

Abstract
Background
Platelets are the cellular mediators of hemostasis and thrombosis, and their function is regulated by a number of molecular mediators, such as small GTPases. These small GTPases are themselves regulated by guanine nucleotide exchange factors such as Arhgefs, several of which are found in platelets, including the highly expressed Arhgef1. However, the role of Arhgef1 in platelets has not yet been investigated.
Methods and Results
We employed mice with genetic deletion of Arhgef1 (ie, Arhgef1−/−) and investigated their platelet phenotype by employing a host of in vivo and in vitro platelet assays. Our results indicate that Arhgef1−/− mice had prolonged carotid artery occlusion and tail bleeding times. Moreover, platelets from these mice exhibited defective aggregation, dense and α granule secretion, αIIbβ3 integrin activation, clot retraction and spreading, in comparison to their wild‐type littermates. Finally, we also found that the mechanism by which Arhgef1 regulates platelets is mediated in part by a defect in the activation of the RhoA–Rho‐associated kinase axis, but not Rap1b.
Conclusions
Our data demonstrate, for the first time, that Arhgef1 plays a critical role in platelet function, in vitro and in vivo.
Keywords: Arhgef1, cardiovascular diseases, platelets, small GTPases, thrombosis
Subject Categories: Basic Science Research, Platelets, Translational Studies
Clinical Perspective
What Is New?
This is the first study to demonstrate that Arhgef1 regulates platelet function, as well as hemostasis and thrombogenesis, in mice.
What Are the Clinical Implications?
Arhgef1 represents a potential therapeutic target for managing thrombotic disorders.
Platelets are the cellular mediators of hemostasis and thrombosis.1, 2, 3 Under normal conditions and upon injury to a blood vessel, platelets undergo drastic shape change, bind to adhesive protein substrates, and aggregate to form a plug, ultimately halting bleeding.3, 4, 5 Unfortunately, hyperactivity of platelets serves as a precursor to thrombotic diseases such as pulmonary embolism and myocardial infarction.6, 7
Platelet activation is regulated by a host of complex molecular events that modulate adhesion, aggregation, and thrombus formation, and include phosphorylation,8, 9, 10 as well as calcium‐based11 and lipid‐based signals.12, 13 These events are coordinated by a number of molecular mediators such as small GTPases,14, 15, 16 which represent a family of low‐molecular‐weight (21–25 kDa) monomeric guanine nucleotide–binding proteins. These proteins work as intracellular “on‐off” signaling switches; coordinating signal transduction and amplification.17, 18 Several subfamilies of small GTPases have been identified in platelets19, 20 and are cardinal regulators of the actin cytoskeleton dynamics, affecting platelet aggregation, secretion, spreading, and thrombus formation.4, 21 They govern the diverse cellular processes through cycling between an inactive GDP‐bound form and an active GTP‐bound form through a process of GDP/GTP replacement.22, 23 Importantly, the GDP/GTP cycling is highly regulated by a group of proteins/protein domains known as guanine nucleotide exchange factors (GEFs) such as Arhgefs.23, 24, 25
While the Rho family of GEFs––a few of which have been identified in platelets26––has been thoroughly studied in other cell systems,27, 28, 29 their role in platelets has not yet been fully understood. To this end, it has been reported that: (1) Arhgef3 is not very important for platelet function, at least in mice30; (2) Arhgef10 regulates platelet activation in vitro (eg, aggregation) and in vivo (eg, hemostasis)31; and (3) Arhegf12 is “essential” for normal platelet function.32 While Arhgef1 is another member of this family that is highly expressed in platelets,26 virtually nothing is known regarding its direct role in platelet function. Given that RhoA is a crucial regulator of platelet responses mainly activated downstream of G12/13 and Gq coupled receptors,33 that these G‐proteins are known to play a vital role in platelet function,34, 35 and that Arhgef1 activates G13,36 we sought to examine the role of Arhgef1 in platelet function in vitro and in vivo.
Materials and Methods
Reagents and Materials
Thrombin, collagen, stir bars, and other disposables were from Chrono‐Log Corporation, whereas U46619 was purchased from Abcam. Fluorescein isothiocyanate–conjugated anti–P‐selectin, anti‐Arhgef1, anti‐actin, anti‐RhoA, anti‐pROCK, and anti‐Rap1b antibodies were purchased from Cell Signaling Technology, Inc, whereas the anti‐β3 and anti‐G13 antibodies were from Sigma Aldrich. The JON/A antibody was from Emfret analytics. Fibrinogen was purchased from Sigma Aldrich. The RhoA and Rap1b Activation Assay Biochem Kits were from Cytoskeleton Inc. Other reagents were of analytical grade.
Animals and Genotyping
Arhgef1−/− mice were obtained from the Mary Lyon Centre at MRC Harwell (www.har.mrc.ac.uk), Oxfordshire, United Kingdom, and genotyped using a polymerase chain reaction (PCR)–based method. PCR was performed using the following primers: Arhgef1‐F (CCTTTGGGCTCTGACCTCTG), Arhgef1‐R (AAGCCGTCAGTCCTCTGTCC), and CAS1 (TCGTGGTATCGTTATGCGCC) with the following PCR condition: denaturation: 94°C for 7 minutes, amplification: 94°C for 1 minute, 58°C for 1 minute, 72°C for 90 seconds for 34 cycles, and finally extension at 72°C for 10 minutes. After completion of the PCR cycle, amplified DNA samples were run in 2% agarose gel and visualized in the gel documentation system. The expected products per genotype are as follows: wild‐type (WT): 520 bp, Het: 362/520 bp, and mutant: 362 bp. Mice were housed in groups of 1 to 5 at 24°C, under 12/12 light/dark cycles, with access to water and food ad libitum.
Methods
All experiments involving animals were performed in compliance with the institutional guidelines and were approved by The University of Texas at El Paso Institutional Animal Care and Use Committee.
The authors declare that all supporting data are available within the article data. Requests for detailed analytic methods will be addressed by the corresponding author. For requests for the Arhgef1 deletion mice, these will be referred by the corresponding author to the Mary Lyon Centre at MRC Harwell (www.har.mrc.ac.uk), Oxfordshire, United Kingdom, and handled as per our Material Transfer Agreement.
Immunoblotting
Immunoblotting was performed as previously described.37, 38 Briefly, and to confirm Arhgef1 deletion and that its deletion did not impact the expression levels of β3 and G13, platelet proteins were separated by SDS‐PAGE and transferred to Immobilon‐P PVDF membranes (Millipore Corp). They were then probed with anti‐Arhgef1, anti‐β3, anti‐G13, or anti‐actin primary antibodies and visualized with an appropriate alkaline phosphatase–coupled secondary antibody using enhanced chemifluorescent substrate (GE Healthcare). For the RhoA–Rho‐associated kinase (ROCK) axis, washed platelets (Arhgef1−/− and WT) were stimulated with thrombin (0.1 U/mL) for 3 minutes, and processed as described above. Membranes were then probed with anti‐RhoA, anti‐pROCKIISer1366, and anti‐actin antibodies, and visualized with an appropriate horseradish peroxidase–coupled secondary antibody using enhanced chemiluminescence substrate (Thermo Scientific). Images were obtained with ChemiDoc MP Imaging System (Bio‐Rad).
Tail Bleeding Time Assay
The tail bleeding time assay was performed as previously described.39, 40, 41 Briefly, mice were anesthetized and placed on a homeothermic blanket (37°C) before the tail was transected 5 mm from the tip using a sterile scalpel. After transection, the tail was immediately immersed in saline (37°C constant temperature), and the time to bleeding cessation was measured. Bleeding stoppage was not considered complete until bleeding had stopped for 1 minute. For statistical analysis purposes, if bleeding did not stop in 10 minutes, the experiment was halted to limit blood loss from the mice and 10 minutes was considered the cutoff bleeding time. The number of WT and knockout (KO) mice used was 8 each.
In Vivo Ferric Chloride Carotid Artery Injury–Induced Thrombosis Model
This assay was performed as previously described.39, 40, 41 Briefly, mice were anesthetized with Avertin (2.5%), and the left carotid artery was exposed and cleaned with normal saline (37°C) before baseline carotid artery blood flow was measured with Transonic Micro‐Flow Probe (Transonic Systems Inc). After stabilizing blood flow, 7.5% ferric chloride was applied to a filter paper (1‐mm diameter) that was immediately placed on top of the carotid artery for 3 minutes. Blood flow was continuously monitored until it stopped completely (stable occlusion). Data were recorded as the time to vessel occlusion and calculated as the difference in time between stable occlusion and removal of the filter paper (with ferric chloride). An occlusion time of 20 minutes was considered as the cutoff time for statistical analysis. The number of WT and KO mice used was 8 each.
Peripheral Blood Cell Counts
Peripheral blood cell counts were performed on whole blood obtained from both Arhgef1−/− and WT mice, using a HEMAVET 950FS Multispecies Hematology System from Erba Diagnostics.
Murine Platelet‐Rich Plasma Preparation
The Arhgef1−/− and WT mice were anesthetized, and blood was collected from the heart. Coagulation was inhibited by 0.38% sodium citrate solution (Fisher Scientific). Blood was centrifuged (237g for 15 minutes) at room temperature (RT), and the platelet‐rich plasma was then collected. Platelets were counted with the HEMAVET 950FS Multispecies Hematology System, and the counts were adjusted to 7×107 platelets per mL before each experiment.
Washed Platelets Preparation
Mouse blood was collected as discussed above, mixed with phosphate‐buffered saline, pH 7.4, and incubated with prostaglandin I2 (10 ng/mL; 5 minutes), followed by centrifugation at 237g for 10 minutes at RT. Platelet‐rich plasma was recovered and platelets were pelleted at 483g for 10 minutes at RT. The pellets were resuspended in HEPES/Tyrode buffer (20 mmol/L HEPES/potassium hydroxide, pH 6.5, 128 mmol/L NaCl, 2.8 mmol/L KCl, 1 mmol/L MgCl2, 0.4 mmol/L NaH2PO4, 12 mmol/L NaHCO3, 5 mmol/L d‐glucose) supplemented with 1 mmol/L EGTA, 0.37 U/mL apyrase and 10 ng/mL prostaglandin I2. Platelets were then washed and resuspended in HEPES/Tyrode (pH 7.4) without EGTA, apyrase, or prostaglandin I2. Platelets were counted using the HEMAVET 950FS Multispecies Hematology System and adjusted to the indicated concentrations.
In Vitro Platelet Aggregation
Platelets from both Arhgef1−/− and WT mice were activated with the thromboxane receptor agonist U46619 (2.5 μmol/L), thrombin (0.05–0.1 U/mL), or collagen (5 μg/mL) and their aggregation response was measured by the turbidometric method using model 700 aggregometer (Chrono‐Log Corporation). Each experiment was repeated 3 times using 3 separate groups, with blood pooled from 8 mice per group. Comparison is based on difference in maximal aggregation.
ATP Release
This assay was performed as previously described.39, 40, 41 Platelets were prepared as described above (250 μL; 7×107/mL) before being placed into siliconized cuvettes and stirred for 5 minutes at 37°C. The luciferase substrate/luciferase mixture (12.5 μL, Chrono‐Log) was then added, followed by the addition of the agonists U46619 (2.5 μmol/L), thrombin (0.05–0.1 U/mL), or collagen (5 μg/mL). Each experiment was repeated 3 times using 3 separate groups, with blood pooled from 8 mice per group. Comparison is based on difference in maximal secretion.
Flow Cytometric Analysis
Flow cytometry analysis was performed as previously described.39, 40, 41 Briefly, platelets (2×107/mL) from Arhgef1−/− and WT mice were stimulated with U46619 (2.5 μmol/L), thrombin (0.1 U/mL), or collagen (5 μg/mL) for 5 minutes. Platelets were then fixed with 2% formaldehyde for 30 minutes at RT and incubated with fluorescein isothiocyanate–conjugated CD62P (P‐selectin) or PE‐conjugated rat anti‐mouse integrin αIIbβ3 (active form) JON/A antibodies at RT for 30 minutes in the dark. The platelet (105 platelets/100 μL) fluorescent intensities were measured using an Accuri C6 Flow Cytometer (BD Biosciences). Results were analyzed using C‐Flow Plus (BD Biosciences). Each experiment was repeated 3 times using 3 separate groups, with blood pooled from 8 mice per group. Comparison is based on difference in mean fluorescent intensity.
Fibrin Clot Retraction Assay
Clot retraction assay was performed as previously described.42 Briefly, whole blood was collected and washed platelets were isolated as discussed above. CaCl2 was added extemporaneously, at a final concentration of 1 mmol/L. The glass tubes that were used for aggregation (Chrono‐Log Corporation) were employed for retraction assays. The washed platelets were resuspended at 1×108/mL in HEPES‐Tyrode buffer (pH 7.4). Fibrinogen (500 μg/mL) was added in 0.5‐mL platelets aliquots, and clot retraction was initiated by quickly adding thrombin (0.1 U/mL). The reaction was transferred to the glass tube and the reaction was set at RT. Pictures were taken at time intervals of 5 minutes up to half an hour using a digital camera. This experiment was repeated at least 3 times, with blood pooled from a group of 8 mice each time. Comparison is based on difference in clot size.
Platelet Spreading
The spreading of the Arhgef1−/− and WT platelets after stimulation with thrombin (0.1 U/mL) was examined as previously described.43 Briefly, sterile glass coverslips were coated with 0.2 μg/mL of fibrinogen for 30 minutes at RT. Washed platelets were placed onto these fibrinogen‐coated coverslips for 5, 30, and 45 minutes before they were fixed with 3.7% (vol/vol) formaldehyde for 15 minutes and quenched with 50 mmol/L ammonium chloride. Cells were rinsed with PBS and incubated with tetramethylrhodamine‐conjugated phalloidin (1 μg/mL) in 10% fetal bovine serum/PBS with 0.2% saponin. Coverslips were mounted and examined and imaged using a Leica DMi8 inverted widefield fluorescence microscope with integrated high‐precision focus drive. Images were processed using LAS X Wizard imaging software. Objective lenses used were a ×63/×100 numeric aperture. Type A immersion oil (Fryer) was used for the ×63 objective. This experiment was repeated at least 3 times, with blood pooled from a group of 8 mice each time. Comparison is based on difference in level of spreading.
Small‐GTPase Pull‐Down Assay
The small GTPase pulldown assay was performed using the small GTPase pulldown kit according to the manufacturer's instructions. Briefly, platelets (Arhgef1−/− and WT) were stimulated with thrombin (0.1 U/mL) for 0, 30, 60, 180, and 300 seconds and reactions were stopped with the lysis buffer. The lysates were cleared by centrifugation, and the supernatants were incubated with either rhotekin RBD for RhoA or GST‐humanRalGDSRBD (788–885) for Rap1b for 1 hour at RT. Next, we collected the protein‐bound bead by centrifugation at 3000g for 1 minute. The bead‐bound complexes were washed 3 times with wash buffer. Finally, bead bound‐complexes were eluted using 2× sample buffer, separated by 12% SDS‐PAGE, and transferred to Immobilon‐P PVDF membranes (Millipore Corp). They were then probed with the indicated primary antibodies and visualized with an appropriate horseradish peroxidase–coupled secondary antibody using enhanced chemiluminescence substrate (Thermo Scientific). Images were obtained with a ChemiDoc MP Imaging System (Bio‐Rad). This experiment was repeated 3 times using 3 separate groups, with blood pooled from 8 mice per group. Comparison is based on difference in RhoA and Rap1b activation.
Statistical Analysis
All experiments were performed 3 times using 3 separate groups, with blood pooled from 8 mice per group, as applicable. Data analysis was performed using GraphPad PRISM statistical software and presented as mean±SD. Mann–Whitney and t tests were used for the evaluation of differences in mean occlusion and bleeding times, whereas t test was used for analysis of the flow cytometry data. Notably, time‐to‐event analyses were used to confirm the bleeding and occlusion time analysis/results, given that the data were censored, and the findings confirm our original results. Significance was accepted at P<0.05.
Results
Confirmation of Arhgef1 Deletion
Given the important contribution of Arhgef1 in the activation of the G13‐RhoA axis44, 45, 46 and G13 in platelet function and thrombogenesis,34 we sought to investigate the direct role Arhgef1 plays in platelet biology. In these studies, we employed mice with Arhgef1 deletion (Arhgef1−/−). We first confirmed the successful deletion of the Arhgef1 gene (genotype) in our mice by PCR analysis of genomic DNA (Figure 1A), as well as by immunoblot (Figure 1B). Notably, the Arhgef1 KO mice appeared healthy and showed no physical differences in comparison to their WT littermates, and produced viable and healthy offspring, nor did the deletion of Arhgef1 impact the expression levels of G13 or the integrin β3 subunit (Figure 1C).
Figure 1.
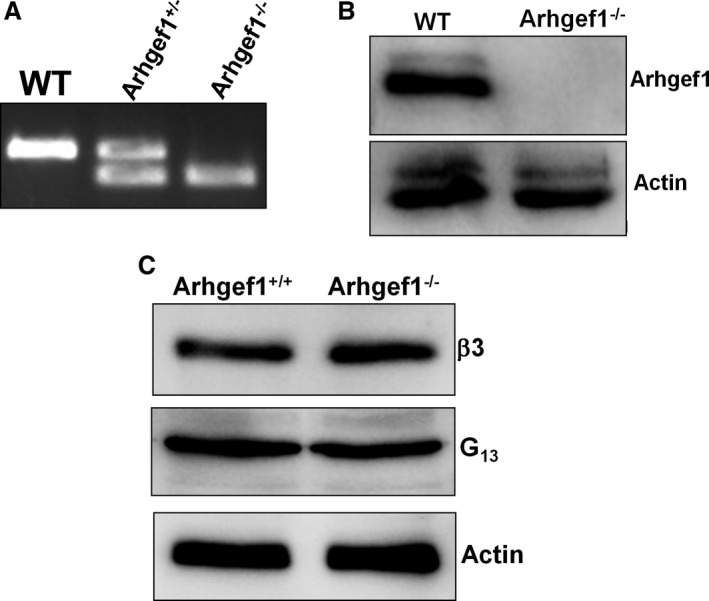
Confirmation of Arhgef1 deletion in mice. A, DNA from Arhgef1 KO and WT mice was isolated and polymerase chain reaction was performed as described in the Methods section. DNA was separated on a 2% agarose gel and visualized using a gel documentation system. B, Arhgef1 protein levels in wild‐type (WT) and Arhgef1−/− mice as determined by Western blotting using platelet extracts (2×108/mL). C, β3 is the subunit of the αIIbβ3 integrin, whereas G13 is the α subunit of the GTPase‐binding protein G13.
Arhgef1 Deletion Alters Physiological Hemostasis and Thrombogenesis
We subsequently investigated whether Arhgef1 regulates thrombogenesis by employing the ferric chloride carotid‐injury model. The Arhgef1−/− mice were found to exhibit dramatically prolonged occlusion times, compared with their WT littermates mice (Figure 2A; median 217.5 and 803, whereas the interquartile range was 73.2 and 193 for WT and KO mice, respectively), indicating that these mice are protected against thrombus formation. In light of this finding, we next sought to examine the role of Arhgef1 in hemostasis by measuring the tail bleeding time. It was observed that the time needed for cessation of bleeding following tail transection was significantly prolonged in the Arhgef1−/− mice, relative to the WT (Figure 2B; median 50 and 363, whereas the interquartile range was 52.5 and 341.5 for WT and KO mice, respectively). These data support the notion that Arhgef1 is essential for normal physiological hemostasis. Together, our findings suggest that Arhgef1 is a key player in in vivo platelet function.
Figure 2.
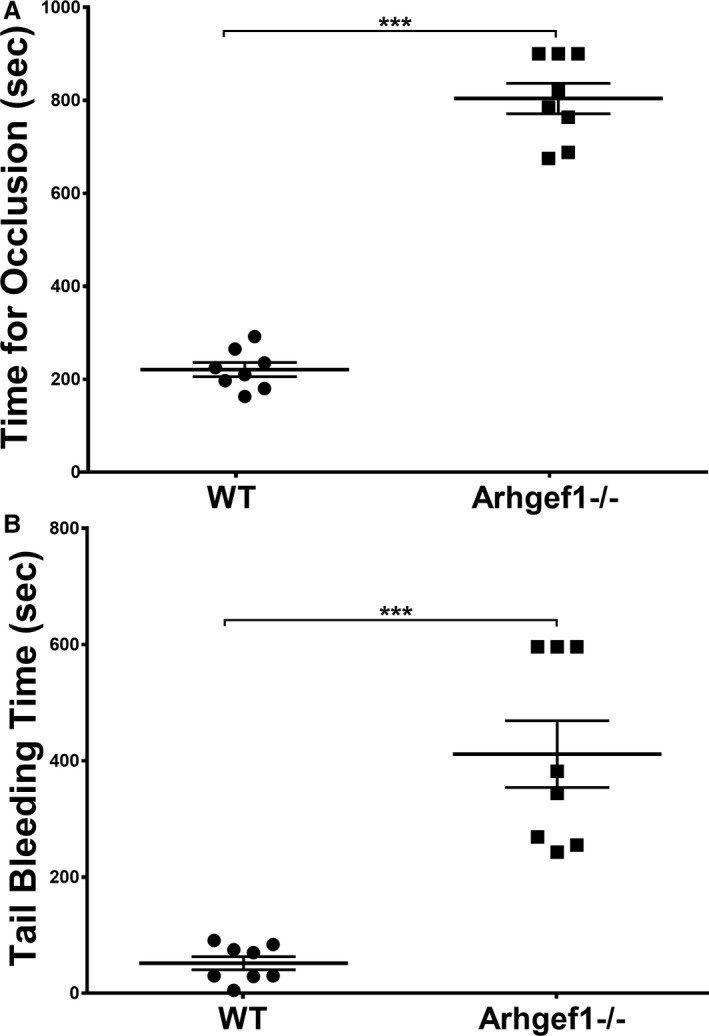
Arhgef1 deletion prolongs thrombus occlusion and bleeding times in mice. (A) Thrombosis was induced in Arhgef1−/− (n=8) and wild‐type (WT; n=8) mice using ferric chloride, as described in the Methods section. Each point represents the occlusion time of a single animal (***P<0.001). B, Bleeding times were measured in Arhgef1−/− (n=8) or WT (n=8) mice as described in the Methods section. Each point represents the bleeding time of a single animal (***P<0.001).
Arhgef1 Deletion Does Not Affect Thrombopoiesis
To ensure that the observed defects in thrombogenesis and hemostasis are not caused by abnormalities in platelet number, the latter were analyzed in the Arhgef1−/− and WT mice in whole blood. Our data revealed no difference in platelet counts between the KO and WT mice (Table), nor were there any differences in the platelet mean volume or other blood cell counts (Table). These data suggest that the phenotype observed in the Arhgef1 deletion mice was caused by defects in platelet function. This issue was next investigated using a host of assays of platelet function.
Table 1.
Peripheral Blood Cell Counts in WT and Arhgef1 KO Mice
| WT | Arhgef1−/− | P Values | |
|---|---|---|---|
| Platelets | 1133±57 | 1081±42 | 0.196 |
| MPV | 4.56±2.66 | 4.74±2.71 | 0.1484 |
| Red blood cells | 8.55±1.28 | 8.65±1.39 | 0.176 |
| Lymphocytes | 6.44±1.58 | 6.75±1.41 | 0.148 |
| Monocytes | 0.46±0.036 | 0.41±0.027 | 0.184 |
| Granulocytes | 2.49±0.23 | 2.32±0.25 | 0.165 |
Blood was collected from the heart and counted as described in the Methods section. All counts are expressed as thousands per microliter, except for red blood cells, which are expressed as millions per microliter. Data are presented as mean±SD. KO indicates knockout; MPV, mean platelet volume; WT, wild‐type.
Arhgef1 Deletion Inhibits Agonist‐Induced Platelet Aggregation
We first examined the role of Arhgef1 in platelet aggregation in response to activation of some of the major G protein–coupled receptors (GPCRs), particularly those coupled to Gq and G13. It was observed that platelets from Arhgef1−/− mice exhibited lower aggregation, in comparison with their WT littermates, in response to the protease‐activated receptor agonist thrombin (0.05–0.1 U/mL; Figure 3A and 3B; inset shows quantification of maximal aggregation), indicating a defective aggregation response. Furthermore, reduced aggregation was also found in response to the thromboxane A2 receptor agonist U46619 (2.5 μmol/L; Figure 3C; inset shows quantification of maximal aggregation). We next extended our studies to the agonist collagen, which acts through the non‐GPCR glycoprotein VI; the latter is not known to directly associate with G13. Interestingly, we again observed impaired aggregation in the Arhgef1 deletion platelets, relative to those from the WT mice (Figure 3D; inset shows quantification of maximal aggregation). Collectively, these findings indicate that Arhgef1 does indeed regulate platelet aggregation, including that which is not GPCR‐mediated, and explains, at least in part, the in vivo phenotype.
Figure 3.
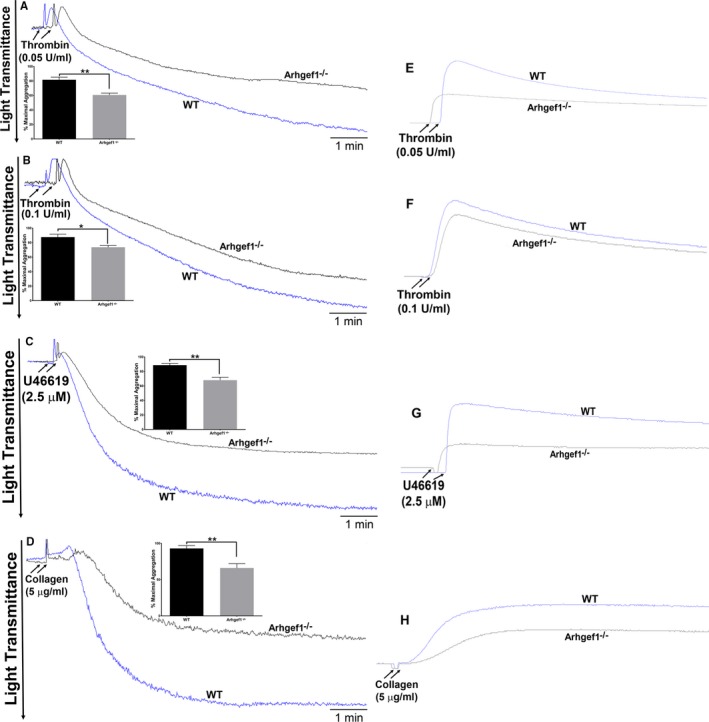
Arhgef1 deletion reduces platelets aggregation and dense granule secretion. A through D, Platelets from Arhgef1−/− and wild‐type (WT) mice were stimulated with thrombin (A and B), U46619 (C), or collagen (D) before their aggregation response was monitored using an aggregometer (inset shows quantification of maximal aggregation). E through H, Platelets from Arhgef1−/− and WT mice were incubated with luciferase luciferin (12.5 μL) before being stimulated with thrombin (E and F), U46619 (G), or collagen (H). ATP release was detected as luminescence and measured by a lumiaggregometer. Each experiment was repeated at least 3 times, with blood pooled from a group of 8 mice each time (*P<0.05; **P<0.01).
Arhgef1 Deletion Inhibits Agonist‐Induced Platelet Secretion
Given the important role of agonist‐induced exocytosis (granules release) in amplifying initial platelet activation events,47, 48 we studied the impact of Arhgef1 deletion on dense (ATP) and α‐granules (P‐selectin) secretion/release. Consistent with the aggregation data, the Arhgef1−/− mice had significantly lower thrombin‐induced ATP secretion (0.05–0.1 U/mL; Figure 3E and 3F) and P‐selectin expression (0.1 U/mL; Figure 4A), as well as reduced U46619‐trigerred ATP secretion (2.5 μmol/L; Figure 3G) and P‐selectin expression (2.5 μmol/L; Figure 4B), compared with the WT mice. Similar results were observed with the non‐GPCR agonist collagen (5 μg/mL; Figures 3H and 4C). These data provide evidence that Arhgef1 is essential for the platelet secretion response.
Figure 4.
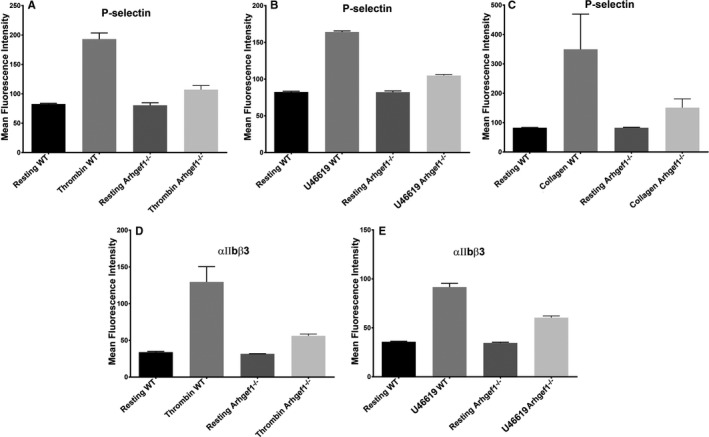
Arhgef1 deletion reduces α‐granule secretion and integrin αIIbβ3 activation. Platelets from Arhgef1−/− and wild‐type (WT) mice were washed before stimulation with thrombin, U46619, or collagen, and incubation with (A through C) fluorescein isothiocyanate–conjugated CD62P antibody (for α granules), or (D through F) fluorescein isothiocyanate–conjugated JON/A antibody, respectively. The fluorescent intensities were measured by flow cytometry. Average mean fluorescence intensities are shown. Each experiment was repeated at least 3 times, with blood pooled from a group of 8 mice each time.
Arhgef1 Deletion Reduces Agonist‐Induced Integrin αIIbβ3 Activation
Activation of integrin GPIIb‐IIIa (αIIbβ3) is required for platelet aggregation49 and involves conformational changes of the latter upon agonist activation.50 Since aggregation was defective in the Arhgef1 deletion platelets, we investigated whether there is a commensurate defect in integrin αIIbβ3 activation. Indeed, as one might expect, it was found that 0.1 U/mL thrombin‐ and 2.5 μmol/L U46619‐induced activation of integrin αIIbβ3 is decreased in Arhgef1−/− platelets, relative to WT (Figure 4D, and 4E, respectively). These observations support the notion that Arhgef1 regulates integrin αIIbβ3 activation in platelets.
Arhgef1 Deletion Impairs Clot Retraction and Inhibits Platelet Spreading
Activation of integrin αIIbβ3 is required for clot retraction and platelet spreading through “outside in signaling,” which triggers an internal signaling cascade.14, 50 In light of the critical function Arhgef1 is found to play in integrin αIIbβ3 activation, we investigated its role in clot retraction and platelet spreading. Our data show defective clot retraction and platelet spreading in the absence of Arhgef1 (Figure 5A and 5B), indicating that it participates in “outside in signaling.”
Figure 5.
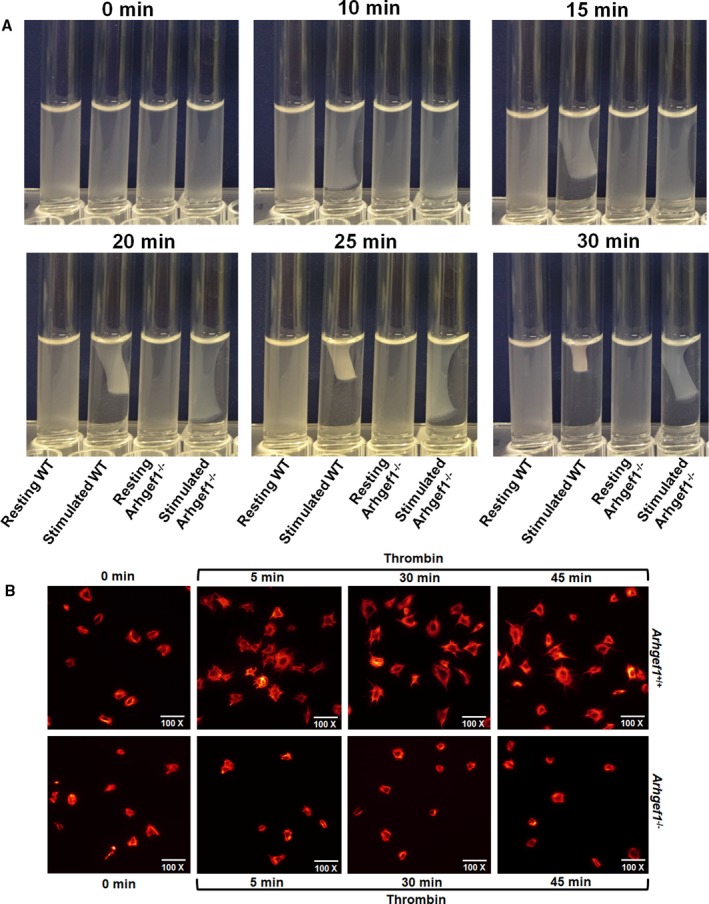
Arhgef1 deletion impairs clot retraction and platelet spreading. A, Whole blood was collected from both Arhgef1−/− and wild‐type (WT) mice and washed platelets were isolated as discussed in the Methods section. Clot retraction was initiated by adding thrombin (0.1 U/mL) and photographed every 5 minutes for 30 minutes. B, Arhgef1−/− and WT platelets were allowed to adhere to fibrinogen‐coated coverslips for 5 minutes after stimulation with thrombin (0.1 U/mL). Tetramethylrhodamine‐conjugated phalloidin in 10% fetal bovine serum/PBS with 0.2% saponin was used to stain for F‐actin and imaged using Leica DMi8 inverted widefield fluorescence microscope with integrated high precision focus drive. Images were processed using LAS X Wizard imaging software. Data are representative of 3 independent experiments. Each experiment was repeated at least 3 times, with blood pooled from a group of 8 mice each time.
Collectively, our studies demonstrate that Arhgef1 is a key regulator of platelet activation in vitro and in vivo.
Arhgef1 Deletion Inhibits RhoA Activation and Phosphorylation of ROCK in Platelets But Has No Effect on Rap1b Activation
To elucidate the mechanism by which Arhgef1 regulates platelet function, we investigated the impact of its deletion on RhoA‐ROCK axis. Our data show that Arhgef1 deletion inhibits RhoA activation in response to thrombin (0.1 U/mL) stimulation (Figure 6A). Notably, RhoA time course activation reveals that “maximal” activation occurs at 60 seconds, with the complete inactivation coinciding with 300 seconds in the WT platelets (Figure 6A). We further investigated the activation of the downstream/RhoA effector ROCK. Consistent with RhoA inhibition, phosphorylation of ROCK (Figure 6B) was also found to be reduced in thrombin (0.1 U/mL) ‐stimulated Arhgef1−/− platelets compared with the WT littermates. We next sought to determine whether the deficiency of Arghef1 affects other small G‐protein activation, namely Rap1b. Interestingly, we did not observe any detectable differences in Rap1b activation between the WT and Arhgef1−/− platelets (Figure 6C). These data suggest that Arhgef1 regulation of small G‐protein activation is specific in nature.
Figure 6.
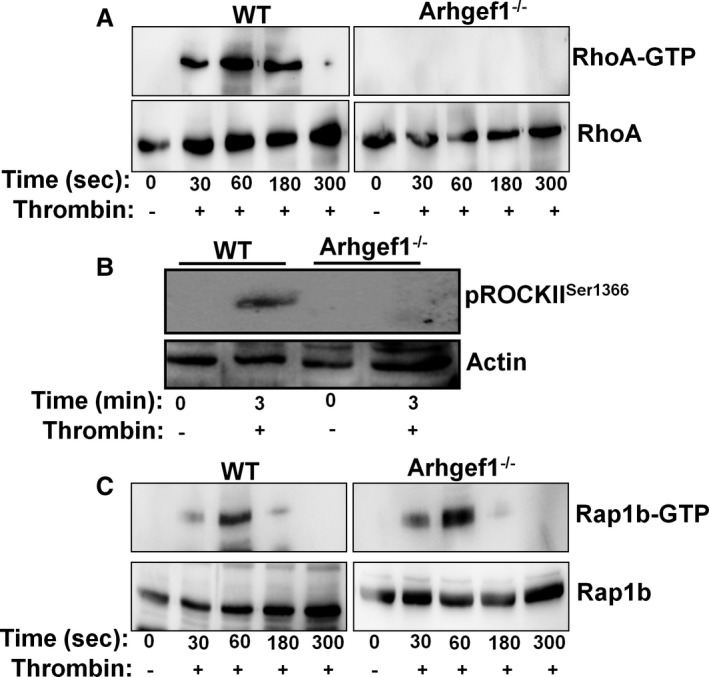
Arhgef1 deletion inhibits RhoA activation and phosphorylation of Rho‐associated kinase (ROCK) in platelets. A, Arhgef1−/− and wild‐type (WT) washed platelets (4×108/mL) were stimulated with thrombin (0.1 U/mL) for 0, 30, 60, 180, and 300 seconds before the RhoA‐GTP pulldown assay was performed. RhoA‐GTP and total RhoA were detected by Western blot using anti‐RhoA antibody. B, Platelets (4×108/mL) from Arhgef1−/− and WT mice were prepared and stimulated with thrombin (0.1 U/mL) for 3 minutes and proteins were lysed using 5× sample buffer. Proteins were separated by SDS‐PAGE and immunoblotted using antibodies to pROCKIISer1366 and actin. C, Arhgef1−/− and WT washed platelets (4×108/mL) were stimulated with thrombin (0.1 U/mL) for 0, 30, 60, 180, and 300 seconds before the Rap1b‐GTP pulldown assay was performed. Rap1b‐GTP and total Rap1b was detected by Western blot using anti‐Rap1b antibody. Western blot data represent 3 individual experiments, with blood pooled from a group of 8 mice each time.
Discussion
Given the importance of platelets in the genesis of cardiovascular disease, their activation mechanisms/signaling have been under intensive investigation for years, which revealed a complex array of pathways and communication at the molecular level.51 In this connection, several soluble agonists were found to activate platelets through binding to their receptors,52 mostly being GPCRs. As for their coupling profile, there is overlap in the G proteins to which these receptors are coupled to,52 with 4 different types identified in platelets including Gq, G13, Gs, and Gi.53
Upon stimulation of their GPCRs, the G proteins’ α‐subunits switch from the inactive GDP bound form, to the GTP54, 55 version, thereby inducing internal signal transduction, such as Rho‐GTPases. The Rho family of GTPases are molecular switches that control some of the most fundamental processes of cell biology, including regulation of gene expressions,56 remodeling of the actin‐based cytoskeleton,57 and platelet function.58, 59 Moreover, G13 and Gq, which are known to activate RhoA34, 60, 61 and bind to GEFs first,62, 63 play a critical role in platelet function. While the role of G13, Gq, and RhoA is well established in platelet function,4, 34, 35 little is known about the function of RhoGEFs. To this end, several RhoGEFs have been identified in platelets26 and found to play a critical role in their function, such as Arhgef3,30 Arhgef10,31 and Arhegf12.32 On the other hand, the role of another highly expressed GEF in platelets, namely Arhgef1, has not been elucidated. Thus in the present study, we aimed at characterizing the role of Arhgef1 in platelet function in vitro and in vivo by employing a genetic deletion mouse model.
The Arhgef1/RhoA signaling pathways have been recently shown to be involved in several physiological/pathological processes; for instance, Arhgef1 plays a role in arterial stiffness64 and atherosclerosis.65 In agreement with these reports, our findings reveal that Arhgef1 directly contributes to thrombogenesis, as its deletion prolonged vessel occlusion time (ie, protected against thrombus formation). This notion was further affirmed by the observation that Arhgef1 is also vital for hemostasis. Similarly, Arhgef1031 and Arhegf1232 were also found to regulate in vivo platelet function, unlike Arhgef3,30 whose deletion was not associated with a platelet phenotype in mice.30 These data provide evidence that Arhgefs play differential yet redundant roles in platelets. Notably, the platelet count and mean platelet volume were found to be normal in Arhgef1 KO mice. These findings indicate that the Arhgef1 phenotype observed––at least thus far––is not a consequence of an abnormality in thrombopoiesis. While this is consistent with what was observed with Arhgef10 and 12 KO mice,31, 66 it is in contrast to the macro thrombocytopenia phenotype observed caused by RhoA deficiency and the increased mean platelet volume associated with Arhgef3 KO mice.30 These data further support the notion that differences do exist in the role of the various Arhgefs in platelet function and their downstream signaling.
We next sought to investigate the mechanism underlying the Arhgef1−/− defects in hemostasis and thrombogenesis. Given the importance of the G13‐RhoA pathway in platelet aggregation34, 62 and secretion,33, 67 we first examined the impact of Arhgef1 deletion on these functional responses. Our data demonstrated that agonist‐induced platelet aggregation, as well as secretion (ie, dense and α granules), were reduced/defective in Arhgef1−/− platelets. These results are consistent with the defective dense and α granules secretion in the RhoA null platelets68 downstream of G13 and Gq.
We also observed defects in integrin αIIbβ3 activation, clot retraction, and platelet spreading in the Arhgef1−/− platelets upon agonist treatment. These findings are not surprising given the role of Arhgef1 in platelet aggregation, and that of RhoA in integrin activation, “outside‐in signaling,”69 and myosin light chain kinase phosphorylation68 (involved in integrin activation), as well as the notion that integrin αIIbβ3 “serves as a noncanonical Gα13‐coupled receptor that provides a mechanism for dynamic regulation of RhoA.”70 While one cannot exclude secondary effects contributing to the in vivo defects, eg, as a result of the function of Arhgef1 in cell types other than platelets, the observed phenotype clearly derives, at least in part, from the defects in a host of platelet functional responses. Interestingly, Arhgef1 appeared to regulate non–GPCR‐mediated platelet activation, namely that downstream of the collagen receptor glycoprotein VI; albeit the latter is not known to be associated with G13. These data indicate that Arhgef1 participates in noncanonical/non‐GPCR signaling. It is noteworthy that Arhgef10 deletion platelets exhibited similar defects to those of Arhgef1,31 whereas there were no apparent defects in the Arhgef12 KO mice66 and the Arhgef3 were without a platelet function phenotype.30 Together, these findings provide further evidence that Arhgefs play differential roles in platelet function.
We also demonstrated that Arghef1 plays an important role in activation of the RhoA signaling pathway. Thus, RhoA activation was impaired and so was the phosphorylation of downstream effectors (namely ROCK) in response to thrombin stimulation, which is consistent with the Arhgef10 deletion phenotype.31 In contrast, Arhgef12 was reported not to have any apparent role in RhoA activation in response to platelet stimulation.66 Interestingly, Arhgef1 was not found to regulate the activation of a separate small GTPase, namely Rap1b, which supports the notion that Athgef1 regulates small GTPases in a specific fashion. Taken together, these findings further indicate that RhoGEFs play distinct roles in platelet function. Finally, while the RhoA pathway clearly plays a vital role in Arhgef1‐mediated platelet activation, one cannot exclude the presence of an alternative pathway and/or a noncanonical role in platelet function (in light of the collagen phenotype), which will be the focus of future experiments.
Conclusions
Our data show that Arhgef1 regulates the RhoA‐ROCK axis and is essential for normal platelet function, as well as for hemostasis and thrombogenesis. These findings may define Arhgef1 as a novel therapeutic target for managing thrombosis‐based cardiovascular disease.
Sources of Funding
This research was supported, in part, by startup funds provided by the School of Pharmacy, The University of Texas at El Paso (to Alshbool). Lozano was supported in part by the National Institute of General Medical Sciences of the National Institutes of Health under award number R25GM123928. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Disclosures
None.
(J Am Heart Assoc. 2019;8:e011712 DOI: 10.1161/JAHA.118.011712.)
References
- 1. Furie B, Furie BC. Thrombus formation in vivo. J Clin Invest. 2005;115:3355–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shin EK, Park H, Noh JY, Lim KM, Chung JH. Platelet shape changes and cytoskeleton dynamics as novel therapeutic targets for anti‐thrombotic drugs. Biomol Ther (Seoul). 2017;25:223–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Golebiewska EM, Poole AW. Platelet secretion: from haemostasis to wound healing and beyond. Blood Rev. 2015;29:153–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Aslan JE, McCarty OJ. Rho GTPases in platelet function. J Thromb Haemost. 2013;11:35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aslan JE, Itakura A, Gertz JM, McCarty OJT. Platelet shape change and spreading In: Gibbins JM, Mahaut‐Smith MP, eds. Platelets and Megakaryocytes. Vol 3, Additional Protocols and Perspectives. New York, NY: Springer New York; 2012:91–100. [DOI] [PubMed] [Google Scholar]
- 6. Bembenek J, Karlinski M, Kobayashi A, Czlonkowska A. Early stroke‐related deep venous thrombosis: risk factors and influence on outcome. J Thromb Thrombolysis. 2011;32:96–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cangemi R, Casciaro M, Rossi E, Calvieri C, Bucci T, Calabrese CM, Taliani G, Falcone M, Palange P, Bertazzoni G, Farcomeni A, Grieco S, Pignatelli P, Violi F; Group SS, Group SS . Platelet activation is associated with myocardial infarction in patients with pneumonia. J Am Coll Cardiol. 2014;64:1917–1925. [DOI] [PubMed] [Google Scholar]
- 8. Xiang B, Zhang G, Liu J, Morris AJ, Smyth SS, Gartner TK, Li Z. A G(i) ‐independent mechanism mediating Akt phosphorylation in platelets. J Thromb Haemost. 2010;8:2032–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li Z, Zhang G, Feil R, Han J, Du X. Sequential activation of p38 and ERK pathways by cGMP‐dependent protein kinase leading to activation of the platelet integrin alphaiib beta3. Blood. 2006;107:965–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kageyama Y, Doi T, Akamatsu S, Kuroyanagi G, Kondo A, Mizutani J, Otsuka T, Tokuda H, Kozawa O, Ogura S. Rac regulates collagen‐induced HSP27 phosphorylation via p44/p42 MAP kinase in human platelets. Int J Mol Med. 2013;32:813–818. [DOI] [PubMed] [Google Scholar]
- 11. Bergmeier W, Stefanini L. Novel molecules in calcium signaling in platelets. J Thromb Haemost. 2009;7(suppl 1):187–190. [DOI] [PubMed] [Google Scholar]
- 12. O'Donnell VB, Murphy RC, Watson SP. Platelet lipidomics: modern day perspective on lipid discovery and characterization in platelets. Circ Res. 2014;114:1185–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ohtsuka H, Iguchi T, Hayashi M, Kaneda M, Iida K, Shimonaka M, Hara T, Arai M, Koike Y, Yamamoto N, Kasahara K. SDF‐1α/CXCR4 signaling in lipid rafts induces platelet aggregation via PI3 kinase‐dependent Akt phosphorylation. PLoS One. 2017;12:e0169609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li Z, Delaney MK, O'Brien KA, Du X. Signaling during platelet adhesion and activation. Arterioscler Thromb Vasc Biol. 2010;30:2341–2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McCarty OJ, Larson MK, Auger JM, Kalia N, Atkinson BT, Pearce AC, Ruf S, Henderson RB, Tybulewicz VL, Machesky LM, Watson SP. Rac1 is essential for platelet lamellipodia formation and aggregate stability under flow. J Biol Chem. 2005;280:39474–39484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pleines I, Eckly A, Elvers M, Hagedorn I, Eliautou S, Bender M, Wu X, Lanza F, Gachet C, Brakebusch C, Nieswandt B. Multiple alterations of platelet functions dominated by increased secretion in mice lacking Cdc42 in platelets. Blood. 2010;115:3364–3373. [DOI] [PubMed] [Google Scholar]
- 17. Yang Z. Small GTPases: versatile signaling switches in plants. Plant Cell. 2002;14(suppl):S375–S388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dütting S, Gaits‐Iacovoni F, Stegner D, Popp M, Antkowiak A, van Eeuwijk JM, Nurden P, Stritt S, Heib T, Aurbach K, Angay O, Cherpokova D, Heinz N, Baig AA, Gorelashvili MG, Gerner F, Heinze KG, Ware J, Krohne G, Ruggeri ZM, Nurden AT, Schulze H, Modlich U, Pleines I, Brakebusch C, Nieswandt B. A Cdc42/Rhoa regulatory circuit downstream of glycoprotein Ib guides transendothelial platelet biogenesis. Nat Commun. 2017;8:15838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Martens L, Van Damme P, Van Damme J, Staes A, Timmerman E, Ghesquiere B, Thomas GR, Vandekerckhove J, Gevaert K. The human platelet proteome mapped by peptide‐centric proteomics: a functional protein profile. Proteomics. 2005;5:3193–3204. [DOI] [PubMed] [Google Scholar]
- 20. Rowley JW, Oler AJ, Tolley ND, Hunter BN, Low EN, Nix DA, Yost CC, Zimmerman GA, Weyrich AS. Genome‐wide RNA‐seq analysis of human and mouse platelet transcriptomes. Blood. 2011;118:e101–e111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cook DR, Rossman KL, Der CJ. Rho guanine nucleotide exchange factors: regulators of Rho GTPase activity in development and disease. Oncogene. 2014;33:4021–4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Garrido A, Brandt M, Djouder N. Transport to rhebpress activity. Small GTPases. 2016;7:12–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bos JL, Rehmann H, Wittinghofer A. GEFs and GAPs: critical elements in the control of small G proteins. Cell. 2007;129:865–877. [DOI] [PubMed] [Google Scholar]
- 24. Cherfils J, Chardin P. GEFs: structural basis for their activation of small GTP‐binding proteins. Trends Biochem Sci. 1999;24:306–311. [DOI] [PubMed] [Google Scholar]
- 25. Vetter IR, Wittinghofer A. The guanine nucleotide‐binding switch in three dimensions. Science. 2001;294:1299–1304. [DOI] [PubMed] [Google Scholar]
- 26. Goggs R, Williams CM, Mellor H, Poole AW. Platelet Rho GTPases‐a focus on novel players, roles and relationships. Biochem J. 2015;466:431–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kher SS, Worthylake RA. Nuanced junctional RhoA activity. Nat Cell Biol. 2012;14:784–786. [DOI] [PubMed] [Google Scholar]
- 28. Pertz O. Spatio‐temporal Rho GTPase signaling—where are we now? J Cell Sci. 2010;123:1841–1850. [DOI] [PubMed] [Google Scholar]
- 29. Buchsbaum RJ, Connolly BA, Feig LA. Interaction of Rac exchange factors Tiam1 and Ras‐GRF1 with a scaffold for the p38 mitogen‐activated protein kinase cascade. Mol Cell Biol. 2002;22:4073–4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zou S, Teixeira AM, Kostadima M, Astle WJ, Radhakrishnan A, Simon LM, Truman L, Fang JS, Hwa J, Zhang PX, van der Harst P, Bray PF, Ouwehand WH, Frontini M, Krause DS. SNP in human ARHGEF3 promoter is associated with DNase hypersensitivity, transcript level and platelet function, and Arhgef3 KO mice have increased mean platelet volume. PLoS One. 2017;12:e0178095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lu DH, Hsu CC, Huang SW, Tu HJ, Huang TF, Liou HC, Liao HM, Chen CH, Fu WM, Gau SS. ARHGEF10 knockout inhibits platelet aggregation and protects mice from thrombus formation. J Thromb Haemost. 2017;15:2053–2064. [DOI] [PubMed] [Google Scholar]
- 32. Zou S, Teixeira AM, Yin M, Xiang Y, Xavier‐Ferrucio J, Zhang PX, Hwa J, Min W, Krause DS. Leukaemia‐associated Rho guanine nucleotide exchange factor (LARG) plays an agonist specific role in platelet function through RhoA activation. Thromb Haemost. 2016;116:506–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jin J, Mao Y, Thomas D, Kim S, Daniel JL, Kunapuli SP. RhoA downstream of G(q) and G(12/13) pathways regulates protease‐activated receptor‐mediated dense granule release in platelets. Biochem Pharmacol. 2009;77:835–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Moers A, Nieswandt B, Massberg S, Wettschureck N, Gruner S, Konrad I, Schulte V, Aktas B, Gratacap MP, Simon MI, Gawaz M, Offermanns S. G13 is an essential mediator of platelet activation in hemostasis and thrombosis. Nat Med. 2003;9:1418–1422. [DOI] [PubMed] [Google Scholar]
- 35. Offermanns S, Toombs CF, Hu YH, Simon MI. Defective platelet activation in G alpha(q)‐deficient mice. Nature. 1997;389:183–186. [DOI] [PubMed] [Google Scholar]
- 36. Kozasa T, Jiang X, Hart MJ, Sternweis PM, Singer WD, Gilman AG, Bollag G, Sternweis PC. p115 RhoGEF, a GTPase activating protein for Galpha12 and Galpha13. Science. 1998;280:2109–2111. [DOI] [PubMed] [Google Scholar]
- 37. Karim ZA, Zhang J, Banerjee M, Chicka MC, Al Hawas R, Hamilton TR, Roche PA, Whiteheart SW. IkappaB kinase phosphorylation of SNAP‐23 controls platelet secretion. Blood. 2013;121:4567–4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Karim ZA, Alshbool FZ, Vemana HP, Conlon C, Druey KM, Khasawneh FT. CXCL12 regulates platelet activation via the regulator of G‐protein signaling 16. Biochim Biophys Acta. 2016;1863:314–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Karim ZA, Alshbool FZ, Vemana HP, Adhami N, Dhall S, Espinosa EV, Martins‐Green M, Khasawneh FT. Third‐hand smoke: impact on hemostasis and thrombogenesis. J Cardiovasc Pharmacol. 2015;66:177–182. [DOI] [PubMed] [Google Scholar]
- 40. Hensch NR, Karim ZA, Pineda J, Mercado N, Alshbool FZ, Khasawneh FT. P2Y12 antibody inhibits platelet activity and protects against thrombogenesis. Biochem Biophys Res Commun. 2017;493:1069–1074. [DOI] [PubMed] [Google Scholar]
- 41. Alshbool FZ, Karim ZA, Vemana HP, Conlon C, Lin OA, Khasawneh FT. The regulator of G‐protein signaling 18 regulates platelet aggregation, hemostasis and thrombosis. Biochem Biophys Res Commun. 2015;462:378–382. [DOI] [PubMed] [Google Scholar]
- 42. Vemana HP, Karim ZA, Conlon C, Khasawneh FT. A critical role for the transient receptor potential channel type 6 in human platelet activation. PLoS One. 2015;10:e0125764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Choi W, Karim ZA, Whiteheart SW. Arf6 plays an early role in platelet activation by collagen and convulxin. Blood. 2006;107:3145–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Scott DW, Tolbert CE, Burridge K. Tension on JAM‐A activates RhoA via GEF‐H1 and p115 RhoGEF. Mol Biol Cell. 2016;27:1420–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dubash AD, Wennerberg K, García‐Mata R, Menold MM, Arthur WT, Burridge K. A novel role for Lsc/p115 RhoGEF and LARG in regulating RhoA activity downstream of adhesion to fibronectin. J Cell Sci. 2007;120:3989–3998. [DOI] [PubMed] [Google Scholar]
- 46. Rouillon C, Mercier N, Lacolley P, Loirand G, Regnault V. Control of coagulation by the RhoA pathway and the exchange factor Arhgef1. Arch Cardiovasc Dis Suppl. 2018;10:113. [Google Scholar]
- 47. Dawood BB, Wilde J, Watson SP. Reference curves for aggregation and ATP secretion to aid diagnose of platelet‐based bleeding disorders: effect of inhibition of ADP and thromboxane A(2) pathways. Platelets. 2007;18:329–345. [DOI] [PubMed] [Google Scholar]
- 48. Storrie B, Whiteheart SW. Editorial: platelet secretion. Platelets. 2017;28:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bennett JS. Structure and function of the platelet integrin alphaIIbbeta3. J Clin Invest. 2005;115:3363–3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shattil SJ, Newman PJ. Integrins: dynamic scaffolds for adhesion and signaling in platelets. Blood. 2004;104:1606–1615. [DOI] [PubMed] [Google Scholar]
- 51. Rivera J, Lozano ML, Navarro‐Nunez L, Vicente V. Platelet receptors and signaling in the dynamics of thrombus formation. Haematologica. 2009;94:700–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Offermanns S. Activation of platelet function through G protein–coupled receptors. Circ Res. 2006;99:1293–1304. [DOI] [PubMed] [Google Scholar]
- 53. Oldham WM, Hamm HE. Heterotrimeric G protein activation by G‐protein‐coupled receptors. Nat Rev Mol Cell Biol. 2008;9:60. [DOI] [PubMed] [Google Scholar]
- 54. Kobilka BK, Deupi X. Conformational complexity of G‐protein‐coupled receptors. Trends Pharmacol Sci. 2007;28:397–406. [DOI] [PubMed] [Google Scholar]
- 55. Venkatakrishnan AJ, Deupi X, Lebon G, Tate CG, Schertler GF, Babu MM. Molecular signatures of G‐protein‐coupled receptors. Nature. 2013;494:185–194. [DOI] [PubMed] [Google Scholar]
- 56. Marinissen MJ, Chiariello M, Gutkind JS. Regulation of gene expression by the small GTPase Rho through the ERK6 (p38 gamma) MAP kinase pathway. Genes Dev. 2001;15:535–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chikumi H, Barac A, Behbahani B, Gao Y, Teramoto H, Zheng Y, Gutkind JS. Homo‐ and hetero‐oligomerization of PDZ‐RhoGEF, LARG and p115RhoGEF by their C‐terminal region regulates their in vivo Rho GEF activity and transforming potential. Oncogene. 2004;23:233. [DOI] [PubMed] [Google Scholar]
- 58. Klages B, Brandt U, Simon MI, Schultz G, Offermanns S. Activation of G12/G13 results in shape change and Rho/Rho‐kinase‐mediated myosin light chain phosphorylation in mouse platelets. J Cell Biol. 1999;144:745–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Suzuki Y, Yamamoto M, Wada H, Ito M, Nakano T, Sasaki Y, Narumiya S, Shiku H, Nishikawa M. Agonist‐induced regulation of myosin phosphatase activity in human platelets through activation of Rho‐kinase. Blood. 1999;93:3408–3417. [PubMed] [Google Scholar]
- 60. Gratacap MP, Payrastre B, Nieswandt B, Offermanns S. Differential regulation of Rho and Rac through heterotrimeric G‐proteins and cyclic nucleotides. J Biol Chem. 2001;276:47906–47913. [DOI] [PubMed] [Google Scholar]
- 61. Moers A, Wettschureck N, Gruner S, Nieswandt B, Offermanns S. Unresponsiveness of platelets lacking both Galpha(q) and Galpha(13). Implications for collagen‐induced platelet activation. J Biol Chem. 2004;279:45354–45359. [DOI] [PubMed] [Google Scholar]
- 62. Huang JS, Dong L, Kozasa T, Le Breton GC. Signaling through G(alpha)13 switch region I is essential for protease‐activated receptor 1‐mediated human platelet shape change, aggregation, and secretion. J Biol Chem. 2007;282:10210–10222. [DOI] [PubMed] [Google Scholar]
- 63. Andrews RK, Gardiner EE. Inside platelets. Blood. 2012;119:907–909. [DOI] [PubMed] [Google Scholar]
- 64. Rouillon C, Mercier N, Lacolley P, Loirand G, Regnault V. Arhgef1/RhoA signaling participate in ageing‐induced arterial stiffness and hypercoagulability. Artery Res. 2017;20:65. [Google Scholar]
- 65. Carbone ML, Chadeuf G, Heurtebise‐Chrétien S, Prieur X, Quillard T, Goueffic Y, Vaillant N, Rio M, Castan L, Durand M, Baron‐Menguy C, Aureille J, Desfrançois J, Tesse A, Torres RM, Loirand G. Leukocyte RhoA exchange factor Arhgef1 mediates vascular inflammation and atherosclerosis. J Clin Invest. 2017;127:4516–4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Williams CM, Harper MT, Goggs R, Walsh TG, Offermanns S, Poole AW. Leukemia‐associated Rho guanine‐nucleotide exchange factor is not critical for RhoA regulation, yet is important for platelet activation and thrombosis in mice. J Thromb Haemost. 2015;13:2102–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Quinton TM, Murugappan S, Kim S, Jin J, Kunapuli SP. Different G protein‐coupled signaling pathways are involved in alpha granule release from human platelets. J Thromb Haemost. 2004;2:978–984. [DOI] [PubMed] [Google Scholar]
- 68. Pleines I, Hagedorn I, Gupta S, May F, Chakarova L, van Hengel J, Offermanns S, Krohne G, Kleinschnitz C, Brakebusch C, Nieswandt B. Megakaryocyte‐specific RhoA deficiency causes macrothrombocytopenia and defective platelet activation in hemostasis and thrombosis. Blood. 2012;119:1054–1063. [DOI] [PubMed] [Google Scholar]
- 69. Chari‐Turaga R, Naik UP. Integrin αIIbβ3: a novel effector of Gα13. Cell Adh Migr. 2011;5:4–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gong H, Shen B, Flevaris P, Chow C, Lam SC, Voyno‐Yasenetskaya TA, Kozasa T, Du X. G protein subunit Galpha13 binds to integrin alphaIIbbeta3 and mediates integrin “outside‐in” signaling. Science. 2010;327:340–343. [DOI] [PMC free article] [PubMed] [Google Scholar]