

Abstract
We investigate the mechanism of time-dependent inhibition (TDI) of human cytochrome P450 2D6 (CYP2D6) by 3,4-methylenedioxymethamphetamine (MDMA, ecstasy), one of the most widespread recreational drugs of abuse. In an effort to unravel the kinetic mechanism of the formation of metabolic inhibitory complex (MIC) of CYP2D6 with MDMA-derived carbene we carried out a series of spectrophotometric studies paralleled with registration of the kinetics of time-dependent inhibition (TDI) in CYP2D6-incorporated proteoliposomes. The high amplitude of spectral signal in this system allowed us to characterize the spectral properties of the formed MIC in details and obtain an accurate spectral signature of MIC formation. This information was then used in the studies with CYP2D6-containing microsomes of insect cells (CYP2D6 Supersomes™). Our results demonstrate that in both systems the formation of the ferrous carbene-derived MIC is relatively slow, reversible and is not associated with the accumulation of the ferric carbene intermediate, as takes place in the case of CYP3A4 and podophylotoxin. Furthermore, the limited amplitude of MIC formation suggests that only a fraction (~50%) of spectrally detectable CYP2D6 in both proteoliposomes and Supersomes participates in the formation of MIC and is therefore involved in the MDMA metabolism. This observation reveals yet another example of a cytochrome P450 that exhibits persistent functional heterogeneity of its population in microsomal membranes. Our study provides a solid methodological background for further mechanistic studies of MIC formation in human liver microsomes and demonstrates that the potency and physiological relevance of MDMA-dependent TDI of CYP2D6 may be overestimated.
Keywords: cytochrome P450 2D6; methylenedioxyphenyl compounds; time-dependent inhibition; 3,4-methylenedioxymethamphetamine; mechanism-based inhibition
Graphical abstract
1. Introduction
The central role the cytochromes P450 (P450) play in human drug metabolism makes these enzymes a major subject for studies of drug disposition, adverse drug effects and drug-drug interactions (DDIs). One of important P450-related problems of high pharmacological importance is the problem of prediction and analysis of DDIs with drugs capable of metabolism-based inhibition (MBI) of P450s. Metabolism of these compounds results in a loss of enzyme activity over time and therefore is associated with time-dependent inhibition (TDI) of the metabolizing P450 enzymes[1–3]. Compounds that show TDI are usually avoided in selection of drug candidates due to complexity and uncertainty associated with DDI prediction. Despite of these reservations, there are quite a few drugs on the market that are known to cause TDI of the P450 enzymes that metabolize them [4–5].
The most complex type of TDI is the one associated with quasi-reversible formation of metabolic intermediate complexes (MIC) [6–7], One of the most representative groups of P450 inhibitors of this type includes methylenedioxyphenyl (MDP) compounds, such as the insecticide synergist piperonyl butoxide and the drugs tadalafil, paroxetine, and podophyllotoxin. This group also includes 3,4-methylenedioxyamphetamine (MDMA) also known as “ecstasy”. This widespread drug of abuse is metabolized by several cytochrome P450 species, of whose CYP2D6 plays the key role [8], The structure of MDMA and the main routes of its transformation by CYP2D6 are shown in Fig. 1.
Fig. 1.
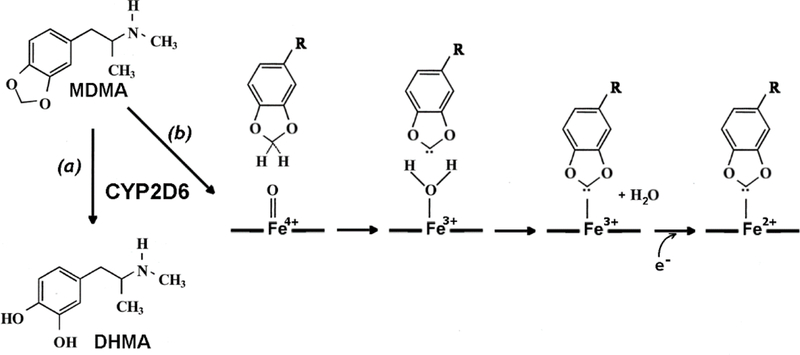
The structure of MDMA and the main routes of its transformation by CYP2D6: the demethylenation path (a) resulting in the formation of 3,4-dihydroxymethamphetamine (DHMA) and the path associated with the in mechanism-based inhibition (b) that involves the formation of MDMI-derived carbene, its binding to the CYP2D6 heme iron(3+) as an axial ligand and the subsequent reduction of the formed MIC.
The mechanism of formation of inhibitory MIC with MDP compounds involves an oxidative conversion of their methylenedioxy group to a carbene. The formed carbene intermediate then binds to the heme iron of ferric cytochrome P450 as an axial ligand. Subsequent reduction stabilizes the carbene-bound porphyrin in ferrous (Fe2+) state [9], The implied mechanism of this process for the case of MDMA-dependent inactivation of CYP2D6 is shown as path (b) in Fig. 1.
An important advantage of studying formation of MICs with MDP compounds is the pronounced changes in the absorbance spectra of P450 caused by the formation of the ferrous carbene-bound intermediate. It is well established that this intermediate has the main Soret band positioned at 455 nm with an additional band at 425–427 nm (“type III” spectrum) [9–13], This is in contrast to the parent low- and high-spin states of ferric P450, where the Soret band is centered at 416–418 and 396–398 nm respectively. The 455 nm absorbance is considered the defining characteristic of MIC formation [9, 14], A pronounced pH dependence of the 425 nm band prompted a hypothesis that this band is due to partial conversion of the 5th axial thiolate ligand of the heme iron to unionized thiol state [9], similar to what is observed in inactivated “cytochrome P420” state of P450 heme proteins [15–17], Moreover, an additional transient peak at 438 nm was attributed to absorbance of a supposed transitory complex of carbene with ferric cytochrome P450 (Fe3+:carbene metabolite) [18].
In our recent study [19] we used these unique spectral properties of carbene-derived MICs to explore the mechanism of TDI of CYP3 A4 by podophylotoxin with UV/VIS absorbance spectroscopy. These studies illustrate the potential of singular value decomposition and/or principal component analysis (PCA) for the studies of the mechanism of MIC formation. Application of these advanced methods of spectral analysis allowed us to demonstrate that the formation of MIC of CYP3 A4 with podophylotoxin goes through a reversible formation of Fe3+:carbene intermediate that undergoes a slow conversion to the Fe2+:carbene complex, which is virtually irreversible.
In the present study, we extend the application of this strategy to CYP2D6 and its interactions with MDMA, which recreational use is known to have potential of causing acute liver failure. MDMA was reported among the most common causes of liver injury in young people [20–22], Hepatotoxicity of MDMA is mediated by its metabolism on cytochrome P450 2D6 (CYP2D6) [8, 23]. Furthermore, CYP2D6-dependent metabolism of MDMA results in a physiologically significant MBI [24–27]. However, the in vitro estimates of the importance of MDMA-dependent TDI were made based on the studies with the classical replot method [24–25]. In previous work, we have shown that the use of this simplistic approach can lead to an overestimation of the potency of TDI and its physiological relevance in the instances of quasi-reversible formation of MIC [6–7]. Therefore, a detailed study of the mechanisms and kinetics of MDMA-dependent MIC formation and the associated TDI of CYP2D6 is warranted.
In an effort to unravel the kinetic mechanism of MIC formation and gain a quantitative assessment of the maximal depth of enzyme conversion to the carbene-derived MIC, we carried out most of our studies in CYP2D6-incorporated proteoliposomes (PLS) containing NADPH-cytochrome P450 reductase (CPR). High amplitude of the spectral signal in this model system allowed us to characterize the spectral properties of the formed MIC in details and obtain an accurate spectral signature of MIC formation. This information was then successfully used in the studies with CYP2D6-containing microsomes of insect cells (CYP2D6 Supersomes™). Besides providing a solid methodological background for further mechanistic studies of MIC formation in human liver microsomes, our results demonstrate that the potency and physiological relevance of MDMA-dependent TDI of CYP2D6 may be overestimated. Furthermore, our study demonstrates that MDMA-dependent MBI affects only a limited fraction (~50%) of spectrally-detectable CYP2D6 in both PLS and Supersomes. The remaining portion of the enzyme is inactive toward MDMA metabolism and remains unaffected by the associated MIC formation. This observation divulges yet another example of cytochrome P450 that exhibits persistent functional heterogeneity of its population in the microsomal membranes [28–29].
2. Methods and Materials
2.1. Materials
Glucose-6-phosphate, protocatechuate 3,4-dioxygenase from Pseudomonas sp., protocatechuic acid, DL-dithiothreitol (DTT) and L-a-phosphatidylcholine (PC) from egg yolk were the products of Sigma-Aldrich (St. Louis, MO). Glucose-6-phosphate dehydrogenase from Leuconostoc mesenteroides and NADPH tetrasodium salt were from EMD Millipore (Billerica, MA). L-a-phosphatidylethanolamine (PE) from bovine liver and 1 -palmitoyl-2-oleoyl-sn-glycero-3-phosphate (phosphatidic acid, PA) were obtained from Avanti Polar Lipids, Inc. (Alabaster, AL). CYP2D6-containing Supersomes™ and 7-hydroxy-4-(aminomethyl)-coumarin (HAMC) were the products of Corning Life Sciences (Corning, NY). Octyl-β-D-glucopyranoside (octylglucoside) was the product of Fluka Honeywell Specialty Chemicals (Seelze, Germany). 4- (2- Hydroxyethyl)-l- piperazine ethanesulfonic acid (HEPES) was obtained from Indofine Chemical Company (Hillsborough, NJ). Dextromethorphan (DXM) hydrobromide hydrate, dextrorphan (DXP) tartrate, 3,4-methylenedioxymethylamphetamine (MDMA) hydrochloride and 3,4-dihydroxymethamphetamine (DHMA) were the products of Cayman Chemical (Ann Arbor, MI). MDMA was obtained as a certified reference material (CRM) solution in methanol (1 mg/ml), which was evaporated and the chemical was re-dissolved in acetone to the concentration of 25 mM. Tris(2-carboxyethyl)phosphine (TCEP) was obtained from Gold Biotechnology (Sent Louis, MO). N-butyl 7-methoxy-4-(aminomethyl)-coumarin (BMAMC) was synthesized from 7-methoxy-4-(bromomethyl)-coumarin (Sigma-Aldrich, St. Louis, MO) and purified as previously described [30], All other chemicals (KC1, NaOH, glycerol, acetone, etc.) were of ACS grade and were used without further purification.
2.2. Protein expression and purification
Recombinant NADPH cytochrome P450 reductase (CPR) from rat liver was expressed in E. coli Topp3 cells and purified as described [31], The N-terminally modified construct of human CYP2D6 [32] was expressed in E. coli DH5a transformed with pGro7 chaperone plasmid (Takara Bio Inc., Kusatsu, Japan) and purified following the procedure described in [33].
2.3. Preparation of proteoliposomes
Proteoliposomes containing NADPH cytochrome P450 reductase were obtained by the octylglucoside dialysis/sorption technique as described earlier [34], Concentrated stock of purified NADPH cytochrome P450 reductase (100–200 μΜ) was added to the octylglucoside-solubilized suspension of 2:1:0.6 mixture of PC, PE and PA to attain the proteimlipid molar ratio of 1:1000 prior to starting dialysis. Incorporation of cytochromes P450 into pre-formed CPR-containing protoliposomes was performed by incubation of a proteoliposomal suspension (2–4 mM by phospholipid content) in 100 mM Na-Hepes buffer, pH 7.4, containing 150 mM KC1, 3 mM TCEP and 20% glycerol (v/v) (LS buffer) with purified cytochromes P450 added as a concentrated stock solutions (100–200 pM) to the P450:lipid ratio of 150:1. Incubation of these mixtures for 16 h at 4 °C at continuous stirring under argon atmosphere resulted in efficient incorporation of the added protein into the membrane. The proteoliposomes were sedimented by centrifugation at 150,000g for 3 h at 4 °C and resuspended in a small volume of the LS buffer.
The content of phospholipids in proteoliposomes was quantified based on the determination of total phosphorus in a chloroform/methanol extract according to Bartlett [35], The resulting preparations were characterized by P450-to-phospholipid molar ratio of 1:180 – 1:250 and contained 7–10 molecules of CYP2D6 per one molecule of CPR active in cytochrome c reduction.
2.4. Activity of NADPH-cytochrome P450 reductase
The concentration of NADPH-cytochrome P450 reductase in microsomal and proteoliposomal membranes was determined based on the rate of NADPH-dependent reduction of cytochrome c at 25°C [36] monitored by an increase in the difference in absorbance at 550 and 541 nm and calculated using the extinction coefficient of 18 mM−1cm−1 [37], Effective molar concentration of CPR was estimated using the turnover number of 3750 min”1, which we determined with our preparations of purified CPR quantified from its absorbance at 456 nm using the extinction coefficient of 21.4 mM−1 cm−1 [38].
2.5. TDI assays and the studies of MDMA metabolism
For TDI assays the 2D6-PLS or 2D6-SS were incubated at CYP2D6 concentrations of ΙμΜ and 0.15 μΜ respectively at various concentration of MDMA (0–330 μΜ). The total volume of the incubation mixture was equal to 420–500 pL and contained 0.1M Na-Hepes buffer, pH 7.4 supplemented with 2mM TCEP and 0.5 mM EDTA, NADPH-regenerating system consisting of 10 mM glucose-6-phosphate and 2 units/ml of glucose-6-phospate dexydrogenase and a reactive oxygen scavenging system containing of superoxide dismutase and catalase at a concentration of 125 U/mL for each. The incubation temperature was set at 30 °C. MDMA metabolism was started by addition of NADPH to the concentration of 0.2 mM. A series 7 to 8 time points were taken at different times during 60 min of incubation. Aliquots of 60 pi were taken and used without any dilution for measuring the rate of enzyme turnover with BMAMC or DXM as probe substrates as described below. The same incubation protocol was used in the studies of the time course of MDMA utilization and DHMA formation. In this case the aliquots were quenched and analyzed with liquid chromatography-mass spectrometry as described below.
2.6. Activity measurements with DXM
Activity measurements with DXM were performed in 0.1 M Na-HEPES buffer pH 7.4 containing 60 mM KCl. DXM was added as a 20mM solution in methanol. The reaction was started by addition of NADPH. The final concentrations of NADPH and DXM were equal to 1 mM and 50 pM, respectively. The probes were incubated in the shaking water bath at 30 °C for 4 min, after which time they were quenched and analyzed with liquid chromatography-mass spectrometry for determination of the amount of formed DXP.
2.7. Determination of DXP, HDMA, and MDMA
The probes obtained in the TDI, MDMA metabolism and DXM turnover assays were quenched with a 1 M solution of formic acid in acetonitrile containing a known concentration of phenacetin as internal standard. After an addition of the above quenching solution in the amount of 25% of the probe volume, the samples were centrifuged at 9,300 g for 10 min and analyzed with liquid chromatography-mass spectrometry. An LC-20AD series high-performance liquid chromatography system (Shimadzu, Columbia, MD) combined with a HTC PAL autosampler (LEAP Technologies, Carrboro, NC) was used to perform chromatography on Kinetex and Synergi reverse-phase columns (100 × 2.1 mm [DXP, Kinetex], 250 mm x 4.6 mm [HDMA, MDMA, Synergi]; Phenomenex, Torrance, CA). Dextrorphan was monitored using a flow rate of 300 pL/min with chromatographic separation initiated by 1 minute of flow of the 90% mobile phase A followed by a linear gradient over the next 7 minutes to 20% mobile phase A. Mobile phase A was then held constant at 20% over 0.5 minutes, followed by a linear gradient back to 90% A over 0.5 minutes. Finally, the column was equilibrated for one minute at initial conditions. The total chromatographic assay time was 10 minutes per sample, and the retention times for the internal standard and DXP were 5.57 and 4.46 minutes, respectively. HDMA and MDMA chromatography was initiated with a flow of 400 μL/min of 95% mobile phase A for 1 minute before a linear ramp to 5% mobile phase A over 7 minutes. Mobile phase A was maintained at 5% for 4 minutes before a linear gradient to 95% over 2.0 minutes and maintenance of the initial conditions for 2.0 minutes to re-equilibrate the column. The total chromatographic assay time was 16.0 minutes per sample, and retention times for HMDA, MDMA, and internal standard were 10.45, 10.78, and 12.89 minutes, respectively.
The quantification of the metabolite was conducted using an API 4000 Q-Trap tandem mass spectrometry system manufactured by Applied Biosystems/MDS Sciex (Foster City, CA) using turbospray ESI operating in positive ion mode. The optimized mass spectrometer tune parameters were as follows: collision gas, medium; curtain gas, 20; ion source gas 1, 35; ion source gas 2, 55; ion spray voltage, 4900; desolvation temperature, 400; declustering potential, 55; entrance potential, 10; collision energy, 60; collision cell exit potential, 10. Analytes were detected using multiple reaction monitoring mode by monitoring the m/z transition from 258.2 to 201.1 (DXP), 182.1 to 123.0 (HDMA), 194.1 to 105.0 (MDMA) and 161.0 to 120.0 (phenacetin). Quantitation of product was achieved with a standard curve ranging from a 1 to 1000 nM concentration of the authentic analytes.
2.8. Fluorometric determination of CYP2D6 activity
Fluorometric determination of CYP2D6 activity were performed using N-butyl 7-methoxy-4- (aminomethyl)-coumarin (BMAMC) as a substrate. This compound was selected in view of a high rate of its CYP2D6-dependent O-demethylation and a high yield of fluorescence of the product, A-butyl 7-hydroxy-4-(aminomethyl)-coumarin (BHAMC) [30, 39]. BMAMC is also characterized with one of the highest affinities to CYP2D6 observed with various fluorogenic substrates of this enzyme [30, 39]. According to literature the KM for BMAMC measured with CYP2D6-containing bacterial membranes is equal to 1.5 μΜ [39]; our measurements in CYP2D6-SS gave a KM estimate of 3.5 ± 0.5μΜ. Although BMAMC is not selective enough for probing CYP2D6 activity in human liver microsomes [30, 40], its properties provide for a high sensitivity and robustness of the real-time monitoring of the CYP2D6 activity in model systems.
For measuring the activity of CYP2D6-PLS and CYP2D6 with BMAMC we used 60 μl of the incubation media containing CYP2D6-SS or CYP2D6-PLS. The sample was placed into a 3 × 3 mm optical cell with continuous stirring and thermostated at 30 °C. An aliquot of a 16 mM stock solution of BMAMC in acetone was added to attain the desired concentration in the range of 1 – 100 μΜ. The measurements made for TDI studies were performed at 50 μΜ BMAMC. If necessary (in the measurements not related to TDI monitoring) the reaction was initiated by addition of a cocktail of NADPH, glucose-6-phosphate, and glucose-6-phosphate dehydrogenase, as described earlier [33], An increase in the concentration of the product (BHAMC) was monitored with a custom-modified PTI QM-1 fluorometer (Photon Technology International, New Brunswick, NJ). The modified PTI instrument was equipped with a Hammamatsu H9059 photon-counting photomultipler module (Hamamatsu Photonics, Japan), a USB-CTR04 highspeed counter/timer module (Measurement Computing Corporation, Norton, MA) and a custom data acquisition software. The excitation was performed with a light emitted by a 405 nm CPS405 laser diode module (Thorlabs Inc, Newton, NJ) combined with a 400 nm long-pass filter. The emission wavelength was set to 470 nm with 20 nm slit. The rate of formation of the fluorescent product was estimated by determining the slope of the linear part of the kinetic curve recorded over a period of 2 – 3 min. Calibration of the assay was done with the use of HAMC, since its fluorescent yield is known to be 6 times lower than that of BHAMC [30], Calibration coefficient for HAMC was determined by measuring the intensity of fluorescence in a series of 4– 5 samples of the reaction mixture containing HAMC at concentrations from 0.05 to 2 μΜ. The coefficient obtained in this way was multiplied by 6 and used to calculate the rate of BMAMC metabolism in our samples.
2.9. Spectral studies of MIC formation
The absorbance spectra were recorded with a MC2000–2 CCD-spectrometer (Ocean Optics, Inc., Dunedin, FL). We used a flashing xenon lamp (PX2 light source from Ocean Optics in the experiments with CYP2D6-PLS and a 6427 light source manufactured by Newport/Oriel Instruments (Stratford, CT) in the experiments with CYP2D6-SS) as a light source. The experiments with CYP2D6-PLS utilized a cylindrical optical cell with 1 cm light path, while in the experiments with CYP2D6-SS the path length of a rectangular cell was equal to 5 mm. The cells were placed into a custom-designed thermostated cell holder with continuous stirring. The total volume of the mixture was 1 and 0.5 ml in the studies with CYP2D6-PLS and CYP2D6-SS respectively. The experiments were carried out at 30 °C. The incubation mixture was similar to that described above for TDI assays, except for the concentration of NADPH, which was decreased to 100 μΜ. The spectra were recorded in 320–700 nm wavelength range. Recording of the baseline was done after addition of all components of the mixture (including NADPH) except for the CYP2D6-containing membranes and MDMA. The CYP2D6-containing membranes were added to the concentration of CYP2D6 of 1 or 0.15 μΜ for CYP2D6-PLS and CYP2D6-SS respectively and the reaction was started by addition of a desired concentration of MDMA (5 – 330 μΜ). The series of spectra depicting the process of MIC formation were recorded over 45 – 90 min of incubation. The time interval between the spectral scans was 3 s during the first 6 min of recording and 30 s thereafter.
2.10. Analysis of spectral series
The series of absorbance spectra were analyzed using principal component analysis (PCA) combined with approximation of the principal component spectra with a combination of the spectral standards of the involved P450 species, as described previously [41–43], Briefly, the PCA procedure was applied to a set of difference spectra obtained by subtraction of the first spectrum in the series (zero time point) from all subsequent spectra. To interpret the resulting series in terms of the changes in concentrations of the low-spin P450, high-spin P450, and the (low spin) P420 states of the ferric heme protein the principal vectors (“spectra of the principal components”) were approximated with a linear combination of the respective spectral standards combined with a low-order (≤4) polynomial function. The latter was introduced to compensate for the baseline fluctuations during the experiment. The high-ranking components characterized by a squared correlation coefficient (R2) above the threshold, which was set to 0.5 in most cases, were considered significant and used to calculate the changes in the concentrations of the P450 species. All spectral analysis procedures were performed using our SPECTRALAB software [41].
2.11. Kinetic modeling.
Kinetic modeling and fitting the data to the TDI model was carried out using Mathematica 11.0.1.0 (Wolfram Research, Champagne, IL) software. Non-linear regression was performed with the NonlinearModelFit routine of the package. As described previously [6–7], association rate constants were fixed and dissociation rate constants were optimized to determine binding constants. The on-rate constant for binding of MDMA was fixed at 1000 μΜ/min, It should be noted, that increasing this estimate to 10,000 μΜ/min or decreasing it to 25 pM/min did not result in any considerable change in the optimized values of the other constants. Models were evaluated using the values of squared correlation coefficient (A2), correlation matrices, and residual plots. The initial parameters for the kinetic model of TDI were obtained from the exponential approximation of the kinetic curves of inactivation, as it will be discussed below.
3. Results
3.1. Obtaining a set of prototypical spectra of CYP2D6 absorbance.
Our approach to the quantitative analysis of the formation of spectral intermediates of cytochromes P450 is based on a combination of principal component analysis (PCA) with approximation of the spectra of principal components with a linear combination of spectral standards (prototypical spectra of absorbance) of the formed intermediate(s) with those of the major initial states. The application of this approach requires the knowledge of the prototypical spectra of all involved states of the heme protein.
In addition to the predominating high- and low-spin P450 states, an important spectrally distinct form of the heme protein present in most (if not all) preparations of purified cytochrome P450 enzymes and membranous P450-containing systems is the so-called “cytochrome P420” [41, 44–45], Conversion of cytochromes P450 into this inactive state is promoted by elevated temperatures, increased hydrostatic pressure, alkaline pH and some other factors [44–47] and involves replacement of the axial thiolate ligand of the heme iron with non-ionized thiol group [15–17], A complementation of the spectral standards of the low- and the high-spin P450 states with the spectrum of absorbance of the ferric P420 state, where the Soret band is positioned at 425–427 nm [41, 48–49], is ultimately required for adequate approximation of any time-dependent spectral series in P450 enzymes due to possible spontaneous P450→P420 conversion upon incubation at ambient or elevated temperatures [41, 45, 50].
In order to obtain a set of prototypical spectra of absorbance of the high-spin P450, low-spin P450 and the (low spin) P420 states of the ferric CYP2D6 enzyme present in our system prior to MDMA-dependent inactivation, we employed the same approach as we used in our studies with other cytochromes P450 [41–42, 51–52], This strategy is based on applying PCA to series of absorbance spectra obtained in the experiments with the temperature- and pressure-induced displacement of the P450 spin equilibrium (spin shift), and the pressure-induced P450→P420 transition. The resulting spectral standards for the P450 high spin, P450 low spin and the (ferric) P420 states of CYP2D6 along with the prototypical differential spectra of the spin shift and the P450→P420 transitions are shown in Fig. 2.
Fig. 2.
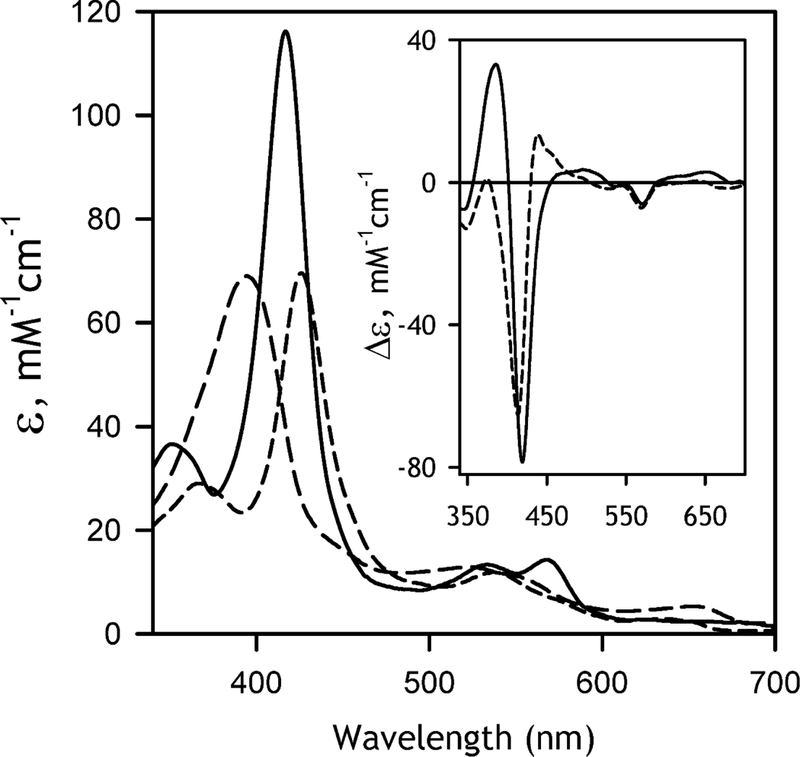
The reconstructed spectra of molar absorbtivity of the pure ferric substrate-free low spin form (solid line), ferric high-spin state (long dash), and ferric pressure-generated cytochrome P420 state (medium dash). The inset shows the differential spectra for the low-to-high spin transition (solid line) and the low-spin P450-to-P420 transition (dashed line).
3.2. Interactions of CYP2D6 with MDMA and DXM.
Application of the newly obtained set of spectral standards for the analysis of substrate-induced spin shift in CYP2D6 is illustrated in Fig. 3. Here we investigate the interactions of purified CYP2D6 in solution and in the membranes of CYP2D6-PLS with MDMA and DXM, a prototypical CYP2D6 substrate. With both compounds and in both systems (in solution and proteoliposomes) we observed a low-amplitude substrate-induced low-to-high spin shift indicative of P450 interactions with a Type-I substrate. In all cases the first principal component (1st PC) obtained with PCA covered over 94% (typically 95–99%) of the observed spectral changes.
Fig. 3.
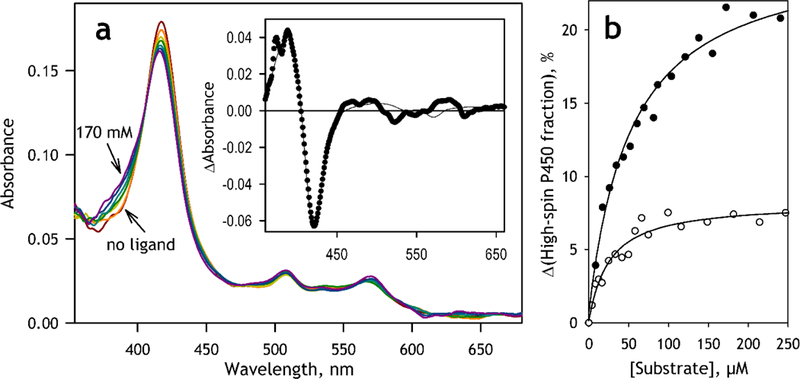
Interactions of CYP2D6-PLS with substrates monitored by substrate-induced spectral changes analyzed with PCA. (a) a series of absorbance spectra obtained in a titration of CYP2D6-PLS (1.7 μM) with DXM The spectra shown in the figure were obtained at no substrate added and at 8.5, 17, 35, 80, 120 and 170 μM DXM. Turbidity contribution was suppressed with a polynomial correction. The band seen at ~507 nm is due to the presence of BODIPY-labeled phosphatidylcholine in the proteoliposomal membrane. The insert shows the spectrum of the first principal component (closed circles) and its approximation with a combination of the standard spectra of absorbance of the pure low-spin, high spin and P420 states of CYP2D6 (solid line). (b) Substrate-induced changes in the content of the high-spin state of CYP2D6-PLS observed in titrations with DXM (closed circles) and MDMA (open circles). Solid lines shows the approximations of the titration results with the equation for the equilibrium of binary association with KD=52 μM, Amax=25 % and KD=24 μM, Amax=8.1 % for the interactions with DXM and MDMA respectively.
The spectra of the first principal component (the first principal vectors) observed with DXM and MDMA were almost identical. In both cases the spectra could be adequately approximated with a combination of our spectral standards with the squared correlation coefficient >0.95 (Fig. 3a, inset). The stoichiometry of the transition derived from this approximation confirms that the substrate binding results in a displacement of the spin equilibrium towards the high spin form. It should be noted, however, that this approximation also suggests some minor (≤20%) involvement of the P420 state of the protein (or a complex with a Type-II ligand, which is spectrally similar to the ferric P420 state) in the substrate-induced transition.
The dependencies of the content of the high spin state of CYP2D6 obtained with PCA could be adequately fitted to the equation for the equilibrium of binary association (“tight binding”) equation ([53], p 73, eq. 11–53). The fitting of the titration curves obtained in solution resulted in the values of the dissociation constant (KD) of 21.8±7.7 and 45.7±3.1 μΜ and the maximal amplitudes of the spin shift of 6.0± 1.5 and 11.9±1.8 % for CYP2D6 interactions with MDMA and DXM, respectively. Incorporation of the enzyme into the proteoliposomes does not result in any considerable change in the KD values, which were equal to 27.9±7.8 and 53.2±1.9 μΜ for the interactions with MDMA and DXM respectively, while increasing the respective amplitudes of the spin shift to 10.9±5.5 and 24.7±0.4 %.
3.3. Initial analysis of the MDMA-dependent spectral changes in CYP2D6 in the presence of NADPH.
A series of absorbance spectra of CYP2D6-PLS obtained in the process of incubation with 64 μΜ MDMA in the presence of NADPH is shown in Fig. 4a. The process of analysis of these spectra with the use of PCA and polynomial correction is illustrated in the panels b -f of the same figure. As seen from the Fig. 4a, these spectra are affected by the turbidity of the proteoliposomal suspension. Some impact from the absorbance of the flavoprotein (CPR) contained in CYP2D6-PLS is also expected. The invariant part of these contributions may be eliminated with the help of approximation of the first spectrum of the series with a combination of the CYP2D6 spectral standards with the prototypical spectra of the CPR absorbance [54] and a low order polynomial. The latter is used to approximate the turbidity component. The approximation of these “background” components found in this way is shown in Fig. 3a with dashed lines.
Fig. 4.
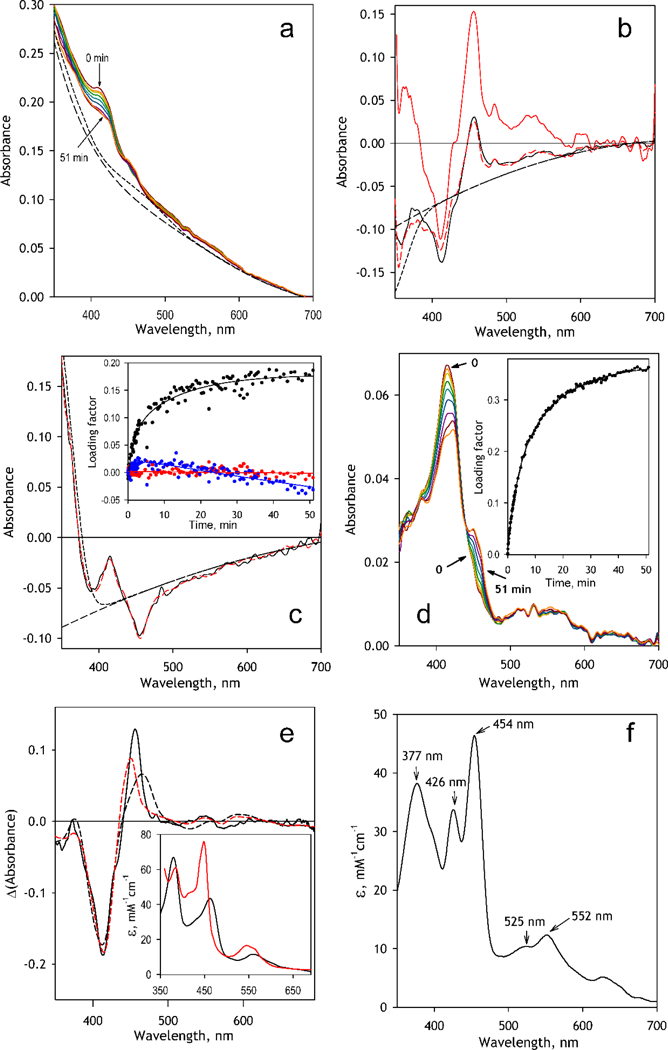
Analysis of the spectral changes observed in the process of incubation of CYP2D6-PLS with MDMA and NADPH. (a) a series of absorbance spectra recorded at 0, 0.3, 0.7, 1.5, 3, 6, 12.5, 25 and 51 min after addition of MDMA (64 μΜ). The dashed lines show the calculated contributions from turbidity alone (long dash) and in combination with absorbance of CPR (short dash); (b) Spectra of the first (black solid line) and the third (red or gray solid line) principal components. Dashed red (or gray) line shows the approximation of the 1st PC with the spectrum of the 3rd PC combined with the approximated contributions from turbidity (long dash) in combination with the changes in NADPH absorbance (short dash). (c) The spectrum of the 2nd PC (black solid line). Dashed red (or gray) line shows its approximation with the combination of the spectra of the 1st and the 3rd PC combined with the approximated contribution from turbidity (long dash) in combination with the changes in NADPH absorbance (short dash). The inset shows the changes in the loading factors for the 1st (black), 2nd (red or light gray) and 3rd (blue or dark gray) PC during incubation. (d) The same series of spectra after correction for the contributions from the turbidity, absorbance of CPR and changes in absorbance of NADPH. Inset shows the changes in the loading factor of the 1st PC; (e) The spectrum of the 1st PC for the series of corrected spectra (solid line) and its approximations with a combination of the CYP2D6 spectral standards and the prototypical spectra of absorbance of the bisthiolate complex of P450cam [59] (black dashed line) or the complex of P450cam(Fe2+) with CH3NO2 (red or gray dashed line). The respective prototypical spectra are shown in the inset. (f) The reconstructed spectrum of molar absorbtivity of the MDMA MIC of CYP2D6.
The first three principal components obtained with PCA are shown in Fig 4b and 4c. A preliminary visual analysis suggests that all three spectra represent different combinations of one and the same P450-originating differential spectrum (with the main minimum and the main maximum at 412 and 456 nm respectively) with different contributions from the changes in turbidity and, probably, in the absorbance of NADPH. The spectrum of the third principal component (Fig. 4b, red trace) appears to be essentially free of these extraneous contributions. Therefore, the changes in the turbidity and the NADPH absorbance contributing to the 1st and the 2nd PC may be found from their approximations with the spectrum of the 3 rd PC combined with the spectrum of NADPH and a second-order polynomial used to approximate the turbidity (dashed lines in Fig 4b and Fig 4c).
Subtraction of the resulting correction traces (short-dashed lines in Fig 4b and Fig 4c) multiplied by the respective loading factors (Fig. 4b, inset) along with suppressing the invariant background absorbance signal (short-dashed line in Fig. 4a) resulted in the spectral series shown in Fig 4d. Application of PCA to this series resulted in the first principal component covering over 98% of the spectral changes in the CYP2D6 absorbance.
The spectrum of the 1st PC found with this analysis (Fig. 4e) represents the major MDMA-induced spectral transition in CYP2D6 observed in these experiments. It exhibits an appearance of the band positioned at 456 nm, which, as we know from the previous studies [9, 55–56], corresponds to the main absorbance band of the ferrous heme adduct with the MDP-derived carbene. Appearance of this band is associated with a decrease in the absorbance (a trough) centered around 412 nm, which reveals a disappearance of the “normal” ferric P450 protein.
In addition to the above main spectral features, which are well known from literature [9, 55– 56], we also observed an appearance of a new absorbance band positioned at 373 nm, which has never been reported before. The presence of this band apparently reflects a split in the Soret absorbance band, which is commonly observed in the co-called “hyperporphyrin” complexes of cytochromes P450 with π-acceptor ligands, such as CO, NO, nitrosoalkanes, etc. [57], Although a hyperporphyrin split of the Soret band in the complexes of cytochromes P450 with π-accepting carbene ligands has been predicted [58], the low-wavelength component of the split Soret band (“blue” Soret) has never been observed before, as all previous studies of P450 inactivation by MDP compounds did not analyze the changes in absorbance below 400 nm [9].
This split of the Soret band with the maxima around 450–460 and 370–380 nm closely resembles the spectral properties of (bis)thiolate complexes of P450 [59], In order to reconstitute the absorbance spectrum of CYP2D6 complex with MDMA-derived carbene we attempted to approximate the spectrum of the 1st principal component found in the above analysis (Fig 4e, solid line) with a combination of the prototypical spectra of absorbance of the CYP2D6 high- and low-spin forms with the spectrum of the complex of P450cam with /;-chlorothiophenol (p-CTP) [59], where the two maxima of the split Soret band are observed at 380 and 463 nm (Fig 4e, insert, black). The result of this approximation is shown in Fig. 4e with a black dashed line. A hypothetical spectrum of CYP2D6 complex with MDMA-derived MIC may now be obtained by adding a combination of the prototypical spectra of absorbance of the high- and low-spin CYP2D6 multiplied by the coefficients found in the above approximation to the spectrum of the 1st principal component. Another possible substitute for the spectrum of carbene-bound intermediate is the spectrum of the complex of ferrous P450cam with nitromethane [57], where the two components of the split Soret band are observed at 384 and 449 nm (Fig 4e, insert, red). Approximation of the first PC with the use of this spectrum is shown in Fig. 4e with the red dashed line. Importantly, the use of either of the two spectra in approximation produced very similar results - the reconstituted spectra obtained with the two substitute prototypes were almost indistinguishable.
The reconstructed spectrum of the MDMA MIC of CYP2D6 obtained by averaging the spectra derived with the strategy described above using three individual datasets is shown in Fig. 4f. As seen from this figure, in addition to the two components of the split Soret band located at 454 and 377 nm, this spectrum also features an auxiliary maximum at 426 nm, which is consistent with the results of earlier studies of P450 inactivation with MDP compounds. This maximum, which is located at 427–428 nm in the differential spectra [9, 55–56], has been hypothesized to reflect a presence of the form of MIC, where the 6th ligand is represented by a thiol group (instead of a thiolate) [9], The positioning of the α-band of absorbance at 552 nm in our spectrum also corroborates with the previous observations, where this band was detected at the position of 556 nm in the differential spectra of MDP-derived MIC [55].
3.4. Spectroscopic studies of MDMA-dependent MIC formation in CYP2D6-PLS.
The set of the prototypical spectra of CYP2D6 and its inhibitory complex with MDMA metabolite obtained above allows analyzing the process of MDMA-dependent MIC formation in quantitative terms. For this purpose we carried out a series of spectroscopic studies of CYP2D6 inactivation at concentrations of MDMA increasing from 5 to 100 μΜ, The resulting series of spectra were analyzed with PC A. In most cases the spectral differences reflected in the 1st principal component covered over 95% (up to 98.6%) of the overall spectral changes. In addition to this main spectral transition, additional spectral differences attributable to P450 transitions were observed in the 2nd PC, which was responsible for 1 – 2% of the total changes.
Importantly, the pairs of the first two principal vectors resulting from the experiments at various concentrations of MDMA were nearly identical. The averaged spectra of the 1st and the 2nd principal components obtained in 8 individual experiments at 6 different concentrations of MDMA are shown in Fig. 5a. The representative kinetics of the changes in the respective loading factors in the process of MIC formation is illustrated in the inset to this panel. As seen from this figure, both of the two PC spectra may be adequately approximated with a combination of the decrease in absorbance of the high- and the low-spin forms of the ferric CYP2D6 with an increase in the absorbance of the MDMA-derived MIC. No appearance of additional spectral bands that may suggest a formation or a transitional presence of other intermediates such as cytochrome P420 or a 438 nm intermediate [18–19] could be detected.
Fig. 5.
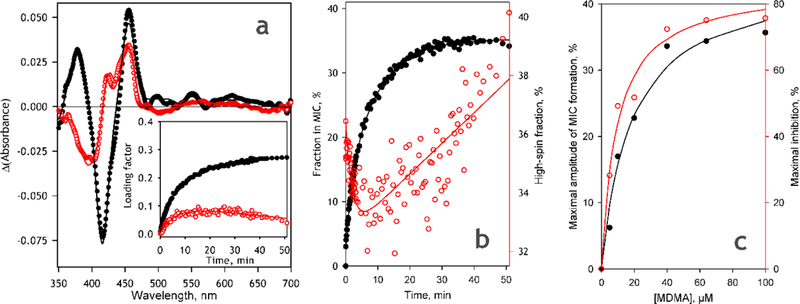
Spectral changes accompanying MDMA-dependent MIC formation in CYP2D6-PLS analyzed by PCA. (a)The averaged spectra of the 1st (black closed circles) and the 2nd (open circles) principal components obtained in 8 individual experiments at 6 different concentrations of MDMA. The inset show the representative kinetics of the changes in the respective loading factors in the process of MIC formation obtained at 64 μM MDMA. (b) Kinetics of the changes in the fraction of CYP2D6 represented by MIC state (black closed circles) and the fraction of non-MIC P450 present in the high spin state (open circles) obtained in the experiment with 64μM MDMA. Solid lines show bi-exponential approximations of the experimental data; (c) Dependence of the maximal amplitude of MIC formation (black closed circles) and the maximal amplitude of TDI monitored by the rate of BMAMC metabolism (open circles) on the concentration of MDMA. Solid lines represent the results of fitting to the Michaelis-Menten equation.
As seen from Fig. 5a, the only difference between the apparent transitions reflected in the 1st and the 2nd PC is the spin state of the heme protein undergoing the transition. If the 1st apparent transition corresponds to a conversion of the low-spin P450 to the MIC, the second PC corresponds to a close-to-stoichiometric formation of the MIC from the high-spin heme protein. While the first apparent process obeys exponential kinetics, the changes in the loading factor of the 2nd PC during TDI are characterized with a convex profile (Fig 5a inset). As a result, the initial phase of MIC formation is marked with a preferential disappearance of the high-spin state, whereas at the latter stages of the process the content of the high-spin P450 state in the non-MIC P450 continuously increases (Fig. 5b).
An important finding made in these experiment concerns the maximal degree of the CYP2D6 conversion to the MDMA-derived MIC (Fig. 5c). As seen from Fig. 5c (dataset shown in black) the maximal amplitude of MIC formation does not exceed 50% of the total P450 content even at the saturating concentrations of MDMA. Dependence of this amplitude on MDMA concentration may be approximated with a hyperbolic (Michaelis-Menten) equation with apparent KI value of 17 ± 4 μΜ and the maximal amplitude of MIC formation of 45 ± 3% of the total P450 content. At the same time, as illustrated with the dataset shown in red in Fig. 5c, the relative amplitude of TDI observed in CYP2D6 activity measurements (see infra) is considerably higher and reaches over 80% at MDMA saturation. This observation suggests that only a fraction (<50%) of the spectrally-detectable CYP2D6 enzyme present in the system is susceptible to MDMA-dependent TDI and is therefore involved in MDMA metabolism.
3.5. MDMA-dependent TDI in CYP2D6-PLS.
In order to investigate how the formation of carbene-derived MIC relates to the activity of the enzyme we investigated the MDMA-dependent TDI in CYP2D6-PLS. In our TDI experiments the role of the probe substrate was played by either DXM, a prototypical substrate of CYP2D6, or a fluorogenic substrate BMAMC. The results obtained with these two substrates were similar. A TDI series obtained with DXM is shown in Fig. 6, while the results of an experiment using BMAMC are exemplified in Fig. 9b (see infra).
Fig. 6.
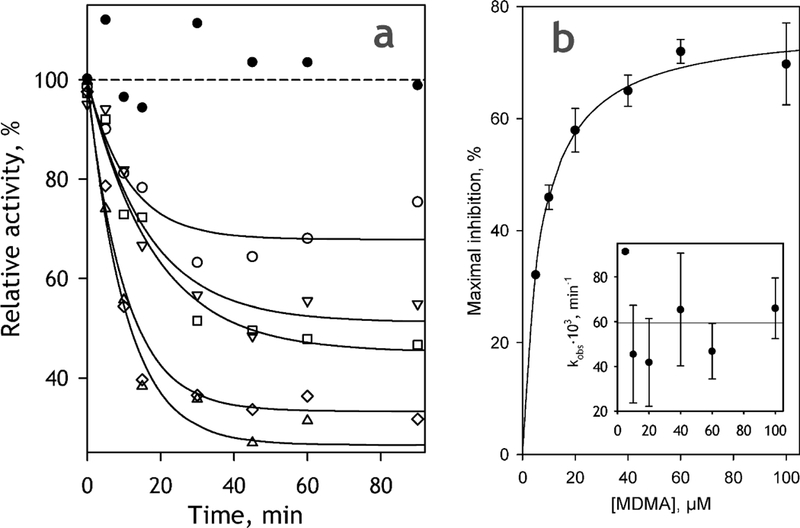
MDMA-dependent TDI of CYP2D6-PLS monitored with the use of DXM as a probe substrate. The curves shown on the panel (a) were obtained at 5 (empty circles), 10 (inverted triangles), 20 (squares), 60 (diamonds) and 100 μM (triangles). Solid line show the approximation of the datasets with a monoexponential equation. The change in the activity observed at no MDMA present are shown with filled circles. (b) Dependence of the maximal amplitude of TDI on the concentration of MDMA. The data points represent the mean values of the results of two experiments and the error bars show the respective standard deviations. Solid line shows the results of data fitting to the Michaelis-Menten equation. The inset shows the effect of MDMA concentration on kobs; horizontal solid line correspond to the averaged value of kobs.
Fig. 9.
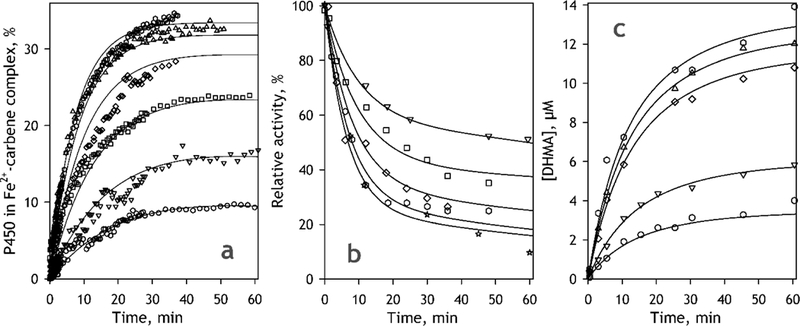
Results of fitting of the kinetics of MIC formation (a), TDI monitored by the rate of BMAMC metabolism (b) and the kinetics of DHMA formation (c) to a numerical solution of the system of differential equations corresponding to the model shown in Fig. 7. The data shown in the figures correspond to MDMA concentrations of 5 (open circles), 10 (inverted triangles) , 20 (squares), 40 (diamonds), 60 (triangles), 100 (hexagons) and 330 μΜ (stars). Solid lines represent the results of global fitting of each dataset.
As expected, the incubation of CYP2D6-PLS with MDMA in the presence of NADPH resulted in a time-dependent loss of the activity of the enzyme. However, contrary to expectations, this time-dependent inactivation was incomplete even at saturating concentrations of MDMA. The dependences of the enzyme activity on incubation time may be approximated by an exponential function (Fig 6a) and the maximal degree of inhibition derived from these approximations exhibit a hyperbolic dependence on MDMA concentration (Fig 6b). According to the results of fitting of this dependence to the Michaelis-Menten equation, the maximal depth of TDI at the enzyme saturation with MDMA is equal to 80±8 %, while the value of kI derived from this approximation is estimated to be of 7.9±2.4 μΜ. It should be noted that this incompleteness of inhibition cannot be explained by MDMA depletion, at least at high MDMA concentrations.
Our measurements of the changes in MDMA concentration showed that, while 1 hour incubation of CYP2D6-PLS with NADPH at 5 μΜ MDMA decreased the concentration of the substrate to 1.5 μΜ, a decrease observed after 1 hour of incubation at ΙΟΟμΜ MDMA did not exceed 30% of the initial concentration (data not shown).
Another important observation is a lack of any pronounced dependence of the effective rate constant of inactivation (kobs) on the concentration MDMA (Fig. 6b inset). Averaging the values obtained at the concentrations changing from 5 to 100 μΜ yields the estimate of kinact of 0.059±0.012 min−1.
These findings were quite unexpected in view of the postulated irreversibility of the formation of the MDMA-derived MIC of CYP2D6 [9], We conclude that, in contrast to the case of inactivation of CYP3 A4 by podophyllotoxin [19], the inactivation of CYP2D6 by MDMA is reversible. The complex of the ferrous CYP2D6 with the substrate-derived carbene, which is generally considered as the cause of MB I by MDP compounds, is therefore hypothesized to undergo a slow dissociation with a release of a carbene-derived metabolite (DHMA) and regeneration of the unmodified enzyme.
3.6. Studies of the MDMA-dependent MIC formation and TDI in CYP2D6-containing Supersomes™.
Summarizing, the above analysis of the MDMA-induced spectral changes and MDMA-dependent TDI in CYP2D6-PLS led us to the following conclusions: (1) Only a fraction (40– 50%) of spectrally detectable P450 in CYP2D6-PLS is involved in MDMA-dependent formation of MIC. The remaining part of the enzyme doesn’t seem to participate in the MDMA metabolism and remains unaffected by thereto related MBI. (2) MDMA-dependent formation of the Fe2+:carbene complex of CYP2D6 appears to be reversible, so that at infinite time of incubation its concentration approaches a steady-state level dictated by the MDMA concentration and the rate constants of the formation and decay of MIC. (3) In contrast to what was reported in our study of the PPT-dependent MBI in CYP3A4 [19], the transition from the Fe3+:carbene intermediate to the Fe2+:carbene complex of CYP2D6 appears to be rapid, so that no accumulation of the Fe3+:carbene intermediate of CYP2D6 is observed.
The above conclusions were made based on the studies in an artificial reconstituted system (PLS). In order to probe if they are relevant to the case of CYP2D6 embedded into a native microsomal membrane, we applied our newly developed approaches to the studies of MDMA-dependent MBI in CYP2D6-containing Supersomes™ (CYP2D6-SS).
Precise studies with absorbance spectroscopy in microsomal system are very challenging. High optical density of the microsomal suspension in the near-UV region, which is further increased by the presence of NADPH, forced us to decrease the path length of the optical cell to 5 mm. However, even with this modification, high optical density of the system did not allow us to work at the CYP2D6 concentrations higher that 0.15 μM. Due to low amplitude of the P450-specific spectral signal and high background turbidity, the procedure of PCA described above for absolute spectra of CYP2D6-PLS was in this case applied to a series of differential spectra. These spectra were obtained by subtraction of the spectrum recorded before addition of MDMA from all other spectra of the series.
A series of turbidity-corrected differential spectra obtained in the experiment with CYP2D6-SS is shown in Fig. 7a. As seen from this figure, the changes in P450 absorbance accompanying MDMA-dpendent MBI in Supersomes are similar to those observed in the CYP2D6-PLS (Fig. 3). A pronounced decrease in optical density in the region of the Soret bands of the high- and low- spin forms of intact P450 (390–420 nm) is accompanied with appearance of two new maxima located at ~455 and ~375 nm. Consequently, the spectrum of the first principal component derived by PCA (Fig. 7b) is very similar to that observed in the PLS system. Although the spectrum of the 2nd PC may also reveal P450-specific bands (Fig. 7b inset) indicative of a transition between the P450 high spin state and a mixture of the P420 and MIC states, its fitting by our set of spectral standards (solid line in Fig. 7b, inset) is loose and the changes in its loading factor (Fig. 7c inset, filled circles) doesn’t reveal any kinetics distinct from that observed with the 1st PC.
Fig. 7.
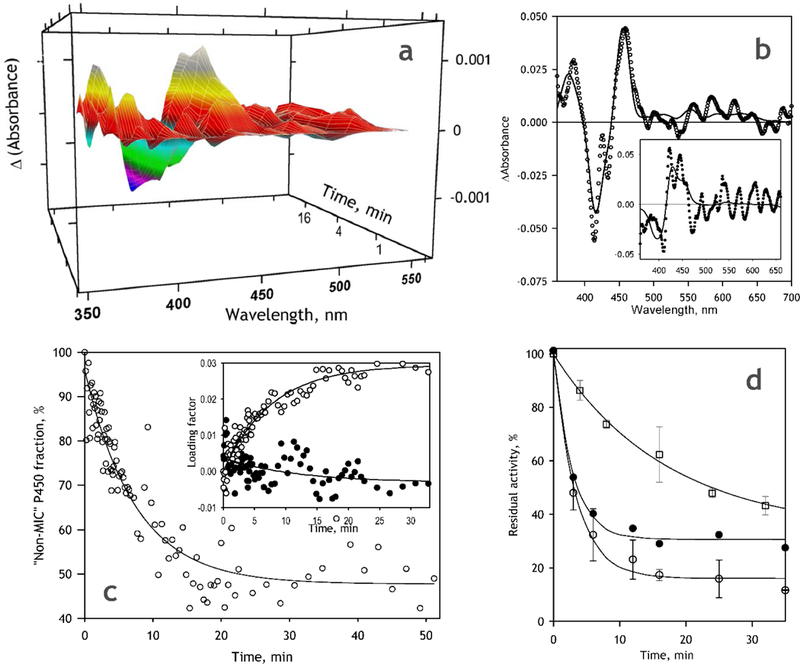
MDMA-dependent MIC formation and the respective TDI in CYP2D6-SS. (a) A series of differential spectra recorded during incubation of CYP2D6-SS in the presence of 100 μM MDMA and NADPH.The spectra shown in the figure are smoothed (3-rd order polynomial, 7-point window) and corrected on the changes in turbidity; (b) The respective spectra of the first (main panel) and the second (inset) principal components. Solid lines show the approximations with a combination of CYP2D6 absorbance standards; (c) Changes in the fraction of CYP2D6 represented by the “intact” (non-MIC) states. The inset shows the respective changes in the loading factors for the 1st (open circles) and the 2nd (closed circles). Solid lines represent exponential approximations of the data sets; (d) Changes in the activity of CYP2D6-SS with BMAMC in the process of incubation with NADPH at no substrate added (open squares) and in the presence of 100 μΜ MDMA (open circles). The data points represent the mean values of the results of two experiments and the error bars show the respective standard deviations. Closed circles correspond to the result of correction of the changes observed with MDMA on MDMA-independent inactivation. Solid lines represent exponential approximations of the respective data.
The kinetics of the changes in the concentration of the MDMA-derived MIC obtained with the PCA procedure is shown in Fig. 7c (main panel). At seen from this figure, similar to that observed in the PLS system, the conversion of CYP2D6 into spectrally-detectable MIC is essentially incomplete and amounts only up to −50% at nearly-saturating MDMA concentration of 100 μΜ.
The results of studying the MDMA-dependent TDI in Supersomes with the use of BMAMC as a probe substrate are shown in Fig. 7d. As seen from this figure, in contrast to the PLS system where no time-dependent inactivation was observed in the absence of MDMA, incubation of Supersomes with NADPH at no MDMA added resulted in a slow but substantial decrease in the enzymatic activity (Fig 7c, squares). Addition of 100 μΜ MDMA makes the inactivation considerably faster and more pronounced (empty circles).
When we compare the values of parameters obtained, using exponential fitting of the kinetic curves of MIC formation and TDI in both CYP2D6-PLS and in CYP2D6-SS (Table 1), the two systems exhibited fairly similar values of both the relative amplitudes and the effective kinetic constants of MIC formation and TDI. In both cases the maximal degree of TDI at 100 μΜ MDMA reaches only about 80% of the functionally active enzyme. In both systems the fraction subjected to MDMA-dependent MIC formation amounts to only about −50% of the spectrally detectable enzyme. Therefore, the main conclusions driven above from the results of our studies in the PLS system appear to be valid for the case of CYP2D6-SS as well.
Table 1.
Parameters of time-dependent inactivation of CYP2D6 in CYP2D6-PLS and CYP2D6-SS obtainedfrom the studies of MIC formation and TDI at 100 μΜ MDMA.*
| CYP2D6-PLS | CYP2D6-SS | |||
|---|---|---|---|---|
| Amax (MIC), % | 37 | ±8 | 49 | ±26 |
| Amax (TDI), % | 80 | ±8 | 84 | ±3 |
| Kobs, (MIC), min−1 | 0.085 | ± 0.028 | 0.19 | ±0.11 |
| Kobs, (TDI), min−1 | 0.15 | ±0.11 | 0.28 | ±0.08 |
The values given in the Table represent the averages of the results of 3–4 individual experiments. The individual estimates of Amax and kobs were obtained from the fitting of the kinetic curves of MIC formation and TDI at 100 μΜ MDMA with an exponential equation. The “±” values show the confidence interval calculated for p = 0.05.
3.7. Kinetic modeling and global data analysis.
A simple kinetic model of the processes involved in MDMA-dependent TDI based on the conclusions driven above is shown in Fig. 8. Addition of MDMA (S) to CYP2D6 (E) results in a reversible formation of the enzyme-substrate complex (ES) governed by the rate constants k1 and k2 . The enzyme turnover observed in the presence of NADPH eventually results in the decay of this complex with the formation of the product of demethylenation (DHMA). The flux through this path is determined by the catalytic constant k3. However, in addition to this productive turnover, there is another path, which leads to the formation of the MDMA-derived carbene and its ligation to the heme iron that results in the formation of the MIC (a Fe2+:carbene complex). The probability that the complex ES will proceed through this path is dictated by the rate constant k4. In order to explain incomplete inhibition of CYP2D6 and hyperbolic dependence of the depth of inhibition on MDMA concentration, a reversibility pathway for enzyme regeneration needs to be included (k5). This pathway results in a release of the carbene from the MIC, its interaction with water and the eventual formation of the demethylenated product (DHMA). We also added an allowance for MDMA-independent inactivation, which may proceed through various mechanisms including H2O2-dependent heme depletion, protein denaturation or inactivation of CPR via loss of FMN. This additional pathway of inactivation is governed by the rate constant k6.
Fig. 8.
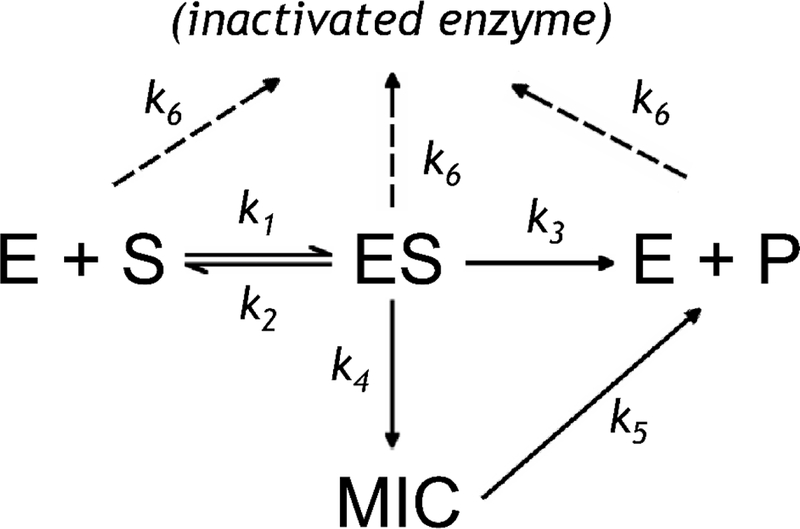
Kinetic model of the processes involved in MDMA-dependent TDI
We employed the numerical integration functionality of the Wolfram Mathematica package (Wolfram Research, Champagne, IL) in probing the applicability of the above model for fitting the series of kinetic curves obtained in our studies of MIC formation with absorbance spectroscopy, the studies of MDMA-dependent TDI with the use of DXM and BMAMC as probe substrates, and in the studies of time-dependent changes in the rate of MDMA metabolism. The fitting was performed with the Levenberg-Marquardt optimization algorithm where we treated all kinetic constants present in the model as the optimization parameters, except for k1 which was fixed at 1000 μM-min−1 (see Materials and Methods). In addition to the set of rate constant, the parameters under optimization also included the parameter Finact which represents the fraction of the total enzyme population that participates in catalysis, formation of MIC and MDMA-dependent TDI. It should be noted, however, that the value of Finact may be reliably determined from the data on MIC formation only. In other two cases (TDI and DHMA formation) the estimate of this parameter was fixed at 0.423, which corresponds to the value found from the analysis of MIC formation. The set of the differential equations used in optimization was written in the way that the acting total concentration of the enzyme ([E]+[ES]+[MIC]) was determined as a product of Fi and the total concentration of spectrally detectable P450 in the system ([E]tot). It should be also noted that the consideration of the pathway of MDMA-independent inactivation (k6) was necessary only in the fitting of the kinetic curves of TDI and DHMA formation, while in the case of the kinetics of MIC formation the best fitting results were obtained with k6=0. Results of these fitting trials are illustrated in Fig. 9 and the resulting parameters of our model are shown in Table 2.
Table 2.
Parameters obtained from approximation of the kinetic curves of MIC formation, TDI and kinetics of MDMA demethylenation in CYP2D6-PLS with a system of differential equations depicting the proposed kinetic scheme of MDMA metabolism (Fig. 5).*
| Parameter | TDI | Kinetics of MIC formation |
Kinetics of DHMA production |
|||
|---|---|---|---|---|---|---|
| Finact | 0.42 | 0.42 | ±0.01 | 0.42 | ||
| kD, μMa | 23.9 | ±1.2 | 55.5 | ±0.4 | 49.5 | ±8.4 |
| k3, min−1 | 3.2 | ±0.8 | 1.7 | ±0.4 | 3.9 | ±0.4 |
| k4, min−1 | 0.085 | ±0.023 | 0.167 | ±0.003 | 0.14 | ±0.04 |
| k5, min−1 | 0.033 | ±0.006 | 0.027 | ±0.003 | 0.047 | ±0.005 |
| k6, min−1 | 0.005 | ±0.002 | N/A | 0.041 | ±0.008 | |
| R2 | >0.9934 | >0.9934 | >0.9921 | |||
The values given for the TDI and DHMA formation kinetics represent the averages of the results obtained with three and two individual datasets, respectively. In the case of TDI two of these data sets were obtained with DXM and another one - with BMAMC as a probe substrate. The “±” values shown for the parameters of TDI and DHMA production correspond to the confidence interval calculated for p = 0.05. In the case of MIC formation the values given in the table represent the averages of the results of a series of individual optimization trials with one and the same data set and the initial estimates for k3 evenly distributed on the interval from 1.0 to 2.25 min’1. Here the “±” values correspond to the standard deviations calculated with the estimates obtained in 7 fitting trials.
Dissociation constant for the complex ES obtained by dividing the optimized value of the off-rate constant (k2) by the value of the on-rate constant (k1), which was fixed at 1,000 μM min−1.
Fig. 9a shows the result of fitting of a series of kinetic curves of the changes in the concentration of MDMA-derived MIC monitored by absorbance spectroscopy. As seen from this figure, the proposed kinetic model provides a satisfactory approximation of the whole data set (R2=0.9935). It should be noted that, when applied to the full data set, where the sampling rate considerably decreases after the first 6 min of incubation (see Materials and Methods), the Levenberg-Marquardt non-linear regression procedure resulted in efficient optimization of all parameters of the model except for k3 The dependence of the sum of square deviations on k3 is extremely weak and the final optimized value of k3 exhibited a pronounced dependence on the initial estimate of this parameter. The values of R2 did not reveal any noticeable changes in the quality of approximation (R2>0.9934) upon increasing the initial estimate of k3 from 0.5 to 2.25 min−1, after which point the R2 value started to decrease gradually. However, resampling the kinetic curves with the constant time interval of 1.5 min (and thereby decreasing the weight of the initial part of the kinetic curves) allowed to find an optimum at k3=1.65 min−1, while having no considerable effect on the other parameters of the model.
The results of fitting of the set of kinetic curves of the decrease in the enzymatic activity of CYP2D6 (TDI series) measured with the use of BMAMC as a probe substrate are shown in Fig. 9b. For this fitting all kinetic curves were normalized on the initial activity of the enzyme measured before the addition of MDMA to the incubation mixture. Similar results were also obtained when our kinetic model was used for fitting of the TDI dataset shown in Fig. 6, which was obtained with the use of DXM as the probe substrate.
In addition to studying the kinetics of MIC formation and monitoring the process of TDI with the use of the probe substrates, we also investigated the kinetics of product (DHMA) formation in the process of incubation of CYP2D6-PLS with NADPH and various concentrations of MDMA. One of the two datasets obtained in these experiments along with the results of its approximation with our kinetic model (Fig. 8) is shown in Fig. 9c. The resulting parameters shown in Table 2 for the kinetics of product formation were obtained by averaging the results of fitting of two datasets obtained in independent experiments.
4. Discussion
In the present study we dissected the mechanism of MDMA-dependent inactivation of CYP2D6 and the pertinent formation of the MDMA-derived MIC with the use of model membranous systems and advanced methods of spectral analysis. High amplitude of the spectral signal in CYP2D6-incorporated PLS allowed us to characterize the spectral properties of the formed MIC in details and obtain an accurate spectral signature of MIC formation. Even a very meticulous analysis of the spectral series documenting the process of MDMA-dependent MIC formation in CYP2D6-PLS did not reveal the presence of any MICs or transient intermediates other than the Fe2+:carbene complex that has the Soret band split between the main peaks located at 454 and 377 nm bands, and an auxiliary maximum found at 426 nm (Fig. 4f). No intermediate with the Soret band at 438 nm, which hypothetically corresponds to a transient Fe3+:carbene intermediate [18–19], could be detected in our studies. Therefore, we can conclude that, in contrast to what was reported in our study of the PPT-dependent MBI in CYP3A4 [19], the transition from the Fe3+:carbene intermediate to the Fe2+:carbene complex of CYP2D6 is rapid. Concequently, no accumulation of the Fe3+:carbene intermediate of CYP2D6 is observed.
In contrast to what we postulated for the PPT-derived Fe2+:carbene complex of CYP3A4, the MDMA-dependent formation of the Fe2+:carbene complex of CYP2D6 appears to be reversible, so that at infinite time of incubation its concentration approaches a steady-state level, which is dictated by the MDMA concentration and the rate constants of the formation and decay of MIC. Taking into account quite mediocre affinity of CYP2D6 to MDMA (KD determined by various methods in our experiments varies from 17 to 56 pM) the steady-state concentration of MDMA-derived MIC of CYP2D6 at physiological concentrations of MDMA may be expected to be relatively low. Thus, the peak concentration of MDMA in blood plasma of healthy volunteers after taking a recreational dose of the drug (100–150 mg) is as low as 1 pM [60–61] and the averaged liver-to-blood ratio of MDMA concentrations based on six cases of MDMA-related fatalities overviewed in [23] amounts to 7.6 +- 4.5. Therefore, the physiological concentration of MDMA in human liver is unlikely to exceed 10 pM, being therefore below the values of KD of CYP2D6-MDMA complexes determined in either spectrophotometric titrations or in MDMA metabolism experiments in this study (Table 2). This estimate of cellular concentration also does not go far above the values of KI estimated for MDMA-dependent inhibition of DXM metabolism in HLM (8.8 – 45.3μΜ, [24]). According to the parameters of MDMA-dependent inactivation determined in the present study, the anticipated amplitude of MIC formation of CYP2D6 at these levels of MDMA concentration does not exceed 15% of the total CYP2D6 content or about 30% of the fraction of the enzyme active in MDMA metabolism.
The above analysis implies that the earlier studies with the use of replot method may overpredict both the rate and the maximal depth of MDMA-dependent inactivation of CYP2D6. The conclusion that the majority of hepatic CYP2D6 is inactivated within an hour after a recreational dose of MDMA derived from the earlier simulations [25] may therefore considerably overestimate the importance of MDMA-dependent MBI of CYP2D6.
Another important finding stemmed from our analysis of MDMA-dependent MIC formation in CYP2D6 concerns the functional heterogeneity of the membrane-bound enzyme that we observed in both the proteolyposomal system (CYP2D6-PLS) and in the insect cell microsomes containing full-length CYP2D6 (CYP2D6-SS). We demonstrated that in both systems the fraction of CYP2D6 involved in MDMA-dependent formation of MIC amounts to no more than 50% of the spectrally detectable P450. The remaining part of the enzyme population remains unaffected by MDMA-dependent modification and therefore is immune from MDMA metabolism and the associated MBI.
This conclusion closely relates to the concept of persistent heterogeneity of microsomal cytochromes P450 introduced in our earlier studies [28–29, 62], According to this concept, the observed functional dissimilarity in the pull of otherwise homogenous P450 enzymes is caused by their oligomerization. It is hypothesized that the divergence of the enzyme between two fractions with different properties may reflect an important difference in the orientation and/or conformation of the subunits constituting the oligomers formed by cytochromes P450 in microsomal membranes [29, 62]. This type of conformational non-equivalence of subunits in oligomer may be termed “positional heterogeneity” [63].
Our recent studies of oligomerization of CYP2D6 demonstrated that its propensity to oligomerize observed in either in CYP2D6-incorporated Supersomes or in human liver microsomes (HLM) is among the highest observed with any human cytochrome P450 studied up to date [33]. According to the above study, the lipid-to-P450 ratio at which the degree of CYP2D6 oligomerization in the microsomal membrane comprises 50% of its total content is as high as −5000:1 [33]. In keeping with this estimate, the protein must be completely oligomerized in our preparations of CYP2D6-PLS, where the lipid-to-P450 was in the range of 180:1 – 250:1. Apparent lack of dynamic equilibrium between the two conformers may be explained by a very slow rate of reorganization of the oligomers through their dissociation and re-association in crowded environment of the membranes [33].
Oligomerization of CYP2D6 in microsomal membranes also involves its interactions with other P450 species. Our recent study with LRET-based approach revealed the formation of mixed oligomers of CYP2D6 with CYP3 A4 and CYP2E1 in the P450-incorporated Supersomes and human liver microsomes (HLM) [33]. Furthermore, the above study also demonstrated a profound effect of MDMA on both the KD and the spatial organization of homo- and heterooligomers of CYP2D6 [33], Along with the results of the present study, these observations indicate that the rate of MDMA metabolism by CYP2D6 and the amplitude of its MDMA-dependent MBI may exhibit a complex dependence on the concentrations of the other P450 species present in HLM. In particular, a pronounced modulation of interactions of CYP2D6 with CYP2E1 in the presence of MDMA [33] suggests that the effects of this ethanol-inducible enzyme on CYP2D6-dependent metabolism of MDMA and the related MBI may be involved in the mechanisms of exacerbation of the MDMA cytotoxicity by alcohol [64–65].
These inferences highlight the need in further extension of the use of the methods developed in this study to the analysis of MDMA-dependent inactivation of CYP2D6 in HLM. The use of the model systems that contains CYP2D6 as the sole P450 enzyme in the present study raises some reservations to the relevance of straight extrapolation of our results to the situation taking place in HLM, where CYP2D6 shares protein partners and directly interacts with multiple other P450 species. The studies of CYP2D6-derived MIC formaition in HLM are methodologically challenging due to low concentration of CYP2D6 and high turbidity of microsomal suspension. However, these difficulties may be circumvented through selective enrichment of HLM membranes with selected P450 species developed in our recent work [33]. The study of MDMA-dependent MIC formation in HLM with the use of these techniques is now underway.
Acknowledgment
This research was supported by NIH grant R01-GM114369.
Footnotes
Conflict of interest
The authors have no conflict of interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].de Montellano PRO, Correia MA. Inhibition of cytochrome P450 enzymes In: de Montellano PRO, editor. Cytochrome P450: Structure, Mechanism, and Biochemistry: Springer, 1995. p. 305–64. [Google Scholar]
- [2].Orr STM, Ripp SL, Ballard TE, Henderson JL, Scott DO, Obach RS, et al. Mechanism-Based Inactivation (MBI) of Cytochrome P450 Enzymes: Structure-Activity Relationships and Discovery Strategies To Mitigate Drug-Drug Interaction Risks. J Med Chem 2012;55:4896–933. [DOI] [PubMed] [Google Scholar]
- [3].Obach RS, Walsky RL, Venkatakrishnan K. Mechanism-based inactivation of human cytochrome P450 enzymes and the prediction of drug-drug interactions. Drug Metab Disp 2007;35:246–55. [DOI] [PubMed] [Google Scholar]
- [4].Riley RJ, Grime K, Weaver R. Time-dependent CYP inhibition. Expert Opin Drug Metab Toxicol 2007;3:51–66. [DOI] [PubMed] [Google Scholar]
- [5].Zimmerlin A, Trunzer M, Faller B. CYP3A Time-Dependent Inhibition Risk Assessment Validated with 400 Reference Drugs. Drug Metab Disp 2011;39:1039–46. [DOI] [PubMed] [Google Scholar]
- [6].Nagar S, Jones JP, Korzekwa K. A Numerical Method for Analysis of In Vitro Time-Dependent Inhibition Data. Part 1. Theoretical Considerations. Drug Metab Disp 2014;42:1575–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Korzekwa K, Tweedie D, Argikar UA, Whitcher-Johnstone A, Bell L, Bickford S, et al. A Numerical Method for Analysis of In Vitro Time-Dependent Inhibition Data. Part 2. Application to Experimental Data. Drug Metab Disp 2014;42:1587–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Antolino-Lobo I, Meulenbelt J, van den Berg M, van Duursen MBM. A mechanistic insight into 3,4-methylenedioxymethamphetamine (“ecstasy”)-mediated hepatotoxicity. Veterinary Quarterly 2011;31:193–205. [DOI] [PubMed] [Google Scholar]
- [9].Murray M Mechanisms of inhibitory and regulatory effects of methylenedioxyphenyl compounds on cytochrome P450-dependent drug oxidation. Curr Drug Metab 2000;1:67–84. [DOI] [PubMed] [Google Scholar]
- [10].Beumel GA, Levi PE, Hodgson E. Spectral interactions of piperonyl butoxide and isocyanides with purified hepatic cytochrome P-450 from uninduced mice. General Pharmacology 1985;16:193–7. [DOI] [PubMed] [Google Scholar]
- [11].Kulkarni AP, Hodgson E. Spectral interactions of insecticide synergists with microsomal cytochrome P-450 from insecticide-resistant and susceptible house-flies. Pesticide Biochemistry and Physiology 1976;6:183–91. [Google Scholar]
- [12].Kulkarni AP, Hodgson E. The metabolism of insecticides - the role of monooxygenase enzymes. Annu Rev Pharm Ther 1984;24:19–42. [DOI] [PubMed] [Google Scholar]
- [13].Kulkarni AP, Hodgson E. Cumene hydroperoxide-generated spectral interactions of piperonyl butoxide and other synergists with microsomes from mammals and insects. Pesticide Biochemistry and Physiology 1978;9:75–83. [Google Scholar]
- [14].Usia T, Watabe T, Kadota S, Tezuka Y. Metabolite-cytochrome P450 complex formation by methylenedioxyphenyl lignans of Piper cubeba: Mechanism-based inhibition. Life Sci 2005;76:2381–91. [DOI] [PubMed] [Google Scholar]
- [15].Martinis SA, Blanke SR, Hager LP, Sligar SG, Hui Bon Hoa G, Rux JJ, et al. Probing ng the heme iron coordination structure of pressure-induced cytochrome P420(cam). Biochemistry 1996;35:14530–6. [DOI] [PubMed] [Google Scholar]
- [16].Perera R, Sono M, Sigman JA, Pfister TD, Lu Y, Dawson JH. Neutral thiol as a proximal ligand to ferrous heme iron: Implications for heme proteins that lose cysteine thiolate ligation on reduction. Proc Natl Acad Sci USA 2003;100:3641–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Dunford AJ, McLean KJ, Sabri M, Seward HE, Heyes DJ, Scrutton NS, et al. Rapid P450 heme iron reduction by laser photoexcitation of Mycobacterium tuberculosis CYP121 and CYP51B1 - Analysis of CO complexation reactions and reversibility of the P450/P420 equilibrium. J Biol Chem 2007;282:24816–24. [DOI] [PubMed] [Google Scholar]
- [18].Dickins M, Elcombe CR, Moloney SJ, Netter KJ, Bridges JW. Further-studies on the dissociation of the isosafrole metabolite-cytochrome-P-450 complex. Biochem Pharm 1979;28:231–8. [DOI] [PubMed] [Google Scholar]
- [19].Barnaba C, Yadav J, Nagar S, Korzekwa K, Jones JP. Mechanism-Based Inhibition of CYP3A4 by Podophyllotoxin: Aging of an Intermediate Is Important for in Vitro/in Vivo Correlations. Mol Pharm 2016;13:2833–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Selim K, Kaplowitz N. Hepatotoxicity of psychotropic drugs. Hepatology 1999;29:1347–51. [DOI] [PubMed] [Google Scholar]
- [21].Carvalho M, Carmo H, Costa VM, Capela JP, Pontes H, Remiao F, et al. Toxicity of amphetamines: an update. Arch Toxycol 2012;86:1167–231. [DOI] [PubMed] [Google Scholar]
- [22].Andreu V, Mas A, Bruguera MB, Salmeron JM, Moreno V, Nogue S, et al. Ecstasy: a common cause of severe acute hepatotoxicity. J Hepatology 1998;29:394–7. [DOI] [PubMed] [Google Scholar]
- [23].Carmo H, Brulport M, Hermes M, Oesch F, Silva R, Ferreira LM, et al. Influence of CYP2D6 polymorphism on 3,4-methylenedioxymethamphetamine (‘ecstasy’) cytotoxicity. Pharmacogenet Genomics 2006;16:789–99. [DOI] [PubMed] [Google Scholar]
- [24].Heydari A, Yeo KR, Lennard MS, Ellis SW, Tucker GT, Rostami-Hodjegan A. Mechanism-based inactivation of CYP2D6 by methylenedioxymethamphetamine. Drug Metab Disp 2004;32:1213–7. [DOI] [PubMed] [Google Scholar]
- [25].Yang J, Jamei M, Heydari A, Yeo KR, de la Torre R, Farre M, et al. Implications of mechanism-based inhibition of CYP2D6 for the pharmacokinetics and toxicity of MDMA. J Psychopharm 2006;20:842–9. [DOI] [PubMed] [Google Scholar]
- [26].Van LM, Heydari A, Yang J, Hargreaves J, Rowland-Yeo K, Lennard MS, et al. The impact of experimental design on assessing mechanism-based inactivation of CYP2D6 by MDMA (Ecstasy). J Psychopharm 2006;20:834–41. [DOI] [PubMed] [Google Scholar]
- [27].O’Mathuna B, Farre M, Rostami-Hodjegan A, Yang J, Cuyas E, Torrens M, et al. The consequences of 3.4-methylenedioxymethamphetamine induced CYP2D6 inhibition in humans. J Clin Psychopharm 2008;28:525–31. [DOI] [PubMed] [Google Scholar]
- [28].Davydov DR, Halpert JR. Allosteric P450 mechanisms: multiple binding sites, multiple conformers or both? Expert Opin Drug Metab Toxicol 2008;4:1523–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Davydov R Molecular Organization of the Microsomal Oxidative System: a New Connotation for an Old Term. Biochemistry Moscow - Suppl B - Biomed Chem 2016;10:10–21. [DOI] [PubMed] [Google Scholar]
- [30].Venhorst J, Onderwater RCA, Meerman JHN, Commandeur JNM, Vermeulen NPE. Influence of N-substitution of 7-methoxy-4-(aminomethyl)-coumarin on cytochrome P450 metabolism and selectivity. Drug Metab Disp 2000;28:1524–32. [PubMed] [Google Scholar]
- [31].Harlow GR, Halpert JR. Analysis of human cytochrome P450 3A4 cooperativity: Construction and characterization of a site-directed mutant that displays hyperbolic steroid hydroxylation kinetics. Proc Natl Acad Sci USA 1998;95:6636–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wang A, Savas U, Hsu MH, Stout CD, Johnson EF. Crystal Structure of Human Cytochrome P450 2D6 with Prinomastat Bound. J Biol Chem 2012;287:10834–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Davydov DR, Davydova NY, Rodgers JT, Rushmore TH, Jones JP. Toward a systems approach to the human cytochrome P450 ensemble: interactions between CYP2D6 and CYP2E1 and their functional consequences. Biochem J 2017;474:3523–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Davydov DR, Davydova NY, Sineva EV, Halpert JR. Interactions among Cytochromes P450 in Microsomal Membranes: Oligomerization of Cytochromes P450 3A4, 3A5 and 2E1 and its Functional Consequences. J Biol Chem 2015;453:219–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Bartlett GR. Phosphorus assay in column chromatography. J Biol Chem 1959;234:466–8. [PubMed] [Google Scholar]
- [36].Shen AL, Porter TD, Wilson TE, Kasper CB. Structural analysis of the FMN binding domain of NADPH-cytochrome P-450 oxidoreductase by site-directed mutagenesis. J Biol Chem 1989;264:7584–9. [PubMed] [Google Scholar]
- [37].Massey V The microestimation of succinate and the extinction coefficient of cytochrome c. Biochim Biophys Acta 1959;34:225–56. [DOI] [PubMed] [Google Scholar]
- [38].French JS, Coon MJ. Properties of NADPH-cytochrome P-450 reductase purified from rabbit liver microsomes. Arch Biochem Biophys 1979;195:565–77. [DOI] [PubMed] [Google Scholar]
- [39].Keizers PHJ, Van Dijk BR, De Graaf C, Van Vugt-Lussenburg BMA, Vermeulen NPE, Commandeur JNM. Metabolism of N-substituted 7-methoxy-4-(aminomethyl)-coumarins by cytochrome P450 2D6 mutants and the indication of additional substrate interaction points. Xenobiotica 2006;36:763–71. [DOI] [PubMed] [Google Scholar]
- [40].Nakamura K, Hanna IH, Cai HL, Nishimura Y, Williams KM, Guengerich FP. Coumarin substrates for cytochrome P450 2D6 fluorescence assays. Anal Biochem 2001;292:280–6. [DOI] [PubMed] [Google Scholar]
- [41].Davydov DR, Deprez E, Hui Bon Hoa G, Knyushko TV, Kuznetsova GP, Koen YM, et al. High-Pressure-Induced Transitions in Microsomal Cytochrome P450 2B4 in Solution -Evidence for Conformational Inhomogeneity in the Oligomers. Arch Biochem Biophys 1995;320:330–44. [DOI] [PubMed] [Google Scholar]
- [42].Renaud JP, Davydov DR, Heirwegh KPM, Mansuy D, Hui Bon Hoa G. Thermodynamic studies of substrate binding and spin transitions in human cytochrome P-450 3A4 expressed in yeast microsomes. Biochem J 1996;319:675–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Davydov DR, Fernando H, Baas BJ, Sligar SG, Halpert JR. Kinetics of dithionite-dependent reduction of cytochrome P450 3A4: Heterogeneity of the enzyme caused by its oligomerization. Biochemistry 2005;44:13902–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Imai Y, Sato R. Conversion of P-450 to P-420 by neutral salts and some other reagents Eur J Biochem 1967; 1:419–26. [DOI] [PubMed] [Google Scholar]
- [45].Yamazaki H, Inoue K, Turvy CG, Guengerich FP, Shimada T. Effects of freezing, thawing, and storage of human liver samples on the microsomal contents and activities of cytochrome P450 enzymes. Drug Metab Disp 1997;25:168–74. [PubMed] [Google Scholar]
- [46].Hui Bon Hoa G, McLean MA, Sligar SG. High pressure, a tool for exploring heme protein active sites. Biochim Biophys Acta 2002;1595:297–308. [DOI] [PubMed] [Google Scholar]
- [47].Manna SK, Mazumdar S. Reversible inactivation of cytochrome P450 by alkaline earth metal ions: Auxiliary metal ion induced conformation change and formation of inactive P420 species in CYP101. J Inorg Biochem 2008; 102:1312–21. [DOI] [PubMed] [Google Scholar]
- [48].Fisher MT, SS F, SS G. High-pressure investigations of cytochrome P-450 spin and substrate binding equilibria. Arch Biochem Biophys 1985;240:456–63. [DOI] [PubMed] [Google Scholar]
- [49].Sun L, Wang ZH, Jiang HL, Tan XS, Huang ZX. Novel Conformational Transitions of Human Cytochrome P450 2C8 during Thermal and Acid-induced Unfolding. Chinese J Chem 2010;28:1491–502. [Google Scholar]
- [50].Davydov DR, Knyushko TV, Hui Bon Hoa G. High pressure induced inactivation of ferrous cytochrome P-450 LM2 (2B4) CO complex: evidence for the presence of two conformers in the oligomer. Biochem Biophys Res Commun 1992;188:216–21. [DOI] [PubMed] [Google Scholar]
- [51].Sineva EV, Davydov DR. Cytochrome P450 from Photobacterium profundum SS9, a piezophilic bacterium, exhibits a tightened control of water access to the active site. Biochemistry 2010;49:10636–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Davydov DR, Kumar S, Halpert JR. Allosteric mechanisms in P450eryF probed with 1-pyrenebutanol, a novel fluorescent substrate. Biochem Biophys Res Commun 2002;294:806–12. [DOI] [PubMed] [Google Scholar]
- [53].Segel IH. Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems. New York: Wiley-Interscience, 1975. [Google Scholar]
- [54].Davydov DR, Sineva EV, Sistla S, Davydova NY, Frank DJ, Sligar SG, et al. Electron transfer in the complex of membrane-bound human cytochrome P450 3A4 with the flavin domain of P450BM-3: the effect of oligomerization of the heme protein and intermittent modulation of the spin equilibrium. Biochim Biophys Acta 2010;1797:378–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Marcus CB, Murray M, Hetnarski K, Wilkinson CF. Methylenedioxyphenyl complexes with microsomal cytochrome-P-450 - invivo complex-formation in rat-liver and in midgut tissues of the southern armyworm (Spodoptera eridania). Pesticide Biochemistry and Physiology 1987;28:140–7. [Google Scholar]
- [56].Murray M, Reidy GF. Invitro formation of an inhibitory complex between an isosafrole metabolite and rat hepatic cytochrome-P-450 PB-b. Drug Metab Disp 1989;17:449–54. [PubMed] [Google Scholar]
- [57].Dawson JH, Andersson LA, Sono M. The diverse spectroscopic properties of ferrous cytochrome-P-450-cam ligand complexes. J Biol Chem 1983;258:3637–45. [PubMed] [Google Scholar]
- [58].Loew G, Goldblum A. Electronic-spectrum of model cytochrome P450 complex with postulated carbene metabolite of halothane. J Am Chem Soc 1980;102:3657–9. [Google Scholar]
- [59].Sono M, Andersson LA, Dawson JH. Sulfur donor ligand-binding to ferric cytochrome P-450-cam and myoglobin - ultraviolet-visible absorption, magnetic circular-dichroism, and electron-paramagnetic resonance spectroscopic investigation of the complexes. J Biol Chem 1982;257:8308–20. [PubMed] [Google Scholar]
- [60].Farre M, de la Torre R, Mathuna BO, Roset PN, Peiro AM, Torrens M, et al. Repeated doses administration of MDMA in humans: pharmacological effects and pharmacokinetics. Psychopharmacology (Berl) 2004;173:364–75. [DOI] [PubMed] [Google Scholar]
- [61].Kalant H The pharmacology and toxicology of “ecstasy” (MDMA) and related drugs. Canadian Med Assoc J 2001;165:917–28. [PMC free article] [PubMed] [Google Scholar]
- [62].Davydov DR. Microsomal monooxygenase as a multienzyme system: the role of P450-P450 interactions. Expert Opin Drug Metab Toxicol 2011;7:543–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Reed JR, Backes WL. Physical Studies of P450-P450 Interactions: Predicting Quaternary Structures of P450 Complexes in Membranes from Their X-ray Crystal Structures. Frontiers Pharmacol 2017;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Carvalho M, Pontes H, Remiao F, Bastos ML, Carvalho F. Mechanisms Underlying the Hepatotoxic Effects of Ecstasy. Curr Pharm Biotechnol 2010;11:476–95. [DOI] [PubMed] [Google Scholar]
- [65].Pontes H, Duarte JA, de Pinho PG, Soares ME, Fernandes E, Dinis-Oliveira RJ, et al. Chronic exposure to ethanol exacerbates MDMA-induced hyperthermia and exposes liver to severe MDMA-induced toxicity in CD1 mice. Toxicology 2008;252:64–71. [DOI] [PubMed] [Google Scholar]