

Abstract
Studies in cervical dysplasia have reported overexpression of the tumour suppressors p14 and p16 – and absence of p53 – in high‐risk human papilloma virus (HPV)‐ associated lesions. In skin carcinogenesis, the relation between these tumour suppressors and HPV remain unclear. We evaluated the expression of the tumour suppressors p14, p16 and p53 in pre‐malignant and malignant squamous skin tumours, and its relation with risk factors for skin carcinogenesis (HPV, immune status and sun exposure). We performed immunohistochemical stainings for p14, p16 and p53 on paraffin embedded material of 71 pre‐malignant squamous skin lesions and 34 squamous cell carcinomas, from 52 renal transplant recipients (RTRs) and 53 immunocompetent individuals. PCR‐based assays were used for detection and genotyping of β‐papilloma virus (β‐PV) types and mucosal HPV types. P14 expression was independent of the expression of p16 and p53, irrespective of immune status and skin site. In 49 of 105 specimens (46.6%), one or more β‐PV types were detected. We found no significant association between p14, p16 or p53 protein expression and overall presence of β‐PV, irrespective of immune status. There was a significant association between presence of β‐PV and lesions from sun‐exposed skin sites in the RTRs (P= 0.002). We conclude that in skin carcinogenesis, relations between the herein studied tumour suppressors and HPV are different from what one would expect based on findings in cervical neoplasia. P14, p16 and p53 expressions are independent of immune status. Our data indicate that in immunosuppressed patients, β‐PV together with ultraviolet radiation act synergetic in promoting carcinogenesis.
Keywords: P14, p16, p53, (pre‐)malignant squamous skin lesions, HPV
Introduction
The tumour suppressor p14 is thought to play a key role in cell cycle control, because p14 provides a cross‐talk between the two main pathways governing cell growth, namely the p14‐MDM2‐p53 and p16‐CDK4/6‐RB pathway. P14 and p16 are products of the INK4a‐ARF locus, which have the unusual capacity to encode two structurally distinct proteins.
Studies on cell lines transfected with a vector encoding the human papilloma virus (HPV) E6 oncoprotein, indicate an inverse relation between p14 and p53 expression [1, 2]. Previous immunohistochemical analysis of cervical dysplasia and squamous cell carcinomas (SCCs) of the uterine cervix demonstrate overexpression of both p14 and p16 in HPV‐associated cases. In these cases, p14 overexpression is attributed to functional inactivation of p53 by the E6 oncoprotein of high‐risk mucosal HPV, and p16 overexpression is attributed to inactivation of pRb by the E7 oncoprotein of high‐risk mucosal HPV [3]. In cutaneous carcinogenesis, in which HPV is also implicated, one might assume that p14 and p16 could be overexpressed in HPV‐associated cases. In addition, a positive relationship between p14 and p16 expression as well as an inverse relationship between p14 and p53 can be inferred.
The presence of HPV DNA, including high‐risk mucosal HPV types, has been reported in non‐melanoma skin cancer afflicting both renal transplant recipients (RTRs) and immunocompetent individuals (ICIs) [4]. To date, the data pertaining to the frequency and types of HPV linked to cutaneous SCC are equivocal. Furthermore, certain HPV types are strongly associated with the development of cutaneous SCC in the inherited disorder epidermodysplasia verruciformis (EV) [5]. These types belong to the β‐papilloma virus (β‐PV) genus and are also known as EV‐HPV types. However, in non‐EV patients, the exact role of β‐PV in non‐melanoma skin cancer is yet to be elucidated.
RTRs have a markedly increased risk to develop keratinocytic intraepidermal neoplasias (KINs) and SCCs of the skin [6]. One could speculate that their enhanced cutaneous carcinogenesis might be caused by a higher frequency of possibly more oncogenic types of HPV, which in turn might be reflected in different expression patterns of p14, p16 and p53. This study aimed to investigate the expression of p14 in relation to p16 and p53 expression in pre‐malignant and malignant squamous skin tumours of RTRs and ICIs, and to assess possible relations with HPV presence and skin site.
Materials and methods
Patients and histopathology
We retrieved 105 formalin fixed and paraffin embedded KIN lesions and SCCs of ICIs and RTRs out of the archives at our department of pathology. The histology was reviewed and categorized by a single dermatopathologist. Pre‐malignant skin lesions were classified according to the KIN grading system by Cockerell [7]. In this grading system, KIN I is histologically characterized by atypia of basal keratinocytes in the lower one third of the epidermis; KIN II lesions demonstrate atypia of keratinocytes in the lower two thirds of the epidermis and KIN III lesions show diffuse atypia involving the full thickness of the epidermis. We considered both KIN I and KIN II lesions as low‐grade KINs (LKINs), corresponding to actinic keratoses. KIN III was considered as high‐grade intraepidermal neoplasia (HKIN), corresponding to Bowen’s disease. This separation was made because clinical behaviour and subsequently treatment of actinic keratoses is generally different from Bowen’s disease. The SCCs were categorized based on the least differentiated region into one of the following groups: ‘well’, ‘moderate’ and ‘poor [8]. The skin lesions were classified into those arising in a sun‐exposed site (head, neck, hands and forearms) and those arising in non sun‐exposed sites.
Immunohistochemistry
Four‐micrometre‐thick paraffin sections were deparaffinized, hydrated and washed in buffered saline phosphate (PBS). Microwave pre‐treatment consisted of 3 min. cooking in citrate buffer (0.01 M, pH 6.0) at 850 W, and 10 min. cooking at 180 W. After pre‐incubation with 20% normal goat serum for 10 min, sections were incubated with primary antibodies for 60 min. at room temperature. For detection of p14, Ab‐2, clone 14PO2, lot no. 850P203 (Neomarkers, Fremont, CA, USA; dilution 1:200) was used, for p53, Ab‐5 (DO‐7), lot no. 186p306 (Neomarkers; dilution 1:200) was used and for p16, we employed p16INK4A Ab‐4, clone 16PO4 or JC2, lot no. 887P301 (Neomarkers; dilution 1:500). After rinsing in PBS, 15 min. after antibody blocking (powervision plus) was performed followed by 30 min. incubation with Poly‐HRP‐antimouse/rabbit/rat IgG (Kit Powervision and Poly‐HRP‐AntiMs/Rb/Rt IgG Biotin‐free, Immunologic, Duiven, the Netherlands, code DPVO‐999HRP, lot no. 30210–410). Diaminobenzidine was used as chromogen. Sections were briefly counterstained with Mayer’s haematoxylin.
Quantification of immunohistochemical results
P14, p16 and p53 immunoreactivity was scored as previously described [9]. In brief, immunoreactivity was scored semi‐quantitatively: 0 (negative), 1+ (up to 10% of lesional cells positive), 2+ (10–50% positive lesional cells) or 3+ (>50% positive lesional cells). Only nuclear staining was considered positive. Scores of 1+, 2+ and 3+ were considered positive. In addition, localization of p16 and p53 immunoreactivity in the epidermal cell layers was assessed: 0 = only basal layer positivity, 1 = positivity confined to basal 1/3 of epidermis, 2 = positivity confined to basal 2/3 of epidermis or 3 = transepidermal positive staining. Scoring was performed without knowledge of patient history. Immunoreactivity was also scored in normal appearing skin, adjacent to the corresponding lesion.
HPV detection and genotyping by SPF10–LiPA25
The combined short PCR fragment–line probe assay (SPF10–LiPA25) system for detection and genotyping of HPV (Labo Bio‐Medical Products, Rijswijk, The Netherlands) has previously been described in detail [10]. Briefly, a single 4‐μm‐thick flanking and sterile cut tissue section was incubated overnight at 56°C in 200 μl of 10 mM Tris‐HCl with 1 mM ethylenediaminetetraacetic acid, 0.2% Tween‐20, and proteinase K (0.3 mg/ml). Proteinase K was inactivated by 10 min. incubation at 100°C. Ten microlitres was used for PCR analysis. A water blank control was processed with each batch of 10 samples. HPV detection was performed using a short PCR fragment (SPF10–PCR) assay [11, 12]. The SPF10–PCR system amplifies a 65 bp fragment of the L1 open reading frame, allowing for detection of at least 43 HPV types. In case of positive PCR result, subsequent HPV genotyping was performed via a reverse hybridization LiPA, allowing for simultaneous typing of the following 25 genotypes of HPV 6, 11, 16, 18, 31, 33, 34, 35, 39, 40, 42, 43, 44, 45, 51, 52, 53, 54, 56, 58, 59, 66, 68, 70 and 74 [10].
β‐PV detection and genotyping by the PM‐PCR reverse hybridization assay (RHA) method
The PM‐PCR RHA method (the skin [β] HPV prototype research assay; Diassay BV, The Netherlands) has previously been described in detail by de Koning et al.[13]. The method was designed for the identification of the 25 established β‐PV types, namely HPV genotypes 5, 8, 9, 12, 14, 15, 17, 19, 20, 21, 22, 23, 24, 25, 36, 37, 38, 47, 49, 75, 76, 80, 92, 93 and 96. The PM‐PCR, generating a biotinylated amplimer of 117 bp from the E1 region, was carried out with precautions to avoid contamination. The RHA was performed according to the kit insert.
Statistics
The association between p14 and p53, and p14 and p16 expression, was assessed with 2 × 2 contingency tables. The expression patterns are statistical independent if the conditional odds ratios do not differ significantly from 1. The Fisher’s exact test was used to test the association between the expression patterns for the separate strata. The Breslow–Day statistic was used to test whether the associations, defined in terms of conditional odds ratios, are equal for the different strata. The Mantel–Haenszel estimator was used to test whether the estimated common odds ratio differed significantly from 1. Independency of p14 expression on p16 or p53 expression was set at P > 0.05 in Breslow–Day and Mantel–Haenszel tests. The difference between LKIN, HKIN, and SCC with regard to p14 expression, was analysed by means of I×J ‐two‐way contingency tables. The Fisher’s exact test was used to test the association between protein expression and HPV status, immune status and sun exposure. Significance was set at P≤ 0.05. The SPSS exact tests, available in SPSS 10.0 for Windows, were used to obtain the exact P‐values.
Results
Patients
Table 1 summarizes the data from all analysed lesions. 75 of the 105 examined lesions came from sun‐exposed sites (71%) and 30 lesions from non‐exposed sites (29%).
Table 1.
Data of all analysed lesions
| LKIN* (n= 22) | HKIN† (n= 49) | SCC† (n= 34) | ||
|---|---|---|---|---|
| ICI§ (n= 52) | Total (%)** | 6 (12) | 30 (58) | 16 (31) |
| Lesions from sun‐exposed sites | 5 | 20 | 13 | |
| RTR# (n= 53) | Total (%)** | 16 (30) | 19 (36) | 18 (34) |
| Lesions from sun‐exposed sites | 12 | 12 | 13 | |
| Total (%)(n= 105) | 22 (21) | 49 (47) | 34 (32) |
*LKIN = low‐grade keratinocytic intraepidermal neoplasia.
†HKIN = high‐grade intraepidermal neoplasia.
†SCC = squamous cell carcinoma.
§ICI = immunocompetent individual.
#RTR = renal transplant recipient.
**Percentage within patient group.
P14 expression in relation to p16 and p53 expression in KINs and SCCs of ICIs and RTRs
Keratinocytes in normal skin were negative for p14. In all cases, p14 staining was nuclear, often in a dotted (‘nucleolar’) pattern (Fig. 1E).
Figure 1.
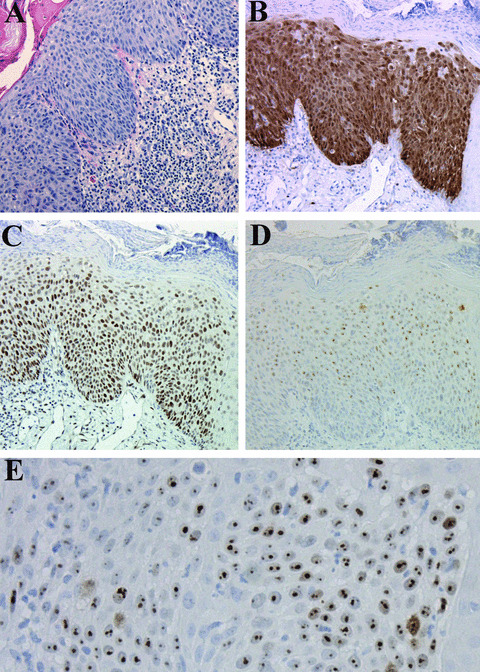
(A) Haematoxylin–eosin staining of a HKIN lesion (100×). (B)–(D). Same lesion with strong transepithelial p16 staining (B), p53 staining (C) and p14 staining (D) (100×). (E). High magnification of the same lesion showing typical nucleolar staining for p14 (400×).
In the patient group as a whole, there was a weakly significant difference in p14 expression (P= 0.05) between the three diagnostic groups (LKIN, HKIN and SCCs).
The percentage of p14+ lesional cells in the RTRs and ICIs are illustrated in Fig. 2. Only 7of 105 cases showed more than 50% p14+ lesional cells; four of these cases were HKIN lesions, and three were SCCs. Between the ICIs and RTRs, there were no significant differences concerning the percentage of p14+ lesions in the three diagnostic groups (Table 2). Normal skin showed only p16 positivity in dendritic melanocytes and sporadic p53 staining in a few basal keratinocytes. KIN grade strongly correlated with localization of p53 and p16 staining in the epidermal layers (r > 0.7, P < 0.0001), with higher‐grade lesions showing higher transepidermal staining (Fig. 1B and C).
Figure 2.
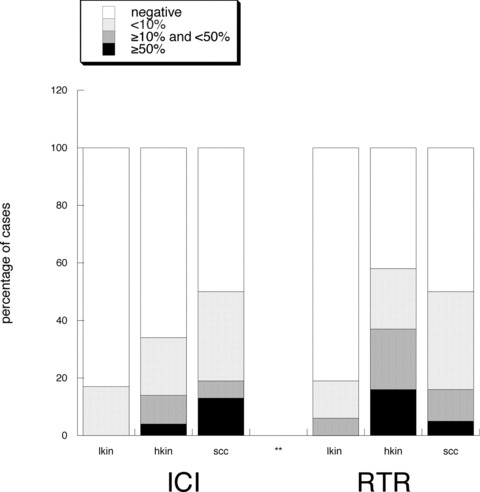
Percentage of lesions in the group of ICIs and RTRs showing absent p14 staining, less than 10% positive cells, between 10–50% positive cells, and >50% p14+ cells.
Table 2.
P14, p16 and p53 expression in relation to immune status
| LKIN n= 6 | ICIs n= 52 n= 52 HKIN n= 30 | SCC n= 16 | LKIN n= 16 | HKIN n= 19 | RTRs n= 53 n= 53 SCC n= 18 | Total n= 105 | P‐value | |
|---|---|---|---|---|---|---|---|---|
| p14+ (%)* | 1 (17) | 10 (33) | 8 (50) | 3 (19) | 11 (58) | 9 (50) | 42 (40) | |
| p14‐ (%) | 5 (83) | 20 (67) | 8 (50) | 13 (81) | 8 (42) | 9 (50) | 63 (60) | |
| p16+ (%) | 4 (67) | 25 (83) | 9 (56) | 11 (69) | 17 (89) | 12 (67) | 78 (74) | |
| p16‐ (%) | 2 (33) | 5 (17) | 7 (44) | 5 (31) | 2 (11) | 6 (33) | 27 (26) | |
| p53+ (%) | 5 (83) | 21 (70) | 16 (100) | 9 (56) | 14 (74) | 12 (67) | 77 (73) | |
| p53‐ (%) | 1 (17) | 9 (30) | 0 (0) | 7 (44) | 5 (26) | 6 (33) | 28 (27) | 0.05 |
*Percentage within diagnosis group.
In the total patient group, we found a weakly significant higher number of p16+ lesions in the HKIN group (P= 0.04). Between the ICIs and RTRs, there were no significant differences concerning the percentage of p16+ lesions in the three diagnostic groups (Table 2).
P53 positivity was always nuclear. In the patient group as a whole, no significant differences were found concerning p53 expression in the three diagnostic groups. Between the RTRs and ICIs, there was a weakly significant difference in p53 positivity in the group of the SCCs, (P= 0.05), due to a higher frequency of p53− SCCs in the RTRs (6/18 cases, 33%), than in the ICIs (0/16 cases, 0%) (Table 2).
The frequencies of the combined expression patterns of p14 and p53, and p14 and p16, are given in Table 3. Both the expressions of p14 and p53, and of p14 and p16, were independent and this was not influenced by skin site or immune status (data not further shown).
Table 3.
The combined expression patterns for p14/p53, and p14/p16, in relation to diagnosis
| Diagnosis | p14−/p53‐ No. (%) | p14−/p53+ No. (%) | p14+/p53− No. (%) | p14+/p53+ No. (%) | Fisher’s test two tailed P‐value |
|---|---|---|---|---|---|
| LKIN (n= 22) | 7 (32) | 11 (50) | 1 (4) | 3 (14) | >0.90 |
| HKIN (n= 49) | 11 (22) | 17 (35) | 3 (6) | 18 (37) | 0.07 |
| SCC (n= 34) | 3 (9) | 14 (41) | 3 (9) | 14 (41) | >0.90 |
| Total (n= 105) | 21 (20) | 42 (40) | 7 (7) | 35 (33) | |
| Diagnosis | p14−/p16− No. (%) | p14−/p16+ No. (%) | p14+/p16− No. (%) | p14+/p16+ No. (%) | Fisher’s test two tailed P‐value |
| LKIN (n= 22) | 7 (32) | 11 (50) | 0 | 4 (18) | 0.30 |
| HKIN (n= 49) | 7 (14) | 21 (43) | 0 | 21 (43) | 0.02 |
| SCC (n= 34) | 6 (18) | 11 (32) | 7 (21) | 10 (29) | >0.90 |
| Total (n= 105) | 20 (19) | 43 (41) | 7 (7) | 35 (33) | |
The expression of p14 and p53 are homogeneous for LKIN, HKIN and SCC. Concerning p14 and p16, the Fisher’s exact test disclosed that in the HKIN group a weakly significant difference was present (P= 0.02).
HPV presence in KINs and SCCs of immunocompetent individuals and renal transplant recipients
In 2 of 105 lesions (1.9%), mucosal HPV DNA was detected by SPF10–PCR (Table 4). Neither one of them contained one of the 25 known mucosal genotypes. Both specimens were HKIN lesions in ICIs, and showed strong p16 staining (>95%) but absent p14 staining. P53 staining was absent or low (15%). Neither the specimens contained β‐PV DNA.
Table 4.
HPV frequencies in skin lesions from RTRs and ICIs
| ICIs n= 52 | RTRs n= 53 | |||
|---|---|---|---|---|
| β‐PV | SPF10–LIPA25 | β‐PV | SPF10–LIPA25 | |
| LKIN* (%)† n= 22 | 1/6 (17) | 0 | 7/16 (44) | 0 |
| HKIN* (%)† n= 49 | 15/30 (50) | 2/30 (7) | 9/19 (47) | 0 |
| SCC (%)† n= 34 | 9/16 (56) | 0 | 8/18 (44) | 0 |
| Total n= 105 | 25/52 (48) | 2/52 (4) | 24/53 (45) | 0 |
*Pre‐malignant lesions = LKIN + HKIN.
†Percentage within diagnosis group.
In 49 of 105 specimens (47%), one or more β‐PV genotypes were detected using the PM PCR RHA method (Table 4). The most common detected types were types 5, 8, 15, 20, 22, 23, 24, 36 and 93. The overall presence of β‐PV DNA did not significantly differ between the RTRs (45.3%) and the ICIs (48.1%). No significant association was found between the overall presence of β‐PV DNA and the type of skin lesion.
Within the RTRs, we found a significant association between the overall presence of β‐PV DNA and lesions from sun‐exposed sites (P= 0.002). This was independent of the type of lesion. Only the presence of HPV type 15 was weakly associated with lesions from sun‐exposed sites (P= 0.018).
No specific β‐PV type was more frequent in either patient groups. The presence of HPV type 8 was weakly associated with SCC in the group of ICIs (P= 0.01), and the presence of HPV type 24 was weakly associated with LKIN in the RTR group (P= 0.03).
The majority of co‐infections (Table 5), in which two or more β‐PV types were co‐detected, contained between two and eight distinct types. The spectrum of β‐PV types within the co‐infections was comparable in both patient groups, with HPV types 15 (10/24, 41.7%) and 23 (9/24, 37.5%) being the most frequent in both groups. Within the RTRs, a significant association was present between the presence of co‐infections and lesions from sun‐exposed sites (P= 0.007). No significant association was found between frequency of co‐infections and type of skin lesion.
Table 5.
Frequencies of co‐infections in skin lesions from RTRs and ICIs
| ICIs n= 52 | RTRs n= 53 | |||||
|---|---|---|---|---|---|---|
| LKIN | HKIN | SCC | LKIN | HKIN | SCC | |
| n= 6 | n= 30 | n= 16 | n= 16 | n= 19 | n= 18 | |
| No infection (%)† | 5 (83) | 15 (50) | 7 (44) | 9 (56) | 10 (53) | 10 (56) |
| Mono‐infection (%)*,† | 1 (17) | 7 (23) | 5 (31) | 2 (13) | 5 (26) | 5 (42) |
| Co‐infection (%)†,† | 0 (0) | 8 (27) | 4 (25) | 5 (31) | 4 (21) | 3 (17) |
| Total number with β‐PV (%)† | 1 (17) | 15 (50) | 9 (56) | 7 (44) | 9 (47) | 8 (44) |
*Mono‐infection = one β‐PV type detected.
†Co‐infection = two or more β‐PV types co‐detected.
†Percentage within diagnosis group.
P14, p16 and p53 expression in relation to HPV presence
The expression of p14, p16 and p53 did not significantly differ between HPV+ and HPV− skin lesions as a total group, and this was not influenced by immune status (P‐values of 0.84, 0.38 and 0.08, respectively). Table 6 summarizes the expression of p14, p16 and p53 in relation to the presence of HPV in the three diagnostic groups. No significant association was detected between the overexpression of p14 or p16 and the presence of HPV in the three diagnostic groups. Although there seemed to be more p14+ lesions in the HPV+ LKIN group (38%) compared to the HPV− LKIN group (7%), this difference was not significant (P= 0.12). Expression of p53 was not significant associated with HPV presence in the three diagnostic groups, although more p53− lesions seemed to be present in the HPV+ LKIN group (62%) compared to the HPV− LKIN group (21%) (P= 0.08) (Table 6).
Table 6.
P14, p16 and p53 expression in relation to HPV
| HPV+*n= 51 | HPV− lesions n= 54 | ||||||
|---|---|---|---|---|---|---|---|
| LKIN | HKIN | SCC | LKIN | HKIN | SCC | P‐value | |
| n= 8 | n= 26 | n= 17 | n= 14 | n= 23 | n= 17 | ||
| p14+ (%)† | 3 (38) | 9 (35) | 9 (53) | 1 (7) | 12 (52) | 8 (47) | 0.12 |
| p14− (%)† | 5 (62) | 17 (65) | 8 (47) | 13 (93) | 11 (48) | 9 (53) | |
| p16+ (%)† | 6 (75) | 23 (88) | 11 (65) | 9 (64) | 19 (83) | 10 (59) | 0.9 |
| p16− (%)† | 2 (25) | 3 (12) | 6 (35) | 5 (36) | 4 (17) | 7 (41) | |
| p53+ (%)† | 3 (38) | 18 (69) | 12 (71) | 11 (79) | 17 (74) | 16 (94) | 0.08 |
| p53− (%)† | 5 (62) | 8 (31) | 5 (29) | 3 (21) | 6 (26) | 1 (6) | |
*β‐PV and mucosal HPV.
†Percentage within diagnosis group.
Discussion
Tumour suppressor expression and HPV
The present study aimed to investigate the expression of p14 in pre‐malignant and malignant squamous skin tumours and its relations to the expression of p16 and p53, and presence of HPV. So far, only Brown et al. studied p14 expression in 40 cutaneous SCCs, of which 30 were derived from immunosuppressed patients [14]; they reported p14 expression in 18/40 cases (45%) of SCCs, which is comparable to our finding of 50% p14+ carcinomas. Interestingly, in KIN lesions, higher‐grade lesions showed higher transepidermal staining for p16 and p53. Similar results are reported by others [15, 16]. This finding supports the view of KIN lesions being precursor lesions of SCCs.
Though previous studies on cell lines have reported an inverse relation between p14 and p53 expression [1, 2], we found in a large group of 105 KIN lesions and SCCs, that the expression of p14 and p53, as well as the expression of p14 and p16, were independent, irrespective of immune status and skin site. This suggests that skin cancer development in RTRs and ICIs is comparable with respect to involvement of cell cycle associated proteins, despite differences in immune status and clinical differences in rate of tumour development between both groups. Most cases (40%) showed a p14−/p53+ pattern, and an inverse relation between p14 and p53 was less prevalent. With respect to p14/p16, a p14−/p16+ pattern was most prevalent (41% of cases).
With respect to the role of HPV and the tumour suppressors p14 and p16 in cutaneous carcinogenesis, we assumed that, in analogue to cervical carcinogenesis, p14 and p16 might be overexpressed in HPV+ pre‐malignant and malignant squamous skin tumours. However, in our study, there was no significant association between p14 or p16 overexpression and HPV presence. Furthermore, the overall presence of β‐PV and mucosal HPV DNA was independent of immune status and the type of skin lesion. Because in cervical carcinogenesis p53 is inactivated by the E6 oncoprotein of high‐risk mucosal HPV, we assumed that HPV+ skin lesions might show less expression of p53. However, in this large study group we did not detect a significant association between p53 expression and HPV presence. In skin carcinogenesis, the relations between expression of these tumour suppressors and HPV presence is different from what one would expect based on in vitro data and findings in cervical neoplasia. We propose three plausible reasons to explain the aforementioned discrepancies.
First, in cutaneous carcinogenesis sun exposure is implicated as a major etiological factor. UV‐induced mutations have been found in p53 and INK4a‐ARF [17, 18] and these mutations can be expected to cause altered protein expression.
Secondly, oncogenic properties of HPV types implicated in cutaneous carcinogenesis seem different from those of high‐risk HPV types in cervical carcinogenesis. In contrast to cervical cancer, in which high‐risk HPV DNA becomes integrated in the host genome, in squamous skin tumours containing presumed oncogenic HPV types, HPV integration is very rare [19]. In addition, it is assumed that in skin carcinogenesis, the E6 oncoprotein mediated degradation of p53 is not as important as in cervical carcinogenesis, because in skin tumours p53 is already often silenced by UV‐induced p53 mutation [20]. Furthermore, in vitro studies show that the E6 oncoprotein of HPV type 38 cannot degrade the p53 protein [21, 22]. This is further supported by evidence that E6 of some HPV types (types 10, 77) may prevent apoptosis in a p53 independent manner, by inactivation of the proapoptotic protein Bak, which is independently of p53, up‐regulated after ultraviolet B damage [23]. This supports a synergetic effect between HPV and ultraviolet radiation in HPV‐associated skin cancer. Based on these data, it can be assumed that UVB‐induced DNA damage can more easily accumulate and persist in HPV‐infected keratinocytes and that this effect will be enhanced by immunosuppression. Our data support this by showing a significant association between the presence of HPV and lesions from sun‐exposed sites in the RTRs (P= 0.002).
Finally, protein expression might not directly reflect changes on the molecular level. For instance for p53, it is known that many of the mutations in the p53 coding region result in a structurally altered, inactive protein that is more stable than its wild‐type counterpart, resulting in higher levels of protein detectable by antibody. The high prevalence of p53 overexpression in our series, 77 of 105 cases (73%), could well reflect cases harbouring p53 mutations as described above. At the moment, the molecular events underlying p16 and p14 overexpression remain to be investigated. In summary, our findings are in accordance with previous studies documenting no straightforward relationships between expression of p14, p16 and p53, and presence of HPV in pre‐malignant and malignant squamous skin tumours.
HPV and (pre‐)malignant skin lesions
The prevalence and spectrum of HPV in pre‐malignant and malignant squamous skin tumours in ICIs and RTRs reported in the literature show considerable variation, depending upon the detection method used [24]. We demonstrated, by PM‐PCR RHA methodology to detect the β‐PV genus, a frequency of β‐PV in SCCs of RTRs of 44%. This result is only slightly lower than that reported by Berkhout et al. (45/81, 55.5%) [25]. Our study detected HPV DNA in 56% of the SCCs in ICIs, which falls within the range reported in the literature, varying between 27.2%[24] and 60%[4].
Herein, we determined that the number of co‐infections did not significantly differ between the ICIs and RTRs (respectively, 48% and 50%). This is in contrast with Harwood et al.[24], who report more co‐infections in the RTRs (39/66, 59.1%) than in the ICIs (3/23, 13%). The spectrum of HPV types in our study did not differ between the RTRs and ICIs and no specific HPV type was more frequent with respect to diagnosis. This suggests that no specific HPV type seems to be strongly associated with immune status or diagnosis, possibly indicating that a large spectrum of HPV types could be involved in skin carcinogenesis. Alternatively, HPV could simply play a coincidental role, because HPV DNA is also detected in normal skin samples of healthy individuals. In fact, De Koning et al. reported the presence of β‐PV in eyebrow hair samples of 14 of 23 (61%) healthy individuals [26] at intake. Over the 2‐year study period all but one individual were found positive for β‐PV.
None of the skin lesions contained one of the 25 mucosal HPV types identified by the SPF10–LiPA25 system. Similar results are reported by de Jong‐Tieben et al.[27] with absence of mucosal HPV types in 96 epithelial skin tumours of RTRs by nested PCR. In contrast, Soler et al.[28] reported a high frequency of the mucosal HPV types 6/11 in benign, pre‐malignant and malignant cutaneous lesions of transplant recipients (30/43, 70%). In this study, we chose for the combined SPF10–LiPA25 system, because this mucosal HPV detection test is highly sensitive, specific and reproducible [11, 12, 29]. Based on this solid technique, we conclude that our data do not support a role for mucosal HPV types in cutaneous carcinogenesis.
Summarizing, we conclude that in pre‐malignant and malignant skin lesions p14 expression is independent of p53 and p16 expression, irrespective of HPV presence, immune status or sun exposure. Our study did not reveal a role for mucosal HPV types in the development of pre‐malignant and malignant squamous skin tumours. Our data indicate that in immunosuppressed patients, β‐PV together with ultraviolet radiation act synergetic in promoting skin carcinogenesis.
Acknowledgement
We would like to thank Professor D.J. Ruiter (Department of Pathology, Radboud University Nijmegen Medical Center, the Netherlands) for his critical comments.
References
- 1. Rodway H, Llanos S, Rowe J, et al . Stability of nucleolar versus non‐nucleolar forms of human p14(ARF). Oncogene . 2004; 23: 6186–92. [DOI] [PubMed] [Google Scholar]
- 2. Stott FJ, Bates S, James MC, et al . The alternative product from the human CDKN2A locus, p14(ARF), participates in a regulatory feedback loop with p53 and MDM2. EMBO J . 1998; 17: 5001–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sano T, Masuda N, Oyama T, et al . Overexpression of p16 and p14ARF is associated with human papillomavirus infection in cervical squamous cell carcinoma and dysplasia. Pathol Int . 2002; 52: 375–83. [DOI] [PubMed] [Google Scholar]
- 4. Iftner A, Klug SJ, Garbe C, et al . The prevalence of human papillomavirus genotypes in nonmelanoma skin cancers of nonimmunosuppressed individuals identifies high‐risk genital types as possible risk factors. Cancer Res . 2003; 63: 7515–9. [PubMed] [Google Scholar]
- 5. Harwood CA, McGregor JM, Proby CM, et al . Human papillomavirus and the development of non‐melanoma skin cancer. J Clin Pathol . 1999; 52: 249–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Berg D, Otley CC. Skin cancer in organ transplant recipients: epidemiology, pathogenesis, and management. J Am Acad Dermatol . 2002; 47: 1–17. [DOI] [PubMed] [Google Scholar]
- 7. Cockerell CJ. Histopathology of incipient intraepidermal squamous cell carcinoma (“actinic keratosis”). J Am Acad Dermatol . 2000; 42: 11–7. [DOI] [PubMed] [Google Scholar]
- 8. McKee PH. Pathology of the Skin. London : 1999. [Google Scholar]
- 9. Blokx WA, de Jong EM, de Wilde PC, et al . P16 and p53 expression in (pre)malignant epidermal tumors of renal transplant recipients and immunocompetent individuals. Mod Pathol . 2003; 16: 869–78. [DOI] [PubMed] [Google Scholar]
- 10. Kleter B, van Doorn LJ, Schrauwen L, et al . Development and clinical evaluation of a highly sensitive PCR‐reverse hybridization line probe assay for detection and identification of anogenital human papillomavirus. J Clin Microbiol . 1999; 37: 2508–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kleter B, van Doorn LJ, ter SJ, et al . Novel short‐fragment PCR assay for highly sensitive broad‐spectrum detection of anogenital human papillomaviruses. Am J Pathol . 1998; 153: 1731–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Melchers WJ, Bakkers JM, Wang J, et al . Short fragment polymerase chain reaction reverse hybridization line probe assay to detect and genotype a broad spectrum of human papillomavirus types. Clinical evaluation and follow‐up. Am J Pathol . 1999; 155: 1473–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. de Koning M, Quint W, Struijk L, et al . Evaluation of a novel highly sensitive, broad‐spectrum PCR‐reverse hybridization assay for detection and identification of beta‐papillomavirus DNA. J Clin Microbiol . 2006; 44: 1792–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brown VL, Harwood CA, Crook T, et al . p16INK4a and p14ARF tumor suppressor genes are commonly inactivated in cutaneous squamous cell carcinoma. J Invest Dermatol . 2004; 122: 1284–92. [DOI] [PubMed] [Google Scholar]
- 15. Kvlividze O, Gogiashvili L, Burkadze G. The characteristics of human papillomavirus expression and cell proliferation in actinic keratosis and Bowen’s disease of the skin. Georgian Med News . 2006; 108–12. [PubMed] [Google Scholar]
- 16. Hodges A, Smoller BR. Immunohisto‐chemical comparison of p16 expression in actinic keratoses and squamous cell carcinomas of the skin. Mod Pathol . 2002; 15: 1121–5. [DOI] [PubMed] [Google Scholar]
- 17. McGregor JM, Berkhout RJ, Rozycka M, et al . p53 mutations implicate sunlight in post‐transplant skin cancer irrespective of human papillomavirus status. Oncogene . 1997; 15: 1737–40. [DOI] [PubMed] [Google Scholar]
- 18. Soufir N, ya‐Grosjean L, de La SP, et al . Association between INK4a‐ARF and p53 mutations in skin carcinomas of xeroderma pigmentosum patients. J Natl Cancer Inst . 2000; 92: 1841–7. [DOI] [PubMed] [Google Scholar]
- 19. Yabe Y, Tanimura Y, Sakai A, et al . Molecular characteristics and physical state of human papillomavirus DNA change with progressing malignancy: studies in a patient with epidermodysplasia verruciformis. Int J Cancer . 1989; 43: 1022–8. [DOI] [PubMed] [Google Scholar]
- 20. McGregor JM, Farthing A, Crook T, et al . Posttransplant skin cancer: a possible role for p53 gene mutation but not for oncogenic human papillomaviruses. J Am Acad Dermatol . 1994; 30: 701–6. [DOI] [PubMed] [Google Scholar]
- 21. Caldeira S, Zehbe I, Accardi R, et al . The E6 and E7 proteins of the cutaneous human papillomavirus type 38 display transforming properties. J Virol . 2003; 77: 2195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Majewski S, Jablonska S. Do epidermodysplasia verruciformis human papillomaviruses contribute to malignant and benign epidermal proliferations Arch Dermatol . 2002; 138: 649–54. [DOI] [PubMed] [Google Scholar]
- 23. Jackson S, Harwood C, Thomas M, et al . Role of Bak in UV‐induced apoptosis in skin cancer and abrogation by HPV E6 proteins. Genes Dev . 2000; 14: 3065–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Harwood CA, Surentheran T, McGregor JM, et al . Human papillomavirus infection and non‐melanoma skin cancer in immunosuppressed and immunocompetent individuals. J Med Virol . 2000; 61: 289–97. [DOI] [PubMed] [Google Scholar]
- 25. Berkhout RJ, Bouwes Bavinck JN, ter SJ. Persistence of human papillomavirus DNA in benign and (pre)malignant skin lesions from renal transplant recipients. J Clin Microbiol . 2000; 38: 2087–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. de Koning MN, Struijk L, Bavinck JN, et al . Betapapillomaviruses frequently persist in the skin of healthy individuals. J Gen Virol . 2007; 88: 1489–95. [DOI] [PubMed] [Google Scholar]
- 27. de Jong‐Tieben LM, Berkhout RJ, Smits HL, et al . High frequency of detection of epidermodysplasia verruciformis‐associated human papillomavirus DNA in biopsies from malignant and premalignant skin lesions from renal transplant recipients. J Invest Dermatol . 1995; 105: 367–71. [DOI] [PubMed] [Google Scholar]
- 28. Soler C, Chardonnet Y, Allibert P, et al . Detection of mucosal human papillomavirus types 6/11 in cutaneous lesions from transplant recipients. J Invest Dermatol . 1993; 101: 286–91. [DOI] [PubMed] [Google Scholar]
- 29. Rubin MA, Kleter B, Zhou M, et al . Detection and typing of human papillomavirus DNA in penile carcinoma: evidence for multiple independent pathways of penile carcinogenesis. Am J Pathol . 2001; 159: 1211–8. [DOI] [PMC free article] [PubMed] [Google Scholar]