

Abstract
Background
Previous studies demonstrated that angiotensin (Ang) III, not Ang II, is the predominant endogenous agonist for Ang type‐2 receptor (AT 2R)‐induced natriuresis in normal rats, and that hypertensive 12‐week‐old spontaneously hypertensive rats (SHR) lack natriuretic responses to Ang III. This study tested whether prehypertensive SHR already have defective Ang III‐induced natriuresis and determined possible mechanisms.
Methods and Results
Female and male normotensive 4‐week‐old SHR and Wistar Kyoto rats were studied after 24‐hour systemic AT 1R blockade. Left kidneys received 30 minute renal interstitial infusions of vehicle followed by Ang III (3.5, 7.0, 14, and 28 nmol/kg per min; each dose for 30 minutes). Right kidneys received vehicle infusions. In 4‐week‐old Wistar Kyoto rats, renal interstitial Ang III increased urine sodium (Na+) excretion but failed to induce natriuresis in 4‐week‐old SHR. Renal Ang III levels were similar between Wistar Kyoto rats and SHR, making increased Ang III degradation as a possible cause for defective natriuresis in SHR unlikely. In Wistar Kyoto rats, renal interstitial Ang III induced translocation of AT 2Rs to apical plasma membranes of renal proximal tubule cells. Simultaneously, Ang III induced retraction of the major Na+ transporter Na+‐H+ exchanger‐3 (NHE‐3) from apical membranes and internalization of Na+/K+ ATPase (NKA) from basolateral membranes of renal proximal tubule cells. Consistent with NHE‐3 and NKA retraction, Ang III increased pSer552‐NHE‐3 and decreased pSer23‐NKA. In contrast, in SHR, intrarenal Ang III failed to induce AT 2R translocation, NHE‐3 or NKA retraction, pSer552‐NHE‐3 phosphorylation, or pSer23‐NKA dephosphorylation.
Conclusions
These results indicate impaired Ang III/AT 2R signaling as a possible primary defect in prehypertensive SHR.
Keywords: angiotensin, angiotensin receptor, hypertension, natriuretic hormone, renal physiology
Subject Categories: ACE/Angiotension Receptors/Renin Angiotensin System, Nephrology and Kidney, Pathophysiology, Hypertension
Short abstract
See Editorial Zhuo and Li
Clinical Perspective
What Is New?
Young prehypertensive spontaneously hypertensive rats have defective natriuresis in response to angiotensin type‐2 receptor activation with predominant endogenous agonist, angiotensin III.
What Are the Clinical Implications?
A renal angiotensin type‐2 receptor signaling defect may, at least in part, trigger the development of hypertension in spontaneously hypertensive rats and potentially in humans.
Introduction
Angiotensin II (Ang II), the primary peptide mediating the effects of the renin–angiotensin system, acts at 2 major receptors: type‐1 (AT1Rs) and type‐2 (AT2Rs).1, 2 Ang II actions via AT1Rs are well known, but the biological signals transduced through AT2Rs have been less clear. Differences in the preferred endogenous Ang peptide agonist for AT2Rs compared with AT1Rs may be, at least in part, responsible for this gap in knowledge. Systematic analysis of Ang peptide binding characteristics demonstrated a 33‐fold higher selectivity of the Ang II heptapeptide metabolite des‐aspartyl1‐Ang II (Ang III) for AT2Rs compared with AT1Rs.3 Ang III has now been confirmed as the preferred AT2R agonist in several tissues.4, 5, 6, 7, 8 Within the kidney, Ang III is the predominant endogenous agonist for AT2R‐mediated natriuresis.9, 10, 11, 12 Equimolar concentration escalation‐response studies in vivo during AT1R blockade produced robust natriuretic responses to Ang III but absent responses to Ang II unless aminopeptidase N, the enzyme degrading Ang III to smaller angiotensin peptide fragments, was concurrently blocked.9, 10, 11 Natriuretic responses to Ang III via AT2Rs are largely mediated through inhibition of sodium (Na+) transport in the renal proximal tubule (RPT) by a nitric oxide (NO)‐ and guanosine cyclic 3′,5′‐monophosphate (cGMP)‐dependent mechanism.12 Natriuresis induced by AT2Rs is also consistently accompanied by receptor recruitment from intracellular sites to the apical plasma membranes of RPT cells (RPTC), a mechanism that could enable and/or reinforce long‐term responses.13
In experimental animal models, as well as humans, hypertension is characterized by impaired pressure‐induced natriuresis. The mechanisms for defective natriuresis are largely unknown.14, 15 In contrast to normotensive Wistar‐Kyoto control rats (WKY), hypertensive 12‐week‐old spontaneously hypertensive rats (SHR) fail to recruit AT2Rs or mount natriuretic responses to Ang III, while the heptapeptide induces AT2R translocation and natriuresis in 12‐week‐old normotensive control WKY.16 These results support a defect in AT2R‐mediated natriuresis in SHR. The present study tested whether the defect may predate hypertension by quantifying Ang III‐induced natriuretic responses in 4‐week‐old prehypertensive female and male SHR and control WKY. Furthermore, we tested possible causes for defective natriuresis as either increased Ang III degradation by measuring kidney Ang III levels, or decreased Ang III action by quantifying Ang III‐induced AT2R translocation to apical plasma membranes and internalization/inactivation of Na+‐H+ exchanger‐3 (NHE‐3) and Na+/K+ ATPase (NKA) in RPTCs of WKY and prehypertensive SHR.
Methods
All experimental protocols were approved by the Animal Care and Use Committee at the University of Virginia and performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The authors will make the data and methods and materials used to conduct the research available to any researcher for purposes of reproducing the results or replicating the procedures.
Animal Preparation
The experiments were conducted on 4‐week‐old female and male WKY and SHR (Envigo) housed in a vivarium under controlled conditions (temperature 21±1°C; humidity 60±10%; light 8:00–20:00) and fed a normal sodium (Na+) diet (0.30% Na+).
For all studies, a 24‐hour subcutaneous osmotic mini‐pump (Alzet Model 2001D) infusing candesartan (0.01 mg/kg per min) was inserted 24 hours before experimentation in order to block systemic AT1Rs. While the rats were under short‐term anesthesia with isoflurane, the pumps were implanted in the interscapular region using sterile technique. On the day of experimentation, the rats were anesthetized with Inactin (thiobutabarbital sodium) (100 mg/kg body weight) and a tracheostomy was performed using polyethylene tubing (PE‐60) to assist respiration. Direct cannulation of the right internal jugular vein using PE‐10 tubing provided intravenous access through which 1% bovine serum albumin made in vehicle 5% dextrose in water (D5W) was infused at 20 μL/min. Direct cannulation of the right carotid artery with PE‐10 tubing provided arterial access for monitoring mean arterial pressure (MAP). Following a midline laparotomy, both ureters were cannulated (PE‐10) to collect urine for the quantification of urine Na+ excretion (UNaV).
Renal Cortical Interstitial Infusion
An open bore micro‐infusion catheter (PE‐10) was inserted under the renal capsule into the cortex of each kidney to ensure renal interstitial (RI) infusion of vehicle D5W or pharmacological agent at 2.5 μL/min with a syringe pump (Harvard; model 55‐222). Vetbond tissue adhesive (3M Animal Care Products) was added to secure the catheter(s) and prevent interstitial pressure loss in the kidney.
Blood Pressure Measurements
MAP was measured by the direct intracarotid method (see above) with the use of a digital blood pressure (BP) analyzer (Micromed, Inc). MAPs were recorded every 5 minutes and averaged for all periods. Experiments were initiated at the same time each day (10:00 am) to prevent any diurnal variation in BP.
Pharmacological Agents
Candesartan (Alomone Labs), a specific, potent, insurmountable inhibitor of AT1Rs (IC50>1×10−5 mol/L and 2.9×10−8 mol/L for AT2Rs and AT1Rs, respectively), was used for systemic AT1R blockade. [des‐Asp1]‐Angiotensin II (Ang III; Bachem; Ki=10.5×10−9 mol/L and 2.2×10−9 mol/L for AT1Rs and AT2Rs, respectively) was used to stimulate intrarenal AT2Rs. PD‐123319 (PD; Parke‐Davis; 10 μg/kg per min), a specific AT2R antagonist, was applied interstitially to block intrarenal AT2Rs.
Total Cortical Cell Membrane Preparation and Western Blot Analysis
Slices of kidney cortex (≈75 mg per kidney) were homogenized (Polytron at setting 6, 3×5‐s pulses) in Mammalian Protein Extraction Reagent (Thermo Scientific) buffer with Halt protease and phosphatase inhibitor cocktail (Thermo Scientific) and then centrifuged at 900g for 10 minutes at 4°C to remove cellular debris. The supernatant was removed and used for total protein quantification with a bicinchoninic acid assay (Pierce). SDS samples were prepared, separated by SDS‐PAGE (10% Tris‐HCl polyacrylamide gels; 40 μg of protein loaded per lane), and transferred onto a nitrocellulose membrane by electroblotting. Membranes were blocked in 5% milk in TBS (Tris‐buffered saline) with 0.1% Tween‐20 (TBST) for 2 hours at 4°C and then incubated overnight at 4°C with the following primary antibodies in 5% milk TBST: AT2R (Millipore; Cat # AB15554; 1:500), NHE‐3 (3H3 monoclonal hybridoma supernatant provided as a generous gift from Dr Peter Aronson, Yale University School of Medicine, 1:15), and α‐NKA (Millipore; Cat # 05‐369; 1:10 000) or in 5% bovine serum albumin in TBST: phospho‐NHE‐3 [Ser 552] (pSer552‐NHE‐3; Thermo Scientific; Cat # MA1‐46464; 1:500) and phospho‐NKA [Ser 23] (pSer23‐NKA; Cell Signaling; Cat # 4006; 1:1000). After 3 washes in TBST, membranes were incubated with their respective horseradish peroxidase–conjugated secondary antibodies in 5% milk TBST (GE Healthcare; Cat # NA934V or NA931V; 1:2500) for 2 hours at room temperature (RT). After 3 washes in TBST, signals were detected using chemiluminescence with the ChemiDoc MP Imaging System, and band densities were measured with Image J software (NIH). The membranes were then stripped for 10 minutes (Restore Western blot stripping buffer; Thermo Scientific) and blocked in 5% milk TBST for 1 hour at 4°C. The membranes were then incubated with the primary antibody β‐tubulin (Millipore; Cat # 05‐661; 1:5000) in 5% milk TBST for 2 hours at RT, followed by the remaining Western procedure as stated above. All signals were normalized to β‐tubulin expression.
RPTC Apical Membrane Isolation and Western Blot Analysis
Slices of kidney cortex (≈75 mg per kidney) were homogenized (Polytron, setting 6, 3×5‐s pulses) in detergent‐free homogenization buffer (10 mmol/L Tris, 200 mmol/L sucrose, 1 mmol/L EDTA, pH 7.4) with Halt protease and phosphatase inhibitor cocktail and then centrifuged at 900g for 10 minutes at 4°C to remove cellular debris. The supernatant was removed and used for total protein quantification with a bicinchoninic acid assay as stated above. One milligram total protein was resuspended in 10 mL detergent‐free homogenization buffer and incubated with 20 μg of biotinylated Lotus tetragonolobus agglutinin lectin (Vector Laboratories; Cat # B‐1325) on a 360° rocker for 2 hours at RT. A 50% vol/vol slurry (20 μL) of Ultralink Neutravidin beads (Pierce) was then added and incubated on a 360° rocker for 30 minutes at RT. The beads were then pelleted and thoroughly washed with PBS using a microcentrifuge spin cup filter. The L tetragonolobus agglutinin affinity‐attached membranes were eluted by incubating the beads in the spin cup filter with 125 μL of 2x sample buffer heated to 70°C for 10 minutes. The samples then underwent the Western blot procedure as described above. The membranes were incubated with the following primary antibodies in 5% milk TBST: AT2R (1:500) and NHE‐3 (1:15) followed by their respective horseradish peroxidase–conjugated secondary antibodies as described above. The membranes were then stripped for 10 minutes, blocked in 5% milk TBST for 1 hour at 4°C, and incubated with a primary antibody against villin (Santa Cruz; Cat # sc‐7672; 1:500) in 5% milk TBST for 2 hours at RT. The remaining Western blotting procedure was the same as stated above. All signals were normalized to the expression of villin, a RPTC brush border membrane marker.
In Vivo Kidney Perfusion and Fixation Procedure
Following the second RI infusion dose of Ang III (7 nmol/kg per min), the rat heart left ventricular cavity was cannulated and the rat was perfused with 20 mL of 4% sucrose in PBS followed by 20 mL of 4% paraformaldehyde in PBS. The kidneys were sliced in half and placed in 4% paraformaldehyde for 2 hours at RT. The slices were rinsed 3× in PBS, immersed in 100 mmol/L Tris‐HCl for 30 minutes, and then rinsed 3× in PBS before being stored in 30% sucrose in PBS overnight at 4°C. The next day, the kidney slices were embedded in Tissue Tek OCT Compound in Cryomold vinyl specimen molds, placed at −20°C until frozen, and then stored at −80°C until processing. Cryostat thin sections (5–8 μm) were placed on Probe On Plus positively charged microscope slides through the University of Virginia Research Histology Core and immediately stained.
Confocal Immunofluorescence Microscopy
After the kidney sections had been spotted onto slides and washed with TBS, they were permeabilized with 0.2% Triton‐X in TBS or 1% SDS in TBS for 10 minutes. The sections were washed in TBS with 0.02% Tween‐20 (TBST) and then blocked in 1% milk TBST or LI‐COR blocking buffer for 1 hour at RT. The kidney sections were incubated with primary antibodies in 1% milk TBST or LI‐COR blocking buffer overnight at 4°C. After washing with TBST, Alexa 647 conjugated donkey anti‐rabbit, Alexa 488 conjugated donkey anti‐mouse secondary antibodies (Invitrogen; Cat # A31573 and A21202; 1:500), and/or Texas‐Red phalloidin (Invitrogen; Cat # A12381; 1:200) were added in 1% milk TBST or LI‐COR blocking buffer for 1 hour at RT. The following antibody pairs were used in 1% milk TBST after permeabilization with 0.2% Triton‐X detergent: (1) AT2R (1:100), adaptor protein 2 (AP‐2; Santa Cruz; Cat # sc‐17771; 1:100), and phalloidin (1:200). (2) NHE‐3 (Millipore; Cat # MAB3136; 1:2000) and AP‐2 (Santa Cruz; Cat # SC‐10761; 1:100). The following primary antibody pair was used in LI‐COR blocking buffer after permeabilization with 0.2% Triton‐X detergent: pSer552‐NHE‐3 (1:100) and phalloidin (1:200). The following antibody pair was used in 1% milk TBST after permeabilization with 1% SDS detergent: α‐NKA (1:2000) and phalloidin (1:200). The following antibody pair was used in LI‐COR blocking buffer after permeabilization with 1% SDS detergent: pSer23‐NKA (Abcam: Cat # ab74069 1:100) and AP‐2 (1:100). Following secondary antibody incubation, the slices were washed in TBST, Fluoromount G (Southern Biotech) was applied, and the specimens were covered with a glass coverslip. Stained tubules were photographed under epifluorescence illumination using an automated Olympus IX81 spinning disk confocal microscope using a 60× plan apo 5 water immersion objective with a numeric aperture of 1.2. The microscope was controlled using Slidebook 5.5 software (3i, Denver, CO) and 5‐μm‐thick z‐stack images were captured using a Hamamatsu EMCCD camera at 0.25‐μm intervals and deconvolved using the autoquant spinning disk deconvolution module. Calculation of immunoreactive AT2R within the RPTC brush border was conducted using F‐actin staining with Alexa 594 phalloidin to create the mask and measuring AT2R fluorescence within that region. Calculation of the immunoreactive NHE‐3 fluorescence intensity at the subapical region of the RPTC was performed by creating a mask using the AP‐2 fluorescence intensity and measuring the NHE‐3 fluorescence within that region. The immunoreactive pSer552‐NHE‐3 fluorescence intensity was measured at the subapical region of the RPTC. The immunoreactive α‐NKA fluorescence intensity was measured in the intracellular region of the RPTC in the S3 segment of the RPT, 4 μm from the basolateral membrane of each cell. Ten boxes were measured per RPTC. Because of the highly convoluted basolateral membrane in the S1 section of the RPT, the pattern of expression of α‐NKA appears to all be internalized and the invaginations cannot be distinguished. Just above the cortical medullary interface, the S3 segment of the RPT was selected for imaging because it has fewer basolateral invaginations. The immunoreactive pSer23‐NKA fluorescence intensity was calculated as total intercellular fluorescence intensity within the RPTC. For each antibody, quantifications were performed on 20 independent measurements of RPTCs from a rat and averaged.
Ang II and Ang III Extraction from Kidney
For whole kidney tissue Ang II and Ang III measurements, kidneys were immersed in cold methanol (100%), minced, and homogenized with a polytron tissue tearer. The homogenates were centrifuged, and the supernatants from the kidney homogenates were dried overnight in a vacuum centrifuge. The dried residue was reconstituted in 1 mL buffer as described in the Enzyme Immunoassay (EIA) Kit (Phoenix Pharmaceutical, Inc). These samples were applied to phenyl‐bonded solid‐phase extraction columns (Bond Elut; Agilent Technologies, Inc) that had been prewashed with 90% methanol followed by water. After sample application, each solid‐phase extraction column was washed sequentially with water, hexane, and chloroform. Ang peptides were eluted from the solid‐phase extraction column with 90% methanol.17 The eluates were collected, evaporated to dryness under vacuum, and stored at −20°C.
Separation of Ang II from Ang III by High‐Performance Liquid Chromatography; Measurement of Kidney Ang II and Ang III Peptide Levels by Fluorescent Immunoassay Kit
Before injecting samples to high‐performance liquid chromatography (HPLC), the retention time and collection interval for Ang II and Ang III were determined after preparation of mobile phase and confirmed daily by injecting 100 pmol of Ang II and Ang III calibrators and determining their elution pattern by monitoring the effluent at 230 nm. To prevent carryover of the calibrators after their injection, we washed the column and the injector valve with 150 and 10 mL, respectively, of wash solution (90% acetonitrile/0.13% HFBA (heptafluorobuteric acid)). Lyophilized kidney samples were resuspended by vortex‐mixing after addition of 300 μL equilibration solvent, which included 65% mobile phase A (0.13% HFBA) and 35% mobile phase B (80% acetonitrile/0.13% HFBA). Before HPLC, insoluble matter in the resuspension was sedimented by centrifugation. A 200‐μL aliquot of supernate was chromatographed by isocratic elution at 1 mL/min on a 25×0.46‐cm, 5 μm Vydac C18 reverse‐phase HPLC column (Advanced Chromatography Technologies Ltd). According to the retention time from high concentration of Ang II and Ang III calibrators, collection time was determined. Ang II eluted at 14 minutes, and Ang III had an elution peak at 16 minutes. During separation of samples, the peaks of Ang II and Ang III could not be observed because of the low amount of Ang peptides. Sample fractions were collected from 13 to 15 minutes for Ang II and from 15 to 17 minutes for Ang III, and evaporated to dryness. The kidney samples and Ang II and Ang III standard fractions were reconstituted in 500 μL enzyme immunoassay buffer. Fifty microliters of standard and sample fractions in duplicate were seeded to an immunoplate, which was incubated with rabbit anti‐peptide IgG antibody and a precoated second antibody overnight. This was followed by addition of 25 μL biotinylated peptide, which interacted with streptavidin–horseradish peroxidase to catalyze the substrate solution. The fluorescence intensity is directly proportional to the amount of biotinylated peptide–streptavidin‐horseradish peroxidase complex but inversely proportional to the amount of the peptide in standard solutions and samples. The fluorescence intensity was measured by a fluorescence microplate reader as described in the EIA kit. The cross‐reactivity for Ang II and Ang III measurement is 100%. The minimum detectable concentration is 8.2 pg/g. The intra‐assay variation was <10% and interassay variation was <15%.
We carefully checked for residual peptide after the calibration solutions were used by measuring the residual Ang II and Ang III in the fractions with just vehicle injection. We also compared the sum of Ang II + Ang III with the total immunoreactive Ang II measured in samples not subjected to HPLC separation. There was no evidence that the peptides from the calibration solutions were contributing to the sample values since the sum of Ang II and Ang III never exceeded the values obtained from the samples not subjected to HPLC.
Specific Protocols
Effects of acute intrarenal Ang III infusion on UNaV and MAP in the presence of systemic AT1R blockade
All studies involved a 2‐kidney model where the right kidney served as a time control receiving RI infusions of vehicle D5W while the left kidney received RI infusions of pharmacological agents. Following a 1‐hour equilibration period (designated control) in which vehicle D5W was infused into the RI space of each kidney, the following groups of rats were studied: (1) Female WKY Ang III (N=6): right kidney received RI infusion of D5W for four 30‐minute periods and the left kidney received RI infusion of Ang III (3.5, 7.0, 14.0, and 28.0 nmol/kg per min; each dose for 30 minutes) following a 30‐minute RI infusion of D5W. (2) Female WKY Ang III + PD (N=6): right kidney received RI infusion of D5W for four 30‐minute periods and the left kidney received RI infusion of Ang III (3.5, 7.0, 14.0, and 28.0 nmol/kg per min; each dose for 30 minutes) + PD (10 μg/kg per min) following a 30‐minute RI infusion of D5W. (3) Female SHR Ang III (N=7): right kidney received RI infusion of D5W for four 30‐minute periods and the left kidney received RI infusion of Ang III (3.5, 7.0, 14.0, and 28.0 nmol/kg per min; each dose for 30 minutes) following a 30‐minute RI infusion of D5W. (4) Male WKY Ang III (N=5): right kidney received RI infusion of D5W for four 30‐minute periods and the left kidney received RI infusion of Ang III (3.5, 7.0, 14.0, and 28.0 nmol/kg per min; each dose for 30 minutes) following a 30‐minute RI infusion of D5W. (5) Male SHR Ang III (N=6): right kidney received RI infusion of D5W for four 30‐minute periods and the left kidney received RI infusion of Ang III (3.5, 7.0, 14.0, and 28.0 nmol/kg per min; each dose for 30 minutes) following a 30‐minute RI infusion of D5W. UNaV and MAP were measured for each period.
Effects of acute intrarenal Ang III infusion on RPTC apical plasma membrane AT2R and NHE‐3 or total cortical homogenate AT2R, NHE‐3, pSer552‐NHE‐3, α‐NKA, and pSer23‐NKA in the presence of systemic AT1R blockade
Female rats followed the same procedure as Protocol 1 (Female WKY Control and Ang III and Female SHR Control and Ang III) and N=6 for each condition. Kidneys were harvested at the end of the study and processed for Western blot analysis. In a separate set of animals, a female WKY and SHR rat were perfused separately following the 7 nmol/kg per min Ang III RI infusion period and processed for confocal microscopy.
Effects of acute intrarenal Ang III infusion on renal tissue levels of Ang II and Ang III in the presence of systemic AT1R blockade
For female rats, the same procedure was followed as for Protocol 1 (Female WKY Control and Ang III and Female SHR Control and Ang III) and N=5 to 8 for each condition. Kidneys were harvested at the end of the study to be processed for Ang II and Ang III peptide levels.
Statistical Analysis
Data are presented as mean±1 SE. Statistical significance was determined by using 2‐way ANOVA followed by multiple comparisons testing with the Student Newman‐Keuls test with 95% confidence. Data for RI angiotensin levels were analyzed by paired t test. The level of significance was set at P<0.05.
Results
Effects of Intrarenal Ang III Infusion ± AT2R antagonist PD on UNaV and MAP in Female WKY and SHR
UNaV (Figure 1A) was unchanged in response to vehicle infusion throughout the experiment in control right kidneys of both WKY and SHR (P=NS). In 4‐week‐old control WKY, UNaV increased immediately in response to 3.5 nmol/kg per min Ang III (P<0.05) and remained elevated (F=5.31; P<0.01) with increasing Ang III infusion rates (7, 14, and 28 nmol/kg per min) compared with control vehicle‐infused kidneys. In contrast, 4‐week‐old SHR did not increase Na+ excretion in response to any dose of Ang III (3.5–28 nmol/kg per min) (P=NS). Consistent with Ang III action through AT2Rs in WKY, natriuretic responses to intrarenal Ang III at all infusion rates were prevented in the presence of concurrent intrarenal infusion of AT2R antagonist PD‐123319 (PD) (P=NS from own control and F=3.25; P<0.05 compared with Ang III). MAP (Figure 1B) in WKY was within the normal range and did not change in response to intrarenal Ang III or PD infusions. MAP was slightly higher in 4‐week‐old SHR compared with 4‐week‐old WKY but remained within the normal range throughout the experiment (P=NS from control and between WKY and SHR).
Figure 1.
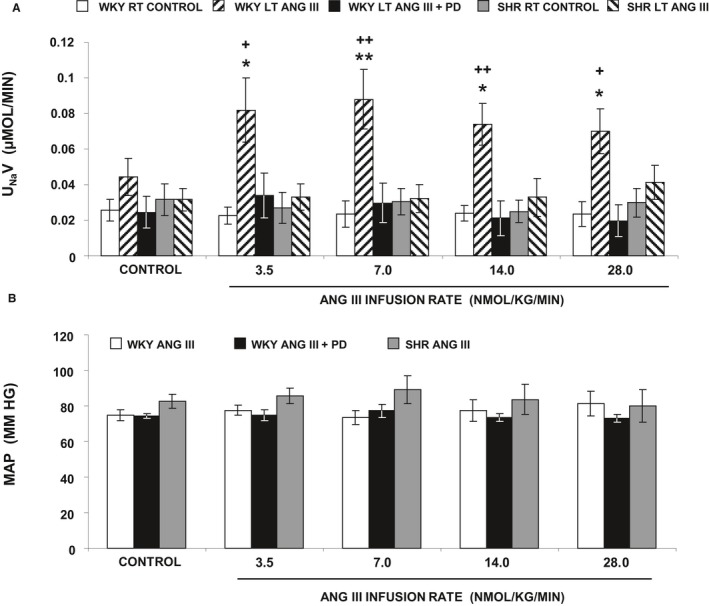
A, Urine Na+ excretion (UN aV) under the following conditions: (clear bar) female Wistar‐Kyoto (WKY) right (RT) kidney renal interstitial (RI) control infusion (N=12), (striped bar) female WKY left (LT) kidney RI angiotensin III (Ang III) (3.5, 7.0, 14.0, and 28.0 nmol/kg per min) infusion (N=6), (black bar) female WKY LT kidney RI Ang III+PD (10 μg/kg per min) infusion (N=6), (gray bar) female spontaneously hypertensive (SHR) RT kidney RI control infusion (N=7), (striped bar) female SHR LT kidney RI Ang III infusion (N=7), all in the presence of systemic AT 1R blockade. B, Mean arterial pressure (MAP) under conditions described in A. Data represent mean±1 SE. *P<0.05 and **P<0.01 from own control and + P<0.05 and ++ P<0.01 from corresponding Ang III+PD periods.
Effects of Intrarenal Ang III Infusion on UNaV and MAP in Male WKY and SHR
To determine whether the natriuretic response induced by Ang III in WKY rats is sex‐specific, we repeated the studies in 4‐week‐old male SHR and WKY control rats (Figure 2). UNaV was unchanged in response to vehicle infusion throughout the experiment in control right kidneys of both WKY (Figure 2A) and SHR (Figure 2B) (P=NS). In 4‐week‐old male control WKY, UNaV dose‐dependently increased in response to Ang III 3.5 nmol/kg per min (P=0.06), and 7, 14, and 28 nmol/kg per min (P<0.01, P<0.05, and P<0.01, respectively) compared with control vehicle‐infused kidneys (F=9.01, P<0.001). In contrast, 4‐week‐old SHR did not increase Na+ excretion in response to any dose of Ang III (3.5–28 nmol/kg per min) (P=NS). MAP (Figure 2C) in male WKY was within the normal range and did not change in response to intrarenal Ang III infusion. MAP was slightly higher in 4‐week‐old male SHR compared with 4‐week‐old male WKY but remained within the normal range throughout the experiment (from control P=NS and between WKY and SHR, F=3.08, P<0.05). Since the responses to intrarenal Ang III infusions were similar for male and female SHR, but female rats may have greater AT2R expression, we conducted the remaining studies with females only.
Figure 2.
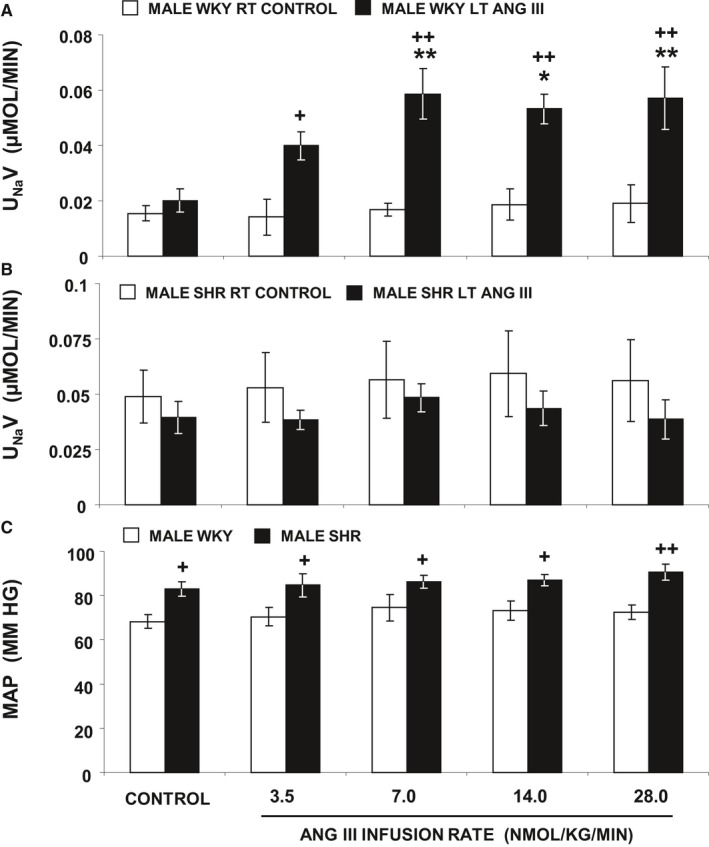
A, Urine Na+ excretion (UN aV) under the following conditions: (white bar) male WKY right (RT) kidney renal interstitial (RI) control infusion (N=5), (black bar) male WKY left (LT) kidney RI Ang III (3.5, 7.0, 14.0, and 28.0 nmol/kg per min) infusion (N=5) all in the presence of systemic AT 1R blockade. B, Male SHR RT and LT kidneys (both N=6) that followed the same procedure as in A. C, Mean arterial pressure (MAP) under conditions described in (A and B). Data represent mean±1 SE. *P<0.05 and **P<0.01 from own control and + P<0.05 and ++ P<0.01 from corresponding period.
Renal Tissue Levels of Ang II and III in Female WKY and SHR
The lack of a response to Ang III in SHR could be because of accelerated degradation of Ang III. To investigate this possibility, we measured whole kidney Ang II and Ang III levels by HPLC and ELISA assay17 in a separate set of 4‐week‐old WKY and SHR (Figure 3A and 3B, respectively). Baseline whole kidney Ang II and Ang III levels were 162±29 and 528±107 pg/g, respectively, in WKY and were 164±29 and 394±68 pg/g, respectively, in SHR. In WKY, Ang III infusion increased whole kidney Ang II (P<0.05) and Ang III levels (P<0.01). In SHR, Ang III infusion also increased Ang III levels (P<0.05), but did not increase Ang II (P=NS). In both WKY and SHR, Ang III infusion resulted in far greater increases in renal tissue Ang III compared with Ang II levels (P<0.01 and P<0.05, respectively). Ang III/Ang II ratios in whole kidney tissue were not significantly different in control vehicle‐infused SHR and WKY and were augmented to a similar degree by Ang III infusion (P<0.01) in both (Figure 3C). A representative chromatogram of Ang II and Ang III peptide separation using HPLC is shown in Figure 3D.
Figure 3.
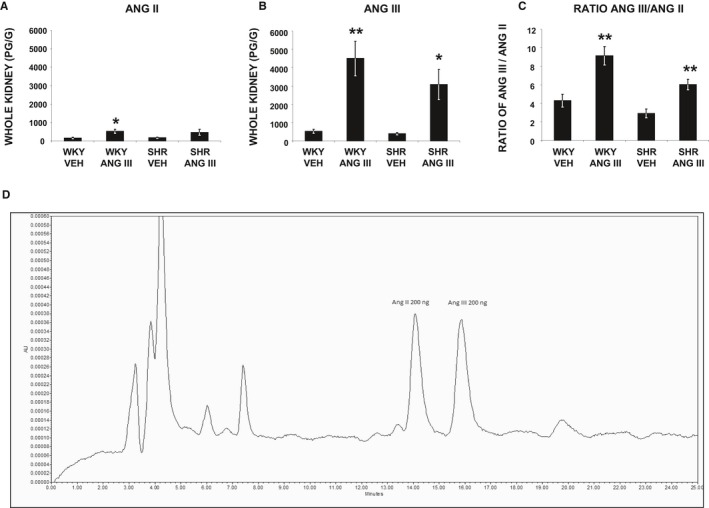
A through C, Angiotensin II (Ang II), angiotensin III (Ang III), and the ratio of Ang III/Ang II peptide levels, respectively, measured in whole kidney tissues of female Wistar‐Kyoto (WKY) and spontaneously hypertensive (SHR) rats (N=5–8) after renal interstitial infusions of vehicle or Ang III (3.5, 7.0, 14.0, and 28.0 nmol/kg per min). Results are reported as pg/g kidney weight. Data represent mean±1 SE. *P<0.05 and **P<0.01 from respective vehicle treatment. D, Representative chromatogram of HPLC showing separation peaks for Ang II and Ang III.
Effects of Intrarenal Ang III on RPTC Apical Plasma Membrane AT2R Density in Female WKY and SHR
To determine whether AT2R activation induces receptor translocation to the apical plasma membranes of RPTCs of female WKY and SHR, we used confocal immunofluorescence microscopy and Western blot analysis. Figure 4A through 4F demonstrates the subcellular distribution of AT2Rs in a representative set of RPTCs from WKY and SHR after intrarenal vehicle or Ang III infusion (7.0 nmol/kg per min) as determined by confocal immunofluorescence microscopy. Figure 4A and 4B depict the RPTC distributions of phalloidin (red) marking the apical brush border and AP‐2 marking the subapical region (blue), respectively. Figure 4C demonstrates the cellular distribution of AT2Rs using an antibody (Millipore; Cat # AB15554) proven to be specific for AT2Rs by immunoblotting AT2R‐null mouse adrenal glands that normally have a high degree of AT2R expression.18 Figure 4D, and 4E at higher magnification, show the merged phalloidin, AP‐2, and AT2R images demonstrating that administration of Ang III in WKY induces a subtle color shift in immunofluorescence from red to orange indicating increased AT2R density in the apical plasma membrane in response to Ang III in WKY. Figure 4F represents a higher power image of merged AP‐2 and AT2R images depicting only the brush border area quantified for AT2R staining. This panel demonstrates a substantial increase in AT2R immunofluorescence staining in the RPTC apical plasma membrane of WKY in response to Ang III. In contrast, Figure 4F shows no change in AT2R density in response to Ang III in SHR. Figure 4G shows the quantitative increase in relative AT2R fluorescence units in response to Ang III (P<0.01) in WKY but not in SHR. As further corroborated by Western blot analysis (Figure 4H and 4I), Ang III increased apical plasma membrane AT2R density (P<0.01) in WKY without changing total cortical homogenate AT2R protein expression. In contrast, in SHR apical plasma membrane AT2R density did not change in response to intrarenal Ang III infusion. AT2R expression was similar in WKY and SHR kidneys during vehicle infusion (Figure 4I).
Figure 4.
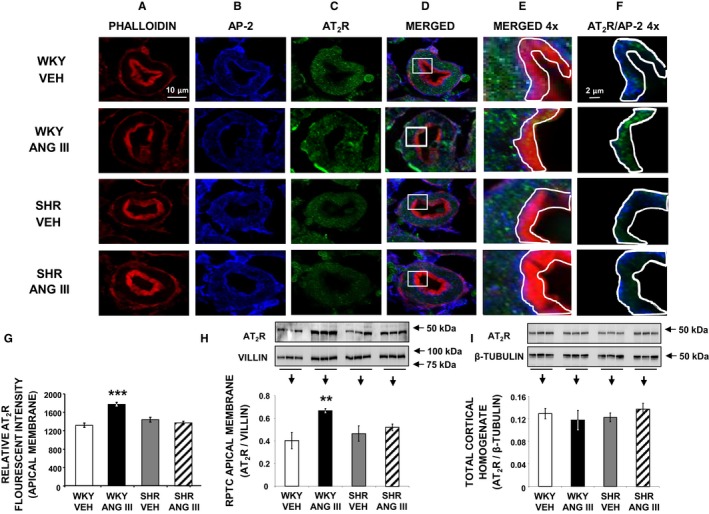
Confocal micrographs (×600 magnifications) showing AT 2R localization in female Wistar Kyoto (WKY) and spontaneously hypertensive (SHR) renal proximal tubule cell (RPTC) thin sections (5–8 μm) after renal interstitial (RI) infusion of vehicle (VEH) or angiotensin III (Ang III; 7.0 nmol/kg per min). As indicated, rows of images show a representative set of RPTCs from (top‐to‐bottom) WKY VEH, WKY Ang III, SHR VEH, and SHR Ang III treatment groups. As indicated, columns (left‐to‐right) depict brush border membrane staining with phalloidin (A), subapical area staining with adaptor protein‐2 (AP‐2) (B), AT 2R staining (C), merged image (D), enlarged merged image (×4) of the square section in (D) (E), and enlarged image with only AT 2R and AP‐2 staining of the brush border membrane area quantified for AT 2R (F). The encircled areas in (E and F) encompass brush border apical membranes quantified for AT 2R intensity. Scale bars in the first and sixth columns represent 10 and 2 μm, respectively. G, Quantification of RPTC apical membrane AT 2R fluorescence intensity performed on 20 independent measurements of RPTCs from 1 rat for WKY VEH (white bar), WKY Ang III (black bar), SHR VEH (gray bar), and SHR Ang III (striped bar) treatments. H and I, Depict Western blot analysis of AT 2R in RPTC apical membranes and total cortical homogenate, respectively, in WKY and SHR after VEH or Ang III (3.5–28.0 nmol/kg per min) infusions (N=6 for each condition). RPTC apical membrane signals were normalized to villin, a brush border apical membrane marker. Total cortical homogenates were normalized to β‐tubulin. Data represent mean±1 SE. **P<0.01 and ***P<0.001 compared with WKY VEH.
Effects of Intrarenal Ang III on RPTC NHE‐3 Apical Plasma Membrane Retraction and Cellular Internalization in Female WKY and SHR
To determine whether AT2R activation with Ang III induces natriuresis by internalizing/inhibiting RPTC Na+ apical membrane transporter NHE‐3, we performed immunofluorescence microscopy and Western blot analysis. Figure 5A through 5F demonstrates the subcellular distribution of NHE‐3 as determined by confocal immunofluorescence microscopy from a representative set of WKY and SHR RPTCs after intrarenal vehicle or Ang III infusion (7.0 nmol/kg per min). Figure 5A depicts RPTC autofluorescence (blue) and Figure 5B and 5C depict AP‐2 (green) and NHE‐3 (red), respectively. Figure 5D and 5E show merged and higher power images of Figure 5A through 5C, respectively. Figure 5F shows a higher power image of autofluorescence and NHE‐3 staining depicting only the subapical region quantified for NHE‐3 staining. In Figure 5F, WKY, but not SHR, demonstrate increased subapical NHE‐3 density in response to Ang III. In Figure 5G, quantification of results shows relative subapical NHE‐3 fluorescence units are increased in WKY (P<0.05) but not in SHR (P=NS). In Figure 5H, Western blot analysis demonstrates a reduction in RPTC apical plasma membrane NHE‐3 density in response to Ang III infusion in WKY, but there is no change in apical membrane NHE‐3 density in response to Ang III in SHR. Figure 5I shows that total cortical homogenate NHE‐3 expression did not change in response to Ang III in either WKY or SHR. However, total cortical NHE‐3 was significantly higher in SHR compared with WKY (P<0.01).
Figure 5.
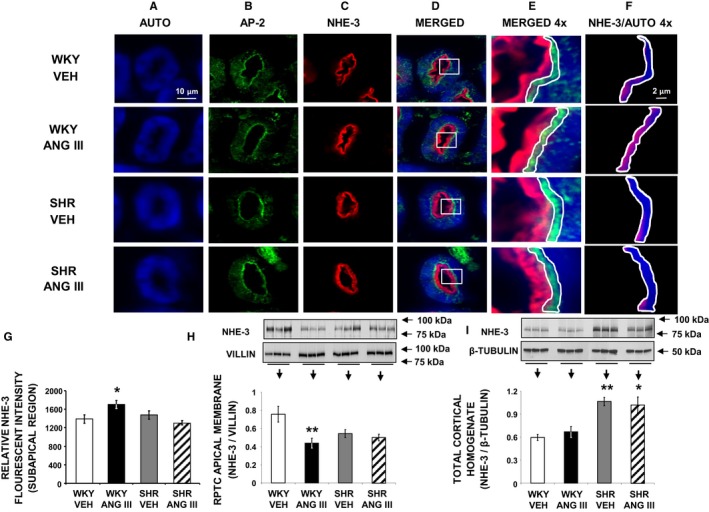
Confocal micrographs (×600 magnification) showing NHE‐3 localization in female Wistar‐Kyoto (WKY) and spontaneously hypertensive (SHR) renal proximal tubule cell (RPTC) thin sections (5–8 μm) after renal interstitial (RI) infusion of vehicle (VEH) or angiotensin III (Ang III; 7.0 nmol/kg per min). As indicated, rows of images show representative sets of RPTCs from (top‐to‐bottom) WKY VEH, WKY Ang III, SHR VEH, and SHR Ang III treatment groups. As indicated, columns (left‐to‐right) depict confocal autofluorescence (A), subapical area staining with adaptor protein‐2 (AP‐2) (B), NHE‐3 staining (C), merged image (D), enlarged image (×4) of the square section in (D) (E), and enlarged image with only NHE‐3 and autofluorescence in the subapical area quantified for NHE‐3 (F). The encircled areas encompass the subapical area that was quantified for NHE‐3 fluorescence intensity. Scale bars in the first and sixth columns represent 10 and 2 μm, respectively. G, The quantification of RPTC subapical area NHE‐3 fluorescence intensity performed on 20 independent measurements of RPTCs from 1 rat for WKY VEH (white bar), WKY Ang III (black bar), SHR VEH (gray bar), and SHR Ang III (striped bar) treatments. H and I, Depict Western blot analysis of RPTC apical membrane and total cortical homogenate NHE‐3 expression, respectively, in female WKY and SHR after VEH or Ang III (3.5–28.0 nmol/kg per min) infusions (N=6 for each condition). RPTC apical membrane signals were normalized to villin, a brush border apical membrane marker. Total cortical homogenates were normalized to β‐tubulin. Data represent mean±1 SE. *P<0.05 and **P<0.01 compared with WKY VEH.
Effects of Intrarenal Ang III on RPTC Phosphorylated NHE‐3 Cellular Internalization in Female WKY and SHR
Phosphorylation of NHE‐3 (pNHE‐3) at serine 552 (pSer552‐NHE‐3) after activation of cyclic adenosine monophosphate (cAMP)‐dependent protein kinase A is required for maximum inhibition of NHE‐3, because mutation of this amino acid reduces the inhibitory effect on NHE‐3 by cAMP.19, 20 Consequently, pNHE‐3 is considered a marker of NHE‐3 retraction/internalization and inactivation.21, 22 The subcellular distribution of pSer552‐NHE‐3 is shown in Figure 6. Figure 6A through 6F show representative confocal micrographs and Figure 6G shows quantitative fluorescence intensity for pSer552‐NHE‐3. Figure 6A through 6C demonstrates RPTC autofluorescence, phalloidin (marking apical plasma membranes), and pSer552‐NHE‐3, respectively. As shown in merged Figure 6D, in WKY Ang III infusion increased pSer552‐NHE‐3 density in the subapical region of the RPTC. This Ang III‐induced increase in pSer552‐NHE‐3 density is best visualized in Figure 6F, a high‐power view of the boxed area in Figure 6E. There was no change in pSer552‐NHE‐3 in response to Ang III in SHR (Figure 6F). As shown in Figure 6G, Ang III increased the relative pSer552NHE‐3 fluorescence intensity (P<0.01) in RPTC subapical regions of Ang III‐infused WKY, but there was no change in response to Ang III in SHR (P=NS). In total cortical homogenate (Figure 6H) Ang III increased pSer552‐NHE‐3 (P<0.01) in WKY, but not in SHR (P=NS). Figure 6I depicts the ratio of total cortical homogenate pSer552‐NHE‐3/NHE‐3 protein expression obtained by Western blot analysis and demonstrates a significant increase in Ang III‐infused WKY (P<0.01), but not SHR kidneys.
Figure 6.
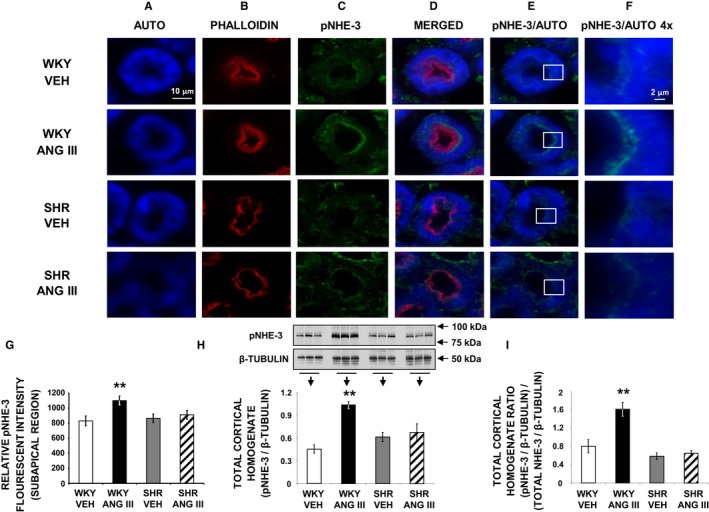
Confocal micrographs (×600 magnification) showing pSer552‐NHE‐3 (pNHE‐3) localization in female Wistar‐Kyoto (WKY) and spontaneously hypertensive (SHR) renal proximal tubule cell (RPTC) thin sections (5–8 μm) after renal interstitial (RI) infusion of vehicle (VEH) or angiotensin III (Ang III; 7.0 nmol/kg per min). As indicated, rows of images show representative sets of RPTCs from (top‐to‐bottom) WKY VEH, WKY Ang III, SHR VEH, and SHR Ang III treatment groups. As indicated, columns (left‐to‐right) depict confocal autofluorescence (A), brush border membrane staining with phalloidin (B), pNHE‐3 staining (C), merged image (D), merged image of only confocal autofluorescence and pNHE‐3 (E), and enlarged images (×4) of the square sections in (E) (F). The scale bars in the first and sixth columns represent 10 and 2 μm, respectively. G, The quantification of RPTC subapical pNHE‐3 fluorescence intensity performed on 20 independent measurements of RPTCs from 1 rat for WKY VEH (white bar), WKY Ang III (black bar), SHR VEH (gray bar), and SHR Ang III (striped bar) treatments. H, Western blot analysis of total cortical homogenate pNHE‐3 protein in female WKY and SHR after VEH or Ang III (3.5–28.0 nmol/kg per min) infusions (N=6 for each condition). All blots were normalized to β‐tubulin. I, Ratio of (pNHE‐3/β‐tubulin)/(NHE‐3/β‐tubulin) for all 4 conditions. Data represent mean±1 SE. **P<0.01 compared with WKY VEH.
Effects of Intrarenal Ang III on RPTC NKA Basolateral Membrane Retraction and Cellular Internalization in Female WKY and SHR
To ascertain that AT2R activation with Ang III also induces natriuresis by internalizing/inhibiting RPTC Na+ basolateral membrane transporter NKA, we performed immunofluorescence microscopy and Western blot analyses for α‐NKA (Figure 7). Figure 7A through 7C show RPTC autofluorescence (blue), apical plasma membrane marker phalloidin (red), and α‐NKA (green), respectively. α‐NKA localizes mainly to the RPTC basolateral membrane in vehicle‐infused WKY (merged Figure 7D), and is internalized in response to Ang III. In vehicle‐infused SHR, α‐NKA is distributed largely in the basolateral membrane and the distribution is unchanged after Ang III infusion. NKA internalization can be best appreciated in high‐power views of merged α‐NKA and phalloidin images (Figure 7F). α‐NKA staining was measured within the small boxes in Figure 7F that were placed 4 μm from the basolateral membrane to ensure measurements of intracellular α‐NKA. For a more detailed explanation of quantitation analysis, please see the Methods section. As quantified in Figure 7G, in WKY Ang III induced a higher intracellular α‐NKA density compared with vehicle‐infused controls (P<0.01), but not in SHR. Figure 7H shows no significant difference in total cellular expression of α‐NKA in WKY or SHR in response to Ang III infusion.
Figure 7.
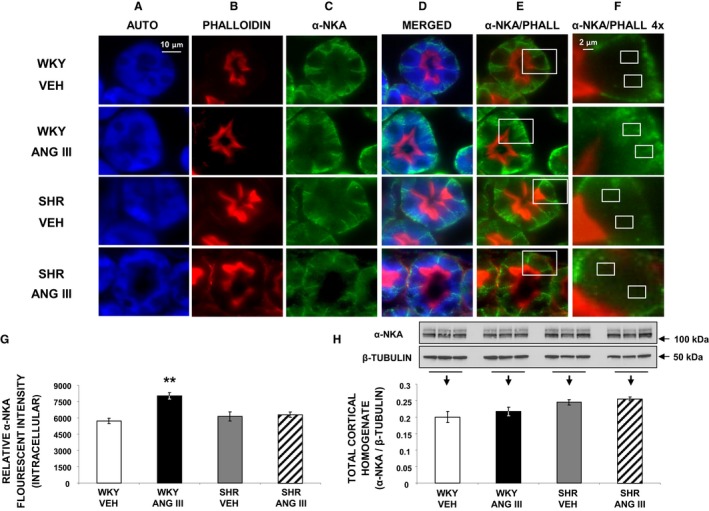
Confocal micrographs (×600 magnification) of α‐NKA localization in female Wistar‐Kyoto (WKY) and spontaneously hypertensive (SHR) renal proximal tubule cell (RPTC) thin sections (5–8 μm) after renal interstitial infusion of vehicle (VEH) or angiotensin III (Ang III; 7.0 nmol/kg per min). As indicated, rows of images show representative sets of RPTCs from (top‐to‐bottom) WKY VEH, WKY Ang III, SHR VEH, and SHR Ang III treatment groups. As indicated, columns (left‐to‐right) depict confocal autofluorescence (A), brush border membrane staining with phalloidin (B), α‐NKA (C), merged image (D), merged image of only phalloidin and α‐NKA (E), and enlarged images (4×) of the square sections in (E) (F). The small white boxes in F encompass the intracellular regions that were quantified for α‐NKA intensity. For a more detailed explanation on quantitation, please see the Methods section. Scale bars in the first and sixth columns represent 10 and 2 μm, respectively. G, The quantification of RPTC intracellular α‐NKA fluorescence intensity performed on 20 independent measurements of RPTCs from 1 rat for WKY VEH (white bar), WKY Ang III (black bar), SHR VEH (gray bar), and SHR Ang III (striped bar) treatments. H, Western blot analysis of total cortical homogenate α‐NKA protein in female WKY and SHR after VEH or Ang III (3.5–28.0 nmol/kg per min) infusions (N=6 for each condition). All blots are normalized to β‐tubulin. Data represent mean±1 SE. **P<0.01 from WKY VEH.
Effects of Intrarenal Ang III on RPTC Phosphorylated NKA Basolateral Membrane Retraction and Cellular Internalization in Female WKY and SHR
Phosphorylation of NKA (pNKA) at serine 23 (pSer23‐NKA) serves as an established indicator of α‐NKA retraction/internalization wherein a reduction signifies retraction.23 The subcellular distribution of pSer23‐NKA is shown for representative confocal micrographs in Figure 8A through 8F, and the relative RPTC pSer23‐NKA fluorescence intensities are shown in Figure 8G. RPTC autofluorescence, AP‐2, and pSer23‐NKA are shown in Figure 8A through 8C, respectively. Merged Figure 8D shows a reduction in RPTC pSer23‐NKA in response to Ang III in WKY but not in SHR. This difference between WKY and SHR is more easily seen in Figure 8F, a high‐power view of the area depicted in the respective box of Figure 8E. The response is quantified in Figure 8G, demonstrating that intracellular pSer23‐NKA fluorescence is reduced by Ang III in WKY (P<0.001) but not in SHR (P=NS). Figure 8H shows that total renal cortical homogenate pSer23‐NKA is reduced by Ang III in WKY (P<0.01), but not in SHR (P=NS). Figure 8I depicts the ratio of total cortical homogenate pSer23‐NKA/α‐NKA protein expression obtained by Western blot analysis and demonstrates a significant decrease in Ang III‐infused WKY (P<0.01) compared with vehicle WKY kidneys, while there was no change in SHR.
Figure 8.
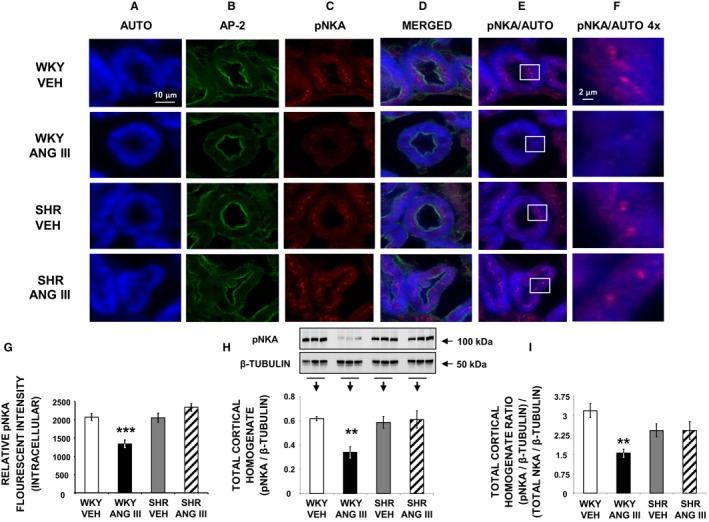
Confocal micrographs (×600 magnification) showing pSer23‐NKA (pNKA) in female WKY and SHR renal proximal tubule cell (RPTC) thin sections (5–8 μm) after renal interstitial infusion of vehicle (VEH) or angiotensin III (Ang III; 7.0 nmol/kg per min). As indicated, rows of images show representative sets of RPTCs from (top‐to‐bottom) WKY VEH, WKY Ang III, SHR VEH, and SHR Ang III treatment groups. As indicated, columns (left‐to‐right) depict confocal autofluorescence (A), subapical area staining with adaptor protein‐2 (AP‐2) (B), pNKA (C), merged image (D), merged image showing only autofluorescence and pNKA (E), and enlarged image (×4) of the square sections in (E) (F). Scale bars in the first and sixth columns represent 10 and 2 μm, respectively. G, The quantification of RPTC intracellular pNKA fluorescence intensity performed on 20 independent measurements of RPTCs from 1 rat for WKY VEH (white bar), WKY Ang III (black bar), SHR VEH (gray bar), and SHR Ang III (striped bar) treatments. H, Western blot analysis for total cortical homogenate pNKA in female WKY and SHR after VEH or Ang III (3.5–28.0 nmol/kg per min) infusions (N=6 for each condition). All blots are normalized to β‐tubulin. I, Ratio of (pNKA/β‐tubulin)/(α‐NKA/β‐tubulin) for all 4 conditions. Data represent mean±1 SE. **P<0.01 and ***P<0.001 compared with WKY VEH.
Discussion
The major new findings of this study are as follows: (1) Natriuretic responses to intrarenal interstitial administration of preferred AT2R agonist Ang III, while robust in 4‐week‐old normotensive male and female WKY controls, are absent in prehypertensive 4‐week‐old male and female SHR; (2) Ang III‐induced natriuretic responses are similar in 4‐week‐old male and female WKY; (3) Ang III‐induced natriuretic responses in 4‐week old WKY are mediated by renal AT2Rs; (4) Renal Ang III levels are normal in SHR compared with WKY at baseline and after intrarenal Ang III administration; (5) Ang III‐induced natriuretic responses in WKY are accompanied by translocation of AT2Rs from intracellular sites to apical plasma membranes of RPTCs, while Ang III‐induced AT2R translocation does not occur in SHR; and (6) Natriuretic responses to Ang III are accompanied by internalization and inactivation of major RPTC Na+ transporters NHE‐3 and NKA in WKY, but not in SHR. These results demonstrate an underlying defect in Ang III‐elicited and AT2R‐mediated natriuresis (Figure 9) in SHR that precedes the rise in BP and may be important in the pathophysiology of hypertension.
Figure 9.
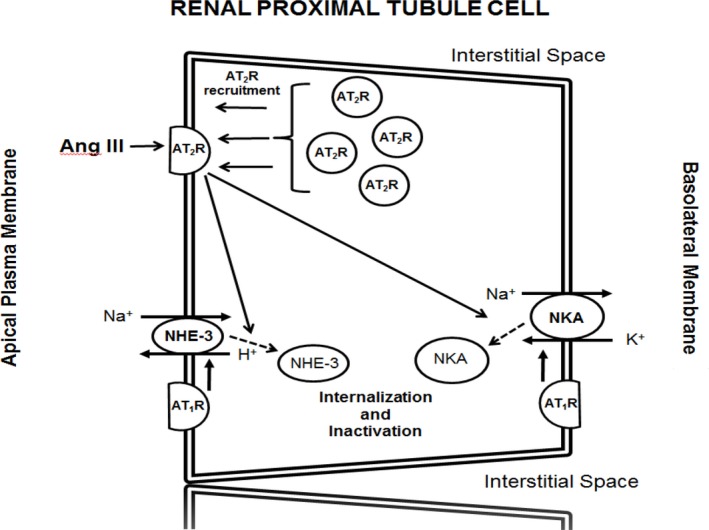
Schematic depiction of the molecular pathways in renal proximal tubule cells by which angiotensin III (Ang III) induces and sustains natriuresis in Wistar‐Kyoto (WKY) but not in spontaneously hypertensive (SHR) rats. Angiotensin type‐1 receptor (AT 1R); angiotensin type‐2 receptor (AT 2R); Na+‐H+ exchanger‐3 (NHE‐3); and Na+/K+ ATPase (NKA).
SHR are an inbred strain of rats that develop hypertension at a relatively early age and are widely used as a model of human primary hypertension.24 Young, prehypertensive SHR exhibit increased RPT Na+ reabsorption, and normal Na+ excretion is achieved only at the expense of elevated renal perfusion pressure.14, 15, 25, 26, 27, 28, 29 Evidence for this pathophysiologic principle is provided by observations that transplantation of prehypertensive kidneys from SHR into WKY induces hypertension in WKY and that both humans with primary hypertension and SHR excrete less Na+ and water than normotensive controls when renal perfusion pressure is lowered to normotensive levels.30, 31, 32 In addition, the chronic relationship between arterial pressure and UNaV is shifted towards higher BP in SHR compared with WKY, reflecting the kidneys’ adaptation to higher perfusion pressures.33
Our previous studies demonstrated defective Ang III‐induced natriuresis in 12‐week‐old hypertensive SHR that was accompanied by failure to translocate AT2Rs to the apical plasma membranes of RPTCs.16 The present study extends this finding to 4‐week‐old prehypertensive SHR and importantly demonstrates for the first time Ang III‐induced internalization and inactivation of major RPTC Na+ transporters NHE‐3 and NKA in WKY, but not in SHR. Activation of the renin–angiotensin system has been suggested as a major contributor to the excess Na+ retention of young SHR.34, 35, 36, 37, 38 Renal AT2Rs oppose the Na+‐retaining actions of AT1Rs,1, 2, 13 and we have previously documented defective Ang III/AT2R‐mediated natriuresis in adult hypertensive SHR.16 The current study further elucidates the mechanism of hypertension in SHR by demonstrating that the Ang III/AT2R defect is a characteristic of the SHR strain per se, independent of BP.
Previous studies have shown that natriuresis mediated by AT2R activation in normal as well as Ang III‐infused Sprague‐Dawley rats is consistently accompanied by receptor recruitment to the apical plasma membranes of RPTCs.16, 18, 39, 40 Our previous studies have shown that AT2R translocation occurs along microtubules via a cyclic AMP‐ and protein phosphatase 2A‐dependent mechanism.40 In the present study, while WKY displayed robust AT2R translocation, this was absent in young prehypertensive SHR. As demonstrated in WKY RPTCs, at least some AT2Rs are expressed on apical membranes at baseline, enabling initial agonist‐receptor binding and natriuresis. Over time, AT2R recruitment is likely to reinforce/sustain the biological response as AT2Rs fail to internalize, desensitize, and undergo degradation.13 The presence of some AT2Rs on apical plasma membranes of SHR at baseline, but failure of RPTCs to translocate AT2R in response to Ang III, suggests a receptor and/or postreceptor signaling defect in SHR. This defect in SHR is not because of reduced renal AT2R expression, because we observed similar levels of AT2R in WKY and SHR in this study.
The present study identifies the Na+ transport mechanism by which Ang III/AT2R induces natriuresis in young WKY as internalization/inactivation of NHE‐3 and NKA in the RPTC, and the absence thereof in SHR. Baseline NHE‐3 expression in renal cortical homogenates was significantly higher in SHR than WKY, consistent with previous observations.38 The internalization of NHE‐3 from the microvilli to the subapical region of the proximal tubule demonstrated here in WKY is consistent with electron microscopy immunocytochemical localization of NHE‐3 in response to AT2R activation in the Sprague‐Dawley rat kidney.18 The specific cell signaling mechanism mediating Na+ transporter inactivation in WKY was not investigated in this study, but a leading candidate is the bradykinin‐NO‐cGMP pathway that mediates natriuretic responses to AT2R activation in Sprague‐Dawley rat kidneys.18 Other potential pathways include inhibition of NAD(P)H oxidase, which potentiates AT2R agonist‐induced natriuresis in Sprague‐Dawley rats.41
Previous studies have demonstrated enhanced renal responses to AT2R activation in female as compared with male rodents.42 However, the results of the current study are consistent with our previous observations12, 18 demonstrating no significant difference between males and females in AT2R‐mediated natriuretic responses in WKY and their absence in SHR. The reasons for these differences are unclear but may be related to differences in rat strain and/or the experimental model with intrarenal rather than systemic infusion, or differences in renal hemodynamic versus tubule responses to AT2R activation. We have previously demonstrated that intrarenal Ang III administration does not alter glomerular filtration rate and that intrarenal AT2R activation induces natriuresis by RPT and not by hemodynamic mechanisms.12, 18 We conducted our Ang III measurements and AT2R and Na+ transporter cellular trafficking studies in female rats because there may be greater AT2R expression in female rats.43
We measured whole kidney tissue Ang II and III levels in WKY and SHR: baseline renal Ang III levels are normal in SHR compared with WKY and rise appropriately with Ang III infusion in SHR and WKY. The increase in renal Ang II during Ang III infusion may be because of, at least in part, product inhibition of aminopeptidase A, the enzyme responsible for cleaving Ang II to Ang III, but this will require further study. Nevertheless, Ang III infusion resulted in far greater increases in Ang III than Ang II in both WKY and SHR. The fact that renal Ang III levels increased in response to Ang III infusion similarly in WKY and SHR, but AT2R‐mediated natriuresis only occurred in WKY, is consistent with our hypothesis that a receptor or postreceptor signaling defect rather than accelerated Ang III metabolism is the cause of impaired natriuresis in SHR.
The present study has limitations. Since the experiments were conducted under AT1R blockade, the physiological importance of AT2R‐mediated natriuresis in WKY cannot be unambiguously discerned. Previous studies from our laboratory have demonstrated that AT2R activation induces natriuresis in the absence of AT1R blockade either when aminopeptidase N is blocked or exogenous nonpeptide AT2R agonist Compound 21 is administered.12, 18, 39
In conclusion, SHR have defective renal Ang III/AT2R signaling manifested by absent AT2R translocation to RPTC apical plasma membranes and lack of internalization/inactivation of major RPTC Na+ transporters NHE‐3 and NKA. The resulting Ang III/AT2R natriuretic defect in SHR is present before hypertension becomes established and does not appear to be because of accelerated Ang III metabolism. These findings provide a potential mechanism for increased RPT Na+ reabsorption and defective pressure‐natriuresis in SHR.
Author Contributions
Carey designed the study. Kemp, Howell, Gildea, and Shao carried out experiments. Carey, Keller, Kemp, and Navar analyzed the data. Kemp and Shao made the figures. Kemp, Carey, Keller, and Navar drafted and revised the paper. All authors approved the final version of the manuscript.
Sources of Funding
This research was supported by NIH grant R01‐HL‐128189 to Dr Carey.
Disclosures
None.
Acknowledgments
We thank Dr Peter Aronson (Yale University School of Medicine) for generously providing the NHE‐3 antibody used for Western blotting in this study. We thank Dr Mark Conaway (University of Virginia School of Medicine) for performing statistical analysis.
(J Am Heart Assoc. 2019;8:e012016 DOI: 10.1161/JAHA.119.012016.)
References
- 1. Karnik SS, Unal H, Kemp JR, Tirupula KC, Eguchi S, Vanderheyden PM, Thomas WE. International Union of Basic and Clinical Pharmacology. XCIX. Angiotensin receptors: interpreters of pathological angiotensinergic stimuli. Pharmacol Rev. 2015;67:754–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Forrester SJ, Booz GW, Sigmund CD, Coffman TM, Kawai T, Rizzo V, Scalia R, Eguchi S. Angiotensin II signal transduction: an update on mechanisms of physiology and pathophysiology. Physiol Rev. 2018;98:1627–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bosnyak S, Jones ES, Christopolous A, Aguilar MI, Thomas WG, Widdop RE. Relative affinity of angiotensin peptides and novel ligands at AT1 and AT2 receptors. Clin Sci (Lond). 2011;121:297–303. [DOI] [PubMed] [Google Scholar]
- 4. Batenburg WW, Garrelds IM, Bunasconi CL, Juillerat‐Jeanneret L, van Kats JP, Saxena PR, Danser AH. Angiotensin II type‐2 receptor‐mediated vasodilation in human coronary arteries. Circulation. 2004;109:2296–2301. [DOI] [PubMed] [Google Scholar]
- 5. Van Esch JH, Oosterveer CR, Batenburg WW, van Veghel R, Danser AH. Effects of angiotensin II and its metabolites in the rat coronary vascular bed: is angiotensin III the preferred ligand of the angiotensin AT2 receptor? Eur J Pharmacol. 2008;588:286–293. [DOI] [PubMed] [Google Scholar]
- 6. Yatabe J, Yoneda M, Yatabe MS, Watanabe T, Felder RA, Jose PA, Sanada H. Angiotensin III stimulates aldosterone secretion from adrenal gland partially via angiotensin II type‐2 receptor but not angiotensin II type‐1 receptor. Endocrinology. 2011;152:1582–1588. [DOI] [PubMed] [Google Scholar]
- 7. Dominska K, Piastowska‐Ciesielska AW, Lachowicz‐Ochedalske T. Similarities and differences between effects of angiotensin III and angiotensin II on human prostate cancer cell migration and proliferation. Peptides. 2012;37:200–206. [DOI] [PubMed] [Google Scholar]
- 8. Park BM, Oh Y‐B, Gao S, Cha SA, Kang KP, Kim SH. Angiotensin III stimulates high stretch‐induced ANP secretion via angiotensin II type 2 receptor. Peptides. 2013;42:131–137. [DOI] [PubMed] [Google Scholar]
- 9. Padia SH, Howell NL, Siragy HM, Carey RM. Renal angiotensin type 2 receptors mediate natriuresis via angiotensin III in the angiotensin type 1 receptor‐blocked rat. Hypertension. 2006;47:537–544. [DOI] [PubMed] [Google Scholar]
- 10. Padia SH, Kemp BA, Howell NL, Siragy HM, Fournie‐Zaluski M‐C, Roques BP, Carey RM. Intrarenal aminopeptidase N inhibition augments natriuretic responses to angiotensin III in angiotensin type 1 receptor‐blocked rats. Hypertension. 2007;49:625–630. [DOI] [PubMed] [Google Scholar]
- 11. Padia SH, Kemp BA, Howell NL, Fournie‐Zaluski M‐C, Roques BP, Carey RM. Conversion of renal angiotensin II to angiotensin III is critical for AT2 receptor‐induced natriuresis in rats. Hypertension. 2008;51:460–465. [DOI] [PubMed] [Google Scholar]
- 12. Kemp BA, Bell JF, Rottkamp DM, Howell NL, Shao W, Navar LG, Padia SH, Carey RM. Intrarenal angiotensin III is the predominant agonist for proximal tubule angiotensin type 2 receptors. Hypertension. 2012;60:387–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Carey RM. AT2 receptors: potential therapeutic targets for hypertension. Am J Hypertens. 2017;30:339–347. [DOI] [PubMed] [Google Scholar]
- 14. Guyton AC, Coleman TG, Young DB, Lohmeier TE, DeClue JW. Salt balance and long‐term blood pressure control. Annu Rev Med. 1908;31:15–27. [DOI] [PubMed] [Google Scholar]
- 15. Guyton AC, Coleman TG, Cowley AV Jr, Scheel KW, Manning RD Jr, Norman RA Jr. Arterial pressure regulation. Overriding dominance of the kidneys in long‐term regulation and in hypertension. Am J Med. 1972;52:584–594. [DOI] [PubMed] [Google Scholar]
- 16. Padia SH, Kemp BA, Howell NL, Gildea JJ, Keller SR, Carey RM. Intrarenal angiotensin III infusion induces natriuresis and AT2 receptor translocation in WKY but not in SHR. Hypertension. 2009;53(part 2):338–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shao W, Seth DM, Prieto MC, Kobori H, Navar LG. Activation of the renin‐angiotensin system by a low‐salt diet does not augment intratubular angiotensinogen and angiotensin II in rats. Am J Physiol Renal Physiol. 2013;304:F505–F514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kemp BA, Howell NL, Gildea JJ, Keller SR, Padia SH, Carey RM. AT2 receptor activation induces natriuresis and lowers blood pressure. Circ Res. 2014;115:388–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Biemesderfer D, Pizzonia J, Abu‐Alfa A, Exner M, Reilly R, Igarashi P, Aronson PS. NHE3: a Na+/H+ exchanger isoform of renal brush border. Am J Physiol Renal Physiol. 1993;265:F736–F742. [DOI] [PubMed] [Google Scholar]
- 20. Riquier AD, Lee DH, McDonough AA. Renal NHE3 and NaPi2 partition into distinct membrane domains. Am J Physiol Cell Physiol. 2009;296:C900–C910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Donowitz M, Li X. Regulatory binding partners and complexes of NHE3. Physiol Rev. 2007;87:825–872. [DOI] [PubMed] [Google Scholar]
- 22. Moe OW, Amemiya M, Yamaji Y. Activation of protein kinase A acutely inhibits and phosphorylates N/H exchanger NHE‐3. J Clin Invest. 1995;96:2187–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Logvinenko NS, Dalubova I, Fedosova N, Larsson SH, Nairn AC, Esmann M, Greengard P, Aperia A. Phosphorylation by protein kinase C of serine‐23 of the alpha‐1 subunit of rat Na+/K+‐ATPase affects its conformational equilibrium. Proc Natl Acad Sci USA. 1996;93:9132–9137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Trippodo NC, Frohlich ED. Similarities of genetic (spontaneous) hypertension: man and rat. Circ Res. 1981;48:309–319. [DOI] [PubMed] [Google Scholar]
- 25. Nagaoka A, Kakihana M, Shibota M, Fujiwara K, Shimakawa K. Reduced sodium excretory ability in spontaneously hypertensive rats. Jpn J Pharmacol. 1982;32:839–844. [DOI] [PubMed] [Google Scholar]
- 26. Heckmann U, Zidek W, Schurek HJ. Sodium reabsorption in the isolated perfused kidney of normotensive and spontaneously hypertensive rats. J Hypertens Suppl. 1989;7:S172–S173. [DOI] [PubMed] [Google Scholar]
- 27. Firth JD, Raine AE, Ledingham JG. Sodium and lithium handling in the isolated hypertensive rat kidney. Clin Sci (Lond). 1989;76:335–341. [DOI] [PubMed] [Google Scholar]
- 28. Christiansen RE, Roald AB, Tenstad O, Iversen BM. Renal hemodynamics during development of hypertension in young spontaneously hypertensive rats. Kidney Blood Press Res. 2002;25:322–328. [DOI] [PubMed] [Google Scholar]
- 29. Beierwaltes WH, Arendshorst WJ, Klemmer PJ. Electrolyte and water balance in young spontaneously hypertensive rats. Hypertension. 1982;4:908–915. [DOI] [PubMed] [Google Scholar]
- 30. Kawabe K, Watanabe TX, Shiono K, Solabe H. Influence on blood pressure of renal isografts between spontaneously hypertensive and normotensive rats. Jpn Heart J. 1978;19:886–894. [DOI] [PubMed] [Google Scholar]
- 31. Omvik P, Tarazi RC, Bravo EL. Regulation of sodium balance in hypertension. Hypertension. 1980;2:515–523. [DOI] [PubMed] [Google Scholar]
- 32. Arendshorst WJ, Beierwaltes WH. Renal tubular reabsorption in spontaneously hypertensive rats. Am J Physiol Renal Physiol. 1979;237:F38–F47. [DOI] [PubMed] [Google Scholar]
- 33. Norman RA Jr, Enobakhare JA, DeClue JW, Douglas BH, Guyton AC. Arterial pressure‐urinary output relationship in hypertensive rats. Am J Physiol. 1978;234:R98–R103. [DOI] [PubMed] [Google Scholar]
- 34. Cheng H‐F, Wang J‐L, Vinson GP, Harris RC. Young SHR express increased type‐1 angiotensin II receptors in renal proximal tubule. Am J Physiol Renal Physiol. 1998;274:F10–F17. [DOI] [PubMed] [Google Scholar]
- 35. Matsushima Y, Kawamura M, Akabane S, Imanishi M, Kuramochi M, Ito K, Omai T. Increases in renal angiotensin II content and tubular angiotensin II receptors in prehypertensive spontaneously hypertensive rats. J Hypertens. 1998;6:791–796. [PubMed] [Google Scholar]
- 36. Correa FM, Viswanathan M, Ciuffo GM, Tsusumi K, Saavedra JM. Kidney angiotensin II receptors and converting enzyme in neonatal and adult Wistar‐Kyoto and spontaneously hypertensive rats. Peptides. 1995;16:19–24. [DOI] [PubMed] [Google Scholar]
- 37. Aldred KL, Harris PJ, Eide E. Increased proximal tubule NHE‐3 and H+‐ATPase activities in spontaneously hypertensive rats. J Hypertens. 2000;18:623–628. [DOI] [PubMed] [Google Scholar]
- 38. LaPointe MS, Sodhi C, Sahai A, Battle D. Na+/H+ exchange activity and NHE‐3 expression in renal tubules from spontaneously hypertensive rats. Kidney Int. 2002;62:157–165. [DOI] [PubMed] [Google Scholar]
- 39. Kemp BA, Howell NL, Keller SR, Gildea JJ, Padia SH, Carey RM. AT2 receptor activation prevents sodium retention and reduces blood pressure in angiotensin II‐dependent hypertension. Circ Res. 2016;119:532–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Padia SH, Kemp BA, Howell NL, Keller SR, Gildea JJ, Carey RM. Mechanism of dopamine D(1) and angiotensin type 2 receptor interaction in natriuresis. Hypertension. 2012;59:437–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sabuhi R, Asghar M, Hussain T. Inhibition of NAD(P)H oxidase potentiates AT2 receptor agonist‐induced natriuresis in Sprague‐Dawley rats. Am J Physiol Renal Physiol. 2010;299:F815–F820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mirabito KM, Hilliard LM, Kett MM, Brown RD, Booth SC, Widdop RE, Moritz KM, Evans RG, Denton KM. Sex‐ and age‐related differences in the chronic pressure‐natriuresis relationship: role of the angiotensin type‐2 receptor. Am J Physiol Renal Physiol. 2014;76:448–452. [DOI] [PubMed] [Google Scholar]
- 43. Peluso AAB, Santos RAS, Unger R, Steckelings UM. The angiotensin type 2 receptor and the kidney. Curr Opin Nephrol Hypertens. 2017;26:36–42. [DOI] [PubMed] [Google Scholar]