

Abstract
Compensating for the loss of extracellular cartilage matrix, as well as counteracting the alterations of the chondrocyte phenotype in osteoarthritis are of key importance to develop effective therapeutic strategies against this disorder. In the present study, we analysed the benefits of applying a potent gene combination to remodel human osteoarthritic (OA) cartilage. We employed the promising recombinant adeno‐associated virus (rAAV) vector to deliver the mitogenic fibroblast growth factor 2 (FGF‐2) factor, alone or simultaneously with the transcription factor Sox9 as a key activator of matrix synthesis, to human normal and OA articular chondrocytes. We evaluated the effects of single (FGF‐2) or combined (FGF‐2/SOX9) transgene expression upon the regenerative activities of chondrocytes in three‐dimensional cultures in vitro and in cartilage explants in situ. Single overexpression of FGF‐2 enhanced the survival and proliferation of both normal and OA chondrocytes, without stimulating the matrix synthetic processes in the increased pools of cells. The mitogenic properties of FGF‐2 were maintained when SOX9 was co‐overexpressed and concomitant with an increase in the production of proteoglycans and type‐II collagen, suggesting that the transcription factor was capable of counterbalancing the effects of FGF‐2 on matrix accumulation. Also important, expression of type‐X collagen, a marker of hypertrophy strongly decreased following treatment by the candidate vectors. Most remarkably, the levels of activities achieved in co‐treated human OA cartilage were similar to or higher than those observed in normal cartilage. The present findings show that combined expression of candidate factors in OA cartilage can re‐establish key features of normal cartilage and prevent the pathological shift of metabolic homeostasis. These data provide further motivation to develop coupled gene transfer approaches via rAAV for the treatment of human OA.
Keywords: articular cartilage, human osteoarthritis, combined gene transfer, FGF‐2, Sox9, rAAV
Introduction
Osteoarthritis (OA) is a progressive degenerative disease of the joint with a gradual degradation of the articular cartilage due to a loss of major matrix components such as proteoglycans and type‐II collagen [1]. The elaboration of new therapeutic regimens against OA is critical, as the current surgical interventions and medical treatments have been unable to restore an original articular cartilage surface to date. The successful approach to manage the development of OA will necessitate counteracting its slow progression and the irreversible, complex processes of degradation by stimulating the regenerative properties of the chondrocytes forming the cartilage.
Application of therapeutic genes to OA cartilage is an attractive strategy to re‐establish the structural integrity of the tissue. Significant advances have been made by applying gene agents that impede the processes of matrix degradation in OA cartilage, in particular those induced by pro‐inflammatory cytokines (interleukin‐1 IL‐1; tumour necrosis factor‐α, TNF‐α) [2]. Among the most effective candidates, the transfer of an IL‐1 receptor antagonist (IL‐1Ra) sequence was reported to protect against cartilage breakdown in experimental models [3, 4] and in vivo[5, 6]. Therapeutic effects have been also ascribed to factors that enhance cartilage anabolism like insulin‐like growth factor I (IGF‐I) [7], fibroblast growth factor 2 (FGF‐2) [8, 9], bone morphogenetic protein 7 (BMP‐7) [10], transforming growth factor β (TGF‐β) [11, 12] and glucuronosyltransferase‐I [13]. Yet, application of these external stimuli has not been sufficient to date to regenerate a normal articular cartilage surface. As the chondrocytes recently became the focus of OA, due to pathological changes in their gene expression patterns, a decreased ability to produce extracellular matrix components and an increased release of matrix‐degrading enzymes [14, 15, 16], candidate agents capable of modulating the cellular phenotype have been tested for their ability to regenerate OA cartilage. Notably, overexpression of SOX9, a transcription factor critical for cartilage formation [17, 18] that is down‐regulated in OA [16, 19] proved beneficial to shift the metabolic balance in OA towards the synthesis of matrix components, thus participating in the reorganization of the cartilage surface [20, 21, 22]. Due to the multiple pathological processes involved in OA, reproduction of an original cartilage architecture might require the action of more than one therapeutic factor [23]. As a matter of fact, Kypriotou et al.[24] and Ikeda et al.[18] suggested that administration of several candidate genes might be more efficient to promote the restoration of the cartilage structure in OA compared with single treatments.
In the present study, we developed a multiple, independent gene transfer approach to investigate the benefits of co‐applying two key modulators of chondrocyte differentiation and cartilage formation upon the reconstruction of human OA cartilage. We delivered the gene sequences for FGF‐2 as a stimulus of cell proliferation [8] and for SOX9, a transcription factor that activates the expression of major cartilage matrix components [17, 18, 20, 21, 22] and might prove beneficial to counterbalance the reported negative effects of FGF‐2 upon the production and accumulation of matrix proteoglycans, in particular in OA cartilage [25, 26]. Gene delivery to human articular chondrocytes was performed in three‐dimensional cultures in vitro and in cartilage explants in situ using the replication‐defective, non‐pathogenic recombinant adeno‐associated virus (rAAV) vector. The rAAV are particularly well suited for these goals, as they have been shown to mediate efficient and sustained transgene expression in the cartilage [27, 28, 29]. This feature could prove important to treat a slow, progressive disease such as OA where the therapeutic effects might be required over prolonged periods of time. Equally important for the study, simultaneous delivery of independent transgenes by separate rAAV vectors has been successfully demonstrated in various systems [30, 31]. These properties, together with a capacity to transduce both dividing and non‐dividing cells like chondrocytes and a reduced immunogenicity, make currently rAAV a favoured gene vehicle to directly transfer candidate factors to experimental models [32, 33, 34].
Materials and methods
All reagents were from Sigma (Munich, Germany) except for the dimethylmethylene blue (DMMB) dye (Serva, Heidelberg, Germany). The anti‐β‐gal (GAL‐13), anti‐FLAG (BioM2 and M1) and anti‐type‐X collagen (COL‐10) antibodies were from Sigma, the anti‐FGF‐2 (C‐18 and N‐19) and anti‐Sox9 (C‐20 and H‐90) from Santa Cruz Biotechnology (Heidelberg, Germany), and the anti‐type‐II collagen (AF‐5710) from Acris (Hiddenhausen, Germany). FGF‐2 secretion was monitored with the human basic fibroblast growth factor (hFGF) basic Quantikine ELISA (DFB50; R&D Systems; Wiesbaden, Germany). Type‐II collagen production was measured with the native type‐II collagen Arthrogen‐CIA Capture ELISA kit (Chondrex, Redmond, WA, USA). Apoptosis was determined using the ApopTag® Plus Peroxidase In Situ Apoptosis Detection Kit (Chemicon‐Millipore GmbH, Schwalbach/Ts., Germany).
Cartilage and cells
Human normal articular cartilage was obtained from unaffected knee joints removed during tumour surgery (10 patients, 67–72 years). OA was excluded on safranin O‐stained sections using the Mankin score [35] (score 1–2). OA cartilage was obtained from joints undergoing total knee arthroplasty (12 patients, 65–78 years) (score 7–9). Explant cultures (6.2‐mm diameter) and chondrocytes were prepared as previously described [28].
Plasmids and rAAV vectors
pAd8 contains AAV‐2 replication and encapsidation functions [36]. rAAV‐lacZ is an AAV‐2‐based plasmid carrying the lacZ gene encoding β‐galactosidase (β‐gal) under the control of the cytomegalovirus immediate‐early (CMV‐IE) promoter [22, 28, 29, 37]. rAAV‐hFGF‐2 carries a 480‐bp hFGF‐2 cDNA fragment [38] that was cloned in rAAV‐lacZ in place of lacZ[29]. rAAV‐FLAG‐hsox9 carries a 1.7‐kb FLAG‐tagged human SOX9 (hsox9) cDNA fragment [39] that was cloned in rAAV‐lacZ in place of lacZ[22]. rAAV vectors were packaged as single‐stranded elements using Adenovirus 5 to provide helper functions in combination with pAd8 [22, 28, 29, 37]. Vector preparations were purified by dialysis and titred by real‐time PCR (about 1010 functional U/ml) [22, 28, 29, 37].
Gene transfer
The vectors were applied to the samples based on concentrations previously tested [22, 28, 29]. Chondrocytes (106) were transduced (300 μl) or co‐transduced (150 μl each vector) with rAAV for 2 days and encapsulated in alginate spheres that were maintained in culture for 26 days to monitor cell number and viability [22, 29]. Explant cultures were transduced (20 or 50 μl) or co‐transduced (20 μl each vector) by direct application of rAAV to the surface of the samples downwards during a 1–2‐min. contact and maintained in culture for 10 days [22, 28].
Analysis of transgene expression
β‐gal activity was detected by X‐Gal staining to determine transduction efficiency [22, 28]. FGF‐2 secretion was monitored by ELISA [29]. Transgene expression was also monitored by indirect immunostaining [22, 29]. Explant cultures were also processed for Western blotting analysis using the same amount of proteins (10 μg) [22]. Briefly, cartilage was lyophilized and then extracted with ice‐cold guanidine buffer (1 M GuHCl, 10 mM CaCl2, 50 mM Tris, protease inhibitors, pH 7.5). Protein concentration was determined with use of the BCA™ Protein Assay Kit (Perbio Science Deutschland GmbH, Bonn, Germany). Revelation was performed with specific antibodies, horseradish peroxidase‐labelled secondary antibodies (Vector Laboratories, Grünberg, Germany), and the ECL Advance Western blotting detection kit (Amersham Biosciences, Freiburg, Germany).
Histological and immunohistochemical analyses
Spheres and explant cultures were processed and paraffin‐embedded sections (5 μm) were stained with safranin O to detect proteoglycans and haematoxylin and eosin to detect cells [8, 22, 28, 29]. Expression of type‐II and type‐X collagen was detected with specific antibodies, biotinylated secondary antibodies (Vector Laboratories), and the ABC method (Vector Laboratories) using diaminobenzidine as the chromogen. Samples were examined under light microscopy (Olympus BX 45; Hamburg, Germany).
Morphometric analyses
Safranin O and type‐II staining intensities and the cell densities were measured at three standardized sites using SIS AnalySIS (Olympus), Adobe Photoshop (Adobe Systems, Unterschleissheim, Germany), and Scion Image (Scion Corporation, Frederick, MD, USA). Safranin O and type‐II collagen staining intensities given as percents were calculated as being the ratio of positively stained tissue surface to the total surface of the site evaluated [22].
Analysis of type‐II collagen and proteoglycan production and DNA contents
Solubilized spheres and explant cultures were digested as previously described [29, 40]. Type‐II collagen production was determined by ELISA [22]. The proteoglycan production was detected by binding to the DMMB dye [22, 29]. The DNA contents were monitored using Hoechst 33258 [22, 29]. The measurements were performed with a GENios spectrophotometer/fluorometer (Tecan, Crailsheim, Germany).
Apoptosis assay
To detect apoptosis in explant cultures, nuclear DNA fragmentation consistent with apoptosis was determined by the terminal deoxynucleotidyl transferase‐mediated dUTP nick end labeling (TUNEL) method. TUNEL staining was performed with the ApopTag® Plus Peroxidase In Situ Apoptosis Detection Kit (Chemicon‐Millipore GmbH) according to the manufacturer's protocol.
Statistical analysis
Each condition was performed in triplicate in three independent experiments with spheres and in triplicate in two independent experiments with explant cultures. Data were obtained by two individuals that were blinded with respect to the treatment groups. Values are expressed as mean ± standard deviation (S.D.). The t‐test and Mann‐Whitney rank sum test were employed where appropriate. P‐values of less than 0.05 were considered statistically significant.
Results
rAAV‐mediated overexpression of FGF‐2 in human articular chondrocytes
The functionality of rAAV‐hFGF‐2, a vector that has been shown to enhance the healing capacity of articular cartilage defects in a rabbit model in vivo[29] was first evaluated in human normal and OA chondrocytes encapsulated in alginate spheres and within articular cartilage explants. In vitro, β‐gal activity noted only in the rAAV‐lacZ‐transduced (control) spheres, was present on the day of encapsulation and for at least 26 days, with constant transduction efficiencies of 76–80% (not shown) [22]. In turn, sustained (26 days) FGF‐2 expression was restricted to the rAAV‐hFGF‐2‐transduced (treated) spheres (Fig. 1A and B).
Figure 1.
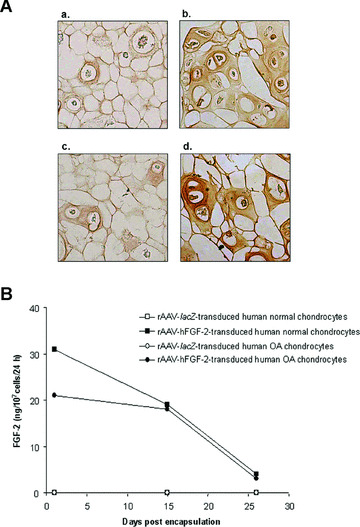
FGF‐2 expression in vitro. (A) Immunocytochemical detection of FGF‐2 in spheres carrying human normal (a and b) and OA chondrocytes (c and d) after 26 days. Cells were transduced with rAAV‐lacZ (a and c) or rAAV‐hFGF‐2 (b and d), encapsulated, and processed after 26 days (n= 9 per condition) to detect FGF‐2 (1:50). Magnification ×100. (B) Time course analysis of FGF‐2 production in culture supernatants from spheres. Conditioned medium was collected at the denoted time points after encapsulation (n= 9 per time point and condition) and FGF‐2 secretion was measured by ELISA (±S.D.).
In situ, β‐gal activity seen only in the rAAV‐lacZ‐transduced (control) cartilage (superficial and middle zones), was present already 5 days after vector administration and for at least 10 days, showing dose‐dependent transduction efficiencies (normal cartilage: from 48 ± 2% to 79 ± 1% using 20 to 50 μl vector, i.e. a 1.6‐fold increase; OA cartilage: from 46 ± 2% to 71 ± 2%, i.e. a 1.5‐fold increase) (always P < 0.001) (not shown). Elevated, dose‐dependent FGF‐2 production levels were achieved in the rAAV‐hFGF‐2‐transduced (treated) cartilage (normal cartilage: from 10.6 ± 0.2 to 19.7 ± 0.6 pg/mg dry weight, i.e. a 1.9‐fold increase; OA cartilage: from 9.6 ± 0.2 to 15.9 ± 0.3 pg/mg dry weight, i.e. a 1.7‐fold increase) (always P < 0.001), with specific immunoreactivity seen in the superficial and middle zones of the cartilage (Fig. 2A). Endogenous FGF‐2 expression was also noted in the control cartilage (Fig. 2A) in contrast to the findings in vitro, possibly due to a difference of sensitivity between the methods of detection, but the control secretion levels were always lower than those achieved in the treated cartilage (about 1.4 ± 0.3 pg/mg dry weight) (always P < 0.001). Accordingly, Western blotting analyses revealed an FGF‐2 immunoreactive band of approximately 18 kD that was two‐ to threefold more intense in the treated cartilage compared with control cartilage (Fig. 2B) [29].
Figure 2.
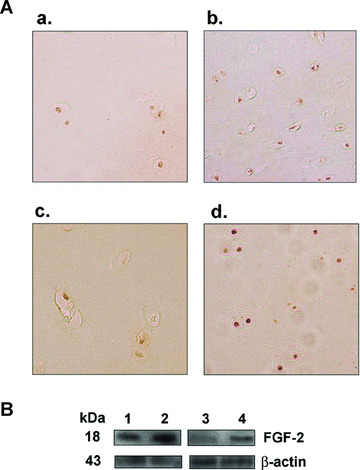
FGF‐2 expression in situ. (A) Immunohistochemical detection of FGF‐2 in explant cultures of human normal (a and b) and OA cartilage (c and d). Explants were transduced with rAAV‐lacZ (a and c) or rAAV‐hFGF‐2 (b and d) (50 μl vector) and processed after 10 days (n= 6 per condition) to detect FGF‐2 (1:50). Magnification ×100 (representative view of the middle zone). (B) Western blotting analysis of FGF‐2 expression: lane 1, rAAV‐lacZ‐transduced normal cartilage; lane 2, rAAV‐hFGF‐2‐transduced normal cartilage; lane 3, rAAV‐lacZ‐transduced OA cartilage; lane 4, rAAV‐hFGF‐2‐transduced OA cartilage.
Effects of rAAV‐hFGF‐2 administration upon the cellular activities of human articular chondrocytes
We next evaluated the ability of rAAV‐mediated FGF‐2 overexpression to stimulate the proliferative and synthetic activities of chondrocytes in the systems described above. In vitro, the cell numbers were always higher in the treated spheres (up to 6.2‐fold), with a significant increase over time (up to 1.8‐fold) compared with control spheres where these numbers decreased (up to 2.2‐fold) (Table 1) (always P < 0.001). Consistent with this, cell viability in the control spheres was only 35% at the end of the evaluation period, much lower than in the treated spheres (92%) and compared with an initial viability of 88% when they were established. The differences in outcome were therefore a combination of increased cell viability and cell division produced by FGF‐2. Similar results were noted when analysing the cell densities on haematoxylin and eosin‐stained sections from spheres (not shown) and the DNA contents (Table 1) (always P < 0.001). In situ, the cell densities in the superficial and middle zones of the cartilage and the DNA contents were always higher in the treated cartilage compared with control cartilage (up to 2.5‐fold), revealing a dose‐dependent effect (up to 1.3‐fold) (Fig. 3 and Table 2) (P≤ 0.029). Importantly, these parameters were higher in the treated OA cartilage compared with normal control cartilage (up to 1.7‐fold), even at the lower candidate vector dose applied (always P < 0.001). A TUNEL analysis revealed that treatment with rAAV‐hFGF‐2 promoted a strong reduction in the numbers of apoptotic cells in OA cartilage that were demonstrated in the damaged control (rAAV‐lacZ‐transduced) explants. Remarkably, the amounts of apoptotic cells observed in the presence of the FGF‐2 transgene were similar to those detected in the normal (control or treated) cartilage (almost no stained cells) (Fig. 3).
Table 1.
Biochemical analyses in transduced chondrocytes. Values are given as mean (S.D.). DNA contents and proteoglycan production are in μg/104 cells and type‐II collagen production in ng/104 cells
| Days | Normal | OA | |||
|---|---|---|---|---|---|
| rAAV‐lacZ | rAAV‐hFGF‐2 | rAAV‐lacZ | rAAV‐hFGF‐2 | ||
| Cells (104/sphere) | 1 | 0.77 (0.02) | 1.23 (0.02)* | 0.66 (0.02) | 1.11 (0.01)* |
| 26 | 0.35 (0.03) | 2.16 (0.01)* | 0.31 (0.02) | 1.83 (0.02)* | |
| DNA contents | 1 | 0.41 (0.02) | 0.82 (0.01)* | 0.38 (0.01) | 0.75 (0.01)* |
| 26 | 0.25 (0.01) | 1.57 (0.02)* | 0.23 (0.01) | 1.46 (0.02)* | |
| Proteoglycan production | 1 | 4.41 (0.01) | 4.44 (0.03) | 4.09 (0.01) | 4.10 (0.04) |
| 26 | 4.19 (0.02) | 4.21 (0.03) | 4.00 (0.02) | 4.01 (0.03) | |
| Type‐II collagen production | 1 | 0.14 (0.01) | 0.15 (0.01) | 0.08 (0.01) | 0.09 (0.01) |
| 26 | 0.10 (0.01) | 0.10 (0.01) | 0.06 (0.01) | 0.06 (0.01) | |
*Significant compared with rAAV‐lacZ.
Figure 3.
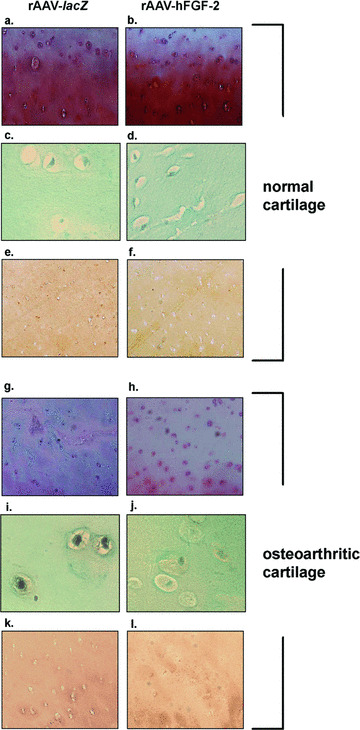
Safranin O staining, detection of apoptosis, and type‐II collagen immunostaining in transduced cartilage. Normal (a–f) and OA cartilage (g–l) were transduced with rAAV‐lacZ (a, c, e, g, i and k) or rAAV‐hFGF‐2 (b, d, f, h, j and l) (50 μl vector) and processed after 10 days (n= 6 per condition) for staining by safranin O and haematoxylin and eosin (a, b, g and h, magnification × 40), for TUNEL assay (c, d, i and j, magnification × 200), and to detect type‐II collagen (1:50) (e, f, k and l, magnification × 40). Representative view of the middle zone.
Table 2.
Morphometric and biochemical analyses in transduced cartilage. Values are given at day 10 as mean (S.D.). DNA contents and proteoglycan production are in μg/mg dry weight, type‐II collagen production in ng/mg dry weight, and Safranin O and type‐II collagen staining in percentage. *Significant compared with rAAV‐lacZ and +with lower vector dose
| Normal | OA | |||||||
|---|---|---|---|---|---|---|---|---|
| rAAV‐lacZ | rAAV‐hFGF‐2 | rAAV‐lacZ | rAAV‐hFGF‐2 | |||||
| 20 μ l | 50 μ l | 20 μ l | 50 μ l | 20 μ l | 50 μ l | 20 μ l | 50 μ l | |
| Cells (/mm2) | 255 (7) | 252 (16) | 475 (20)* | 633 (12)*,+ | 238 (11) | 245 (11) | 338 (9)* | 419 (14)*,+ |
| DNA contents | 1.28 | 1.32 | 1.47 | 1.73 | 1.24 | 1.25 | 1.38 | 1.51 |
| (0.01) | (0.01) | (0.03)* | (0.03)*,+ | (0.01) | (0.02) | (0.03)* | (0.04)*,+ | |
| Proteoglycan production | 2.68 | 2.68 | 2.67 | 2.68 | 2.46 | 2.47 | 2.47 | 2.45 |
| (0.01) | (0.02) | (0.03) | (0.04) | (0.01) | (0.03) | (0.04) | (0.03) | |
| Safranin O staining | 47 | 47 | 47 | 48 | 31 | 31 | 30 | 32 |
| (2) | (1) | (3) | (3) | (1) | (2) | (2) | (2) | |
| Type‐II collagen production | 0.022 | 0.021 | 0.021 | 0.023 | 0.017 | 0.018 | 0.017 | 0.019 |
| (0.001) | (0.003) | (0.005) | (0.002) | (0.001) | (0.001) | (0.001) | (0.001) | |
| Type‐II collagen staining | 43 | 41 | 41 | 40 | 38 | 36 | 37 | 37 |
| (2) | (1) | (2) | (3) | (1) | (2) | (1) | (2) | |
In contrast, there was no difference in the proteoglycan and type‐II collagen production between the treated (rAAV‐hFGF‐2) and control (rAAV‐lacZ) spheres in vitro (Table 1) (P≥ 0.125), with a decrease over time in both cases (up to 1.5‐fold) (P≤ 0.029). Similar results were noted when analysing the immunoreactivity to type‐II collagen on sections from spheres (not shown). There was also no difference in the proteoglycan production, safranin O staining intensity, type‐II collagen production and immunoreactivity between the treated and control cartilage in situ (Fig. 3 and Table 2) (P≥ 0.107).
Efficient FGF‐2/Sox9 co‐overexpression in human articular chondrocytes
rAAV‐FLAG‐hsox9, a vector that promotes the synthesis of extracellular matrix components in human OA cartilage [22] was next co‐delivered with rAAV‐hFGF‐2 (rAAV‐FLAG‐hsox9/rAAV‐hFGF‐2) in encapsulated human normal and OA chondrocytes and in human normal and OA cartilage explants compared with control transductions (rAAV‐lacZ and rAAV‐lacZ/rAAV‐hFGF‐2). Single SOX9 application was not performed in these experiments, as it has been already characterized in these systems [22]. In vitro, β‐gal activity was seen only when rAAV‐lacZ was administered, starting on the day of encapsulation and for at least 26 days with constant transduction efficiencies of up to 82% (not shown). Sustained FGF‐2 expression was noted only when rAAV‐hFGF‐2 was applied, i.e. in the rAAV‐lacZ/rAAV‐hFGF‐2‐ and rAAV‐FLAG‐hsox9/rAAV‐hFGF‐2‐transduced cells (normal chondrocytes: from 4.69 ± 0.04 to 1.20 ± 0.02 and from 5.61 ± 0.08 to 1.47 ± 0.04 ng/107 cells/24 hrs over time, respectively; OA chondrocytes: from 3.54 ± 0.06 to 0.91 ± 0.02 and from 3.65 ± 0.04 to 1.10 ± 0.03 ng/107 cells/24 hrs) compared with rAAV‐lacZ transduction (below the limit of detection) (see also Fig. 4A). The FLAG tag was detected in spheres transduced with rAAV‐FLAG‐hsox9 (rAAV‐FLAG‐hsox9/rAAV‐hFGF‐2) (Fig. 4A). Sox9 immunoreactivity was noted in all types of spheres (Fig. 4A), in good agreement with the endogenous expression of SOX9 in chondrocytes [22], yet specific Sox9 immunostaining was more intense when rAAV‐FLAG‐hsox9 was applied. Some reactivity for Sox9 was also seen extracellularly as already noted by us when applying the SOX9 vector alone [22] and by others in experimental conditions in situ[19, 41], possibly due to the quality of the antibodies employed. Similar results were noted in OA chondrocytes (not shown).
Figure 4.
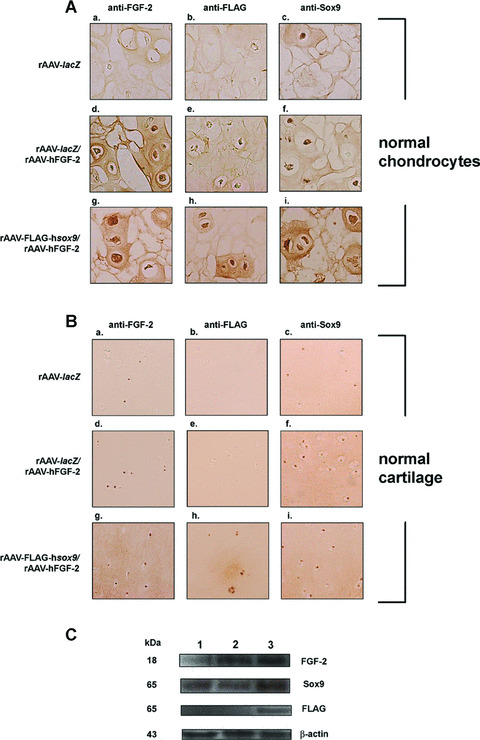
rAAV‐mediated transgene co‐overexpression in vitro and in situ. (A) Immunocytochemical analyses in spheres carrying human normal chondrocytes after 26 days. Cells were transduced with rAAV‐lacZ (a–c), rAAV‐lacZ/rAAV‐hFGF‐2 (d–f), or rAAV‐FLAG‐hsox9/rAAV‐hFGF‐2 (g–i), encapsulated, and processed after 26 days (n= 9 per condition) to detect FGF‐2 (1:50) (a, d, and g), the FLAG tag (1:80) (b, e and h), and Sox9 (1:100) (c, f and i). Magnification ×100. (B) Immunohistochemical analyses in human normal cartilage. Explants were transduced with rAAV‐lacZ (a–c), rAAV‐lacZ/rAAV‐hFGF‐2 (d–f), or rAAV‐FLAG‐hsox9/rAAV‐hFGF‐2 (g–i) and processed after 10 days (n= 6 per condition) to detect FGF‐2 (1:50) (a, d and g), the FLAG tag (1:80) (b, e and h), and Sox9 (1:100) (c, f and i). Magnification ×100 (representative view of the middle zone). (C) Western blotting analysis of transgene expression in cartilage (representative results in normal cartilage): lane 1, rAAV‐lacZ‐transduced cartilage; lane 2, rAAV‐lacZ/rAAV‐hFGF‐2‐transduced cartilage; lane 3, rAAV‐FLAG‐hsox9/rAAV‐hFGF‐2‐transduced cartilage.
In situ, β‐gal activity was detected only when rAAV‐lacZ was administered, both in the superficial and middle zones of the cartilage (not shown). High levels of FGF‐2 production were noted in the presence of rAAV‐hFGF‐2, i.e. in the rAAV‐lacZ/rAAV‐hFGF‐2‐ and rAAV‐FLAG‐hsox9/rAAV‐hFGF‐2‐transduced explants (normal cartilage: between 10.50 ± 0.71 and 11.50 ± 0.70 pg/mg dry weight; OA cartilage: between 8.50 ± 0.70 and 9.00 ± 1.41 pg/mg dry weight), with specific immunoreactivity seen both in the superficial and middle zones of the cartilage (Fig. 4B). Endogenous FGF‐2 expression was also noted in the rAAV‐lacZ‐transduced cartilage (Fig. 4B), but the control secretion levels were always lower than those achieved when rAAV‐hFGF‐2 was applied (about 1.2 ± 0.1 pg/mg dry weight) (P < 0.001). Accordingly, Western blotting analyses revealed an 18 kD FGF‐2 immunoreactive band that was about threefold more intense in the cartilage treated by rAAV‐hFGF‐2 compared with rAAV‐lacZ transduction (Fig. 4C) [29]. The FLAG tag was detected when rAAV‐FLAG‐hsox9 was provided (rAAV‐FLAG‐hsox9/rAAV‐hFGF‐2) (Fig. 4B and C). Sox9 immunoreactivity was present in all types of explants (Fig. 4B) both in the superficial and middle zones, in good agreement with previous findings showing endogenous expression of SOX9 in the cartilage [22]. Consistent with this, Western blotting analyses revealed a Sox9 immunoreactive band (approximately 65 kD) [22] that was about twofold higher in the cartilage when rAAV‐FLAG‐hsox9 was applied compared with control transductions (Fig. 4C) [22]. Similar results were obtained in OA cartilage (not shown).
Biological effects of the FGF‐2/SOX9 combination in human articular chondrocytes
Stimulation of the proliferative and synthetic properties in chondrocytes by FGF‐2/SOX9 co‐overexpression was next tested as described in the single rAAV transduction experiments. In vitro, the cell numbers were always higher in spheres when rAAV‐hFGF‐2 was applied (rAAV‐lacZ/rAAV‐hFGF‐2 and rAAV‐FLAG‐hsox9/rAAV‐hFGF‐2) (up to 5.5‐fold), with a significant increase over time (up to 1.9‐fold) compared with rAAV‐lacZ‐transduced spheres where the cell numbers decreased (up to 2.1‐fold) (Table 3) (P≤ 0.029). Consistent with this, viability in rAAV‐lacZ‐transduced spheres was only 30% at the end of the evaluation period compared with 93% in the other spheres and with an initial viability of 87% when they were established. The differences in outcome were here again a combination of increased cell viability and cell division produced by FGF‐2. Similar results were noted for the cell densities on haematoxylin and eosin‐stained sections from spheres (not shown) and the DNA contents (Table 3) (always P < 0.001). Yet, there was no additive effect of Sox9 upon the stimulation of cell proliferation (cell numbers and DNA contents) promoted by FGF‐2 (rAAV‐FLAG‐hsox9/rAAV‐hFGF‐2 compared with rAAV‐lacZ/rAAV‐hFGF‐2 treatment) (P≥ 0.081). In situ, the cell densities in the superficial and middle zones of the cartilage and the DNA contents were higher when rAAV‐hFGF‐2 was applied (up to 1.8‐fold) (Fig. 5 and Table 4) (always P < 0.001). Importantly, these parameters were higher in OA cartilage treated by rAAV‐hFGF‐2 compared with normal rAAV‐lacZ‐transduced cartilage (up to 1.4‐fold) (always P < 0.001). However, there was no synergistic effect between Sox9 and FGF‐2 upon the stimulation of cell proliferation (cell numbers and DNA contents) (P≥ 0.176).
Table 3.
Biochemical analyses in co‐transduced chondrocytes. Values are given as mean (S.D.). DNA contents and proteoglycan production are in μg/104 cells and type‐II collagen production in ng/104 cells. *Significant compared with rAAV‐lacZ and +with rAAV‐lacZ/rAAV‐hFGF‐2
| Days | Normal | OA | |||||
|---|---|---|---|---|---|---|---|
| rAAV‐lacZ | rAAV‐lacZ+ rAAV‐hFGF‐2 | rAAV‐FLAG‐hsox9+ rAAV‐hFGF‐2 | rAAV‐lacZ | rAAV‐lacZ+ rAAV‐hFGF‐2 | rAAV‐FLAG‐hsox9+ rAAV‐hFGF‐2 | ||
| Cells (104/sphere) | 1 | 0.73 | 0.99 | 1.01 | 0.64 | 0.93 | 0.96 |
| (0.01) | (0.03)* | (0.03)* | (0.02) | (0.09)* | (0.10)* | ||
| 26 | 0.35 | 1.88 | 1.89 | 0.33 | 1.79 | 1.82 | |
| (0.02) | (0.03)* | (0.03)* | (0.02) | (0.04)* | (0.03)* | ||
| DNA contents | 1 | 0.39 | 0.56 | 0.57 | 0.36 | 0.52 | 0.53 |
| (0.01) | (0.01)* | (0.02)* | (0.01) | (0.02)* | (0.03)* | ||
| 26 | 0.22 | 0.79 | 0.81 | 0.21 | 0.72 | 0.75 | |
| (0.02) | (0.01)* | (0.01)* | (0.02) | (0.02)* | (0.02)* | ||
| Proteoglycan production | 1 | 3.63 | 3.62 | 4.00 | 3.52 | 3.52 | 3.65 |
| (0.01) | (0.01) | (0.02)*,+ | (0.01) | (0.02) | (0.02)*,+ | ||
| 26 | 3.42 | 3.42 | 4.84 | 3.30 | 3.32 | 4.40 | |
| (0.02) | (0.02) | (0.04)*,+ | (0.01) | (0.01) | (0.03)*,+ | ||
| Type‐II collagen production | 1 | 0.12 | 0.12 | 0.22 | 0.08 | 0.08 | 0.19 |
| (0.01) | (0.01) | (0.01)*,+ | (0.01) | (0.01) | (0.01)*,+ | ||
| 26 | 0.08 | 0.08 | 0.43 | 0.05 | 0.05 | 0.30 | |
| (0.01) | (0.01) | (0.02)*,+ | (0.01) | (0.01) | (0.02)*,+ | ||
Figure 5.
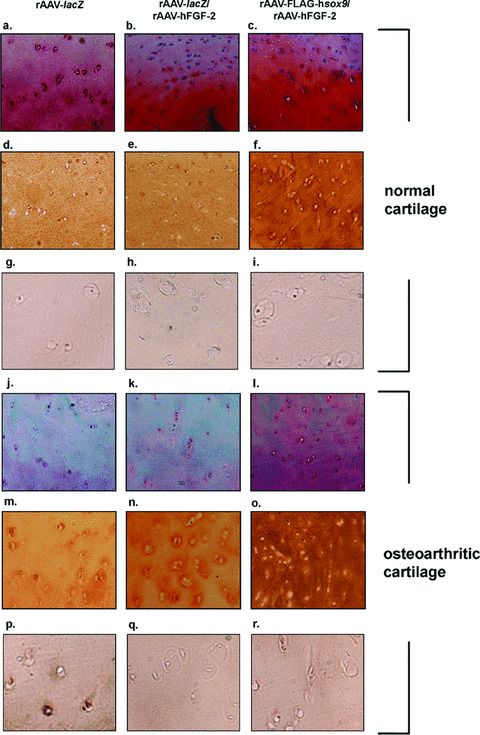
Safranin O staining and type‐II and type‐X collagen immunostaining in co‐transduced cartilage. Human normal (a–i) and OA cartilage (j–r) were transduced with rAAV‐lacZ (a, d, g, j, m and p), rAAV‐lacZ/rAAV‐hFGF‐2 (b, e, h, k, n, and q), or rAAV‐FLAG‐hsox9/rAAV‐hFGF‐2 (c, f, i, l, o and r) and processed after 10 days (n= 6 per condition) for staining by safranin O and haematoxylin and eosin (a–c and j–l, magnification ×40) and to detect type‐II collagen (1:50) (d–f and m–o, magnification ×40) and type‐X collagen (1:200) (g–i and p–r, magnification ×200). Representative view of the middle zone.
Table 4.
Morphometric and biochemical analyses in co‐transduced cartilage. Values are given at day 10 as mean (S.D.). DNA contents and proteoglycan production are in μg/mg dry weight, type‐II collagen production in ng/mg dry weight, and safranin O and type‐II collagen staining in percentage. *Significant compared with rAAV‐lacZ and +with rAAV‐lacZ/rAAV‐hFGF‐2.
| Normal | OA | |||||
|---|---|---|---|---|---|---|
| rAAV‐lacZ | rAAV‐lacZ+ rAAV‐ hFGF‐2 | rAAV‐FLAG‐hsox9+ rAAV‐ hFGF‐2 | rAAV‐lacZ | rAAV‐lacZ+ rAAV‐ hFGF‐2 | rAAV‐FLAG‐Hsox9+ rAAV‐ hFGF‐2 | |
| Cells (/mm2) | 273 | 480 | 481 | 255 | 378 | 378 |
| (2) | (4)* | (6)* | (3) | (4)* | (5)* | |
| DNA contents | 1.24 | 1.41 | 1.40 | 1.22 | 1.34 | 1.33 |
| (0.01) | (0.01)* | (0.01)* | (0.01) | (0.01)* | (0.02)* | |
| Proteoglycans production | 2.60 | 2.60 | 2.76 | 2.44 | 2.44 | 2.58 |
| (0.02) | (0.01) | (0.02)*,+ | (0.02) | (0.03) | (0.02)*,+ | |
| Safranin O stainin | 44 | 43 | 56 | 28 | 29 | 36 |
| (1) | (2) | (1)*,+ | (1) | (1) | (1)*,+ | |
| Type‐II collagen production | 0.021 | 0.022 | 0.034 | 0.019 | 0.018 | 0.029 |
| (0.002) | (0.002) | (0.003)*,+ | (0.002) | (0.002) | (0.002)*,+ | |
| Type‐II collagen staining | 42 | 41 | 56 | 39 | 38 | 55 |
| (1) | (2) | (2)*,+ | (1) | (2) | (2)*,+ | |
There was no difference in the proteoglycan and type‐II collagen production (Table 3) and type‐II immunoreactivity (not shown) between the rAAV‐lacZ‐ and rAAV‐lacZ/rAAV‐hFGF‐2‐transduced spheres in vitro (P≥ 0.171), with a decrease over time in both cases (up to 1.6‐fold) (P≤ 0.009). In contrast, these parameters were higher in the presence of rAAV‐FLAG‐hsox9 compared with control transductions (up to 2.4‐fold) (P≤ 0.029), with an increase over time (up to 1.9‐fold) (P≤ 0.029). There was also no difference in the proteoglycan production, safranin O staining intensity, type‐II collagen production and immunoreactivity between the rAAV‐lacZ‐ and rAAV‐lacZ/rAAV‐hFGF‐2‐transduced cartilage in situ (Fig. 5 and Table 4) (P≥ 0.522). In contrast, these parameters were higher in the presence of rAAV‐FLAG‐hsox9 compared with control transductions (up to 1.6‐fold) (always P < 0.001) both in the superficial and middle zones of the cartilage. As previously noted [22], type‐II collagen immunostaining was uniformly distributed in the normal control cartilage, whereas typical pericellular deposits of type‐II collagen representative of an active synthesis were observed in the OA cartilage or following application of the SOX9 vector. Notably, these parameters in the OA cartilage treated by rAAV‐FLAG‐hsox9 were similar to (P= 0.147) or higher (up to 1.4‐fold) than (P < 0.001) those measured in the normal rAAV‐lacZ‐transduced cartilage, although the vector dose applied in these experiments was not sufficient to restore the native safranin O staining intensity (P < 0.001). Interestingly, focal type‐X collagen immunostaining that was noted in the control (rAAV‐lacZ‐transduced) OA cartilage (middle and deep zones) strongly decreased when the rAAV‐hFGF‐2 vector was administered (≥70%) (Fig. 5). This effect was maintained when co‐applying the rAAV‐FLAG‐hsox9 vector. In contrast, immunoreactivity to type‐X collagen in normal cartilage transduced by either test condition remained undetectable.
Discussion
A difficulty to treat human OA lies in the identification of agents capable of counterbalancing the slow and irreversible progression of the disease. Elaboration of an effective therapy against OA will most likely necessitate managing the various processes implicated in its outcome in order to rebuild an original articular cartilage surface. Gene‐based transfer of therapeutic candidates is an attractive approach to achieve this goal. Encouraging results to restore OA cartilage were obtained by applying external chondroprotective and chondroregenerative stimuli including IL‐1Ra [3, 4, 5, 6], IGF‐I [7], FGF‐2 [8, 9], BMP‐7 [10], and TGF‐β[11, 12] or components capable of modulating the chondrocyte phenotype such as the Sox9 transcription factor [20, 21, 22]. Despite remarkable advances, complete reproduction of an original architecture in OA cartilage has not been reported to date. As often emphasized [18, 23, 24], combining key properties of particular agents might be more beneficial to reconstruct OA cartilage compared with single treatments. In the present study, we investigated the ability of two factors that are crucial for chondrocyte differentiation and cartilage formation to remodel the architecture of human OA cartilage compared with normal cartilage. Specifically, we focused on delivering the gene sequences for FGF‐2 to provide proliferative stimuli to chondrocytes [8] and for SOX9, a transcription factor that plays key roles in increasing matrix synthesis [17, 18, 20, 21, 22]. Concomitant expression of SOX9 might also prove beneficial to counteract the putative effects of FGF‐2 upon the synthesis and accumulation of matrix proteoglycans, in particular in OA cartilage [25, 26, 42, 43]. To achieve these goals, we employed rAAV to deliver the therapeutic candidates, as these vectors can mediate high levels of transgene expression throughout the whole depth of the cartilage over extended periods of time [27, 28] and are capable of delivering separate transgenes in target tissues [30, 31].
Consistent with our findings using rabbit chondrocytes [29], efficient transgene expression was achieved in human chondrocytes via transduction by the candidate constructs with an early onset of expression probably due to the high permissivity of these cells to rAAV [28], followed by relatively high (for about 15 days) and sustained detection levels for up to day 26, the longest time point examined. These features of rAAV might be well suited to treat over time a slow, progressive disease like OA. In agreement with the well‐established mitogenic activities of FGF‐2 [8, 29, 44], application of rAAV‐hFGF‐2 enhanced the proliferation of human normal and OA chondrocytes in vitro. This effect was maintained when the SOX9 vector was co‐administered, relatively to the dose employed in single rAAV‐hFGF‐2 transduction, but not further increased by the transcription factor, as expected [45, 46] and consistent with our findings using rAAV‐FLAG‐hsox9 alone [22]. In contrast, treatment by rAAV‐hFGF‐2 did not enhance matrix synthesis in vitro[29, 44], suggesting that the increases in cell numbers were not accompanied by a stimulation of matrix production, concordant with the demonstration that FGF‐2 can inhibit the synthesis and accumulation of proteoglycans in cartilage [25, 26]. Yet, these processes could be further activated in the presence of rAAV‐FLAG‐hsox9 proportionally to the dose of vector applied [22] in good agreement with the properties of the transcription factor [20, 47, 48, 49], suggesting that Sox9 could compensate for the effects of FGF‐2 on the metabolic activities of chondrocytes.
In situ, high levels of transgene expression were achieved early on and for up to 10 days, the longest time point examined following direct application of the vectors to human cartilage explants, as previously reported using this class of vectors [22]. It remains to be seen if expression of the transgenes in situ will be maintained over extended periods of time for the development of an effective, long‐term therapy against OA. Interestingly, there was no difference in the transduction efficiencies between normal and OA cartilage in contrast with the findings of Goater et al.[50] who reported that injured cartilage might be more susceptible to rAAV gene transfer, probably due to a better diffusion of the vectors through the impeded matrix. Yet, compared with these authors, we delivered much higher (10‐fold) vector doses that are known to transduce similarly normal and OA cartilage [22, 28]. Endogenous transgene (FGF‐2) expression was noted in situ in contrast to the findings in vitro, probably reflecting a difference of sensitivity between the detection methods employed. Specific immunoreactivity was seen throughout the thickness of the explant cultures, probably due to the ability of the small rAAV to penetrate the dense cartilage matrix [22, 27, 28]. This feature of rAAV may be therefore advantageous to deliver therapeutic candidates in‐depth compared with large vectors that have more value for ex vivo applications [3, 10, 51]. Consistent with the findings in vitro, our data indicate that administration of rAAV‐hFGF‐2 promoted an increase in the cell densities and DNA contents in cartilage in situ. Again, these effects were maintained but not further enhanced by overexpression of SOX9[22, 45, 46]. Remarkably, these properties of FGF‐2 were concomitant with a strong reduction in the apoptotic cell population in the treated OA cartilage, in good agreement with previous findings using recombinant FGF‐2 [52]. As also seen in vitro, treatment by the FGF‐2 vector did not stimulate matrix synthesis [29, 44], suggesting that the anabolic pathways might have been affected in the enhanced pools of cells in the explants [25, 26]. These observations, especially with respect to the decrease in type‐X collagen production compared with control OA cartilage [53, 54, 55] are compatible with the reported effects of the growth factor upon expression of this matrix molecule [42, 43]. Yet, synthesis of the major components of the natural cartilage matrix (proteoglycans, type‐II collagen) could be activated in the presence of the SOX9 vector, corroborating the hypothesis that the transcription factor might be capable of counterbalancing the effects of FGF‐2. Importantly, down‐regulation of type‐X collagen expression was maintained when rAAV‐FLAG‐hsox9 was added, concordant with previous findings showing that this factor can suppress markers of hypertrophic differentiation [56, 57]. Most remarkably, co‐treatment of OA cartilage by the candidate vectors induced both cell proliferation and matrix synthesis to levels similar to or higher than those noted in the normal control cartilage. This report is the first evidence showing that, even in the absence of additive effects a therapeutic gene combination can activate key regenerative events, prevent the pathological shift of metabolic homeostasis, and inhibit undesirable hypertrophy in human OA cartilage over time. For comparison, only partial and short‐term effects of Sox9 were noted in OA chondrocytes using adenoviral and retroviral vectors [20, 21], contrasting with the stability of rAAV transgenes [32]. It remains to be seen if modified (different from normal) rates of cell division and matrix production caused by overexpression of FGF‐2 and SOX9 might alter the integrity of the cartilage over time. Kypriotou et al.[24] showed that too elevated Sox9 concentrations in chondrocytes may disturb the balance between transcription factors and inhibit type‐II collagen expression. This is even more important in view of the effects of FGF‐2 upon cartilage homeostasis, as this factor has been shown to not only stimulate the production of matrix metalloproteinases (MMP‐1, MMP‐3, and MMP‐13) [58, 59, 60] but to also activate that of tissue inhibitor of metalloproteinases (TIMP‐1) [58, 59]. It is likely that a tight regulation of transgene expression will be necessary to generate an appropriate treatment for OA. This might be practicable by designing vectors where the candidate sequence is placed under the control of regulatable (tetracycline‐sensitive) or tissue‐specific (SOX9, type‐II collagen, Cartilage Oligomeric Matrix Protein) promoters.
Despite improvements compared with control and single rAAV transductions [22], complete reproduction of an original surface in OA cartilage was not afforded by co‐overexpression of two factors determining for chondrocyte differentiation and cartilage formation. Although not tested here, repeated administration of the combination might improve the structural organization of the cartilage over time. Yet, it is possible that regeneration of a native phenotype in OA will require the action of additional candidates, as chondrogenesis is a complex process controlled by several regulatory agents such as secreted proteins and peptides, signalling molecules, and other transcription factors. Beside the factors employed here, they include TGF‐β, the BMPs, IGF‐I, the parathyroid hormone‐related peptide, cartilage‐derived morphogenetic proteins, the growth/differentiation factor 5, the cartilage‐derived retinoic acid sensitive protein, members of the Wnt and hedgehog families, SOX5 and SOX6, Cbfa‐1/Runx‐2, Cart‐1, and the Ets factors [61, 62]. Although no evidence was provided in OA cartilage, Ikeda et al.[18] reported that administration of a SOX5/SOX6/SOX9 trio to mesenchymal stem cells was more efficient to promote chondrocyte differentiation compared with SOX9 alone. In summary, the results of this study indicate that direct, prolonged rAAV‐mediated co‐overexpression of FGF‐2 and SOX9 can simultaneously modulate chondrocyte proliferation and compensate for the loss of matrix components in human OA cartilage, contributing to the regeneration of an original structure in OA cartilage. Evaluation of this approach in animal models of OA [5, 6] will allow determining the benefits of co‐applying these factors to OA cartilage in vivo. The present findings support the concept of developing coupled gene transfer approaches using rAAV for the treatment of human OA, at least for early stages when some cartilage surface is still maintained.
Acknowledgements
This work was supported by the German Research Society (Deutsche Forschungsgemeinschaft) (grants DFG CU 55/1–1, /1–2, and /1–3 of M.C. and H.M.) and the German Osteoarthritis Foundation (Deutsche Arthrose‐Hilfe) (grants DAH of M.C., D.K., and H.M.). We thank T. Thurn, S. Schetting, and A. Weimer for technical assistance, R. J. Samulski (The Gene Therapy Center, University of North Carolina, Chapel Hill, NC, USA) and X. Xiao (The Gene Therapy Center, University of Pittsburgh, Pittsburgh, PA, USA) for providing the genomic AAV‐2 plasmid clones and the 293 cell line, M. Seno (Department of Bioscience and Biotechnology, Faculty of Engineering, Okayama University, Japan) for the human FGF‐2 cDNA, and G. Scherer (Institute for Human Genetics and Anthropology, Albert‐Ludwig University, Freiburg, Germany) for the human SOX9 cDNA.
References
- 1. Poole AR. An introduction to the pathophysiology of osteoarthritis. Front Biosci . 1999; 4: D662–70. [DOI] [PubMed] [Google Scholar]
- 2. Pelletier JP, DiBattista JA, Roughley P, et al . Cytokines and inflammation in cartilage degradation. Rheum Dis Clin North Am . 1993; 19: 545–68. [PubMed] [Google Scholar]
- 3. Baragi VM, Renkiewicz RR, Jordan H, et al . Transplantation of transduced chondrocytes protects articular cartilage from interleukin 1‐induced extracellular matrix degradation. J Clin Invest . 1995; 96: 2454–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nixon AJ, Haupt JL, Frisbie DD, et al . Gene‐mediated restoration of cartilage matrix by combination insulin‐like growth factor‐I(interleukin‐1 receptor antagonist therapy. Gene Ther . 2005; 12: 177–86. [DOI] [PubMed] [Google Scholar]
- 5. Pelletier JP, Caron JP, Evans C, et al . In vivo suppression of early experimental osteoarthritis by interleukin‐1 receptor antagonist using gene therapy. Arthritis Rheum . 1997; 40: 1012–9. [DOI] [PubMed] [Google Scholar]
- 6. Frisbie DD, Ghivizzani SC, Robbins PD, et al . Treatment of experimental equine osteoarthritis by in vivo delivery of the equine interleukin‐1 receptor antagonist gene. Gene Ther . 2002; 9: 12–20. [DOI] [PubMed] [Google Scholar]
- 7. Madry H, Zurakowski D, Trippel SB. Overexpression of human insulin‐like growth factor‐I promotes new tissue formation in an ex vivo model of articular chondrocyte transplantation. Gene Ther . 2001; 8: 1443–9. [DOI] [PubMed] [Google Scholar]
- 8. Madry H, Emkey G, Zurakowski D, et al . Overexpression of human fibroblast growth factor 2 stimulates cell proliferation in an ex vivo model of articular chondrocyte transplantation. J Gene Med . 2004; 6: 238–45. [DOI] [PubMed] [Google Scholar]
- 9. Inoue A, Takahashi KA, Arai Y, et al . The therapeutic effects of basic fibroblast growth factor contained in gelatin hydrogel microspheres on experimental osteoarthritis in the rabbit knee. Arthritis Rheum . 2006; 54: 264–70. [DOI] [PubMed] [Google Scholar]
- 10. Hidaka C, Quitoriano M, Warren RF, et al . Enhanced matrix synthesis and in vitro formation of cartilage‐like tissue by genetically modified chondrocytes expressing BMP‐7. J Orthop Res . 2001; 19: 751–8. [DOI] [PubMed] [Google Scholar]
- 11. Ulrich‐Vinther M, Stengaard C, Schwarz EM, et al . Adeno‐associated vector mediated gene transfer of transforming growth factor‐beta1 to normal and osteoarthritic human chondrocytes stimulates cartilage anabolism. Eur Cell Mater . 2005; 10: 40–59. [DOI] [PubMed] [Google Scholar]
- 12. Blaney Davidson EN, Vitters EL, Van Den Berg WB, et al . TGF beta‐induced cartilage repair is maintained but fibrosis is blocked in the presence of Smad7. Arthritis Res Ther . 2006; 8: R65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Venkatesan N, Barre L, Benani A, et al . Stimulation of proteoglycan synthesis by glucuronosyltransferase‐I gene delivery: a strategy to promote cartilage repair. Proc Natl Acad Sci USA . 2004; 101: 18087–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Aigner T, Vornehm SI, Zeiler G, et al . Suppression of cartilage matrix gene expression in upper zone chondrocytes of osteoarthritic cartilage. Arthritis Rheum . 1997; 40: 562–9. [DOI] [PubMed] [Google Scholar]
- 15. Smith RL. Degradative enzymes in osteoarthritis. Front Biosci . 1999; 4: D704–12. [DOI] [PubMed] [Google Scholar]
- 16. Aigner T, Gebhard PM, Schmid E, et al . SOX9 expression does not correlate with type II collagen expression in adult articular chondrocytes. Matrix Biol . 2003; 22: 363–72. [DOI] [PubMed] [Google Scholar]
- 17. Bi W, Deng JM, Zhang Z, et al . SOX9 is required for cartilage formation. Nat Genet . 1999; 22: 85–9. [DOI] [PubMed] [Google Scholar]
- 18. Ikeda T, Kamekura S, Mabuchi A, et al . The combination of SOX5, SOX6, and SOX9 (the SOX trio) provides signals sufficient for induction of permanent cartilage. Arthritis Rheum . 2004; 50: 3561–73. [DOI] [PubMed] [Google Scholar]
- 19. Salminen H, Vuorio E, Saamanen AM. Expression of SOX9 and type IIA procollagen during attempted repair of articular cartilage damage in a transgenic mouse model of osteoarthritis. Arthritis Rheum . 2001; 44: 947–55. [DOI] [PubMed] [Google Scholar]
- 20. Tew SR, Li Y, Pothacharoen P, et al . Retroviral transduction with SOX9 enhances re‐expression of the chondrocyte phenotype in passaged osteoarthritic human articular chondrocytes. Osteoarthritis Cartilage . 2005; 13: 80–9. [DOI] [PubMed] [Google Scholar]
- 21. Li Y, Tew SR, Russell AM, et al . Transduction of passaged human articular chondrocytes with adenoviral, retroviral, and lentiviral vectors and the effects of enhanced expression of SOX9. Tissue Eng . 2004; 10: 575–84. [DOI] [PubMed] [Google Scholar]
- 22. Cucchiarini M, Thurn T, Weimer A, et al . Restoration of the extracellular matrix in human osteoarthritic articular cartilage by overexpression of the transcription factor SOX9. Arthritis Rheum . 2007; 56: 158–67. [DOI] [PubMed] [Google Scholar]
- 23. Loeser RF, Pacione CA, Chubinskaya S. The combination of insulin‐like growth factor 1 and osteogenic protein 1 promotes increased survival of matrix synthesis by normal and osteoarthritic human articular chondrocytes. Arthritis Rheum . 2003; 48: 2188–96. [DOI] [PubMed] [Google Scholar]
- 24. Kypriotou M, Fossard‐Demoor M, Chadjichristos C, et al . SOX9 exerts a bifunctional effect on type II collagen gene (COL2A1) expression in chondrocytes depending on the differentiation state. DNA Cell Biol . 2003; 22: 119–29. [DOI] [PubMed] [Google Scholar]
- 25. Posever J, Phillips FM, Pottenger LA. Effects of basic fibroblast growth factor, transforming growth factor‐beta 1, insulin‐like growth factor‐1, and insulin on human osteoarthritic articular cartilage explants. J Orthop Res . 1995; 13: 832–7. [DOI] [PubMed] [Google Scholar]
- 26. Loeser RF, Chubinskaya S, Pacione C, et al . Basic fibroblast growth factor inhibits the anabolic activity of insulin‐like growth factor 1 and osteogenic protein 1 in adult human articular chondrocytes. Arthritis Rheum . 2005; 52: 3910–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Arai Y, Kubo T, Fushiki S, et al . Gene delivery to human chondrocytes by an adeno associated virus vector. J Rheumatol . 2000; 27: 979–82. [PubMed] [Google Scholar]
- 28. Madry H, Cucchiarini M, Terwilliger EF, et al . Recombinant adeno‐associated virus vectors efficiently and persistently transduce chondrocytes in normal and osteoarthritic human articular cartilage. Hum Gene Ther . 2003; 14: 393–402. [DOI] [PubMed] [Google Scholar]
- 29. Cucchiarini M, Madry H, Ma C, et al . Improved tissue repair in articular cartilage defects in vivo by rAAV‐mediated overexpression of human fibroblast growth factor 2. Mol Ther . 2005; 12: 229–38. [DOI] [PubMed] [Google Scholar]
- 30. Rendahl KG, Leff SE, Otten GR, et al . Regulation of gene expression in vivo following transduction by two separate rAAV vectors. Nat Biotechnol . 1998; 16: 757–61. [DOI] [PubMed] [Google Scholar]
- 31. Muramatsu S, Fujimoto K, Ikeguchi K, et al . Behavioral recovery in a primate model of Parkinson's disease by triple transduction of striatal cells with adeno‐associated viral vectors expressing dopamine‐synthesizing enzymes. Hum Gene Ther . 2002; 13: 345–54. [DOI] [PubMed] [Google Scholar]
- 32. Xiao X, Li J, Samulski RJ. Efficient long‐term gene transfer into muscle tissue of immunocompetent mice by adeno‐associated virus vector. J Virol . 1996; 70: 8098–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mason JM, Grande DA, Barcia M, et al . Expression of human bone morphogenic protein 7 in primary rabbit periosteal cells: potential utility in gene therapy for osteochondral repair. Gene Ther . 1998; 5: 1098–104. [DOI] [PubMed] [Google Scholar]
- 34. Mi Z, Ghivizzani SC, Lechman ER, et al . Adenovirus‐mediated gene transfer of insulin‐like growth factor 1 stimulates proteoglycan synthesis in rabbit joints. Arthritis Rheum . 2000; 43: 2563–70. [DOI] [PubMed] [Google Scholar]
- 35. Mankin HJ, Dorfman H, Lippiello L, et al . Biochemical and metabolic abnormalities in articular cartilage from osteo‐arthritic human hips. II. Correlation of morphology with biochemical and metabolic data. J Bone Joint Surg Am . 1971; 53: 523–37. [PubMed] [Google Scholar]
- 36. Samulski RJ, Chang LS, Shenk T. Helper‐free stocks of recombinant adeno‐associated viruses: normal integration does not require viral gene expression. J Virol . 1989; 63: 3822–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cucchiarini M, Ren XL, Perides G, et al . Selective gene expression in brain microglia mediated via adeno‐associated virus type 2 and type 5 vectors. Gene Ther . 2003; 10: 657–67. [DOI] [PubMed] [Google Scholar]
- 38. Seno M, Masago A, Nishimura A, et al . BALB(c 3T3 cells co‐expressing FGF‐2 and soluble FGF receptor acquire tumorigenicity. Cytokine . 1998; 10: 290–4. [DOI] [PubMed] [Google Scholar]
- 39. Sudbeck P, Schmitz ML, Baeuerle PA, et al . Sex reversal by loss of the C‐terminal transactivation domain of human SOX9. Nat Genet . 1996; 13: 230–2. [DOI] [PubMed] [Google Scholar]
- 40. Madry H, Cucchiarini M, Stein U, et al . Sustained transgene expression in cartilage defects in vivo after transplantation of articular chondrocytes modified by lipid‐mediated gene transfer in a gel suspension delivery system. J Gene Med . 2003; 5: 502–9. [DOI] [PubMed] [Google Scholar]
- 41. Uusitalo H, Hiltunen A, Ahonen M, et al . Accelerated up‐regulation of L‐Sox5, Sox6, and Sox9 by BMP‐2 gene transfer during murine fracture healing. J Bone Miner Res . 2001; 16: 1837–45. [DOI] [PubMed] [Google Scholar]
- 42. Iwasaki M, Nakahara H, Nakata K, et al . Regulation of proliferation and osteochondrogenic differentiation of periosteum‐derived cells by transforming growth factor‐beta and basic fibroblast growth factor. J Bone Joint Surg Am . 1995; 77: 543–54. [DOI] [PubMed] [Google Scholar]
- 43. Ogawa T, Shimokawa H, Fukada K, et al . Localization and inhibitory effect of basic fibroblast growth factor on chondrogenesis in cultured mouse mandibular condyle. J Bone Miner Metab . 2003; 21: 145–53. [DOI] [PubMed] [Google Scholar]
- 44. Trippel SB, Wroblewski J, Makower AM, et al . Regulation of growth‐plate chondrocytes by insulin‐like growth‐factor I and basic fibroblast growth factor. J Bone Joint Surg Am . 1993; 75: 177–89. [DOI] [PubMed] [Google Scholar]
- 45. Panda DK, Miao D, Lefebvre V, et al . The transcription factor SOX9 regulates cell cycle and differentiation genes in chondrocytic CFK2 cells. J Biol Chem . 2001; 276: 41229–36. [DOI] [PubMed] [Google Scholar]
- 46. Drivdahl R, Haugh KH, Sprenger CC, et al . Suppression of growth and tumorigenicity in the prostate tumor cell line M12 by overexpression of the transcription factor SOX9. Oncogene . 2004; 23: 4584–93. [DOI] [PubMed] [Google Scholar]
- 47. Bell DM, Leung KK, Wheatley SC, et al . SOX9 directly regulates the type‐II collagen gene. Nat Genet . 1997; 16: 174–8. [DOI] [PubMed] [Google Scholar]
- 48. Lefebvre V, Huang W, Harley VR, et al . SOX9 is a potent activator of the chondrocyte‐specific enhancer of the pro alpha1(II) collagen gene. Mol Cell Biol . 1997; 17: 2336–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ng LJ, Wheatley S, Muscat GE, et al . SOX9 binds DNA, activates transcription, and coexpresses with type II collagen during chondrogenesis in the mouse. Dev Biol . 1997; 183: 108–21. [DOI] [PubMed] [Google Scholar]
- 50. Goater J, Müller R, Kollias G, et al . Empirical advantages of adeno associated viral vectors for in vivo gene therapy for arthritis. J. Rheumatol . 2000; 27: 983–9. [PubMed] [Google Scholar]
- 51. Doherty PJ, Zhang H, Tremblay L, et al . Resurfacing of articular cartilage explants with genetically‐modified human chondrocytes in vitro . Osteoarthritis Cartilage . 1998; 6: 153–9. [DOI] [PubMed] [Google Scholar]
- 52. Schmal H, Zwingmann J, Fehrenbach M, et al . bFGF influences human articular chondrocyte differentiation. Cytotherapy . 2007; 9: 184–93. [DOI] [PubMed] [Google Scholar]
- 53. Hoyland JA, Thomas JT, Donn R, et al . Distribution of type X collagen mRNA in normal and osteoarthritic human cartilage. Bone Miner . 1991; 15: 151–63. [DOI] [PubMed] [Google Scholar]
- 54. von der Mark K, Kirsch T, Nerlich A, et al . Type X collagen synthesis in human osteoarthritic cartilage. Indication of chondrocyte hypertrophy. Arthritis Rheum . 1992; 35: 806–11. [DOI] [PubMed] [Google Scholar]
- 55. Kirsch T, Swoboda B, Nah H. Activation of annexin II and V expression, terminal differentiation, mineralization and apoptosis in human osteoarthritic cartilage. Osteoarthritis Cartilage . 2000; 8: 294–302. [DOI] [PubMed] [Google Scholar]
- 56. Tsuchiya H, Kitoh H, Sugiura F, et al . Chondrogenesis enhanced by overexpression of sox9 gene in mouse bone marrow‐derived mesenchymal stem cells. Biochem Biophys Res Comm . 2003; 301: 338–43. [DOI] [PubMed] [Google Scholar]
- 57. Hardingham TE, Oldershaw RA, Tew SR. Cartilage, SOX9 and Notch signals in chondrogenesis. J Anat . 2006; 209: 469–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Vincent T, Hermansson M, Bolton M, et al . Basic FGF mediates an immediate response of articular cartilage to mechanical injury. Proc Natl Acad Sci USA . 2002; 99: 8259–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Vincent TL, Hermansson MA, Hansen UN, et al . Basic fibroblast growth factor mediates transduction of mechanical signals when articular cartilage is loaded. Arthritis Rheum . 2004; 50: 526–33. [DOI] [PubMed] [Google Scholar]
- 60. Wang X, Manner PA, Horner A, et al . Regulation of MMP‐13 expression by RUNX2 and FGF2 in osteoarthritic cartilage. Osteoarthritis Cartilage . 2004; 12: 963–73. [DOI] [PubMed] [Google Scholar]
- 61. Smits P, Li P, Mandel J, et al . The transcription factors L‐SOX5 and SOX6 are essential for cartilage formation. Dev Cell . 2001; 1: 277–90. [DOI] [PubMed] [Google Scholar]
- 62. Cucchiarini M, Madry H. Gene therapy for cartilage defects. J Gene Med . 2005; 7: 1495–509. [DOI] [PubMed] [Google Scholar]