

Abstract
Premature, sudden death is devastating. Certain patient populations are at greater risk to succumb to sudden death. For instance, infants under one year of age are at risk for sudden infant death syndrome (SIDS), and patients with epilepsy are at risk for sudden unexpected death in epilepsy (SUDEP). Deaths are attributed to these syndromic entities in these select populations when other diagnoses have been excluded. There are a number of similarities between these syndromes, and the commonalities suggest that the two syndromes may share certain etiological features. One such feature may be deficiency of arousal to carbon dioxide (CO2). Under normal conditions, CO2 is a potent arousal stimulus. Circumstances surrounding SIDS and SUDEP deaths often facilitate CO2 elevation, and faulty CO2 arousal mechanisms could, at least in part, contribute to death.
Keywords: serotonin, arousal, sudden death, chemoreception
Towards a pathophysiological understanding of sudden, unexpected death
Sudden, premature death of individuals takes a heavy toll on their families, circle of acquaintances, and society at large. Often, there is an evident cause for premature death, such as accidental injury, inflicted injury (e.g. suicide, homicide), or critical illness. Unfortunately, too often, there is no immediately identifiable cause for death. In some respects, these cases can be even more devastating for loved ones, as the latter are left with the unsettling questions of why their loved one died, and whether anything could have been done to prevent the loss.
For certain patient populations, syndromic sudden death entities have been described and are attributed as the cause of death when other possible diagnoses for death causes have been excluded. Two such entities that have been gaining considerable attention recently are the sudden infant death syndrome (SIDS; see Glossary) and sudden unexpected death in epilepsy (SUDEP). Despite their notoriety, the pathophysiology of each is poorly understood. In both entities the actual death is typically unwitnessed and the victim is found already dead, often in bed. The fact that death is commonly unwitnessed makes it challenging to retrospectively piece together circumstances of death. Based on this incomplete information, measures have been proposed and implemented in an attempt to identify high risk individuals and mitigate death. Yet, the incidence of SIDS and SUDEP is still too high.
SIDS and SUDEP share a number of common features, including potential pathophysiological mechanisms. Among these, dysregulation of certain arousal mechanisms may be particularly pertinent. The objective of this article is to present relevant features of SIDS and SUDEP, highlighting commonalities, and to outline the hypothesis that impaired CO2-induced arousal is a key common feature among these entities.
Sudden infant death syndrome
SIDS is defined as the sudden and unexpected death of an infant under the age of one year that remains unexplained after thorough review of the clinical history, death scene investigation, and complete autopsy [1]. It is the leading cause of sudden death in infants under one year of age. Considerable work has been done in attempt to understand the risk factors for SIDS and its pathophysiological features. Collectively, this work produced the triple risk model for SIDS. This model fits into the broad framework of triple risk models, whereby an individual with an underlying susceptibility is exposed to a detrimental exogenous factor during a critical period (Figure 1). In the case of SIDS, the critical period is the first year of life, , particularly the first six months or before infants are able to roll over on their own. While considerable work is still ongoing to better define the at-risk individuals, some genetic, pathophysiological, and environmental features have been identified from babies that died of SIDS compared to babies that died for other reasons [2].
Figure 1. Triple risk models for SIDS and SUDEP.
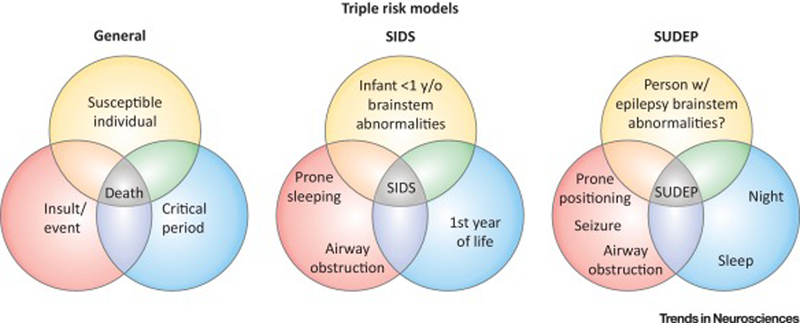
In a general triple risk model for death (left), risk of death is increased when a susceptible individual (yellow) is subjected to an insult or event (red) during a critical period (blue). Applying such a model to SIDS (middle), the risk of dying from SIDS is increased when an infant during the first year of life with specific brainstem abnormalities (especially in the 5-HT system) sleeps prone or otherwise has its airway obstructed during sleep. To apply the model to SUDEP (right), there is an increased risk of dying from SUDEP when a person with epilepsy (especially refractory epilepsy with generalized tonic-clonic seizures) and perhaps brainstem abnormalities has a generalized tonic-clonic seizure during the night and/or during sleep. SUDEP victims are additionally often found prone with evidence of airway obstructions. There are of course nuances to application of these models as alluded to in the text, and details are still emerging for the SUDEP model.
A major exogenous factor that has been identified is airway occlusion or obstruction, such as could occur if the infant was lying prone in the crib. As such, the “Back to Sleep” campaign was implemented in which it was strongly suggested that infants be placed on their backs to sleep [3–5]. Along with this, it was suggested that other items such as stuffed animals, pillows, and blankets be removed from the crib, and the infant be dressed in tight fitting clothes. This campaign contributed to reduction in the incidence of SIDS from 1.2 in 1000 live births to approximately 0.6 in 1000 live births in the U.S., where it remains today [3,6].
Sudden unexpected death in epilepsy
SUDEP is defined as the “sudden, unexpected, witnessed or unwitnessed nontraumatic and non-drowning death in patients with epilepsy, with or without evidence for a seizure and excluding documented status epilepticus, in which postmortem examination does not reveal a toxicological or anatomic cause of death” [7]. It is the leading cause of death in patients with medically refractory epilepsy [8]. It is second only to stroke among neurological diseases in terms of years of potential life lost to disease, owing in part to the fact that it occurs commonly in younger individuals [9]. SUDEP is thought to occur most commonly in patients with mesial temporal lobe epilepsy, but certain epilepsy populations, such as children with the severe myoclonic epilepsy of infancy, or Dravet syndrome [10], have an especially high rate of SUDEP [11]. In 2017, the American Academy of Neurology, estimated the overall global incidence of SUDEP to be 0.58/1000 patient-years, and incidence of 0.22 and 1.2/1000 patient-years in children and adults, respectively, based on review and analysis of literature published before April 2015 [8]. Incidence estimates in more discreet patient populations in which all epilepsy deaths are captured during a defined time window suggest the incidence may be more comparable between children and adults. For instance in Sweden, examination of all epilepsy deaths during 2008 revealed an incidence of 1.2/1000 patient-years [12]. In Canada, review of all epilepsy death in children in 2014–2015 revealed an incidence of 1.17/1000 patient-years [13]. These discreet population-based studies suggest that the incidence is comparable across age groups, whereas an earlier large meta-analysis indicated a higher incidence in the third and fourth decades of life [14].
SUDEP deaths have been subcategorized into definite, probable, and possible SUDEP. In definite SUDEP there is an autopsy that confirms there is no competing cause of death. In probable SUDEP, there is no autopsy. A “plus” designation can be added to definite and probable SUDEP if there was a comorbidity that could have contributed to death, but was not thought to have contributed. In possible SUDEP, there is a comorbidity that could have contributed to death. If it is perceived that a person would have likely died following a seizure, but survived at least for one hour because of resuscitative efforts, this is termed a near SUDEP. While in each of these subcategories the definition of SUDEP is applied slightly differently, they can largely be grouped together when considering anatomical, pathological, physiological, and electrophysiological features that portend increased risk for SUDEP.
Similarities between SIDS and SUDEP
There are a number of similarities between SIDS and SUDEP [15] (Table 1). Both are essentially diagnoses of exclusion. In both, the individual is usually healthy (except for epilepsy in SUDEP). In both, autopsy is normal. Similar etiological factors have been proposed for both, including respiratory demise, cardiac demise, and impairment of arousal. In both, the death is commonly unwitnessed, with the individual being found dead in bed/crib and in the prone position. The incidence is comparable between the two entities. And in both entities dysregulation of the serotonin (5-HT) system has been identified or proposed. 5-HT is an attractive candidate in the pathophysiology of SIDS and SUDEP because it is involved in modulating many associated factors implicated in these disease entities, including breathing, cardiovascular control, sleep and wakefulness, arousal, circadian rhythms (i.e. both entities occur more commonly at night), and seizures [11,16]. Due to these similarities, especially the association with prone positioning, a back to sleep campaign, similar to that which was so successful in reducing the risk for SIDS, has been proposed for SUDEP as well; however, while it is easy to have a patient with epilepsy start the night sleeping on their back, it is rather difficult to keep them that way throughout the night [17].
Table 1.
Similarities between SIDS and SUDEP.
| SIDS | SUDEP | |
|---|---|---|
| Diagnostic method | Diagnosis of exclusion | Diagnosis of exclusion |
| Baseline health | Normal | Normal expect seizures |
| Routine autopsy findings | Normal | Normal; pulmonary edema |
| Incidence | ~0.6 per 1000 live births | 0.2–1.2 in 1000 persons with epilepsy |
| Proposed mechanism of death | Respiratory Cardiac Arousal impairment Thermoregulatory dysregulation |
Respiratory Cardiac Arousal impairment |
| Circumstances of death | Often found prone | Often found prone |
| Link to 5-HT | Yes | Yes |
Adapted from [15] with permission.
Interestingly, there are disease entities that may lie on a continuum of sorts bookended by SIDS and SUDEP. These include: apparent life threatening events (ALTEs) in infancy/childhood (which may be the SIDS analogue to near-SUDEP, that is, an event where the victim would have likely died but survived at least for one hour because of resuscitative efforts); sudden unexplained death in childhood (SUDC) [18] or sudden unexplained death in the young (SUDY); and near-SUDEP [2,7]. SIDS is a specific subcategory of sudden unexpected infant death (SUID) which encompasses all sudden deaths in infants [19,20]. There are case reports of witnessed seizures in infants that have ultimately died as a consequence of the seizure [21]. If these seizures had not been witnessed then it is likely that the baby would have died, been found dead in the crib in the morning, and SIDS would have been listed as cause of death on the death certificate. This raises the question of how many SIDS deaths may actually be SUDEP, with the baby either experiencing a first and fatal seizure, or the baby having had unrecognized seizures and then having a final fatal seizure. Indeed, seizures in infants can often be bland and unrecognized. Also recently, mutations in the SCN1A gene that encode the voltage gated sodium channel (Nav1.1) have been identified in two babies whose deaths were attributed to SIDS [22]. Such mutations occur in Dravet syndrome, the epileptic encephalopathy with a particularly high incidence of SUDEP [10]. At the time of their death, these babies were too young to have begun to display the seizures and other features of Dravet syndrome.
Anatomic abnormalities in the hippocampus and temporal lobe have been identified in many cases of SIDS and SUDC [18,23–27]. Similar such abnormalities, including mesial temporal or hippocampal sclerosis are among the hallmarks of temporal lobe epilepsy (TLE), a common form of epilepsy associated with SUDEP. Recently, a distinct clinicopathological entity was described in a cohort of children 1–6 years of age that died in association with a febrile seizure. This has been dubbed hippocampal maldevelopment associated with sudden death (HMASD), and is characterized by sudden, sleep-associated death in a child that had been born at term and was found in the prone position [18,26]. Specific abnormalities seen in these cases include bilamination of the dentate gyrus [26] which can also be seen in SIDS [25] and TLE [28,29].
CO2-induced arousal from sleep
Among the proposed etiologies for SIDS and SUDEP, one that is especially intriguing, and that has garnered relatively little attention, is impaired arousal, especially to carbon dioxide (CO2). CO2 is expelled by the lungs as one breathes. Drive to breathe is dictated in large part by the serum CO2 concentration which is exquisitely tightly regulated. In addition to potently driving breathing, CO2 is a powerful arousal stimulus [30]. As can be imagined, acute rises in CO2 occur when an individual is unable to expel CO2, such as in the setting of an airway obstruction that might occur when an individual is lying prone in a crib or bed perhaps with a pillow and bedclothes covering the nose and mouth. It has been proposed that such a rise in CO2 would activate arousal circuitry in a normal baby to wake the baby up, cause them to cry out, summoning a caregiver who would come to their aid, and ostensibly correct the airway blockage to allow resumption of normal breathing [16,20,31]. It has been proposed, among other possibilities, that there is an impaired CO2-arousal system in SIDS-susceptible babies such that when they rebreathe CO2 as described above, they do not arouse, and thus do not cry out, and the blockage is not corrected [16,32]. They thus become acidotic and hypoxic and ultimately succumb. We posit that similar such mechanisms could be at play in SUDEP. CO2 rises in association with a convulsive seizure, especially a convulsive or generalized tonic-clonic seizure, with which SUDEP is more commonly associated [8]. It has been suggested that the rise in CO2 is part of the seizure cessation mechanism [33]. In patients at risk for SUDEP, it is possible that there is a dysfunctional CO2 arousal system contributing to seizure-related demise. This would be exacerbated by situations that cause airway obstruction, such as ending up in the prone position or having a physical barrier, such as a pillow, present [34–36]. In patients with epilepsy, seizures can also lead to laryngospasm which could cause airway obstruction to lead to SUDEP [37–39].
Mechanisms of CO2-induced arousal from sleep have been somewhat controversial (Figure 2, Key Figure). For quite some time, the general thinking was that arousal from sleep in response to CO2 required stimulation of breathing, which subsequently activated stretch and mechanoreceptors that in turn activate pathways through the nucleus tractus solitarius (NTS) and other sites to cause arousal [40,41]. More recently, central mechanisms whereby CO2 can be directly sensed by neurons that feed into and influence arousal networks have been proposed [42–45]. A number of possible sites have chemosensory properties and have been implicated in sleep wake regulation, including 5-HT neurons in the midbrain dorsal raphe nucleus (DRN) or medullary raphe, glutamatergic neurons in the retrotrapezoid nucleus (RTN, or lateral parafacial nucleus), glutamatergic neurons in the NTS, noradrenergic neurons in the locus coeruleus (LC), and orexinergic neurons in the lateral hypothalamus [42–44,46–49]. Whether or how each of these is involved in CO2-induced arousal is unclear. and which site might be the primary sensor of CO2 to incite arousal is unknown.
Figure 2. Schematic depicting possible mechanisms for CO2 induced arousal.
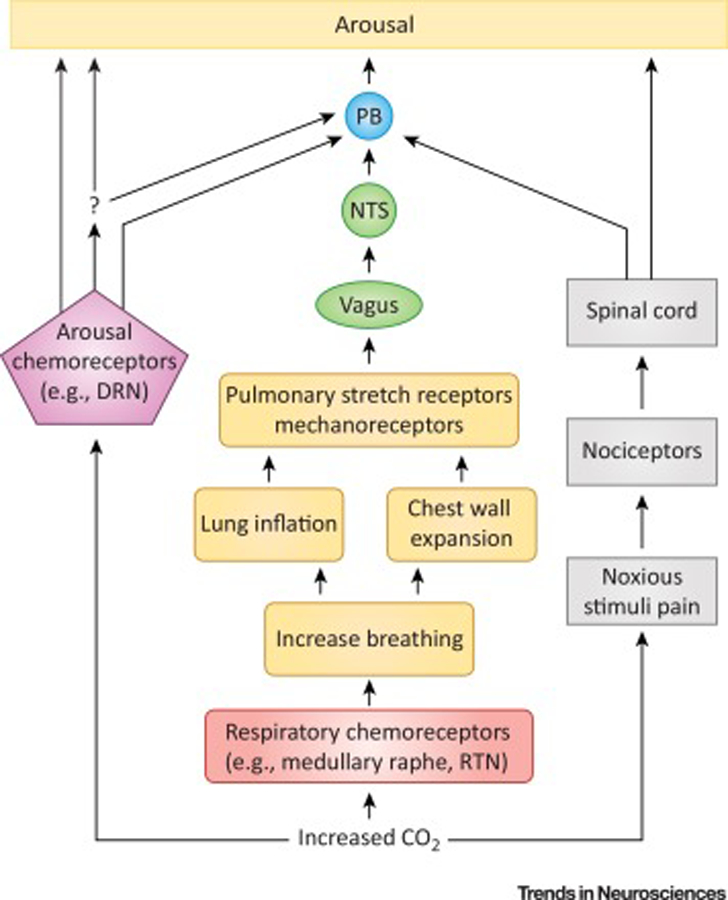
Inspired CO2 rapidly induces a rise in serum CO2 concentration (bottom), which is detected by peripheral (e.g. carotid bodies; not depicted) and central respiratory (red) and arousal (purple) chemosensors. Evidence implicates medullary raphe 5-HT neurons and glutamatergic RTN neurons as important respiratory chemosensors. In a long-standing proposed mechanism for CO2 arousal (middle) activation of respiratory chemosensors increases breathing, causing lung inflation and chest wall expansion, which activates stretch and mechanoreceptors in these structures (orange). This activation is transmitted via the vagus nerve to the NTS (green) and ultimately to PB (blue) which leads to arousal (yellow). The PB may be the final common mediator of arousal to a number of different stimuli. Arousal pathways downstream of PB were analyzed by Kaur et al. [45] and not depicted here. In the more recently emerging proposed CO2 arousal mechanism (left), changes in serum CO2 are detected directly by arousal chemosensors which cause arousal, perhaps through a PB mediated mechanism, either directly, or indirectly through some yet to be determined intermediary (question mark). A leading candidate for the arousal chemosensors is DRN 5-HT neurons; however, other structures such as norepinephrine (NE) neurons in LC and orexin neurons in the lateral hypothalamus (LH) have been implicated. In addition, the NTS is itself chemosensitive. There can also be a noxious component to inspiration of CO2 which can cause arousal through spinally mediated nociceptive pathways (right; black). DRN, dorsal raphe nucleus; NTS, nucleus tractus solitarius; PB, parabrachial nucleus; RTN, retrotrapezoid nucleus
Serotonin in SIDS, SUDEP, and CO2-induced arousal
One candidate for acting as the primary sensor of CO2 in the context of CO2-induced arousal is 5-HT neurons in the DRN (Figure 2). These neurons are robustly chemosensitive in vitro [50] and in vivo [51]. In vitro, most DRN 5-HT neurons tested are chemosensitive and responded with a four-fold increase in firing in response to a pH change from 7.4 to 7.2, well within physiological range [50]. Genetic elimination of 5-HT neurons in the CNS attenuates arousal to inspired CO2 in mice [43]. Direct application of CO2-enriched artificial cerebrospinal fluid to the DRN, but not the medullary raphe, causes arousal from sleep in wild type mice [42]. Stimulation of the DRN in 5-HT neuron deficient mice does not cause arousal [42], suggesting that these neurons are important sensors of the pH changes associated with the rise in CO2. Acute inactivation of DRN 5-HT neurons either pharmacologically or optogenetically also attenuates CO2-induced arousal [42]. Mechanisms of CO2 induced arousal downstream of DRN 5-HT neuron activation are unknown. Presumably, these inputs feed into known arousal circuitry, but the specific nature of these connections remains to be clarified [42]. A series of experiments have implicated the parabrachial nucleus (PB), especially the exterolateral PB (PBel) in CO2-induced arousal [45,52,53]. The PB is well situated to activate arousal in response to a variety of arousing stimuli, including increased CO2 concentration. The PB is not known to be chemosensitive itself, thus some other site might be the primary sensor of CO2 and cause arousal through downstream activation of PB. While the evidence for DRN 5-HT neurons as the primary CO2 sensors is strong, the DRN is not known to project heavily to the PB. Thus, if these two structures are part of a circuit in CO2-induced arousal, either direct connection between the structures are yet to be elucidated, or they may be connected through an intermediary site.
A number of abnormalities in the brainstem serotonergic system have been identified in brains from babies whose deaths were attributed to SIDS. These include reduced binding of ligands to the 5HT1A receptor, an inhibitory autoreceptor in the brainstem raphe nuclei; an increased proportion of immature appearing 5-HT neurons in the raphe; decreased concentration of 5-HT in the medulla; and elevated serum 5-HT [54–61]. However, no differences were found in the concentrations of the 5-HT metabolite 5-hyroxyindoleacetic acid (5-HIAA) or the 5-HT precursor tryptophan in CSF between SIDS cases and age-matched controls [62]. These studies have mainly focused on the caudal serotonergic neurons in the medulla that are thought to be involved in the regulation of breathing. Closer examination of rostral brainstem structures such as the DRN is warranted.
A preponderance of evidence has also implicated 5-HT and 5-HT system abnormalities in SUDEP. Until recently this has been more circumstantial evidence including reduced oxygen desaturation following seizures in patients taking selective serotonin reuptake inhibitors (SSRI) [63], and prevention of seizure related respiratory dysregulation in animal seizure models [64–73]. Emerging data has identified anatomic abnormalities, including in serotonergic nuclei, in brains of patients who died of SUDEP. These include reduced functional connectivity networks involving thalamus, anterior cingulate, brainstem, putamen, and amygdala [74]; excess volume loss in the medulla including in the raphe [75] and other brainstem regions [76]; reduced tryptophan hydroxylase staining in the ventrolateral medulla and reduced 5-HT transporter expression in the medullary raphe in SUDEP cases compared to control [77]; increased grey matter volume assessed by magnetic resonance imaging (MRI) in anterior hippocampus, amygdala, and parahippocampal gyrus in SUDEP cases and high risk individuals compared to controls and low risk individuals [78]; and alterations in expression of a variety of markers in brainstem [79]. These abnormalities have been identified largely from post-mortem analyses. Whether patients at risk for SUDEP display altered arousal or ventilatory responses to CO2 is unknown. Likewise, whether the identified anatomic abnormalities translate into impaired CO2 responses is yet to be determined. It has been demonstrated that seizures can impair breathing by impinging upon circuitry that passes through the amygdala [80,81]. It has been proposed through animal studies that such impingement might occur through spreading depolarization that is incited by the seizure and propagates into brainstem [82–84]. One could speculate that these could extend to CO2-arousal systems, and by silencing neural activity in these areas impair CO2-induced awakening. Alternatively, as shown in animal models, seizure can directly impair firing of both medullary and midbrain 5-HT neurons that are involved in ventilatory and arousal responses to CO2 [73].
Concluding remarks and future perspectives
Discovery of biomarkers to more easily identify individuals at risk for SIDS and SUDEP is underway [8,54,62]. Once etiological features are clearly defined and at risk individuals reliably ascertained, prophylactic strategies can be deployed. Converging lines of evidence, as discussed in this article, point at dysregulation of CO2-induced arousal as a shared candidate pathophysiological mechanism in both SIDS and SUDEP. According to this proposed mechanism, prophylactic strategies may include methods to temper the rise in CO2 that may occur in the peri-death period, in addition to ways to lessen the impairment in CO2-arousal mechanisms in susceptible individuals. Before this can be accomplished, however, a clearer understanding of the clinical-pathophysiological deficiencies in these entities and a more precise understanding of the mechanisms underlying CO2-induced arousal is required. Perhaps an appropriate place to start is with a more concerted comparative analysis of cases representing each aforementioned sudden death entity. Complementarily, modern experimental technology including, but not limited to high field strength magnetic resonance imaging, ultra-small scale microscopy, optogenetics, chemogenetics, and calcium-imaging, permitting more detailed anatomical and circuit analysis will aid in dissecting mechanisms. These techniques can be first applied in animal models. Insights gained can then be tested and translated for use in patient tissue samples. In the clinical setting, continued efforts are needed to make respiratory assessments more routine in the long-term monitoring units, such as those used in epilepsy care and in care of high risk infants. These monitoring practices could be used not only to assess respiratory responses to CO2 and hypoxia, but also arousal responses. This will allow more comprehensive assessment of changes in sensitivity of these systems in patients at high risk for SIDS and SUDEP, and could be extended to examine certain other at-risk populations if and when better biomarkers for other types of sudden and unexpected death are identified.
Highlights.
SIDS and SUDEP are among the leading causes of death in their respective patient populations (i.e. infants under one year of age and patients with refractory epilepsy), but their pathophysiology is poorly understood.
Some common features of SIDS and SUDEP have been identified, including impaired cardio-respiratory function and dysfunctional serotonin signaling.
A possible common pathophysiological mechanism is impaired arousal in response to elevated CO2. Individuals at risk for both diseases experience situations in which serum CO2 concentrations rise (e.g. due to physical airway obstruction, or as a consequence of a seizure). However, the physiological mechanisms of CO2-induced arousal from sleep are not fully understood, and whether a definitive link to SIDS or SUDEP exists remains to be validated.
Better understanding of the mechanisms of CO2-induced arousal, and how these mechanisms are impaired to possibly contribute to death in SIDS and SUDEP, may help address the pressing need for effective prevention strategies in these conditions.
Outstanding Questions.
Does SIDS lie on a continuum with SUDEP, and other disease entities such as ALTE, SUDC, and SUDY?
Do some cases of SIDS represent a first presentation of epilepsy, with seizures being previously unappreciated and the fatal seizure being unwitnessed?
Are there ways to identify susceptible individuals? What are the biomarkers (e.g. serum/CSF measures, imaging features, etc) for increased risk of death from SIDS and SUDEP? Once effective biomarkers are identified, what prophylactic measures can be deployed in the relevant individuals?
Are there comparable anatomical and/or pathophysiological changes in brains from people who die from SUDEP and those from babies who die from SIDS?
How is CO2 arousal circuitry impaired in the first place in individuals at risk for SIDS and/or SUDEP?
Is serotonin key in these processes? Is the DRN the primary CO2 sensor for CO2 induced arousal? What role do other chemosensitive structures (e.g. LC, RTN, medullary raphe, orexin neurons, carotid bodies, etc) play in this context?
How are the DRN and PB connected to each other in mediating CO2 induced arousal from sleep?
In the setting of airway obstruction and/or respiratory arrest following a seizure, the rise in CO2 does not occur in isolation, but rather is associated with hypoxia. In fact, the hypoxic component may be more critical in leading to death. Hypoxia can also be a potent arousal stimulus, especially in rodent models. What are the relative roles of hypercapnia and hypoxia in arousal? Will prophylactic measures have to address both hypercapnia and hypoxia?
Acknowledgements
Dr. Buchanan is supported by the NIH/NINDS R01NS095842; the Pappajohn Biomedical Institute and Iowa Neuroscience Institute at the University of Iowa; and the Beth Levitt Tross Professorship in Epilepsy Research.
Glossary
- Epilepsy
disease in which the person is subject to spontaneous, unprovoked seizures
- Medically refractory epilepsy
epilepsy in which seizures cannot be controlled after adequate trials of two or more anti-seizures medications
- Near-Sudden unexpected death in epilepsy (near-SUDEP)
event from which it is perceived that a person would have died following a seizure, but survived at least for one hour because of resuscitative efforts
- Sudden infant death syndrome (SIDS)
sudden and unexpected death of an infant under the age of one year that remains unexplained after thorough review of the clinic history, death scene investigation, and complete autopsy
- Sudden unexplained death in childhood (SUDC)
sudden death of a child over the age of 12 months that remains unexplained after an autopsy and thorough investigation
- Sudden unexpected death in epilepsy (SUDEP)
sudden, unexpected, witnessed or unwitnessed nontraumatic and non-drowning death in patients with epilepsy, with or without evidence for a seizure and excluding documented status epilepticus, in which postmortem examination does not reveal a toxicological or anatomic cause of death
- Sudden unexpected infant death (SUID)
sudden and unexpected death in an infant; includes SIDS and all other causes of death
- Sudden unexplained death in the young (SUDY)
sudden death in a young person between 1–35 years of age
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Willinger M et al. (1991) Defining the sudden infant death syndrome (SIDS): deliberations of an expert panel convened by the National Institute of Child Health and Human Development. Pediatr. Pathol 11, 677–684 [DOI] [PubMed] [Google Scholar]
- 2.Goldstein RD et al. (2016) Sudden unexpected death in fetal life through early childhood. Pediatrics 137, pii: e20154661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kinney HC and Thach BT (2009) The sudden infant death syndrome. N. Engl. J. Med 361, 795–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kattwinkel J et al. (1994) Infant sleep position and sudden infant death syndrome (SIDS) in the United States: joint commentary from the American Academy of Pediatrics and selected agencies of the Federal Government. Pediatrics 93, 820. [PubMed] [Google Scholar]
- 5.AAP Task Force on Infant Positioning and SIDS (1992) Positioning and SIDS. Pediatrics 89, 1120–1126 [PubMed] [Google Scholar]
- 6.Hauck FR and Tanabe KO (2008) International trends in sudden infant death syndrome: stabilization of rates requires further action. Pediatrics 122, 660–666 [DOI] [PubMed] [Google Scholar]
- 7.Nashef L et al. (2012) Unifying the definitions of sudden unexpected death in epilepsy. Epilepsia 53, 227–233 [DOI] [PubMed] [Google Scholar]
- 8.Harden C et al. (2017) Practice guideline summary: Sudden unexpected death in epilepsy incidence rates and risk factors: Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology 88, 1674–1680 [DOI] [PubMed] [Google Scholar]
- 9.Thurman D (2013) The epidemiology of SUDEP: a public health perspective. Epilepsy Currents 13 (Suppl 2), 923447727 [Google Scholar]
- 10.Dravet C et al. (2011) Severe myoclonic epilepsy in infancy (Dravet syndrome) 30 years later. Epilepsia 52 Suppl 2, 1–2 [DOI] [PubMed] [Google Scholar]
- 11.Devinsky O et al. (2016) Sudden unexpected death in epilepsy: epidemiology, mechanisms, and prevention. Lancet Neurol 15, 1075–1088 [DOI] [PubMed] [Google Scholar]
- 12.Sveinsson O et al. (2017) The incidence of SUDEP: A nationwide population-based cohort study. Neurology 89, 170–177 [DOI] [PubMed] [Google Scholar]
- 13.Keller AE et al. (2018) Incidence of sudden unexpected death in epilepsy in children is similar to adults. Neurology 91, e107–e111 [DOI] [PubMed] [Google Scholar]
- 14.Thurman DJ et al. (2014) Sudden unexpected death in epilepsy: assessing the public health burden. Epilepsia 55, 1479–1485 [DOI] [PubMed] [Google Scholar]
- 15.Richerson GB and Buchanan GF (2011) The serotonin axis: Shared mechanisms in seizures, depression, and SUDEP. Epilepsia 52 Suppl 1, 28–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kinney HC et al. (2009) The brainstem and serotonin in the sudden infant death syndrome. Annu. Rev. Pathol 4, 517–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tao JX et al. (2015) Should the “Back to Sleep” campaign be advocated for SUDEP prevention? Epilepsy Behav 45, 79–80 [DOI] [PubMed] [Google Scholar]
- 18.Hefti MM et al. (2016) Sudden unexpected death in early childhood: general observations in a series of 151 cases: Part 1 of the investigations of the San Diego SUDC Research Project. Forensic Sci. Med. Pathol 12, 4–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krous HF et al. (2004) Sudden infant death syndrome and unclassified sudden infant deaths: a definitional and diagnostic approach. Pediatrics 114, 234–238 [DOI] [PubMed] [Google Scholar]
- 20.Thach BT (2015) Potential Central Nervous System Involvement in Sudden Unexpected Infant Deaths and the Sudden Infant Death Syndrome. Compr. Physiol 5, 1061–1068 [DOI] [PubMed] [Google Scholar]
- 21.Kinney HC et al. (2013) Witnessed sleep-related seizure and sudden unexpected death in infancy: a case report. Forensic Sci. Med. Pathol 9, 418–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brownstein CA et al. (2018) The Genetics of Sudden Infant Death Syndrome. In: Duncan JR, Byard RW, eds. SIDS, Sudden Infant, and Childhood Death: The Past, the Present, and the Future Adelaide (AU: ): University of Adelaide Press; pp. 711–730. [PubMed] [Google Scholar]
- 23.Kinney HC et al. (2009) Sudden death, febrile seizures, and hippocampal and temporal lobe maldevelopment in toddlers: a new entity. Pediatr. Dev. Pathol 12, 455–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Edlow BL et al. (2016) The Structural Connectome of the Human Central Homeostatic Network. Brain Connect 6, 187–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kinney HC et al. (2015) Dentate gyrus abnormalities in sudden unexplained death in infants: morphological marker of underlying brain vulnerability. Acta Neuropathol 129, 65–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hefti MM et al. (2016) Hippocampal malformation associated with sudden death in early childhood: a neuropathologic study: Part 2 of the investigations of The San Diego SUDC Research Project. Forensic Sci. Med. Pathol 12, 14–25 [DOI] [PubMed] [Google Scholar]
- 27.Kinney HC et al. (2007) Sudden death in toddlers associated with developmental abnormalities of the hippocampus: a report of five cases. Pediatr. Dev. Pathol 10, 208–223 [DOI] [PubMed] [Google Scholar]
- 28.Houser CR (1990) Granule cell dispersion in the dentate gyrus of humans with temporal lobe epilepsy. Brain Res 535, 195–204 [DOI] [PubMed] [Google Scholar]
- 29.Armstrong DD (1993) The neuropathology of temporal lobe epilepsy. J Neuropathol. Exp. Neurol 52, 433–443 [DOI] [PubMed] [Google Scholar]
- 30.Berthon-Jones M and Sullivan CE (1984) Ventilation and arousal responses to hypercapnia in normal sleeping humans. J. Appl. Physiol 57, 59–67 [DOI] [PubMed] [Google Scholar]
- 31.Lijowska AS et al. (1997) Sequential arousal and airway-defensive behavior of infants in asphyxial sleep environments. J Appl. Physiol (1985. ) 83, 219–228 [DOI] [PubMed] [Google Scholar]
- 32.Hunt CE (1981) Abnormal hypercarbic and hypoxic sleep arousal responses in near-miss SIDS infants. Pediatr Res 15, 1462–1464 [DOI] [PubMed] [Google Scholar]
- 33.Tolner EA et al. (2011) Five percent CO(2) is a potent, fast-acting inhalation anticonvulsant. Epilepsia 52, 104–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang S et al. (2016) The incidence of peri-ictal prone position in patients with generalized convulsive seizures. Epilepsy Behav 61, 158–161 [DOI] [PubMed] [Google Scholar]
- 35.Liebenthal JA et al. (2015) Association of prone position with sudden unexpected death in epilepsy. Neurology 84, 703–709 [DOI] [PubMed] [Google Scholar]
- 36.Purnell BS et al. (2018) Dead in the night: Sleep-wake and time-of-day influences on sudden unexpected death in epilepsy. Front Neurol 9, 1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lacuey N et al. (2018) Ictal laryngospasm monitored by video-EEG and polygraphy: a potential SUDEP mechanism. Epileptic. Disord 20, 146–150 [DOI] [PubMed] [Google Scholar]
- 38.Stewart M (2018) An explanation for sudden death in epilepsy (SUDEP). J Physiol Sci 68, 307–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakase K et al. (2016) Laryngospasm, central and obstructive apnea during seizures: Defining pathophysiology for sudden death in a rat model. Epilepsy Res 128, 126–139 [DOI] [PubMed] [Google Scholar]
- 40.Berry RB and Gleeson K (1997) Respiratory arousal from sleep: mechanisms and significance. Sleep 20, 654–675 [DOI] [PubMed] [Google Scholar]
- 41.Gleeson K et al. (1990) The influence of increasing ventilatory effort on arousal from sleep. Am. Rev. Respir. Dis 142, 295–300 [DOI] [PubMed] [Google Scholar]
- 42.Smith HR et al. (2018) Dorsal raphe serotonin neurons mediate CO2-induced arousal from sleep. J Neurosci 38, 1915–1925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Buchanan GF and Richerson GB (2010) Central serotonin neurons are required for arousal to CO2. Proc. Natl. Acad. Sci. USA 107, 16354–16359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guyenet PG and Abbott SB (2013) Chemoreception and asphyxia-induced arousal. Respir. Physiol Neurobiol 188, 333–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaur S et al. (2017) A genetically defined circuit for arousal from sleep during hypercapnia. Neuron 96, 1153–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Buchanan GF et al. (2015) 5-HT2A receptor activation is necessary for CO2-induced arousal. J. Neurophysiol 114, 233–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Abbott SB et al. (2013) Optogenetic stimulation of c1 and retrotrapezoid nucleus neurons causes sleep state-dependent cardiorespiratory stimulation and arousal in rats. Hypertension 61, 835–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pineda J and Aghajanian GK (1997) Carbon dioxide regulates the tonic activity of locus coeruleus neurons by modulating a proton- and polyamine-sensitive inward rectifier potassium current. Neuroscience 77, 723–743 [DOI] [PubMed] [Google Scholar]
- 49.Williams RH et al. (2007) Control of hypothalamic orexin neurons by acid and CO2. Proc. Natl. Acad. Sci. USA 104, 10685–10690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Severson CA et al. (2003) Midbrain serotonergic neurons are central pH chemoreceptors. Nat. Neurosci 6, 1139–1140 [DOI] [PubMed] [Google Scholar]
- 51.Veasey SC et al. (1997) Single-unit responses of serotonergic dorsal raphe neurons to specific motor challenges in freely moving cats. Neuroscience 79, 161–169 [DOI] [PubMed] [Google Scholar]
- 52.Kaur S et al. (2013) Glutamatergic signaling from the parabrachial nucleus plays a critical role in hypercapnic arousal. J Neurosci 33, 7627–7640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yokota S et al. (2015) Respiratory-related outputs of glutamatergic, hypercapnia-responsive parabrachial neurons in mice. J Comp Neurol 523, 907–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Haynes RL et al. (2017) High serum serotonin in sudden infant death syndrome. Proc. Natl. Acad. Sci. USA 114, 7695–7700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Duncan JR et al. (2010) Brainstem serotonergic deficiency in sudden infant death syndrome. JAMA 303, 430–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Paterson DS et al. (2006) Multiple serotonergic brainstem abnormalities in sudden infant death syndrome. JAMA 296, 2124–2132 [DOI] [PubMed] [Google Scholar]
- 57.Paterson DS et al. (2006) Serotonergic and glutamatergic neurons at the ventral medullary surface of the human infant: observations relevant to central chemosensitivity in early human life. Autonom Neurosci 124, 112–124 [DOI] [PubMed] [Google Scholar]
- 58.Kinney HC et al. (2003) Serotonergic brainstem abnormalities in northern plains Indians with the sudden infant death syndrome. J Neuropathol Exp Neurol 62, 1178–1191 [DOI] [PubMed] [Google Scholar]
- 59.Kinney HC et al. (2001) Medullary serotonergic network deficiency in the sudden infant death syndrome: review of a 15-year study of a single dataset. J Neuropathol Exp Neurol 60, 228–247 [DOI] [PubMed] [Google Scholar]
- 60.Panigraphy A et al. (2000) Decreased serotonergic receptor binding in rhombic lip-derived regions of the medulla oblongata in the sudden infant death syndrome. J. Neuropathol. Exp. Neurol 59, 377–384 [DOI] [PubMed] [Google Scholar]
- 61.Bright FM et al. (2017) Medullary Serotonin Neuron Abnormalities in an Australian Cohort of Sudden Infant Death Syndrome. J Neuropathol. Exp. Neurol 76, 864–873 [DOI] [PubMed] [Google Scholar]
- 62.Rognum IJ et al. (2014) Serotonin metabolites in the cerebrospinal fluid in sudden infant death syndrome. J Neuropathol. Exp. Neurol 73, 115–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bateman LM et al. (2010) Serotonin reuptake inhibitors are associated with reduced severity of ictal hypoxemia in medically refractory partial epilepsy. Epilepsia 51, 2211–2214 [DOI] [PubMed] [Google Scholar]
- 64.Tupal S and Faingold CL (2006) Evidence supporting a role of serotonin in modulation of sudden death induced by seizures in DBA/2 mice. Epilepsia 47, 21–26 [DOI] [PubMed] [Google Scholar]
- 65.Uteshev VV et al. (2010) Abnormal serotonin receptor expression in DBA/2 mice associated with susceptibility to sudden death due to respiratory arrest. Epilepsy Res 88, 183–188 [DOI] [PubMed] [Google Scholar]
- 66.Faingold CL et al. (2010) DBA/1 mice exhibit chronic susceptibility to audiogenic seizures followed by sudden death associated with respiratory arrest. Epilepsy Behav 17, 436–440 [DOI] [PubMed] [Google Scholar]
- 67.Faingold CL et al. (2011) Differences in serotonin receptor expression in the brainstem may explain the differential ability of a serotonin agonist to block seizure-induced sudden death in DBA/2 vs. DBA/1 mice. Brain Res 1418, 104–110 [DOI] [PubMed] [Google Scholar]
- 68.Faingold CL et al. (2011) Prevention of seizure-induced sudden death in a chronic SUDEP model by semichronic administration of a selective serotonin reuptake inhibitor. Epilepsy Behav 22, 186–190 [DOI] [PubMed] [Google Scholar]
- 69.Faingold CL et al. (2016) Serotonergic agents act on 5-HT3 receptors in the brain to block seizure-induced respiratory arrest in the DBA/1 mouse model of SUDEP. Epilepsy Behav 64, 166–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Feng HJ and Faingold CL (2017) Abnormalities of serotonergic neurotransmission in animal models of SUDEP. Epilepsy Behav 71, 174–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kommajosyula SP et al. (2017) Specific subcortical structures are activated during seizure-induced death in a model of sudden unexpected death in epilepsy (SUDEP): A manganese-enhanced magnetic resonance imaging study. Epilepsy Res 135, 87–94 [DOI] [PubMed] [Google Scholar]
- 72.Buchanan GF et al. (2014) Serotonin neurones have anti-convulsant effects and reduce seizure-induced mortality. J Physiol 592, 4395–4410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhan Q et al. (2016) Impaired Serotonergic Brainstem Function during and after Seizures. J Neurosci 36, 2711–2722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Allen LA et al. (2017) Dysfunctional Brain Networking among Autonomic Regulatory Structures in Temporal Lobe Epilepsy Patients at High Risk of Sudden Unexpected Death in Epilepsy. Front Neurol 8, 544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mueller SG et al. (2018) Brainstem network disruption: A pathway to sudden unexplained death in epilepsy? Hum. Brain Mapp, August 10. doi: 10.1002/hbm.24325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mueller SG et al. (2014) Evidence for brainstem network disruption in temporal lobe epilepsy and sudden unexplained death in epilepsy. Neuroimage. Clin 5, 208–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Patodia S et al. (2018) The ventrolateral medulla and medullary raphe in sudden unexpected death in epilepsy. Brain 141, 1719–1733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wandschneider B et al. (2015) Structural imaging biomarkers of sudden unexpected death in epilepsy. Brain 138, 2907–2919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Michalak Z et al. (2017) Neuropathology of SUDEP: Role of inflammation, blood-brain barrier impairment, and hypoxia. Neurology 88, 551–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dlouhy BJ et al. (2015) Breathing Inhibited When Seizures Spread to the Amygdala and upon Amygdala Stimulation. J Neurosci 35, 10281–10289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nobis WP et al. (2018) Amygdala-stimulation-induced apnea is attention and nasal-breathing dependent. Ann. Neurol 83, 460–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Aiba I and Noebels JL (2015) Spreading depolarization in the brainstem mediates sudden cardiorespiratory arrest in mouse SUDEP models. Sci. Transl. Med 7, 282ra46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Aiba I et al. (2016) Leaky RyR2 channels unleash a brainstem spreading depolarization mechanism of sudden cardiac death. Proc. Natl. Acad. Sci. U. S. A 113, E4895–E4903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Aiba I and Shuttleworth CW (2014) Characterization of inhibitory GABA-A receptor activation during spreading depolarization in brain slice. PLoS. One 9, e110849. [DOI] [PMC free article] [PubMed] [Google Scholar]