

Abstract
Transcription factor E2F1 is a key regulator of cell proliferation and apoptosis. Its activity is strictly controlled by the pRB/E2F pathway. In the majority of cancer cells, however, this pathway is frequently found deregulated, and the underlying mechanism involving transcriptional control by E2F1 has not yet been fully elucidated. Here we report the identification of two putative E2F1‐binding sites located upstream from Siah1 transcription start site (+1). Chromatin immunoprecipitation assay reveals that transcription factor E2F1 is capable of binding to the putative sites, and luciferase reporter assay shows that E2F1 can activate transcription from the Siah1 promoter. Ectopic expression of E2F1 elevates the Siah1 level, hence suppressing the β‐catenin/TCF activity. Consistently, knock‐down of endogenous E2F1 by a shRNA strategy results in reduced expression of Siah1. Moreover, repression of β‐catenin/TCF activity by E2F1 can be attenuated by shRNA‐based repression of endogenous Siah1, implying that Siah1 is a bona fide E2F1 target gene, which at least partly, mediates the suppression of β‐catenin/TCF signalling pathway.
Keywords: E2F1, Siah1, transcription, β‐catenin/TCF
Introduction
The E2F1, a member of the E2F transcription factor family, is a regulator of cell proliferation and apoptosis and was first identified as a factor bound to the promoter of adenovirus E2 gene. E2Fs control the cell cycle by regulating the expression of a number of genes, whose products are required for the S‐phase entry and cell cycle progression [1]. Most of the E2F family proteins can associate with and are regulated by pocket protein family proteins in a cell cycle progression‐dependent manner. Among eight E2F family members identified so far, E2F1 is best characterized. It strongly promotes cell cycle progression by regulating critical regulator genes involved in the DNA replication and G1/S transition. In addition, numerous studies have suggested that ectopic expression of E2F1 induces apoptosis through different mechanisms [2, 3, 4]. Many important genes involved in apoptosis have been identified as E2F1 targets and in addition, E2F1 has been reported to induce apoptosis by stabilizing and activating p53 activity. Moreover, E2F1‐deficient mice exhibit a defect in thymocyte apoptosis and an increasing susceptibility to the development of tumours [5, 6], which implies the ability of E2F1 to suppress tumorigenesis under physiological circumstances.
Seven in absentia homologue 1 (Siah‐1), a member of the RING‐finger‐containing E3 ubiquitin ligases, is a highly related mammalian homologue of Drosophila Sina, which targets the transcriptional repressor Tramtrack for ubiquitination and proteasome‐mediated degradation [7, 8]. Like their Drosophila homologue, mammalian Siah proteins can bind E2 conjugating enzyme (E2s) via an amino‐terminal ring domain and promotes the targets for proteasomal degradation [9, 10]. Siah has two human homologues, Siah1 and Siah2, which share significant amino acid sequence homology. They have been implicated in the ubiquitination and proteasome‐dependent degradation for many important protein targets such as OBF1 [11, 12], Kid [13], DCC [14], Bag‐1 [15], NcoR [16], Numb [17] and α‐synuclein [18]. Recently, Siah1 was reported to potentiate the proteasomal degradation of β‐catenin through Siah‐1 interacting protein (SIP)/Skp1/Ebi [19] or APC pathway [20], and then inhibits cell proliferation by repressing the activation of T‐cell factor/lymphoid enhancer factor (TCF/LEF) family transcription factors.
Here we show that E2F1 can repress TCF/LEF activity by directly transactivating the expression of Siah1. This pathway suggests a novel effect of E2F1 on suppressing cell cycle progression via a E2F1‐Siah1‐β‐catenin/TCF pathway.
Materials and methods
Reagents and antibodies
The following antibodies were used in this study: Siah1 goat polyclonal antibody P‐18 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA); E2F1 rabbit polyclonal antibody C‐20 (Santa Cruz Biotechnology, Inc.); β‐actin mouse monoclonal antibody (Santa Cruz Biotechnology, Inc.); the majority of reagents used for this study were purchased from Sigma‐Aldrich, Inc (St Louis, MO, USA). Restriction enzymes were ordered from New England Biolabs (Beverly, MA, USA).
Plasmids
The UCSC Genome Browser Database and the Genomatix Suite of sequence analysis tools MatInspector (professional version 6.2.2) were used for analysing the Siah1 promoter. Siah1 promoter constructs were described previously [21]. A primer pair used for pSiah‐BglII was: 5′> GAA GAT CTC TAG CTT CTC GCG GGC CAG AG <3′ and 5′> CCA AGC TTC TCG AGA GCG CGC CCC GCA <3′. The promoter fragments were subcloned into the XhoI/HindIII sites of pGL3‐Basic plasmid (Promega Corporation, Madison, WI, USA). The primers used for site‐specific deletion of E2F‐binding sites are: 5′>CGC CTG CGC GCG CCC CGC CCC CCC AGG CTC<3′ and 5′>GGG GGG GCG GGG CGC GCG CAG GCG CAC AGG CGT TG<3′ for E2F‐BS1; 5′> GGC GGG CTC GAG GAC CGG CGG CGG CGG CGG CCA GGG <3′ and 5′>CCC TGG CCG CCG CCG CCG CCG GTC CTC GAG CCC GCC GCG C<3′ for E2F‐BS2. The detailed procedure of PCR‐mediated mutagenesis, E2F expression plasmids and luciferase reporter plasmid pApaf1‐Luc were described previously [22, 23]. pU6‐E2F1 shRNA and control vector were gifted by Dr. W. Douglas Cress [24]; pSuper‐Siah1 shRNA and control vector were kindly provided by Dr. Solomon H. Snyder (Department of Pharmacology and Molecular Sciences, Johns Hopkins University School of Medicine, Baltimore, MD, USA) [25].
Cell culture, transfection and RNAi
Human non‐small cell lung carcinoma cells H1299 were cultured in RPMI‐1640 medium containing 10% (v/v) fetal bovine serum (FBS) with 2 mM L‐glutamine adjusted to contain 1.5 g/l sodium bicarbonate, 4.5 g/l glucose, 10 mM HEPES and 1.0 mM sodium pyruvate. Culture medium was purchased from Invitrogen Corporation (GIBCO, Grand Island, NY, USA). Cells were maintained at 37°C in a humidified 5% CO2–containing atmosphere. Transfection of cells with various mammalian expression constructs by Lipofectamine™ 2000 (Invitrogen, Carlsbad, CA, USA) was performed according to the methods provided by manufacturer’s specification. For RNAi assay, H1299 cells were transfected with appropriate shRNA or control vector and cultured for 48 hrs before cells were harvested to examine the effectiveness of RNA interference.
Semi‐quantitative RT‐PCR and Western blot analysis
For semi‐quantitative RT‐PCR, H1299 cells were first transfected with E2F1 expression plasmid or empty vector and were cultured for additional 24 hrs. Total RNA was then isolated using SV Total RNA Isolation System (Promega Corporation) and 0.1 μg RNA from each preparation was used as PCR template. RT‐PCR was performed by TaKaRa One‐step RNA PCR Kit (AMV) (TaKaRa Bio, Inc., Otsu, Shiga, Japan) using the following specific primer pairs: Siah‐1: 5′> GC TCC ATT CGC AAC TTG GCT ATG and Siah‐2: 5′> GC TCA ACA CAT GGA AAT AGT TAC <3′; β‐actin: 5′‐GAC CTG ACT GAC TAC CTC ATG AAG AT‐3′ and 5′‐GTC ACA CTT CAT GAT GGA GTT AAG G ‐3′; Western blotting was performed according to the protocol described previously [22]. Protein concentration in each sample was measured by BCA Protein Assay Kit (Pierce Biotechnology, Inc., Rockford, IL, USA).
Chromotin immunoprecipitation assay (ChIP)
ChIP assays were performed as described previously [23]. Briefly, H1299 cells (2 × l07 cells) were fixed with 1% formaldehyde and then neutralized by adding 0.125 M glycine. Cells were collected and lysed in cell lysis buffer (5 mM PIPES, pH 8.0, 85 mM KCl, 0.5% NP40, protease inhibitor cocktail). Nuclei pellet was obtained by centrifuge and subsequently lysed in Nuclei lysis buffer (50 mM Tris‐Cl, pH 8.1, 10 mM ethylenediaminetetraacetic acid (EDTA), 1% SDS, protease inhibitor cocktail). The nuclei lysate was sonicated to obtain soluble chromatin with average length of 1000 bp. After 1:10 dilution by dilution buffer (10 mM Tris‐Cl, pH 8.1, 2 mM EDTA, 150 mM NaCl, 1% Trition X‐100), chromatin solutions were pre‐cleared and then incubated with or without anti‐E2F1 antibody (sc‐193X, Santa Cruz Biotechnology, Inc.) and the mixture was incubated at 4°C overnight on a rotating platform. The same immunocomplexes were recovered with protein A‐Sepharose beads. After extensive washing, the bound DNA fragments were eluted, and the resulting DNAs were subjected to PCR reactions using the following primer pair: 5′> CGC TCG AGC AGC AAC GGT AGC CGA GTA G <3′ and 5′> GCA GAT CTT GGC CGC CGC CGC CGC CGT TTC GC <3′. PCR products were separated by gel electrophoresis on 2% agarose gel and visualized.
FACS cell cycle analysis
H1299 cells were harvested and washed with phosphate buffered saline (PBS). After brief centrifugation (200 ×g), cell were collected and fixed overnight in 70% ethanol at 4°. Then cells were collected by centrifugation for 5 min. at 200 ×g and then incubated in ice‐cold PBS for 30 min. After incubation, cells were collected, washed and then resuspended in PBS containing 5 mg/ml propidium iodide and 50 μg/ml RNAse A. The cells were incubated at room temperature for 20 min., and then applied to measure fluorescence intensity using a flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA).
Dual‐luciferase reporter assays
H1299 cells were seeded into 12‐well plates (5 × 105 cells per well) and cultured for 20 hrs in RPMI‐1640 medium plus 10% FBS before transfection. E2F1 expression plasmid (50–200 ng) or empty vector was individually cotransfected into H1299 cells, together with appropriate Siah1 promoter reporter plasmids (200 ng) using Lipofectamine™ 2000 (Invitrogen). In each transfection, cells were also cotransfected with Renilla luciferase reporter plasmid. For TCF activity assay, equal amounts of pTop‐flash reporter plasmid (100 ng) and pRL‐CMV (5 ng) were cotransfected into each cell sample. Firefly and Renilla luciferase activity were assayed using Dual‐Luciferase Reporter Assay System according to manufacturer’s instructions (Promega Corporation). Fold‐activation values were measured relative to the levels of luciferase activity in cells transfected with empty vectors and normalized by Renilla luciferase activities.
Results
Identification of E2F1‐binding sites at Siah1 gene locus
To explore how Siah1 gene is regulated in p53‐independent manners, which may reflect the real situation for most cancer types with null or mutated p53, We used MatInspector (professional version 6.2.2), which is designed to inspect possible transcription‐factor‐binding sites in a given sequence, to scan the genomic sequence upstream of Siah1 coding region. Two putative E2F‐binding sites (BS) were found within the near upstream region, one (BS1) is at –124 bp to –117 bp, the other (BS2) is at –15 bp to –8 bp upstream of Siah1 transcription initiation site, respectively (Fig. 1A). Sequence alignment between the consensus E2F1‐binding site (C/G)CG(C/G)(C/G)AAA and the two predicted E2F1‐binding sites shows very high identity with each other (Fig. 1B).
Figure 1.
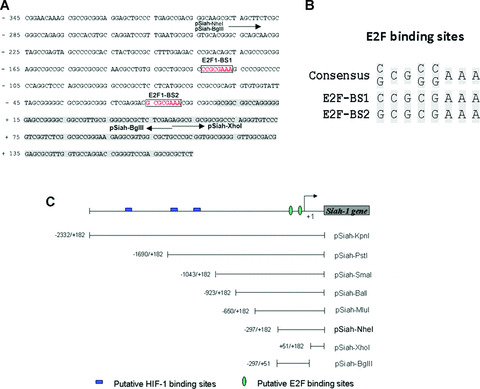
Putative E2F1‐binding sites are located in Siah1 promoter (A) Human genomic sequence of Siah1 gene from –345 to +174 is shown. Shaded part represents the first exon of Siah1. The boxed are the predicted E2F1‐binding sites. The arrow under the DNA sequence represents the start/end position of some indicated promoters. (B) Alignment of the putative E2F‐binding sites with the consensus sequence. (C) Schematic representation of various Siah1 promoter deletion constructs. Putative E2F‐binding sites are indicated. The constructs harbouring different fragment of Siah1 promoter were cloned into pGL3‐Basic reporter vector (Promega Corporation).
E2F1 activates Siah1 promoter
To examine whether Siah1 promoter could be activated by E2F1, luciferase reporter assays were performed with a series of Siah1 promoter deletion mutants. The schematic representation of the constructs used in this assay is shown in Fig. 1C. A p53‐null cell line H1299 was employed to exclude the unanticipated influence directly or indirectly due to p53. E2F1 expression plasmid and empty vector were transfected into H1299 cells together with each Siah1 promoter reporter plasmid indicated in Fig. 2A, respectively. Equal concentrations of Renilla reporter plasmid pRL‐CMV were also cotransfected as an internal control. Luciferase activity was measured and the relative activation fold of each Siah1 promoter normalized to the pRL‐CMV internal standard was presented in the histogram with arbitrary units. As shown in Fig. 2A, all Siah1 promoter fragments containing the two putative E2F1‐binding sites displayed significant activation fold under ectopic E2F1 expression. Moreover, as the Siah1 promoter undergoes continuous deduction from 5′ to 3′, the responsiveness to E2F1 did not display significant reduction until the two putative E2F1‐binding sites has been deleted (lane: XhoI), suggesting that the Siah1 promoter region pSiah‐BglII (–297 –+51) may represent the Siah1 minimal response element to E2F1(lane: BglII). Then we compared the responsiveness of the pSiah‐BglII to pApaf1‐Luc (–396/+208), one of the known E2F1‐responsive promoters described elsewhere [22]. As shown in Fig. 2B, two promoters exhibited similar patterns and comparable fold increases upon E2F1 activation.
Figure 2.

Activation of Siah1 promoter by E2F1. (A) Determination of minimal promoter region of Siah1 that can be activated by E2F1. H1299 cells were individually cotransfected with equal amount (100 ng) of eight indicated Siah1 promoter deletion constructs of Luc‐reporter, together with either pcDNA3 empty vector (200 ng) or E2F1 expressing vector pcDNA3‐E2F1 (200 ng). Renilla luciferase plasmid pRL‐CMV was also introduced into each transfected cell samples as internal control. Cells were harvested 24 hrs after transfection and luciferase activities were assayed using the dual luciferase assay system. Fold activation values were measured relative to the levels of Renilla luciferase activity and were listed under their respective bars. (B) pSiah‐BglII is sufficient to activate Siah1 promoter activity in response to E2F1. H1299 cells transfected with either reporter plasmids pSiah‐BglII (100 ng) or pApaf‐Luc (pGL3‐Apaf‐1[–396/+208])(100 ng) were further cotransfected in combination with pcDNA3 expression vectors (200 ng) or pcDNA3‐E2F1 (200 ng). Renilla luciferase plasmid pRL‐CMV was also introduced into each transfected cells as internal control. Fold activation values were measured relative to the levels of Renilla activity. (C) Schematic diagrams of E2F deletion mutants of Siah1 promoter. (D) H1299 cells were transfected with either reporter plasmids pSiah‐BglII (100ng) or one of E2F1 deletion mutants (pSiah‐BglII‐M1, pSiah‐BglII‐M2 and pSiah‐BglII‐MM, 100 ng) along with pcDNA3 expression vectors (200 ng) or pcDNA3‐E2F1 (200 ng). Renilla luciferase plasmid pRL‐CMV was also introduced into each transfected cells as internal control. Fold activation values were measured relative to the levels of Renilla activity. (E) Activation of Siah1 promoter by E2F1 is dose‐dependent. H1299 cells were cotransfected with pSiah‐BglII (200 ng), together with either pcDNA3 vector (200 ng), pcDNA3‐E2F1(1–363) (200 ng, an E2F1 deletion mutant) or increasing concentrations of pcDNA3‐E2F1 (from 50 ng, 100 ng to 200 ng) separately. Renilla luciferase plasmid pRL‐CMV was introduced into each cotransfection cell sample as internal control. Luciferase activity was measured and plotted. (F) Siah1 promoter specifically responds to E2F1. H1299 cells were cotransfected with reporter plasmid pSiah‐BglII (200 ng), together with one of the indicated activator plasmids: pcDNA3, pcDNA3‐E2F1, pcDNA3‐E2F2 and pcDNA3‐E2F3 at equal amount. Renilla luciferase plasmid pRL‐CMV was introduced into each cotransfected cell samples as internal control. Luciferase activity was measured and plotted after normalizing with respect to Renilla luciferase activity. In (A)–(F), all cells were harvested 24 hrs after transfection and Luciferase and Renilla activity assayed using the Dual‐Luciferase Reporter Assay System (Promega Corporation). Luciferase values were corrected for transfection efficiency with Renilla activity. All experiments were independently performed in triplicate. Vertical error bars are the standard deviations from the mean of the values within three standard deviations.
Because there exist putative‐binding motifs of other transcriptional factors in this minimal E2F1‐responsive promoter, we generated three mutant promoter constructs with deletion of either E2F1‐binding site (pSiah‐BglII‐M1 and pSiah‐BglII‐M2) or double deletion of E2F1‐binding sites (E2F1‐BS1 and E2F1‐BS2) to examine the importance of the two putative E2F‐binding sites. The schematic representations are presented in Fig. 2C. As shown in Fig. 2D, the deletion of either or both E2F1‐binding sites significantly impaired the responsiveness of Siah1 promoter to E2F1, which demonstrates that both E2F1‐binding sites are crucial for the activation of Siah1 promoter.
Next, H1299 cells were cotransfected with pSiah‐BglII, together with either pcDNA3 empty vector, pcDNA3‐E2F1 (1–363) (an E2F1 transactivation domain‐deficient variant) [26] or increasing amount of pcDNA3‐E2F1 (50–200 ng). As shown in Fig. 2E, increasing amount of wild‐type E2F1 resulted in correspondingly augmented activation fold of pSiah‐BglII (lanes 2, 3 and 4), whereas the mutated E2F1 (lane 5) or the empty vector (lane 1) failed to do so. To examine whether the responsiveness of Siah1 promoter is limited to E2F1, E2F family members E2F2 and E2F3 were tested along with E2F1 under the same condition. As shown in Fig. 2F, E2F2 and E2F3 exhibited much less activation than E2F1, suggesting that Siah1 promoter responds specifically and significantly to E2F1.
E2F1 binds to Siah1 promoter in vivo
To determine if endogenous E2F1 protein directly interacts with Siah1 promoter, we performed the ChIP assay, which allows the detection of proteins bound to specific regions of DNA in vivo. We designed a pair of primers flanking Siah promoter region –293 ∼+51bp harbouring the two putative E2F1‐binding sites. H1299 cells were transfected with or without pcDNA‐HA‐E2F1 prior to ChIP assay. As shown in Fig. 3, the promoter regions were found enriched by antibody against E2F1 in H1299 cells both transfected or untransfected with pcDNA‐HA‐E2F1, (the upper panel, lanes 4 and 8). The enrichment was not detected after the amplification of an unrelated genomic DNA fragment β‐actin (the lower panel, lane 3 versus lane 4; lane 7 versus lane 8). This result implied that E2F1 could interact with Siah1 promoter directly in vivo. Taken together, our results demonstrated that E2F1 indeed interacts with and activates Siah1 promoter.
Figure 3.
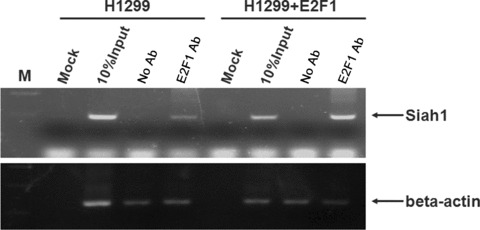
E2F1 binds to Siah promoter in vivo. H1299 cells were transfected with or without pcDNA‐HA‐E2F1 expression vector. Twenty‐four hours after transfection, cells were harvested and subjected to ChIP assay. ChIP products were amplified by PCR reaction. β‐actin was used as a negative control. M: DNA marker; Mock: control IP without nuclear lysate; No Ab: IP without E2F1 antibody; E2F1 Ab: IP with E2F1 antibody. For the right four lanes, cells were expressing with pcDNA3‐E2F1.
E2F1 induces Siah1 expression
To examine whether E2F1 could regulate expression of Siah1, H1299 cells were starved in 0.5% serum for 48 hrs, which allows the cells to be synchronized at G0/G1‐phase and keeps endogenous E2F1 activity at a low level. Starved H1299 cells were then transfected with E2F1 expression plasmid or empty vector. Twenty‐four hours after transfection, cells were harvested and subject to Western analysis using antibodies specific to E2F1, Siah1 and β‐actin, respectively. As shown in Fig. 4A, the Siah1 level was up‐regulated in pcDNA‐E2F1‐transfected cells compared to that in empty vector‐transfected cells.
Figure 4.
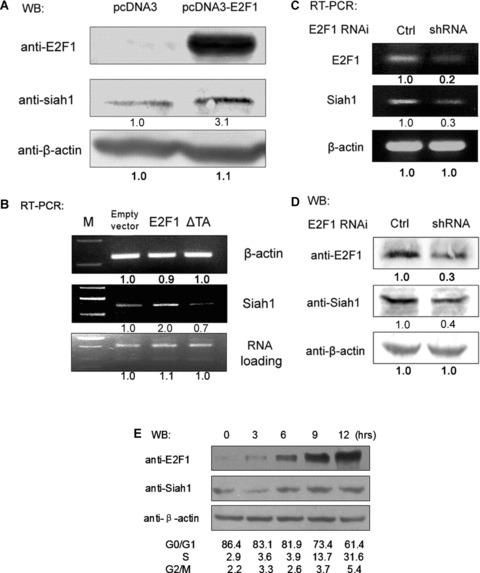
E2F1 up‐regulates the expression of endogenous Siah1. (A) H1299 cells were incubated in medium containing 0.5% serum for 48 hrs Treated cells were then transfected with either pcDNA3 (200 ng) or pcDNA3‐E2F1 (200 ng). Thirty‐six hours after transfection, cells were harvested and subject to Western analysis with indicated antibodies. (B) H1299 cells were treated under same conditions as described in (A). Thirty‐six hours after transfection, cells were harvested for RNA isolation and equal amounts of total RNA were reverse transcribed and amplified. PCR amplification of β‐actin was performed simultaneously as negative control. Total RNA used for each PCR reaction was shown as loading control (bottom panel). (C) H1299 cells were transfected with 1ug of E2F1 shRNA construct pU6‐E2F1 or pU6 empty vector. Forty‐eight hours after transfection, cells were harvested for preparation of total RNA and followed by semi‐quantitative RT‐PCR analysis with primer pairs specific to E2F1, Siah1 and β‐actin. (D) H1299 cells were treated the same way as described in (C) and then subject to Western analysis using antibodies against E2F1, Siah1 and β‐actin, respectively. (E) H1299 cells were incubated in medium containing 0.5% serum for 48 hrs and then released in medium containing 20% serum. Cells were harvested at the indicated time‐points and applied to Western blots and FACS analysis.
To confirm the Western analysis results (Fig. 4A), total RNA was prepared from cells pre‐treated under the same conditions as described above. This time we included a sample of cells expressing E2F1deltaTA, an E2F1 dominant negative construct deficient for the C‐terminal transactivation (TA) domain. Equal amounts of total RNA were used to perform semi‐quantitative RT‐PCR with primer pairs specific to Siah1 and β‐actin (control). As shown in Fig. 4B, up‐regulation of Siah1 mRNA level in H1299 expressing ectopic E2F1 was observed (lane 2). As expected, cells expressing either E2F1deltaTA (lane 3) or empty vector (lane 1) did not show significant elevation of Siah1 expression.
To further validate that the endogenous E2F1 regulates Siah expression, H1299 cells were transfected with either E2F1 shRNA or empty control construct. After 48 hrs, cells were harvested for semi‐quantitative RT‐PCR and Western analysis. As shown in Fig. 4, repression of endogenous E2F1 effectively reduced both the endogenous Siah1 mRNA (Fig. 4C) and protein level (Fig. 4D). Taken together, we demonstrated that E2F1 is able to transcriptionally up‐regulate the expression of Siah1.
To examine the relationship between Siah1 expression and cell cycle progression, we starved H1299 cells in medium containing 0.5% serum for 48 hrs and then released in medium containing 20% serum. Cells were harvested at different time‐points and applied to FACS cell cycle analysis and Western blots using antibodies against E2F1 and Siah1. As shown in Fig. 4E, under 20% serum stimulation, synchronized cells re‐entered cell cycle as demonstrated by the fact that more than 30% of cells entered S‐phase after 12 hrs. Correspondingly, levels of Siah1 were clearly up‐regulated along with the increase in E2F1, indicating that the expression of Siah1 was cell cycle‐dependent and Siah1 is likely to be regulated by E2F1.
Siah1 mediates the repression of β‐catenin/TCF activity by E2F1
Siah1 was reported to mediate the proteasomal degradation of β‐catenin and accordingly represses the activation of downstream transcription factor TCF/LEF. We then examined whether E2F1 could interfere with the TCF/LEF activity through Siah1. H1299 cells were transfected with either pSuper‐Siah1 shRNA or pSuper empty vector. Forty‐eight hours after transfection, both shRNA and mock treated cells were harvested for preparation of total RNA and analysed by semi‐quantitative RT‐PCR to examine Siah1 expression. As shown in Fig. 5A, the pSuper‐Siah1 shRNA significantly repressed the expression of the endogenous Siah1 at its mRNA level. To evaluate the effect of E2F1 on β‐catenin/TCF activity by elimination of Siah1, H1299 cells were transfected with either pSuper‐Siah1 shRNA or pSuper empty vector. Forty‐eight hours later, shRNA‐ or mock‐treated cells were further cotransfected with E2F1 expression vector or control vector, together with equal amounts of pRL‐CMV and pTop‐flash plasmid, which contains the wild‐type binding site of TCF that serves as a reporter for endogenous TCF Activity. An additional 24 hrs later, cells were harvested to measure the luciferase activity. The relative luciferase activity of pTop‐flash promoter normalized to the pRL‐CMV internal standard was presented in the histogram with arbitrary units (Fig. 5B). Comparing the two columns both transfected with pSuper vector (control), the TCF activity was obviously repressed by ectopically expressed E2F1 (lane 1 versus lane 2). From the two columns both transfected with E2F1 expression vector, the suppressed TCF activity by ectopic E2F1 expression was significantly derepressed through the repression of Siah1 (lane 2 [0.56] versus lane 4 [0.71]). Taken together, these results demonstrated that Siah1 mediated the repression of TCF activity by E2F1.
Figure 5.

Repression of Siah1 expression attenuates E2F1‐repressed β‐catenin/TCF activity. (A) H1299 cells were transfected with gene‐specific shRNA construct pSuper‐Siah1 (1ug) or pSuper empty vector (1ug). Forty‐eight hours after transfection, cells were harvested for preparation of total RNA and semi‐quantitative RT‐PCR analysis with primer pairs specific to Siah1 and β‐actin. Actin (control) was then performed. (B) H1299 cells were transfected with either Siah1 shRNA construct pSuper‐Siah1 (200 ng) or pSuper empty vector (200 ng). Forty‐eight hours after transfection, 200 ng of pTop‐flash and 5 ng of pRL‐CMV were further introduced into each transfected cells. An additional 24 hrs later, cells were harvested for measuring dual luciferase activity as described in Fig. 2. Luciferase values were corrected for transfection efficiency with Renilla activity. All the experiments were independently performed in triplicate. The data of triplicate experiments and their average were indicated in the figure. Error bars indicate S.D. (standard deviation). ***P < 0.001; **P < 0.01.
Discussion
In this study, we found two highly conserved E2F1‐binding sites existing in the promoter region of Siah1 gene. E2F1 was found to bind to and activate Siah1 promoter directly. Semi‐quantitative RT‐PCR and Western analysis demonstrated that ectopic E2F1 expression up‐regulated endogenous Siah1 at both the mRNA and protein levels. Repression of endogenous E2F1 by E2F1‐specific shRNA led to a reduction of Siah1 expression. Finally we found that although ectopic E2F1 expression could suppress the activity of β‐catenin/TCF, this suppression, however, could be attenuated by knocking‐down of endogenous Siah1. Based on these results, we conclude that E2F1 represses the β‐catenin/TCF activity by transcriptionally up‐regulating the expression of Siah1.
E2F1 is a key transcription factor that effectively drives cell cycle progression from G0/G1 into S‐phase. Many E2F1 target genes are known to be involved in cell cycle progression and DNA replication [1]. In normal cells, activity of E2F1 is strictly controlled by pRB/E2F1 pathway and kept at a relatively low level [27, 28, 29, 30]; however, in most tumour cells, pRB/E2F1 pathway is usually disrupted at multiple points and leads to unrestrained activity of E2F1, which keeps the cells in a proliferative state and is thought to be one of the important reason for the carcinogenic effects. Recently cumulative evidence suggests that ectopic expression of E2F1 could lead to cell apoptosis and knockout of endogenous E2F1 makes mice vulnerable to tumorigenesis [3, 5, 6]. Therefore, in healthy cells, E2F1 activity must be kept at a low level. E2F1 was known to be ubiquitinated by an E3 ligase complex Skp, Cullin, F‐box containing complex (SCF complex)skp2 and degraded via the ubiquitin‐mediated proteasome pathway [31]. Our finding in this study demonstrated that E2F1 could elevate Siah1 level. The latter was reported to degrade β‐catenin via a proteasome‐mediated proteolysis by forming a complex with SIP and SCFskp2[19]. Thus, one needs to test a hypothesis whether there exists a negative feedback loop involving E2F1‐up‐regulated Siah1, by forming a ubiquitination complex with SIP/SCFskp2, which in turn causes E2F1 degradation to maintain a relatively lower level of E2F1 within the cell. Furthermore, β‐catenin/TCF is implicated in the downstream of Wnt signalling pathway, and its deregulation is thought to be related to tumorigenesis in many cancers [32, 33]. Our finding also suggests a novel tumour suppression function of E2F1 in Wnt signalling pathway, in which E2F1 up‐regulates Siah1 expression to restrict the β‐catenin/TCF activity. A proposed model is shown in Fig. 6, in which Siah1 serves as the mediator between pRB/E2F1 and Wnt/[β]‐catenin pathways. A recent study also reported that E2F1 can up‐regulate protein Axin2 to suppress Wnt/β‐catenin/TCF activity [34]. Whether there exists some other hierarchical regulation in these signalling pathways remains to be investigated.
Figure 6.

Hypothetical model for illustrating the proposed pathway through which E2F1 represses β‐cantenin/TCF activity by elevating the level of Siah1.
Because hypoxia circumstances exist in most tumours, and under this condition, transcription factor HIF‐1α (hypoxia inducible factor‐1α) is stabilized for the inhibition of oxygen‐dependent degradation. The targets of HIF‐1α include the genes involved in cell cycle control, cell proliferation, metabolism and angiogenesis [35, 36, 37, 38, 39, 40]. Recently, Siah1 has been reported to enhance HIF‐α stability by promoting the degradation of two E3 ligases (FIH and PHD1/3) targeted for HIF‐α[41, 42], implying that E2F1 may have promoting effect on HIF‐αvia un‐regulated Siah1; meanwhile, there is a report that β‐catenin could associate with HIF‐1α to potentiate its stabilization and transcriptional activity [43], this suggests that E2F1 may have an inhibitory effect on HIF‐1‐αvia suppressing β‐catenin/TCF/LEF activity. In summary, our finding that E2F1 can suppress β‐catenin/TCF/LEF activity via up‐regulation of Siah1 may shed new light on the relationship between two important transcription factors E2F1 and HIF‐1α.
Acknowledgements
We are grateful to Dr. Solomon H. Snyder (Johns Hopkins University School of Medicine) for pSuper & pSuper‐Siah1 plasmids; and Dr. W. Douglas Cress (H. Lee Moffitt Cancer Center and Research Institute, FL, USA) for pU6 & pU6‐E2F1 plasmids. This research was supported by grants from the National Natural Science Foundation of China (30530200), grants from the Ministry of Science and Technology of China (2002CB713702, 2006AA02Z101 and 2006CB910300), a grant from the Chinese Academy of Sciences (KSCX1‐YW‐R‐57) and a grant from MOE China NO706035.
References
- 1. Helin K. Regulation of cell proliferation by the E2F transcription factors. Curr Opin Genet Dev. 1998; 8: 28–35. [DOI] [PubMed] [Google Scholar]
- 2. Kowalik TF, DeGregori J, Schwarz JK, et al . E2F1 overexpression in quiescent fibroblasts leads to induction of cellular DNA synthesis and apoptosis. J Virol. 1995; 69: 2491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shan B, Lee WH. Deregulated expression of E2F‐1 induces S‐phase entry and leads to apoptosis. Mol Cell Biol. 1994; 14: 8166–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ginsberg D. E2F1 pathways to apoptosis. FEBS Lett. 2002; 529: 122–5. [DOI] [PubMed] [Google Scholar]
- 5. Yamasaki L, Jacks T, Bronson R, et al . Tumor induction and tissue atrophy in mice lacking E2F‐1. Cell. 1996; 85: 537–48. [DOI] [PubMed] [Google Scholar]
- 6. Field SJ, Tsai FY, Kuo F, et al . E2F‐1 functions in mice to promote apoptosis and suppress proliferation. Cell. 1996; 85: 549–61. [DOI] [PubMed] [Google Scholar]
- 7. Della NG, Senior PV, Bowtell DD. Isolation and characterisation of murine homologues of the Drosophila seven in absentia gene (sina). Development. 1993; 117: 1333–43. [DOI] [PubMed] [Google Scholar]
- 8. Hu G, Chung YL, Glover T, et al . Characterization of human homologs of the Drosophila seven in absentia (sina) gene. Genomics. 1997; 46: 103–11. [DOI] [PubMed] [Google Scholar]
- 9. Hu G, Fearon ER. Siah‐1 N‐terminal RING domain is required for proteolysis function, and C‐terminal sequences regulate oligomerization and binding to target proteins. Mol Cell Biol. 1999; 19: 724–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. House CM, Frew IJ, Huang HL, et al . A binding motif for Siah ubiquitin ligase. Proc Natl Acad Sci USA. 2003; 100: 3101–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tiedt R, Bartholdy BA, Matthias G, et al . The RING finger protein Siah‐1 regulates the level of the transcriptional coactivator OBF‐1. EMBO J. 2001; 20: 4143–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Boehm J, He Y, Greiner A, et al . Regulation of BOB.1/OBF.1 stability by SIAH. EMBO J. 2001; 20: 4153–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Germani A, Bruzzoni‐Giovanelli H, Fellous A, et al . SIAH‐1 interacts with alpha‐tubulin and degrades the kinesin Kid by the proteasome pathway during mitosis. Oncogene. 2000; 19: 5997–6006. [DOI] [PubMed] [Google Scholar]
- 14. Hu G, Zhang S, Vidal M, et al . Mammalian homologs of seven in absentia regulate DCC via the ubiquitin‐proteasome pathway. Genes Dev. 1997; 11: 2701–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sourisseau T, Desbois C, Debure L, et al . Alteration of the stability of Bag‐1 protein in the control of olfactory neuronal apoptosis. J Cell Sci. 2001; 114: 1409–16. [DOI] [PubMed] [Google Scholar]
- 16. Zhang J, Guenther MG, Carthew RW, et al . Proteasomal regulation of nuclear receptor corepressor‐mediated repression. Genes Dev. 1998; 12: 1775–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Susini L, Passer BJ, Amzallag‐Elbaz N, et al . Siah‐1 binds and regulates the function of Numb. Proc Natl Acad Sci USA. 2001; 98: 15067–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liani E, Eyal A, Avraham E, et al . Ubiquitylation of synphilin‐1 and alpha‐synuclein by SIAH and its presence in cellular inclusions and Lewy bodies imply a role in Parkinson’s disease. Proc Natl Acad Sci USA. 2004; 101: 5500–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Matsuzawa SI, Reed JC. Siah‐1, SIP, and Ebi collaborate in a novel pathway for beta‐catenin degradation linked to p53 responses. Mol Cell. 2001; 7: 915–26. [DOI] [PubMed] [Google Scholar]
- 20. Liu J, Stevens J, Rote CA, et al . Siah‐1 mediates a novel beta‐catenin degradation pathway linking p53 to the adenomatous polyposis coli protein. Mol Cell. 2001; 7: 927–36. [DOI] [PubMed] [Google Scholar]
- 21. Maeda A, Yoshida T, Kusuzaki K, et al . The characterization of the human Siah‐1 promoter(1). FEBS Lett. 2002; 512: 223–6. [DOI] [PubMed] [Google Scholar]
- 22. Song ZY, Yao XB, Wu M. Direct interaction between Survivin and Smac/DIABLO Is essential for the anti‐apoptotic activity of Survivin during taxol‐induced apoptosis. J Biol Chem. 2003; 278: 23130–40. [DOI] [PubMed] [Google Scholar]
- 23. Xie W, Jiang P, Miao L, et al . Novel link between E2F1 and Smac/DIABLO: proapoptotic Smac/DIABLO is transcriptionally upregulated by E2F1. Nucleic Acids Res. 2006; 34: 2046–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ma YH, Cress WD, Haura EB. Flavopiridol‐induced apoptosis is mediated through up‐regulation of E2F1 and repression of Mcl‐1. Mol Cancer Ther. 2003; 2: 78–81. [PubMed] [Google Scholar]
- 25. Hara MR, Agrawal N, Kim SF, et al . S‐nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat Cell Biol. 2005; 7: 665–74. [DOI] [PubMed] [Google Scholar]
- 26. Hofmann F, Martelli F, Livingston DM, et al . The retinoblastoma gene product protects E2F‐1 from degradation by the ubiquitin‐proteasome pathway. Genes Dev. 1996; 10: 2949–59. [DOI] [PubMed] [Google Scholar]
- 27. Chellappan SP, Hiebert S, Mudryj M, et al . The E2F transcription factor is a cellular target for the RB protein. Cell. 1991; 65: 1053–61. [DOI] [PubMed] [Google Scholar]
- 28. Nevins JR, Chellappan SP, Mudryj M, et al . E2F transcription factor is a target for the RB protein and the cyclin A protein. Cold Spring Harb Symp Quant Biol. 1991; 56: 157–62. [DOI] [PubMed] [Google Scholar]
- 29. Hiebert SW, Chellappan SP, Horowitz JM, et al . The interaction of RB with E2F coincides with an inhibition of the transcriptional activity of E2F. Genes Dev. 1992; 6: 177–85. [DOI] [PubMed] [Google Scholar]
- 30. Nevins JR. E2F: a link between the Rb tumor suppressor protein and viral oncoproteins. Science. 1992; 258: 424–29. [DOI] [PubMed] [Google Scholar]
- 31. Marti A, Wirbelauer C, Scheffner M, et al . Interaction between ubiquitin‐protein ligase SCFSKP2 and E2F‐1 underlies the regulation of E2F‐1 degradation. Nat Cell Biol. 1999; 1: 14–9. [DOI] [PubMed] [Google Scholar]
- 32. Polakis P. Wnt signaling and cancer. Genes Dev. 2000; 14: 1837–51. [PubMed] [Google Scholar]
- 33. Bienz M, Clevers H. Linking colorectal cancer to Wnt signaling. Cell. 2000; 103: 311–20. [DOI] [PubMed] [Google Scholar]
- 34. Hughes TA, Brady HJ. Cross‐talk between pRb/E2F and Wnt/beta‐catenin pathways: E2F1 induces axin2 leading to repression of Wnt signalling and to increased cell death. Exp Cell Res. 2005; 303: 32–46. [DOI] [PubMed] [Google Scholar]
- 35. Ryan HE, Lo J, Johnson RS. HIF‐1 alpha is required for solid tumor formation and embryonic vascularization. EMBO J. 1998; 17: 3005–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Semenza GL. HIF‐1: mediator of physiological and pathophysiological responses to hypoxia. J Appl Physiol. 2000; 88: 1474–80. [DOI] [PubMed] [Google Scholar]
- 37. Semenza GL. Targeting HIF‐1 for cancer therapy. Nat Rev Cancer. 2003; 3: 721–32. [DOI] [PubMed] [Google Scholar]
- 38. Jaakkola P, Mole DR, Tian YM, et al . Targeting of HIF‐alpha to the von Hippel‐Lindau ubiquitylation complex by O2‐ regulated prolyl hydroxylation. Science. 2001; 292: 468–72. [DOI] [PubMed] [Google Scholar]
- 39. Semenza GL. HIF‐1 and tumor progression: pathophysiology and therapeutics. Trends Mol Med . 2002; 8: S62–7. [DOI] [PubMed] [Google Scholar]
- 40. Semenza GL. HIF‐1 and human disease: one highly involved factor. Genes Dev. 2000; 14: 1983–91. [PubMed] [Google Scholar]
- 41. Nakayama K, Frew IJ, Hagensen M, et al . Siah2 regulates stability of prolyl‐hydroxylases, controls HIF1alpha abundance, and modulates physiological responses to hypoxia. Cell. 2004; 117: 941–52. [DOI] [PubMed] [Google Scholar]
- 42. Fukuba H, Yamashita H, Nagano Y, et al . Siah‐1 facilitates ubiquitination and degradation of factor inhibiting HIF‐1alpha (FIH). Biochem Biophys Res Commun. 2007; 353: 324–9. [DOI] [PubMed] [Google Scholar]
- 43. Kaidi A, Williams AC, Paraskeva C. Interaction between beta‐catenin and HIF‐1 promotes cellular adaptation to hypoxia. Nat Cell Biol. 2007; 9: 210–7. [DOI] [PubMed] [Google Scholar]