

Abstract
This study aims to develop a new animal model of vulnerable plaques and investigate the potential mechanisms of exogenous p53‐induced plaque instability. Forty rabbits underwent aortic balloon injury, were fed a 1% cholesterol diet for 10 weeks and then normal chow for 6 weeks. Rabbits were divided into Ad5‐CMV.p53‐treated group (n= 16), Ad5‐CMV.lac Z‐treated group (n= 16) and blank control group (n= 8). Under the guidance of intravascular ultrasound, a 50‐μl suspension of adenovirus containing p53 or lac Z was injected into the largest plaque of the first two groups, respectively, and these rabbits received pharmacological triggering 2 weeks later. In 76.9% of rabbits with p53 transfection, plaque rupture was found, which was significantly (P < 0.05) higher than that in the Ad5‐CMV.lac Z‐treated plaques (23.1%), or blank controls plaques (0%). Increased apoptotic cells, and subsequently, decreased vascular smooth muscle cells and collagen content, enhanced intima macrophage accumulation, increased C‐reactive protein (CRP) and matrix metalloproteinases staining and high serum levels of high sensitive CRP (hs‐CRP) and monocyte chemoattractant protein‐1 (MCP‐1) were observed in Ad5‐CMV.p53‐treated rabbits. However, a binary logistic regression model revealed that hs‐CRP concentration rather than apoptosis rate played an independent role in plaque rupture with an odds ratio as 1.314 (95% CI: 1.041–1.657, P= 0.021), and there were high positive correlations between inflammatory biomarkers (hs‐CRP or MCP‐1) and apoptosis (R 2= 0.761, and R 2= 0.557, respectively, both P < 0.01). Intraplaque injection of p53 gene provides a safe and effective method for inducing plaque vulnerability in rabbits. The destabilizing effect of p53 overexpression is mediated mainly through apoptosis‐enhanced inflammation rather than cell apoptosis itself.
Keywords: atherosclerosis, vulnerable plaque, p53, apoptosis, inflammation
The tumour suppressor gene p53 codes for a transcription factor that activates genes involved in cell growth arrest (p21, GADD45) and apoptosis (e.g. Bax, Fas, IGF‐bp3), which are up‐regulated in a variety of pathological settings such as DNA damage, enhanced oxidative stress and accumulation of oxidized lipoproteins [1]. It has been demonstrated that p53 expression is increased in human atherosclerotic lesions, both in lipid‐laden macrophages and vascular smooth muscle cells (VSMCs) [2]. Exogenous p53 gene (Ad5‐CMV.p53) has been transfected into the abdominal aorta in rabbits or the carotid artery in mice to induce plaque instability [3, 4]. However, the exact mechanism of p53‐induced plaque rupture is poorly understood.
It has been well established that a vulnerable plaque is the major cause of acute sequelae of atherosclerosis [5, 6]. A plaque with a thin cap, a large lipid core and abundant activated macrophages have been generally regarded as a plaque vulnerable to rupture. However, an animal model of vulnerable plaques mimicking human lesions is still lacking, which has limited the progress of basic research in plaque instability. In our previous studies, we used a catheter to inject Ad5‐CMV.p53 into an abdominal aortic segment which was rich in plaques and ligated at its both ends for 10 min. Although we achieved a high rate of plaque rupture in rabbits [3, 7], this model has several limitations that require further improvement. First, aortic ligation may lead to abdominal organ ischemia and reperfusion injury contributing to a relatively high mortality rate; second, the transfection efficiency is difficult to control during 10 min. contact of Ad5‐CMV.p53 suspension with the endothelium; third, p53 suspension spreads to the whole cardiovascular system after release of the aortic ligation, which may cause systemic adverse effects. Therefore, the present study was carried out to develop a new animal model of vulnerable plaques and investigate the possible mechanisms of exogenous p53‐induced plaque instability using this new model.
Materials and methods
Rabbit housing and diet
Adult New Zealand white male rabbits weighing 1.7–2.3 kg (n= 40) were housed at the Animal Center of Shandong University Qilu Hospital. The Principle of Laboratory Animal Care published by the National Institute of Health in 1996 was followed, and the experimental protocol was approved by the Animal Care Committee of Shandong University.
All rabbits underwent balloon‐induced endothelial injury of the abdominal aorta before they were fed a diet of 1% cholesterol (120–140 g/day) for 10 weeks and then a normal diet for 6 weeks. Thereafter these rabbits were divided into three groups: Ad5‐CMV.p53‐treated group (n= 16), Ad5‐CMV.lac Z‐treated group (n= 16) and blank control group (n= 8). Rabbits of blank control group were killed for histological and immunohistochemical studies at the end of week 16. Body weight was monitored throughout the experiment.
Balloon endothelial injury
Rabbits were anaesthetized with 3% sodium pentobarbital (30 mg/kg given intravenously, with additional 4 mg/kg doses as needed). Rabbits underwent balloon injury of the abdominal aorta with a balloon catheter introduced through the right femoral artery. When the catheter was advanced into the thoracic aorta, the balloon was inflated with 0.3 ml saline and the catheter was gently retracted to the iliofemoral artery. After this process was repeated for three times, the catheter was removed and the incision closed.
IVUS imaging and adenoviral suspension transduction protocol
Intravascular ultrasound (IVUS, Galaxy, Boston Scientific Corporation, Natick, MA, USA) studies involved use of a commercially available system (Galaxy, Boston Scientific Corporation, USA) at weeks 10 and 16 since the beginning of the experiment. The 40‐MHz IVUS catheter was advanced into the thoracic aorta through the left femoral artery and the ultrasonic images were recorded during automatic pullback of the catheter to the iliofemoral artery at a speed of 0.5 mm/sec. After the largest plaque in the abdominal aorta was imaged with IVUS, a median incision in the abdomen was made and the corresponding portion of the abdominal aorta was isolated. Under the guidance of IVUS, a 50‐μl suspension of adenovirus containing either p53 (8 × 109 plaque forming unit (pfu)/ml, Shanghai Shuiyuan Shengwu Technology Co. Ltd., Shanghai, China) or lac Z (8 × 109 pfu/ml, Shanghai Shuiyuan Shengwu Technology Co. Ltd.) was injected from the adventitia of the aorta into the plaque being imaged. The injected site was marked by an iliopsoas stitch. The abdominal cavity was closed and the animals were given antibiotics intravenously to prevent infection. One day later, three rabbits each in the two treatment groups were killed and their aortas were removed.
Pharmacological triggering of plaque disruption
Two weeks after transfection, plaque disruption was triggered by administration of Russell's viper venom (RVV, 0.15 mg/kg intraperitoneally, Sigma, St. Louis, MO, USA), followed 30 min. later by histamine (0.02 mg/kg intravenously). Rabbits were killed 24–48 hrs after the above procedure by bolus injection of sodium pentobarbital (100 mg/kg intravenously), and the effects on plaque integrity were assessed.
Biochemical studies
Fasting serum total cholesterol (TC) and low‐density lipoprotein cholesterol (LDL‐C) levels were measured at weeks 10, 16 and 18, respectively. Serum TC and LDL‐C was obtained by enzymatic assays using an automated biochemical analyser (Roche Hitachi 917, Block Scientific, Inc., NY, USA). At the same time, C‐reactive protein (CRP) was assayed using a highly sensitive ELISA kit (Adlitteram Diagnostic Laboratories, San Diego, CA, USA), and monocyte chemoattractant protein‐1 (MCP‐1) was performed using ELISA kits (Adlitteram Diagnostic Laboratories).
Quantification of apoptosis
Apoptosis was assessed by terminal deoxynucleotidyl transferase end‐labelling (TUNEL, Calbiochem, EMD Bioscences, Darmstadt, Germany) staining. Only TUNEL+ nuclei with morphological features of apoptosis, including cell shrinkage, nuclear pyknosis, chromatin condensation and nuclear fragmentation were included. Counterstaining of the nucleus involved methyl green. The apoptosis rate was expressed as the proportion of apoptotic cells to total number of cells in a given area.
Histological and immunohistochemical staining
Removed aortas were cut into 2.5‐mm serial sections, fixed overnight in 4% paraformaldehyde, with a second overnight tissue processing and dehydration and embedded in paraffin. Serial cross‐sections underwent general histological staining with haematoxylin eosin, Masson's trichome and picrosirius red, and specific immunohistochemical staining. Slides were stained with antibodies against recombinant human p53 (Labvision, Lab Vision NeoMarkers, Fremont, CA, USA, 1:100), a macrophage‐specific antibody (RAM‐11, Lab Vision NeoMarkers, 1:800), α‐smooth muscle cell actin (Chemicon, Chemicon International, Boston, MA, USA, 1:200), matrix metalloproteinase (MMP) 2 (Chemicon, Chemicon International, 1:100), MMP9 (Chemicon, Chemicon International, 1:1200) and CRP (R&D, R&D Systems, Minneapolis, MI, USA, 1:30). Immunohistochemical staining was visualized by use of a DAB kit according to the manufacturer's instructions. To clarify whether MMP‐2 and MMP‐9 were secreted from smooth muscle cells or macrophages, serial cross‐sections were stained consecutively with antibodies against α‐actin, antibodies against MMP2 or MMP9 and then antibodies against RAM11. Nucleus counterstaining involved haematoxylin (blue) or methyl green (green). The remaining specimens were embedded in optimum cutting temperature (OCT) compound (Tissue‐Tek, Fisher Scientific), snap‐frozen, and stored at −70°C. X‐Gal staining was performed on 7‐μm‐thick cryostat sections using a β‐galactosidase activity assay kit (Genmed, Genmed Scientifics, Inc., Washington, DC, USA). After incubation with x‐gal at 37°C for 24 hrs, sites exhibiting characteristic X‐Gal blue colour were considered to have positive expression of lac Z product.
Histological analysis
Sections stained with haematoxylin and eosin were analysed for disrupted plaques, histologically defined as loss of fibrous‐cap continuity with overlying luminal thrombosis. Six cross‐sections from the histological sections of aorta were used for analysis of cap thickness, intima thickness and cap/intima ratio, and the values averaged. Slides were scanned by microscope (Olympus BX51, Olympus Corporation, Tokyo, Japan) for computerized image analysis with Image‐Pro Plus 5.0 (Media Cybernetics, Washington, DC, USA). Positive staining of collagen, VSMCs and macrophages was expressed as a percentage of the staining area divided by the plaque area in at least 10 high power fields (×400) and the values were averaged.
Statistical analysis
Values were expressed as mean ± S.D. and assessed by one‐sample Kolmogorov–Smirnov test to check if it is normal distribution. Differences in continuous variables between two groups were assessed by unpaired t‐test, and comparison among multiple groups was performed by analysis of variance with ANOVA. Categorical variables were analysed by Fisher's exact test. A bivariate correlation was used to choose quantitative variables which demonstrated significant correlations with plaque rupture, and these variables were then introduced into a binary logistic regression model to select variables which had independent effects on plaque rupture. Subsequently, linear regression analyses were performed to evaluate the relationship between apoptosis and inflammatory markers including hs‐CRP and MCP‐1. A P‐value < 0.05 was considered statistically significant.
Results
Body weights and biochemical studies
All animals underwent initial balloon injury and the next abdominal operation without complications and showed full recovery. The weight of rabbits at baseline was 1.97 ± 0.22 kg, but increased to 3.23 ± 0.21 kg and 3.31 ± 0.18 kg at weeks 10 and 16, respectively (both P < 0.01). The serum TC and LDL‐C levels of all rabbits increased significantly after the high‐cholesterol diet and were reduced 6 and 8 weeks later (P < 0.01, Fig. 1A and B). However, values did not significantly differ among the three groups (P > 0.05, Fig. 1A and B).
Figure 1.

Biochemical measurements in three groups of rabbits. Groups A, B and C represent Ad5‐CMV.p53‐treated group, Ad5‐CMV.lac Z‐treated group and blank control group, respectively. (A, B, C and D) showed levels of serum TC, LDL‐C, hs‐CRP and MCP‐1 in three groups at baseline, weeks 10, 16 and 18, respectively. **P < 0.01 versus baseline, # P < 0.05 versus week 10, ## P < 0.01 versus week 10, P < 0.01 versus week 16, $$ P < 0.01 versus group A.
Levels of hs‐CRP and MCP‐1 were not significantly different among rabbits in the three groups at baseline, weeks 10 and 16 before gene transfection (P > 0.05). However, as shown in Fig. 1C and D, the two inflammatory biomarkers were significantly higher in rabbits with Ad5‐CMV.p53‐treated group than those in rabbits with Ad5‐CMV.lac Z‐treated group and the blank control group at week 18 (all P < 0.01).
IVUS imaging
By IVUS imaging, all plaques were characterized as fibrofatty and eccentric (Fig. 2A and B). No ruptured plaque and thrombosis could be detected by IVUS before p53 gene transfection and pharmacological triggering. The site of adenovirus injection was displayed by IVUS as the dent at the side of the vessel wall (Fig. 2C).
Figure 2.
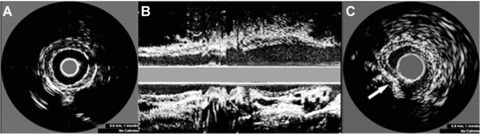
IVUS images of plaques in the abdominal aorta at week 16. (A) Circumferential plaque distribution in a short‐axis view. (B) Longitudinal plaque distribution in a long axis view. (C) The method of intraplaque injection. Arrow indicates the position of a needle.
Apoptosis
One day after transfection, TUNEL staining showed apoptosis in 1.87%± 0.17% of Ad5‐CMV.lac Z‐treated plaque cells as compared with 10.99%± 0.78% of Ad5‐CMV.p53‐treated cells. However, 14 days after transfection, the apoptosis rate was 0.97%± 0.13% and 2.71%± 0.42% in the Ad5‐CMV.lac Z‐treated group and Ad5‐CMV.p53‐treated group, respectively. Cell apoptosis was significantly increased in Ad5‐CMV.p53‐treated plaques, compared with Ad5‐CMV.lac Z‐treated plaques at days 1 and 14, although TUNEL+ cells were reduced at day 14 in both groups (Fig. 6A). In Ad5‐CMV.p53‐treated plaques, TUNEL+ cells were located mainly in the fibrous cap and the intima (Fig. 3A), while TUNEL staining was observed in the lipid core of Ad5‐CMV. Lac Z‐treated plaques which seemed to be more dispersed (Fig. 3B). Few apoptotic cells in the blank control group were found which were located mainly in the lipid core (0.15%± 0.07%).
Figure 6.
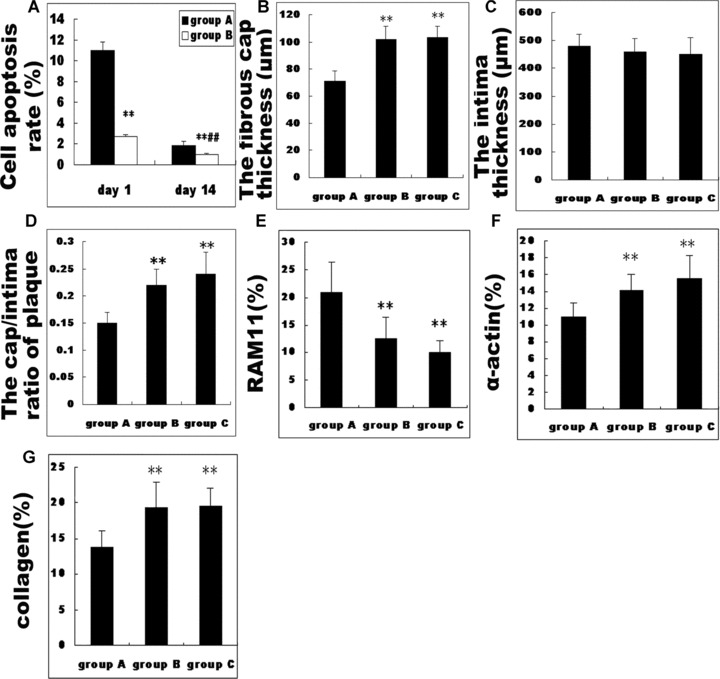
Pathologic measurements in three groups of rabbits. Groups A, B and C represent Ad5‐CMV.p53‐treated group, Ad5‐CMV.lac Z‐treated group and blank control group, respectively. (A) showed the comparison in apoptosis rate between Ad5‐CMV.p53‐treated and Ad5‐CMV.lac Z‐treated groups. **P < 0.01 versus group A, ## P < 0.01 versus day 1. (B, C and D) showed the comparison in intima thickness, cap thickness and the cap/intima ratio among three groups. **P < 0.01 versus group A. (E, F and G) showed the comparison in the relative content of macrophages, α‐actin and collagen in plaques among three groups. **P < 0.01 versus group A.
Figure 3.

TUNEL, p53 and lac Z staining. (A and B) TUNEL staining at day 1 after transfection with Ad5‐CMV.p53 (A) and Ad5‐CMV.lac Z (B). Bars = 50μm. (C and D) x‐gal staining of transfected Ad5‐CMV.lac Z 1 day and 14 days after injection, respectively. Efficient transfection occurred in the intima. Bars = 100 μm. (E and F) p53 staining in Ad5‐CMV.p53‐treated group 1 day and 14 days after injection, respectively. p53+ cells were located mainly in the superficial layer of the plaques. A substantial number of p53+ cells were identified in the intima at day 1 after transfection but these cells became sparse by day 14. (G) p53 staining in Ad5‐CMV.lac Z‐treated group 14 days after injection. Only scanty p53+ cells were found mainly in the lipid core of the plaques. Bars = 50 μm.
Adenoviral expression pattern
To evaluate gene transfection efficiency and expression duration, Ad5‐CMV.lac Z β‐galactosidase activity was measured and detected 1 day after transfection, both in the lesion core and fibrous cap (Fig. 3C), which was still detectable 14 days after transfection (Fig. 3D). In Ad5‐CMV.p53‐treated animals, p53+ cells were localized mainly in the superficial, smooth muscle cell‐rich layer of plaques. One day after Ad5‐CMV.p53 transfection, a substantial number of p53+ cells were identified in the intima (Fig. 3E). However, by day 14, p53 staining was minimal (Fig. 3F). Scanty p53+ cells were identified mainly in the lipid core of the Ad5‐CMV.lac Z‐treated plaques (Fig. 3G), while no p53 expression was found in plaques of the blank control group.
Morphometry and immunohistochemical staining
Total cross‐sectional plaque area did not differ among the three groups. The intima thickness of plaques with Ad5‐CMV.p53 treatment did not differ from that with Ad5‐CMV.lac Z treatment and that of blank control group (Fig. 6C). In comparison with Ad5‐CMV.lac Z‐treated plaques and blank control plaques, the decrease in the fibrous cap thickness in Ad5‐CMV.p53‐treated plaques (Fig. 6B) was confirmed by decreased VSMC number (Fig. 6F) and collagen‐rich matrix (Fig. 6G) on staining with Masson trichrome (Fig. 4), α‐actin staining (Fig. 5) and picrosirius red staining viewed under polarized light (Fig. 4). This effect was seen to translate to a significant decrease in the cap/intima ratio in Ad5‐CMV.p53‐treated arteries (0.16 ± 0.01), compared with that in Ad5‐CMV.lac Z‐treated plaques and blank control plaques (0.21 ± 0.02 and 0.24 ± 0.04, respectively, all P < 0.01) (Fig. 6D).
Figure 4.
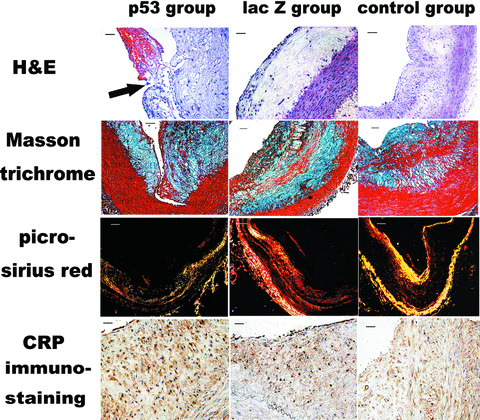
Histopathological assay showing haematoxylin and eosin staining, Masson trichrome staining, picrosirius red staining of collagens visualized under polarized light and CRP immunostaining in Ad5‐CMV.p53 transfected group, Ad5‐CMV.lac Z transfected group and blank control group. Haematoxylin and eosin staining demonstrated that the thickness of the aortic intima was significantly increased in all three groups of animals and the arrow indicated an intraluminal thrombus attached to a disrupted fibrous cap in Ad5‐CMV.p53‐treated group (Bar = 50 μm). In contrast, the relative content of SMCs showed by Masson trichrome staining and the relative content of collagen displayed by picrosirius red staining was markedly reduced in Ad5‐CMV.p53 transfected group than the Ad5‐CMV.lac Z transfected group and blank control group. Bars = 100 μm. However, more positive CRP staining was found in Ad5‐CMV.p53‐ treated group than in the Ad5‐CMV.lac Z‐treated group (Bars = 50 μm).
Figure 5.

Immunohistochemical staining showing α‐actin, RAM11, MMP‐2 and MMP‐9 immunostaining in Ad5‐CMV.p53 transfected group and Ad5‐CMV.lac Z transfected group. To clarify whether MMP‐2 and MMP‐9 were secreted from smooth muscle cells or macrophages, serial cross sections were stained with antibodies against α‐actin, MMP‐2 or MMP‐9 and the RAM11 consecutively. Although the positive staining of MMP‐2 and MMP‐9 was found in both macrophages and smooth muscle cells, most of these staining corresponded to RAM11 staining area in the Ad5‐CMV.p53‐ and Ad‐5‐ CMV.lac Z‐treated plaques. The relative content of α‐actin was dramatically decreased in the Ad5‐CMV.p53 transfected group than in the Ad5‐CMV.lac Z transfected group and blank control group (bars = 100 μm). Conversely, the relative content of macrophages was dramatically increased in the Ad5‐CMV.p53 transfected group than in Ad5‐CMV.lac Z transfected group (bars = 50 μm). Likewise, more positive MMP‐2 and MMP‐9 staining was found in Ad5‐CMV.p53 transfected group than in the Ad5‐CMV.lac Z transfected group and blank control group (bars = 100 μm).
In both Ad5‐CMV.lac Z‐treated and blank control rabbits, plaques contained a thick VSMC‐rich fibrous cap (Figs 4 and 5) with abundant collagen and matrix, with definite macrophages scattered throughout the lesions (Fig. 5). While in the Ad5‐CMV.p53‐treated rabbits, there was a thinner fibrous cap (Figs 4 and 5), with focal accumulation of cap macrophages (Figs 5 and 6E) as confirmed by RAM11 immunostaining. CRP, MMP‐9 and MMP‐2 staining showed an increase in lesions treated with Ad5‐CMV.p53, compared with plaques treated with Ad5‐CMV.lac Z and blank plaques (Figs 4 and 5). Although both macrophages and smooth muscle cells showed positive MMPs staining, MMP‐2 and MMP‐9 staining was mainly found in RAM11+ cells in plaques of the Ad5‐CMV.p53‐ and Ad5‐CMV.lac Z‐treated groups (Fig. 5).
Induction of plaque rupture
To study the vulnerability of plaques to rupture after haemodynamic challenge and enhance the development of rupture‐related thrombosis, we triggered plaque disruption and thrombosis by intraperitoneal injection of Russell's viper venom, a pro‐coagulant and endothelial toxin, followed by intravenous injection of histamine, a vasopressor. Some cross‐sections showed thrombus superimposed on an atherosclerotic plaque (Fig. 4A). The fibrous cap was disrupted, exposing the thrombogenic core to the blood. The content of the atheromatous plaque have seeped through the gap in the cap into the lumen, which suggests that plaque rupture preceded thrombosis. After pharmacological triggering, of 13 Ad5‐CMV.p53‐treated rabbits, 10 showed plaque rupture (76.9%) as compared with only 3 of 13 Ad5‐CMV.lac Z‐treated rabbits (23.1%, P < 0.01) or none in the blank control group (0%, P < 0.01).
Role of apoptosis and inflammation in plaque vulnerability
Bivariate correlation analysis showed good positive correlations between hs‐CRP, MCP‐1 or apoptosis rate and plaque rupture (r= 0.758, 0.681, 0.603, respectively, all P < 0.01). In contrast, no correlation was found between TC or LDL‐C and plaque rupture (both P > 0.05). In order to compare the relative importance of apoptosis and inflammation in mediating plaque instability, a logistic regression model was applied in which the status of plaque rupture was the dependent variable and the apoptosis rate and inflammatory markers were entered as the independent covariables. The results showed that only serum level of hs‐CRP had an independent effect on plaque rupture with odds ratios as 1.314 (95% CI: 1.041–1.657, P= 0.021), suggesting that inflammation played a more important role in plaque instability than apoptosis.
Correlation between inflammatory markers and apoptosis rate
A linear regression analysis was performed to reveal the relationship between apoptosis rate and inflammatory markers (Fig. 7). The results demonstrated good positive correlations between hs‐CRP or MCP‐1 and apoptosis rate (R 2= 0.761 and R 2= 0.557, respectively, both P < 0.01).
Figure 7.

Results of linear regression analysis. (A) There was a good positive correlation between apoptosis rate and serum levels of hs‐CRP. (B) There was a good positive correlation between apoptosis rate and serum levels of MCP‐1.
Discussion
In the present study, we developed a new animal model of vulnerable plaques by targeted injection of Ad5‐CMV.p53 into plaques of balloon‐injured abdominal aortas under IVUS guidance in rabbits fed high‐fat diet. In comparison with other imaging techniques, IVUS is superior in providing real‐time high‐resolution images of not only plaque characteristics but also the entire vascular wall [8, 9], which allowed us to inject adenovirus suspension from the adventitia into the plaques of the abdominal aorta precisely. After pharmacological triggering, a plaque rupture rate of 76.9% was achieved in Ad5‐CMV.p53‐treated rabbits, which was similar to our previous results. Compared with existing models, however, our current animal model has the advantages of targeted gene delivery, more accurate dosing, higher transfection efficiency, higher safety profile and lower mortality rate. Although unproved, this model may be suitable for targeted drug or gene therapy of vulnerable plaques as well.
A high‐cholesterol diet for 10 weeks in balloon‐injured rabbits resulted in large and eccentric plaques seen on IVUS. Since hypercholesterolaemia per se may enhance plaque instability which makes it difficult to elucidate the specific mechanisms of p53‐induced plaque rupture, all rabbits were fed a normal chow for 6 weeks after atherosclerotic plaques were established to reduce the serum lipids to a relatively normal level. Pathological studies in the blank control group revealed characterizations of stable lesions with a thick fibrous cap, a small lipid core, a plenty of VSMCs and few macrophages.
After Ad5‐CMV.p53 injection in the present study, the fibrous cap of balloon‐injured plaque showed efficient p53 transfection in the endothelium and a superficial layer, but the expression of transfected p53 in the plaque seemed to be lower than that of transfected β‐galactosidase. In the vessel wall, a high p53 expression may lead to enhanced apoptosis and rapid clearance of transfected cells and consequently a possible underestimation of the transfection efficiency. Alternatively, the finding may also be related to the discrepancy in intracellular half‐life of β‐galactosidase and wild‐type p53, estimated to be at least 1 to 2 days [10] and only about 10 min. [11], respectively. The process of apoptosis in cells and their phagocytosis by surrounding macrophages and VSMCs is estimated to be extremely rapid, lasting from 2 to 4 hrs [12, 13, 14]. These notions were supported by our finding of more p53 staining in plaques at day 1 after p53 transfection than positive staining at day 14.
The vulnerable plaque is characterized by an atrophic fibrous cap [6], a lipid‐rich necrotic core, accumulation of inflammatory cells [15], and imbalance between extracellular matrix synthesis and degradation [16]. In the present study, we observed that p53 expression appeared mainly in the superficial layer of plaques after Ad5‐CMV.p53 injection. Since SMCs from atherosclerotic plaques are more susceptible to p53‐mediated apoptosis than normal SMCs [12, 13], and the number of SMCs in plaques decreased after gene transfection, it is likely that p53‐mediated apoptosis mainly occurs in SMCs. The increase in apoptotic VSMCs may have contributed to the reduction of collagen synthesis, leading to an imbalance between extracellular matrix synthesis and degradation, the basis for plaque rupture [17].
An important finding in this study was that there were high correlations between apoptosis rate and serum levels of hs‐CRP or MCP‐1. Cell apoptosis may mediate the inflammatory process by enhancing intimal accumulation of macrophages and increasing the production and release of inflammatory molecules including hs‐CRP, MCP‐1 and proteolytic enzymes. Proteolytic activity of MMPs is assumed to be driven by inflammatory activity in the plaque and responsible for the degradation and thinning of the fibrous cap, thereby favouring plaque rupture. Clarke et al. [19] showed that apoptosis alone does not recapitulate multiple features of vulnerable plaque in normal arteries, but VSMC apoptosis within established atherosclerotic plaques induces inflammation, remodelling, thrombosis or calcification, thus resulting in plaque instability. Phagocytosis of apoptotic cells normally inhibits macrophage release of IL‐6 [20], but VSMCs death and failed apoptotic body clearance may cause local MCP‐1 release, attracting macrophages with subsequent activation and release of cytokines such as IL‐6 [19], ICAM‐1 and VCAM‐1 [7]. Acting synergistically, these cytokines may in turn stimulate further macrophage recruitment and production of many inflammatory and cytotoxic molecules from macrophages and vascular cells that promote plaque instability and thrombosis. Our results demonstrated that among apoptotic and inflammatory parameters, only serum levels of hs‐CRP had independent effects on plaque rupture, indicating that inflammation plays a pivotal role in plaque instability.
It is well accepted that CRP is produced mainly in the liver and reflects a systemic inflammatory reaction. Although previous animal and human studies virtually denied the presence of extrahepatic CRP production, recent studies have identified CRP expression in atherosclerotic plaques [21, 22]. In our study, CRP staining was positive in plaques of all three groups and was more prominent in Ad5‐CMV.p53‐treated plaques than that in Ad5‐CMV.lac Z‐treated and blank plaques. It is possible that these locally produced proteins may act as an inflammation promoter rather than an innocent biomarker in vulnerable plaques.
In this study, we found that MMP‐2 and MMP‐9 expression was mostly located in macrophages in atherosclerotic plaques. The increased secretion of MMPs by macrophages may degrade plaque matrix, weaken the fibrous cap and contribute to destabilization and rupture of atherosclerosis plaques, as demonstrated in the present study. Similar results were obtained in previous studies. Focal degradation of the fibrous cap collagen by MMPs produced by macrophages was found in human atheroma [23] and was associated with plaque rupture in a rabbit model of atherosclerosis [24].
Our findings may contradict previous research work [25, 26, 27], in which p53 deficiency was seen to lead to accelerated atherosclerosis. In particular, recent study has identified a role for endogenous p53 in protecting VSMCs from apoptosis, trans‐differentiation of bone marrow stromal cells into VSMCs in atherosclerosis, and altering the mode of cell death in plaque [28]. However, we believe that different expression levels of p53 may be responsible for the contradictory results. The cellular response to p53 depends on the cell type, the level of p53 expression, and the presence of other apoptotic stimuli [29, 30, 31]. A low‐level expression of p53 gene often induces cell growth arrest, while a high‐level expression of p53 gene induces only cell apoptosis. Thus, p53 overexpression may exert a destabilizing effect that involves apoptosis‐induced inflammation and matrix turnover in plaque progression and rupture in atherosclerosis rabbits.
There were some limitations in our study. A better control virus in this model might be an adenovirus containing an inactive form of p53, such as a dominant‐negative form of p53 to stabilize plaque. Because no p53 expression was detected in control group, we used Ad5‐CMV.lac Z instead as a control gene. As well, Newman et al. [32] has pointed out that adenoviral vector alone, to a certain extent, results in vascular inflammation. In our study, we used a relative low viral dose (∼4 × 108 pfu per animal) and used Ad5‐CMV.lac Z for comparison in the experiments. Further studies, such as transactivation of p53 gene targets, are needed to better elucidate the mechanism underlying the effect of p53 on plaque instability. In addition, we used pharmacological triggering to simulate haemodynamic stress in patients with acute coronary syndrome. RVV contains proteases that activate coagulation factors V and X. It has been demonstrated that in the absence of arterial wall abnormalities, a low dose of RVV rarely induces arterial thrombosis [33]. In the present study, we found that most thrombi were attached to the sites of ruptured or eroded plaques in the abdominal aorta, which lends support to the idea that arterial thrombosis promoted by RVV in our rabbits was initiated by plaque disruption. Histamine may contribute to plaque disruption by raising blood pressure and inducing vasospasm in rabbits. This effect is mediated by histamine receptor 1 that regulates release of norepinephrine at the pre‐synaptic sites [34].
In conclusion, we have developed a new animal model of vulnerable plaques by direct injection of Ad5‐CMV.p53 into atherosclerotic plaques, which offers advantages of targeted gene delivery, accurate dosing, high transfection efficiency, high plaque rupture rate and low mortality rate. The mechanisms of p53‐mediated plaque instability may involve cell apoptosis in the fibrous cap, and the resultant apoptotic bodies may induce focal accumulation of macrophages and aggravate release of inflammatory molecules. Thus, intraplaque injection of p53 gene may provide a useful method for establishing an animal model of vulnerable plaques.
Acknowledgements
This study was supported by the National 973 Basic Research Program of China (no. 2005CB523301), the National High‐tech Research and Development Program of China (no. 2006AA02A406), the Program of Introducing Talents of Discipline to Universities (No.B07035) grants from the National Natural Science Foundation of China (nos. 0470701, 30470702, 30570747 and 30670873) and a grant of Natural Science Foundation of Shandong Province (no. Y2007C064).
References
- 1. Ko LJ, Prives C. p53: puzzle and paradigm. Genes Dev . 1996; 10: 1054–72. [DOI] [PubMed] [Google Scholar]
- 2. Ihling C, Menzel G, Wellens E, et al . Topographical association between the cyclin‐dependent kinases inhibitor P21, p53 accumulation, and cellular proliferation in human atherosclerotic tissue. Arterioscler Thromb Vasc Biol . 1997; 17: 2218–24. [DOI] [PubMed] [Google Scholar]
- 3. Chen WQ, Zhang Y, Zhang M, et al . Establishing an animal model of unstable atherosclerotic plaques. Chin Med J . 2004; 117: 1293–8. [PubMed] [Google Scholar]
- 4. von der Thüsen JH, van Vlijmen BJ, Hoeben RC, et al . Induction of atherosclerotic plaque rupture in apolipoprotein E−/− mice after adenovirus‐mediated transfer of p53. Circulation . 2002; 105: 2064–70. [DOI] [PubMed] [Google Scholar]
- 5. Burke AP, Farb A, Malcom GT, et al . Coronary risk factors and plaque morphology in men with coronary disease who died suddenly. N Engl J Med. 1997, 336: 1276–82. [DOI] [PubMed] [Google Scholar]
- 6. Davies MJ. The pathophysiology of acute coronary syndromes. Heart. 2000, 83: 361–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen WQ, Zhang L, Liu YF, et al . Prediction of atherosclerotic plaque ruptures with high‐frequency ultrasound imaging and serum inflammatory markers. Am J Physiol Heart Circ Physiol . 2007; 293: H2836–44. [DOI] [PubMed] [Google Scholar]
- 8. de Korte CL, Woutman HA, van der Steen AF, et al . Vascular tissue characterisation with IVUS elastography. Ultrasonics . 2000; 38: 387–90. [DOI] [PubMed] [Google Scholar]
- 9. de Korte CL, Sierevogel MJ, Mastik F, et al . Identification of atherosclerotic plaque components with intravascular ultrasound elastography in vivo a Yucatan pig study. Circulation . 2002; 105: 1627–30. [DOI] [PubMed] [Google Scholar]
- 10. Smith RL, Geller AI, Escudero KW, et al . Long‐term expression in sensory neurons in tissue culture from herpes simplex virus type 1 (HSV‐1) promoters in an HSV‐1‐derived vector. J Virol . 1995; 69: 4593–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Soussi T. The p53 tumor suppressor gene: from molecular biology to clinical investigation. Ann NY Acad Sci . 2000; 910: 121–37. [DOI] [PubMed] [Google Scholar]
- 12. Bennett MR, Evan GI, Schwartz SM. Apoptosis of human vascular smooth muscle cells derived from normal vessels and coronary atherosclerotic plaques. J Clin Invest . 1995; 95: 2266–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bennett MR, Littlewood TD, Schwartz SM, et al . Increased sensitivity of human vascular smooth muscle cells from atherosclerotic plaques to p53‐mediated apoptosis. Circ Res . 1997; 81: 591–9. [DOI] [PubMed] [Google Scholar]
- 14. Bennett MR, Gibson DF, Schwartz SM, et al . Binding and phagocytosis of apoptotic vascular smooth muscle cells is mediated in part by exposure of phosphatidylserine. Circ Res . 1995; 77: 1136–42. [DOI] [PubMed] [Google Scholar]
- 15. Moreno PR, Falk E, Palacios IF, et al . Macrophages infiltration in acute coronary syndromes: Implications for plaque rupture. Circulation . 1994; 90: 775–8. [DOI] [PubMed] [Google Scholar]
- 16. Lindstedt KA, Leskinen MJ, Kovanen PT. Proteolysis of the pericellular matrix: a novel element determining cell survival and death in the pathogenesis of plaque erosion and rupture. Arterioscler Thromb Vasc Biol . 2004; 24: 1350–8. [DOI] [PubMed] [Google Scholar]
- 17. Falk E. Why do plaques rupture Circulation. 1992; 86: III30–42. [PubMed] [Google Scholar]
- 18. Kolodgie FD, Narula J, Burke AP, et al . Localization of apoptotic macrophages at the site of plaque rupture in sudden coronary death. Am J Pathol . 2000; 157: 1259–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Clarke MC, Figg N, Maguire JJ, et al . Apoptosis of vascular smooth muscle cells induces features of plaque vulnerability in atherosclerosis. Nat Med . 2006; 12: 1075–80. [DOI] [PubMed] [Google Scholar]
- 20. Xu W, Roos A, Schlagwein N, et al . IL‐10‐producing macrophages preferentially clear early apoptotic cells. Blood . 2006; 107: 4930–7. [DOI] [PubMed] [Google Scholar]
- 21. Ishikawa T, Hatakeyama K, Imamura T, et al . Involvement of C‐reactive protein obtained by directional coronary atherectomy in plaque instability and developing restenosis in patients with stable or unstable angina pectoris. Am J Cardiol 2003; 91: 287–92. [DOI] [PubMed] [Google Scholar]
- 22. Inoue T, Kato T, Uchida T, et al . Local release of C‐reactive protein from vulnerable plaque or coronary arterial wall injured by stenting. J Am Coll Cardiol. 2005; 46:239–45. [DOI] [PubMed] [Google Scholar]
- 23. Shah PK, Falk E, Badimon JJ, et al . Human monocytederived macrophages induce collagen breakdown in fibrous caps of atherosclerotic plaques: potential role of matrix‐degrading metalloproteinases and implications for plaque rupture. Circulation . 1995; 92: 1565–9. [PubMed] [Google Scholar]
- 24. Rekhter MD, Hicks GW, Brammer DW, et al . Hypercholesterolemia causes mechanical weakening of rabbit atheroma: local collagen loss as a prerequisite of plaque rupture. Circ Res . 2000; 86: 101–10. [DOI] [PubMed] [Google Scholar]
- 25. Guevara NV, Kim HS, Antonova EI, et al . The absence of p53 accelerates atherosclerosis by increasing cell proliferation in vivo. Nat Med . 1999; 5: 335–9. [DOI] [PubMed] [Google Scholar]
- 26. van Vlijmen BJ, Gerritsen G, Franken AL, et al . Macrophage p53 deficiency leads to enhanced atherosclerosis in APOE*3‐Leiden transgenic mice. Circ Res . 2001; 88: 780–6. [DOI] [PubMed] [Google Scholar]
- 27. Merched AJ, Williams E, Chan L. Macrophage‐specific p53 expression plays a crucial role in atherosclerosis development and plaque remodeling. Arterioscler Thromb Vasc Biol . 2003; 23: 1608–14. [DOI] [PubMed] [Google Scholar]
- 28. Mercer J, Figg N, Stoneman V, et al . Endogenous p53 protects vascular smooth muscle cells from apoptosis and reduces atherosclerosis in ApoE knockout mice. Circ Res. 2005; 96; 667–74. [DOI] [PubMed] [Google Scholar]
- 29. Muller M, Strand S, Hug H, et al . Drug‐induced apoptosis in hepatoma cells is mediated by the CD95 (APO‐1/Fas) receptor/ligand system and involves activation of wild‐type p53. J Clin Invest . 1997; 99: 403–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Aragane Y, Kulms D, Metze D, et al . Ultraviolet light induces apoptosis via direct activation of CD95 (Fas/APO‐1) independently of its ligand CD95L. J Cell Biol . 1998; 140: 171–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sheikh MS, Burns TF, Huang Y, et al . p53‐dependent and ‐independent regulation of the death receptor KILLER/DR5 gene expression in response to genotoxic stress and tumor necrosis factor alpha. Cancer Res . 1998; 58: 1593–8. [PubMed] [Google Scholar]
- 32. Newman KD, Dunn PF, Owens JW, et al . Adenovirus‐mediated gene transfer into normal rabbit arteries results in prolonged vascular cell activation, inflammation, and neointimal hyperplasia. J Clin Invest . 1995; 96: 2955–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Constantinides P. Plaque fissures in human coronary thrombosis. J Atheroscler Res . 1966; 6: 1–17. [Google Scholar]
- 34. Awano K, Yokoyama M, Fukuzaki H. Role of serotonin, histamine and thromboxane A2 in platelet‐induced contractions of coronary arteries and aortae from rabbits. J Cardiovasc Pharmacol . 1989; 13: 781–92. [PubMed] [Google Scholar]