

Abstract
The standardised Ginkgo biloba extract EGb® 761 (Dr. Willmar Schwabe Pharmaceuticals, Karlsruhe, Germany) is one of the most widely used herbal remedies. Indications for this extract range from dementia to peripheral vascular disease, based on well‐documented vascular effects. Surprisingly, the actions of EGb® 761 on angiogenesis as a function of vascular cells have not been investigated to date. The anti‐cancer activity of EGb® 761 in vitro and epidemiological data showing reduced risk for ovarian cancer in regular users have prompted us to investigate this issue. We show an anti‐angiogenic profile of EGb® 761 in vitro (inhibited proliferation, migration and tube formation of endothelial cells) and in vivo in the chicken chorio‐allantoic membrane (CAM) assay. An analysis of the underlying mechanisms indicates inhibition of growth factor‐induced extracellular signal‐regulated kinase (ERK) phosphorylation by EGb® 761. Inhibitory effects of EGb® 761 on ERK as well as of the upstream kinases map‐erk‐kinase (MEK) and rapidly growing fibrosarcoma (Raf)‐1 could be completely reversed by pre‐treatment with sodium vanadate (inhibitor of tyrosine phosphatases). Sodium vanadate also reversed the EGb® 761‐induced inhibition of endothelial cell migration. Focusing on tyrosine phosphatases upstream of the Raf‐MEK‐ERK cascade, we identified the tyrosine phosphatase Src homology‐2 domain‐containing phosphatase 1 (SHP‐1) as one target of EGb® 761. SHP‐1 was rapidly activated by EGb® 761, and silencing SHP‐1 (siRNA) abrogated reduction of endothelial proliferation by EGb® 761. In summary, we identify EGb® 761 as a potent anti‐angiogenic drug. The underlying mechanism is the activation of protein tyrosine phosphatases, leading to inhibition of the Raf‐MEK‐ERK pathway. These findings provide a rational basis for using EGb® 761 for an additional therapeutic indication: anti‐angiogenesis‐based tumour prevention and adjuvant therapy.
Keywords: Ginkgo biloba, angiogenesis, ERK, tyrosine phosphatase, SHP‐1
Introduction
The defined and standardised Ginkgo biloba (G. biloba) extract EGb® 761 (Dr. Willmar Schwabe Pharmaceuticals, Karlsruhe, Germany) is a herbal remedy that is clinically well established for the treatment of dementia of various origins and peripheral vascular diseases (e.g. Raynaud's syndrome, tinnitus and peripheral artery occlusion disease). The following two points have prompted us to investigate a possible anti‐angiogenic action of the extract EGb® 761 and identify the underlying molecular mechanisms and target molecules: (1) the action of this extract on endothelial cells (e.g. up‐regulation of endothelial nitric oxide production via an increase of endothelial nitric oxide synthase (eNOS) expression and phosphoinositol‐3 dependent kinase (PI(3)K) protein kinase B (Akt)‐driven activity [1]) and (2) epidemiological data supporting an association between regular use of G. biloba and a decreased risk for ovarian cancer [2]. We deemed this of interest because no data on this issue exist to date, and, since a major challenge to modern phytomedicine is to provide a rational basis and the molecular mechanisms for herbal drugs, which receive growing popularity as a ‘mild and natural’ alternative to ‘chemical’ drugs.
The focus of the present study lies on the influence of EGb® 761 on growth factor‐signalling pathways in endothelial cells, concentrating on the ERK‐cascade and the role of phosphatases for the following reasons: Tumour development and progression beyond a size of few millimetres‐cubed crucially depends on the onset of angiogenesis, that is, the ‘angiogenic switch’[3]. Angiogenesis is the formation of new blood vessels by sprouting from pre‐existing capillaries, and is a pre‐requisite for the supply of fresh nutrients and oxygen responsible for tumour growth. Inhibition of angiogenesis, therefore, represents a valid approach for cancer therapy or even prevention and is successfully employed in clinical treatment. Amongst the drugs used are small molecular compounds targeting receptor tyrosine kinases (RTKs), such as gefitinib (Iressa®, AstraZeneca, Wilmington, DE, USA), as well as therapeutic antibody‐binding growth factors or their receptors, such as bevacizumab (Avastin®, Roche, Basel, Switzerland). Angiogenesis is mainly triggered by the secretion of pro‐angiogenic factors, such as vascular endothelial growth factor (VEGF) and fibroblast growth factor (FGF). These growth factors bind and activate specific RTKs in endothelial cells, leading to the activation of signalling cascades. Activation of the extracellular signal‐regulated kinase (ERK) plays an important role in the regulation of angiogenesis, including the steps of cell proliferation, migration and tube formation [4]. The classical pathway of ERK activation is mediated by the Ras‐Raf‐MEK signal transduction cascade, which is regulated by a number of signalling molecules, most notably, nitric oxide, protein kinase B/Akt and cyclic AMP [5, 6, 7, 8]. Furthermore, ERK phosphorylation is regulated by a delicate balance between the action of protein kinases and protein phosphatases. Negative regulation of RTKs through rapid dephosphorylation by tyrosine‐specific phosphatases is important for limiting the respective response. Recent evidence has shown that numerous phosphatases exist that counteract the activity of VEGF and FGF receptor signalling and additionally function as tumour suppressors [9]. In the present work, we demonstrate that the standardised G. biloba extract EGb® 761 activates protein tyrosine phosphatases (amongst others, Src homology‐2 domain‐containing phosphatase 1 [SHP‐1]) in endothelial cells, resulting in an inhibition of the Raf‐MEK‐ERK pathway. This action is linked to an anti‐angiogenic response, both in vitro and in vivo.
Materials and methods
Cell culture and EGb 761
The HMEC‐1 cell line (immortalised human microvascular dermal endothelial cells [HMEC]) was kindly provided from the Center for Disease Control and Prevention (Atlanta, GA, USA) [10]. Cells were cultured in an endothelial cell growth medium (ECGM) (Promocell, Heidelberg, Germany). Human umbilical vein endothelial cells (HUVECs) were isolated and cultured by a standard procedure, as described previously [11].
EGb 761 has been developed and has become commercially available as drops and tablets under the trade name Tebonin® (Dr. Willmar Schwabe Pharmaceuticals, Karlsruhe, Germany). EGb 761 is a water‐acetone extract that has been stringently standardised to ensure the consistency of its composition and reliable safety and efficacy profiles. The standard is a ratio of 35–67:1 (dried G. biloba leaves to final extract) containing approximately 24% flavonoid glycosides, 6% terpene trilactones, 7% proanthocyanidins and certain low‐molecular‐weight organic acids. Terpene trilactones are unique to G. biloba and can be divided into different subgroups: ginkgolides A, B, C and J (diterpenoids) as well as the bilobalide (sesquiterpene). Moreover, EGb 761 is standardised to contain less than 5 p.p.m. ginkgolic acids. EGb 761 was always freshly dissolved in growth medium.
Proliferation
In total, 1.5 × 103 HMECs were cultured for 24 hrs and treated with EGb® 761 for 72 hrs. The cells were stained with crystal violet solution (0.5%[w/v] crystal violet, 20%[v/v] methanol) for 10 min. The bound dye was solubilised with a dissolving buffer (50% sodium citrate 0.1 M, 50% ethanol). Absorbance was measured at 540 nm in a photometer (SPECTRAFluor Plus; Tecan, Crailsheim, Germany).
Quantification of apoptosis
Staining of cells with propidium iodide was performed according to Nicoletti et al.[12]. Briefly, HMECs were incubated in a fluorochrome solution (0.1%[m/v] sodium citrate, 0.1%[v/v] Triton X‐100 and 50 μg/ml propidium iodide in PBS) overnight at 4°C. The samples were analysed on a FACSCalibur (Becton Dickinson, Heidelberg, Germany), as described previously [13].
Migration
Confluent HUVEC monolayers were scratched and immediately treated either with M199 (negative control; PAN Biotech, Aidenbach, Germany) or with ECGM containing different stimuli. After 16 hrs, images were taken with a TILLvisiON system (TILL Photonics, Lochham, Germany) connected to an Axiovert 200 microscope (Zeiss, Göttingen, Germany). Migration was expressed as the ratio of pixels covered by cells to the total number of pixels in the wound area (S.CO LifeScience, Garching, Germany).
Tube formation
ibidi μ‐slides (18‐well; ibidi GmbH, Munich, Germany) were coated with Matrigel® (Schubert & Weiss‐OMNILAB, Munich, Germany). A medium containing 1 × 104 HMECs and EGb® 761 was added to the wells. After 16 hrs, images were taken using the TILLvisiON system.
CAM (chorio‐allantois membrane) assay
Fertilised White Leghorn chicken eggs from Lohmann Tierzucht (Cuxhaven, Germany) were incubated at 37°C for 72 hrs. The eggs were transferred into dishes, incubated for further 72 hrs and stimulated with either FGF (1 ng/dish) alone or EGb® 761 (1.25, 2.5 and 3.75 μg/disk, respectively) and FGF. On the next day, CAMs were photographed using a stereomicroscope (Olympus, Munich, Germany).
Western blot analysis
Confluent HMECs or HUVECs were starved in M199 for 4 hrs and subsequently exposed to EGb® 761 (500 μg/ml, 30 min.) before FGF (5 ng/ml, 30 min.) or VEGF (50 ng/ml, 10 min.) treatment. Western blot analysis was performed as described previously [1]. Protein bands of interest were detected using an Odyssey imaging system (Li‐Cor Biosciences, Lincoln, NE, USA).
Raf‐1 kinase assay
Raf‐1 kinase activity was analysed with non‐radioactive Raf kinase assay kit (Upstate, Lake Placid, NY, USA), according to the manufacturer's instructions. HUVECs were lysed with M‐Per® (mammalian protein extraction reagent), additionally containing Complete® 8% (Roche, Mannheim, Germany), phenylmethylsulphonyl fluoride (PMSF) 2 mM, NaF 2 mM, activated Na3VO4 2 mM, sodium pyrophosphate 5 mM and sodium glycerophosphate 4 mM. Proteins were incubated with 1 μg inactive MEK‐1, Mg2+ (75 mM) and ATP (500 mM) (30°C, 30 min.). Laemmli buffer was added, samples were heated (95°C, 5 min.) and phosphorylated MEK‐1, a parameter for Raf‐1 kinase activity, was detected by Western blot analysis.
SHP‐1 phosphatase assay
After incubation with EGb 761 for 30 min., HUVECs were washed twice with ice‐cold PBS to remove the extract and subsequently lysed with SHP‐1 lysis buffer containing 20 mM Tris‐HCl (pH 7.35), 1 mM EDTA, 1% Triton X‐100, 0.5% deoxycholat, 1 mM pefabloc, 1 μM leupeptin, 1 μM pepstatin and 1 μM aprotinin. SHP‐1 was immunoprecipitated with μMACS protein G micro‐beads (Miltenyi Biotec, Bergisch Gladbach, Germany) and equilibrated in the phosphatase assay buffer (HEPES 20 mM, NaCl 100 mM and MgCl2 5 mM, pH 5). One hundred millimolar para‐nitrophenyl phosphate (pNPP; Sigma‐Aldrich, Taufkirchen, Germany) was added for 1 hr (37°C) to allow formation of p‐nitrophenylene anion by dephosphorylation of pNPP by SHP‐1. The reaction was terminated with 1 M NaOH. Absorbance was measured at 405 nm.
SHP‐1 siRNA
HUVECs (2 × 106 cells) were transfected with 3.0 μg ON‐TARGETplus SHP‐1 siRNA (J‐009778–09: 1.5 μg 5′‐PUAUGGGACGCAUUUGUUCCUU‐3′ and J‐009778–10: 1.5 μg 5′‐PUAAGGUACGGAGUUUGUAUUU‐3′, respectively; Dharmacon, Lafayette, CO, USA) by electroporation with the Nucleofector™ II (Amaxa, Cologne, Germany), according to the manufacture's protocol. Concurrently, HUVECs were transfected with non‐targeting siRNA (D‐001810–01, sequence: 5′‐UGGUUUACAUGUCGACUAA‐3′), using it as transfection control. Experiments were started 24 hrs after transfection. For the proliferation assay, 1.0 × 104 transfected cells were seeded into flat‐bottomed 96‐well plates. Furthermore, transfection efficiency was determined by Western blot analysis using antibodies against rabbit polyclonal anti‐SH‐PTP1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA).
Data analysis
Data are presented as the mean ± standard error of mean (SEM) of three independent experiments. To compare three or more groups, one‐way ANOVA followed by the Student–Newman–Keuls post‐hoc test was performed. The differences between groups were considered significant if P≤ 0.05.
Results
EGb® 761 exerts anti‐angiogenic effects in vitro and in vivo
EGb® 761 (50–500 μg/ml, 72 hrs) reduces endothelial cell proliferation (Fig. 1A) to 56% of untreated controls after 72 hrs (500 μg/ml) in a concentration‐dependent manner. Furthermore, EGb® 761 does not possess considerable apoptotic activity on endothelial cells, as it does not affect confluent endothelial cells and affects proliferating cells only at the highest concentration of 500 μg/ml (3.6‐fold, data not shown). These findings indicate that the reduced number of cells is due to a strong anti‐proliferative effect, rather than an induction of apoptosis. Endothelial cell migration as examined in the scratch assay was significantly inhibited (Fig. 1B). In comparison to untreated controls (endothelial cell growth medium), the percentage of cell‐covered area in relation to the total image area was reduced to 77% and 72% with EGb® 100 μg/ml and 500 μg/ml, respectively. Incubation of HMECs with G. biloba extract on Matrigel® (100 μg/ml and 500 μg/ml, respectively) led to a significant reduction in tube formation (Fig. 1C). Most importantly, these anti‐angiogenic properties were confirmed in vivo, performed with the CAM assay. In this assay, we observed a strong induction of angiogenesis with FGF and a complete inhibition of this effect by EGb® 761 (Fig. 1D). These data for the first time report an anti‐angiogenic effect of EGb® 761 in vitro and in vivo.
Figure 1.
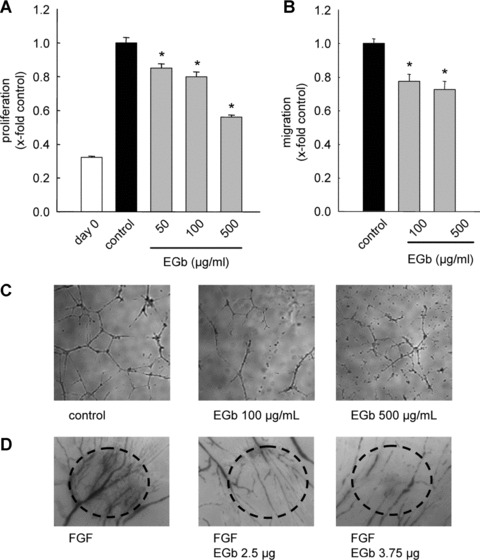
EGb® 761 has anti‐angiogenic properties in vitro and in vivo. EGb® 761 inhibits proliferation (A), migration (B), tube formation (C) and angiogenesis in the CAM assay (D). (A) HMECs were stimulated with different concentrations of EGb® 761 for 72 hrs. (B) A confluent HUVEC monolayer was scratched and exposed to ECGM, starvation medium (M199) or ECGM containing EGb® 761 for 16 hrs. (C) HMECs were seeded on Matrigel® and were either left untreated (control) or treated with EGb® 761 for 16 hrs. (D) Fertilised eggs were stimulated with FGF (1 ng/dish) alone or FGF and EGb® 761. The dotted circles indicate the position of the dish carrying the growth factor and EGb® 761.
ERK inhibition abrogates angiogenesis
In order to confirm the influence of ERK on angiogenic parameters in our setting, we investigated whether U0126, a specific MEK‐1/2 inhibitor, shows an anti‐angiogenic profile. Indeed, U0126 (10 μM, 72 hrs) lowers cell proliferation in HMEC‐1 by 32% (Fig. 2A). Furthermore, U0126 (10 μM, 16 hrs) showed a reduction in both angiogenic parameters cell migration (Fig. 2B) and tube formation (Fig. 2C). In addition to these in vitro experiments, ERK inhibition by U0126 led to a diminished number of vascular structures in the CAM assay (Fig. 2D). These data support the hypothesis that ERK indeed plays a crucial role in angiogenesis.
Figure 2.
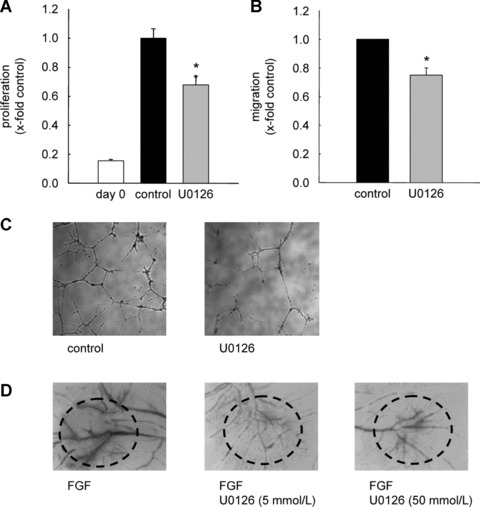
ERK inhibition exerts anti‐angiogenic effects. (A) HMECs were either kept untreated (control) or stimulated with U0126 (10 μM). (B) HUVEC monolayers were wounded and stimulated with ECGM alone or a medium containing U0126 (10 μM) for 16 hrs. *P < 0.05 versus control, Kruskal–Wallis one‐way ANOVA on ranks (Dunn's method). (C) HMECs were incubated with U0126 (10 μM). After 16 hrs, tube formation was observed using the TILLvisON system. (D) One representative image of the chicken CAM is shown. Fertilised eggs were stimulated either with FGF (1ng/dish) alone or with FGF and U0126.
EGb® 761 inhibits growth factor‐induced ERK phosphorylation
To investigate the influence of EGb® 761 on ERK phosphorylation, endothelial cells were pre‐treated with EGb® 761 (500 μg/ml, 30 min.) before exposure to FGF (5 ng/ml, 30 min.). EGb® 761 completely abolished FGF‐induced ERK phosphorylation both in HMECs (Fig. 3A) and in HUVECs (Fig. 3B). Moreover, EGb® 761 also suppressed VEGF‐induced ERK phosphorylation (Fig. 3C and D, respectively). Under basal conditions, EGb® 761 had no effect (Fig. 3A–D). These results suggest that EGb® 761 abrogates ERK phosphorylation induced by a number of growth factors.
Figure 3.
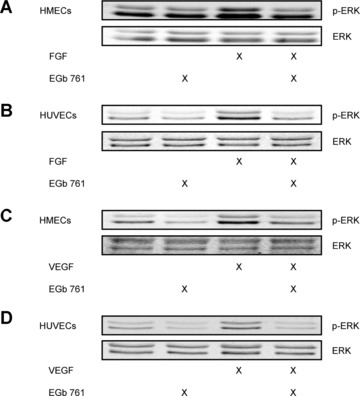
EGb® 761 abrogates growth factor‐induced ERK phosphorylation in ECs. HMECs (A) or HUVECs (B) were pre‐treated with EGb® 761 (500 μg/ml, 30 min.) and subsequently exposed to FGF (5 ng/ml, 30 min.). HMECs (C) or HUVECs (D) were pre‐treated with EGb® 761 (500 μg/ml, 30 min.) before VEGF (50 ng/ml, 10 min.). The levels of phospho‐ERK and total ERK protein were determined by Western blot analysis.
EGb® 761 blocks the Raf‐MEK‐ERK‐pathway via activation of tyrosine phosphatases
The reduction of ERK phosphorylation by EGb® 761 could be explained either by an inhibition of upstream kinases or by an activation of respective phosphatases. In order to investigate an activation of phosphatases by EGb® 761, phosphatase inhibitors were used. Pre‐treatment of HMECs with NaF (serine/threonine phosphatase inhibitor) did not reverse the effects of EGb® 761 on ERK phosphorylation (Fig. 4A), whereas incubation with Na3VO4 (tyrosine phosphatase inhibitor) before exposure to EGb® 761 and FGF blocked the effect of G. biloba extract (Fig. 4B). Thus, EGb® 761 abolished growth factor‐induced ERK phosphorylation in endothelial cells via induction of tyrosine phosphatase activity.
Figure 4.
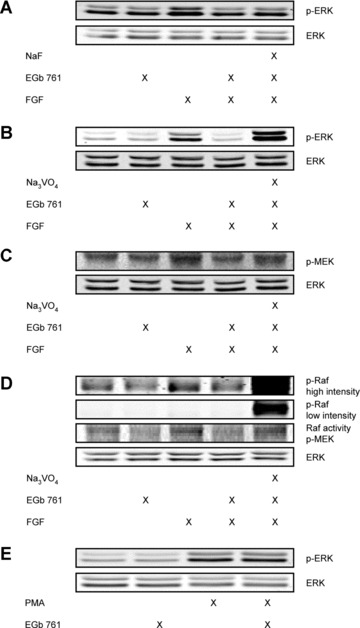
EGb® 761 suppresses ERK, MEK and Raf‐1 kinase activity via activation of tyrosine phosphatases. HMECs were pre‐treated with NaF (1 mM, 30 min.; A) or Na3VO4 (100 μM, 30 min.; B). Subsequently, the cells were stimulated with EGb® 761 (500 μg/ml, 30 min.) and FGF (5 ng/ml, 30 min.). (C) HUVECs were pre‐incubated with Na3VO4 and stimulated with EGb® 761 and FGF. (D) The stimulation scheme was the same as in (C). Representative Western blots are shown for phospho‐Raf‐1 (Ser338/Tyr341) at both high and low intensity. Additionally, Raf kinase activity (indicated as phospho‐MEK) was determined. (E) HMECs were pre‐treated with EGb® 761 and subsequently exposed to PMA (50 nM, 10 min.).
To identify potential targets of phosphatases in ERK signalling, we examined the activation of the upstream kinases MEK‐1/2 and Raf‐1 using Western blot analysis and a kinase activity assay, respectively. EGb® 761 completely abolished FGF‐induced MEK‐1/2 phosphorylation at Ser217/221 in HUVECs. Moreover, the effect of EGb® 761 on MEK‐1/2 phosphorylation was reversed by pre‐incubation with the tyrosine phosphatase inhibitor sodium vanadate (Fig. 4C). Furthermore, we observed a reduction of FGF‐induced Raf‐1 activity and phosphorylation at Ser338/Tyr341 by EGb® 761. Again, pre‐treatment with sodium vanadate blocked Raf‐1 activation as well as phosphorylation (Fig. 4D). These data indicate that G. biloba extract inhibits the growth factor signalling upstream of the classical Raf‐MEK‐ERK pathway.
To confirm an involvement of protein tyrosine phosphatases linked with VEGF and FGF receptors, the cells were treated with EGb® 761 and phorbol‐12 myristate‐13‐acetate (PMA). PMA leads to a growth factor‐independent activation of the MAPK signalling cascade Raf‐MEK‐ERK. As expected, EGb® 761 did not suppress the PMA‐induced ERK phosphorylation (Fig. 4E). These results suggest that EGb® 761 terminates ERK phosphorylation by activation of one or more tyrosine phosphatases linked with GF receptors, but not by influencing the receptor activities themselves.
The anti‐angiogenic effect of EGb® 761 depends on the activation of protein tyrosine phosphatases
To causally link the activation of tyrosine phosphatases to the anti‐angiogenic effects of EGb® 761, endothelial cell migration was analysed in the presence of sodium vanadate. Pre‐treatment of endothelial cells with sodium vanadate (10 nM, 30 min.) before exposure to EGb® 761 (100 and 500 μg/ml, respectively) for 16 hrs reversed the inhibitory effects of EGb® 761 on endothelial cell migration (Fig. 5). These data provide further evidence that EGb® 761 has its anti‐angiogenic properties via an activation of tyrosine phosphatases.
Figure 5.
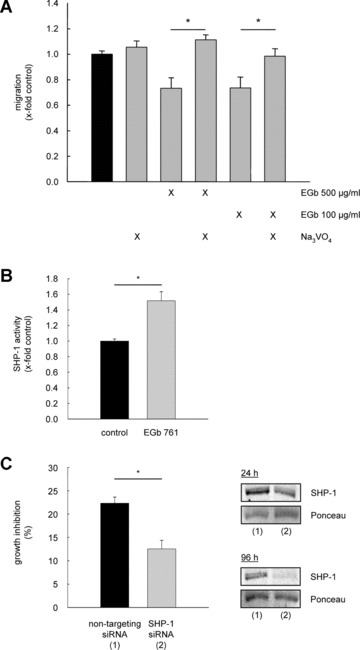
Induction of tyrosine phosphatase activity by EGb® 761 leads to the inhibitory effect on angiogenic parameters. (A) HUVEC monolayers were wounded and pre‐treated with Na3VO4 (10 mM, 30 min.) before stimulation with EGb® 761 for further 16 hrs. *P < 0.05 versus EGb® 761, with a concentration of 100 and 500 μg/ml, respectively. (B) HUVECs were either left untreated (co) or treated with EGb® 761 for 30 min. and subsequently washed and lysed. SHP‐1 activity was determined as described in Materials and Methods. *P < 0.05 versus control, Kruskal–Wallis one‐way ANOVA on ranks (Dunn's method). (C) HUVECs were either transfected with non‐targeting siRNA (1) or SHP‐1 siRNA (2). After 24 hrs, the cells were stimulated with EGb® 761 for 72 hrs. *P < 0.001 for SHP‐1 siRNA versus non‐targeting siRNA, Student's t‐test. The right panel shows reduced SHP‐1 protein levels in transfected cells after 24 and 96 hrs.
EGb® 761 inhibits endothelial proliferation via activation of SHP‐1
An important protein tyrosine phosphatase in FGF and VEGF signalling is the cytoplasmic SHP‐1. This phosphatase has been shown to interact with FGF and VEGF receptors and to play a negative regulatory role in the ERK phosphorylation. To analyse a potential involvement of SHP‐1 in the inhibitory effects of EGb® 761 on ERK phosphorylation, we examined whether G. biloba extract influences SHP‐1 activity. Indeed, SHP‐1 activity was induced by EGb® 761 (500 μg/ml, 30 min.) to approximately 1.5‐fold (Fig. 5B). siRNA experiments were performed to clarify whether SHP‐1 leads to the reduced endothelial proliferation by EGb® 761, and, indeed, HUVEC proliferation was not reduced after treatment with EGb® 761 (500 μg/ml) for 72 hrs in cells lacking SHP‐1. In contrast, HUVECs transfected with non‐targeting siRNA showed the known suppressed endothelial cell proliferation (Fig. 5C).
Discussion
In recent years, the interest in herbal remedies has grown rapidly in the industrialised world, since these drugs are increasingly considered as effective and safe alternatives to synthetic drugs. Consequently, the discovery of their underlying signalling mechanisms has gained importance, but it is still in its infancy. Major limitations for the clinical use of herbal remedies are the constancy of the formulation and the quality of the preparation. One of the few well‐established plant products supported by clinical trials is the standardised special extract EGb® 761 from the leaves of G. biloba. This extract has been proven effective in clinical studies on dementia of various origins [14] and diseases with a vascular background (intermittent claudication [15], Raynaud's disease [16] and tinnitus [17]). In part, the underlying mechanisms in endothelial cells have already been identified [1]. It therefore lied close at hand to also investigate the actions of EGb® 761 on angiogenesis. This is of special interest in the tumour context, as EGb® 761 has been shown to inhibit breast cancer cell proliferation both in vitro and in vivo[18], an effect that was recently extended to glioma, hepatocellular carcinoma and oral cavity cancer cells, suggesting a widespread applicability of EGb® 761 for tumour growth control [19, 20, 21]. Epidemiological studies have even shown a negative correlation between regular consumption of Ginkgo extract and ovarian cancer occurrence [2].
Inhibition of angiogenesis restricts tumour growth and therefore serves as one major approach for anticancer therapy. Since some descriptive studies exist, reporting anti‐angiogenic properties of Ginkgo preparations [22, 23], we studied this phenomenon on a mechanistic level. This study for the first time shows a molecular background of anti‐angiogenic properties of EGb® 761 in vitro as well as in vivo and suggests that inhibition of angiogenesis by EGb® 761 is mediated via inhibition of the ERK signalling pathway. The ERK pathway, activated through growth factors like FGF and VEGF in endothelial cells, is of particular relevance to angiogenesis [4]. Accordingly, we found that blockade of ERK activation by U0126, a specific MEK1/2 inhibitor, attenuates endothelial angiogenesis including proliferation, migration and tube formation to about the same degree as did EGb® 761. Along this line, EGb® 761 reduces ERK phosphorylation induced either by VEGF or by FGF in endothelial cells. This argues against an effect on a distinct growth factor receptor or its respective tyrosine kinase activity. Downstream of the respective growth factor receptors, activation of ERK is regulated by a three‐step signalling module comprising the upstream MAPK kinase kinase Raf‐1 and MAPK kinase MEK. The pathway can be negatively modulated by various mechanisms at almost every step of the Ras‐Raf‐MEK‐ERK cascade. We found that activation, that is, phosphorylation, of the upstream kinases MEK and Raf‐1 following growth factor stimulation was abrogated by EGb® 761. Consequently, we searched for the underlying mechanisms:
To this end, we have previously shown that EGb® 761 activates the signalling cascades of nitric oxide/cyclic GMP as well as PI3K/Akt [1]. Both cascades lead to an inhibition of the serine/threonine kinase Raf‐1 via phosphorylation at Ser259, which keeps the kinase in an auto‐inhibited state [5, 6, 7, 8]. In addition, EGb® 761 clearly increased cyclic AMP levels (data not shown). Cyclic AMP activates protein kinase A (PKA), which has been shown to phosphorylate Raf‐1 at Ser259, thereby inactivating the Raf‐MEK‐ERK pathway [24]. Therefore, the involvement of nitric oxide/cyclic GMP, PI3K/Akt as well as cyclic AMP/PKA in the reduction of ERK phosphorylation by EGb® 761 was investigated using specific inhibitors [L‐Nitro‐arginine‐methylene‐ester (L‐NAME), wortmannin and Rp‐adenosine‐3′,5′‐cyclic mono‐phosphorothioate triethylamine (cAMPS‐Rp), respectively]. None of these cascades appeared to be involved in the inhibitory effect of EGb® 761 on ERK phosphorylation (data not shown), though the important role of nitric oxide for angiogenesis as such is not debatable and has been well established [25].
This leaves two possible explanations for the inhibitory effect of EGb® 761 on the ERK pathway: inhibition of kinases even more upstream (i.e. RTKs) or phosphatase activation. The lack of inhibitory effects of EGb® 761 on ERK phosphorylation after stimulation with PMA and the fact that EGb® 761 had effects after stimulation of the cells with both VEGF and FGF argue against the RTK hypothesis. In contrast, we for the first time supply evidence that EGb® 761 influences the activity of tyrosine phosphatases (PTPs), which were identified as the crucial regulators of the reduced ERK phosphorylation and were causally linked with the anti‐angiogenic effect of EGb® 761. Recently, evidence has been given that PTPs function as tumour suppressors, and that deregulation of PTP function is associated with tumourigenesis in different types of human cancer [9]. Furthermore, individual PTPs exert an antagonistic role in growth factor signalling either by direct dephosphorylation of RTKs or by dephosphorylation of a member of the MAP kinase cascade. Regarding the ERK cascade, PTPs such as MKP‐1, SHP‐1, PTP1B and HC PTP‐A have been discussed amongst others. We focused on MKP‐1 and SHP‐1, which have both been shown to be of functional relevance in endothelial cells [11, 26]. MKP‐1 is a dual‐specificity MAPK phosphatase that inactivates ERK by dephosphorylation of the two MAPK residues (Thr202/Tyr204) critical for its functionality. MKP‐1 is mainly regulated via protein expression, which was not affected by EGb® 761 (analysed by Western blot, data not shown). We, therefore, excluded MKP‐1 as the putatively responsible phosphatase. Next, we focused on the SHP‐1, which is a cytoplasmic PTP. SHP‐1 was shown to suppress cellular proliferation and angiogenesis by directly dephosphorylating the FGF and VEGF receptor. Therefore, we hypothesized that EGb® 761 may act on SHP‐1. In a tyrosine phosphatase activity assay, SHP‐1 was rapidly activated by EGb® 761 within 30 min. Furthermore, using SHP‐1‐targeting siRNA, we could confirm the role of SHP‐1 activation in suppression of endothelial proliferation by the G. biloba extract. Thus, SHP‐1 was identified to be one target molecule for G. biloba extract, conferring its anti‐angiogenic activity.
In conclusion, the standardised G. biloba extract EGb® 761 activates protein tyrosine phosphatases (such as SHP‐1), resulting in an inhibition of the Raf/MEK/ERK‐pathway. This action causes an anti‐angiogenic response, both in vitro and in vivo (Fig. 6). These findings provide a rational basis for further investigating the chemo‐preventive and anti‐cancer effects of the drug EGb® 761. One big advantage of EGb® 761 that makes broader clinical studies in the tumour context feasible is the proven clinical safety of this drug and the fact that it can be chronically consumed without side effects. This is of special interest, considering new therapeutic concepts (metronomic scheme of drug application for anti‐angiogenesis) and changing paradigms in viewing cancer as a chronic disease instead of a death sentence. The present study adds G. biloba to the important list of rational herbal remedies with a known molecular basis of action.
Figure 6.
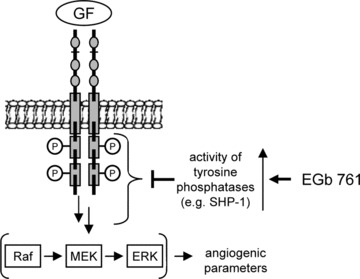
Hypothesized mechanisms for the anti‐angiogenic properties of EGb® 761. Ginkgo biloba extract EGb® 761 activates tyrosine phosphatases acting upstream of the Raf‐MEK‐ERK pathway.
Acknowledgements
We thank Cornelia Niemann and Jana Peliskova for excellent technical assistance and Dr. Willmar Schwabe Pharmaceuticals, Karlsruhe, Germany, for providing EGb® 761. This work was supported by the ‘Deutsche Forschungsgemeinschaft’ (DFG, GRK438).
References
- 1. Koltermann A, Hartkorn A, Koch E, et al . Ginkgo biloba extract EGb((R)) 761 increases endothelial nitric oxide production in vitro and in vivo. Cell Mol Life Sci . 2007; 64: 1715–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ye B, Aponte M, Dai Y, et al . Ginkgo biloba and ovarian cancer prevention: epidemiological and biological evidence. Cancer Lett . 2007; 251: 43–52. [DOI] [PubMed] [Google Scholar]
- 3. Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer . 2003; 3: 401–10. [DOI] [PubMed] [Google Scholar]
- 4. Gollob JA, Wilhelm S, Carter C, et al . Role of Raf kinase in cancer: therapeutic potential of targeting the Raf/MEK/ERK signal transduction pathway. Semin Oncol. 2006; 33: 392–406. [DOI] [PubMed] [Google Scholar]
- 5. Chong H, Vikis HG, Guan KL. Mechanisms of regulating the Raf kinase family. Cell Signal . 2003; 15: 463–9. [DOI] [PubMed] [Google Scholar]
- 6. Suhasini M, Li H, Lohmann SM, et al . Cyclic‐GMP‐dependent protein kinase inhibits the Ras/mitogen‐activated protein kinase pathway. Mol Cell Biol . 1998; 18: 6983–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Villalobo A. Nitric oxide and cell proliferation. FEBS J . 2006; 273: 2329–44. [DOI] [PubMed] [Google Scholar]
- 8. Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nat Rev Mol Cell Biol . 2004; 5: 875–85. [DOI] [PubMed] [Google Scholar]
- 9. Ostman A, Hellberg C, Bohmer FD. Protein‐tyrosine phosphatases and cancer. Nat Rev Cancer . 2006; 6: 307–20. [DOI] [PubMed] [Google Scholar]
- 10. Ades EW, Candal FJ, Swerlick RA, et al . HMEC‐1: establishment of an immortalized human microvascular endothelial cell line. J Invest Dermatol . 1992; 99: 683–90. [DOI] [PubMed] [Google Scholar]
- 11. Furst R, Schroeder T, Eilken HM, et al . MAPK phosphatase‐1 represents a novel anti‐inflammatory target of glucocorticoids in the human endothelium. FASEB J . 2007; 21: 74–80. [DOI] [PubMed] [Google Scholar]
- 12. Nicoletti I, Migliorati G, Pagliacci MC, et al . A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J Immunol Methods . 1991; 139: 271–9. [DOI] [PubMed] [Google Scholar]
- 13. Lopez‐Anton N, Rudy A, Barth N, et al . The marine product cephalostatin 1 activates an endoplasmic reticulum stress‐specific and apoptosome‐independent apoptotic signaling pathway. J Biol Chem . 2006; 281: 33078–86. [DOI] [PubMed] [Google Scholar]
- 14. Ernst E, Pittler MH. Ginkgo biloba for dementia: a systematic review of double‐blind, placebo‐controlled trials. Clin Drug Invest . 1999; 17: 301–8. [Google Scholar]
- 15. Pittler MH, Ernst E. Ginkgo biloba extract for the treatment of intermittent claudication: a meta‐analysis of randomized trials. Am J Med . 2000; 108: 276–81. [DOI] [PubMed] [Google Scholar]
- 16. Muir AH, Robb R, McLaren M, et al . The use of Ginkgo biloba in Raynaud's disease: a double‐blind placebo‐controlled trial. Vasc Med . 2002; 7: 265–7. [DOI] [PubMed] [Google Scholar]
- 17. Morgenstern C, Biermann E. The efficacy of Ginkgo special extract EGb 761 in patients with tinnitus. Int J Clin Pharmacol Ther . 2002; 40: 188–97. [DOI] [PubMed] [Google Scholar]
- 18. Papadopoulos V, Kapsis A, Li H, et al . Drug‐induced inhibition of the peripheral‐type benzodiazepine receptor expression and cell proliferation in human breast cancer cells. Anticancer Res . 2000; 20: 2835–47. [PubMed] [Google Scholar]
- 19. Chao JC, Chu CC. Effects of Ginkgo biloba extract on cell proliferation and cytotoxicity in human hepatocellular carcinoma cells. World J Gastroenterol . 2004; 10: 37–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim KS, Rhee KH, Yoon JH, et al . Ginkgo biloba extract (EGb 761) induces apoptosis by the activation of caspase‐3 in oral cavity cancer cells. Oral Oncol . 2005; 41: 383–9. [DOI] [PubMed] [Google Scholar]
- 21. Li W, Pretner E, Shen L, et al . Common gene targets of Ginkgo biloba extract (EGb 761) in human tumor cells: relation to cell growth. Cell Mol Biol . 2002; 48: 655–62. [PubMed] [Google Scholar]
- 22. Juarez CP, Muino JC, Guglielmone H, et al . Experimental retinopathy of prematurity: angiostatic inhibition by nimodipine, Ginkgo biloba, and dipyridamole, and response to different growth factors. Eur J Ophthalmol . 2000; 10: 51–9. [DOI] [PubMed] [Google Scholar]
- 23. Monte M, Davel LE, de Lustig ES. Inhibition of lymphocyte‐induced angiogenesis by free radical scavengers. Free Radic Biol Med . 1994; 17: 259–66. [DOI] [PubMed] [Google Scholar]
- 24. Dumaz N, Marais R. Integrating signals between cAMP and the RAS/RAF/MEK/ ERK signalling pathways. Based on the anniversary prize of the Gesellschaft fur Biochemie und Molekularbiologie Lecture delivered on 5 July 2003 at the Special FEBS Meeting in Brussels. FEBS J . 2005; 272: 3491–504. [DOI] [PubMed] [Google Scholar]
- 25. Morbidelli L, Donnini S, Ziche M. Role of nitric oxide in the modulation of angiogenesis. Curr Pharm Des . 2003; 9: 521–30. [DOI] [PubMed] [Google Scholar]
- 26. Krotz F, Engelbrecht B, Buerkle MA, et al . The tyrosine phosphatase, SHP‐1, is a negative regulator of endothelial superoxide formation. J Am Coll Cardiol . 2005; 45: 1700–6. [DOI] [PubMed] [Google Scholar]