

Abstract
Disease-modifying therapies are needed for Fragile X Syndrome (FXS), as at present there are no effective treatments or cures. Herein, we report on a tetrahydroquinoline-based selective histone deacetylase 6 (HDAC6) inhibitor SW-100, its pharmacological and ADMET properties, and its ability to improve upon memory performance in a mouse model of FXS, Fmr1−/− mice. This small molecule demonstrates good brain penetrance, low-nanomolar potency for the inhibition of HDAC6 (IC50 = 2.3 nM), with at least a thousand-fold selectivity over all other class I, II, and IV HDAC isoforms. Moreover, through its inhibition of the α-tubulin deacetylase domain of HDAC6 (CD2), in cells SW-100 upregulates α-tubulin acetylation with no effect on histone acetylation and selectively restores the impaired acetylated α-tubulin levels in the hippocampus of Fmr1−/− mice. Lastly, SW-100 ameliorates several memory and learning impairments in Fmr1−/− mice, thus modeling the intellectual deficiencies associated with FXS, and hence providing a strong rationale for pursuing HDAC6-based therapies for the treatment of this rare disease.
Keywords: Phenylhydroxamate, permeability, Ames negative, acetylated α-tubulin, memory and learning impairments
Graphical Abstract
INTRODUCTION
Fragile X Syndrome (FXS) is the most common form of inherited severe cognitive impairment in males (1 in 3000–4000 individuals) and a significant cause of intellectual disability in females (1 in 7000–8000 individuals).1 This condition causes an array of developmental problems including learning disability, cognitive impairment, and behavioral characteristics. Other signs and symptoms may include autism spectrum disorders, seizures, and characteristic physical features. In most cases, FXS is caused by transcriptional silencing of the fragile X mental retardation 1 (FMR1) gene due to the methylation of a cytosine-guanine-guanine (CGG) trinucleotide repeat expansion (number of repeats in FXS > 200, normal < 50),2 resulting in the absence of the gene product, fragile X mental retardation protein (FMRP) which regulates mRNA trafficking and dendritic translation. FMRP deficiency affects the translational regulation of multiple proteins, including several that are important for learning and memory.3–5 At present, there are no effective treatments or cures for FXS. Metabotropic glutamate receptor 5 (mGlu5) inhibitors were considered as a potential therapy for FXS based on the “mGluR theory of FXS”.6–8 Recent phase II clinical trials, however, with two different mGlu5 negative allosteric modulators (basimglurant and mavoglurant) failed to induce significant improvement over placebo for primary end points.9,10 In addition to mGluR5, a diverse set of alternative therapeutic targets such as GSK-3β, ERK1/2, IGF1, and MMP9 have been investigated in animal studies or early clinical trials.11 Among them, the pan-histone deacetylase (HDAC) inhibitor valproic acid (VPA) was found to ameliorate the symptoms of attention deficit hyperactivity disorder (ADHD) in FXS boys.12 After VPA treatment in FXS lymphoblastoid cells, however, FMR1-mRNA levels remained low, FMRP protein was undetectable, and the gene remained methylated, although a significant increase in histone acetylation was observed.13 Additionally, there is accumulating evidence that the downregulation of class I and II HDAC levels leads to neuroprotection or improvement of learning and memory in rodent models of various brain diseases.14–18 Some research studies have demonstrated, for example, that the administration of siRNA targeting class I HDACs 1–3, which are predominantly responsible for regulating histone acetylation, improves cognitive impairments in the FXS intellectual deficiency mouse model.19 However, to date no research has been published on the activity of selective HDAC6 inhibitors in FXS animal models.
Histone deacetylases (HDACs) are a family of proteins responsible for catalyzing the hydrolysis of acetylated histone lysine residues to produce deacylated products. There are 18 known mammalian HDACs, which are divided into four classes, based on their sequence similarity to yeast homologues: class I (HDAC1, 2, 3, and 8), class IIa (HDAC4, 5, 7, and 9), class IIb (HDAC6 and 10), class III (SIRT1–7), and class IV (HDAC11).20 Among these isoforms, HDAC6 has attracted particular attention due to its relative uniqueness within its family: (a) unlike its related class members, HDAC6 contains two tandem protein deacetylase motifs (CD1 and CD2) and has an N-terminal nuclear export sequence and a C-terminal cytoplasmic retention sequence;21,22 (b) unlike other HDAC members, which are present in the nucleus, HDAC6 is found primarily within the cytosol and is responsible for regulating the acetylation state of specific cytosolic proteins, although it is also able to shuttle into the nucleus and thus can also potentially regulate transcription.23 Its regulation of α-tubulin acetylation has sparked considerable interest because microtubule-dependent transport rates are facilitated by higher levels of acetylated α-tubulin.24–27 This effect presumably stems from the increased association of the motor proteins kinesin-1 and dynein with acetylated α-tubulin, which in turn regulates both anterograde and retrograde transport activities.28 In addition to facilitating anterograde transport of new cargo to synaptic zones, acetylated α-tubulin also increases the ability of damaged organelles or misfolded proteins to leave synaptic zones.29 Selective inhibition of HDAC6 thus appears to offer a potential therapeutic approach for treating a number of neurological disorders such as depression,30 stroke,31 Parkinson’s disease,32 Alzheimer’s disease,33–35 Charcot-Marie-Tooth disease,36–38 Rett Syndrome,33,39 and amyotrophic lateral sclerosis (ALS).40 Intriguingly, significantly fewer mitochondria and greatly reduced mitochondrial mobility have been observed in hippocampal neurites of Fragile-X-associated tremor/ataxia syndrome (FXTAS, 50 < number of CGG repeats < 200).41,42 Since HDAC6 inhibitors have been shown to increase mitochondrial movement including vesicular trafficking of brain-derived neurotrophic factor (BDNF),43–45 we postulated that HDAC6 inhibition could provide a useful therapeutic intervention for FXS. Therefore, an early aim of the present study was to investigate whether selective inhibition of HDAC6 could increase the acetylation of α-tubulin in brain, and if so whether that modification would improve the FXS-like phenotypes in Fmr1−/− mice.
A number of broad-spectrum HDAC inhibitors, such as vorinostat (SAHA), romidepsin, panobinostat, and belinostat, are being used as anticancer agents due to their ability to enhance gene expression by increasing histone acetylation, and in turn promoting cell-cycle arrest, differentiation, and apoptosis. However, significant concerns have emerged that pan-HDAC inhibitors may be too broadly acting for clinical use beyond oncology, especially with respect to their potential neurotoxicity.46,47 In the past decade, selective HDAC6 inhibitors (HDAC6is) have been reported,48 and two (poorly selective) HDAC6is (rocilinostat and citarinostat) are in clinical trials for several types of cancer.49–51 However, only a handful of HDAC6is have been reported that are brain-penetrant and display appropriate in vitro and in vivo activities in models of neurological diseases.30,52–55
One selective HDAC6i discovered by our group, tubastatin A (TubA), has been investigated for its potential to be used in central nervous system (CNS) disorders.30–35,39,44 TubA exhibits four of the six desirable physicochemical properties for CNS drugs within the predicted optimal ranges: tPSA (40 < tPSA ≤ 90 Å2), pKa (≤ 8.0, basic moiety), clogP (≤ 3), and MW (≤ 360), whereas two parameters including its HBD (optimally = 0) and clogD (optimally ≤ 2) are in less favorable ranges. As such, TubA receives a relatively high CNS multiparameter optimization (MPO) desirability score of 5.33 (> 5).56–58 Moreover, the positive calculated logBB value of TubA (0.18, determined by ACD/Percepta software) suggests that TubA should cross the blood-brain barrier (BBB) effectively. However, its measured ratio of brain to plasma levels has been reported to be only modest (AUCbrain/ AUCplasma = 0.18).30 Thus, prior to exploring the effects of an HDAC6i in the FXS mouse model, we needed to identify a compound with improved brain penetrance.
To better understand the reasons for the poor brain penetrance of TubA, and to further guide the identification of future novel brain penetrant analogues, we present here a set of HDAC6i analogues selected from our compound library. Using a series of design modifications and in vitro experiments we were able to identify the novel brain-penetrant HDAC6i, designated SW-100. This compound is easy to synthesize, and it possesses low nanomolar potency against HDAC6, excellent isozyme selectivity over other HDACs, selective engagement of HDAC6-CD2 as measured in cells, as well as α-tubulin/ histone acetylation selectivity in neurons. Moreover, we were able to show that selective HDAC6i increases the levels of acetylated α-tubulin in the hippocampus of Fmr1−/− mice, and that this correlates with improved learning and memory performances in this animal model of FXS.
RESULTS AND DISCUSSION
SW-100 Is a Highly Selective and Potent HDAC6 Inhibitor.
The structure of a typical HDACi consists of a cap that interacts with the surface of the catalytic cavity, a linker that occupies the hydrophobic channel, and a zinc binding group (ZBG) that coordinates with the zinc ion (Zn2+) at the deep bottom of the pocket. In general, the phenylhydroxamate motif has been demonstrated to impart HDAC6 selectivity to a host of ligands, as first revealed by the invention of TubA. X-ray crystal structures of several related compounds in complex with HDAC6 typically display similar monodentate phenyl-hydroxamate-Zn2+ coordination geometry, while a canonical bidentate Zn2+ coordination was observed for the hydroxamate inhibitors that bear an alkyl chain in the linker region, as exemplified by the pan-HDAC inhibitor TSA and the partially selective, HDAC6i ricolinostat.59 The two residues Phe583 and Phe643 located within the hydrophobic channel engage in a double π-stacking interaction with the phenyl group present in the linker.59 A previous publication has reported that even the capless parent compound, phenylhydroxamate (1) (Figure 1a), shows an IC50 of 115 nM against HDAC6 and more than 10-fold selectivity over class I HDACs.60 In this case, the crystal structure of the enzyme—inhibitor complex containing phenylhydroxamate also displays bidentate zinc coordination, which is believed to underlie its high potency.61 Thus, the capping group attached to the phenylhydroxamate moiety is able to affect its degree of coordination with the catalytic Zn2+ ion leading to either bidentate or monodentate chelation. It has been calculated that the monodentate coordination mode is slightly less stable by ∼0.5 kcal/mol compared to bidentate coordination.59 Appendage of an N,N-diethylaminomethylene group or a pyrrolidinomethylene group on the phenyl-hydroxamic acid core leads to compounds 2 and 3, respectively, both of which show reduced HDAC6 activity. On the other hand, the heteroaromatic pyrrolomethylene (4) and indolylmethylene (5) derivatives, which can be viewed as structurally simplified analogues of TubA, provide analogues of nanomolar activity against HDAC6 and high selectivity over HDAC1 (Figure 1a).62 Docking simulations of compounds 2–5 and TubA to the active site of HDAC6-CD2 (Figure 1b–f) have been carried out, and these studies reveal that 2, 4, and TubA engage in monodentate coordination with Zn2+, with binding being further enhanced by hydrogen bonding interactions with Gly582 and Tyr745 (2, Figure 1c); Gly582 and His614 (4, Figure 1d); or Gly582, Cys584, and Tyr745 (TubA, Figure 1e), respectively. Within the narrow hydrophobic channel of HDAC6-CD2, Phe583 and Phe643 engage in double π-stacking interactions with the bulky phenyl group present in the linker; this binding mode is consistent with the interactions observed from the X-ray structure of the phenylhydroxamate 1 in complex with HDAC6.60 The N,N-diethylamino group of 2 did not show any hydrophobic interactions with the rim region, while the pyrrole cap of 4 was found to engage in a hydrophobic interaction with the Pro464 side chain (Figure 1c), which is similar to that observed for the tricyclic moiety of TubA (Figure 1e). Overall, hydrophobic interactions between the aromatic caps and the residues present on the rim region of HDAC6 provide additional binding energies, regardless of the size of these cap groups. This information may be helpful in the design of other novel ligands that may in turn show an improved ability to penetrate the blood-brain barrier.
Figure 1.

Hydrophobic caps are valuable for enhancing HDAC6 potency. (a) HDAC1/6 enzyme potencies and structures of compounds 1–5. (b) Empirical conformation of 1 (green) in HDAC6-CD2 (PDB: 6CSR). (c–e) Docked pose of 2 (yellow), 4 (magenta), and TubA (orange) in HDAC6-CD2; (f) Superimposed structures of 2, 4, and TubA in the catalytic site of HDAC6-CD2. Interatomic distances of both polar interactions and coordination are depicted in green dashed lines (distance in Å). Zn2+ ions are depicted as dark gray spheres. The mesh molecular surface is depicted in orange (wires) and pale green (surface).
Next we selected several HDAC6is containing different types of monocyclic or bicyclic aromatic caps from our work for further investigation (Figure 2a).62,63 The majority of these compounds exhibit desirable calculated CNS physicochemical properties and MPO scores that would suggest ready brain permeability (Table S1).58 TubA and another well-known HDAC6i NexA64 were included as positive controls. A number of these potent HDAC6is including TubA and NexA exhibit high selectivity against the class I HDACs 1, 2, and 3 but poorer selectivity against HDAC8 (Figure 2b and c). The benzimidazole analogues 8 and 9 were also found to show strong activity against the class IIa isoforms. On the other hand, based upon these data and other structure—activity relationship data available to us, we designed the tetrahydroquinoline (THQ)-based analogue SW-100. The synthetic route to SW-100 starts from 4-chloroaniline (13) which was first converted to the N-phenylpropanamide 14 by treatment with 3-chloropropanoyl chloride. Next, intramolecular Friedel—Crafts cyclization using aluminum trichloride followed by reduction of the intermediate lactam with LiAlH4 afforded the tetrahydroquinoline 15. The building block 15 was then alkylated with commercially available methyl 4-(bromomethyl)benzoate to provide the corresponding ester, which upon treatment with an aqueous NH2OH/NaOH solution afforded the desired hydroxamate analogue SW-100 (Scheme 1). The chemical route has been used to prepare multigram amounts of this HDAC6i.
Figure 2.

SW-100 is a highly potent and selective HDAC6 inhibitor. (a) Structures of compounds 6–12, and SW-100. (b) Heat map summary of HDAC activities. (c) Full HDAC potency table of 6–12, SW-100, TubA, and NexA. IC50 values are the mean of two experiments ± SD obtained from curve-fitting of a 10-point enzymatic assay starting from 30 μM with 3-fold serial dilution against all HDAC isoform; TSA was tested in singlet 10-dose IC50 mode with 3-fold serial dilution starting at a concentration of 10 μM against HDAC class I, IIb, and IV isoforms; TMP269, a selective HDAC class IIa inhibitor, was tested in singlet 10-dose IC50 mode with 3-fold serial dilution starting at a concentration of 10 μM against HDAC class IIa isoforms.
Scheme 1.
a
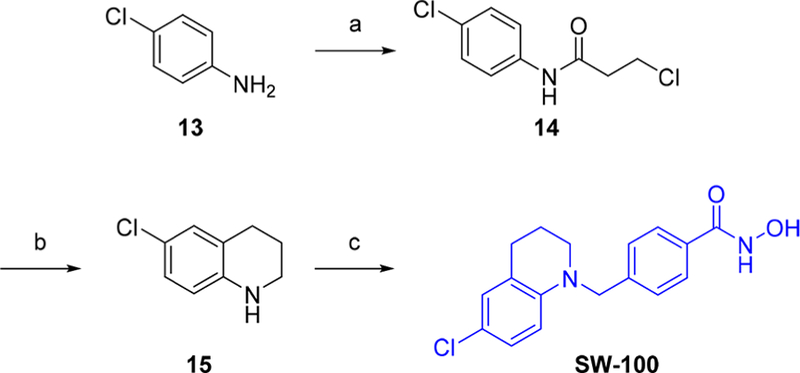
aReagents and conditions. (a) 3-chloropropanoyl chloride, acetone, reflux; (b) (i) AlCl3, 140 °C (neat), (ii) LiAlH4, THF, 0 °C to reflux; (c) (i) methyl 4-(bromomethyl)benzoate, K2CO3, DMF, 100 °C, (ii) NaOH, 50 wt % aq. NH2OH, THF/MeOH (1:1), 0 °C to rt.
As apparent from the data presented in Figure 2, SW-100 exhibits a 1000-fold to 10000-fold selectivity for HDAC6 relative to all other HDAC isozymes, including HDAC8 (SI: 1280-fold; data from Reaction Biology Corp.).
The docking results of SW-100 with HDAC6-CD2 shown in Figure 3 indicate that the hydroxamate group engages in the expected monodentate interaction with the Zn2+ ion, which is further reinforced by hydrogen bonding between its NH and the Gly582 backbone as well as the carbonyl and Tyr745/ His614, respectively. Both residues Phe583 and Phe643 located in the hydrophobic channel engage in double π-stacking interactions with the phenyl ring present in the linker of SW-100. Also, an additional interaction was observed between the chlorine atom on the THQ capand Asn645 present on the rim of the HDAC6-CD2 pocket (Figure 3a). Superimposed and surface views of SW-100 and TubA in Figure 3b—d show that TubA engages in hydrophobic interactions with Leu712 while SW-100 is turned toward residues Phe642 and Asn645. From these static images, it is clear that the cap groups add to inhibitor potency through interactions with different residues comprising the large rim area. Moreover, in comparison with the parent THQ analogue 16 [blue ligand in Figure 3b which lacks the chlorine atom; structure shown in Figure S2, IC50 (HDAC6) = 15 nM],62 SW-100 rotates within the binding pocket to allow for the interaction with Asn645 at the rim surface which may in turn be responsible for the improved potency of SW-100.
Figure 3.

(a) Binding interactions of SW-100 (orange) within the HDAC6-CD2. (b) Superimposed structures of SW-100 (orange), 16 (blue), and TubA (cyan) with HDAC6-CD2. (c) Surface pose of SW-100 (orange) within HDAC6-CD2. (d) Surface pose of TubA (cyan) within HDAC6-CD2. The Zn2+ ion is depicted as a gray sphere. Molecular surfaces are depicted in in gray, yellow (hydrophobic interaction area), and green (polar interaction area).
It is well appreciated that the ability of a small molecule to inhibit an enzyme in its pure/isolated state may not properly translate to its predicted outcomes in the cellular context. It thus became of interest to further assess the potency and selectivity of SW-100 in live cells using the NanoBRET target engagement assay, which is based on the competitive displacement of a luminescent reporter complex utilizing a cell-permeable SAHA tracer.65 These results are summarized in Figure 4, and demonstrate that SW-100 maintains high selectivity over HDAC1 in cells. The measured HDAC6 potency of SW-100 was slightly higher when the α-tubulin preferring deacetylase domain of HDAC6, the CD2 domain (IC50 = 97 nM) was used in place of full-length HDAC6 (IC50 = 279 nM). Results are also provided for SAHA for comparison purposes (Figure 4).
Figure 4.

SW-100 selectively inhibits HDAC6-CD2 and enhances α-tubulin acetylation in cells. (a–d) HDAC1/6 NanoBRET target engagement assay for compound SW-100 in HEK293. IC50 values are the mean of four experiments ± SEM obtained from curve-fitting of a 12-point engagement assay starting from a concentration of 20 μM with 3-fold serial dilution. SAHA was used as a positive control.
SW-100 Displays a Significantly Improved Ability To Cross the Blood-Brain Barrier.
A compound’s brain uptake is largely determined by its ability to diffuse over cellular membranes and its affinity for drug efflux transporters like P-glycoprotein (P-gp), which is extensively expressed at the BBB.66 Compounds with permeability coefficients [Papp(A−B)] higher than 3 × 10−6 cm/s generally show high brain uptake, while compounds with Papp(A−B) less than 1 × 10−6 cm/s in the MDCK-MDR1 cell assay are less likely to penetrate the BBB.67 The data in Table 1 reveal that all compounds studied can be classified as moderately permeable (2 × 10−6 cm/s < Papp(A−B) < 20 × 10−6 cm/s) except for the benzimidazole analogue 12 that shows both a modest Papp(A−B) and Papp(B−A). All three benzimidazole analogues (8, 9, and 12)display unfavorable efflux ratios (>2.5) for CNS access as results from their higher Papp(B−A).68 Notably, the extremely high Papp(B−A) of TubA results in an efflux ratio of 9.83, while compounds 6, 7, 10, 11, and SW-100 exhibit more favorable efflux ratios (<1.0). The predicted physicochemical properties and desirable CNS-MPO scores suggest that all these compounds should exhibit similar permeability properties (Table S1). However, diverse P-gp efflux ratios were obtained which may be related to the amphoteric properties of these inhibitors. The calculated pKa values of the acidic hydroxamate groups are in the range of 9.0–9.2, while the pKa values of basic functional group in the cap groups range from 0.84 to 7.14 (Table 1 and Figure S1). A zwitterionic form is a preferred protonation form for many amphoteric drugs at physiological pH, while a neutral status may become predominant when the pKa values of the basic moiety are lower than 3.69 Moreover, the zwitterionic character of a compound usually leads to poor membrane permeability. However, this is amenable to improvement by replacing a more basic moiety with a less basic one.70,71 We thus plotted the efflux ratios against the cap group’s pKa, and the observed correlation is consistent with the foregoing postulate (Figure S1). In light of the favorable cell line permeability data for SW-100, we next carried out brain/plasma PK studies in vivo. As is apparent from the data presented in Table 2, SW-100 affords a reasonably good brain/plasma ratio of 2.44 at 1 h and 4.54 at 4 h upon IP administration in wild-type (WT) mice, data that are consistent with its low efflux ratio. In contrast, TubA displays a poor brain/plasma ratio of 0.15 at 8 min and 0.86 at 60 min after IV administration in WT mice which is consistent with its permeability properties (Table S2).
Table 1.
Permeability Determination for 6–12, SW-100, and TubAa
| pKa |
MDCK-MDR1 cell line |
||||
|---|---|---|---|---|---|
| compd | acidic | basic |
Papp(A−B) (10−6, cm/s) |
Papp(B−A) (10−6, cm/s) |
efflux ratio |
| SW-100 | 9.05 | 2.91 | 4.58 | 2.33 | 0.51 |
| 6 | 9.20 | 2.07 | 7.35 | 5.24 | 0.70 |
| 7 | 9.20 | 5.12 | 17.61 | 11.04 | 0.63 |
| 8 | 9.20 | 5.95 | 4.44 | 13.45 | 3.23 |
| 9 | 9.00 | 5.86 | 3.89 | 13.17 | 4.23 |
| 10 | 9.05 | 0.99 | 9.14 | 5.54 | 0.62 |
| 11 | 9.05 | 0.84 | 10.46 | 11.08 | 1.05 |
| 12 | 9.05 | 6.55 | 0.66 | 2.05 | 2.95 |
| TubA | 9.20 | 7.14 | 2.00 | 19.66 | 9.83 |
pKa values were obtained by Marvin Sketch version 18.5. MDCK-MDR1 cell line assays were carried out by Pharmaron, Inc. (Irvine, CA).
Table 2.
Brain/Plasma Pilot PK Studies and Initial ADMET Evaluation of SW-100a
| brain/plasma PK studies | |||||
|---|---|---|---|---|---|
| route | dose (mg/kg) |
time (h) |
brain concn (ng/mL) |
plasma concn (ng/mL) |
brain/ plasma ratio |
| IP | 20 | 1 | 141.8 ± 52.3 | 58.2 ± 24.9 | 2.44 |
| IP | 20 | 4 | 11.5 ± 0.30 | 2.52 ± 0.13 | 4.54 |
| ADMET parameters | |||||
| liver microsomal stability (t1/2 min) | human | 23 | |||
| mouse | 23 | ||||
| hepatocyte stability (t1/2, min) | human | 30 | |||
| mouse | 10 | ||||
| hERG test (IC50, μM) | HEK293 cells | 12.23 | |||
| CYP inhibition (% @10 μM) | 1A2 | 20.53 | |||
| 2C9 | 18.84 | ||||
| 2C19 | 83.21 | ||||
| 2D6 | 2.51 | ||||
| 3A4 | −0.63 | ||||
| Ames test | TA98, TA100, TA1535, TA1537, WP2 uvrA | negative | |||
SW-100 was administered to C57BL/6 male mice by IP administration at the dose of 20 mg/kg. Blood and brain samples were collected at 1 and 4 h time points. Brain tissues were homogenized at a 1:4 ratio of tissue weight (g) to volume of PBS (mL). The data found (ng/mL) was multiplied by 5 to obtain the concentration (ng/mL) in brain tissues. PK studies and ADMET profiles were conducted by Pharmaron, Inc., Irvine, CA.
SW-100 was also evaluated in both liver microsomal and hepatocyte stability assays, as well as for hERG and CYP inhibition. The data in Table 2 indicate that its metabolic stability is low, while inhibition of the hERG and CYPs are generally in an acceptable range. In addition, to investigate the possible mutagenicity of SW-100,72 the compound was incubated with four strains of Salmonella typhimurium (TA98, TA100, TA1535, and TA1537) and one strain of Escherichia coli (WP2 uvrA) in the presence and absence of mammalian microsomal enzymes (S9 mix). The results in Table 2 show that SW-100 is negative under the conditions of this Ames assay.
SW-100 Selectively Enhances the Levels of Acetylated α-Tubulin in Vitro.
Compounds 6, 7, 10, 11, and SW-100, having reasonable efflux ratios, were further evaluated for their ability to induce α-tubulin acetylation (Figure 5a–c) in cells. HEK293 cells were treated with the HDAC6i at concentrations of 0.01, 0.1, 1, and 10 μM. As is apparent from the data displayed in Figure 5a the monocylic analogues 6 and 7 were able to elevate the levels of acetylated α-tubulin only at concentrations of 10 μM (one-way ANOVA F(10,32) = 8.425, p < 0.0001), while the indole analogues 10 and 11 began to show reasonable activity at 1 μM (one-way ANOVA F(10,22) = 5.973, p = 0.0002, Figure 5b). In contrast, SW-100 treatment led to a more obvious increase in the acetylated α-tubulin levels in a dose-dependent manner that was similar to the results shown by the comparator compound NexA (one-way ANOVA F(10,45) = 4.537, p = 0.0002, Figure 5c). Further, in order to ascertain the functional selectivity of SW-100, we assessed its ability to induce histone acetylation in comparison to tubulin acetylation in the N2a cells. As is apparent from the bar graphs in Figure 5d–f, SW-100 increased the ratio of acetylated α-tubulin to α-tubulin (>8-fold compared to vehicle, p = 0.07) while inducing only modest changes in the levels of acetylated histone 3 to histone 4 (<1.5-fold compared to vehicle). These effects are similar to those found for the reference HDAC6i TubA (one-way ANOVA, F(3,12) = 3.272, p = 0.05) at 1 μM. In contrast, the pan-active HDACi TSA increased levels of both acetylated α-tubulin and acetylated histone H3 significantly compared to vehicle (p < 0.01, Figure 5a and c).
Figure 5.

SW-100 increases the levels of acetylated α-tubulin in a dose-dependent manner in HEK293 cells (a–c) and selectively increases the levels of acetylated α-tubulin in N2a cells (d–f). (a–c) The α-tubulin acetylation assay was carried out in HEK293 cells treated with the indicated drug and dose for a period of 48 h. Densitometric analysis of the acetyl-α-tubulin to total tubulin in these HEK293 cells was performed and the resulting ratios normalized to the 10 μM dose of TubA. Graphs are depicted as mean ± SEM of the resulting ratiometric values (test compounds: n = 3 or 4; TubA: n = 4; vehicle: n = 4). Statistical significance was analyzed by one-way ANOVA in comparison with the vehicle group. *p < 0.05, **p < 0.01, ***p < 0.001. (d–f) α-Tubulin/histone acetylation assays in N2a cells. N2a cells were treated with 1 μM or 10 nM SW-100. TubA and TSA were used as positive controls. The ratios of acetylated α-tubulin to total α-tubulin and acetylated histone 3 to histone 4 were quantified by Western blot. Tubulin acetylation and histone acetylation were repeated and normalized to the vehicle group. Graphs represent mean ± SEM n = 4. Statistical significance was analyzed by one-way ANOVA in comparison with the vehicle group. **p < 0.01. n/d, not determined.
Efficacy Studies of HDAC6i SW100 in Fmr1−/− Mice Modeling the Learning and Memory Deficits of FXS.
Given its excellent potency, target selectivity, as well as its improved brain penetrance, SW-100 was deemed a suitable candidate for studies in animal models of CNS disorders such as the FXS mouse model. We advanced SW-100 into preliminary behavioral experiments to assess if this class of HDAC6i could show the potential for ameliorating learning and memory impairments in Fmr1−/− mice that display several phenotypes characteristic of FXS.73,74 Fmr1−/− mice were previously shown to have learning disability and cognitive impairments in a variety of tasks including novel object recognition, temporal ordering, and coordinate and categorical spatial processing.75 Furthermore, these mice also exhibit elevated locomotor activity, but, this did not interfere with the ability of Fmr1−/− mice to explore the objects.76 SW-100, 20 mg/kg, was administered twice a day in order to compensate for its short metabolic half-life.
In the coordinate spatial processing task, the distance between two identical objects is altered between the habituation and testing periods. The categorical spatial processing task involves interchanging the positions of two new identical objects following the habituation phase without altering the distance between them. Exploration ratios were calculated as time exploring during the 5 min test session divided by time exploring during the 5 min test session plus the last 5 min of the habituation session.
No significant differences between groups was found in total time exploring objects, when first exposed to the objects, in all behavioral tests (data not shown). Fmr1−/− mice displayed reduced exploration ratios in both the coordinate [F(3,31) = 2.92, p < 0.05; Figure 6a] and categorical [F(3,31) = 4.22, p < 0.05; Figure 6b] spatial processing tasks compared to WT mice. While administration of SW-100 did not significantly alter the behavior of WT mice in these tests, it tended to reverse the impairment in Fmr1−/− mice in coordinate spatial processing and in categorical spatial processing, so after SW-100 treatment the Fmr1−/− mice no longer exhibited deficits compared with vehicle-treated WT mice.
Figure 6.

SW-100 effects on performance of Fmr1−/− mice in four cognitive tasks. (a) Performance of WT mice (n = 17) and Fmr1−/− mice (n = 18) in the coordinate spatial processing task (means ± SEM; *p < 0.05 compared to vehicle-treated WT mice). (b) Performance of WT mice (n = 17) and Fmr1−/− mice (n = 18) in the categorical spatial processing task (means ± SEM; *p < 0.05 compared to vehicle-treated WT mice). (c, d) Performance of WT mice (n = 18) and Fmr1−/− mice (n = 16) in novel object recognition. (c) Percent time spent exploring the novel (N) and familiar (F) object (means ± SEM; *p < 0.01 compared to time spent with familiar object). (d) Discrimination index (means ± SEM; *p < 0.01 compared to vehicle-treated WT mice). (e, f) Performance of WT mice (n = 17) and Fmr1−/− mice (n = 18) on the temporal order task. (e) Percent time spent exploring the first (1) and last (3) object presented. (means ± SEM; *p < 0.05 compared to time spent with object 1); (f) Discrimination index (means ± SEM; *p < 0.05 compared to vehicle-treated WT). (g) Scheme of treatment and testing. Arrows indicate SW-100 treatment.
In the novel object recognition test, WT littermate mice spent significantly more time exploring the novel object than the familiar object, [t(14) = 7.50, p < 0.01; Figure 6c]. In contrast, Fmr1−/− mice spent less time exploring the novel object, indicating that Fmr1−/− mice do not remember the familiar object [t(14) = 8.40, p < 0.01]. The exploration ratio (calculated by dividing the difference between the time spent with the novel object versus the familiar object divided by total time exploring) was significantly lower in Fmr1−/− mice than WT mice [F(3,31) = 22.49, p < 0.01; Figure 6d]. Administration of SW-100 did not alter the novel object recognition of WT mice [t(16) = 6.43, p < 0.01], but improved the abilities of Fmr1−/− mice, which after treatment spent significantly more time exploring the novel object than the familiar object [t(18) = 7.24, p < 0.01; Figure 6c]. Furthermore, the exploration ratio was significantly increased in Fmr1−/− mice treated with SW-100 [F(3,31) = 22.49, p < 0.01; Figure 6d], demonstrating that HDAC6 inhibition effectively reversed the novel object recognition deficit in Fmr1−/− mice.
WT mice displayed temporal order recognition as more time was spent exploring the initial object [t(14) = 7.10, p < 0.01; Figure 6e], whereas Fmr1−/− mice displayed a deficit in temporal order recognition [t(14) = 2.20, p = 0.05; Figure 6e]. Thus, the object exploration ratio (calculated by dividing the difference between the time spent with the initial object (object 1) versus the more recent object (object 3) by total time exploring), differed significantly between Fmr1−/− and WT mice, revealing a temporal order recognition deficit in Fmr1−/− mice [F(3,31) = 9.24, p < 0.01; Figure 6f]. Fmr1−/− mice treated with SW-100 spent significantly more time exploring the first object compared to the most recent object presented, similarly to untreated or treated WT mice [WT: t(16) = 5.00, p < 0.01; Fmr1−/−: t(18) = 7.53, p < 0.01; Figure 6e]. Furthermore, the exploration ratio was significantly increased in Fmr1−/− mice treated with SW-100 [F(3,31) = 9.24, p < 0.05; Figure 6f]. These results demonstrate that temporal ordering is impaired in Fmr1−/− mice and that this deficit is corrected by inhibition of HDAC6. Overall, we observed that Fmr1−/− mice displayed significant deficits in these four cognitive tasks, and that administration of the HDAC6i SW-100 ameliorated all of these cognitive deficits.
α-Tubulin Acetylation Effects of SW-100 in Fmr1−/− Mice.
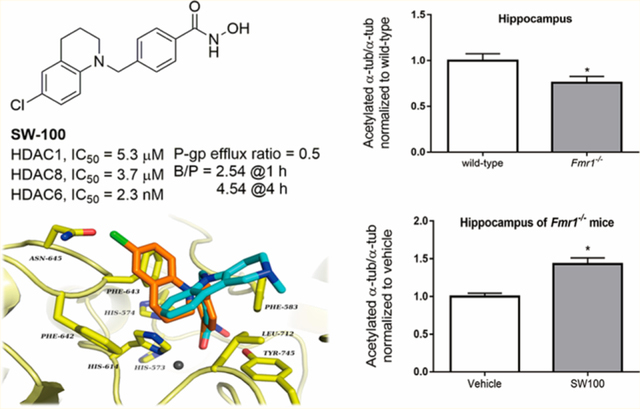
We examined the levels of acetylated α-tubulin levels in the hippocampus of the Fmr1−/− mice and WT mice. We found that the acetylated α-tubulin levels were 30% lower in the hippocampus of Fmr1−/− mice than that of WT mice [t(18) = 2.72, p < 0.05; Figure 7a]. The impaired acetylated α-tubulin levels were significantly increased in Fmr1−/− mice [t(16) = 4.38, p < 0.01; Figure 7c] by treatment with SW-100, while no significant influence on hippocampal α-tubulin acetylation was found in the WT mice [t(15) = 0.08 p = 0.93; Figure 7b]. The lack of any significant changes in the acetylated α-tubulin levels in the WT mice results from the fact that estimated acetylated α-tubulin levels in WT brains are approximately 75% of the total tubulin (i.e., only 25% of brain α-tubulin is nonacetylated). Consequently, any putative increase (of theoretical maximum of 25% to reach 100% acetylated α-tubulin) would be only marginal and difficult to quantify accurately. To the contrary, lower basal levels of acetylated α-tubulin in Fmr1−/− brains increase the dynamic range of the assay and allow for more accurate quantification upon inhibitor treatment.
Figure 7.
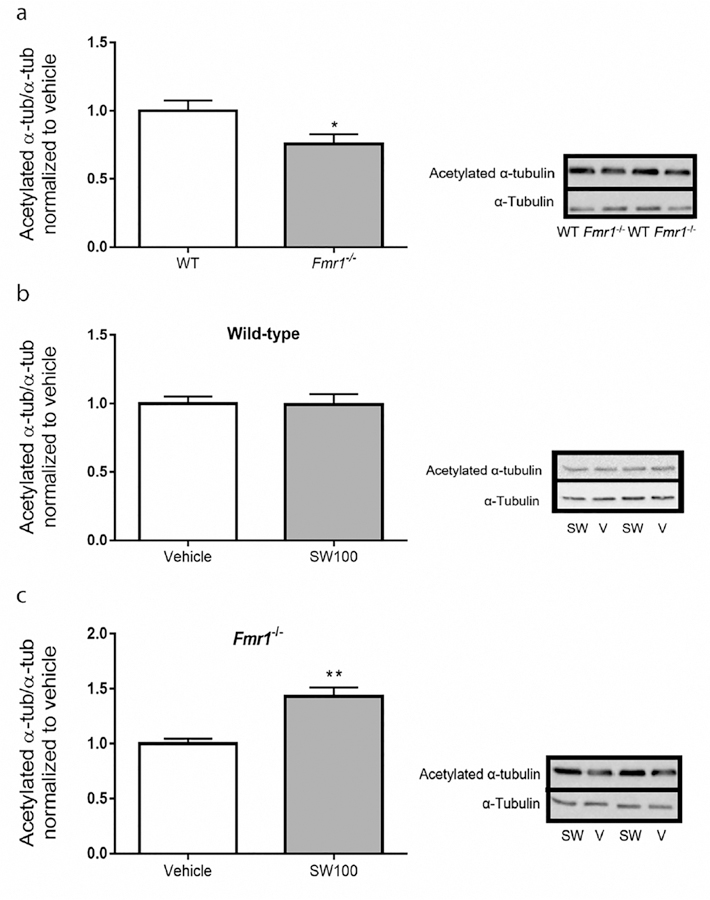
SW-100 selectively increases the levels acetylated α-tubulin in the hippocampus of Fmr1−/− mice. Twenty mg/kg SW-100 was administered twice a day on days 1 and 2 and an additional injection was given on day 3 (1 h prior to cognition assessments) and another injection was given on day 4 (1 h prior to cognition assessments). (a) Basal levels of acetylated α-tubulin in the hippocampus of WT and Fmr1−/− mice. Data were normalized relative to the WT mice. Immunoblots from representative mice in each group, at 2 h after treatment, showing acetylated α-tubulin in the hippocampus of (b) WT mice and (c) Fmr1−/− mice. Data were normalized to the vehicle group (0 mg/kg). Graphs represent mean ± SEM; *p < 0.05 (a), **p < 0.01. (c) Compared to control condition (n = 6–10 per group).
CONCLUSION
The main goal of this study was to identify a potent HDAC6 inhibitor with excellent isoform selectivity and improved BBB permeability, and then to test its ability to improve a cardinal phenotype in Fmr1−/− mice that model intellectual deficits present in FXS patients.
In general, it would appear reasonable that a compound claimed to be selective for HDAC6 would exhibit selectivity for HDAC6 over HDAC1 in the range of 100–1000-fold. However, many of the published HDAC6is exhibit relatively low selectivity versus other HDACs, and in particular against HDAC8 and the class IIa isozymes.77 In contrast, the novel compound SW-100, displayed at least 1000-fold selectivity against all class I, II, and IV HDAC isoforms. These results position SW-100 as one of the most selective HDAC6is reported to date. Moreover, cellular target engagement assays revealed that SW-100 is able to displace a SAHA-based tracer from HDAC6-CD2 at low nanomolar concentrations, the key catalytic site that has been linked to the tubulin deacetylase function of HDAC6. The cellular selectivity of compound SW-100 was further investigated through α-tubulin and histone acetylation assays, and the results obtained are consistent with its HDAC isoform selectivity determined from the isolated enzyme assays.
Brain penetrance is one major challenges for the development of HDAC6is as potential therapeutics for CNS disorders due in part to the polarity of hydroxamate ZBG. Only a limited number of brain penetrable selective HDAC6is have been published in studies with animals.30,33,53,55 While the tool compound TubA displays excellent HDAC6 selectivity, it is not particularly useful for study in animal models of CNS disorders, for as we have found it exhibits a high efflux ratio (9.83) in the MDCK-MDR1 cell line assay and thus a poor brain/plasma ratio. To overcome this weakness, we identified SW-100 that has a higher lipophilicity but lower pKa value and thus an acceptable brain/plasma ratio.
Defects in α-tubulin acetylation are associated with age-related neurodegenerative diseases such as Alzheimer’s disease, Huntington’s disease, and Parkinson’s disease, among others.78 Increasing the levels of acetylated α-tubulin by Hdac6 deletion were beneficial in a mouse model of ALS treatment,79 while other articles have reported that HDAC6 inhibition in mouse models of Alzheimer’s disease can improve performance in various memory tasks.33,54 Although the exact role of α-tubulin acetylation in these diseases is still being studied, the hypothesis has emerged that the defect in α-tubulin acetylation is associated with dysfunctional microtubule-mediated axonal and/or dendritic transport.80 As Fernandez-Barreda et al. stated,80 proteins that modulate the levels of microtubules are potential pharmacological targets for therapeutic strategies aimed at correcting the defects in α-tubulin acetylation found in a variety of neurodegenerative diseases, because alterations in α-tubulin acetylation can lead to disorganization of neuronal networks.81
The mouse model of FXS, Fmr1−/− mice, display cognitive impairments that can be detected in measurements of novel object recognition, temporal ordering of objects, and coordinate and categorical spatial processing.75,82–85 Intellectual disability may be the most difficult characteristic of FXS to treat, but it is critical for patients. Treatment of Fmr1−/− mice with the HDAC6i SW-100 improved their performance in all four of these tasks but did not alter the performance of WT mice in any of these tasks. These findings demonstrate that cognitive impairments are not irreversible in this mouse model of FXS and raise the possibility that HDAC6 inhibition may be a feasible therapeutic intervention for FXS.
Overall, we report the development of a new HDAC6i, SW-100, that is highly selective, is potent, and penetrates the CNS. In vivo treatment with SW-100 to Fmr1−/− mice reversed deficits in the levels of acetylated α-tubulin in the hippocampus and ameliorated several cognitive impairments. We believe that this research offers a truly innovative approach to the treatment of FXS, and that further improvements in compound design will lead to drug candidates that can be tested in humans.
METHODS
Chemistry.
General Information.
1H NMR and 13C NMR spectra were obtained on 400/100 or 300 MHz Bruker spectrometers, except where noted otherwise, using the solvent residual peak as the internal reference (chemical shifts: CD3OD, 3.31/49.15; DMSO-d6, 2.50/39.52; D2O, 4.80). The following abbreviations for multiplicities were used: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, dd = double doublet, dt = double triplet, td = triple doublet, ddd = double double doublet, and br s= broad singlet. TLC plates (Merck silica gel 60 F254, 250 μm thickness) were used to monitor reaction progress, and spots were visualized under UV (254 nm). Low resolution mass spectrometry (LRMS) was carried out on a Shimadzu LCMS2020-PDA+ELSD+MS, and high-resolution mass spectrometry (HRMS) on a Shimadzu IT-TOF instrument under the following conditions: column, ACE 3AQ (50 × 2.1 mm i.d.); mobile phase, 8– 100% CH3CN/H2O containing 0.1% formic acid at a flow rate of 0.5 mL/min for 4 min. Flash chromatography was performed on a Combi-Flash Rf system (Teledyne ISCO) with silica gel cartridges. Preparative HPLC was performed using a Shimadzu preparative LC under the following conditions: column, ACE 5AQ (150 × 21.2 mm i.d.); mobile phase: 8–100% MeOH (CH3CN)/H2O containing 0.05% TFA at a flow rate of 17 mL/min for 30 min; UV detection at 254 and 280 nm. Analytical HPLC was carried out on an Agilent 1260 series instrument under the following conditions: column, ACE 3 (150 × 4.6 mm i.d.); mobile phase, 8–100% MeOH (CH3CN)/H2O containing 0.05% TFA at a flow rate of 1.0 mL/min for 25 min; UV detection at 254 nm. The purities of all tested compounds were >98%, as determined by analytical HPLC.
4-[(Diethylamino)methyl]-N-hydroxybenzamide (2).
(i) General procedure A: To KOtBu (536.5 mg, 4.79 mmol) in anhydrous DMF (15 mL) under argon was added diethylamine (350 mg, 4.79 mmol) dissolved in anhydrous DMF (10 mL). The reaction was stirred for 15 min at 0 °C, then methyl 4-(bromomethyl)benzoate (1.097 g, 4.79 mmol) was added as a solution in anhydrous DMF (8 mL). The reaction was stirred for 2 h at 70 °C, and then quenched and neutralized with 2 N HCl. The residue was extracted with EtOAc (3 × 30 mL), and the combined organic layers were washed with H2O (2 × 30 mL) and brine (15 mL), dried over sodium sulfate, filtered, and concentrated under vacuum. The crude product was purified via Combi-Flash column chromatography (EtOAc/hexane = 0–100%) to afford methyl 4-[(diethylamino)methyl]benzoate as a yellow oil (790 mg, 75%). (ii) General Procedure B: Solid NaOH (374 mg, 9.36 mmol) was dissolved in a 50% aqueous solution of NH2OH (4 mL) at 0 °C. A solution of methyl 4-[(diethylamino)methyl]benzoate (518 mg, 2.34 mmol) in 1:1 THF/MeOH (6 mL) was added dropwise with vigorous stirring. Upon complete addition, the ice bath was removed, and the reaction was allowed to stir for 15 min. The reaction was then neutralized with 2 N HCl solution, and the mixture was concentrated under vacuum. The crude product was purified by preparative HPLC to yield the title compound 2 as a white solid after neutralization and lyophilization (448 mg, 83%). 1H NMR (400 MHz, CD3OD) δ 7.72 and 7.42 (AA′XX′ multiplet, JAX + JAX′ = 8.2 Hz, 4H), 3.65 (s, 2H), 2.55 (q, J = 7.2 Hz, 4H), 1.08 (t, J = 5.0 Hz, 6H). 13C NMR (100 MHz, CD3OD) δ 166.3, 139.5, 131.9, 129.7 (2C), 127.0 (2C), 56.1, 46.4 (2C), 9.3 (2C). ESI HRMS calcd for C12H19N2O2: [M + H]+, m/z 233.1441; found: 233.1441.
N-Hydroxy-4-(pyrrolidin-1-ylmethyl)benzamide (3).
(i) The ester intermediate was synthesized according to General Procedure A from pyrrolidine (0.12 mL, 1.52 mmol) and methyl 4-(bromomethyl)-benzoate (336 mg, 1.52 mmol) and isolated as a viscous clear oil (166 mg, 50%). (ii) The title compound 3 was synthesized according to General Procedure B from methyl 4-(pyrrolidin-1-ylmethyl)benzoate (120 mg, 0.55 mmol) and afforded as an off-white solid after preparative-HPLC purification and lyophilization (38 mg, 31%). 1H NMR (400 MHz, DMSO-d6) δ 11.33 (s, 1H), 10.69 (br s, 1H), 7.81 and 7.58 (AA′XX′ multiplet, JAX + JAX′ = 8.0 Hz, 4H), 4.39 (s, 2H), 3.22 (br s, 4H), 1.94 (br s, 4H). 13C NMR (100 MHz, CD3OD) δ 165.6, 134.2, 133.6, 130.3 (2C), 127.6 (2C), 57.2, 53.6 (2C), 22.4 (2C). ESI HRMS calcd for C12H17N2O2: [M + H]+, m/z 221.1285; found: 221.1286.
4-(1-Pyrrolylmethyl)-N-hydroxybenzamide (4).62
(i) The ester intermediate was synthesized from pyrrole (150 mg, 2.24 mmol) and methyl 4-(bromomethyl)benzoate (495 mg, 1.52 mmol) according to General Procedure A and isolated as a viscous clear oil (432 mg, 90%). (ii) The title compound 4 was synthesized from methyl 4-(1-pyrrolylmethyl)benzoate (400 mg, 1.86 mmol) according to General Procedure B and afforded as a white solid after preparative-HPLC purification and lyophilization (225 mg, 56%). 1H NMR (400 MHz, CD3OD) δ 7.69 and 7.19 (AA′XX′ multiplet, JAX + JAX′= 8.3 Hz, 4H), 6.74–6.72 (m, 2H), 6.12–6.11 (m, 2H), 5.16 (s, 2H). 13C NMR (100 MHz, CD3OD) δ 168.0, 144.6, 132.8, 128.5 (2C), 128.2 (2C), 122.3 (2C), 109.6 (2C), 53.6. ESI HRMS calcd for C12H13N2O2: [M+ H]+, m/z 217.0972; found: 217.0974.
4-(1-Indolylmethyl)-N-hydroxybenzamide (5).62
(i) The ester intermediate was synthesized according to General Procedure A (substituting NaH for KOtBu) from methyl 4-(bromomethyl)-benzoate (978 mg, 4.27 mmol) and indole (500 mg, 4.27 mmol) and isolated as a white solid (860 mg, 76%). (ii) The title compound 5 was synthesized according to General Procedure B from methyl 4-(1-indolylmethyl)benzoate (840 mg, 3.17 mmol) and afforded as a white solid after preparative-HPLC purification and lyophilization (443 mg, 53%). 1H NMR (400 MHz, DMSO-d6) δ 11.14 (s, 1H), 9.0 (br s, 1H), 7.67, 7.23 (AA′XX′ multiplet, JAX + JAX′= 8.1 Hz, 4H), 7.56 (d, J = 7.8 Hz, 1H), 7.52 (d, J = 3.1 Hz, 1H), 7.42 (d, J = 8.1 Hz, 1H), 7.09 (t, J = 7.4 Hz, 1H), 7.02 (t, J = 7.3 Hz, 1H), 6.50 (d, J = 3.0 Hz, 1H), 5.48 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 163.9, 141.4, 135.7, 129.2 (2C), 128.3, 127.1 (2C), 126.8 (2C), 121.2, 120.5, 119.2, 110.1, 101.1, 48.7. ESI HRMS calcd for C16H15N2O2: [M+ H]+, m/z 267.1128; found: 267.1137.
4-(1-Pyrazolylmethyl)-N-hydroxybenzamide (6).62
(i) The ester intermediate was synthesized according to General Procedure A from pyrazole (8.85 g, 0.13 mol) and methyl 4-(bromomethyl)benzoate (30 g, 0.13 mol), and then isolated as a viscous yellow oil (24.5 g, 86%). (ii) The title compound 6 was synthesized according to General Procedure B from methyl 4-(1-pyrazolylmethyl)benzoate (20 g, 0.09 mmol) and afforded as a white solid after column chromatography and recrystallization (11.17 g, 56%). 1H NMR (300 MHz, DMSO-d6) δ 11.19 (br s, 1H), 9.09 (br s, 1H), 7.84 (d, J = 1.8 Hz, 1H), 7.70 and 7.24 (AA′XX′ multiplet, JAX + JAX’= 8.1 Hz, 4H), 7.48 (d, J = 1.2 Hz), 6.29 (t, J = 2.1 Hz, 1H), 5.38 (s, 2H). ESI HRMS calcd for C11H12N3O2: [M + H]+, m/z 218.0924; found: 218.0917. Purity: 99.86% (254 nm), 99.45% (ELSD).
4-[4-(Dimethylamino)benzyl]-N-hydroxybenzamide (7).62
(i) To a solution of 4-(dimethylamino)phenylboronic acid (12.4 g, 75.0 mmol) and methyl 4-(bromomethyl)benzoate (20 g, 90 mmol) in acetonitrile/water (500/50 mL) were added Pd(PPh3)4 (866 mg, 0.75 mmol) and K2CO3 (20.7 g, 150 mmol) under an argon atmosphere. The mixture was heated to reflux and stirred for 6 h. Then, the mixture was diluted with H2O (200 mL), and the organic products were extracted with EtOAc (3 × 200 mL). The combined organic layers were washed with brine (200 mL), dried over sodium sulfate, filtered, and concentrated under vacuum. The residue was purified by flash chromatography (0–10% MeOH/DCM) to yield methyl 4-[4-(dimethylamino)benzyl]benzoate as an orange oil (9.6 g, 48%). (ii) The title compound 7 was synthesized according to General Procedure B from methyl 4-[4-(dimethylamino)benzyl]benzoate (7.6 g, 28.3 mmol) and afforded as a white solid after column chromatography and recrystallization (6.7 g, 88%). 1H NMR (300 MHz, DMSO-d6) δ 11.11 (s), 8.95 (s), 7.64, 7.25 (AA′XX′ multiplet, JAX + JAX′ = 8.1 Hz, 4H), 7.03, 6.65 (AA′XX′ multiplet, JAX + JAX′ = 8.7 Hz, 4H), 3.84 (s, 2H), 2.83 (s, 6H). ESI HRMS calcd for C16H19N2O2: [M + H]+, m/z 271.1441; found: 271.1448. Purity: 98.66% (254 nm), 99.35% (ELSD).
4-[(6-Fluoro-2-methyl-1-benzimidazolyl)methyl]-N-hydroxybenzamide (8).62
To a round-bottom flask charged with 4-fluoro-2-nitroacetanilide (10 g, 50.5 mmol) in MeOH (200 mL) under an atmosphere of argon was added Pd/C (10 weight%, 1.0 g). Then a hydrogen atmosphere (balloon pressure) was established by three vacuum/purge cycles. The mixture was allowed to stir at room temperature overnight, then concentrated under vacuum to approximately one-third of its volume, filtered through a plug of Celite, and concentrated. The crude aniline intermediate was taken up in 10% AcOH in DCE (200 mL), and methyl 4-formylbenzoate (8.28 g, 50.5 mmol) and NaBH(OAc)3 (16.7 g, 75.7 mmol) were added sequentially under an argon atmosphere. The resulting suspension was allowed to stir for 24 h at room temperature. The reaction was quenched with water (100 mL), and the solution was extracted with DCM (2 × 100 mL). The combined organic layers were washed with brine, dried over sodium sulfate, and concentrated. The crude reductive amination product was taken up in toluene (200 mL), and p-TsOH·H2O (950 mg, 5.0 mmol) was added. The mixture was allowed to stir under reflux for 2 h. After cooling to room temperature, saturated aqueous sodium bicarbonate solution (100 mL) was added, and the organics were extracted into EtOAc (2 × 100 mL). The combined organic layers were washed with brine, dried over sodium sulfate, and concentrated under vacuum. Combi-Flash column chromatography (0–10% MeOH/DCM) afforded the ester intermediate as a light-yellow viscous oil (5.1 g, 34% over 3 steps). (ii) The title compound 8 was synthesized according to General Procedure B from the ester intermediate (1.5 g, 5.3 mmol) and afforded as an off-white powder after preparative-HPLC purification and lyophilization (1.2 g, 80%). 1H NMR (400 MHz, DMSO-d6) δ 11.18 (s, 1H), 9.05 (s, 1H), 7.90, 7.18 (AA′XX′ multiplet, JAX + JAX′ = 8.4 Hz, 4H), 7.56 (dd, J = 8.7, 4.9 Hz, 1H), 7.40 (dd, J = 9.2 Hz, 2.4 Hz, 1H), 7.01 (td, J = 10, 2.4 Hz, 1H), 5.51 (s, 2H), 2.49 (s, 3H). ESI HRMS calcd for C16H13FN3O2: [M − H]−, m/z 298.0997; found: 298.0992. Purity: 98.49% (254 nm), 100% (ELSD).
3-Fluoro-N-hydroxy-4-[(2-methyl-1-benzimidazolyl)methyl]-benzamide (9).62
(i) The ester intermediate was synthesized according to General Procedure A (substituting K2CO3 for KOtBu) from methyl 4-(bromomethyl)-3-fluorobenzoate (6 g, 24.3 mmol) and 2-methylbenzimidazole and isolated as a viscous orange oil (5.3 g, 73%). (ii) The title compound 9 was synthesized according to General Procedure B from the ester intermediate (4.0 g, 13.4 mmol) and afforded as an off-white powder after column chromatography and recrystallization (3.0 g, 75%). 1H NMR (400 MHz, DMSO-d6) δ 11.5 (br s, 1H), 9.21 (br s, 1H), 7.85–7.79 (m, 2H), 7.67–7.60 (m, 2H), 7.58–7.50 (m, 2H), 7.46 (m, 1H), 5.85 (s, 2H), 2.89 (s, 3H). ESI HRMS calcd for C16H13FN3O2: [M − H]−, m/z 298.0997; found: 298.0998. Purity: 98.17% (254 nm), 100% (ELSD).
4-[(6-Chloroindolin-1-yl)methyl]-N-hydroxybenzamide (10).63
(i) The ester intermediate was synthesized according to General Procedure A (substituting K2CO3 for KOtBu) from methyl 4-(bromomethyl)benzoate (34.3 g, 150 mmol) and 6-chloroindoline (23 g, 150 mmol) and isolated as a clear oil (40 g, 89%). (ii) The title compound 10 was synthesized according to General Procedure B from methyl 4-[(6-chloroindolin-1-yl)methyl]benzoate (20 g, 66.4 mmol) and afforded as a white solid after column chromatography and recrystallization (6.08 g, 30%). 1H NMR (CD3OD, 300 MHz) δ 7.70 and 7.46 (AA′XX′ multiplet, JAX + JAX′ = 8.0 Hz, 4H), 6.98 (d, J = 7.8 Hz, 1H), 6.58 (dd, J = 7.5, 1.8 Hz, 1H), 6.44 (d, J = 1.5 Hz, 1H), 4.32 (s, 2H), 3.33 (t, J = 8.6 Hz, 2H), 2.91 (t, J = 8.6 Hz, 2H). ESI HRMS calcd for C16H16ClN2O2: [M + H]+, 303.0895; found: 303.0892. Purity: 98.50% (254 nm), 100% (ELSD).
4-[(6-Fluoroindolin-1-yl)methyl]-N-hydroxybenzamide (11).63
(i) The ester intermediate was synthesized according to General Procedure A (substituting K2CO3 for KOtBu) from methyl 4-(bromomethyl)benzoate (50.3 g, 220 mmol) and 6-fluoroindoline (30 g, 220 mmol) and isolated as a clear oil (23 g, 37%). (ii) The title compound 11 was synthesized according to General Procedure B from methyl 4-[(6-fluoroindolin-1-yl)methyl]benzoate (11 g, 38.6 mmol) and afforded as a white solid after column chromatography and recrystallization (6.46 g, 59%). 1H NMR (DMSO-d6, 300 MHz) δ 11.18 (s, 1H), 9.01 (s, 1H), 7.73 and 7.40 (AA′XX′ multiplet, JAX + JAX′ = 8.4 Hz, 4H), 7.00 (m, 1H), 6.40 (dd, J = 10.8, 2.1 Hz, 1H), 6.29 (td, J = 8.4, 2.4 Hz, 1H), 4.38 (s, 2H), 3.35 (t, J = 8.2 Hz, 2H), 2.87 (t, J = 8.2 Hz, 2H). ESI HRMS calcd for C16H16FN2O2 [M + H]+: 287.1190; found: 287.1197. Purity: 99.44% (254 nm), 100% (ELSD).
N-Hydroxy-4-[[(1-methyl-1H-benz[d]imidazol-2-yl)amino]-methyl]benzamide (12).63
(i) A mixture of 1-methyl-1H-benz[d]-imidazol-2-amine (1.2 g, 8.2 mmol) and methyl 4-formylbenzoate (1.3 g, 8.2 mmol) in toluene (40 mL) was refluxed for 6 h. The excess solvent was removed, and the residue was redissolved in a mixture of MeOH (10 mL) and THF (10 mL). Then sodium borohydride (0.47 g, 12.2 mmol) was added at 0 °C. After stirring for 30 min, the reaction was quenched with ice water and extracted with EtOAc (2 × 30 mL). Combined organic layers were washed with brine, dried over Na2SO4, and concentrated under vacuum. The crude ester intermediate, a yellow oil (1.5 g, 62%), was used in next step without further purification. (ii) The title compound 12 was synthesized according to General Procedure B from methyl 4-[[(1-methyl-1H-benz[d]imidazol-2-yl)amino]methylbenzoate (1.5 g, 5.1 mmol) and afforded as a white solid after column chromatography and recrystallization (800 mg, 53%). 1H NMR (D2O, 300 MHz) δ 7.65 and 7.46 (AA′XX′ multiplet, JAX + JAX′ = 8.3 Hz, 4H), 6.92 (m, 2H), 4.71 (s, 2H), 3.59 (s, 3H). ESI HRMS calcd for C16H17N4O3 [M + H]+: 297.1346; found: 297.1352. Purity: 99.30% (254 nm), 100% (ELSD).
3-Chloro-N-(4-chlorophenyl)propanamide (14).
To a round-bottom flask charged with 4-chloroaniline (13, 5.0 g, 39.37 mmol) in acetone (50 mL) was added 3-chloropropanoyl chloride (1.9 mL, 19.69 mmol) at room temperature. The resulting mixture was allowed to stir for 1 h under reflux condition. Then the reaction was cooled to room temperature and quenched with 2 N HCl (30 mL), and then extracted with EtOAc (3 × 25 mL). The combined organic extracts were washed with brine (20 mL), dried over Na2SO4, and concentrated under vacuum. The crude product was used directly without further purification, and the title compound 14 was obtained as off-white powder (4.6 g, 54%). 1H NMR (CDCl3) δ 7.56 (br s, 1H), 7.46 (d, J = 8.7 Hz, 2H), 7.28 (d, J = 8.8 Hz, 2H), 3.87 (t, J = 6.3 Hz, 2H), 2.81 (t, J = 6.3 Hz, 2H). 13C NMR (CDCl3) δ 167.9, 136.0, 129.8, 129.1 (2C), 121.4 (2C), 40.4, 39.8.
6-Chloro-1,2,3,4-tetrahydroquinoline (15).
(i) In a three-necked flask charged with 14 (4.6 g, 21.1 mmol) under an argon atmosphere was added AlCl3 (5.47 g, 42.2 mmol) at 140 °C. The resulting mixture was stirring at the same temperature for 12 h. Then the reaction was quenched with 1 N HCl (20 mL) carefully at 0 °C and extracted with EtOAc (3 × 30 mL). The combined organic extracts were washed with brine (20 mL), dried over Na2SO4, and concentrated under vacuum. The crude product was purified by flash chromatography (0–80% EtOAc/hexane) to afford as white powder (3.1 g, 17.1 mmol). (ii) To a stirred solution of LiAlH4 (1.952 g, 51.4 mmol) in THF (30 mL) were dropwise added lactam intermediate (3.1 g, 17.1 mmol) in THF (20 mL) at 0 °C. The resulting mixture was stirred at same temperature for 20 min, then heated at reflux condition for additional 1 h. The reaction was subsequently quenched with water (2.0 mL), 5 N NaOH (2.0 mL), and water (10 mL). The afforded precipitate was filtered off and washed with EtOAc (3 × 20 mL). The filtrate was washed with brine (20 mL), dried over Na2SO4, and concentrated under vacuum. The product 6a was obtained as colorless oil (2.43 g, 69.5% for two steps) and used directly into next step without further purification. 1H NMR (CDCl3) δ 6.93–6.86 (m, 2H), 6.38 (d, J = 8.0 Hz, 1H), 3.77 (br s, 1H), 3.33–3.15 (m, 2H), 2.72 (t, J = 6.4 Hz, 2H), 1.94–1.88 (m, 2H). 13C NMR (CDCl3) δ 143.2, 128.8, 126.3, 122.7, 120.9, 114.9, 41.7, 26.7, 21.6.
4-((6-Chloro-3,4-dihydroquinolin-1(2H)-yl)methyl)-N-hydroxybenzamide (SW-100).
(i) To a round-bottom flask charged with 6-chloro-1,2,3,4-tetrahydroquinoline (15, 2.43 g, 14.6 mmol) and methyl 4-(bromomethyl)benzoate (4.99 g, 21.9 mmol) in DMF (30 mL) was added K2CO3 (4.03 g, 29.2 mmol). The resulting mixture was allowed to stir at 100 °C for 2 h. After completion of the reaction, the solution was cooled to room temperature, quenched with H2O (30 mL), and extracted with EtOAc (3 × 20 mL). The combined organic extracts were washed with brine (20 mL), dried over Na2SO4, and concentrated under vacuum. The crude product was purified by flash chromatography (0–20% EtOAc/Hexane) to afford ester intermediate as white solid (3.6 g, 78%). (ii) The title compound SW-100 was synthesized according to General Procedure B from methyl 4-((6-chloro-3,4-dihydroquinolin-1(2H)-yl)methyl)benzoate (3.6 g, 11.4 mmol) and afforded as a white solid (1.8 g, 50%) after preparative-HPLC purification and lyophilization. 1H NMR (DMSO-d6) δ 11.16 (s, 1H), 8.94 (br s, 1H), 7.70 (AA′XX′ multiplet, JAX + JAX′ = 8.2 Hz, 2H), 7.28 (AA’XX’ multiplet, JAX + JAX’ = 8.1 Hz, 2H), 6.94 (d, J = 2.4 Hz, 1H), 6.87 (dd, J = 8.8, 2.5 Hz, 1H), 6.36 (d, J = 8.8 Hz, 1H), 4.51 (s, 2H), 3.39–3.35 (m, 3H), 2.73 (t, J = 6.1 Hz, 2H), 1.96–1.82 (m, 2H). 13C NMR (DMSO-d6) δ 164.2, 143.7, 142.0, 131.4, 128.2, 127.2 (2C), 126.5 (2C), 126.4, 123.9, 118.7, 111.9, 53.9, 27.4, 21.4. ESI HRMS calcd. for C17H16ClN2O2: [M−H]−, m/z 316.0906; found: 316.0899. Purity: 99.39% (254 nm), 100% (ELSD).
Docking Simulation.
The crystallographic structure of HDAC6 was obtained from the Protein Data Bank (PDB) with the access code 5G0I (resolution of 1.99 Å).86 The three-dimensional (3D) structures of compounds 1–5, 16, SW100, and TubA were built up in their neutral forms by molecular mechanics using Merck molecular force field (MMFF94) using Spartan’14 software (Wavefuction, Inc.). The equilibrium geometry at ground state were calculated with the Density Functional Theory (DFT), method Becke, three-parameter, Lee—Yang—Parr (B3LYP) with the standard 6–31G* basis set available in Spartan’14. All docking simulations were performed with the GOLD 5.4 software (CCDC), in the catalytic domain 2 (CD2) of HDAC6. GOLD 5.4 has four fitness functions available: GoldScore, Chem-Score, ASP, and ChemPLP. All fitness functions were evaluated by redocking of the cocrystallized ligand to identify the most suitable fitness function for the docking into HDAC6. Crystallographic waters were removed during the docking runs. Hydrogen atoms were added to the protein according to the data inferred by the program on the ionization and tautomeric states. The set of amino acid residues selected as the binding cavity was delimited by a 10 Å radius from the cocrystallized ligand. The Zn2+ ion was maintained at the catalytic site in order to evaluate its coordination with each ligand. After redocking, the root-mean-square deviation (RMSD) between the best result for each fitness function, and the experimental conformation of each cocrystallized ligands were calculated. The fitness function with the lower value of RMSD (<2.0 Å), and the best performance in the redocking was used for the docking of compounds 1–5, 16, SW-100, and TubA. ChemPLP was the best fitness function found (higher RMSD obtained equal to 0.357 Å). The number of genetic operations (crossover, migration, mutation) in each docking run used in the searching procedure was set to 100,000. The program optimizes hydrogen-bond geometries by rotating hydroxyl and amino groups of the amino acid side chains. The score of each pose identified is calculated as the negative of the sum of a series of energy terms involved in the protein—ligand interaction process, thus the more positive the score is, the better the interaction. The interaction between 1 and HDAC6-CD2 was obtained from PDB with the access code 6CSR.61
HDAC1–11 Enzyme Assays.
HDAC inhibition assays were performed by the Reaction Biology Corporation (Malvern, PA) using human full-length recombinant HDAC1–11 isoforms. Compounds 6–12, SW-100, TubA, and NexA were tested in duplicate 10-dose IC50 mode with 3-fold serial dilution starting at 30 μM against all HDAC isoforms. Trichostatin A (TSA) and TMP269 were tested as references in a 10-dose IC50 with 3-fold serial dilution starting at 10 μM. A fluorogenic peptide derived from residues 379–382 of p53 (RHKK(Ac)AMC) was used as the substrate for HDAC1, 2, 3, 6, 10, and 11; a fluorogenic peptide containing a trifluoroacetylated lysine, Ac-LGK(TFA)-AMC, was used as the substrate for HDAC4, 5, 7, and 9; and a fluorogenic peptide derived from p53 residues 379–382 (RHK(Ac)K(Ac)AMC) was used as the substrate for HDAC8. IC50 values were calculated using the GraphPad Prism 4 program based on a sigmoidal dose—response equation. The blank (DMSO) value was entered as 1.0 × 10−12 of concentration for curve-fitting. The reaction buffer contained: 50 mM Tris·HCl pH 8.0, 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, 1 mg/mL BSA, and a final concentration of 1% DMSO. The enzyme was added into wells of the reaction plate, and stock solutions of compounds were distributed into the enzyme mixture by acoustic technology (Echo550 instrument; nanoliter range). The plates were spun down and preincubated for 5–10 min. The substrate was then delivered to all reaction wells to initiate the reaction, which was incubated for 2 h at 30 °C. After incubation, developer and tested compound were added to quench the reaction and generate fluorescence. Kinetic measurements were then taken for 1.5 h at 15 min intervals to ensure that development was complete. End point readings were taken for analysis after the development reached a plateau.
HDAC6 and 11 data of SW100 were repeated and confirmed in Dr. Cyril Bařinka’s lab.
Permeability Studies in MDCK-MDR1 cells.
Permeability studies were performed by Pharmaron, Inc. (Irvine, CA). MDCK-MDR1 cells were cultured in DMEM medium supplemented with 20% FBS and maintained at 37 °C and 5% CO2. Cell monolayers were prepared by culturing MDCK-MDR1 cells in 96-well HTS transwell plates at a starting cell density of 1 × 105 cells/m2 for 4–8 days. Integrity of the monolayer was confirmed by measuring TEER (transepithelial electric resistance) and TEER value >300 Ω*cm2 were used for assay. Test compounds were added into the donor chambers (both apical and basolateral), and the transwell plates were incubated at 37 °C for 2 h. Transwells were separated, and samples from donor and receiver chambers were collected. Compound concentrations were determined by LC-MS/MS (LC system: Shimadzu; MS analysis: API 4000). Apparent permeability (Papp) was calculated according to the equation:
where VA is the volume in the acceptor well.
Pilot Brain and Plasma Pharmacokinetic Studies.
Pharmaco-kinetics (PK) studies were performed by Pharmaron, Inc. (Irvine, CA). To generate a stock solution, 4.14 mg of compound SW-100 was dissolved in 0.104 mL of DMSO. Then 0.414 mL of PEG-400 and 0.518 mL of saline were added to this solution with vortexing to obtain a final volume of 1.036 mL. The final concentration of the formulation solution (10% DMSO:40% PEG400:50% saline) was 4 mg/mL. According to our previous experience with TubA,31,35 a drug dosage of 20 mg/kg was administered into C57BL/6 male mice via a single intraperitoneal (IP) injection.
Approximately 0.025 mL of blood was collected from 2 mice/group at two points (60 and 240 min) after injection. All blood samples were transferred into plastic microcentrifuge tubes containing heparin-Na as anticoagulant. Collection tubes with blood samples and anticoagulant were inverted several times for proper mixing of the tube contents and then placed on wet ice prior to centrifugation for plasma. Blood samples were centrifuged at 4000g for 5 min at 4 °C to obtain plasma. Mice were sacrificed to collect brain samples from other two groups (2 mice/group) at two points (60 and 240 min) after injection. Brain tissues were weighed accurately and were homogenized at a 1:4 ratio of tissue weight (g) to volume of PBS (mL). The data found (ng/mL) was multiplied with five to obtain concentration (ng/mL) in brain tissues. Plasma samples and brain homogenates were stored below −70 °C until analysis. Thirty μL Plasma samples or brain homogenate were diluted with 3 μL of blank matrix, and then 200 μL of acetonitrile containing internal standard was added for precipitating protein. The mixture was vortexed for 2 min and centrifuged at 4000 × g for 15 min at 4 °C. Ten μL of the supernatant after diluting 3 times with water was taken for LC-MS/ MS analysis (LC system: Shimadzu; MS analysis: API 4000). All samples were processed on ice.
α-Tubulin Acetylation Studies in HEK293 Cells.
HEK293 cells were grown in DMEM containing 4.5 g/L d-glucose, l-glutamine, 1 mg/L sodium pyruvate, supplemented with 10% FBS and 1% penicillin/streptomycin. At 24 h prior to treatment, cells were plated on 12-well plates. Once 70% confluency was reached, the media was aspirated and replaced with media containing HDAC6i at concentrations of 10 nM, 100 nM, 1 μM, and 10 μM. Cells were treated for 24 h and then were washed with DPBS and harvested. Cells were lysed in Mammalian Protein Extraction Reagent (MPER) lysis buffer with added protease inhibitors, and protein concentrations determined for the lysates using the DC Protein Concentration Assay. Loading buffer was added to 2 μg of protein, and samples were incubated in a boiling water bath for 5 min to ensure denaturation. Electrophoresis of these samples was performed on 5% stacking and 12% resolving polyacrylamide gels in Tris-glycine running buffer (25 mM Tris-base, 192 mM glycine, 0.1% SDS), and then electro-transferred to a PVDF membrane in transfer buffer (25 mM Tris-base, 192 mM glycine, 20% methanol) overnight at 4 °C. The membranes were then blocked in 5% skim milk powder dissolved in TBST (10 mM Tris, 150 mM NaCl, 0.05% Tween) for 2 h at room temperature, then treated with mouse anti-acetyl-tubulin primary antibody at a 1:3000 dilution in blocking solution overnight at 4 °C. The membranes were washed thoroughly in TBST and then probed with HRP-tagged goat anti-mouse secondary antibody, diluted in blocking solution (1:5000) for 2 h at room temperature. Upon further washing in TBST, membranes were treated with ECL detection reagents and then developed on film. To account for total cellular α-tubulin in addition to protein loads, the membrane was stripped and reprobed with rabbit anti-α-tubulin primary (1:1000) and goat anti-rabbit (1:5000) secondary antibodies in a similar fashion. Signal was assessed through densitometry using ImageJ software.
Cell Transfection, Treatments, and BRET Measurements.
NanoBRET target engagement was performed against HDAC6 (CD2) and HDAC1 according to the manufacturer’s protocol (Promega) in HEK293 cells (ATCC). Plasmid constructs encoding NanoLuc-HDAC6 (CD2) and HDAC1-NanoLuc encoded HDAC open reading frames matching previous work.65 HDAC6 (CD2) encoded a GSSGAIA linker between Nanoluc and HDAC6 (CD2) and HDAC1-NanoLuc encoded a SWTWEGNKWTWK linker between HDAC1 and NanoLuc. For target engagement analysis for HDAC6, a HEK293 cell line stably expressing full-length NanoLuc-HDAC6 was used (Promega). NanoBRET HDAC Tracer (Promega) was added to a final concentration of 250 nM, 1000 nM, or 100 nM for HDAC6 (CD2), HDAC1, and HDAC6 (respectively) immediately prior to test compound addition. Tracer concentrations were selected for each HDAC such that tracer occupancy did not impart a shift in observed compound IC50 value. Serially diluted test compounds were then added to the cells and allowed to equilibrate for 2 h prior to BRET measurements. To measure BRET, NanoBRET NanoGlo Substrate-(Promega) and Extracellular NanoLuc inhibitor was added per the manufacturer’s protocol, and filtered luminescence was measure on GloMax Discover luminometer equipped with 450 nm BP filter (donor) and 610 nm LP filter (acceptor), using 0.5 s integration time. Milli-BRET units (mBU) are the BRET values ×1000. Competitive displacement data were then graphed with GraphPad Prism software using a three-parameter curve-fitting with the following equation:
α-Tubulin/Histone Acetylation Studies in N2a Cells.
Cells were washed with phosphate-buffered saline (PBS) and collected using the EpiQuik Total Histone Extraction Kit (EpiGentek). Protein concentrations were determined using the micro BCA kit (Thermo-Fisher Scientific) according to manufacturer’s protocol. Protein samples were supplemented with reducing sample buffer (Thermo Scientific) and denaturized by incubating at 95 °C for 5 min. The nonspecific binding was blocked by incubation of the PVDF membrane in 5% bovine serum albumin (BSA), diluted in Tris Buffered Saline Tween (TBST, 50 mM TRIS, 150 mM NaCl, 0.1% Tween-20 (Applichem) for 1 h at room, temperature followed by incubation with primary antibodies overnight. The antibodies, diluted in TBST, were directed against α-tubulin (1:5000, T6199, Sigma-Aldrich), acetylated α-tubulin (1:5000, T6793 monoclonal, Sigma-Aldrich), histone H3 acetyl k9+k14 (1:1000, 9677L, Cell Signaling), and histone 4 (1:1000, ab10158, Abcam). Secondary antibodies conjugated with horseradish peroxidase (Dako, 1:5000, 1 h, RT) where used prior to detection with ECL substrate (life technologies) with a LAS 4000 Image Analyzer (GE Healthcare). Luminescent signals were quantified with ImageQuant TL 7.0 software (GE Healthcare) and then graphed with GraphPad Prism software. A mild reblotting buffer (Merck Millipore Corp.) was applied to strip the blots.
Behavior Amelioration Study in Fmr1−/− Mice.
Male adult (8–10 weeks old) C57BL/6 WT mice, and C57BL/6 mice with a disruption of the Fmr1 gene (originally kindly provided with matched controls by Dr. W. Greenough, University of Illinois) were used. Mice were housed in groups of 3–5 in standard cages in light and temperature-controlled rooms and were treated in accordance with NIH and the University of Miami Institutional Animal Care and Use Committee regulations. Mice were treated with drugs through IP administration twice a day (20 mg/kg) for 2 days and 1 h prior to cognition testing on the third and fourth days.
Four cognitive tasks were tested: coordinate and categorical spatial processing, novel object recognition, and temporal order memory, carried out as previously described.75,85,87 Mice were habituated to the testing room with a white noise generator (55 dB) for 1 h. Behavioral tests were conducted during 2 consecutive days, two tests every 24 h (shown in Figure 6g). 70% ethanol was used to clean each apparatus and object used between each test session. Test sessions were filmed, and films were scored by an investigator blind to the genotype and treatment. Mice were assessed in the novel object recognition task and coordinate spatial processing task on day 3 and were assessed in the temporal ordering task and categorical spatial processing task on day 4.
For the coordinate spatial processing task, each mouse was allowed to explore two novel objects that were 45 cm apart for 15 min. After 5 min in an opaque chamber, each mouse was allowed to explore the same two objects that had been moved closer together (30 cm) for 5 min. Increased exploration of the objects during the test session compared with the last 5 min of the habituation phase was considered a measure of memory of the distance between objects. The exploration ratio was calculated as time (exploring during the 5 min test session)/(exploring during the 5 min test session plus the last 5 min of the habituation session).
For the categorical spatial processing task, each mouse was allowed to explore two novel objects that are 45 cm apart for 15 min. After 5 min in an opaque chamber, each mouse was allowed to explore the same two objects that had been transposed for 5 min. Increased exploration of the objects during the test session compared with the last 5 min of the habituation phase is considered a measure of memory of the distance between objects. The exploration ratio was calculated as time (exploring during the 5 min test session)/ (exploring during the 5 min test session plus the last 5 min of the habituation session).
For the novel object recognition task, time spent exploring an object only includes the mouse sniffing or touching the object with its nose, vibrissa, mouth, or forepaws. As described previously,87 novel object recognition was measured by allowing each mouse to explore two identical objects for 5 min, and after a 5 min period in an opaque chamber, mice were allowed to explore an unused familiar object and a novel object for 5 min. The discrimination index was calculated as the times ((exploring object 2 − exploring object 1)/total time of object exploration) × 100). To measure temporal order memory, each mouse underwent three sessions exploring three new sets of objects (objects 1, 2, 3). During the test session, the mouse was allowed to explore an unused copy of object 1 and an unused copy of object 3 for 5 min. Intact temporal order memory was evident when mice spent more time exploring the first object presented (object 1) than the most recent object presented (object 3). A discrimination index was calculated as the times ((exploring object 3 − exploring object 1)/ total time of object exploration) × 100).
Immunoblot Analysis of Mice Samples.
Mouse hippocampi were rapidly dissected in ice-cold phosphate-buffered saline and homogenized in ice-cold lysis buffer containing 20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, 10% glycerol, 1 μg/mL leupeptin, 1 μg/mL aprotinin, 1 μg/mL pepstatin, 1 mM phenylmethanesulfonyl fluoride, 50 mM NaF, 1 mM sodium orthovanadate, and 100 nM okadaic acid. The lysates were centrifuged at 20,800 × g (14,000 rpm) for 10 min. Protein concentrations in the supernatants were determined using the Bradford protein assay.88 Lysates were mixed with Laemmli sample buffer (2% SDS) and placed in a boiling water bath for 5 min. Proteins (10 μg) were resolved in SDS-polyacrylamide gels, transferred to nitrocellulose, and incubated with primary antibodies to acetylated α-tubulin (1:4000, T7451, Sigma-Aldrich) or α-tubulin (1:4000, T5168, Sigma-Aldrich). Double loading was used to measure ratio for each protein. Immunoblots were developed using horseradish peroxidase-conjugated goat anti-mouse, followed by detection with enhanced chemiluminescence.
Statistical Analysis.
Statistical significance was assessed by one-way ANOVA or by Student’s t test when indicated. GraphPad Prism 6 or 7 software was used. All data were expressed as mean ± SEM, and significance was set at p < 0.05.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank B. Havlinova for excellent technical assistance.
Funding
This work was supported by grants from the NIH (NS079183, to A.P.K. and L.V.D.B) and the CIHR (PJT-153015, to J.H.E), and a NARSAD Distinguished Investigator Grant to R.S.J. from the Brain & Behavior Research Foundation. This work was also in part supported by the Czech Science Foundation (15–19640S), the CAS (RVO: 86652036), and the project “BIOCEV” (CZ.1.05/1.1.00/02.0109) from the ERDF.
ABBREVIATIONS
- ADMET
absorption, distribution, metabolism, excretion, and toxicity
- GSK-3β
glycogen synthase kinase-3beta
- ERK1/2
extracellular signal-regulated kinase 1 and 2
- IGF1
insulin-like growth factor 1
- MMP9
matrix metallopeptidase 9
- SIRT
sirtuins
- DMF
N,N-dimethylformamide
- DMSO
dimethyl sulfoxide
- DCM
dichloromethane
- DCE
1,2-dichloroethene
- THF
tetrahydrofuran
- TLC
thin layer chromatography
- HPLC
high performance liquid chromatography
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschemneuro.8b00600.
Physicochemical properties of analogues 6–12, SW100 and TubA; brain/plasma pilot PK studies of TubA; comparision of pKa values and P-gp efflux ratios; structure of compound 16; 1H and 13C NMR spectra and purity reports for compounds 6–12, SW-100, TubA, and NexA (PDF)
The authors declare the following competing financial interest(s): A.P.K. is the owner of StarWise Therapeutics LLC and thus declares potential financial interests as does G.Z. who is employed by StarWise Therapeutics.
REFERENCES
- (1).Prevalence of Fragile X Syndrome: https://fragilex.org/understanding-fragile-x/fragile-x-101/prevalence/ (accessed Oct. 20, 2018).
- (2).Penagarikano O, Mulle JG, and Warren ST (2007) The pathophysiology of fragile x syndrome. Annu. Rev. Genomics Hum. Genet 8, 109–29. [DOI] [PubMed] [Google Scholar]
- (3).Khandjian EW, Corbin F, Woerly S, and Rousseau F (1996) The fragile X mental retardation protein is associated with ribosomes. Nat. Genet 12, 91–3. [DOI] [PubMed] [Google Scholar]
- (4).Laggerbauer B, Ostareck D, Keidel EM, Ostareck-Lederer A, and Fischer U (2001) Evidence that fragile X mental retardation protein is a negative regulator of translation. Hum. Mol. Genet 10, 329–38. [DOI] [PubMed] [Google Scholar]
- (5).Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, Licatalosi DD, Richter JD, and Darnell RB (2011) FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Bear MF, Huber KM, and Warren ST (2004) The mGluR theory of fragile X mental retardation. Trends Neurosci 27, 370–7. [DOI] [PubMed] [Google Scholar]
- (7).Michalon A, Sidorov M, Ballard TM, Ozmen L, Spooren W, Wettstein JG, Jaeschke G, Bear MF, and Lindemann L (2012) Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice. Neuron 74, 49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Krueger DD, and Bear MF (2011) Toward fulfilling the promise of molecular medicine in fragile X syndrome. Annu. Rev. Med 62, 411–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Youssef EA, Berry-Kravis E, Czech C, Hagerman RJ, Hessl D, Wong CY, Rabbia M, Deptula D, John A, Kinch R, Drewitt P, Lindemann L, Marcinowski M, Langland R, Horn C, Fontoura P, Santarelli L, Quiroz JA, and FragXis Study, G. (2018) Effect of the mGluR5-NAM Basimglurant on Behavior in Adolescents and Adults with Fragile X Syndrome in a Randomized, Double-Blind, Placebo-Controlled Trial: FragXis Phase 2 Results. Neuropsychopharmacology 43, 503–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Scharf SH, Jaeschke G, Wettstein JG, and Lindemann L (2015) Metabotropic glutamate receptor 5 as drug target for Fragile X syndrome. Curr. Opin. Pharmacol 20, 124–34. [DOI] [PubMed] [Google Scholar]
- (11).Davenport MH, Schaefer TL, Friedmann KJ, Fitzpatrick SE, and Erickson CA (2016) Pharmacotherapy for Fragile X Syndrome: Progress to Date. Drugs 76, 431–45. [DOI] [PubMed] [Google Scholar]
- (12).Torrioli M, Vernacotola S, Setini C, Bevilacqua F, Martinelli D, Snape M, Hutchison JA, Di Raimo FR, Tabolacci E, and Neri G (2010) Treatment with valproic acid ameliorates ADHD symptoms in fragile X syndrome boys. Am. J. Med. Genet., Part A 152A, 1420–7. [DOI] [PubMed] [Google Scholar]
- (13).Tabolacci E, De Pascalis I, Accadia M, Terracciano A, Moscato U, Chiurazzi P, and Neri G (2008) Modest reactivation of the mutant FMR1 gene by valproic acid is accompanied by histone modifications but not DNA demethylation. Pharmacogenet. Genomics 18, 738–41. [DOI] [PubMed] [Google Scholar]
- (14).Penney J, and Tsai LH (2014) Histone deacetylases in memory and cognition. Sci. Signaling 7, re12. [DOI] [PubMed] [Google Scholar]
- (15).Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, Nieland TJ, Zhou Y, Wang X, Mazitschek R, Bradner JE, DePinho RA, Jaenisch R, and Tsai LH (2009) HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 459, 55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Kim MS, Akhtar MW, Adachi M, Mahgoub M, Bassel-Duby R, Kavalali ET, Olson EN, and Monteggia LM (2012) An essential role for histone deacetylase 4 in synaptic plasticity and memory formation. J. Neurosci 32, 10879–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).McQuown SC, Barrett RM, Matheos DP, Post RJ, Rogge GA, Alenghat T, Mullican SE, Jones S, Rusche JR, Lazar MA, and Wood MA (2011) HDAC3 is a critical negative regulator of long-term memory formation. J. Neurosci 31, 764–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Tang BL (2014) Class II HDACs and neuronal regeneration. J. Cell. Biochem 115, 1225–33. [DOI] [PubMed] [Google Scholar]
- (19).Pardo M, Cheng Y, Velmeshev D, Magistri M, Eldar-Finkelman H, Martinez A, Faghihi MA, Jope RS, and Beurel E (2017) Intranasal siRNA administration reveals IGF2 deficiency contributes to impaired cognition in Fragile X syndrome mice. JCI insight 2, No. e91782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, and van Kuilenburg AB (2003) Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem. J 370, 737–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Boyault C, Sadoul K, Pabion M, and Khochbin S (2007) HDAC6, at the crossroads between cytoskeleton and cell signaling by acetylation and ubiquitination. Oncogene 26, 5468–76. [DOI] [PubMed] [Google Scholar]
- (22).Matthias P, Yoshida M, and Khochbin S (2008) HDAC6 a new cellular stress surveillance factor. Cell Cycle 7, 7–10. [DOI] [PubMed] [Google Scholar]
- (23).Li T, Zhang C, Hassan S, Liu X, Song F, Chen K, Zhang W, and Yang J (2018) Histone deacetylase 6 in cancer. J. Hematol. Oncol 11, 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Perdiz D, Mackeh R, Pous C, and Baillet A (2011) The ins and outs of tubulin acetylation: more than just a post-translational modification? Cell. Signalling 23, 763–71. [DOI] [PubMed] [Google Scholar]
- (25).Arce CA, Casale CH, and Barra HS (2008) Submembraneous microtubule cytoskeleton: regulation of ATPases by interaction with acetylated tubulin. FEBS J 275, 4664–74. [DOI] [PubMed] [Google Scholar]
- (26).Hammond JW, Huang CF, Kaech S, Jacobson C, Banker G, and Verhey KJ (2010) Posttranslational modifications of tubulin and the polarized transport of kinesin-1 in neurons. Mol. Biol. Cell 21, 572–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Hirokawa N, Niwa S, and Tanaka Y (2010) Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron 68, 610–38. [DOI] [PubMed] [Google Scholar]
- (28).Gardiner J, Barton D, Marc J, and Overall R (2007) Potential role of tubulin acetylation and microtubule-based protein trafficking in familial dysautonomia. Traffic 8, 1145–9. [DOI] [PubMed] [Google Scholar]
- (29).Liu XA, Rizzo V, and Puthanveettil SV (2012) Pathologies of Axonal Transport in Neurodegenerative Diseases. Transl. Neurosci 3, 355–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Jochems J, Boulden J, Lee BG, Blendy JA, Jarpe M, Mazitschek R, Van Duzer JH, Jones S, and Berton O (2014) Antidepressant-like properties of novel HDAC6-selective inhibitors with improved brain bioavailability. Neuropsychopharmacology 39, 389–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Wang Z, Leng Y, Wang J, Liao HM, Bergman J, Leeds P, Kozikowski A, and Chuang DM (2016) Tubastatin A, an HDAC6 inhibitor, alleviates stroke-induced brain infarction and functional deficits: potential roles of alpha-tubulin acetylation and FGF-21 up-regulation. Sci. Rep 6, 19626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Pinho BR, Reis SD, Guedes-Dias P, Leitao-Rocha A, Quintas C, Valentao P, Andrade PB, Santos MM, and Oliveira JM (2016) Pharmacological modulation of HDAC1 and HDAC6 in vivo in a zebrafish model: Therapeutic implications for Parkinson’s disease. Pharmacol. Res 103, 328–39. [DOI] [PubMed] [Google Scholar]
- (33).Zhang L, Liu C, Wu J, Tao JJ, Sui XL, Yao ZG, Xu YF, Huang L, Zhu H, Sheng SL, and Qin C (2014) Tubastatin A/ACY-1215 improves cognition in Alzheimer’s disease transgenic mice. J. Alzheimer’s Dis 41, 1193–205. [DOI] [PubMed] [Google Scholar]
- (34).Zhang L, Sheng S, and Qin C (2013) The role of HDAC6 in Alzheimer’s disease. J. Alzheimer’s Dis 33, 283–95. [DOI] [PubMed] [Google Scholar]
- (35).Selenica ML, Benner L, Housley SB, Manchec B, Lee DC, Nash KR, Kalin J, Bergman JA, Kozikowski A, Gordon MN, and Morgan D (2014) Histone deacetylase 6 inhibition improves memory and reduces total tau levels in a mouse model of tau deposition. Alzheimer’s Res. Ther 6, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).d’Ydewalle C, Krishnan J, Chiheb DM, Van Damme P, Irobi J, Kozikowski AP, Vanden Berghe P, Timmerman V, Robberecht W, and Van Den Bosch L (2011) HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease. Nat. Med 17, 968–74. [DOI] [PubMed] [Google Scholar]
- (37).Kim JY, Woo SY, Hong YB, Choi H, Kim J, Choi H, Mook-Jung I, Ha N, Kyung J, Koo SK, Jung SC, and Choi BO (2016) HDAC6 Inhibitors Rescued the Defective Axonal Mitochondrial Movement in Motor Neurons Derived from the Induced Pluripotent Stem Cells of Peripheral Neuropathy Patients with HSPB1Mutation. Stem Cells Int 2016, 9475981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Benoy V, Vanden Berghe P, Jarpe M, Van Damme P, Robberecht W, and Van Den Bosch L (2017) Development of Improved HDAC6 Inhibitors as Pharmacological Therapy for Axonal Charcot-Marie-Tooth Disease. Neurotherapeutics 14, 417–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Gold WA, Lacina TA, Cantrill LC, and Christodoulou J (2015) MeCP2 deficiency is associated with reduced levels of tubulin acetylation and can be restored using HDAC6 inhibitors. J. Mol. Med 93, 63–72. [DOI] [PubMed] [Google Scholar]
- (40).Guo W, Naujock M, Fumagalli L, Vandoorne T, Baatsen P, Boon R, Ordovas L, Patel A, Welters M, Vanwelden T, Geens N, Tricot T, Benoy V, Steyaert J, Lefebvre-Omar C, Boesmans W, Jarpe M, Sterneckert J, Wegner F, Petri S, Bohl D, Vanden Berghe P, Robberecht W, Van Damme P, Verfaillie C, and Van Den Bosch L (2017) HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients. Nat. Commun 8, 861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Ross-Inta C, Omanska-Klusek A, Wong S, Barrow C, Garcia-Arocena D, Iwahashi C, Berry-Kravis E, Hagerman RJ, Hagerman PJ, and Giulivi C (2010) Evidence of mitochondrial dysfunction in fragile X-associated tremor/ataxia syndrome. Biochem. J 429, 545–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Kaplan ES, Cao Z, Hulsizer S, Tassone F, Berman RF, Hagerman PJ, and Pessah IN (2012) Early mitochondrial abnormalities in hippocampal neurons cultured from Fmr1 pre-mutation mouse model. J. Neurochem 123, 613–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Chen S, Owens GC, Makarenkova H, and Edelman DB (2010) HDAC6 regulates mitochondrial transport in hippocampal neurons. PLoS One 5, No. e10848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Xu X, Kozikowski AP, and Pozzo-Miller L (2014) A selective histone deacetylase-6 inhibitor improves BDNF trafficking in hippocampal neurons from Mecp2 knockout mice: implications for Rett syndrome. Front. Cell. Neurosci 8, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Uutela M, Lindholm J, Louhivuori V, Wei H, Louhivuori LM, Pertovaara A, Akerman K, Castren E, and Castren ML (2012) Reduction of BDNF expression in Fmr1 knockout mice worsens cognitive deficits but improves hyperactivity and sensorimotor deficits. Genes Brain Behav 11, 513–23. [DOI] [PubMed] [Google Scholar]
- (46).Langley B, D’Annibale MA, Suh K, Ayoub I, Tolhurst A, Bastan B, Yang L, Ko B, Fisher M, Cho S, Beal MF, and Ratan RR (2008) Pulse inhibition of histone deacetylases induces complete resistance to oxidative death in cortical neurons without toxicity and reveals a role for cytoplasmic p21(waf1/cip1) in cell cycle-independent neuroprotection. J. Neurosci 28, 163–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Rivieccio MA, Brochier C, Willis DE, Walker BA, D’Annibale MA, McLaughlin K, Siddiq A, Kozikowski AP, Jaffrey SR, Twiss JL, Ratan RR, and Langley B (2009) HDAC6 is a target for protection and regeneration following injury in the nervous system. Proc. Natl. Acad. Sci. U. S. A 106, 19599–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Wang XX, Wan RZ, and Liu ZP (2018) Recent advances in the discovery of potent and selective HDAC6 inhibitors. Eur. J. Med. Chem 143, 1406–18. [DOI] [PubMed] [Google Scholar]
- (49).Yee AJ, Bensinger WI, Supko JG, Voorhees PM, Berdeja JG, Richardson PG, Libby EN, Wallace EE, Birrer NE, Burke JN, Tamang DL, Yang M, Jones SS, Wheeler CA, Markelewicz RJ, and Raje NS (2016) Ricolinostat plus lenalidomide, and dexamethasone in relapsed or refractory multiple myeloma: a multicentre phase 1b trial. Lancet Oncol 17, 1569–78. [DOI] [PubMed] [Google Scholar]
- (50).Clinical trials for ACY-1215 https://clinicaltrials.gov/ct2/results?term=ACY-1215&Search=Search (accessed Sep. 28, 2018).
- (51).Clinical trials for ACY-241 https://clinicaltrials.gov/ct2/results?term=ACY-241&Search=Search (accessed Sep. 28, 2018).
- (52).Lee HY, Fan SJ, Huang FI, Chao HY, Hsu KC, Lin TE, Yeh TK, Lai MJ, Li YH, Huang HL, Yang CR, and Liou JP (2018) 5-Aroylindoles Act as Selective Histone Deacetylase 6 Inhibitors Ameliorating Alzheimer’s Disease Phenotypes. J. Med. Chem 61, 7087–102. [DOI] [PubMed] [Google Scholar]
- (53).Fan SJ, Huang FI, Liou JP, and Yang CR (2018) The novel histone de acetylase 6 inhibitor, MPT0G211, ameliorates tau phosphorylation and cognitive deficits in an Alzheimer’s disease model. Cell Death Dis 9, 655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Yu CW, Chang PT, Hsin LW, and Chern JW (2013) Quinazolin-4-one derivatives as selective histone deacetylase-6 inhibitors for the treatment of Alzheimer’s disease. J. Med. Chem 56, 6775–91. [DOI] [PubMed] [Google Scholar]
- (55).Strebl MG, Campbell AJ, Zhao WN, Schroeder FA, Riley MM, Chindavong PS, Morin TM, Haggarty SJ, Wagner FF, Ritter T, and Hooker JM (2017) HDAC6 Brain Mapping with [(18)F]Bavarostat Enabled by a Ru-Mediated Deoxyfluorination. ACS Cent. Sci 3, 1006–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Wager TT, Chandrasekaran RY, Hou X, Troutman MD, Verhoest PR, Villalobos A, and Will Y (2010) Defining desirable central nervous system drug space through the alignment of molecular properties, in vitro ADME, and safety attributes. ACS Chem. Neurosci 1, 420–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Wager TT, Hou X, Verhoest PR, and Villalobos A (2010) Moving beyond rules: the development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem. Neurosci 1, 435–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Wager TT, Hou X, Verhoest PR, and Villalobos A (2016) Central Nervous System Multiparameter Optimization Desirability: Application in Drug Discovery. ACS Chem. Neurosci 7, 767–75. [DOI] [PubMed] [Google Scholar]
- (59).Porter NJ, Mahendran A, Breslow R, and Christianson DW (2017) Unusual zinc-binding mode of HDAC6-selective hydroxamate inhibitors. Proc. Natl. Acad. Sci. U. S. A 114, 13459–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Wagner FF, Olson DE, Gale JP, Kaya T, Weiwer M, Aidoud N, Thomas M, Davoine EL, Lemercier BC, Zhang YL, and Holson EB (2013) Potent and selective inhibition of histone deacetylase 6 (HDAC6) does not require a surface-binding motif. J. Med. Chem 56, 1772–6. [DOI] [PubMed] [Google Scholar]
- (61).Porter NJ, Wagner FF, and Christianson DW (2018) Entropy as a Driver of Selectivity for Inhibitor Binding to Histone Deacetylase 6. Biochemistry 57, 3916–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Shen S, Benoy V, Bergman JA, Kalin JH, Frojuello M, Vistoli G, Haeck W, Van Den Bosch L, and Kozikowski AP (2016) Bicyclic-Capped Histone Deacetylase 6 Inhibitors with Improved Activity in a Model of Axonal Charcot-Marie-Tooth Disease. ACS Chem. Neurosci 7, 240–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Gaisina IN, Lee SH, Kaidery NA, Ben Aissa M, Ahuja M, Smirnova NN, Wakade S, Gaisin A, Bourassa MW, Ratan RR, Nikulin SV, Poloznikov AA, Thomas B, Thatcher GRJ, and Gazaryan IG (2018) Activation of Nrf2 and Hypoxic Adaptive Response Contribute to Neuroprotection Elicited by Phenyl-hydroxamic Acid Selective HDAC6 Inhibitors. ACS Chem. Neurosci 9, 894–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Bergman JA, Woan K, Perez-Villarroel P, Villagra A, Sotomayor EM, and Kozikowski AP (2012) Selective histone deacetylase 6 inhibitors bearing substituted urea linkers inhibit melanoma cell growth. J. Med. Chem 55, 9891–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Robers MB, Dart ML, Woodroofe CC, Zimprich CA, Kirkland TA, Machleidt T, Kupcho KR, Levin S, Hartnett JR, Zimmerman K, Niles AL, Ohana RF, Daniels DL, Slater M, Wood MG, Cong M, Cheng YQ, and Wood KV (2015) Target engagement and drug residence time can be observed in living cells with BRET. Nat. Commun 6, 10091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Jin X, Luong TL, Reese N, Gaona H, Collazo-Velez V, Vuong C, Potter B, Sousa JC, Olmeda R, Li Q, Xie L, Zhang J, Zhang P, Reichard G, Melendez V, Marcsisin SR, and Pybus BS (2014) Comparison of MDCK-MDR1 and Caco-2 cell based permeability assays for anti-malarial drug screening and drug investigations. J. Pharmacol. Toxicol. Methods 70, 188–94. [DOI] [PubMed] [Google Scholar]
- (67).Wang Q, Rager JD, Weinstein K, Kardos PS, Dobson GL, Li J, and Hidalgo IJ (2005) Evaluation of the MDR-MDCK cell line as a permeability screen for the blood-brain barrier. Int. J. Pharm 288, 349–59. [DOI] [PubMed] [Google Scholar]
- (68).Ghose AK, Herbertz T, Hudkins RL, Dorsey BD, and Mallamo JP (2012) Knowledge-Based, Central Nervous System (CNS) Lead Selection and Lead Optimization for CNS Drug Discovery. ACS Chem. Neurosci 3, 50–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Tam KY, Avdeef A, Tsinman O, and Sun N (2010) The permeation of amphoteric drugs through artificial membranes—an in combo absorption model based on paracellular and transmembrane permeability. J. Med. Chem 53, 392–401. [DOI] [PubMed] [Google Scholar]
- (70).Wu YJ, Guernon J, McClure A, Luo G, Rajamani R, Ng A, Easton A, Newton A, Bourin C, Parker D, Mosure K, Barnaby O, Soars MG, Knox RJ, Matchett M, Pieschl R, Herrington J, Chen P, Sivarao DV, Bristow LJ, Meanwell NA, Bronson J, Olson R, Thompson LA, and Dzierba C (2017) Discovery of non-zwitterionic aryl sulfonamides as Nav1.7 inhibitors with efficacy in preclinical behavioral models and translational measures of nociceptive neuron activation. Bioorg. Med. Chem 25, 5490–505. [DOI] [PubMed] [Google Scholar]
- (71).Hamdouchi C, Maiti P, Warshawsky AM, DeBaillie AC, Otto KA, Wilbur KL, Kahl SD, Patel Lewis A, Cardona GR, Zink RW, Chen K, Cr S, Lineswala JP, Neathery GL, Bouaichi C, Diseroad BA, Campbell AN, Sweetana SA, Adams LA, Cabrera O, Ma X, Yumibe NP, Montrose-Rafizadeh C, Chen Y, and Miller AR (2018) Discovery of LY3104607: A Potent and Selective G Protein-Coupled Receptor 40 (GPR40) Agonist with Optimized Pharmacokinetic Properties to Support Once Daily Oral Treatment in Patients with Type 2 Diabetes Mellitus. J. Med. Chem 61, 934–45. [DOI] [PubMed] [Google Scholar]
- (72).Shen S, and Kozikowski AP (2016) Why Hydroxamates May Not Be the Best Histone Deacetylase Inhibitors—What Some May Have Forgotten or Would Rather Forget? ChemMedChem 11, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Pieretti M, Zhang FP, Fu YH, Warren ST, Oostra BA, Caskey CT, and Nelson DL (1991) Absence of expression of the FMR-1 gene in fragile X syndrome. Cell 66, 817–22. [DOI] [PubMed] [Google Scholar]
- (74).The Dutch-Belgian Fragile X Consortium, Bakker CE, Verheij C, Willemsen R, et al. (1994) Fmr1 knockout mice: a model to study fragile X mental retardation. Cell 78, 23–33. [PubMed] [Google Scholar]
- (75).Franklin AV, King MK, Palomo V, Martinez A, McMahon LL, and Jope RS (2014) Glycogen synthase kinase-3 inhibitors reverse deficits in long-term potentiation and cognition in fragile X mice. Biol. Psychiatry 75, 198–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Yuskaitis CJ, Mines MA, King MK, Sweatt JD, Miller CA, and Jope RS (2010) Lithium ameliorates altered glycogen synthase kinase-3 and behavior in a mouse model of fragile X syndrome. Biochem. Pharmacol 79, 632–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Wang XX, Wan RZ, and Liu ZP (2018) Recent advances in the discovery of potent and selective HDAC6 inhibitors. Eur. J. Med. Chem 143, 1406–18. [DOI] [PubMed] [Google Scholar]
- (78).Li L, and Yang XJ (2015) Tubulin acetylation: responsible enzymes, biological functions and human diseases. Cell. Mol. Life Sci 72, 4237–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Taes I, Timmers M, Hersmus N, Bento-Abreu A, Van Den Bosch L, Van Damme P, Auwerx J, and Robberecht W (2013) Hdac6 deletion delays disease progression in the SOD1G93A mouse model of ALS. Hum. Mol. Genet 22, 1783–90. [DOI] [PubMed] [Google Scholar]
- (80).Fernandez-Barrera J, Correas I, and Alonso MA (2018) Age-related neuropathies and tubulin acetylation. Aging 10, 524–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Erck C, Peris L, Andrieux A, Meissirel C, Gruber AD, Vernet M, Schweitzer A, Saoudi Y, Pointu H, Bosc C, Salin PA, Job D, and Wehland J (2005) A vital role of tubulin-tyrosine-ligase for neuronal organization. Proc. Natl. Acad. Sci. U. S. A 102, 7853–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Ventura R, Pascucci T, Catania MV, Musumeci SA, and Puglisi-Allegra S (2004) Object recognition impairment in Fmr1 knockout mice is reversed by amphetamine: involvement of dopamine in the medial prefrontal cortex. Behav. Pharmacol 15, 433–42. [DOI] [PubMed] [Google Scholar]
- (83).Pacey LK, Tharmalingam S, and Hampson DR (2011) Subchronic administration and combination metabotropic glutamate and GABAB receptor drug therapy in fragile X syndrome. J. Pharmacol. Exp. Ther 338, 897–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Eadie BD, Cushman J, Kannangara TS, Fanselow MS, and Christie BR (2012) NMDA receptor hypofunction in the dentate gyrus and impaired context discrimination in adult Fmr1 knockout mice. Hippocampus 22, 241–54. [DOI] [PubMed] [Google Scholar]
- (85).King MK, and Jope RS (2013) Lithium treatment alleviates impaired cognition in a mouse model of fragile X syndrome. Genes Brain Behav 12, 723–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Miyake Y, Keusch JJ, Wang L, Saito M, Hess D, Wang X, Melancon BJ, Helquist P, Gut H, and Matthias P (2016) Structural insights into HDAC6 tubulin deacetylation and its selective inhibition. Nat. Chem. Biol 12, 748–54. [DOI] [PubMed] [Google Scholar]
- (87).Pardo M, King MK, Perez-Costas E, Melendez-Ferro M, Martinez A, Beurel E, and Jope RS (2015) Impairments in cognition and neural precursor cell proliferation in mice expressing constitutively active glycogen synthase kinase-3. Front. Behav. Neurosci 9, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem 72, 248–54. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.