activation. Taken together, these data suggest that mTORC1 signaling can generate a pro‐oxidant gene program in both endothelial cells and vascular rings and this contributes to the vascular endothelial dysfunction induced by mTORC1 signaling activation.
In addition to oxidative stress, proinflammatory cytokines can influence vascular endothelial function.26 A link between the mTORC1 pathway and inflammatory signaling has been demonstrated where gene silencing of S6‐kinase signaling attenuates tumor necrosis factor‐α–induced expression of E‐selectin, I‐CAM1, and V‐CAM1 in endothelial cells.27 In a model of diabetic peripheral artery disease, endothelial‐specific knockout of mTORC1 signaling reduces interleukin 1β (IL‐1β) expression.4 In our current study, expression of IL‐1β and the anti‐inflammatory interleukin 10 (IL‐10) was unaltered in endothelial cells with elevated mTORC1 signaling suggesting that elevated mTORC1 signaling is not sufficient to alter the expression of these particular cytokines.
As noted above, ROS signaling can impair endothelial‐mediated vasorelaxation responses through oxidative degradation of NO and eNOS uncoupling. A link between uncoupling of eNOS and elevated mTORC1 has been suggested to connect endothelial dysfunction with vascular aging.28 This is based on the observation that inhibition of mTORC1 signaling with rapamycin resulted in improved eNOS coupling5 and mTORC1 signaling has been recently demonstrated to regulate the phosphorylation of eNOS at its activating Ser1177 site in endothelial cells.29 In this study, we found no change in the expression of eNOS protein in response to mTORC1 signaling activation in endothelial cells. However, Ad‐S6KCA infection of aortic rings showed a paradoxical increase in eNOS expression (phosphorylated and total) in response to increased mTORC1 activity. The reason for this increase in eNOS expression is unclear but may represent a compensatory mechanism to counteract the pro‐oxidant gene environment in response to increased mTORC1 signaling. ROS induced uncoupling of eNOS has been demonstrated in a variety of cardiovascular diseases.30, 31 Indeed, in the context of aging where vascular mTORC1 signaling is elevated, eNOS protein and activity levels are up‐regulated while NO bioavailability is reduced and endothelial function is impaired.32 Additionally, dietary treatment of aged mice with rapamycin reduced ROS signaling and improved endothelial function12 suggesting improved eNOS homeostasis. Furthermore, a recent study demonstrated increased eNOS expression in cultured endothelial cells stimulated with branched‐chained amino acids33 further suggesting a potential compensatory effect in vascular cells and tissues. We did not measure NO or peroxynitrite levels or investigate eNOS uncoupling in this study. Therefore, additional studies are warranted to elucidate the connection between mTORC1 signaling and eNOS/NO activity in vascular rings.
As a link between mTORC1 signaling and ROS generation in endothelial cells, we investigated the NFκB transcriptional complex, a group of transcription factors involved in redox sensitive transcriptional programming which can regulate a variety of downstream targets that determine endothelial function.34, 35, 36 In endothelial cells, NFκB activation results in increased ROS production and NO dysfunction.37, 38 In this study we demonstrate a physical interaction between the mTOR catalytic subunit and both the IKKβ and p65 subunits of the NFκB complex in endothelial cells. Furthermore, blockade of IKKβ subunit activity in endothelial cells abolishes the ROS generation in response to elevated mTORC1 signaling demonstrating that mTORC1 signaling through its interaction with NFκB can regulate ROS in endothelial cells. However, blockade of IKKβ signaling in aortic rings with activation of mTORC1 signaling ex vivo did not improve endothelial function to acetylcholine where ROS signaling is elevated. These data suggest that the ROS derived from NFκB alone is not sufficient to improve vascular endothelial function and contribution of other ROS sources (such as NADPH and xanthine oxidases, cyclooxygenases, uncoupled eNOS, or mitochondrial respiration complexes) will need to be further investigated in mediating the endothelial dysfunction associated with mTORC1 signaling activation.
We recognize several limitations to our current study. First, leucine‐induced increase in mTORC1 signaling yielded a robust activation in the smooth muscle layer in addition to the endothelium of aortic rings via immunohistochemistry. Interestingly, we found no effect of increased mTORC1 signaling on smooth muscle relaxation responses to SNP or both receptor‐dependent (PGF2α) and ‐independent (KCl‐induced depolarization) activation of the smooth muscle contractile apparatus. These data suggest that the smooth muscle in response to elevated mTORC1 signaling is able to trigger a similar increase in calcium. However, a recent study suggests altered adrenergic‐mediated contractility of vascular rings in response to a high concentration of a branched‐chained amino acid cocktail.33 Therefore, additional studies are warranted to more thoroughly assess different contractile agents and signaling pathways in response to mTORC1 signaling. Second, we used an immortalized lung endothelial cell line as opposed to endothelial cells derived from the aorta which was used to assess vascular functions. Further studies are warranted in aortic endothelial cells. Finally, despite its high sensitivity, dihydroethidium staining as used in our current study is not the ideal method to assess ROS production.39 However, we used complimentary molecular and pharmacological approaches to refine the role of ROS in the effects evoked by mTORC1 activation. Despite this, more specific methods such as cyclic hydroxylamines or cytoplasmic redox probes39 will be needed to confirm our findings.
Conclusion
The current study identifies a critical role for mTORC1 signaling in the regulation of vascular endothelial function through ROS signaling. Studies in both endothelial cells and vascular rings demonstrate that elevated mTORC1 activity increases pro‐oxidant gene expression and ROS generation and this contributes to reduced endothelial‐mediated vasorelaxation. Furthermore, blockade of mTORC1 signaling or ROS production protects endothelial‐mediated vasorelaxation responses in settings where mTORC1 signaling is altered. Inhibition of the NFκB complex at the level of the IKKβ subunit prevents mTORC1‐induced ROS generation in endothelial cells improving endothelial cell function but not improving endothelial function measured ex vivo suggesting other ROS sources playing a role in the vascular endothelial function. These findings highlight the importance of mTORC1 signaling in the regulation of vascular function and provide a potential target for therapeutic approaches aimed at preventing the endothelial dysfunction associated with diseases of altered energy balance such as obesity and related conditions. Future studies are warranted to examine the role of altered endothelial mTORC1 signaling using genetic and pharmacological approaches to demonstrate its pathophysiological relevance for vascular endothelial dysfunction in various disease states such as obesity.
Sources of Funding
This work was supported by National Institutes of Health grant HL084207, American Heart Association grants 14EIA18860041 and 16POST30830004, the University of Iowa Fraternal Order of Eagles Diabetes Research Center, and the University of Iowa Center for Hypertension Research.
Disclosures
None.
Supporting information
Table S1. qPCR Primer Sequences
Table S2. Aortic Ring EC50 and Emax Values
Figure S1. Valine does not activate mTORC1 signaling in aortic rings ex vivo.
Figure S2. mTOR physically interacts with the p65 subunit of the NFκB transcriptional complex.
Acknowledgments
The authors thank Dr Justin Grobe (University of Iowa) for statistical assistance and Andrew Olson (University of Iowa) for technical assistance.
(J Am Heart Assoc. 2019;8:e010662 DOI: 10.1161/JAHA.118.010662.)
This article was handled independently by Hossein Ardehali, MD, PhD as a guest editor. The editors had no role in the evaluation of the manuscript or in the decision about its acceptance.
References
- 1. Reho JJ, Rahmouni K. Oxidative and inflammatory signals in obesity‐associated vascular abnormalities. Clin Sci (Lond). 2017;131:1689–1700. [DOI] [PubMed] [Google Scholar]
- 2. Davignon J, Ganz P. Role of endothelial dysfunction in atherosclerosis. Circulation. 2004;109:III27–III32. [DOI] [PubMed] [Google Scholar]
- 3. Kim JA, Jang HJ, Martinez‐Lemus LA, Sowers JR. Activation of mTOR/p70S6 kinase by ANG II inhibits insulin‐stimulated endothelial nitric oxide synthase and vasodilation. Am J Physiol Endocrinol Metab. 2012;302:E201–E208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fan W, Han D, Sun Z, Ma S, Gao L, Chen J, Li X, Li X, Fan M, Li C, Hu D, Wang Y, Cao F. Endothelial deletion of mTORC1 protects against hindlimb ischemia in diabetic mice via activation of autophagy, attenuation of oxidative stress and alleviation of inflammation. Free Radic Biol Med. 2017;108:725–740. [DOI] [PubMed] [Google Scholar]
- 5. Rajapakse AG, Yepuri G, Carvas JM, Stein S, Matter CM, Scerri I, Ruffieux J, Montani JP, Ming XF, Yang Z. Hyperactive S6K1 mediates oxidative stress and endothelial dysfunction in aging: inhibition by resveratrol. PloS One. 2011;6:e19237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Harlan SM, Guo DF, Morgan DA, Fernandes‐Santos C, Rahmouni K. Hypothalamic mTORC1 signaling controls sympathetic nerve activity and arterial pressure and mediates leptin effects. Cell Metab. 2013;17:599–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ono H, Pocai A, Wang Y, Sakoda H, Asano T, Backer JM, Schwartz GJ, Rossetti L. Activation of hypothalamic S6 kinase mediates diet‐induced hepatic insulin resistance in rats. J Clin Invest. 2008;118:2959–2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ackah E, Yu J, Zoellner S, Iwakiri Y, Skurk C, Shibata R, Ouchi N, Easton RM, Galasso G, Birnbaum MJ, Walsh K, Sessa WC. Akt1/protein kinase Balpha is critical for ischemic and VEGF‐mediated angiogenesis. J Clin Invest. 2005;115:2119–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Quilley J. Oxidative stress and inflammation in the endothelial dysfunction of obesity: a role for nuclear factor kappa B? J Hypertens. 2010;28:2010–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sciarretta S, Forte M, Frati G, Sadoshima J. New insights into the role of mTOR signaling in the cardiovascular system. Circ Res. 2018;122:489–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hafizi S, Wang X, Chester AH, Yacoub MH, Proud CG. ANG II activates effectors of mTOR via PI3‐K signaling in human coronary smooth muscle cells. Am J Physiol Heart Circ Physiol. 2004;287:H1232–H1238. [DOI] [PubMed] [Google Scholar]
- 12. Lesniewski LA, Seals DR, Walker AE, Henson GD, Blimline MW, Trott DW, Bosshardt GC, LaRocca TJ, Lawson BR, Zigler MC, Donato AJ. Dietary rapamycin supplementation reverses age‐related vascular dysfunction and oxidative stress, while modulating nutrient‐sensing, cell cycle, and senescence pathways. Aging Cell. 2017;16:17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Long C, Cook LG, Hamilton SL, Wu GY, Mitchell BM. FK506 binding protein 12/12.6 depletion increases endothelial nitric oxide synthase threonine 495 phosphorylation and blood pressure. Hypertension. 2007;49:569–576. [DOI] [PubMed] [Google Scholar]
- 14. Teutonico A, Schena PF, Di Paolo S. Glucose metabolism in renal transplant recipients: effect of calcineurin inhibitor withdrawal and conversion to sirolimus. J Am Soc Nephrol. 2005;16:3128–3135. [DOI] [PubMed] [Google Scholar]
- 15. Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–168. [DOI] [PubMed] [Google Scholar]
- 16. Lindenfeld J, Miller GG, Shakar SF, Zolty R, Lowes BD, Wolfel EE, Mestroni L, Page RL II, Kobashigawa J. Drug therapy in the heart transplant recipient: part II: immunosuppressive drugs. Circulation. 2004;110:3858–3865. [DOI] [PubMed] [Google Scholar]
- 17. Newgard CB, An J, Bain JR, Muehlbauer MJ, Stevens RD, Lien LF, Haqq AM, Shah SH, Arlotto M, Slentz CA, Rochon J, Gallup D, Ilkayeva O, Wenner BR, Yancy WS Jr, Eisenson H, Musante G, Surwit RS, Millington DS, Butler MD, Svetkey LP. A branched‐chain amino acid‐related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009;9:311–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shah SH, Bain JR, Muehlbauer MJ, Stevens RD, Crosslin DR, Haynes C, Dungan J, Newby LK, Hauser ER, Ginsburg GS, Newgard CB, Kraus WE. Association of a peripheral blood metabolic profile with coronary artery disease and risk of subsequent cardiovascular events. Circ Cardiovasc Genet. 2010;3:207–214. [DOI] [PubMed] [Google Scholar]
- 19. Shah SH, Sun JL, Stevens RD, Bain JR, Muehlbauer MJ, Pieper KS, Haynes C, Hauser ER, Kraus WE, Granger CB, Newgard CB, Califf RM, Newby LK. Baseline metabolomic profiles predict cardiovascular events in patients at risk for coronary artery disease. Am Heart J. 2012;163:844–850.e841. [DOI] [PubMed] [Google Scholar]
- 20. Tobias DK, Lawler PR, Harada PH, Demler OV, Ridker PM, Manson JE, Cheng S, Mora S. Circulating branched‐chain amino acids and incident cardiovascular disease in a prospective cohort of US women. Circ Genom Precis Med. 2018;11:e002157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang Y, Wu Z, Meininger CJ, Wu G. L‐leucine and NO‐mediated cardiovascular function. Amino Acids. 2015;47:435–447. [DOI] [PubMed] [Google Scholar]
- 22. Wang CY, Kim HH, Hiroi Y, Sawada N, Salomone S, Benjamin LE, Walsh K, Moskowitz MA, Liao JK. Obesity increases vascular senescence and susceptibility to ischemic injury through chronic activation of Akt and mTOR. Sci Signal. 2009;2:ra11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ma L, Ma S, He H, Yang D, Chen X, Luo Z, Liu D, Zhu Z. Perivascular fat‐mediated vascular dysfunction and remodeling through the AMPK/mTOR pathway in high‐fat diet‐induced obese rats. Hypertens Res. 2010;33:446–453. [DOI] [PubMed] [Google Scholar]
- 24. Paravicini TM, Touyz RM. NADPH oxidases, reactive oxygen species, and hypertension: clinical implications and therapeutic possibilities. Diabetes Care. 2008;31(suppl 2):S170–S180. [DOI] [PubMed] [Google Scholar]
- 25. Lynch CM, Kinzenbaw DA, Chen X, Zhan S, Mezzetti E, Filosa J, Ergul A, Faulkner JL, Faraci FM, Didion SP. Nox2‐derived superoxide contributes to cerebral vascular dysfunction in diet‐induced obesity. Stroke. 2013;44:3195–3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK. Reactive oxygen species in metabolic and inflammatory signaling. Circ Res. 2018;122:877–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ming XF, Rajapakse AG, Carvas JM, Ruffieux J, Yang Z. Inhibition of S6K1 accounts partially for the anti‐inflammatory effects of the arginase inhibitor L‐norvaline. BMC Cardiovasc Disord. 2009;9:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Donato AJ, Walker AE, Magerko KA, Bramwell RC, Black AD, Henson GD, Lawson BR, Lesniewski LA, Seals DR. Life‐long caloric restriction reduces oxidative stress and preserves nitric oxide bioavailability and function in arteries of old mice. Aging Cell. 2013;12:772–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Decker B, Pumiglia K. mTORc1 activity is necessary and sufficient for phosphorylation of eNOS(S1177). Physiol Rep. 2018;6:e13733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li Q, Youn JY, Cai H. Mechanisms and consequences of endothelial nitric oxide synthase dysfunction in hypertension. J Hypertens. 2015;33:1128–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Donato AJ, Machin DR, Lesniewski LA. Mechanisms of dysfunction in the aging vasculature and role in age‐related disease. Circ Res. 2018;123:825–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. van der Loo B, Labugger R, Skepper JN, Bachschmid M, Kilo J, Powell JM, Palacios‐Callender M, Erusalimsky JD, Quaschning T, Malinski T, Gygi D, Ullrich V, Luscher TF. Enhanced peroxynitrite formation is associated with vascular aging. J Exp Med. 2000;192:1731–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhenyukh O, Gonzalez‐Amor M, Rodrigues‐Diez RR, Esteban V, Ruiz‐Ortega M, Salaices M, Mas S, Briones AM, Egido J. Branched‐chain amino acids promote endothelial dysfunction through increased reactive oxygen species generation and inflammation. J Cell Mol Med. 2018;22:4948–4962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Perkins ND. Integrating cell‐signalling pathways with NF‐kappaB and IKK function. Nat Rev Mol Cell Biol. 2007;8:49–62. [DOI] [PubMed] [Google Scholar]
- 35. Read MA, Whitley MZ, Williams AJ, Collins T. NF‐kappa B and I kappa B alpha: an inducible regulatory system in endothelial activation. J Exp Med. 1994;179:503–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hajra L, Evans AI, Chen M, Hyduk SJ, Collins T, Cybulsky MI. The NF‐kappa B signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation. Proc Natl Acad Sci USA. 2000;97:9052–9057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Csiszar A, Wang M, Lakatta EG, Ungvari Z. Inflammation and endothelial dysfunction during aging: role of NF‐kappaB. J Appl Physiol (1985). 2008;105:1333–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Donato AJ, Eskurza I, Silver AE, Levy AS, Pierce GL, Gates PE, Seals DR. Direct evidence of endothelial oxidative stress with aging in humans: relation to impaired endothelium‐dependent dilation and upregulation of nuclear factor‐kappaB. Circ Res. 2007;100:1659–1666. [DOI] [PubMed] [Google Scholar]
- 39. Dikalov SI, Harrison DG. Methods for detection of mitochondrial and cellular reactive oxygen species. Antioxid Redox Signal. 2014;20:372–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. qPCR Primer Sequences
Table S2. Aortic Ring EC50 and Emax Values
Figure S1. Valine does not activate mTORC1 signaling in aortic rings ex vivo.
Figure S2. mTOR physically interacts with the p65 subunit of the NFκB transcriptional complex.