

Abstract
BCR/ABL can cause chronic myelogenous leukaemia (CML) in part by altering the transcription of specific genes with growth‐ and/or survival‐promoting functions. Recently, BCR/ABL has been shown to activate survivin, an important regulator of cell growth and survival, but the precise molecular mechanisms behind its expression and consequences thereof in CML cells remain unclear. Here, we reported that BCR/ABL promotes survivin expression and its cytoplasmic accumulation. The increase of survivin was largely controlled at the transcriptional level through a mechanism mediated by JAK2/PI3K signal pathways that activated c‐Myc, leading to transactivation of survivin promoter. Dynamic down‐regulation of survivin was a key event involved in imatinib‐induced cell death while forced expression of survivin partially counteracted imatinib's effect on cell survival. Additionally, shRNA‐mediated silencing of survivin or c‐Myc eradicated colony formation of K562 cells in semi‐solid culture system, implying an essential role for this transcriptional network in BCR/ABL‐mediated cell transformation and survival. Finally, interruption of c‐Myc activity by 10058‐F4 exerted an anti‐leukaemia effect with a synergistic interaction with imatinib and overcame the anti‐apoptosis rescued by IL‐3 supplement. In conclusion, we have identified JAK2/PI3K‐mediated and c‐Myc‐dependent transactivation of survivin as a novel pathway in the transcriptional network orchestrated by BCR/ABL. These results suggest that the interference with this circuitry might be a potential utility for CML treatment.
Keywords: BCR/ABL, leukaemia, survivin, c‐Myc, transcription, signal transduction
Introduction
The oncogenic kinase BCR/ABL, generated by the reciprocal chromosomal translocation t (9; 22)q(34; 11), is responsible for CML induction and maintenance [1, 2]. Molecular signalling in CML is highly complex, and several different mechanisms have been implicated in BCR/ABL‐dependent growth and accumulation of leukaemic cells in CML [1, 2]. One important mechanism is the regulation of survival‐ and apoptosis‐related genes. A number of signal transducers and transcription factors have been associated with the malignant phenotype of CML cells [2], some of which lead to the expression and/or activation of members of Bcl‐2 family of apoptosis modulator including Bcl‐xL, Mcl‐1 and A1 [3, 4, 5, 6, 7]. Recently, other molecules, such as haem oxygenase‐1 [8, 9] and WT1 [10], have been identified as novel BCR/ABL‐dependent survival factors. Most of these factors can confer resistance to imatinib, suggesting that the survival signals downstream of BCR/ABL may serve as a kinase‐independent mechanism for development of drug resistance. In coincident with these observations, microarray analysis manifests that transcripts from gene with anti‐apoptotic and with involvement in signal transduction/transcriptional regulation are overexpressed in CML cells innatively resistant to imatinib [11]. However, these factors cannot take full responsibilities to the anti‐apoptotic phenotype of CML because some of them may only be detected in leukaemic cells of a subgroup of patients [12]. Further elucidation of the molecular network in BCR/ABL‐dependent survival and apoptosis no doubt will allow comprehensive insights into the mechanisms of disease progression and drug resistance in CML.
Survivin is a promising target for cancer therapeutics that is differentially expressed in cancer and intersects multiple pathways for tumour maintenance [13, 14]. Our previous understanding in this field indicates that it may be involved in the development and progression in CML [15]. Clinical reports from small sample sizes demonstrate that survivin expression is higher in patients with BCR/ABLPos than in patients with BCR/ABLNeg CML, with such an increase being more pronounced in the blast crisis than in the chronic phase [16, 17]. Moreover, elevated survivin is found in adriamycin‐resistant K562 cells [18]. Recent studies also suggest that strategies to reduce survivin expression hold a great promise for CML treatment, both imatinib‐sensitive and ‐resistant [19, 20]. Although these studies have related to some aspects about its regulation, the accurate molecular mechanisms governing survivin expression and its actual roles in CML cells still remain undefined. In this report, we have outlined a novel transcriptional circuitry consisting of JAK2/PI3K/c‐Myc/survivin in BCR/ABL signal network, suggesting that targeting its effector, c‐Myc, was feasible to CML treatment.
Materials and methods
Cells, culture conditions and reagents
The IL‐3‐dependent 32Dcl3 myeloid precursor and 32D‐BCR/ABL cells (from laboratory of Perrotti D; The Ohio State University, Columbus, OH, USA) were maintained in IMDM (Gibco, Carlsbad, CA, USA) supplemented with 10% heat‐inactivated FBS and 2 mM L‐glutamine. WEHI‐conditioned medium (10%) was used as source of mIL‐3 for 32Dcl3 cells. K562 cells were cultured in RPMI 1640 with 10% heat‐inactivated FBS and 2 mM L‐glutamine. 293T cells and the amphotropic‐packaging cell line Phoenix Ampho (Nolan GP, Stanford University School of Medicine) (ATCC SD3444) were maintained in DMEM with 10% heat‐inactivated FBS and 2 mM L‐glutamine. Bone marrow from CML patients and healthy donors was obtained after informed consent in accordance with the Institutional Review Board of CAMS & PUMC. Mononuclear cells were purified by Ficoll‐Hypaque density‐gradient centrifugation as described [21]. In inhibition experiments, the following reagents were used: imatinib (Novartis, Basel, Switzerland), PI3K inhibitor LY294002 (50 uM), MEK inhibitor PD98059 (40 uM), JAK2 inhibitor AG490 (100 uM), actinomycin D (all from Sigma, St. Louis, MO, USA), c‐myc inhibitor 10058‐F4 (60 uM) (Calbiochem, San Diego, CA, USA).
Polymerase chain reaction analysis
Total RNA was extracted using Trizol (Invitrogen, Carlsbad, CA, USA) according to manufacture's instructions. Reverse transcription was performed on 1 μg total RNA, with 0.5 μg oligo‐dT, 2.5 mmol/L dNTP, 25 units Ribonuclease inhibitor and 200 units of murine Moloney leukaemia virus reverse transcriptase (Promega, Madison, WI, USA) in a total volume of 25 μl for 90 min. at 37°C. PCR was performed on 2 μl of the reverse transcription products, in the presence of 20 pmol of the specific primers, 2.5 units of Taq polymerase (TaKaRa, Shiga, Japan), and 0.4 mmol/L dNTP, in a final volume of 50 μl. The following primers were used: survivin: 5′‐CTGGCAGCTGTACCTCAA‐3′ and 5′‐CGGGTAGTCTTTGCAGTCT‐3′; vegf: 5′‐CGATGAAGCCCTGGAGTG‐3′ and 5′‐GACGATGATGGCGTGGTG‐3′; β‐actin: 5′‐CTGTCCCTGTATGCCTCTG‐3′ and 5′‐ATGTCACGCACGATTTCC‐3′. Thermal cycle conditions included holding the reaction at 94°C for 5 min. followed by 30 cycles of 30 sec. at 94°C, 30 sec. at 60°C and 60 sec. at 72°C for survivin or vegf, and 25 cycles for β‐actin under same condition. The PCR products were electrophoresed on a 1.5% agarose gel visualized by ethidium bromide staining. For mRNA stability assay, 5 μg of total RNA were used as loading controls, intensities of signals were determined by densitometric analysis using Quantity One software (Bio‐Rad, version 4.6.2, Hercules, CA, USA) and standardized to 28 s and 18 s rRNA. Half‐time was calculated by using regression analysis.
Cell extracts and Western blot
Antibodies against survivin (R&D system, Minneapolis, USA), caspase‐3, pAkt(Ser473),Akt, pStat5(Tyr694), pErk1/2(Thr202/Tyr204),Erk1/2 (Cell Signaling Technology, Danvers, MA, USA), Stat5,c‐myc, β‐actin, nucleolin and GADPH (Santa Cruz Biotechnology, CA, USA) were used in this study. Whole cell extracts were prepared by lysing cells at 1 × 106/100 μl in lysis buffer (20 mM Tris‐Cl, pH8.0, 1% NP‐40, 137 mM NaCl, 20 mM DTT, 2 mM Sodium Vanadate containing protease inhibitor cocktail) for 30 min. on ice. Cytosolic and nuclear proteins were prepared using a cytosol/nuclear fractionation kit (Beyotime, Suzhou, China). The protein concentration was determined using BCA protein Assay Kit (Pierce, Rockford, IL, USA). Immunoblotting was carried out by Tricine‐SDS‐PAGE. In brief, 20 μl total cell lysate or up to 75 μg of subcellular extracts were denatured in SDS buffer for 5 min. at 95°C, separated on 10% gels, and electrotransferred to PVDF membranes (Millipore, Bedford, MA, USA) using submerged electrophoretic transfer. After blocking with 5% skimmed milk in TBST buffer (20 mM Tris‐HCl, pH 7.5, 0.137 M NaCl, and 0.01% Tween 20) for 2 hrs at room temperature with constant shaking, the membranes were incubated in TBST buffer containing the relevant primary antibody (1:500–2000) and 5% skimmed milk overnight at 4°C. After washing in TBST buffer, the membranes were incubated in 5% skimmed milk in TBST buffer containing the appropriate anti‐IgG HRP‐conjugated second antibodies (1:10,000) at room temperature for 1 hr with constant shaking. The expression of the target protein was detected by the ECL protein detection kit (Pierce) following the manufacturer's instructions. For normalization of protein loading, the same membranes were processed by the same procedure with an antibody against β‐actin.
Construction of reporter plasmids and luciferase assays
A 1.788‐Kb fragment from −1764 to +24 relative to the transcription initiation site of human survivin promoter was amplified and subcloned into the BglII and HindIII sites of pGL3‐basic vector (Promega). The final full‐length reporter plasmid was designated to pGL3‐S1764. Its deletion and site‐directed mutation reporters were generated as follows. All constructs were confirmed by sequencing. To generate progressive 5′‐unidirectional deletion mutants, a series of forward primers were used in combination with the same reverse primer in a PCR with pGL3‐S1764 as template. The reverse primer with the HindIII site was 5′‐GCGAAGCTTCAAATCTGGCGGTTAATG‐3′, the forward primers, each of which bears a BglII site, were 5′‐GGAGATCTGGGTAGTATGGTAATGCCTTC‐3′(pGL3‐S1764), 5′‐GCAGATCTTGGGCGATAGAGCGAGAC‐3′(pGL3‐S1138), 5′‐GGAGATCTGTCCTTCATGCCCGTCTG‐3′(pGL3‐S580), 5′‐TTAGATCTCAAGCGATTCTCCTGCCTC‐3′(pGL3‐S415), 5′‐GGCAGATCTTTGTATTTTTAGTAGAGAC‐3′(pGL3‐S343), 5′‐AAAGATCTACGCGGCGGGAGGACTA‐3′(pGL3‐S116), 5′‐TTGAAGATCTCTCTACTCCCAGAAG‐3′(pGL3‐S69). The resulting PCR products were gel‐purified, digested with BglII and HindIII and subcloned into the BglII/ HindIII sites of the pGL3‐basic vector. For pGL3‐S19, the oligonucleotides of 5′‐GATCTCCCGACATGCCCCGCGGCGCGCCATTAACCGCCAGATTTGA‐3 and 5′‐AGCTTCAAATCTGGCGGTTAATGGCGCGCCGCGGGGCATGTCGGGA‐3 were annealed and ligated into the pGL3‐basic vector. Site‐directed mutagenesis of survivin promoter core region was carried out by PCR with primers as follows (each pair only shows sense chain): 5′‐GGCGCTAGGTGTGGTTAGGGACGAGCTGGC‐3′(mut1), 5‐ACCGCGACCACGGGATTAGCCACGCGGCGGG‐3′(mut2), 5‐CCTGCACGCGCTCTTTGGTAGCAGTCGAGG‐3′(mut4), 5‐TGCACCCGGCCTGTTCGAGTTCTTTGAAAGC‐3(mut5), 5′‐ACGGGCAGAGCTTCGATGCGGGAGGACTAC‐3′(mut6). Mut3 and Mut 7 were generated by using Mut1 and Mut 5 as template, respectively.
Luciferase assays were performed by using the Dual‐Luciferase reporter system (Promega), in which transfection efficiency was monitored by cotransfected with pRL, an expression vector of renilla reniformis luciferase. In short, 6∼8 × 105 K562 cells in 24‐well plate were transiently transfected with 1 μg of an appropriate reporter gene together with 20 ng of pRL by Lipofectamine 2000 (Invitrogen) according to manufacture's instructions. After an 8‐hr incubated period in the culture medium, the cells were treated with indicated inhibitors or DMSO for 16 hrs; at this time‐point, the cell viability of each group was similar and more than 90% as determined by trypan blue staining (not shown). Then they were lysed in lysis buffer supplied by the manufacturer, followed by the measurement of firefly and renilla luciferase activities on luminometer. The relative firefly luciferase activities were calculated by normalizing transfection efficiency according to the renilla luciferase activities.
Chromatin immunoprecipitation (ChIP) assay
K562 cells were treated with DMSO, 2.5 uM imatinib, and 60 uM 10058‐F4, respectively. ChIP was performed using an EZ‐ChIP assay kit (Upstate, Charlottesville, VA, USA) following the manufacturer's instructions. Then the association of c‐Myc with surviving promoter was measured by PCR. PCR was performed using primers that amplify a region containing c‐Myc binding sites (forward primer:5′‐GTTGGGATTACAGGCGTGAG‐3′; reverse primer: 5′‐CTCTTAGGCGGTCCACCC ‐3′). A pair of primers (forward primer: 5′‐TGCCTTCAACTTACAAACG‐3; reverse primer: 5′‐CTCTGCTTCTTCCTCCCT‐3′) amplifying a distal region upstream c‐Myc binding sites served as a control. Amplification conditions were as follows: 5 min. at 94°, followed by 35 cycles of 1 min. at 94°, 30 sec. at 59° and 30 sec. at 72° with a final extension of 7 min. at 72°. The samples were run on a 2% agarose gel visualized by ethidium bromide staining. Parallel ChIP analyses were performed using an antibody against RNA‐polymerase as positive control, and an isotype IgG as negative control.
Cell‐death analysis
Cell viability was determined by trypan blue exclusion. At least 500 cells of each sample were counted. The percentage of viable cell was calculated with the formula of % viability = total viable cell numbers/total cell numbers ×100. Apoptotic cell death was analysed by measuring externalized phosphatidyl serine with an Annexin V‐FITC staining kit or by trypan blue staining as described [22].
Retrovirus production and transduction
Survivin cDNA was amplified using the primers 5′‐GTAAGATCTATGGGTGCCCGACGTTG‐3′ and 5′‐AAAAGTCGACTCAATCCATGGCAGCCAG ‐3′, then subcloned into the Bgl II and Sal I sites of the retroviral plasmid pLEGFP‐N1 (Clontech, Mountain View, CA, USA). Retroviruses containing survivin and pLEGFP‐N1 backbone were produced using Phoenix Ampho cells in accordance with a procedure described in Nolan lab website (http://www.stanford.edu/group/nolan/). The cells were exposed to retrovirus supernatants in 1640 medium in the presence of 4 μg/ml polybrene (Sigma) and centrifuged at 2000 rpm for 1 hr at 32°C. The efficiency of transduction was 50∼60% as measuring GFP expression under an Olympus digital fluorescence microscope at 72 hrs. The stable transduction cells were selected in the present of 800 μg/ml G418 (Promega).
RNA interference and colony formation assay
shRNA sequences against Fluc, survivin [23] and c‐Myc [24] were subcloned into a shRNA stable expression vector pGE‐1 (Stratagene, La Jolla, CA, USA). To test the efficiency of RNA interference, the constructs were transfected into 293T cells using Lipofectamine 2000 and the expression of survivin and c‐Myc were determined by Western blot after 48 hrs. For colony formation assay, K562 cells were transfected in the same way and plated in 0.9% methylcellulose containing 800 μg/ml G418 at 48‐hr post‐transfection. The number of colony was counted after 2 weeks as previously described [25].
Statistics
All experiments were performed at least three times and the results are expressed as mean ± standard error. The Student's t‐test was used to compare the data from each group. P < 0.05 was considered to be statistically significant.
Results
BCR/ABL increases survivin production and its subcellular accumulation
To explore the mechanisms regulating survivin expression in BCR/ABL‐expressing cells, we first examined survivin expression in 32D‐BCR/ABL cells and its parent 32Dcl3 cells. As expected, both mRNA and protein levels of survivin were higher in 32D‐BCR/ABL cells than 32Dcl3 cells. The induction of survivin dropped off at levels similar to that of parent cells upon imatinib treatment (Fig. 1A and B). However, survivin expression was similar in imatinib‐treated and untreated 32Dcl3 cells (Fig. 1A and B), suggesting that imatinib‐induced down‐regulation of survivin is BCR/ABL kinase‐specific. This further supported the notion that survivin is regulated by BCR/ABL tyrosine kinase in CML cells [19, 20].
Figure 1.
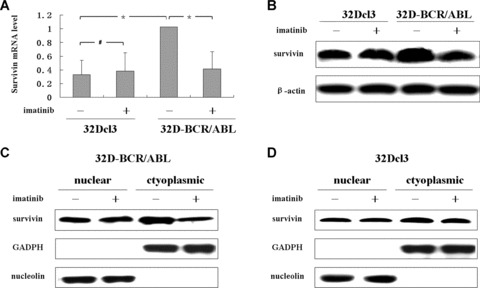
BCR/ABL increases survivin expression and its subcellular accumulation. 32D‐BCR/A BL and 32Dcl3 cells were treated or untreated for 12 hrs with 2 uM imatinib. (A) The survivin mRNA was detected by RT‐PCR. Expression of survivin, normalized to the level of untreated 32D‐BCR/ABL cells, is shown. Data are mean ± S.D. of triplicates assays. *P < 0.001, # no significance. (B) Western blot analysis of the protein level of survivin, β‐actin was measured as a loading control. (C, D) Western blot analysis of subcellular survivin levels. Aliquots of cytoplasmic and nuclear fractions from indicated cells were analysed, GADPH and nucleolin were used as markers for cytoplasmic and nuclear fractions, respectively.
Because survivin exists in multiple subcellular pools and plays distinct roles [26, 27], we next investigated whether BCR/ABL could influence its intracellular distribution. As expected, survivin was detected in both cytoplasmic and nuclear compartment (Fig. 1C and D). Of particular importance, inhibition of BCR/ABL activity by imatinib decreased cytoplasmic survivin without manifestly affecting nuclear survivin (Fig. 1C). However, imatinib has no effects on cytoplasmic or nuclear surivivin of 32Dcl3 cells (Fig. 1D). These results implied that BCR/ABL not only modulated survivin expression but also influenced its cytoplasmic accumulation, which might involve a post‐translational mechanism, i.e. protein degradation or transportation [20, 28].
Regulation of the steady‐state level of survivin transcript
The steady‐state level of mRNA expression has been shown to be dynamically regulated by many factors at both transcriptional and post‐transcriptional levels and determined by the transcription rate (transcription activation) and transcript degradation rate (mRNA stability), respectively. In the present study, we first determined whether increased survivin transcript in BCR/ABL‐transformed cells was a consequence of strengthened mRNA stability by measuring the survivin mRNA half‐life. Actinomycin D was utilized to block de novo mRNA synthesis and then semi‐quantitative PCR was carried out in parental and BCR/ABL‐expressing 32Dcl3 cells at different time. As a control, VEGF mRNA level, another important target of BCR/ABL [22, 29], was shown in delayed degradation in BCR/ABL‐expressing cells (half‐time about 2.4 hrs for 32Dcl3 versus 5.4 hrs for 32D‐BCR/ABL). No discrepancy of survivin mRNA stability was found among 32Dcl3 and 32D‐BCR/ABL cells (half‐time: 13.5 hrs for 32Dcl3 versus 12.5 hrs for 32D‐BCR/ABL) (Fig. 2A). Therefore, stability did not appear to be the major factor for differential expression level of survivin mRNA, indicating that the observed effect of BCR/ABL on survivin expression is rather dependent on transcription activation.
Figure 2.

Regulation of the steady‐state level of survivin gene transcript. (A) BCR/ABL does not affect survivin mRNA stability. 32D‐BCR/ABL and 32Dcl3 cells were incubated with 5 μg/ml actinomycin D and then harvested at indicated time. RNA was extracted and RT‐PCR analysis of survivin and vegf mRNA levels was performed. The amount of mRNA at each time‐point was normalized to that of the initial stages. The half‐time was calculated by drawing the best‐fit linear curve with mRNA versus time. One representative result from two independent experiments is shown. (B) Inhibition of BCR/ABL diminishes survivin promoter activity. K562 cells were transfected with indicated promoter reporter vectors and cultured for 8 hrs, after which cells were incubated for 16 hrs in the absence or presence of imatinib. The promoter activity was measured by normalizing firefly to renilla luciferase activity. Mean values ± S.D. from three independent experiments in triplicate are shown. *P≤ 0.001, #no significance as compared to untreated group.
To detect the effect of BCR/ABL on survivin promoter activity, we used luciferase reporter plasmids in which 5′‐flanking sequences of survivin promoter were fused to the firefly luciferase coding sequences in the pGL3‐basic vector. Additionally, VEGF promoter (−1176/+55) [29] as well as pGL3‐control vector was used as controls. As shown in Fig. 2B, imatinib strongly inhibited VEGF promoter activity in K562 cells to a similar extent reported by other group [29]. The inhibition effect of imatinib on survivin promoter activity was also evident as shown with a dose‐dependent way (Fig. 2B). In contrast, imatinib has no effect on pGL3‐control promoter. These data demonstrated that transcription activation was a major contributing factor to enhanced survivin expression in BCR/ABL‐expressing cells, confirming and extending previous data [19].
C‐Myc binding sites in the proximal survivin promoter are essential for transactivation by BCR/ABL kinase
To determine the essential responsive elements for activation of survivin promoter activity in BCR/ABL‐expressing cells, progressive deletion mutants from the entire survivin promoter (designated pGL3‐S1764) were generated and cotransfected into K562 cells. There was no significant loss of survivin promoter activity with deletions from −1764 to −343, whereas further deletion to much less sequences gradually eliminated its activity (Fig. 3A). Importantly, imatinib inhibited pGL3‐S343 promoter activity to a similar extent observed from its upstream precursors and this inhibitive activity was totally abrogated when further deletion to −116 position (Fig. 3A). These results suggested that the core region for survivin promoter activity is approximately within −343 upward the transcription start site, and that the sequence from −343 to −116 is required for response to BCR/ABL kinase.
Figure 3.

Characterization of c‐Myc as a transcription factor regulating survivin expression. (A) Progressive deletion mutation analysis of survivin promoter activity and its response to imatinib. K562 cells were transfected with pGL3‐S1764 and its progressive deletion mutants and cultured for 8 hrs, after which cells were incubated for 16 hrs in the absence or presence of 2.5 uM imatinib. The promoter activity was measured by normalizing firefly to renilla luciferase activity. Mean values ± S.D. from three independent experiments in triplicate are shown.*P≤ 0.001,#no significance. (B) A schematic diagram of the proximal survivin promoter indicating potential transcription factor binding sites. (C) Site‐directed mutation analysis of survivin promoter activity and its response to imatinib. Constructs of Sp1, STAT/TCF and c‐Myc binding sites mutation described in the illustration above were generated and transfected into K562 cells. The promoter activity was measured by normalizing firefly to renilla luciferase activity. Mean values ± S.D. from three independent experiments in triplicate are shown. *P < 0.01, #no significance. (D) ChIP assay analysed in vivo binding activity between c‐Myc and survivin promoter. Treated K562 cells were subjected to the ChIP protocol. The immunoprecipitations were performed with antibody against c‐Myc, and an isotype IgG as negative control. A parallel analysis were performed using an antibody against RNA‐polymerase as systematic control.
According to previous reports and analysis by TRANSFAC software (http://www.gene‐regulation.com), there are a cluster of binding sites for c‐Myc along with STAT/TCF, two Sp1‐like binding sites and another c‐Myc binding site (E‐box) within the imatinib responsive region (Fig. 3B). To determine whether these cis‐elements were essential for BCR/ABL to activate survivin gene, site‐directed mutants were generated (Fig. 3C). As compared with the wild‐type construct, mutations of STAT/TCF or Sp1 binding sites did not significantly affect the action of imatinib. Surprisingly, an unexpected increase activity was observed in STAT/TCF element mutant, which presumably results from the introduction of an unclear enhancer‐binding site by mutagenesis. In contrast, mutations in individual or both c‐Myc putative sites impaired imatinib‐mediated reduction of the survivin promoter activity (Fig. 3C), suggesting that c‐Myc‐binding sites are functionally responsive to BCR/ABL kinase.
To further determine that c‐Myc is involved in BCR/ABL‐induced survivin expression, ChIP assays were carried out (Fig. 3D). The cross‐linked chromatins were fragmented and immunoprecipitated with antibody against c‐Myc. Our results indicated that c‐Myc exhibited binding to the region containing E‐box but not a distal upstream in the survivin promoter. However, the binding activity was decreased either in the presence of imatinib or 10058‐F4. These results indicated that expression of survivin gene involves interaction of c‐Myc with the E‐Box region within the survivin promoter, consistent with the report from growth factor‐medicated survivin expression in breast cancer cells [30].
These results showed that c‐Myc is crucial for survivin expression in BCR/ABL transformed cells. Since then, we sought to determine the correlation between c‐Myc and survivin expression. Of interest, the expression way of c‐Myc in 32D‐BCR/ABL and 32Dcl3 cells was consistent with that of survivin (Fig. 4A). To further confirm the in vitro result, we collected samples from normal and patients to test if c‐Myc is deregulated in primary CML cells. A significantly higher expression of survivin was detected in all five CML cells but not normal cells with correlative to that of c‐Myc (Fig. 4B).
Figure 4.
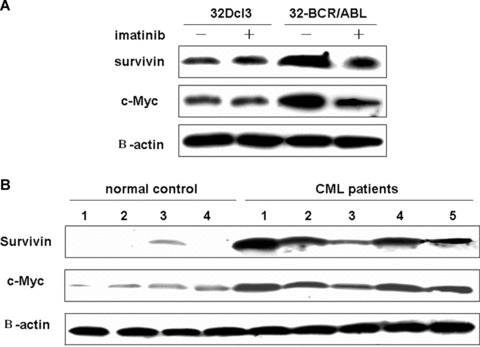
The correlation between c‐Myc and survivin expression in CML cells. (A) 32D‐BCR/A BL and 32Dcl3 cells were treated or untreated for 12 hrs with 2 uM imatinib. Western blot was carried out to determine the expression of c‐Myc and survivin. (B) Western blot analysis of c‐Myc and survivin expression in primary normal and CML cells.
Both JAK2 and PI3K signals contribute to transactivation of the survivin gene
Three major signal pathways have been implicated in the malignant transformation by BCR/ABL, namely MAPK/MEK, PI3K/AKT and JAK2/STAT [2]. Here, we employed selective chemical inhibitors of specific signalling components to test which of the molecule signals are responsible for survivin expression. First, the specificity of these inhibitors was tested (Fig. 5A). Also, pGL3‐control vector was used as a control to exclude the possibility of unspecific effect. We found that each of these inhibitors does not affect the activity of pGL3‐control obviously (Fig. 5B). Survivin promoter activity was sharply reduced by PI3‐kinase inhibitor LY294002 and JAK2 inhibitor AG490 separately, but not altered by the presence of MEK1/2 inhibitor PD98059 (Fig. 5C). Although combinative treatment of two of them reduced promoter activity to a similar extent observed in singular treatment (PD98059 excepted), we could not rule out the possible cross‐link between these signals on survivin promoter activity, as a cross‐talk between JAK2 and PI3K did exist in CML cells (see ‘Discussion’).
Figure 5.
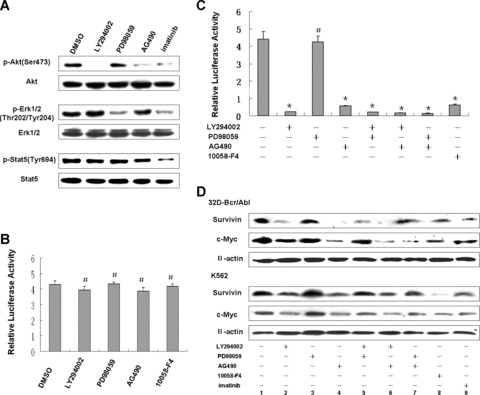
Survivin expression is dependent upon PI3K and JAK2, but not ERK1/2. (A) 32D‐BCR/ABL and K562 cells were treated with indicated signalling inhibitors as well as imatinib for 4 hrs. Fifty μg of total cell lysate protein was used for Western blot with antibodies against phosphorylation (upper panel) and its homologous proteins (lower panel). One of results from K562 cells was shown. (B, C) Effects of inhibitors on promoter activities. K562 cells transfected with pGL3‐control (B) and survivin promoter construct (C) were treated with indicated inhibitors for 16 hrs. The promoter activity was determined as described in Fig. 3. Mean values ± S.D. from three independent experiments in triplicate are shown. *P < 0.001, #no significance as compared to untreated control. (D) Western blot analysis of c‐Myc and survivin expression after treatment with aforementioned signalling inhibitors.
Consistent with results from the promoter activity assay, we found that both LY294002 and AG490 solely diminished c‐Myc and survivin protein, whereas no obvious change was observed in PD98059 treatment (Fig. 5D). Combination of LY294002 and AG490 did not further reduce survivin and c‐Myc protein. Surprisingly, PD98059 has a tendency to rescue the reduction induced by LY294002 or AG490 (Fig. 5D, lane 2 versus lane 5; lane 4 versus lane 7), with unknown mechanisms. Anyway, these data provided evidence that regulation of survivin expression in BCR/ABL‐expressing cells depends on JAK2 and PI3K signalling pathways.
Survivin is essential for BCR/ABL‐mediated survival and transformation
To elucidate whether survivin participates in the signalling that controls apoptosis and survival in CML cells, we first treated cells with different concentration of imatinib for different period. As shown in Fig. 6, imatinib treatment could diminish survivin expression and initiated caspase3 activation, leading to cell death in a dose‐dependent manner in BCR/ABL‐expressing cells (Fig. 6A). In control 32Dcl cells, neither survivin expression nor cell viability varied when treated with imatinib. Additionally, the kinetic relation between survivin reduction and cell apoptosis was also analysed. We found an earlier decrease of survivin preceded to the onset of caspase3 and the emergence of cell death upon imatinib treatment (Fig. 6B), suggestive of survivin as an important factor downstream the survival signalling in CML cells.
Figure 6.
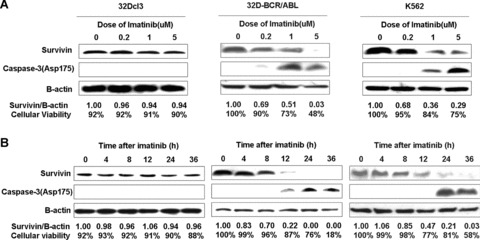
Decrease of survivin and introduction of cell death by imatinib treatment. Cells were treated for 24 hrs with imatinib in different concentrations (A) or with 2 uM imatinib for different time (B). Western blots show survivin expression and caspase‐3 activation upon imatinib treatment. The relative‐fold decrease of survivin protein after normalization to β‐actin internal control and the percentage of cell viability determined by trypan blue exclusion staining for each treatment are indicated.
Subsequently, we stably expressed survivin using retroviral transduction to observe whether forced expression of survivin can protect against cytotoxic effect of imatinib. Overexpression of survivin neither affected the proliferation nor the viability in K562 cells cultured in the normal condition (Fig. 7Ai). However, survivin conferred a modestly preserved effect on the viability in the present of imatinib (Fig. 7Aii), suggesting that survivin can only partially replace the survival promotion of BCR/ABL. To substantiate that, we also stably transduced survivin into 32Dcl3 cells and compared their growth factor‐independent survival with 32D‐BCR/ABL cells. Comparing with BCR/ABL, survivin did only partially protect 32 Dcl3 cells against cell death induced by IL‐3 deprivation (Fig. 7B).
Figure 7.
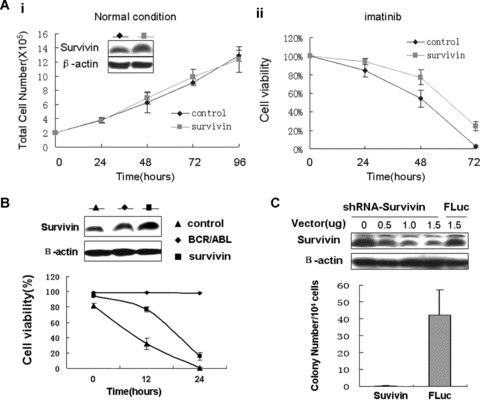
Roles of survivin in BCR/ABL‐mediated survival and transformation. (A) Enforced expression of survivin protects against imatinib‐induced cell death without affecting normal proliferation. Transfected cells were cultured in normal condition and in the presence of 1 uM imatinib and then cell number and viability were determined by trypan blue exclusion staining at indicated time‐points. Mean values ± S.D. from three independent experiments in triplicate are shown. Inset, survivin protein expression in K562 cells transfected with empty vector and survivin. (B) Comparison of survivin and BCR/ABL effects on the viability of 32Dcl3 cells after IL‐3 withdraw. Transfected cells were cultured without IL‐3 supplement and viability was determined by trypan blue exclusion staining. Mean values ± S.D. from three independent experiments in triplicate are shown. Upper panel, survivin protein expression in 32D‐BCR/ABL and 32Dcl3 cells transfected with empty vector and survivin. (C) Knockdown survivin expression inhibits the capacity of colony formation. K562 cells were transfected with shRNA‐survivin or the control shRNA‐Fluc. Thereafter, cells were cultured in methylcellulose containing G418. Twelve to 14 days later, colonies were counted. Mean values ± S.D. from three independent experiments in triplicate are shown. The efficiency of RNAi was carried out in 293T cell and shown in the upper panel.
In contrast with most non‐transformed haematopoietic cells, one of the transformed properties of K562 cells is their ability to grow in semi‐solid media [25]. To determine whether survivin is involved in this property of K562 cells, we used a stable shRNA expression vector to achieve a sustained silence of survivin expression in semi‐solid culture system. A powerful and specific shRNA sequence against survivin and a control sequence against firefly luciferase (described in Yang's studies [23]) were subcloned into stable expression vector and showed an efficient silencing (Fig. 7C). Stable knockdown of survivin expression almost completely diminished the colony formation of K562cell in methylcellulose (Fig. 7C), suggesting that survivin is involved in this aspect of cellular transformation by BCR/ABL. As a control, K562 cells transfected with shRNA‐Fluc grew efficiently in methylcellulose (Fig. 7C).
Inhibition of c‐Myc activity by 10058‐F4 enhances imatinib‐induced cell death
Because 10058‐F4 could block the binding activity of c‐Myc to survivin promoter (Fig. 3D), leading to reduction of survivin expression (Fig. 5D, lane 8), we examined whether 10058‐F4 would enhance the effect of imatinib. In fact, the combination of 10058‐F4 and imatinib was more effective in inducing cell death than was either agent alone in both 32D‐BCR/ABL and K562 cells (Fig. 8A). Previous studies demonstrated that IL‐3 can reverse the induction of apoptosis by imatinib via activation JAK2 pathway [31]. These studies and our present findings supported that c‐Myc has an important role in the cell survival effecting as a downstream mediator of JAK2 in CML cells. Therefore, we next determined whether 10058‐F4 would circumvent the IL‐3‐mediated rescue. As shown in Fig. 8B, 10058‐F4‐induced apoptosis in 32D‐BCR/ABL could not be reversed by addition of IL‐3 containing WEHI medium; in contrast, the induction of apoptosis by imatinib was markedly reversed by WEHI medium. Finally, we ask whether stable knockdown of c‐Myc has a same effect as survivin on the colony formation. As expected, the colony forming ability was disabled when K562 cells were subjected to c‐Myc disruption by shRNA‐mediated silencing (Fig. 8C).
Figure 8.

Interruption of c‐Myc activity enhances imatinib‐induced cell death and inhibits colony formation. (A) 10058‐F4 induced cell apoptosis with a synergistic effect with imatinib. 32D‐BCR/ABL and K562 cells were exposed to 2 uM imatinib, 60 uM 10058‐F4, or both for 24 hrs and 48 hrs, respectively. Apoptosis was assayed by Annexin V/PI staining and trypan blue staining. One representative result is presented. (B) 10058‐F4 overcame the anti‐apoptosis rescued by IL‐3. 32D‐BCR/ABL cells were incubated with 2 uM imatinib or 60 uM 10058‐F4 either in standard medium or IL‐3 (WEHI)‐supplemented medium, and cell death measured by trypan blue staining. Mean values ± S.D. from three independent experiments in triplicate are shown. (C) Knockdown c‐Myc expression inhibited the capacity of colony formation of K562 cells. Cells were transfected with shRNA‐c‐Myc and shRNA‐Fluc. Colonies were counted after cultured in methylcellulose for 14 days. Mean values ± S.D. from three independent experiments in triplicate are shown. The efficiency of RNAi was carried out in 293T cell and shown in the upper panel.
Discussion
BCR/ABL tyrosine kinase asserts its biological effect at least in part through inhibition of apoptosis [2, 3]. The network controlling apoptosis and survival in CML is very intricate and not fully understood. Many of anti‐apoptotic signals and its downstream effectors have been outlined during the past few years. Stat5 is a well‐known pathway responsible for activation of several Bcl‐2 family members [4, 5, 6, 7]. Apart from Stat5 pathway, our current study characterized JAK2/PI3K‐mediated and c‐Myc‐dependent transcriptional regulation of survivin expression as a novel molecular signalling of apoptosis regulation in BCR/ABL‐transformed cells.
Survivin has been implicated in multiple essential functions, including cell division, resistance to apoptosis, surveillance checkpoints of genomic integrity and the cellular stress response in many human cancers [13, 14]. This survivin ‘network’ is recently exploited in CML cells [19, 20], but many facets, especially with respect to its deregulated mechanisms and how it links with those known survival signals in CML, are ill‐defined. Our findings herein suggested that the survivin expression in CML cells is regulated at transcriptional and post‐translational levels as BCR/ABL increased survivin mRNA transcript and its cytoplasm accumulation (Fig. 1). The cellular pattern of survivin distribution has been linked with cancer development and prognosis [32]. It has been shown that mitochondrial survivin inhibits apoptosis and promotes tumourigenesis [27]. The nucleocytoplasmic transport is considered to be essential for such biological activities of survivin [28]. The nuclear survivin‐mediated mitosis is essential for normal cell proliferation [23], but whether it is indispensable to cancer cells is controversial [31]. Our findings that cytoplasmic survivin was responded well to imatinib (Fig. 1) recapitulated the important role of cytoplasmic survivin in CML cells. Heretofore, the underlying mechanisms by which BCR/ABL regulates its cytoplasmic store are totally unknown. According to others and our present findings, at least three mechanisms could be involved (Fig. 9): first, BCR/ABL transcriptionally activates survivin expression; second, BCR/ABL inhibits proteasome‐mediated degradation of survivin [20]; third, BCR/ABL regulates its nucleocytoplasmic transport through Crm1‐dependent nuclear export signal in survivin [28].
Figure 9.

A working model for BCR/ABL‐mediated induction of survivin expression. BCR/ABL kinase increases c‐Myc expression and activity through the JAK2/PI3K/AKT pathway, then c‐Myc directly interacts with survivin promter, activating survivin gene transcription. Perhaps, BCR/ABL also regulates nucleo‐cytoplasmic transport and degradation of survivin. The elevation of survivin and its intercellular distribution results in resistance to apoptosis and accelerated proliferation.
Because transcription deregulation is a general consequence of BCR/ABL transformation, our present work has preferentially focused on the elucidation of its transcriptional regulation in detail. Mutational analyses and ChIP assay revealed that c‐Myc is a bona fide transcription regulator responding to imatinib (Fig. 3). Using specific signal inhibitors, we found that regulation of survivin by c‐Myc is dependent on the JAK2 and PI3K signalling pathway but not the MAPK pathway (Fig. 5), which is inconsistent with the study from other group where the regulation of survivin expression by BCR/ABL is thought to via MAPK pathway [20]. We noticed that in that study survivin expression was examined at 48 hrs after drug treatment, a time‐point a majority of cells underwent apoptosis. So, the reduced survivin expression may be a consequence of cell death. Despite that, it should be noted that we from present data cannot exclude a contribution from MAPK signalling to modulating survivin expression in BCR/ABL‐expressing cells, because the effect of MAPK on survivin expression varied among cancer cell lines even with a same tissue origin [33]. Moreover, MAPK pathway enhances IRES‐dependent Myc protein synthesis through the HNRPK translation‐stimulatory activity [34]. However, we could not observe a decrease of c‐Myc expression upon PD98059, which may be due to a compensation by Cap‐dependent protein synthesis once IRES‐mediated mechanism is shut off. Our results also implied an existence of cross‐talk between JAK2 and PI3K regarding to regulation of c‐Myc (Fig. 5). This cross‐link has previously been described by Arlinghaus groups, who found that JAK2 is linked to PI3K via phosphorylating Gab2, an adaptor molecule coupling BCR/ABL tyrosine kinase to PI3K recruitment [35]. It is also notable that in this case, JAK2 and Stat5 are not linked as they are in cytokine‐induced signalling pathways but are two separate pathways activated by BCR/ABL [31, 36]. Moreover, blocking PI3K or JAK2 kinase does not interfere with Stat5 activation [10, 31, 36], which is reinforced in our present study (Fig. 5A). Therefore, we can exclude the possibility that JAK2 or PI3K inhibitor reduced survivin expression by repressing Stat5 activation. This suggested that Stat5 is not involved in the transcriptional regulation of survivin in CML cells, which is further upheld by the finding that mutation of STAT binding site in survivin promoter did not impair its responsivity to imatinib (Fig. 3C).
The region responsible for BCR/ABL kinase identified in our study is different to a possible region from −1194 to −1078 as characterized in BCR/ABL‐transduced Mo7e cells [19]. The explanation for this differentiation is unclear, but could be related to the diversity of pathways and mechanisms for BCR/ABL to transform cells of different type, or the different methods used to the deletion mutation. In addition to c‐Myc, several other factors can directly interact with their corresponding cis‐elements in human survivin promoter, namely, β‐catenin/TCF [37, 38], Stat3 [39], Rb/E2F [40, 41], HRE [33], p53 [41, 42], Sp1/KLF5 [42, 43]. Consistent with abundant expression of survivin in cancer, the activity of these factors is frequently disordered in various cancers [37, 38, 39, 40, 41, 42]. In CML cells, mutations of p53 and Rb genes frequently occur at the accelerated phase(AP) and blast crisis (BC) [1], which may contribute to a further elevation of survivin in those cases with AP and BC [16, 17, 18]. Loss of repressive functions of p53 and Rb in K562 cells can also explain why mutation or deletion of their upstream elements did not completely repress survivin promoter activity (Fig. 3A). Moreover, BCR/ABL has been supposed to regulate VEGF via Sp1 and HIF‐1α[29, 44]. Sp1 is responsible for basal and cell cycle‐dependent regulation of survivin expression [43], and HIF‐1α for epidermal growth factor receptor regulation of survivin expression [33]. Our deletion mutation analysis revealed that these elements seem required for a basal level of survivin promoter activity in K562 cells, but not for response to imatinib (Fig. 3A). Several NF‐kappaB binding sites have been identified in mouse survivin promoter [45, 46]. However, human survivin promoter does not possess such elements in its corresponding loci [43, 45]. Of interest, NF‐kappaB is considered to transactivate c‐Myc expression in CML cells [35], so it possibly regulates human survivin expression through activation c‐Myc. Together, we concluded that BCR/ABL‐induced survivin expression is, if not all, at least in part dependent on the transcription factor c‐Myc.
Our present findings together with other reports [19, 20] strongly suggested that survivin is an important gene functioning as regulating cell survival and apoptosis in CML. We also found that overexpression of survivin could only partially protect against cell death upon imatinib or IL‐3 deprivation (Fig. 7A and B). This is reasonable because there are many anti‐apoptotic factors involved [3, 4, 5, 6, 7, 8, 9, 10], and only supplement with one of them could not fully replace other factors’ functions. Although stably repressing survivin expression almost totally impaired the colony forming ability of K562 (Fig. 7C), it remains to determine whether silencing survivin inhibits in vivo leukaemogenesis and whether survivin is associated with other malignant features of CML cells, i.e. differentiation arrest and failures of genome surveillance [1].
c‐Myc is a well‐characterized transcription factor that heterodimerizes with Max and plays a pivotal role in the regulation of proliferation, differentiation and apoptosis [47]. In line with its multiple functions, c‐Myc is required for and cooperates with BCR/ABL in transformation of haematopoiesis [48], which is further substantiated in our ex vivo colony formation assay (Fig. 8C). However, little is known about its downstream target network related to CML development [2]. It is possible that c‐Myc may constitute an anti‐apoptotic signal equipped by the survivin network [14]. More recently, aberrant miRNA expression regulated by c‐Myc has been described in CML, where overexpression of miR‐17–92 polycistron was found in CP CML but not in BC CML [24]. In addition, c‐Myc‐regulated miRNAs have been found to modulate its transcriptional targets, through which c‐Myc simultaneously activates its targets and limits its translation [49]. It will be exciting to examine whether c‐Myc regulates differential expression of survivin observed in the transition from chronic stage to blast crisis [16, 17, 18] through this mechanism.
Previous crystal analysis of survivin structure reveals no obvious opportunity for drug design [50]. As a consequence, no truly survivin‐directed antagonists are available now [14]. Our current findings add c‐Myc as a candidate to design drug against survivin. Since small‐molecule antagonists that selectively disrupt c‐Myc activity have been identified [51], it is rational to use such agents to antagonize survivin. Our ex vivo studies support this proposal by finding that 10058‐F4 induced apopotosis with a synergistic interaction with imatinib, and more importantly, it could overcome IL‐3‐induced resistance to imatinib (Fig. 8A and B). Growth factors‐mediated resistance in CML is a recently identified mechanism for the development of resistance to imatinib and nilotinib [52]. In this regard, specific inhibition of c‐Myc activity by small‐molecule antagonists could enhance the effects of imatinib as well as overcome its resistance. Additionally, the small‐molecule drugs seem more feasible for clinical testing than strategies using anti‐sense oligonucleotides or dominant negative mutants [19, 20].
In summary, our findings highlight a novel molecular signalling (Fig. 9) whereby BCR/ABL controls survivin expression and suggest an attractive therapeutic strategy by interference with its effector c‐Myc.
Acknowledgements
We gratefully thank Dr. D. Perrotti for providing cell lines and Drs. D. Yang, L. Venturini for an offer of shRNA sequences. This study was supported by National Natural Science Foundation of China (30570357 and 30600238) and Tianjin Municipal Science and Technology Commission (06YFSZSF01300 and 05YFJZJC01500).
References
- 1. Melo JV, Barnes DJ. Chronic myeloid leukaemia as a model of disease evolution in human cancer. Nat Rev Cancer . 2007; 7: 441–53. [DOI] [PubMed] [Google Scholar]
- 2. Deininger MW, Goldman JM, Melo JV. The molecular biology of chronic myeloid leukemia. Blood . 2000; 96: 3343–56. [PubMed] [Google Scholar]
- 3. Fernandez‐Luna JL. Bcr‐Abl and inhibition of apoptosis in chronic myelogenous leukemia cells. Apoptosis . 2000; 5: 315–8. [DOI] [PubMed] [Google Scholar]
- 4. Horita M, Andreu EJ, Benito A, et al . Blockade of the Bcr‐Abl kinase activity induces apoptosis of chronic myelogenous leukemia cells by suppressing signal transducer and activator of transcription 5‐dependent expression of Bcl‐xL. J Exp Med . 2000; 191: 977–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gesbert F, Griffin JD. Bcr/Abl activates transcription of the Bcl‐X gene through STAT5. Blood . 2000; 96: 2269–76. [PubMed] [Google Scholar]
- 6. Aichberger KJ, Mayerhofer M, Krauth MT, et al . Identification of mcl‐1 as a BCR/ABL‐dependent target in chronic myeloid leukemia (CML): evidence for cooperative antileukemic effects of imatinib and mcl‐1 antisense oligonucleotides. Blood . 2005; 105: 3303–11. [DOI] [PubMed] [Google Scholar]
- 7. Nieborowska‐Skorska M, Hoser G, Kossev P, et al . Complementary functions of the antiapoptotic protein A1 and serine/ threonine kinase pim‐1 in the BCR/ABL‐mediated leukemogenesis. Blood . 2002; 99: 4531–9. [DOI] [PubMed] [Google Scholar]
- 8. Mayerhofer M, Florian S, Krauth MT, et al . Identification of heme oxygenase‐1 as a novel BCR/ABL‐dependent survival factor in chronic myeloid leukemia. Cancer Res . 2004; 64: 3148–54. [DOI] [PubMed] [Google Scholar]
- 9. Yoshida C, Yoshida F, Sears DE, et al . Bcr‐Abl signaling through the PI‐3/S6 kinase pathway inhibits nuclear translocation of the transcription factor Bach2, which represses the antiapoptotic factor heme oxygenase‐1. Blood . 2007; 109: 1211–9. [DOI] [PubMed] [Google Scholar]
- 10. Svensson E, Vidovic K, Lassen C, et al . Deregulation of the Wilms’ tumour gene 1 protein (WT1) by BCR/ABL1 mediates resistance to imatinib in human leukaemia cells. Leukemia . 2007; 21: 2485–94. [DOI] [PubMed] [Google Scholar]
- 11. Tipping AJ, Deininger MW, Goldman JM, et al . Comparative gene expression profile of chronic myeloid leukemia cells innately resistant to imatinib mesylate. Exp Hematol . 2003; 31: 1073–80. [PubMed] [Google Scholar]
- 12. Ravandi F, Kantarjian HM, Talpaz M, et al . Expression of apoptosis proteins in chronic myelogenous leukemia: associations and significance. Cancer . 2001; 91: 1964–72. [DOI] [PubMed] [Google Scholar]
- 13. Altieri DC. Validating survivin as a cancer therapeutic target. Nat Rev Cancer . 2003; 3: 46–54. [DOI] [PubMed] [Google Scholar]
- 14. Altieri DC. Survivin, cancer networks and pathway‐directed drug discovery. Nat Rev Cancer . 2008; 8: 61–70. [DOI] [PubMed] [Google Scholar]
- 15. Cong XL, Han ZC. Survivin and leukemia. Int J Hematol . 2004; 80: 232–8. [DOI] [PubMed] [Google Scholar]
- 16. Badran A, Yoshida A, Wano Y, et al . Expression of the antiapoptotic gene survivin in chronic myeloid leukemia. Anticancer Res . 2003; 23: 589–92. [PubMed] [Google Scholar]
- 17. Hernández‐Boluda JC, Bellosillo B, Vela MC, et al . Survivin expression in the progression of chronic myeloid leukemia: a sequential study in 16 patients. Leuk Lymphoma . 2005; 46: 717–22. [DOI] [PubMed] [Google Scholar]
- 18. Moriai R, Asanuma K, Kobayashi D, et al . Quantitative analysis of the anti‐apoptotic gene survivin expression in malignant haematopoietic cells. Anticancer Res . 2001; 21: 595–600. [PubMed] [Google Scholar]
- 19. Wang Z, Sampath J, Fukuda S, et al . Disruption of the inhibitor of apoptosis protein survivin sensitizes Bcr‐abl‐positive cells to STI571‐induced apoptosis. Cancer Res . 2005; 65: 8224–32. [DOI] [PubMed] [Google Scholar]
- 20. Carter BZ, Mak DH, Schober WD, et al . Regulation of survivin expression through Bcr‐Abl/MAPK cascade: targeting survivin overcomes imatinib resistance and increases imatinib sensitivity in imatinib‐responsive CML cells. Blood . 2006; 107: 1555–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhou B, Bi YY, Han ZB, et al . G‐CSF‐mobilized peripheral blood mononuclear cells from diabetic patients augment neovascularization in ischemic limbs but with impaired capability. J Thromb Haemost . 2006; 4: 993–1002. [DOI] [PubMed] [Google Scholar]
- 22. Cong XL, Li B, Yang RC, et al . Enhanced growth suppression of Philadephia1 leukemia cells by targeting bcr3/abl2 and VEGF through antisense strategy. Leukemia. 2005; 19: 1517–24. [DOI] [PubMed] [Google Scholar]
- 23. Yang D, Welm A, Bishop JM. Cell division and cell survival in the absence of survivin. Proc Natl Acad Sci USA . 2004; 101: 15100–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Venturini L, Battmer K, Castoldi M, et al . Expression of the miR‐17–92 polycistron in chronic myeloid leukemia (CML) CD34+ cells. Blood . 2007; 109: 4399–405. [DOI] [PubMed] [Google Scholar]
- 25. de Groot RP, Raaijmakers JA, Lammers JW, et al . STAT5 activation by BCR‐Abl contributes to transformation of K562 leukemia cells. Blood . 1999; 94: 1108–12. [PubMed] [Google Scholar]
- 26. Fortugno P, Wall NR, Giodini A, et al . Survivin exists in immunochemically distinct subcellular pools and is involved in spindle microtubule function. J Cell Sci . 2002; 115: 575–85. [DOI] [PubMed] [Google Scholar]
- 27. Dohi T, Beltrami E, Wall NR, et al . Mitochondrial survivin inhibits apoptosis and promotes tumorigenesis. J Clin Invest . 2004; 114: 1117–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Knauer SK, Krämer OH, Knösel T, et al . Nuclear export is essential for the tumor‐promoting activity of survivin. FASEB J . 2007; 21: 207–16. [DOI] [PubMed] [Google Scholar]
- 29. Legros L, Bourcier C, Jacquel A, et al . Imatinib mesylate (STI571) decreases the vascular endothelial growth factor plasma concentration in patients with chronic myeloid leukemia. Blood . 2004; 104: 495–501. [DOI] [PubMed] [Google Scholar]
- 30. Cosgrave N, Hill AD, Young LS. Growth factor‐dependent regulation of survivin by c‐myc in human breast cancer. J Mol Endocrinol . 2006; 37: 377–90. [DOI] [PubMed] [Google Scholar]
- 31. Xie S, Lin H, Sun T, et al . Jak2 is involved in c‐Myc induction by Bcr‐Abl. Oncogene . 2002; 21: 7137–46. [DOI] [PubMed] [Google Scholar]
- 32. Li F, Yang J, Ramnath N, et al . Nuclear or cytoplasmic expression of survivin: what is the significance Int J Cancer . 2005; 114: 509–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Peng XH, Karna P, Cao Z, et al . Cross‐talk between epidermal growth factor receptor and hypoxia‐inducible factor‐1alpha signal pathways increases resistance to apoptosis by up‐regulating survivin gene expression. J Biol Chem . 2006; 281: 25903–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Notari M, Neviani P, Santhanam R, et al . A MAPK/HNRPK pathway controls BCR/ABL oncogenic potential by regulating MYC mRNA translation. Blood . 2006; 107: 2507–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Samanta AK, Lin H, Sun T, et al . Janus kinase 2: a critical target in chronic myelogenous leukemia. Cancer Res . 2006; 66: 6468–72. [DOI] [PubMed] [Google Scholar]
- 36. Xie S, Wang Y, Liu J, et al . Involvement of Jak2 tyrosine phosphorylation in Bcr‐Abl transformation. Oncogene . 2001; 20: 6188–95. [DOI] [PubMed] [Google Scholar]
- 37. Kim PJ, Plescia J, Clevers H, et al . Survivin and molecular pathogenesis of colorectal cancer. Lancet . 2003; 362: 205–9. [DOI] [PubMed] [Google Scholar]
- 38. Ma H, Nguyen C, Lee KS, et al . Differential roles for the coactivators CBP and p300 on TCF/beta‐catenin‐mediated survivin gene expression. Oncogene . 2005; 24: 3619–31. [DOI] [PubMed] [Google Scholar]
- 39. Gu L, Chiang KY, Zhu N, et al . Contribution of STAT3 to the activation of survivin by GM‐CSF in CD34+ cell lines. Exp Hematol . 2007; 35: 957–66. [DOI] [PubMed] [Google Scholar]
- 40. Jiang Y, Saavedra HI, Holloway MP, et al . Aberrant regulation of survivin by the RB/E2F family of proteins. J Biol Chem . 2004; 279: 40511–20. [DOI] [PubMed] [Google Scholar]
- 41. Hoffman WH, Biade S, Zilfou JT, et al . Transcriptional repression of the anti‐apoptotic survivin gene by wild type p53. J Biol Chem . 2002; 277: 3247–57. [DOI] [PubMed] [Google Scholar]
- 42. Zhu N, Gu L, Findley HW, et al . KLF5 interacts with p53 in regulating survivin expression in acute lymphoblastic leukemia. J Biol Chem . 2006; 281: 14711–8. [DOI] [PubMed] [Google Scholar]
- 43. Li F, Altieri DC. Transcriptional analysis of human survivin gene expression. Biochem J . 1999; 344: 305–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mayerhofer M, Valent P, Sperr WR, et al . BCR/ABL induces expression of vascular endothelial growth factor and its transcriptional activator, hypoxia inducible factor‐1alpha, through a pathway involving phosphoinositide 3‐kinase and the mammalian target of rapamycin. Blood . 2002; 100: 3767–75. [DOI] [PubMed] [Google Scholar]
- 45. Li F, Altieri DC. The cancer antiapoptosis mouse survivin gene: characterization of locus and transcriptional requirements of basal and cell cycle‐dependent expression. Cancer Res . 1999; 59: 3143–51. [PubMed] [Google Scholar]
- 46. Kawakami H, Tomita M, Matsuda T, et al . Transcriptional activation of survivin through the NF‐kappaB pathway by human T‐cell leukemia virus type I tax. Int J Cancer . 2005; 115: 967–74. [DOI] [PubMed] [Google Scholar]
- 47. Levens D. Disentangling the MYC web. Proc Natl Acad Sci USA . 2002; 99: 5757–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sawyers CL, Callahan W, Witte ON. Dominant negative MYC blocks transformation by ABL oncogenes. Cell . 1992; 70: 901–10. [DOI] [PubMed] [Google Scholar]
- 49. O’Donnell KA, Wentzel EA, Zeller KI, et al . c‐Myc‐regulated microRNAs modulate E2F1 expression. Nature . 2005; 435: 839–43. [DOI] [PubMed] [Google Scholar]
- 50. Muchmore SW, Chen J, Jakob C, et al . Crystal structure and mutagenic analysis of the inhibitor‐of‐apoptosis protein survivin. Mol Cell . 2000; 6: 173–82. [PubMed] [Google Scholar]
- 51. Wang H, Hammoudeh DI, Follis AV, et al . Improved low molecular weight Myc‐Max inhibitors. Mol Cancer Ther . 2007; 6: 2399–408. [DOI] [PubMed] [Google Scholar]
- 52. Wang Y, Cai D, Brendel C, et al . Adaptive secretion of granulocyte‐macrophage colony‐stimulating factor (GM‐CSF) mediates imatinib and nilotinib resistance in BCR/ABL+ progenitors via JAK‐2/STAT‐5 pathway activation. Blood . 2007; 109: 2147–55. [DOI] [PubMed] [Google Scholar]