

Abstract
Cryo-electron microscopy (cryo-EM) is rapidly emerging as a powerful tool for protein structure determination at high resolution. Here, we report the structure of a complex between Escherichia coli β-galactosidase and the cell-permeant inhibitor phenylethyl β-d-thiogalactopyranoside (PETG), determined by cryo-EM at a resolution of ~2.2 Å. Besides the PETG ligand, we identified densities in the map for ~800 water molecules and for magnesium and sodium ions. While it is likely that continued advances in detector technology may further enhance resolution, our findings demonstrate that preparation of specimens of adequate quality and intrinsic protein flexibility, rather than imaging or image processing technologies, now represent the major bottlenecks to achieving resolutions close to 2 Å using single particle cryo-EM.
Icosahedral viruses were the first biological assemblies whose structures were determined at near-atomic resolution using cryo-electron microscopy (cryo-EM) combined with methods for image averaging (1–10). Over the last two years, structures for a variety of non-viral assemblies have been reported using cryo-EM at resolutions between ~2.8 Å and ~4.5 Å (11–20). Four of these instances have been of complexes with sizes below 1 MDa: the 700 kDa proteasome at 3.3 Å (16) and 2.8 Å resolution (17), the 465 kDa E. coli β-galactosidase at 3.2 Å resolution (18), the 440 kDa anthrax protective antigen pore at 2.9 Å (19), and the 300 kDa TrpVl ion channel at 3.4 Å resolution (20). Because these structures are of complexes that are dispersed in the aqueous phase, the peripheral regions of the proteins are less ordered, and are at lower resolution than the more central regions; nevertheless, most side-chain densities are clearly delineated in the well-ordered regions of the maps. In crystallographically determined structures of proteins at resolutions of 2.3 Å or better, features such as protein-ligand hydrogen bonding, salt bridges and location of key structured water molecules can be ascertained with a high degree of confidence (21). There is great potential for the use of cryo-EM methods in applications such as drug discovery and development if similar resolutions could be achieved without crystallization. Whether there are fundamental limitations with currently available methods for specimen preparation, microscope hardware, inelastic scattering from the ice layer, inaccuracies in microscope alignment, detector technology, data collection procedures or image processing software to achieve resolutions approaching 2 Å is a question that remains unanswered in the current context of rapid advances in the cryo-EM field (22). This is especially relevant for smaller protein complexes (<1 MDa) with low symmetry where the errors in alignment of the projection images make the analysis more challenging than for larger or more symmetric complexes such as ribosomes and ordered viruses (23).
We recently reported the structure of E. coli β-galactosidase at 3.2 Å resolution (18). Comparing the cryo-EM derived structure with that derived by x-ray crystallography, we identified regions such as the periphery of the protein and crystal contact zones where there were measurable deviations between crystal and solution structures. To test whether we could further improve map resolution, we explored a range of experimental conditions including variations in specimen preparation, imaging and steps in data processing. We analysed the structure of β-galactosidase bound to phenylethyl β-d-thiogalactosidase (PETG), a potent inhibitor that blocks enzyme activity by replacing the oxygen in the O-glycosidic bond with a sulfur atom. Although no crystal structure is available for the complex formed between E. coli β-galactosidase and PETG, a crystal structure is available for PETG bound to T. reesei β-galactosidase (24). There is, however, very little sequence similarity (sequence identity of 12.8% determined by Clustal 12.1) between the two variants, with the Trichoderma reesei variant displaying a completely different fold and crystallizing as a monomer instead of a tetramer (fig. S1).
Cryo-EM images recorded from plunge-frozen specimens of the β-galactosidase PETG complex and the corresponding radially averaged power spectra were analyzed to select images displaying signal at high resolution (fig. S2, A to C). For each recorded image, we also assessed the extent of movement during the course of the ~8 s exposure (fig. S2D). From a dataset of 1487 images, which displayed detectable signal in the power spectra extending beyond 3 Å and had low amounts of beam-induced movement during the exposure, we extracted 93,686 projection images using automated particle selection procedures using a Gaussian disk as a template. We used various combinations and subsets of the frames collected from each region and iteratively evaluated their contribution to map quality (fig. S3A). The final map, which we assessed as having the highest overall map quality, was obtained using the information in the images collected from ~12 e−/Å2-of each exposure (Fig. 1A). We estimate the overall average resolution of the map to be ~2.2 Å using both the 0.143 Fourier Shell Correlation (FSC) criterion, as well as the resolution at which an FSC obtained between the experimental cryo-EM map and the map computed from the map-derived cryo-EM atomic model has a value of 0.5 (fig. S3B). This was further supported by visual inspection of map quality (movie SI). The 2.2 Å mean resolution of our map indicates that some regions such as at the periphery are at lower resolution than 2.2 Å, while other regions closer to the center are at higher resolution, displaying features consistent with electron density maps from x-ray structures determined at resolutions of ~2 Å (fig. S4).
Fig. 1. Cryo-EM density map of β-galactosidase-PETG complex at 2.2 Å resolution.
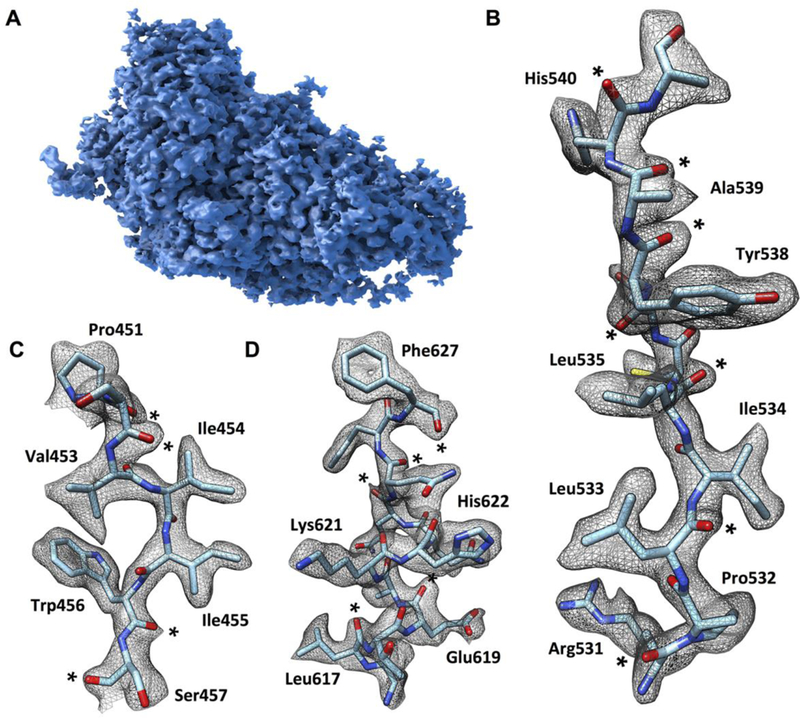
(A) Surface representation of the density map of one of the four protomers in the tetrameric complex. (B to D) Visualization of selected map regions showing delineation of secondary structural elements, amino acid densities and carbonyl moieties (indicated by *). The density for Phe 627 is thinned out in the center of the aromatic ring, revealing the presence of a “hole” in the ring, a feature typically observed in structures determined by x-ray crystallography at resolutions of ~2 Å.
An overview of the density map for one of the four equivalent chains in the β-galactosidase complex and densities for regions from different portions of the molecule are shown in Fig. 1. The path of the polypeptide chain is well-delineated, enabling placement of the sequence into the density map (Fig. 1, B to D). Examination of the map also shows clear densities for several backbone carbonyl groups and several ordered water molecules in the structure. We identified 194 densities in each protomer where we could place water molecules with confidence based on the shape of the local density, map value and location at the right distance range for hydrogen bonding to polar groups in the vicinity. In the vast majority of instances, these water molecules are at locations also identified in the 1.7 Å crystal structure of β-galactosidase (PDB 1DP0), providing independent validation of their assignment. Selected examples of tightly bound water molecules in the structure are shown in Fig. 2, illustrating instances where they are present in connected chains, coordinated to multiple polar residues, coordinated to the polypeptide backbone, or coordinated to the Mg2+ ion in the active site. The fact that water molecules can be placed with confidence in a structure of a 465 kDa complex determined by single particle cryo-EM is an exciting advance that bodes well for the use of cryo-EM in drug discovery applications.
Fig. 2. Visualization of tightly bound water molecules in the structure of the β-galactosidase-PETG complex.
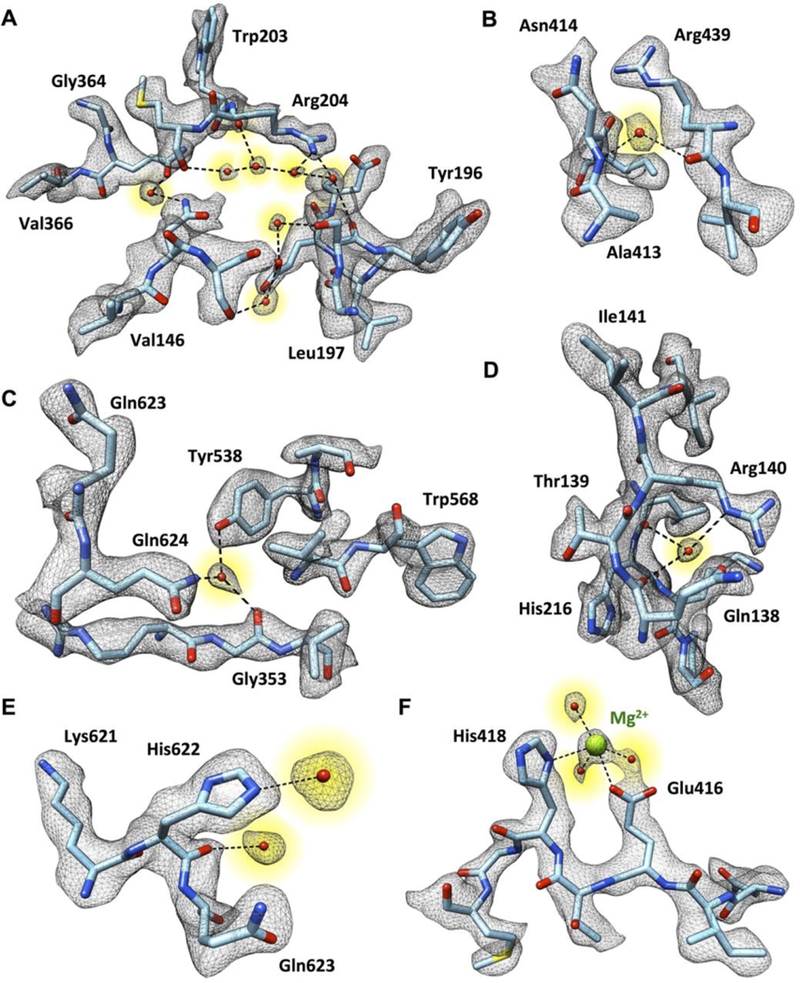
(A to F) Selected examples of densities for water molecules (highlighted in yellow) hydrogen-bonded in pearl-string-like chains (A), connected to the polypeptide backbone and multiple amino acid side chains (B to E), or interacting with the Mg2+ ion in the active site (F).
The cryo-EM map includes density for PETG in the active site (Fig. 3A). The key catalytic residues Glu461 (general base for acid catalysis) coordinating to the Mg2+ ion and Glu537 (nucleophile) are in close proximity to the ligand, with density for a structured water molecule visible in the binding pocket. In addition, other residues that stabilize the binding of the inhibitor: Asn102, Asp201, Met502, Tyr503, Trp568, His540, Phe601 and Trp999, also show appropriate steric dispositions as shown in Fig. 3B. There are substantial differences in the orientation and location of PETG molecule in the T. reesei enzyme (Fig. 3C), with rotation of the benzyl moiety around the S-C7 single bond by almost 180 degrees. This is perhaps not unexpected given that there is very little overall structural similarity between the enzymes from these two species (fig. SI), and the pattern of residues that are involved in H-bonding to the ligand are also different (Fig. 3, B and D). Stereochemical parameters of the key conserved catalytic residues (Glu200 and Glu298) in the T. reesei complex are different from those in the E. coli complex, as are the general distribution of non-polar residues that stabilize the inhibitor in the pocket, establishing the value of direct determination of the actual structure of the ligand in a protein complex even when a related structure is available.
Fig. 3. Active site structure in PETG-liganded E. coli β-galactosidase.
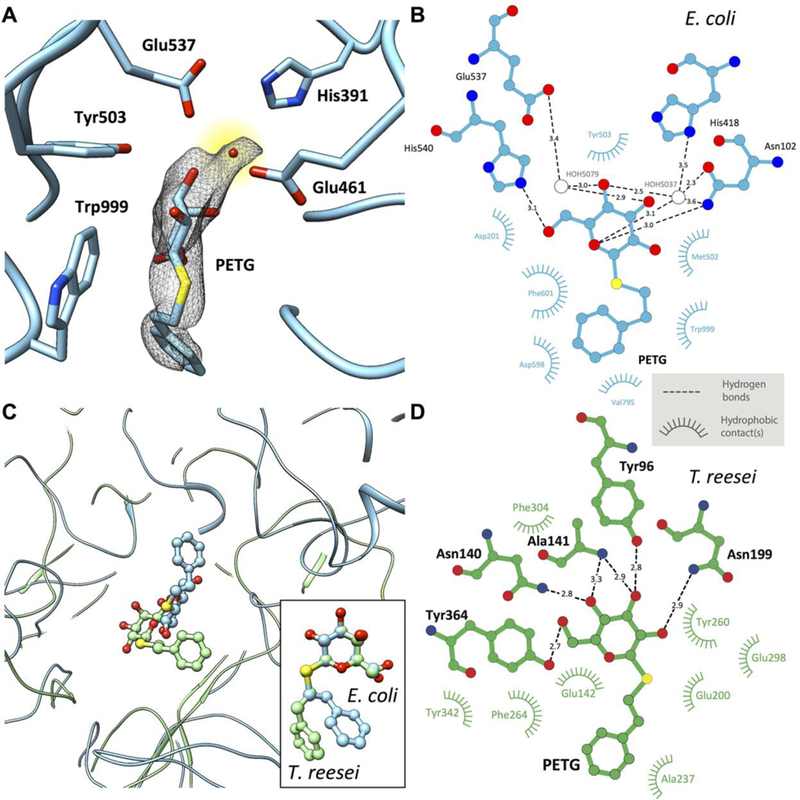
(A) Uncorrected cryo-EM density map showing density for PETG, an associated water molecule and five of the amino acids that line the binding pocket. The density for the PETG ligand appears to be at lower resolution than the surrounding amino acids, presumably because of some wobble in the binding pocket. (B) Plot of distances of various parts of PETG to residues in the vicinity of (β-galactosidase from E. coli determined using LIGPLOT (www.ebi.ac.uk/thornton-srv/software/LIGPLOT/). (C) Superposition of the ligand binding pocket structures in (β-galactosidase from E. coli (cyan, determined by cryo-EM at 2.2 Å resolution) and T. reesei (green, determined by x-ray crystallography at 1.4 Å resolution) illustrating the differences in protein and ligand structures, and comparison between the corresponding configurations of PETG (inset). (D) Plot of distances of various parts of PETG to amino acids in the vicinity of β-galactosidase from T. reesei determined using LIGPLOT.
In Fig. 4A, we show examples of the densities observed for each of the 20 standard amino acids, where the level of detail at which individual C, N and O atoms are observed is consistent with maps derived from x-ray crystallography at nominal resolutions of ~2 Å (fig. S4). However, in contrast to a 2Fo-Fc map obtained by x-ray crystallography, both phases and amplitudes of cryo-EM density maps are derived experimentally from the images, eliminating the need to assign phases derived from the atomic model as is customarily done in x-ray crystallography. As a consequence, density contours in cryo-EM maps are subject to inaccuracies from a number of resolution-lowering distortions (instances of which are visible in Fig. 4A) that can arise at various stages of data collection and processing. Factors that can contribute to distortions include inaccuracies in determination of the contrast transfer function for each image, errors in orientation determination during refinement, unique patterns of radiation damage in each of the molecular images used for reconstruction and the changes introduced from applying a uniform temperature factor correction to scale the map. Despite these distortions, which appear to be random, the overall shape of the residues can nevertheless be distinguished clearly. As more structures are determined at these higher resolutions, it is possible that there may be enough statistical basis to study these distortions quantitatively, and perhaps exploit patterns that may emerge from this analysis to improve refinement strategies in order to achieve even higher resolution.
Fig. 4. Illustration of map quality at the level of amino acids.
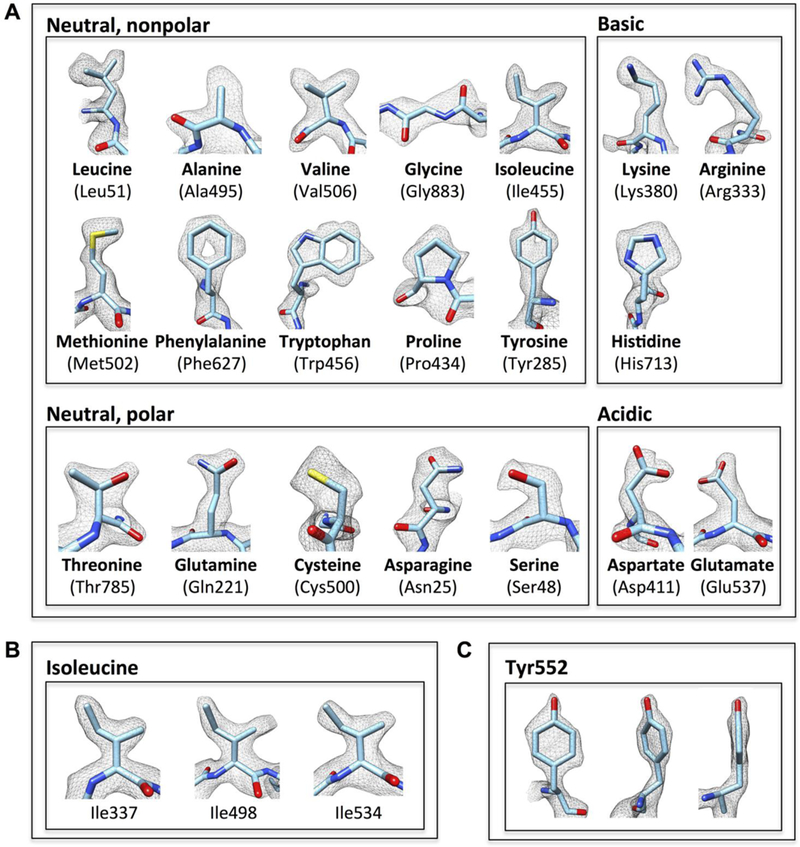
(A) Visualization of map density for examples of each of the 20 standard amino acids, which are grouped into neutral (nonpolar and polar), basic and acidic categories. (B and C) Illustration of contours of densities for multiple lie residues (B), and front, tilted and edge views for Tyr552 (C). In each case, the density contours are consistent with the 2.2 Å resolution we report.
The data collection schemes currently used in cryo-EM with direct electron detectors enables the use of numerous combinations in which the dose can be fractionated during the exposure, and a number of ways in which different subsets of the frames collected for each exposure can be combined to generate a 3D reconstruction. The map we present in Figs. 1 to 4 was obtained from a subset that excludes the very early portion of the exposure, uses the next 12 e-/Å2, and excludes the latter part of the exposure. In the course of our studies, we analyzed many different maps constructed by using different subsets of the exposure. The highest resolution features such as holes in the rings of the aromatic residues (Fig. 4 and fig. S4) were better resolved in maps constructed using the interval of the exposure containing the highest resolution information (fig. S2A).
Based on our present analysis, and its comparison with our earlier cryo-EM structure of β-galactosidase at 3.2 Å resolution, we can now articulate our best understanding of all the changes that we introduced that enabled us to improve the resolution to ~2.2 Å. Perhaps the most important is the much more careful selection of regions where the ice was thin enough to obtain the highest detectable signals, yet thick enough to allow a spread of orientations (as judged by the distribution of orientations assigned to each molecular image used to construct the final map). Second, the use of a lower dose rate to minimize the effects of coincidence loss of the detector, and the use of a finer pixel size resulted in improved image contrast and maximization of amplitudes at low resolution (11), which allowed us to go to closer to focus and still be able to correctly pick and align particles. Third, we carried out 3D classification throughout the iterative refinement cycle, which we did not do in the case of the structure at 3.2 Å resolution. Finally, we believe that the use of nearest neighbor interpolation during motion correction, coupled with better quality data than we had before allowed improved recovery of higher resolution information in the final reconstruction.
X-ray crystallographic methods have led to the deposition of almost 95,000 atomic resolution protein and protein-nucleic acid structures over the last few decades. There have been impressive advances in speed, resolution and in the development of highly automated workflows over the years. Relative to those of the x-ray field, cryo-EM methods are still in an early phase of development, with only about 30 deposited models for coordinates derived from electron microscopic analysis at near-atomic resolution. The spectacular recent progress by many groups worldwide suggests that this number will increase rapidly, and extend to specimens that may not be easily amenable to crystallization. Our demonstration here that the structure of a ligand-protein complex can be determined in the solution phase at resolutions close to 2 Å suggests that cryo-EM is positioned to become an indispensable tool in structural biology and for drug discovery applications.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by funds from the Center for Cancer Research, National Cancer Institute, and the IATAP program at NIH, Bethesda, MD. We thank J. J. Fernandez for providing the code to run TOMOCTFFIND. This study utilized the high-performance computational capabilities of the Biowulf Linux cluster at the National Institutes of Health, Bethesda, MD (http://biowulf.nih.gov). We thank V. Falconieri with expert assistance in preparation of figures and the supplementary movie. The density map and refined atomic model have been deposited with the Electron Microscopy Data Bank (accession number EMD-2984) and Protein Data Bank (entry code 5a1a), respectively.
Footnotes
SUPPLEMENTARY MATERIALS
REFERENCES AND NOTES
- 1.Grigorieff N, Harrison SC, Near-atomic resolution reconstructions of icosahedral viruses from electron cryo-microscopy. Curr. Opin. Struct Biol. 21, 265–273 (2011). Medline doi: 10.1016/j.sbi.2011.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guo F, Liu Z, Fang PA, Zhang Q, Wright ET, Wu W, Zhang C, Vago F, Ren Y, Jakana J, Chiu W, Serwer P, Jiang W, Capsid expansion mechanism of bacteriophage T7 revealed by multistate atomic models derived from cryo-EM reconstructions. Proc. Natl. Acad. Sci. U.S.A. 111, E4606–E4614 (2014). Medline doi: 10.1073/pnas.1407020111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu H, Jin L, Koh SB, Atanasov I, Schein S, Wu L, Zhou ZH, Atomic structure of human adenovirus by cryo-EM reveals interactions among protein networks. Science 329,1038–1043 (2010). Medline doi: 10.1126/science.1187433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu X, Zhang Q, Murata K, Baker ML, Sullivan MB, Fu C, Dougherty MT, Schmid MF, Osburne MS, Chisholm SW, Chiu W, Structural changes in a marine podovirus associated with release of its genome into Prochlorococcus. Nat. Struct. Mol. Biol. 17, 830–836 (2010). Medline doi: 10.1038/nsmh.1823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Settembre EC, Chen JZ, Dormitzer PR, Grigorieff N, Harrison SC, Atomic model of an infectious rotavirus particle. EMB0 J. 30, 408–416 (2011). Medline doi: 10.1038/emboj.2010.322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Z, Hryc CF, Bammes B, Afonine PV, Jakana J, Chen DH, Liu X, Baker ML, Kao C, Ludtke SJ, Schmid MF, Adams PD, Chiu W, An atomic model of brome mosaic virus using direct electron detection and real-space optimization. Nat. Commun. 5,4808 (2014). Medline doi: 10.1038/ncomms5808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wolf M, Garcea RL, Grigorieff N, Harrison SC, Subunit interactions in bovine papillomavirus. Proc. Natl. Acad. Sci. U.S.A. 107, 6298–6303 (2010). Medline doi: 10.1073/pnas.0914604107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu X, Ge P, Jiang J, Atanasov I, Zhou ZH, Atomic model of CPV reveals the mechanism used by this single-shelled virus to economically carry out functions conserved in multishelled reoviruses. Structure 19, 652–661 (2011). Medline doi: 10.1016/j.str.2011.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang X, Ge P, Yu X, Brannan JM, Bi G, Zhang Q, Schein S, Zhou ZH, Cryo-EM structure of the mature dengue virus at 3.5-Å resolution. Nat. Struct. Mol. Biol. 20,105–110 (2013). Medline doi: 10.1038/nsmb.2463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang X, Jin L, Fang Q, Hui WH, Zhou ZH, 3.3 Å cryo-EM structure of a nonenveloped virus reveals a priming mechanism for cell entry. Cell 141, 472–482 (2010). Medline doi: 10.1016/j.cell.2010.03.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bai XC, Fernandez IS, McMullan G, Scheres SH, Ribosome structures to near-atomic resolution from thirty thousand cryo-EM particles. eLife 2, e00461 (2013). Medline doi: 10.7554/eLife.00461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amunts A, Brown A, Bai XC, Llácer JL, Hussain T, Emsley P, Long F, Murshudov G, Scheres SH, Ramakrishnan V, Structure of the yeast mitochondrial large ribosomal subunit. Science 343,1485–1489 (2014). Medline doi: 10.1126/science.1249410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greber BJ, Boehringer D, Leibundgut M, Bieri P, Leitner A, Schmitz N, Aebersold R, Ban N, The complete structure of the large subunit of the mammalian mitochondrial ribosome. Nature 515, 283–286 (2014). Medline [DOI] [PubMed] [Google Scholar]
- 14.Lu P, Bai XC, Ma D, Xie T, Yan C, Sun L, Yang G, Zhao Y, Zhou R, Scheres SH, Shi Y, Three-dimensional structure of human γ-secretase. Nature 512, 166–170 (2014). Medline doi: 10.1038/nature13567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fischer N, Neumann P, Konevega AL, Bock LV, Ficner R, Rodnina MV, Stark H, Structure of the E. coli ribosome-EF-Tu complex at <3 Å resolution by Cs-corrected cryo-EM. Nature 520, 567–570 (2015). Medline doi: 10.1038/nature14275 [DOI] [PubMed] [Google Scholar]
- 16.Li X, Mooney P, Zheng S, Booth CR, Braunfeld MB, Gubbens S, Agard DA, Cheng Y, Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat. Methods 10, 584–590 (2013). Medline doi: 10.1038/nmeth.2472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Campbell MG, Veesler D, Cheng A, Potter CS, Carragher B, 2.8 Å resolution reconstruction of the Thermoplasma acldophilum 20S proteasome using cryo-electron microscopy. eLife 4, e06380 (2015). Medline doi: 10.7554/eLife.06380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bartesaghi A, Matthies D, Banerjee S, Merk A, Subramaniam S, Structure of β-galactosidase at 3.2-Å resolution obtained by cryo-electron microscopy. Proc. Natl. Acad. Sci. U.S.A. 111, 11709–11714 (2014). Medline doi: 10.1073/pnas.1402809111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang J, Pentelute BL, Collier RJ, Zhou ZH, Atomic structure of anthrax protective antigen pore elucidates toxin translocation. Nature 10.1038/nature14247 (2015). Medline doi: 10.1038/nature14247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liao M. Cao E, Julius D. Cheng Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 504,107–112 (2013). Medline doi: 10.1038/nature12822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blundell TL, Jhoti H, Abell C, High-throughput crystallography for lead discovery in drug design. Nat. Rev. Drug Discov. 1, 45–54 (2002). Medline doi: 10.1038/nrd706 [DOI] [PubMed] [Google Scholar]
- 22.Agard D, Cheng Y, Glaeser RM, Subramaniam S, in Advances in Imaging and Electron Physics, Hawkes PW, Ed. (Elsevier, 2014), chap. 2, pp. 113–137. [Google Scholar]
- 23.Henderson R, The potential and limitations of neutrons, electrons and x-rays for atomic resolution microscopy of unstained biological molecules. Q. Rev. Biophys. 28,171–193 (1995). Medline doi: 10.1017/S003358350000305X [DOI] [PubMed] [Google Scholar]
- 24.Maksimainen M, Hakulinen N, Kallio JM, Timoharju T, Turunen O, Rouvinen J, Crystal structures of Trichoderma reesei β-galactosidase reveal conformational changes in the active site. J. Struct. Biol. 174, 156–163 (2011). Medline doi: 10.1016/j.jsb.2010.11.024 [DOI] [PubMed] [Google Scholar]
- 25.Grigorieff N, FREALIGN: High-resolution refinement of single particle structures. J. Struct. Biol. 157,117–125 (2007). Medline doi: 10.1016/j.jsb.2006.05.004 [DOI] [PubMed] [Google Scholar]
- 26.Lyumkis D, Brilot AF, Theobald DL, Grigorieff N, Likelihood-based classification of cryo-EM images using FREALIGN. J. Struct. Biol. 183 377–388 (2013). Medline doi: 10.1016/j.jsb.2013.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Emsley P, Lohkamp B, Scott WG, Cowtan K, Features and development of Coot. Acta Crystallogr. D 66,486–501 (2010). Medline [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zwart PH, Afonine PV, Grosse-Kunstleve RW, Hung LW, loerger TR, McCoy AJ, McKee E, Moriarity NW, Read RJ, Sacchettini JC, Sauter NK, Storoni LC, Terwilliger TC, Adams PD, Automated structure solution with the PHENIX suite. Methods Mol Biol. 426, 419–435 (2008)._Medline doi: 10.1007/978-1-60327-058-8_28 [DOI] [PubMed] [Google Scholar]
- 29.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE, UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004). Medline doi: 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- 30.Coles CH, Shen Y, Tenney AP, Siebold C, Sutton GC, Lu W, Gallagher JT, Jones EY, Flanagan JG, Aricescu AR, Proteoglycan-specific molecular switch for RPTPσ clustering and neuronal extension. Science 332, 484–488 (2011). Medline doi: 10.1126/science.1200840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bommer M, Kunze C, Fesseler J, Schubert T, Diekert G, Dobbek H, Structural basis for organohalide respiration. Science 346, 455–458 (2014). Medline doi: 10.1126/science.1258118 [DOI] [PubMed] [Google Scholar]
- 32.Du J, Zhou Y, Su X, Yu JJ, Khan S, Jiang H, Kim J, Woo J, Kim JH, Choi BH, He B, Chen W, Zhang S, Cerione RA, Auwerx J, Hao Q, Lin H, Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 334, 806–809 (2011). Medline doi: 10.1126/science.1207861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dreyfus C, Laursen NS, Kwaks T, Zuijdgeest D, Khayat R, Ekiert DC, Lee JH, Maetlagel Z, Bujny MV, Jongeneelen M, van der Vlugt R, Lamrani M, Korse HJ, Geelen E, Sahin Ö, Sieuwerts M, Brakenhoff JP, Vogels R, Li OT, Poon LL, Peiris M, Koudstaal W, Ward AB, Wilson IA, Goudsmit J, Friesen RH, Highly conserved protective epitopes on influenza B viruses. Science 337,1343–1348(2012). Medline [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.