

Abstract
Extracts from Pygeum africanum are used in the treatment of prostatitis, benign prostatic hyperplasia and prostate cancer (Pca), major health problems of men in Western countries. The ligand‐activated human androgen receptor (AR) supports the growth of the prostate gland. Inhibition of human AR by androgen ablation therapy and by applying synthetic anti‐androgens is therefore the primary goal in treatment of patients. Here, we show that atraric acid (AA) isolated from bark material of Pygeum africanum has anti‐androgenic activity, inhibiting the transactivation mediated by the ligand‐activated human AR. This androgen antagonistic activity is receptor specific and does not inhibit the closely related glucocorticoid or progesterone receptors. Mechanistically, AA inhibits nuclear transport of AR. Importantly, AA is able to efficiently repress the growth of both the androgen‐dependent LNCaP and also the androgen‐independent C4–2 Pca cells but not that of PC3 or CV1 cells lacking AR. In line with this, AA inhibits the expression of the endogenous prostate specific antigen gene in both LNCaP und C4–2 cells. Analyses of cell invasion revealed that AA inhibits the invasiveness of LNCaP cells through extracellular matrix. Thus, this study provides a molecular insight for AA as a natural anti‐androgenic compound and may serve as a basis for AA derivatives as a new chemical lead structure for novel therapeutic compounds as AR antagonists, that can be used for prophylaxis or treatment of prostatic diseases.
Keywords: antihormone, androgen antagonist, natural compound, Pygeum africanum, androgen receptor, prostate hyperplasia
Introduction
The prostate gland is a part of the male reproductive system and is, normally, about the size of a walnut. Aging men will eventually develop a prostate disease at some stage of life. The most common of them are benign prostatic hyperplasia (BPH) and prostate cancer (PCa) [1]. BPH is associated with bothersome lower urinary tract symptoms (LUTS) that affect quality of life, by interfering with normal daily activities and sleep patterns [2]. PCa is one of the most commonly diagnosed cancers and one of the leading causes of cancer mortality of men in Western countries [3]. Evidence shows that androgens have a leading role by promoting the development of BPH and PCa [4, 5]. The most important androgens in adult males are testosterone, the main circulating androgen, and dihydrotestosterone (DHT), the most potent and major androgen in tissue. Both of them exert their androgenic effects in the prostate through the androgen receptor (AR) protein [6].
AR is the major drug target of PCa therapy [7] and is a member of the nuclear hormone receptor superfamily, a large group of ligand‐dependent transcription factors. Nuclear receptors can bind to steroids (e.g. androgens, glucocorticoids, progestins and oestrogens) and non‐steroids (e.g. thyroid hormone) [8]. AR consists of an amino (N)‐terminal domain harbouring a potent transactivation function, followed by a DNA‐binding domain and a carboxy (C)‐terminal ligand‐binding domain (LBD). Binding of androgens induces a conformational change in the AR that induces the translocation into the nucleus and the binding to androgen response elements in the promoter region of target genes. The binding of the co‐regulatory proteins co‐activators or co‐repressors to the AR, regulates the transcriptional properties of the receptor in the context of the general transcriptional machinery and chromatin to either stimulate or to inhibit target gene transcription [9, 10, 11, 12].
Anti‐androgens (androgen antagonists) can block the AR‐mediated gene transactivation. Therefore, in current treatments the steroidal anti‐androgen cyproterone acetate and non‐steroidal hydroxyflutamide (OH‐F) and bicalutamide are applied for patient hormone therapies [7, 13].
Initially, PCa cells are growing androgen dependently; however, tumours develop eventually therapy resistance. Hereby the growth of the PCa becomes androgen independent and may no longer be inhibited by anti‐androgens or androgen ablation. How the androgen independence arises is not fully understood and suggests an LBD‐independent pathway. Activation of signalling pathways might contribute to androgen resistance. Also, there are some evidence that AR gene mutations can contribute to this independence. The mutated AR decreases the specificity of ligand binding and responses to other steroids and anti‐androgens and can once again activate its response genes [7, 13, 14, 15, 16, 17, 18].
The model cell line for human PCa LNCaP expresses such an AR mutation, which renders the anti‐androgen OH‐F into a potent agonist [19, 20]. Therefore, developing or identification of novel androgen antagonists that inhibit AR despite these mutations and therapy resistance would advance our knowledge and may lead to additional hormone treatments for patients that can be applied when PCa exhibits resistance to the known anti‐androgens.
For centuries phytotherapy is considered a first line treatment for BPH in Asia, Africa and India. Also in Europe and in the United States medicinal plants have been used even more extensively the last decades [21].
Extracts from the bark of Pygeum africanum is one of the most popular phytotherapeutic preparations used. Tadenan®, a medication that contains an extract from the bark of this African plum tree, is broadly and increasingly used in Europe and in the United States as dietary supplement in order to treat LUTS and BPH and to prevent PCa [22]. The bark of the African plum tree is licensed in France and other European countries for symptomatic BPH. In Africa, phytotherapy by P. africanum is known since centuries in order to treat the ‘old men disease’. It was demonstrated that Tadenan® suppresses the effects of androgens on micturition and inhibits the growth of prostate gland in rats [23] and recently that extracts of P. africanum suppresses PCa growth in vitro and in vivo[24]. However, the exact mechanism of how P. africanum can contribute to treatment and to prevention of BPH and PCa remains largely unknown.
In order to identify anti‐androgenic activities of P. africanum we isolated recently the natural compound atraric acid (AA) from its bark extracts [25]. In this study we wanted to gain some insights into the action of AA. The data reveal that the novel plant derived androgen antagonist AA binds to the human AR and inhibits agonist‐induced nuclear translocation of AR and thus providing a new chemical platform for AR antagonists. Moreover, the data suggest that AA exhibits a high specificity for AR and inhibits PCa cell growth of both androgen‐dependent and androgen‐independent cells. Furthermore, AA inhibits LNCaP cell invasion through extracellular matrix (ECM). Thus, AA might help in understanding AR antagonism and provide a novel platform for anti‐androgens to inhibit AR and PCa proliferation.
Materials and methods
Chemicals and hormones
Dexamethason, progesterone, 3,5,3′‐triiodothyronine (T3) DHT, estradiol (E2), AA and xanthoxylin were obtained from Sigma‐Aldrich (Taufkirchen, Germany). Methyltrienolone (R1881) was obtained from Perkin Elmer (Waltham, MA, USA). All test compounds were dissolved in ethanol or/and dimethylsulfoxide (DMSO), except T3, which was dissolved in 50 mM NaOH. These compounds were added to the culturing medium such that the final concentration of ethanol and/or DMSO did not exceed 0.1%. Control incubations (no test compounds) were performed in the presence of only 0.1% ethanol and/or DMSO.
Plasmids
The plasmid pMMTV‐luc responsive to androgens is described in Gast et al.[26]. The expression vector for the human AR, pSG‐hAR and the AR mutants are described in Dotzlaw et al.[27], for human GR in Schulz et al.[28], and for human TR‐β in Baniahmad et al.[29]. Human PR1 and PR2 expression vectors were kindly provided by Pierre Chambon (Strasbourg, France), and human oestrogen receptor (ER)‐α and ER‐β expression vectors from L.C. Murphy (Manitoba, Canada). The plasmid p(DR4)2‐tk‐luc is described in Leers et al.[30] and the reporter p3ERE‐TATA‐luc was kindly provided by D.M. Heery (Nottingham, UK) [31].
For transfection experiments, CV1 cells were seeded onto six‐well tissue culture plates (Nunc, Roskilde, Denmark) at 1.2 × 105 cells per well and grown in Dulbecco’s modified eagle medium (DMEM) medium supplemented with 10% (v/v) dextran‐coated charcoal stripped serum [32]. Six hours later, cells were transfected by using the CaPO4 method [27, 32]. The DNA mixture for transfections consisted of 1μg of the appropriated luciferase reporter constructs, 0.2 μg of the appropriated mammalian steroid receptor expression vector and 0.2 μg of the cytomegalovirus‐driven β‐galactosidase expressions vector, as internal control for transfection efficiency. After 14 hrs, media were replaced either with or without the addition of the appropriate hormones together with the indicated compounds.
For the transient transfection experiments with the ER receptors, the culture medium of the stock cells was changed to phenol‐red‐free DMEM (Invitrogen, Karlsruhe, Germany) supplemented with 10% (v/v) dextran‐coated charcoal stripped serum, 1% (v/v) glutamine, 1% (v/v) sodium pyruvate and penicillin streptomycin. One week later, cells were seeded into six‐well plates at 1.5 × 105 per well. Twenty‐four hours later cells were transfected by using the CaPO4 method [27] with 2 μg reporter plasmid, 0.2 μg receptor expressions vector and 0.2 μg of the cytomegalovirus‐driven β‐galactosidase expressions.
After additional 48 hrs cells were harvested and assayed for luciferase and β‐galactosidase activity. All transfection assays were performed in duplicate and were repeated three times. The deviations of the mean are indicated. A twofold difference is highly statistical significant.
For green fluorescent protein‐androgen receptor (GFP‐AR) detection HeLa cells were grown in DMEM with 10% heat‐inactivated foetal bovine serum (FBS, Invitrogen). Transfection experiments were performed using the calcium phosphate method as described previously (Moehren et al. 2007). Cells with 20% confluency were seeded out in 10‐cm dishes using DMEM supplemented with 10% charcoal‐stripped FBS and transiently transfected 24 hrs later with 31.8 μg of the GFP‐hAR expression plasmid (a generous gift from Dr. O. Jänne, University of Helsinki, Finland). After 1 day the cells were washed twice with phosphate buffered saline (PBS) and fresh medium containing 10% charcoal‐stripped FBS was added. Forty‐eight hours after transfection the cells were treated with AA (10–5 M) and/or R1881 (methyltrienolone) (3 × 10–11 M) for 1 hr. Cells were then analysed using a fluorescence microscopy with a fluorescine isothiocyanate filter. As control, cell morphology with the fluorescence overlay is shown.
Cell growth assays
Human prostate carcinoma LNCaP cells were kindly provided by Alexei Protopopov (Microbiology and Tumor Biology Center, Karolinska Institute, Stockholm, Sweden) [33]. The cells were cultured in Roswell Park Memorial Institute medium (RPMI)‐1640 medium, supplemented with 10% (v/v) foetal calf serum (FCS) (Invitrogen), 1% (v/v) penicillin and streptomycin (Invitrogen), 1% (v/v) L‐glutamine (Invitrogen), 1% (v/v) sodium pyruvate (Sigma), C4–2 cells [34] were cultured in DMEM medium supplemented with 10% (v/v) FCS (Invitrogen), 20% F12 (Sigma) 5 μg/ml insulin 13.6 pg/ml T3 (Sigma), 5 μg/ml apotransferrin (Sigma), 0.25 μg/ml Biotin (Sigma) 1% (v/v) penicillin and streptomycin (Invitrogen). Cells were seeded onto 24‐well tissue culture plates (Nunc) at 5 × 103 cells per well in appropriated medium containing 5% FCS. On day 2, cells were fed with fresh medium and treated with ethanol/DMSO or with the indicated hormones or compounds. Every second day the media were replaced with fresh media together with freshly added compounds. The cells were trypsinized and counted using a counting cell chamber (Double Neubauer, Brand, Germany) at the indicated times.
Nuclear extract preparation
LNCaP cells reaching 50% confluency were washed three times in PBS and RPMI‐medium supplemented with 2% (v/v) charcoal‐dextran‐stripped FBS was added. After 3 days of cultivation the cells were treated with the indicated hormones for 1 hr.
For extract preparation LNCaP cells were washed twice with ice cold PBS, harvested with 1 ml PBS and centrifuged at 1000 ×g for 5 min. at 4°C. The cell pellet was resuspended in three volumes of hypotonic buffer A (20 mM HEPES, pH 7.9, 10% glycerol, 0.2% Nonidet P‐40, 10 mM KCl, 1 mM ethylenediaminetetraacetic acid [EDTA], 0.1 mM PMSF, 1 mM DTT) for 7 min. on ice followed by centrifugation at 3000 ×g for 10 min. at 4°C. The cytoplasmatic supernatant was removed and the nuclei pellet resuspended in two volumes of buffer B (420 mM NaCl, 20 mM HEPES, pH 7.9, 10 mM KCl, 1 mM EDTA, 0.1 mM PMSF), followed by rotational incubation for 40 min. at 4°C. To remove cell debris, the extract was centrifuged at 12,000 ×g for 15 min. at 4°C. The supernatant was transferred into a new tube and diluted with three volumes of buffer C (10 mM KCl, 20 mM HEPES, pH 7.9, 1 mM EDTA, 0.1 mM PMSF). The cytosolic and nuclear extracts were separated on SDS‐PAGE and blotted onto a PVDF‐membrane (Millipore, Schwalbach, Germany). Western blot analysis was performed by using a mouse anti‐AR antibody (F39.4.1, BioGenex Laboratories, San Ramon, CA, USA) and the enhanced chemiluminescence detection method (Amersham Pharmacia Biotech, Munich, Germany). The rabbit anti‐GAPDH antibody was obtained from Abcam (ab9485). Actin (H‐196) and nucleophosmin (H‐106) antibodies were purchased from Santa Cruz (Heidelburg, Germany).
Whole cell binding assay
Whole cell binding assays using COS‐7 cells and (H3)‐mibolerone were described earlier [35]. Specific bound mibolerone was calculated as the difference between total and non‐specific binding. The reported values are averages of three independent assays, with each condition performed in triplicate. Data were plotted as percentage (H3)‐ mibolerone specifically bound.
Quantitative real‐time PCR (qRT‐PCR) for detection of mRNA levels
The detection of the prostate‐specific antigen (PSA) gene expression has been described previously [36]. Shortly, 106 LNCaP or C4–2 cells were seeded out per 10‐cm dish in appropriate medium containing 10% FCS. Forty hours later the cells were washed twice with PBS and the medium was replaced with fresh RPMI (10% charcoal‐stripped FCS). After further incubation for 48 hrs the medium was changed to fresh RPMI (10% charcoal‐stripped FCS) and the indicated hormones and/or compounds were added. RNA was isolated 24 hrs after hormone induction using peqGOLD TriFast (PEQLAB Biotechnologie GMBH, Erlangen, Germany) according to the manufacturer’s protocol. 1 μg of RNA were used per sample in an one‐step qRT‐PCR reaction using the SuperScript III Platinum SYBR Green One‐Step qRT‐PCR Kit (Invitrogen) with these primer sequences:
PSA‐forw: 5′‐ACTGCATCAGGAACAAAAGCGTGA‐3′
PSA‐rev: 5′‐CGCACACACGTCATTGGAAATAAC‐3′
β‐actin‐forw: 5′‐ACAGAGCCTCGCCTTTGCCGA‐3′
β‐actin‐rev: 5′‐CACGATGGAGGGGAAGACG‐3′
Invasion assay
Cell invasion through ECM was analysed using the QCM™ 24‐well Cell Invasion Assay kit (Millipore, ECM 554) according to manufactures’ protocol. In detail, LNCaP cells were incubated in serum‐free DMEM without HEPES and antibiotics for 24 hrs. Cells were washed two times with 1× PBS, harvested with trypsin and re‐suspended in DMEM with 0.1% charcoal‐stripped FCS without HEPES and antibiotics. Subsequently, cells were pelleted (800 rpm, 7 min.), resuspended in 5‐ml DMEM with 0.1% charcoal‐stripped FCS (without HEPES and antibiotics) and were brought to a volume that gives 0.5 × 106 cells per millilitre. The pre‐warmed ECM was re‐hydrated with 300 μl pre‐warmed serum‐free DMEM for 30 min. Media were removed and replaced with 300 μl cell suspension on top of the ECM. R1881 (3 × 10–11 M), AA (10−4 M or 10−5 M), or Casodex (10−7 M) were added to the cell suspension. To the lower chamber 500 μl DMEM with 10% charcoal‐stripped FCS (with HEPES, penicillin and streptomycin) was added. The cells were incubated for 48 or 72 hrs at 37°C under 5% CO2 atmosphere. Migrated cells on the bottom of the lower chamber were counted with a microscope.
Results
AA as a receptor‐specific androgen antagonist that inhibits PSA expression
Previously, we have described the isolation of AA from a P. africanum methylene chloride extract [25]. To investigate the potential anti‐androgenic activity of AA, we performed transient transfection assays in CV1 cells with cotransfecting human AR with the mouse mammary tumour virus (MMTV)‐luc reporter gene, a well‐known AR responsive promoter [26]. CV1 cells were used because they lack functional AR, GR or PR that are able to bind to the same response element and thus interfere with the functional assays. Also, CV1 cells lack functional thyroid hormone and oestrogen receptors. Initial experiments revealed that AA effectively inhibited the AR‐mediated transaction at 10–5 M (supplementary data). Cells were treated without or with increasing concentrations of the agonists DHT (Fig. 1A) or methyltrieneolone (R1881; Fig. 1B). The latter is a synthetic AR‐specific agonist that is not metabolized, whereas DHT can be degraded and may serve as ligands for other nuclear receptors [37]. Co‐treatment of androgens with AA leads to the inhibition of the ligand‐activated human AR whereas treatment of AA alone did not affect the reporter activity. Thus, the data reveal that AA inhibits the ligand‐induced transactivation of AR and suggest that AA has an anti‐androgenic activity. Concentration series of the natural androgen DHT were performed to determine the efficacy to inhibit DHT‐activated AR (Fig. 1A). At 10 nM DHT, AA (10 μM) was able to strongly inhibit AR mediated transactivation. As control, treatment of cells with the solvent itself did not affect the reporter. AA also potently inhibited AR‐mediated transactivation in a dose‐dependent manner using the synthetic AR‐ligand R1881 (Fig. 1B). This provides an indication that AA competes with DHT or R1881 for binding to AR.
Figure 1.
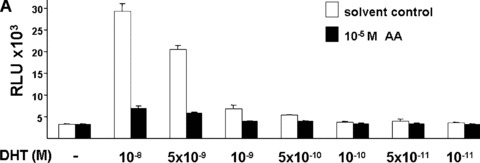
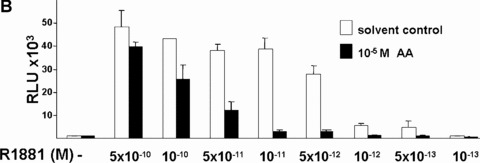
Inhibition of transactivation function of ligand‐activated human AR by AA isolated from P. africanum bark extracts. CV1 cells, lacking functional endogenous AR were transfected with the expression vector for human AR and the androgen responsive reporter MMTV‐luciferase treated with the indicated concentration series of the androgen (white bars) DHT (A) or with the synthetic AR ligand R1881 (B). In addition cells were treated with 10 μM AA (black bars), dissolved in ethanol/DMSO (1:1) or as control with this solvent alone. Luciferase values obtained were normalized to the cotransfected internal control and indicated as relative light units. The error bars indicate the variation of the mean.
Taken together, the data suggest that AA is able to repress the transactivation function mediated by agonist‐induced human AR.
To analyse the receptor specificity of AA, various members of the nuclear hormone receptor superfamily were used for reporter assays (Fig. 2). The closely related members are GR and PR, for which the MMTV reporter is also a suitable responsive reporter. Ten and 100 μM of AA did not affect the glucocorticoid‐induced transactivation of human GR significantly. In case of human PR, both isoforms were tested, the PR‐B isoform with a more potent transactivation function and the PR‐A isoform, which is generated by an internal translation start site. The progesterone‐induced transactivation of both PR forms were unaffected by co‐treatment with 10 μM AA, whereas the transactivation mediated predominately by the PR‐B isoform was strongly inhibited at 10‐fold higher AA concentrations (100 μM; Fig. 2A). Using TR with a TR responsive reporter, p(DR4)2‐tk‐luc, the addition of AA did not affect significantly the thyroid hormone (T3)‐induced transactivation of TR (Fig. 2B). Furthermore, AA was tested for its influence on the oestrogen receptors ER‐α‐ and ER‐β‐mediated transactivation. In contrast to AR and GR, the ER is localized predominantly in the nucleus in the absence of ligand [38, 39, 40]. Interestingly, the hormone‐induced transactivation of both ERs was not affected by AA. However, in the absence of oestrogens both ERs were only weakly induced by 10 μM and more efficiently by 100 μM AA in a dose‐ and receptor‐dependent manner (Fig. 2C and not shown). This suggests that AA at higher concentrations serves as ER agonist. Taking together, this indicates that AA is at lower concentrations specific for AR and affects only at higher concentrations PR and ER significantly.
Figure 2.
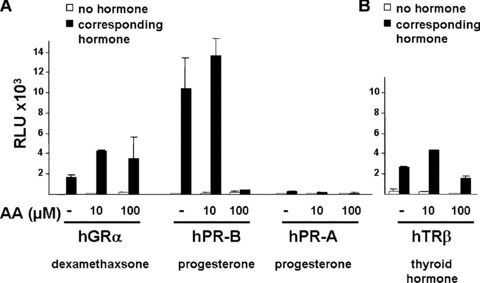
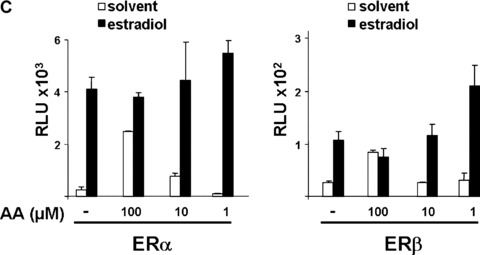
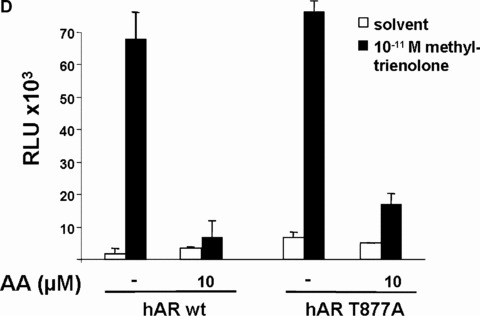
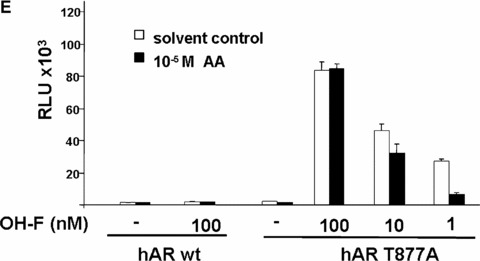
AA at lower concentrations inhibits specifically ligand‐activated human AR. Closely related members of the nuclear hormone receptor superfamily were selected to test for their inhibition by AA. The human glucocorticoid receptor (hGR), both human progesterone receptors (PR‐A and ‐B forms), the human thyroid hormone receptor β (human TR‐β), and both human oestrogen receptors (ER‐α and ER‐β) were tested with the appropriate reporters (MMTV‐luciferase for hGR and both PR forms (A), p(DR4)2‐tk‐luc for TR (B), and p3ERE‐TATA‐luc for the ERs (C), respectively) in CV1 cells, lacking functional endogenous GR, PR, ER or TR. Indicated concentrations of AA were tested for inhibition of glucocorticoid (dexamethasone) induced hGR, progesterone‐induced PR‐A and ‐B isoforms, estradiol for both ERs, and of thyroid hormone for TRβ. (D) The mutant AR T877A, which is endogenously expressed in LNCaP cells was compared to the wild‐type AR for inhibition by AA. (E) AA inhibits the transactivation mediated by the OH‐F treated mutant AR T877A.
This suggests that at lower concentrations of 10 μM AA has a strong preference for the AR and is an AR‐specific antagonist.
LNCaP cells are used as a model cell line for analysing human PCa. For that purpose we used the human LNCaP PCa cell line that is known to grow androgen dependently. In addition, we employed the C4–2 PCa cell line that also expresses functional endogenous AR and exhibits an androgen‐independent growth [34] making them an interesting model representing the transition of the initial androgen‐dependent disease to an androgen‐independent state. Both cell lines express the mutant AR, AR T877A, which can be activated by the complete androgen antagonist OH‐F [19]. The activation of this AR mutant by the complete antagonist OH‐F represents a mechanism by which the cancer becomes resistant to hormone therapy treatment. Because AA (for structure see supplemental data) has more structural similarities to OH‐F compared to other AR antagonists used in PCa therapy, we focused on comparing AA with OH‐F activity.
Therefore, first pre‐tests were performed to test whether AA is able to inhibit the transactivation of the mutant AR T877A. The effect of AA with 10 mM AA with or without androgen agonist on wild‐type and the mutant AR T877A was compared (Fig. 2D). In contrast, OH‐F activated the mutant AR effectively in the absence of agonist and poorly inhibited AR transactivation in the presence of R1881 (supplementary data). As expected, AA inhibited also the OH‐F‐induced transactivation of the mutant AR T877A (supplementary data). Taken together, the obtained data reveal that AA inhibits the agonist‐induced transactivation of both wild‐type and mutant AR T877A.
To analyse whether AA represses an endogenous AR‐regulated gene, the well‐known AR target gene PSA was analysed. Androgens induce the expression PSA through the AR that binds to androgen response elements in the promoter and enhancer region of PSA. Therefore, to confirm the AR antagonism of AA also for an endogenous AR target gene, the endogenous PSA expression of both LNCaP and C4–2 cells were analysed by treatment with or without DHT or R1881 in the absence or presence of AA at 10−4 or 10−5 M. The RNA was extracted and analysed by quantitative RT‐PCR for PSA expression levels (Fig. 3). Treatment of both cell types with either agonist induced the PSA mRNA expression. For LNCaP cells, AA inhibited the expression of PSA at 10−5 M. The inhibition was more pronounced using 10−4 M AA (Fig. 3A). Treatment of C4–2 cells with 10−4 M AA inhibited the expression of PSA also in the absence of agonist (Fig. 3B), which might reflect the notion that AR is partially active in these cells [41]. At 10−4 M AA, both DHT‐ and R1881‐induced PSA expression was potently inhibited. Notably, this was also observed using the lower 10−5 M AA concentration. Thus, these findings suggest that AA inhibits the expression of the PSA gene in both androgen‐dependent and androgen‐independent PCa cells.
Figure 3.
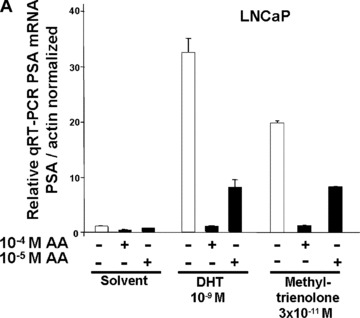
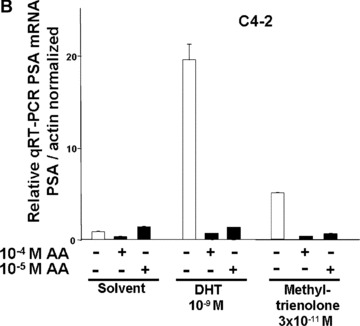
AA inhibits the endogenous PSA gene expression of androgen‐dependent and androgen‐independent PCa cells. The androgen‐dependent LNCaP (A) or the androgen‐independent C4–2 cells (B) were treated with the indicated androgens DHT or R1881 (methyltrienolone) with and without AA. Quantitative real‐time RT‐PCR was performed for PSA mRNA and β‐actin mRNA for normalization.
Compared to OH‐F, which has among the AR antagonists used in PCa therapy structurally the most similarities to AA and is an agonist of the AR mutant T877A, the obtained data using AA suggest that AA appears to act distinct from OH‐F.
Taken together, AA can inhibit the AR‐mediated transactivation and endogenous androgen‐induced gene expression. Thus, these findings suggest that AA is a novel AR antagonist.
Growth inhibition of human prostate cancer cells by AA
The growth of prostate cells and PCa cells is initially dependent on androgens [11, 13]. To analyse whether the androgen antagonism of AA affects cell growth, LNCaP and C4–2 cells were used. Although AA represents a novel structural platform for AR antagonists (for structure see supplementary data), AA has some structural similarities to OH‐F. Therefore, we compared the effect of AA with that of OH‐F on cell growth using the human PCa cell lines LNCaP and C4–2, expressing endogenously the human AR, with the PC3 as well as with CV1 kidney cell lines, that lack endogenously expressed AR. Because growth of LNCaP cells in long‐term cultures is impaired with charcoal‐stripped serum to remove hormones, we used normal serum that is known to contain androgens naturally and represents a similar situation as in patients with PCa. For that purpose an equal number of cells were seeded out in 5% untreated serum together with or without addition of OH‐F or AA. As expected, treatment of LNCaP and C4–2 cells with OH‐F revealed no significant inhibition of LNCaP cell growth (Fig. 4A, 4B). Interestingly, treatment of LNCaP cells with AA inhibited potently LNCaP proliferation. Growth inhibition was observed after 4 to 6 days of treatment with AA. We did not observe enhanced de‐attachment or an increase in the apoptosis rate by AA. The growth inhibitory effect was more prominent after day 10 of treatment. This strongly indicates that AA is mechanistically different to OH‐F for mediating inactivation of AR.
Figure 4.
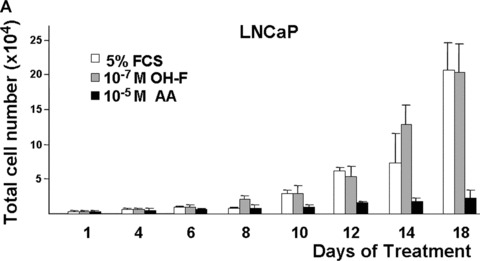
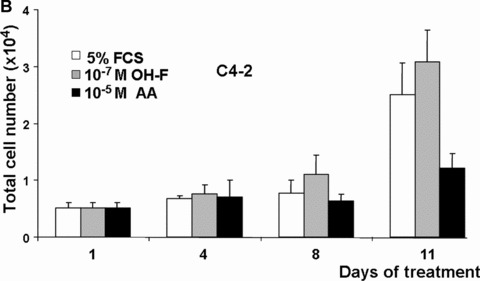
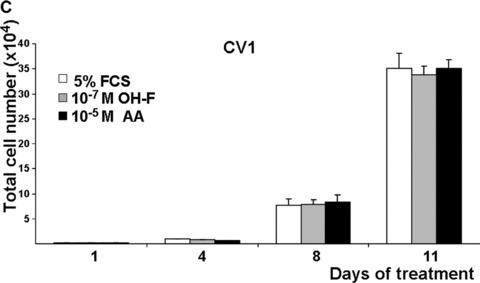
AA inhibits the growth of the prostate cancer (PCa) cell lines LNCaP and C4–2. (A) The model human Pca cell line LNCaP cells were treated with 10 μM AA. Equal amounts of cells were seeded out and were either treated with AA, the known androgen antagonist hydroxyflutamide (OH‐F) or solvent control. The number of cells was counted at the indicated days. (B and C) Same experimental set‐up except the human androgen‐independent growing PCa cell line C4–2 (B) or the monkey kidney cell line CV1 (C) was used.
Treatment of the androgen‐independent PCa cell line C4–2 also led to a growth reduction by AA and not by OH‐F (Fig. 4B) without observing toxic effects. Even after treatment with 10−4 M AA no detectable cell toxicity was observed. In contrast to C4–2 cells, using CV1 cells that lack endogenously expressed AR, no significant change in growth was observed by treatment with either OH‐F or AA (Fig. 4C). Similarly, the growth rate of the AR‐deficient PC3 PCa cell line was not affected by AA (supplementary data). The lack of influence on the growth of both AR negative CV1 and PC3 cells suggests that AA has no significant toxicity and implies that AA inhibits the AR.
Taken together, these data suggest that the androgen antagonism of AA is able to inhibit growth of human PCa cells.
AA inhibits cell invasion through ECM
The AR agonist R1881, besides promoting PCa cell proliferation, also enhances cell invasion through ECM, thought to be an important step towards cancer metastasis [42]. In line with previous observations, R1881 enhances cellular invasion of LNCaP cells through ECM (Fig. 5). Interestingly, addition of AA at 10 μM abrogated this promoting effect. Further, using 100 μM AA cell invasion was further inhibited. AA itself had no significant effect on cell invasion (supplementary data). These findings suggest that AA is an AR antagonist also at the level of cellular invasion.
Figure 5.
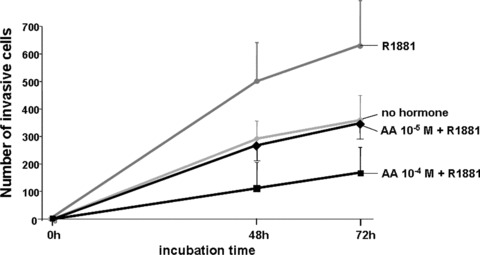
AA inhibits cellular invasion of LNCaP cells through extracellular matrix (ECM). Cell invasion through ECM was analysed using LNCaP cells. The cells were seeded out with 0.1% charcoal‐treated FCS with R1881 (3 × 10−11 M), with and without AA (10−4 or 10−5 M) and incubated for 48 or 72 hrs at 37°C under 5% CO2 atmosphere. Migrated cells through ECM were counted with a microscope.
Thus, these observations suggest that AA inhibits cellular invasion through ECM.
AA inhibits ligand‐induced nuclear localization
To get insights into the molecular mechanisms of AA‐mediated inhibition of AR, we first analysed whether the LBD of AR is target of inhibition by AA. As expected, deletion of the LBD (AR –ΔC) leads to a hormone‐independent transactivation (Fig. 6). However, compared with the wild‐type AR, the AR –ΔC activity was unaffected by AA (Fig. 6). This suggests that the LBD is the target of AA‐mediated repression. The N‐terminal deletion of AR (AR –ΔC) exhibited no significant transactivation function and was not affected by AA, indicating that AA treatment has no significant influence on this mutant and does not unspecifically influence the promoter activity. Further, because the SUMO‐mutant of AR, that was shown to lack binding of the corepressor SMRT [27] and Alien [36], was also inhibited by AA (Fig. 6A), it appears that corepressor binding to AR is not involved as a molecular mechanism of repression of AR by AA. Lack of interaction of various corepressors with AR in mammalian cells treated with of AA suggests this notion (supplementary data).
Figure 6.
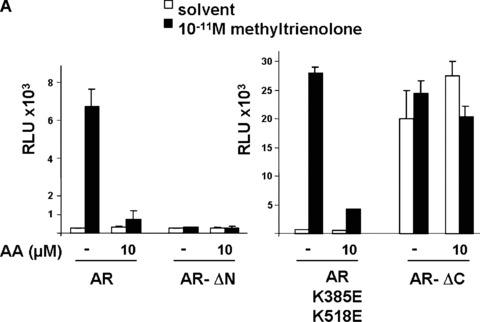
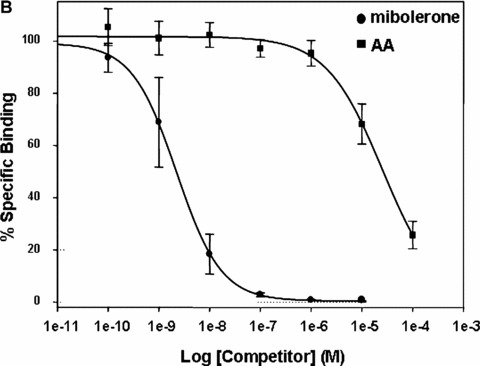
The ligand‐binding domain of AR is target for AA‐mediated inhibition. (A) The indicated AR deletion mutants were tested for their ability to be repressed by AA. AR –ΔN, N‐terminal deletion of AR (aa 1–504 are deleted); AR –ΔC, C‐terminal deletion (aa 628–919 are deleted); AR K385E and K518E represent amino acid exchanges at these positions. (B) Competitive whole cell binding assays were performed using pSG5‐flag‐wtAR transfected COS‐7 cells incubated with 1 nM (3H)‐mibolerone in the absence and presence of increasing concentrations of either unlabelled mibolerone (circle) or AA (rectangles) for 90 min. Competition for binding is illustrated by the percentage of [3H] mibolerone specifically bound to the AR. Results are averages of three independent experiments with each condition in triplicate (±S.E.M.).
To further investigate the molecular mechanism of AA‐mediated inhibition of AR, corepressor recruitment and AR stability were analysed. It is known that androgen antagonists recruit corepressors, such as NCoR, SMRT or Alien to inactivate the AR [27, 32, 36, 43, 44, 45, 46]. However, using our previously established modified two‐hybrid test system we were unable to detect corepressor interaction with AR in the presence of AA (supplementary data).
Because the deletion analysis suggests that AA acts through the hormone‐binding domain of AR, competitive hormone binding assays were performed to detect whether AA is able to compete for androgen binding to AR (Fig. 6B). Using increasing amounts of AA, competition of the androgen mibolerone was observed at the concentration of 10 μM that was effective in both inhibition of transactivation and growth proliferation. Thus, these data suggest that AA binds to the LBD of AR.
Another potential possibility to inhibit AR‐mediated transactivation is to decrease AR protein levels by AA. Pre‐tests of control experiments of AR levels in transfected CV1 cells used for the reporter assays suggested that AA did not reveal a change of AR protein level (supplemental data). To investigate the protein levels of endogenously expressed AR, LNCaP cells were stimulated with or without AA in the presence of 5% FCS prior preparation of whole cell extracts. Subsequent Western analyses did also not reveal an AA‐induced decrease of AR protein levels (Fig. 7A).
Figure 7.
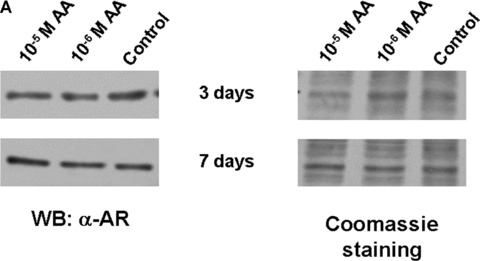
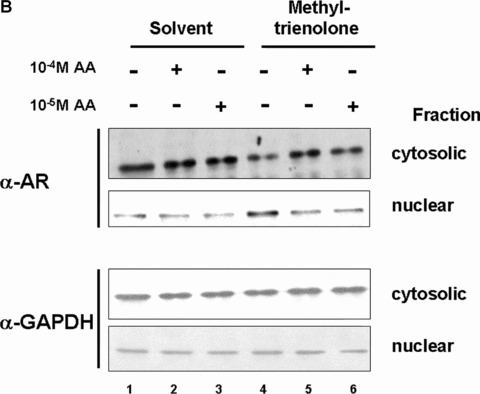

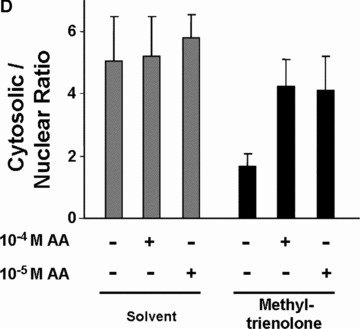
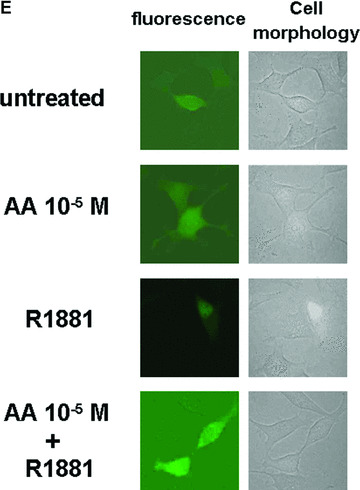
The ligand‐induced nuclear trans‐location of AR is inhibited by AA. (A) AR protein levels with or without treatment of LNCaP cells with AA detected by Western analysis. (B) AR levels in the cytosolic and nuclear fractions were detected by Western with treatment of AA alone or with methyltrienolone in combination. Detection of GAPDH levels serves as internal loading control. (C) As internal controls for fractionation actin and nucleophosmin were detected in cytosolic and nuclear fractions. (D) Quantification of the cytosolic and nuclear ratio of AR dependent on the indicated treatment with AA and methyltrienolone. The variation of the mean of six experiments is shown. (E) HeLa cells transiently transfected with expression vector of GFP‐AR fusion were treated with AA (10−5 M) and/or R1881 (3 × 10−11 M) for 1 hr. Unfixed cells were then analysed using a fluorescence microscopy with a fluorescine isothiocyanate filter. As control, cell morphology with the fluorescence overlay is shown.
To analyse the possibility that AA inhibits the ligand‐induced translocation of AR to the nucleus LNCaP cells were treated with or without androgen and AA prior cell fractionation to yield a cytosolic and nuclear fraction (Fig. 7B and C). Ligand‐induced nuclear localization of AR was observed with methyltrienolone treatment decreasing the amount of cytosolic AR and increasing the nuclear AR (Fig. 7B, compare lane 1 with lane 4). As controls for loading the detection of glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH; Fig. 7B) and for cytosolic and nuclear fractionation that of actin and nucleophosmin were used (Fig. 7C). Interestingly, addition of AA abrogated the methyltrienolone‐induced nuclear translocation of AR (Fig. 7B and D, lane 5 and 6) at the effective concentrations that lead to inhibition of AR‐mediated transactivation and cell growth. To confirm the inhibition of the AR translocation a GFP‐AR fusion was employed. Treatment of cells with R1881 resulted in predominantly nuclear staining of living cells whereas AA alone or AA in combination with R1881 resulted in predominant cytosolic staining (Fig. 7). These data confirm the observed inhibition of agonist‐induced nuclear translocation of AR.
Thus, these data suggest that AA competes with androgens for the binding to AR and inhibits the nuclear translocation of AR.
Discussion
One of the most common diseases of aging men are BPH and PCa. PCa developed in recent years as the second leading cause of cancer death [3]. Anti‐androgens are used in the current treatment of both diseases, as they can block the AR‐mediated gene transactivation [7, 13]. The commonly used anti‐androgens exhibit a much lower affinity for AR compared to the known agonists. Cyproterone acetate, OH‐F and Casodex have an affinity for AR that is reported to be more than a magnitude lower compared to that of agonists [47, 48, 49]. The reasons for that are unknown.
Estrogens have been suggested to inhibit PCa cell growth [50]. Estrogens bind to ERs whereas ER‐β is the predominant ER in prostate cells. However, the roles of ERs are still controversial in influencing prostate cell growth [51]. Because AA is agonistic for ERs at a much higher concentration compared to the AR antagonism, the suitability of AA at these high concentrations on PCa cell growth cannot be predicted.
Extracts from the bark of P. africanum (Tadenan®) are one of the most famous phytotherapeutic agents, which have been used since centuries in Africa and earn increasing popularity in Europe and in United States for the treatment of BPH [22]. Recently, the extracts were shown to inhibit PCa growth [24]. In the present study, we have characterized the natural compound AA, identified from P. africanum, as an androgen antagonist. AA is very effective at 10 μM concentration both in inhibiting the ligand‐activated human AR and in inhibiting potently the growth of the AR expressing human PCa cell lines LNCaP and C4–2.
The currently used anti‐androgens in PCa therapy have been shown not only to promote the translocation of AR from the cytoplasm to the nucleus but also to mediate the recruitment of the receptor to the target genes [45, 52, 53]. A molecular mechanism of the anti‐androgen‐mediated inhibition of AR of the known anti‐androgens used in therapy was shown to involve corepressors [27, 36, 45, 46, 47]. As a molecular basis for the AR‐antagonism by AA, we propose that AA inhibits the androgen‐induced translocation of AR into the cell nucleus. This also suggests that AA inhibits AR transactivation not necessarily through corepressor recruitment.
Treatment with AA did not significantly affect the proliferation of CV1 and PC3 cells (Fig. 3 and supplemental data), which indicates that AA inhibits growth not through a general unspecific toxic effect and appears to be more specific for AR expressing cells. This does not rule out that other components of the extracts of P. africanum also influence cell growth or could synergize with AA to inhibit PCa growth.
In this report we also demonstrate that AA displays its anti‐androgenic potential due to the competition of androgens for the binding to the AR and by inhibition of its nuclear translocation. AA is able to inhibit the transactivation mediated by both DHT and R1881 bound human AR (Fig. 1). Whereas DHT is subject to enzymatic degradation and thus inactivation in cells, the synthetic R1881 is very stable. Therefore, we conclude that AA does not act through pathways destabilizing DHT through activation of androgen degrading enzymes, rather AA possesses other antagonistic mechanism. In addition, our findings have the notion that AA mediates AR‐antagonism also through inhibition of androgen‐induced cellular invasion through ECM suggesting that AA is important to inhibit growth and might also to reduce metastatic potential of PCa.
Furthermore, the ability of AA at the concentration of 10 μM to inhibit the androgen‐mediated transactivation appears to be specific for AR, because AA is not able to repress the activity of the other tested nuclear receptors at this concentration.
Resistance to antihormone therapy is a major problem for long‐term treatment of patients with PCa. Thus, one goal to overcome therapy resistance is to make use of different androgen antagonists that inactivate the AR at different stages during its activation. Several mechanisms, including mutations of AR, are known to contribute to therapy resistance. Both LNCaP and C4–2 cell lines express a mutant AR (AR T877A) that can be activated by OH‐F and renders the cells resistant to growth inhibition [19, 20]. Although, AA shares some structural similarities with OH‐F, based on its chemical structure, AA represents a novel lead structure and a platform for an anti‐androgen that may be fruitfully used for AA‐based derivatives. The transcriptional and growth analysis suggest that AA inactivates the AR‐based on a different mechanism as OH‐F, which activates the mutant AR T877A and does not inhibit LNCaP or C4–2 cell growth. Notably, this threonine at position 877 is frequently mutated in PCa cells [15]. Taken together, it suggests that AA uses a different underlying antagonistic mechanism as compared to OH‐F and may serve as a novel lead structure and chemical platform for natural antagonists.
References
- 1. Sampson N, Untergasser G, Plas E, et al. The ageing male reproductive tract. J Pathol. 2007; 211: 206–18. [DOI] [PubMed] [Google Scholar]
- 2. Skolarikos A, Thorpe AC, Neal DE. Lower urinary tract symptoms and benign prostatic hyperplasia. Minerva Urol Nefrol. 2004; 56: 109–22. [PubMed] [Google Scholar]
- 3. Bray F, Sankila R, Ferlay J, et al. Estimates of cancer incidence and mortality in Europe in 1995. Eur J Cancer. 2002; 38: 99–166. [DOI] [PubMed] [Google Scholar]
- 4. Andriole G, Bruchovsky N, Chung LW, et al. Dihydrotestosterone and the prostate: the scientific rationale for 5alpha‐reductase inhibitors in the treatment of benign prostatic hyperplasia. J Urol. 2004; 172: 1399–403. [DOI] [PubMed] [Google Scholar]
- 5. Ross R, Bernstein L, Judd H, et al. Serum testosterone levels in healthy young black and white men. J Natl Cancer Inst. 1986; 76: 45–8. [PubMed] [Google Scholar]
- 6. Ntais C, Polycarpou A, Tsatsoulis A. Molecular epidemiology of prostate cancer: androgens and polymorphisms in androgen‐related genes. Eur J Endocrinol. 2003; 149: 469–77. [DOI] [PubMed] [Google Scholar]
- 7. Balk SP. Androgen receptor as a target in androgen‐independent prostate cancer. Urology. 2002; 60: 132–8; discussion 138–9. [DOI] [PubMed] [Google Scholar]
- 8. Tenbaum S, Baniahmad A. Nuclear receptors: structure, function and involvement in disease. Int J Biochem Cell Biol. 1997; 29: 1325–41. [DOI] [PubMed] [Google Scholar]
- 9. Burke LJ, Baniahmad A. Co‐repressors 2000. FASB J. 2000; 14: 1876–88. [DOI] [PubMed] [Google Scholar]
- 10. Rosenfeld MG, Glass CK. Coregulator codes of transcriptional regulation by nuclear receptors. J Biol Chem. 2001; 276: 36865–8. [DOI] [PubMed] [Google Scholar]
- 11. Feldman BJ, Feldman D. The development of androgen‐independent prostate cancer. Nat Rev Cancer. 2001; 1: 34–45. [DOI] [PubMed] [Google Scholar]
- 12. Brinkmann AO, Blok LJ, de Ruiter PE, et al. Mechanisms of androgen receptor activation and function. J Steroid Biochem Mol Biol. 1999; 69: 307–13. [DOI] [PubMed] [Google Scholar]
- 13. Culig Z, Klocker H, Bartsch G, et al. Androgen receptors in prostate cancer. Endocr Relat Cancer. 2002; 9: 155–70. [DOI] [PubMed] [Google Scholar]
- 14. Taplin ME. Drug insight: role of the androgen receptor in the development and progression of prostate cancer. Nat Clin Pract Oncol. 2007; 4: 236–44. [DOI] [PubMed] [Google Scholar]
- 15. Taplin ME, Bubley GJ, Ko YJ, et al. Selection for androgen receptor mutations in prostate cancers treated with and‐ rogen antagonist. Cancer Res. 1999; 59: 2511–5. [PubMed] [Google Scholar]
- 16. Tindall DJ. Pursuing the androgen pathway on the quest to control prostate cancer. Cancer Biol Ther. 2007; 6; 805–10. [DOI] [PubMed] [Google Scholar]
- 17. Zhao XY, Malloy PJ, Krishnan AV, et al. Glucocorticoids can promote androgen‐independent growth of prostate cancer cells through a mutated androgen receptor. Nat Med. 2000; 6: 703–6. [DOI] [PubMed] [Google Scholar]
- 18. Dehm SM, Tindall DJ. Molecular regulation of androgen action in prostate cancer. J Cell Biochem. 2006, 99: 333–44. [DOI] [PubMed] [Google Scholar]
- 19. Culig Z, Hoffmann J, Erdel M, et al. Switch from antagonist to agonist of the androgen receptor bicalutamide is associated with prostate tumour progression in a new model system. Br J Cancer. 1999; 81: 242–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Veldscholte J, Berrevoets CA, Brinkmann AO, et al. Anti‐androgens and the mutated androgen receptor of LNCaP cells: differential effects on binding affinity, heat‐shock protein interaction, and transcription activation. Biochemistry. 1992; 31: 2393–9. [DOI] [PubMed] [Google Scholar]
- 21. Levin RM, Das AK. A scientific basis for the therapeutic effects of Pygeum africa‐num and Serenoa repens. Urol Res. 2000; 28: 201–9. [DOI] [PubMed] [Google Scholar]
- 22. Dreikorn K. The role of phytotherapy in treating lower urinary tract symptoms and benign prostatic hyperplasia. World J Urol. 2002; 19: 426–35. [DOI] [PubMed] [Google Scholar]
- 23. Yoshimura Y, Yamaguchi O, Bellamy F, et al. Effect of Pygeum africanum tadenan on micturition and prostate growth of the rat secondary to coadministered treatment and post‐treatment with dihydrotestosterone. Urology. 2003; 61: 474–8. [DOI] [PubMed] [Google Scholar]
- 24. Shenouda NS, Sakla MS, Newton LG, et al. Phytosterol Pygeum africanum regulates prostate cancer in vitro and in vivo . Endocrine. 2007; 31: 72–81. [DOI] [PubMed] [Google Scholar]
- 25. Schleich S, Papaioannou M, Baniahmad A, et al. Extracts from Pygeum africanum and other ethnobotanical species with antiandrogenic activity. Planta Med. 2006; 72: 807–13. [DOI] [PubMed] [Google Scholar]
- 26. Gast A, Schneikert J, Cato AC. N‐terminal sequences of the human androgen receptor in DNA binding and transrepressing functions. J Steroid Biochem Mol Biol. 1998; 65: 117–23. [DOI] [PubMed] [Google Scholar]
- 27. Dotzlaw H, Moehren U, Mink S, et al. The amino terminus of the human AR is target for corepressor action and antihormone agonism. Mol Endocrinol. 2002; 16: 661–73. [DOI] [PubMed] [Google Scholar]
- 28. Schulz M, Eggert M, Baniahmad A, et al. RU486‐induced glucocorticoid receptor agonism is controlled by the receptor N terminus and by corepressor binding. J Biol Chem. 2002; 277: 26238–43. [DOI] [PubMed] [Google Scholar]
- 29. Baniahmad A, Tsai SY, O’Malley BW, et al. Kindred S thyroid hormone receptor is an active and constitutive silencer and a repressor for thyroid hormone and retinoic acid responses. Proc Natl Acad Sci USA. 1992; 89: 10633–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Leers J, Steiner C, Renkawitz R, et al. A thyroid hormone receptor‐dependent glucocorticoid induction. Mol Endocrinol. 1994; 8: 440–7. [DOI] [PubMed] [Google Scholar]
- 31. Kalkhoven E, Valentine JE, Heery DM, et al. Isoforms of steroid receptor co‐activator 1 differ in their ability to potentiate transcription by the oestrogen receptor. EMBO J. 1998; 17: 232–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dotzlaw H, Papaioannou M, Moehren U, et al. Agonist‐antagonist induced coactivator and corepressor interplay on the human androgen receptor. Mol Cell Endocrinol. 2003; 213: 79–85. [DOI] [PubMed] [Google Scholar]
- 33. Protopopov AI, Li J, Winberg G, et al. Human cell lines engineered for tetracycline‐regulated expression of tumor suppressor candidate genes from a frequently affected chromosomal region, 3p21. J Gene Med. 2002; 4: 397–406. [DOI] [PubMed] [Google Scholar]
- 34. Thalmann GN, Anezinis PE, Chang SM, et al. Androgen‐independent cancer progression and bone metastasis in the LNCaP model of human prostate cancer. Cancer Res. 1994; 54: 2577–81. [PubMed] [Google Scholar]
- 35. Tanner TM, Verrijdt G, Rombauts W, et al. Anti‐androgenic properties of Compound A, an analog of a non‐steroidal plant compound. Mol Cell Endocrinol. 2003; 201: 155–64. [DOI] [PubMed] [Google Scholar]
- 36. Moehren U, Papaioannou M, Reeb CA, et al. Alien interacts with the human androgen receptor and inhibits prostate cancer cell growth. Mol Endocrinol. 2007; 21: 1039–48. [DOI] [PubMed] [Google Scholar]
- 37. Negri‐Cesi P, Motta M. Androgen metabolism in the human prostatic cancer cell line LNCaP. J Steroid Biochem Mol Biol. 1994; 51: 89–96. [DOI] [PubMed] [Google Scholar]
- 38. Htun H, Holth LT, Walker D, et al. Direct visualization of the human estrogen receptor alpha reveals a role for ligand in the nuclear distribution of the receptor. Mol Biol Cell. 1999; 10: 471–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Welshons WV, Lieberman ME, Gorski J. Nuclear localization of unoccupied oestrogen receptors. Nature. 1984; 307: 747–9. [DOI] [PubMed] [Google Scholar]
- 40. Yamashita S. Intranuclear localization of hormone‐occupied and ‐unoccupied estrogen receptors in the mouse uterus: application of 1 nm immunogold‐silver enhancement procedure to ultrathin frozen sections. J Electron Microsc. 1995; 44: 22–9. [PubMed] [Google Scholar]
- 41. Chen S, Xu Y, Yuan X, et al. Androgen receptor phosphorylation and stabilization in prostate cancer by cyclin‐dependent kinase 1. Proc Natl Acad Sci USA. 2006; 103: 15969–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chuan YC, Pang ST, Cedazo‐Minguez A, et al. Androgen induction of prostate cancer cell invasion is mediated by ezrin. J Biol Chem. 2006; 281: 29938–48. [DOI] [PubMed] [Google Scholar]
- 43. Liao G, Chen LY, Zhang A, et al. Regulation of androgen receptor activity by the nuclear receptor corepressor SMRT. J Biol Chem. 2003; 278: 5052–61. [DOI] [PubMed] [Google Scholar]
- 44. Shang Y, Myers M, Brown M. Formation of the androgen receptor transcription complex. Mol Cell. 2002; 9: 601–10. [DOI] [PubMed] [Google Scholar]
- 45. Zhu P, Baek SH, Bourk EM, et al. Macrophage/cancer cell interactions mediate hormone resistance by a nuclear receptor derepression pathway. Cell. 2006; 124: 615–29. [DOI] [PubMed] [Google Scholar]
- 46. Chen CD, Welsbie DS, Tran C, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004; 10: 33–9. [DOI] [PubMed] [Google Scholar]
- 47. Buzek SW, Sanborn BM. Nuclear androgen receptor dynamics in testicular peritubular and Sertoli cells. J Androl. 1990; 11: 514–20. [PubMed] [Google Scholar]
- 48. Mukherjee A, Kirkovsky L, Yao XT, et al. Enantioselective binding of Casodex to the androgen receptor. Xenobiotica. 1996; 26: 117–22. [DOI] [PubMed] [Google Scholar]
- 49. He Y, Yin D, Perera M, et al. Novel nonsteroidal ligands with high binding affinity and potent functional activity for the androgen receptor. Eur J Med Chem. 2002; 37: 619–34. [DOI] [PubMed] [Google Scholar]
- 50. Heldring N, Pike A, Andersson S, et al. Estrogen receptors: how do they signal and what are their targets. Physiol Rev. 2007; 87: 905–31. [DOI] [PubMed] [Google Scholar]
- 51. Prins GS, Korach KS. The role of estrogens and estrogen receptors in normal prostate growth and disease. Steroids. 2008; 73: 233–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jenster G, Trapman J, Brinkmann AO. Nuclear import of the human androgen receptor. Biochem J. 1993; 293: 761–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Masiello D, Cheng S, Bubley GJ, et al. Bicalutamide functions as an androgen receptor antagonist by assembly of a transcriptionally inactive receptor. J Biol Chem. 2002; 277: 26321–6. [DOI] [PubMed] [Google Scholar]