

Abstract
ADAM15 belongs to a family of transmembrane multi‐domain proteins implicated in proteolysis, cell–cell and cell–matrix interactions in various disease conditions. In osteoarthritis (OA), ADAM15 is up‐regulated in the chondrocytes already at early stages of cartilage degeneration where it seems to exert homeostatic effects likely associated with its ability to enhance integrin‐mediated chondrocyte adhesion to the surrounding collagen matrix. The aim of our present study was, therefore, to characterize functional domains of ADAM15 involved in collagen II (CII) interaction and to analyse associated outside‐in signalling events. Accordingly, ADAM15 and respective deletion mutants were stably transfected into the chondrocyte cell line T/C28a4. Transfected cells were adhered to CII and phosphoproteins analysed by Western blotting. Co‐immunoprecipitation served to identify protein binding to ADAM15. Our results elucidate the prodomain as critical for the capacity of ADAM15 to enhance CII adhesion, thereby identifying for the first time a cell‐adhesive role of a metalloproteinase prodomain. Moreover, the cytoplasmic tail of ADAM15 confers a modulatory effect on the autophosphorylation site Y397 of the focal adhesion kinase (FAK) during chondrocyte–collagen interaction. In conclusion, the newly uncovered impact of ADAM15 on signalling events that arise from chondrocyte interactions with its collagen matrix might contribute to the elucidation of the mechanism underlying its proposed chondroprotective role in degenerative cartilage disease.
Keywords: chondrocyte, osteoarthritis, ADAM15, collagen adhesion, focal adhesion kinase
Introduction
The disintegrin and metalloproteinase ADAM15 belongs to a large family of about 40 homologous transmembrane glycoproteins that share a common modular ectodomain architecture [1, 2]. The extracellular components consist of a cysteine‐rich region and a disintegrin domain, which contributes to cell–cell interactions via its capability to interact with alphavβ3, alpha5β1 and the alpha9 integrins [3, 4, 5], followed by a metalloproteinase module that is capped by a large prodomain of about 200 amino acid residues [1, 2]. The cytoplasmic domain of ADAM15 contains SH3‐binding sites, which can potentially activate kinases of the src‐kinase family [6]. Although ADAM15 is not present in normal healthy tissues, it gets up‐regulated in tumourigenesis [7] as well as in other non‐neoplastic conditions of tissue remodelling such as atherosclerosis [8] and inflammatory as well as degenerative joint diseases [9, 10]. In the latter rheumatic conditions, high expression of ADAM15 has been demonstrated in synovial fibroblasts, especially in rheumatoid arthritis (RA), whereas chondrocytes exhibited ADAM15 up‐regulation already at very early stages of osteoarthritis (OA) development [9, 10].
The precise role of ADAM15 in the pathogenesis of human OA has remained enigmatic so far. However, some basic mechanisms of potential relevance have already been elucidated. Thus, ADAM15 contains in its metalloproteinase (MP) domain a common metalloproteinase consensus motif. It is thought that this metalloproteinase is kept in an inactive conformation by the adjacent prodomain, which upon its own cleavage finally liberates the active protease from latency [reviewed in 1]. Initial in vitro experiments suggest that ADAM15 is indeed capable to cleave collagen type IV [11], thereby indicating the potential of its catalytic domain to contribute to pathologic extracellular matrix (ECM) remodelling. However, more direct experimental support for such a role of ADAM15 in proteolytic cartilage‐degradation in vivo is missing. Theoretically, the MP domain could also contribute to cartilage pathology via the proteolytic shedding of membrane‐bound receptor ligands in analogy to the proinflammatory effects of ADAM17 (TNFalpha converting enzyme) [reviewed in 12]. Although studies of EGF‐receptor (EGFR) transsignalling in tumour cells suggest a respective potential of the ADAM15 MP domain to act as a sheddase of EGFR ligands [13], corresponding evidence for a similar role in degenerative cartilage disease is entirely missing. Although the function of the ADAM15 MP domain remains enigmatic, the accelerated development of murine osteoarthritic lesions in ADAM15‐deficient mice compared with wild‐type rather suggests a homeostatic than a destructive role of ADAM15 in cartilage remodelling [14]. Accordingly, the more recently uncovered reinforcement of integrin‐dependent collagen II (CII) adhesion by ADAM15 overexpression in the human chondrocyte cell line T/C28a4 associated with cell survival promoting effects in vitro[14] has gained importance for a better understanding of ADAM15 up‐regulation in early OA cartilage. Therefore, the aim of the present study was to identify the functional domain(s) of ADAM15 involved in the enhanced CII binding by transfecting T/C28a4 chondrocytes with full‐length ADAM15 cDNA and its respective deletion mutants and to characterize concomittant alterations of the outside‐in signalling during collagen adhesion. The results of our investigation uncover the critical role of the prodomain for the capacity of ADAM15 to reinforce chondrocyte‐binding to CII thereby identifying for the first time a direct function of a MP‐prodomain for cell adhesion to ECM. The cytoplasmic tail of ADAM15, although dispensible for the enhanced CII adhesion, however, confers a modulatory effect on outside‐in signalling during chondrocyte–matrix interaction. Thus, our results demonstrate that ADAM15 overexpression results in a reduced phosphorylation at the critical autophosphorylation site at Y397 in the non‐receptor tyrosine kinase focal adhesion kinase (FAK) localized to focal adhesions.
Materials and methods
Materials
Several antibodies directed against ADAM15 were used: a goat polyclonal and a mouse monoclonal from R&D Systems (Wiesbaden, Germany), a rabbit polyclonal against the prodomain and against the cytoplasmatic domain from Chemicon/Millipore (Schwalbach, Germany). Anti‐pTyr99 antibody was from Santa Cruz Biotechnology (Heidelberg, Germany). Anti‐phospho Y397 FAK was from BioSource (Solingen, Germany), mouse monoclonal anti‐FAK (clone 4.47, Upstate‐Millipore), rabbit anti‐actin and mouse monoclonal anti‐Gst (clone Gst‐2) from Sigma‐Aldrich (Taufkirchen, Germany). Isotype‐specific control antibodies were from AbD Serotec (Düsseldorf, Germany). Bovine type II collagen was from Mdbiosciences (Egg, Switzerland), human plasma fibronectin from Chemicon/Millipore. The chondrocyte cell line T/C28a4 [15] was kindly provided by Dr. M.B. Goldring (Hospital for Special Surgery, New York, NY, USA). Primary, non‐passaged human chondrocytes from either normal or osteoarthritic (OA) cartilage were kindly provided by Dr. Thomas Aigner (Institute of Pathology, Leipzig, Germany) [16]. Although the normal cartilage was obtained from donors at autopsy not later than 48 hrs post‐mortem, the human OA specimens were derived from the hip or knee joints of patients who had undergone endoprosthetic joint surgery for severe osteoarthritis. Prior to use, the categorization of cartilage specimens into normal or osteoarthritic had been validated by microscopic assessments of an experienced pathologist (Dr. T. Aigner).
Cloning of full‐length ADAM15 and deletion mutants
Full‐length ADAM15 was subcloned from the previously used pcDNA vector containing the full‐length sequence [14] into the pexchange‐1 vector and a puromycin cassette was inserted using the Cre recombinase according to the supplier’s instructions (Stratagene, Amsterdam, The Netherlands). The ADAM15 mutants were cloned accordingly into the pexchange‐1 vector using the full‐length ADAM15 plasmid as template. Transmembrane‐anchored ADAM15 lacking the complete cytoplasmic domain (Δcyto) was cloned using the primer pair 5′TCGGATCCCCCGGGCTGCAGGAATT3′ and 5′ACTCGAGTCAGTACCAGTAGCCGGCACCAA 3′ for amplification. ADAM15 mutants either lacking the prodomain (ΔPro), or the pro‐ and MP‐domain (ΔProMP) were cloned using fusion PCR (polymerase chain reaction) and the full‐length ADAM15 plasmid as a template: in the first PCR step, the nucleotide sequence encoding the signal peptide was amplified using primer I 5′TCGGATCCCCCGGGCTGCAGGAATT3′ and primer II 5′CACAATCACCAACTCCACAGTCTTAGGCAGAGGGCTGCCCC3′. In the second PCR, a sequence starting at the 5′start of the MP‐domain (bp 639) was amplified using primer III 5′AAGACTGTGGAGTTGGTGATTGTG3′ and primer IV 5′CTCGAGGTCGACGGTATCGA3′. In the following third step, the PCR products from step 1 and 2 were combined, denatured at 94°C and annealed for 10 min. to ligate the overlapping complementary nucleotides from the 3′ end of the signal peptide and the 5′ start of the MP domain and subsequently amplified by PCR using primer I and IV. The ΔProMP mutant was cloned accordingly, with primer I and 5′GCCCGGCTCCACAAACATAGGCAGAGGGCTGCCC 3′ for the (i) PCR of the signal peptide, and a primer starting at the disintegrin domain 5′ATGTTTGTGGAGCCGGGC 3′ and primer (IV) for the (ii) PCR. The empty pexchange vector was transfected into the chondrocytic T/C28a4 to serve as control. All plasmids were sequenced completely in both directions on an automated sequencer (ABI model 373, Applied Biosystems, Weiterstadt, Germany).
Generation of permanent cell lines transfected with ADAM15 and domain deletion mutants
The transfection and selection procedures were performed as described previously [14]. Briefly, T/C28a4 chondrocytic cells were transfected in 6‐well tissue culture plates using JetPei (Biomol, Hamburg, Germany) according to the manufacturer’s instructions. Twenty‐four hours after transfection, cells were treated with 10 μg/ml puromycin and cell foci surviving the antibiotic pressure were pooled, grown to confluency and checked for protein expression using Western blotting. Cells that were transfected with the empty pexchange‐1 vector served as a control.
Cell culture
Cells were kept in Dulbecco’s modified Eagle’s medium supplemented with 10% heat‐inactivated foetal calf serum (FCS) and kept under constant antibiotic culture using 3 μg/ml puromycin, All tissue culture reagents were from Gibco/Invitrogen, Karlsruhe, Germany. For all subsequent tests, cells were grown to subconfluency to a cell density of about 4 × 106 cells/75 cm2 tissue culture flask.
Generation of the recombinant prodomain of ADAM15
The nucleotide sequence 61–642 (amino acid 21–214 encoding the entire prodomain without the signal peptide) was amplified using pexchange‐1 plasmid containing the ADAM15 sequence as template with primers 5′CAGGATCCTGGCCGCTCCCAAATAT3′ and 5′AACTCGAGGGTCTCTGTTACCACATCCCG3′. The correctness of the amplified nucleotide sequence was confirmed by sequencing. After digestion with BamHI/XhoI, the PCR product was cloned into the pGEX 3P vector and expressed as a Gst (glutathione S‐transferase) fusion protein in BL21 E. coli (Amersham, GE Healthcare, Munich, Germany). Protein expression was induced using 10 mM IPTG for 3 hrs at room temperature and the harvested E. coli were lysed with Bug Buster protein extraction reagent and benzonase nuclease according to the supplier’s instructions (Novagen/Merck, Darmstadt, Germany). The supernatant containing the Prodomain‐Gst fusion protein was incubated with Gst‐sepharose (Amersham) for 30 min. at room temperature, thoroughly washed with PBS (150 mM NaCl, pH 7.4, 1 mM KH2PO4, 3 mM Na2HPO4) containing 1% Triton X‐100. Prodomain‐Gst was eluted with 10 mM reduced glutathione in 50 mM Tris/HCl, pH 8.0, containing a protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany). In order to isolate the Gst‐protein, the Gst‐sepharose with the bound Prodomain‐Gst fusion protein was treated with Precission Protease according to the manufacturer’s instructions (Amersham), thoroughly washed with PBS/Triton X‐100, and subsequently eluted with the reduced glutathione buffer. Protein concentration was determined using bicinchoninic acid (BCA) reagent (Pierce/Thermo Fisher Scientific, Schwerte, Germany).
Far Western with recombinant prodomain
Collagen type II (2.5 and 5 μg) and bovine serum album (BSA, 2.5 μg) were separated by 10% SDS/PAGE (sodium dodecyl sulfate/polyacrylamide gel electrophoresis), transferred to nitrocellulose filter, blocked in PBS containing 0.05% Tween 20, 1% BSA, and incubated with soluble Pro‐Gst fusion protein or the Gst‐tag (1 μg/ml) overnight at 4°C, incubated with either anti‐Gst‐ (1:2000) or anti‐prodomain‐IgG (1:2000) for 2 hrs at room temperature, followed by detection with HRP (horseradish peroxidase)‐conjugated secondary antibodies (Dako, Hamburg, Germany) and developed using a chemiluminiscent reagent (ECL Plus Western Blotting Detection System, Amersham, Freiburg, Germany).
Binding assay of recombinant Gst‐tagged prodomain using ELISA technique
Ninety‐six‐well Maxisorp plates (Nunc, Wiesbaden, Germany) were coated with 10 μg/ml native type II collagen (CII) or heat‐denatured CII, incubated overnight at 4°C, and blocked with heat‐inactivated 0.01% BSA (Fraction V, Sigma‐Aldrich) for 1 hr at 37°C. Denatured CII was prepared by heating at 80°C for 15 min., quickly added to the wells and kept at 37°C overnight. Soluble recombinant Pro‐Gst fusion protein and Gst were added to the wells at various concentrations from 0 to 250 nM in PBS (volume 100 μl) and incubated overnight at 4°C. Plates were washed 3 times with PBS, incubated with either anti‐Gst (1:2000) or antibodies directed against the prodomain of ADAM15 (1:2000) for 2 hrs at room temperature, washed again, incubated with HRP‐conjugated antibodies (1:2000) for 1 hr at room temperature and developed with ABTS solution (Roche Diagnostics). Absorbance was measured at 405 nm. Mean values and standard deviations were calculated from triplicate determinations and corrected for non‐specific binding of the primary and secondary antibodies on BSA‐coated wells.
Cell adhesion assay
Adhesion assays were performed as described previously [15]. Briefly, ninety‐six‐well Maxisorp plates (Nunc) were coated with collagen type II (5 μg/ml) and fibronectin (5 μg/ml) overnight at 4°C and blocked with heat denatured 1% BSA for 1 hr at 37°C. After trypsinization, cells were adhered on collagen type II for 1 hr, washed twice with PBS, fixed with 0.1% glutaraldehyde and stained with 0.1% crystalviolet solution. The optical density (OD) was read at 595 nm. Adhesion assays were performed in quadruplicates and the results shown are representative of least five independently performed experiments.
Signal transduction during adhesion to collagen type II
Twenty‐four‐well tissue culture plates (Greiner Bio‐One, Frickenhausen, Germany) were coated with 300 μl of a 100 μg/ml CII solution, completely dried under a sterile fume hood and stored overnight at 4°C. Cells transfected with vector, entire ADAM15 or an ADAM15‐mutant lacking the cytoplasmic domain, were gently trypsinized, washed twice with PBS to remove all traces of FCS, counted and resuspended in FCS‐free DMEM medium at a cell density of 1 × 106 cells/ml. The CII‐coated wells were blocked with PBS/1% BSA for 30 min. at 37°C. 7 × 105 cells were added to one well (2 cm2) and adhered to CII for 15, 30 and 60 min. at 37°C. Cells start to attach to CII after 30–40 min., and are hardly spread after 1 hr adhesion to CII, so that they can be easily collected from the well. After each time‐point, the plates were put on ice and the cells scraped off, centrifuged for 2 min., 13,000 ×g at 4°C, lysed and immunoblotted using phosphotyrosine‐specific antibodies as described below. Experiments were performed at least three times by two different investigators.
Preparation of cell lysates and Western blotting
Transfected cells and primary human chondrocytes were washed with ice‐cold PBS, scraped off the tissue culture flask and lysed in RIPA buffer for 1 hr at 4°C (150 mM NaCl, 50 mM Tris/HCl, pH 7.2, 1% Nonidet P40, 1% Triton X‐100, 0.25% sodium desoxycholate, 5 mM EDTA containing a proteinase inhibitor cocktail (according to the supplier's formulation and instruction: 1 tablet/10 ml, Roche Diagnostics) and phosphatase inhibitor cocktail I and II (10 μl/1 ml lysis buffer, Sigma‐Aldrich). After centrifugation for 5 min at 4°C, protein concentration was determined using BCA reagent. Samples were separated by 10% SDS/PAGE, transferred to nitrocellulose filter and incubated with the respective antibodies overnight at 4°C. Blots were incubated with appropriate HRP‐conjugated antibodies (1:5000, Dako) and developed using the ECL Plus chemiluminescent reagent. Signals were exposed to X‐rays films, scanned and signal density was measured with ImageJ program (Rasband, W.S., ImageJ, U.S. National Institutes of Health, Bethesda, MD, USA, http://rsb.info.nih.gov/ij/, 1997–2007). Before reprobing filters, blots were stripped with 62.5 mM Tris/HCl, pH 6.8, 2% SDS, 0.1 M β‐Mercaptoethanol for 30 min. at 50°C.
Preparation of cell lysates and co‐immunoprecipitation
Cells transfected with vector or full‐length ADAM15 were washed with ice‐cold PBS, scraped off the tissue culture flask and lysed in 10 mM HEPES, pH 7.0, 150 mM NaCl, 5 mM EDTA containing 1% Triton X‐100 and the proteinase and phosphatase inhibitor cocktails (10 μl/ml lysis buffer) as described above for 1 hr at 4°C. Protein concentration was determined and diluted to a concentration of 4 mg/ml. Human primary late stage (Mankin grades >7) OA chondrocytes that were pooled from five different donors were lysed as mentioned above. For co‐immunoprecipitation experiments, the mouse monoclonal anti‐ADAM15, the goat polyclonal anti‐ADAM15 that co‐immunoprecipitated identical bands and the antibodies against FAK (clone 4.47) were incubated with the freshly prepared cell lysates and Immunopure immobilized Protein G agarose beads (Pierce) under constant agitation for 90 min. at 4°C. The beads were washed 4 times with lysis buffer, boiled in sample buffer (125 mM Tris/HCl, pH 6,8, 4% SDS, 40% Glycerol, 10%β‐Mercaptoethanol, 20 μg/ml Bromophenol blue) for 5 min. at 95°C and subjected to SDS/PAGE. Subsequent Western blotting was performed as described above.
Results
Generation of permanent cell lines expressing ADAM15 and domain deletion mutants
The multi‐domain structure of ADAM15 was analysed using SMART [17, Simple Modular Architecture Research Tool at http://smart.embl‐heidelberg.de]. The prodomain comprises amino acids 21–213, whereas the MP‐domain localizes to aa 225–405 and the cytoplasmic region covers the carboxyterminal end between aa 718–814. In order to analyse the functional impact of the different ADAM15 domains on collagen adhesion or signal transduction, respective deletion mutants were cloned and stably expressed in the chondrocyte cell line T/C28a4 as shown in Fig. 1A. The truncated constructs either lack the aminoterminal prodomain (ΔPro), the pro‐and MP‐domain (ΔProMP) or consist of an intact extracellular portion that is still transmembrane‐anchored, however, devoid of its cytoplasmic tail (Δcyto).
Figure 1.
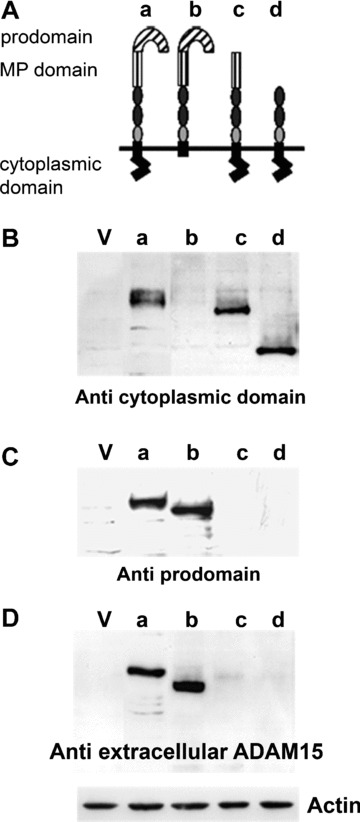
Generation of stably transfected cell lines expressing ADAM15 and deletion mutants lacking certain domains. (A) Schematic overview of the multi‐domain structure of ADAM15 (a) and architecture of the generated deletions mutants (b–d). (B–D) show immunoblots of cells transfected with vector (lane V), full‐length ADAM15 (lane a), transmembrane‐anchored ADAM15 lacking the cytoplasmic domain (Δ cyto, lane b) and ADAM15 lacking the prodomain and both the pro‐ and metalloproteinase (MP) domain (lanes c, d) using antibodies against the cytoplasmic domain (B), against the prodomain (C) and a mouse monoclonal antibody directed to the extracellular recombinant ADAM15 (D). Blots were stripped and reprobed with anti‐actin IgG (shown exemplarily for one blot).
Figure 1B–D shows the expression of the various mutants detected by Western blotting using four different antibodies. A polyclonal antibody directed to the cytoplasmic domain of ADAM15 revealed a strong expression of the full‐length ADAM15 at 100 kD (Fig. 1B). The ΔPro, which lacks the amino acid residues 21‐213, exhibits a reduced electrophoretic mobility corresponding to 78 kD and the additional removal of the MP‐domain yields a protein of 50 kD (ΔProMP). As expected, the Δcyto‐mutant remained undetectable by the antibody specific for the cytoplasmic domain that had been deleted in the respective transfected construct (Fig. 1B). The polyclonal antibody with specificity for the prodomain detected only the full‐length ADAM15 and the Δcyto at 85 kD (Fig. 1C). The commercially available monoclonal as well as the polyclonal goat antibodies both originally raised against the entire recombinant extracellular part of human ADAM15 only detected the full‐length ADAM15 and ADAM15 Δcyto, thus allowing for the conclusion that their epitope/s localize to the prodomain (aa 21–213). The result is shown for the monoclonal antibody (Fig. 1D).
The prodomain binds to native collagen type II
The analysis of the different ADAM15 domains using the SMART program resulted in the prediction of a type II fibronectin collagen‐binding domain (aa 107–134), and a fibronectin type III repeat (aa 65–130), thereby suggesting that the prodomain might confer binding capabilities to ECM molecules such as collagen. To test this hypothesis, the prodomain was recombinantly expressed in E. coli as a fusion protein composed of the prodomain and a Gst‐tag (Pro‐Gst). Figure 2A shows a SDS‐PAGE and the corresponding Western blot developed with an anti‐Gst antibody that stains the Pro‐Gst fusion protein as a 49 kD band and the cleaved off Gst‐tag at 26 kD in the expected molecular weight. Binding of the Pro‐Gst was performed in a Far Western by incubating a solution of the fusion protein with a nitrocellulose filter, to which CII and BSA have been blotted. Strong binding of Pro‐Gst to CII could be detected using antibodies either directed against the prodomain or the Gst‐tag (Fig. 2B and C). However, the detection signal of the CII band using the antibody against the prodomain was somewhat weaker compared with the signal detected by the anti‐Gst, presumably because binding of the prodomain to collagen might affect the accessibility of the anti‐prodomain antibody to its epitope. In addition, binding studies of Pro‐Gst were performed in 96‐well plates coated with native triple helical CII using either anti‐prodomain or anti‐Gst antibodies for detection of the bound fusion protein (Fig. 2B and C). Binding of Pro‐Gst was concentration‐dependent and reached a plateau at 125 nM when developed with anti‐Gst, or 250 nM when detected with anti‐prodomain antibodies. The destruction of the triple helical conformation of CII by heat‐denaturation abrogated the binding of Pro‐Gst completely.
Figure 2.
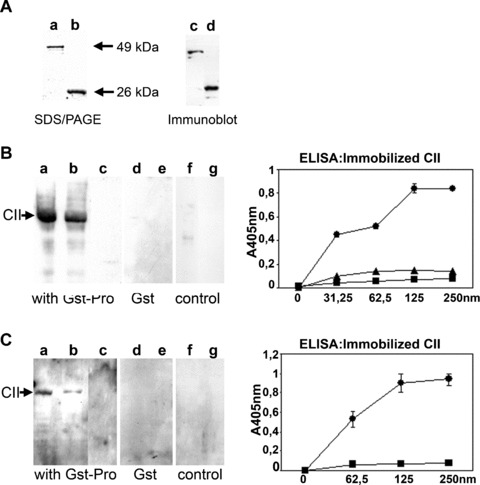
Binding of Pro‐Gst fusion protein to native and denatured collagen type II (CII). (A) SDS/PAGE of the purified Pro‐Gst at 49 kD and Gst‐tag at 26 kD (lanes a, b) and the corresponding immunoblot using anti‐Gst antibodies (lanes c, d). (B, C) Binding of Pro‐Gst to CII and BSA was performed using Far Western Technique. After electrophoretic separation on a 10% polyacrylamide gel, CII (5, 2.5 μg, lanes a, b; 5 μg, lanes d, f) and BSA (5 μg, lanes c, e, g) were transferred to a nitrocellulose filter, incubated with Pro‐Gst and detected with anti‐Gst (B) and anti‐prodomain antibodies (C). Incubation with the Gst‐tag alone is shown in (B, C; lanes d, e). Controls were performed without addition of Pro‐Gst, but using the detection antibodies (B, C; lanes f, g). Using ELISA technique, soluble Pro‐Gst (0–250 nM) was incubated on immobilized native CII (•) or denatured CII (▪) and detected with anti‐Gst (B) or anti‐prodomain antibodies (C). B, ▴ incubation of the Gst‐tag with native CII. Mean values and standard deviation from triplicates are shown.
The prodomain is critical for the reinforcement of collagen adhesion by ADAM15
Chondrocytic cells overexpressing the entire ADAM15 molecule attach to CII significantly better than vector‐transfected control cells [Ref. 14 and Fig. 3A]. This 2–2.5‐fold enhanced binding compared with vector control also applies to chondrocytes transfected with an ADAM15 mutant lacking the complete cytoplasmic domain, indicating that this intracellular part is dispensible for the proadhesive effect of ADAM15 on chondrocyte interaction with CII (Fig. 3B). However, cells expressing a truncated ADAM15 mutant devoid of its prodomain lose their capability of an enhanced collagen binding. The additional deletion of the MP domain (ΔProMP) resulted in a further impairment of CII binding of 25–30% as compared with the vector control (Fig. 3C). All transfected cells did not exhibit any statistically significant difference in their binding ability to fibronectin. No attachment to BSA was observed.
Figure 3.
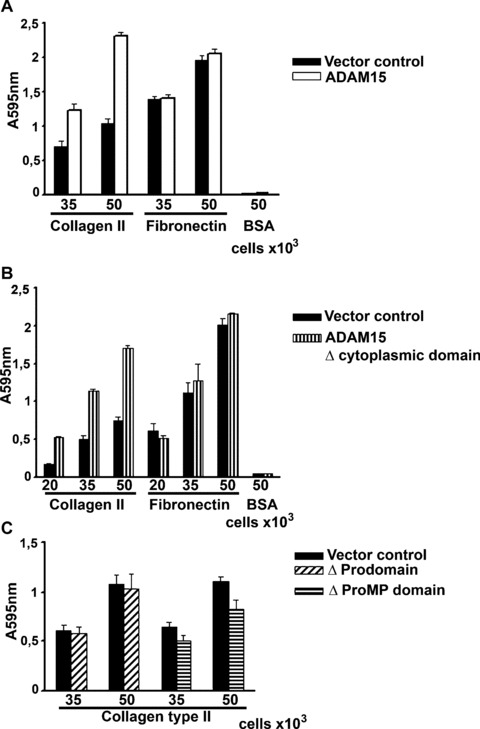
ADAM15 and ADAM15 Δcyto exhibit reinforced binding to collagen type II (CII). Cells were adhered at various densities to CII, fibronectin and BSA. (A) Shown are results of representative adhesion experiments using vector‐transfected control cells (solid bar) and ADAM15‐transfected cells (open bar). (B) Cells expressing only the transmembrane‐anchored extracellular part of ADAM15 (Δcyto) also show stronger binding to CII compared with the control cells. (C) Cells expressing ADAM15 without the prodomain (Δ pro) exhibited the same binding to CII as control, and the additional removal of the MP‐domain (Δ ProMP) consistently reduced cell adhesion to about 25% below the level of vector control cells. All cells transfected with ADAM15 and the deletion constructs did not exhibit enhanced binding to fibronectin compared with the vector control cells and none of the cells exhibited binding to BSA (shown exemplarily for ADAM15‐ and Δcyto‐transfected cells). Values are the mean and standard deviations of quadruplicates.
The cytoplasmic domain is involved in ADAM15‐dependent signal transduction during adhesion to collagen type II
Vector‐, ADAM15‐ and ADAM15 Δ cyto‐transfected cells were plated on CII for 15, 30, 45 and 60 min under strict serum‐free conditions and analysed for tyrosine‐phosphorylated proteins by Western blot with a pan tyr‐phospho antibody. Under these adhesion conditions, the T/C28a4 chondrocytes attach very loosely to the collagen‐coated wells, stay round‐shaped up to 30–40 min. and start to spread very slightly after 60 min. of adhesion (not shown). Differences of staining intensities of phosphoprotein bands between vector control and the ADAM15‐transfected cells were discernible very prominently in the molecular weight ranges of 120–130 kD as well as 60–80 kD (Fig. 4A). The phosphorylation pattern of the cells expressing ADAM15 without the cytoplasmic tail, however, remained unchanged compared with vector control, thus indicating an involvement of the cytoplasmic domain in the transduction of ADAM15‐dependent signals during collagen adhesion (Fig. 4B).
Figure 4.
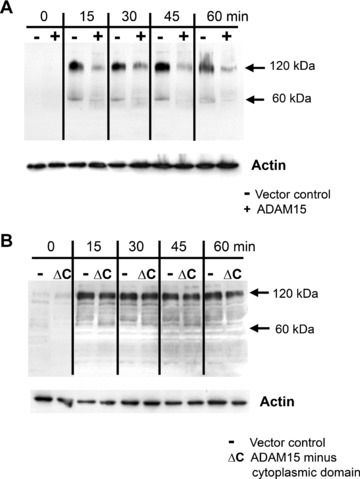
Protein tyrosine phosphorylation during adhesion to collagen type II. ADAM15 and ADAM15 Δcyto and vector‐transfected cells were adhered under serum‐free conditions to CII and lysed after 15, 30, 45 and 60 min. Cell lysates (50 μg) were electrophoresed, blotted and developed using a pan tyrosine phosphorylation antibody (pTyr99). (A) ADAM15‐transfected cells (+) exhibit a prominently reduced phosphorylation signal in the molecular weight range of 120–130 kD compared with vector‐transfected cells (−), whereas (B) ADAM15 Δcyto cells (ΔC) show no different phosphorylation signals intensities in this molecular weight range. Actin served as a loading control. The experiments were repeated at least three times.
FAK is less phosphorylated at its autophosphorylation site in ADAM15‐transfected cells during adhesion to CII
We analysed the possibility of a modulation of the phophorylation state of FAK by ADAM15 overexpression, because FAK plays a pivotal role in adhesion processes and has a known molecular weight of 125 kD corresponding to the above mentioned molecular weight range of ADAM15‐dependent differences in phospho signal intensities. Chondrocytic cells expressing ADAM15, ADAM15 Δ cyto and the vector control were adhered to immobilized CII and the phosphorylation of FAK at its autophosphorylation site Y397 [18] was analysed after 15, 30 and 60 min. of attachment. Both the vector‐ and ADAM15‐transfected cells did activate FAK in the adhesion process, but the phosphorylation signal at Y397 was two‐ to three‐fold lower in the ADAM15‐transfected cells than in the vector control (Fig. 5A). The same set of experiments was performed on cells transfected with the ADAM15 Δ cyto and the signal intensities of the phosphorylated Y397 of FAK remained unchanged compared with the vector control cells (Fig. 5B), indicating in agreement with the above‐described results a critical role of the cytoplasmic domain in ADAM15‐dependent signals during collagen adhesion. To control equal loading, blots were reprobed with anti‐FAK antibodies and the expression of ADAM15 and/or ADAM15 Δ cyto was verified with ADAM15‐specific antibodies.
Figure 5.
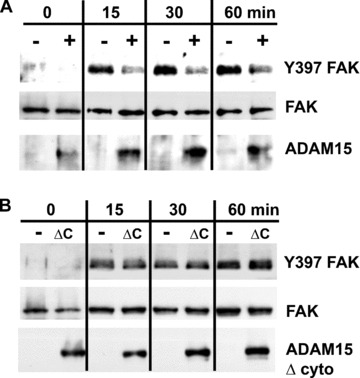
Adhesion to CII results in a reduced phosphorylation of focal adhesion kinase (FAK) at Y397 in ADAM15 expressing cells. Cells were adhered to CII and lysed after 15, 30 and 60 min. Cell lysates (40 μg) were applied to SDS/PAGE and immunoblotted using anti‐pY397 FAK antibodies. (A) ADAM15 (+)‐transfected cells exhibit a 2–3 times reduced phosphorylation of FAK compared with vector control (−) cells, whereas (B) ADAM15 Δ cyto expressing cells (ΔC) show an identical activation of FAK during adhesion. As loading control, all filters were stripped and reprobed with anti‐FAK antibodies and expression of ADAM15 and ADAM15 Δ cyto was controlled using anti‐ADAM15 antibodies. Optical density of the bands was analysed using ImageJ software and normalized to non‐phosphorylated FAK expression.
ADAM15 and FAK co‐immunoprecipitate with each other
Based on the modulatory effect of ADAM15 on FAK phosphorylation, we further analysed the possibility of an underlying protein–protein interaction between both molecules. Accordingly, co‐immunoprecipitation studies were performed on ADAM15‐ and vector‐transfected T/C28a4 chondrocytes using ADAM15‐ and FAK‐specific antibodies for selective protein precipitation and subsequent Western blot analyses. ADAM15 protein can be precipitated specifically only in the ADAM15 expressing cells (+) using the mouse monoclonal anti‐ADAM15 antibody (Fig. 6A, left panel) and accordingly, FAK co‐immunoprecipitates in ADAM15‐ (+), but not in the vector‐transfected cells (–) (Fig. 6A, right panel). Conversely, co‐immunoprecipitation was also performed by capturing FAK with a specific monoclonal antibody in the first step followed by the detection of the FAK‐associated ADAM15 protein in ADAM15‐transfected (+) only, but not in the control cells (–) (Fig. 6B, right panel). The anti‐FAK monoclonal antibody can precipitate FAK from both vector and ADAM15‐transfected cells (Fig. 6B, left panel). Additional experiments demonstrate that the interaction of ADAM15 with FAK is not restricted to the human cell line, but is also detectable in primary human OA chondrocytes obtained from late‐stage disease (Fig. 7B). Although FAK is expressed in normal as well as in OA chondrocytes, ADAM15 expression remains clearly confined to OA chondrocytes (Fig. 7A and ref [9]).
Figure 6.
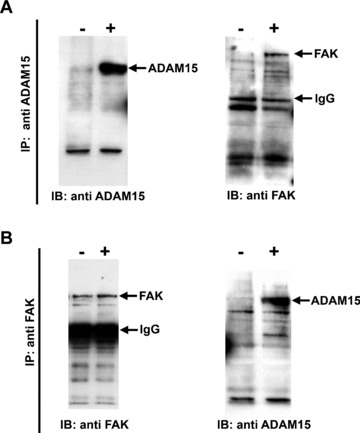
ADAM15 interacts with FAK. Co‐immunoprecipitation (IP) experiments were performed in vector‐ (−) and ADAM15‐transfected cells (+) by using either anti‐ADAM15 or anti‐FAK antibodies for selective protein precipitation followed by immunoblotting (IB) with a polyclonal anti‐ADAM15 antibody directed against the cytoplasmic tail or a monoclonal anti‐FAK. (A) ADAM15 precipitates specifically with the mouse monoclonal anti‐ADAM15 in the ADAM15 expressing cells only (left panel) and FAK is co‐immunoprecipitated with ADAM15 (right panel). (B) Using anti‐FAK antibodies, FAK is precipitated in cell lysates from vector‐ and ADAM15‐transfected cells (left panel) and ADAM15 co‐immunoprecipitates with FAK in ADAM15 expressing cells only (right panel).
Figure 7.
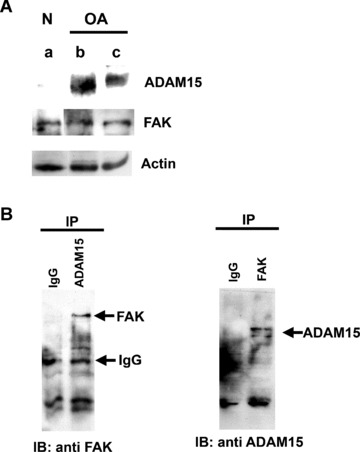
In primary cells from human OA cartilage FAK interacts with ADAM15 that is not expressed in normal human chondrocytes. (A) Cell lysates (50 μg) of normal (lane a) and OA chondrocytes from 2 different donors (lanes b, c) were applied to SDS/PAGE and immunodetected using anti‐ADAM15 and anti‐FAK antibodies. Actin served as a loading control. (B) Left panel, FAK could be co‐immunoprecipitated from pooled lysates of OA‐chondrocytes using anti‐ADAM15 antibodies, right panel, ADAM15 co‐precipitates specifically with FAK. As control isotype specific IgG were used.
Discussion
The recently uncovered proadhesive effects of ADAM15 in chondrocyte binding to CII associated with an anti‐apoptotic function [14] have provoked the present study aiming at the characterization of functional domains that might confer such homeostatic effects of ADAM15 in cell–matrix interactions. The stable expression of ADAM15 as well as certain deletion mutants lacking selected subdomains of the multi‐modular molecule in T/C28a4 chondrocytes allowed us to identify the prodomain as critical for the reinforcement of cell adhesion to CII. Moreover, upon expression as a recombinant bacterial protein a direct interaction with CII could be demonstrated. This result was not a priori predictable despite its accordance with our preceding elucidation of so far unknown potential ECM‐interaction sites by an in silico analysis of the prodomain sequence. Thus, the involvement in cell adhesion is an unexpected new facet of prodomain function because its established role in ADAM family homologues as well as in the closely related matrix metalloproteinases is to keep the MP domain in a catalytically inactive conformation [1]. In addition, there is clear evidence for a role of prodomains in proper protein folding in the endoplasmatic reticulum and for ADAM12 and 17 [19, 20] but not ADAM10 [21]. This chaperone function is even required for cell membrane expression. Although the potential role of the ADAM15‐prodomain as a chaperone is unclear so far, it is however not imperative for ADAM15 cell surface expression. Thus, we could show by cell surface biotinylation that ADAM15 mutants lacking the prodomain (ΔPro and ΔProMP) are expressed on the cell surface (data not shown) in accordance with the previously published data on ADAM10 [21]. Of course, the surface expressions of the ΔPro and ΔProMP constructs do not prove the lack of any chaperone function of the ADAM15 prodomain because our experiments cannot exclude an unproper folding of the mutant proteins in the cell membrane. Accordingly, the ADAM15−MP domain expressed on the surface of cells transfected with the ΔPro constructs could be affected by the misfolding as well. Because an unproperly folded MP domain might also contribute to the observed loss of enhanced collagen binding in the respective transfectants, our results do not allow for the conclusion that the collagen‐interacting properties of ADAM15 reside exclusively in its prodomain. However, in accordance with the adhesion experiments in the ΔPro‐transfected chondrocytes the direct collagen binding of the recombinant prodomain clearly supports its functional role in matrix interactions. The prodomain in ADAM15 spans a region of approximately 200 amino acid residues and sequence comparison with the prodomains of other ADAMs reveals only a minor degree of homology (about 30% sequence identity and a maximum of 40–50% homology), suggesting that the newly unravelled function in cell adhesion might remain a unique ADAM15‐specific property. A likewise new function to the well‐known role in preserving the inactive conformation of the MP domain has more recently been described for the ADAM10 prodomain. Thus, the soluble recombinant ADAM10 prodomain was shown to efficiently and specifically inhibit ADAM10 proteolytic activity thereby proving its regulatory capacity on cellular shedding events even after removal from the catalytic domain upon activation [22]. Although our results using the overexpression of deletion mutants that lack the prodomain provide unequivocal evidence for its critical role in the reinforcement of adhesion to CII, analogous studies on the cytoplasmic tail revealed its dispensibility in this respect. It is therefore indicated that an outside‐in signal via the cytoplasmic domain is not required for the proadhesive effect of ADAM15 that is conferred by its ectodomain. However, the cytoplasmic tail that harbours signalling motifs, such as src homology 3 (SH3) ligand domains, has already been characterized for its potential to participate in phosphorylation‐dependent interactions with src family protein tyrosine kinases [23, 24]. Accordingly, our studies clearly show that during chondrocyte adhesion to CII the pattern of protein tyrosine phosphorylation is altered in ADAM15 overexpressing T/C28a4 cells in dependency on the presence of the cytoplasmic domain. Moreover, our studies provide first experimental evidence for the modulation of CII adhesion‐dependent FAK phosphorylation by ADAM15. FAK is ubiquitously expressed and central to the integration of growth factor receptor and ECM signals by forming a signalling scaffold that is involved in growth, differentiation and survival pathways in many different cell types [18, 25]. FAK is maintained in an inactive and autoinhibited state and gets activated via autophosphorylation at tyrosine 397 (Y397) by the engagement of integrins with the ECM [26]. Phosphorylated Y397 provides a binding site for c‐Src, and the subsequent interaction of Src with FAK sustains both protein kinases in their activated states thereby giving rise to the initiation of further downstream signalling cascades [27]. Contrary to the expectations based on the well‐established FAK activation by integrin‐mediated ECM engagement, the enhanced binding of ADAM15‐transfected chondrocytes to CII is not mirrored by an enhanced phosphorylation of FAK during CII‐attachment. Our results provide clear evidence for the down‐regulation of the phosphorylation signal at the critical autophosphorylation site Y397 of FAK by ADAM15 during CII adhesion thereby indicating a decoupling of cell attachment from FAK signalling. The reciprocal co‐immunoprecipitation of ADAM15 and FAK not only in T/C28a4 cells but also in primary human chondrocytes from OA cartilage suggests their presence in a protein complex that is likely formed during CII adhesion in the focal contacts thereby leaving the possibility of a direct protein–protein interaction an open option although it cannot be excluded that one or more bridging intermediate protein(s) might be involved as well.
Because FAK phosphorylation is a part of the integrin‐mediated mechanotransduction [28], it is conceivable that the newly uncovered modulatory effect of ADAM15 might affect the perception of the mechanical forces in the cartilage matrix by the chondrocyte. Thus, it remains an intriguing possibility that the up‐regulation of ADAM15 in degenerative joint disease [10] might contribute to an adaptation of the outside‐in signalling in the chondrocytes to the elevated mechanical forces that arise from the increased catabolism in the ECM already at early stages of OA [29]. Although this scenario is conceivable to apply to the attenuation of the FAK‐Y397 phosphorylation signal in integrin‐dependent collagen adhesion, it is unlikely that this hypothesis could explain all facets of ADAM15‐mediated cell protective effects, e.g. the enhanced resistance to growth factor starvation and will have to be addressed in future studies. In addition, functional studies of genetically manipulated chondrocytes imply that not all phenotypic characteristics of articular chondrocytes remain conserved under such experimental conditions. Accordingly, the respective conclusions on OA‐pathogenesis inferred by the present investigations in a human chondrocyte line remain somehow restrained by the respective limitations of the in vitro model.
The present investigation, however, provides unequivocal evidence for a so far unknown CII‐binding property of the ADAM15 prodomain and its crucial role in the reinforced collagen adhesion of ADAM15 overexpressing chondrocytes. Moreover, our studies uncover in these cells an uncoupling between the enhanced CII adhesion and a concomitantly reduced phosphorylation signal at the critical FAK‐autophosphorylation site Y397. The proadhesive effect associated with the prodomain and the alteration of outside‐in signalling conferred by the cytoplasmic tail are likely contributing to the earlier described [14] homeostatic functions of ADAM15 in the matrix metabolism of OA cartilage.
Acknowledgements
The authors are grateful to Dr. M.B. Goldring (Hospital for Special Surgery, New York, NY, USA) for providing the chondrocyte cell line T/C28a4 and Dr. Thomas Aigner for his support with the experiments on primary human chondrocytes. We thank Barbara Roy for excellent technical assistance. Supported by a grant from the Interdisciplinary Clinical Research Center at the Medical Faculty of the Friedrich‐Alexander‐University Erlangen– Nuremberg, Project C3.
References
- 1. Tousseyn T, Jorissen E, Reiss K, et al . (Make) Stick and cut loose‐disintegrin metalloproteinases in development and disease. Birth Defects Res . 2006; 78: 24–46. [DOI] [PubMed] [Google Scholar]
- 2. Blobel CP. ADAMs: key components in EGFR signalling and development. Nat Rev Mol Cell Biol . 2005; 6: 32–43. [DOI] [PubMed] [Google Scholar]
- 3. White JM. ADAMs: Modulators of cell‐cell and cell‐matrix interactions. Curr Opin Cell Biol . 2003; 15: 598–606. [DOI] [PubMed] [Google Scholar]
- 4. Nath D, Slocombe PM, Stephens PE, et al . Interaction of metargidin (ADAM‐15) with alphavbeta3 and alpha5beta1 integrins on different haemopoietic cells. J Cell Sci . 1999; 112: 579–87. [DOI] [PubMed] [Google Scholar]
- 5. Eto K, Huet C, Tarui T, et al . Functional classification of ADAMs based on a conserved motif for binding to integrin alpha 9beta 1: implications for sperm‐egg binding and other cell interactions. J Biol Chem . 2002; 277: 17804–10. [DOI] [PubMed] [Google Scholar]
- 6. Seals DF, Courtneidge SA. The ADAMs family of metalloproteinases: multidomain proteins with multiple functions. Genes Dev . 2003; 17: 7–30. [DOI] [PubMed] [Google Scholar]
- 7. Mochizuki S, Okada Y. ADAMs in cancer cell proliferation and progression. Cancer Sci . 2007; 98: 621–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Herren B, Raines EW, Ross R. Expression of a disintegrin‐like protein in cultured human vascular cells and in vivo . FASEB J . 1997; 11: 173–80. [DOI] [PubMed] [Google Scholar]
- 9. Böhm BB, Aigner T, Gehrsitz A, et al . Up‐regulation of MDC15 (metargidin) messenger RNA in human osteoarthritic cartilage. Arthritis Rheum . 1999; 42: 1946–50. [DOI] [PubMed] [Google Scholar]
- 10. Böhm BB, Aigner T, Blobel CP, et al . The expression of the disintegrin‐metalloproteinase MDC15 (metargidin) is highly enhanced in rheumatoid synovial tissue. Arthritis Rheum . 2001; 44: 2046–54. [DOI] [PubMed] [Google Scholar]
- 11. Martin J, Eynstone LV, Davies M, et al . The role of ADAM15 in glomerular mesangial cell migration. J Biol Chem . 2002; 277: 33683–9. [DOI] [PubMed] [Google Scholar]
- 12. Huovila AP, Turner AJ, Pelto‐Huikko M, et al . Shedding light on ADAM metalloproteinases. Trends Biochem Sci . 2005; 30: 413–22. [DOI] [PubMed] [Google Scholar]
- 13. Hart S, Fischer OM, Prenzel N, et al . GPCR‐induced migration of breast carcinoma cells depends on both EGFR signal transactivation and EGFR‐independent pathways. Biol Chem . 2005; 386: 845–55. [DOI] [PubMed] [Google Scholar]
- 14. Böhm BB, Aigner T, Roy B, et al . Homeostatic effects of the metalloproteinase disintegrin ADAM15 in degenerative cartilage remodeling. Arthritis Rheum . 2005; 52: 1100–9. [DOI] [PubMed] [Google Scholar]
- 15. Goldring MB, Birkhead JR, Suen LF, et al . Interleukin‐1‐β‐modulated gene expression in immortalized human chondrocytes. J Clin Invest . 1994; 94: 2307–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhiyong F, Bau B, Yang H, et al . Freshly isolated osteoarthritic chondrocytes are catabolically more active than normal chondrocytes, but less responsive to catabolic stimulation with interleukin‐1b. Arthritis Rheum . 2005; 52: 136–43. [DOI] [PubMed] [Google Scholar]
- 17. Schultz J, Milpetz F, Bork P, et al . SMART, a simple modular architecture research tool: identification of signaling domains. Proc Natl Acad Sci USA . 1998; 95: 5857–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Parsons JT. Focal adhesion kinase: the first ten years. J Cell Sci . 2003; 116: 1409–16. [DOI] [PubMed] [Google Scholar]
- 19. Milla ME, Leesnitzer MA, Moss ML, et al . Specific sequence determinants are required for the expression of functional tumor necrosis factor‐α converting enzyme (TACE). J Biol Chem . 1999; 274: 30563–70. [DOI] [PubMed] [Google Scholar]
- 20. Loechel F, Overgaard MT, Oxvig C, et al . Regulation of human ADAM 12 protease by the prodomain. J Biol Chem . 1999; 274: 13427–33. [DOI] [PubMed] [Google Scholar]
- 21. Anders A, Gilbert S, Garten W, et al . Regulation of the alpha‐secretase ADAM10 by its prodomain and proprotein convertases. FASEB J . 2001; 15: 1837–9. [DOI] [PubMed] [Google Scholar]
- 22. Moss ML, Bomar M, Liu Q, et al . The ADAM10 prodomain is a specific inhibitor of ADAM10 proteolytic activity and inhibits cellular shedding events. J Biol Chem . 2007; 282: 35712–21. [DOI] [PubMed] [Google Scholar]
- 23. Poghosyan Z, Robbins SM, Houslay MD, et al . Phosphorylation‐dependent interactions between ADAM15 cytoplasmic domain and Src family protein‐tyrosine kinases. J Biol Chem . 2002; 277: 4999–5007. [DOI] [PubMed] [Google Scholar]
- 24. Howard L, Nelson KK, Maciewicz RA, et al . Interaction of the metalloproteinase disintegrins MDC9 and MDC15 with two SH3 domain‐containing proteins, endophilin I and SH3PX1. J Biol Chem . 1999; 274: 31693–9. [DOI] [PubMed] [Google Scholar]
- 25. Wozniak MA, Modzelewska K, Kwong L, et al . Focal adhesion regulation of cell behaviour. Biochimica et Biophysica Acta . 2004; 1692: 103–19. [DOI] [PubMed] [Google Scholar]
- 26. Kornberg L, Earp HS, Parsons JT, et al . Cell adhesion or integrin clustering increases phosphorylation of a focal adhesion‐associated tyrosine kinase. J Biol Chem . 1992; 33: 23439–42. [PubMed] [Google Scholar]
- 27. Calalb MB, Polte TR, Hanks SK. Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: a role for Src family kinases. Mol Cell Biol . 1995; 1: 954–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Millward‐Sadler SJ, Salter DM. Integrin‐dependent signal cascades in chondrocyte mechanotransduction. Ann Biomed Engin . 2004; 32: 435–46. [DOI] [PubMed] [Google Scholar]
- 29. Loeser RF. Molecular mechanisms of cartilage destruction: mechanics, inflammatory mediators, and aging collide. Arthritis Rheum . 2006; 54: 1357–60. [DOI] [PMC free article] [PubMed] [Google Scholar]