

Abstract
Objective
To investigate the efficacy and safety of aceneuramic acid extended-release (Ace-ER), a treatment intended to replace deficient sialic acid, in patients with GNE myopathy.
Methods
UX001-CL301 was a phase 3, double-blind, placebo-controlled, randomized, international study evaluating the efficacy and safety of Ace-ER in patients with GNE myopathy. Participants who could walk ≥200 meters in a 6-minute walk test at screening were randomized 1:1, and stratified by sex, to receive Ace-ER 6 g/d or placebo for 48 weeks and assessed every 8 weeks. The primary endpoint was change in muscle strength over 48 weeks measured by upper extremity composite (UEC) score. Key secondary endpoints included change in lower extremity composite (LEC) score, knee extensor strength, and GNE myopathy–Functional Activity Scale (GNEM-FAS) mobility domain score. Safety assessments included adverse events (AEs), vital signs, and clinical laboratory results.
Results
Eighty-nine patients were randomized (Ace-ER n = 45; placebo n = 44). Change from baseline to week 48 for UEC score between treatments did not differ (least square mean [LSM] Ace-ER −2.25 kg vs placebo −2.99 kg; LSM difference confidence interval [CI] 0.74 [−1.61 to 3.09]; p = 0.5387). At week 48, there was no significant difference between treatments for the change in key secondary endpoints: LEC LSM difference (CI) −1.49 (−5.83 to 2.86); knee extension strength −0.40 (−2.38 to 1.58); and GNEM-FAS mobility domain score −0.72 (−2.01 to 0.57). Gastrointestinal events were the most common AEs.
Conclusions
Ace-ER was not superior to placebo in improving muscle strength and function in patients with GNE myopathy.
Classification of evidence
This study provides Class I evidence that for patients with GNE myopathy, Ace-ER does not improve muscle strength compared to placebo.
GNE myopathy (hereditary inclusion body myopathy) is a rare, severely debilitating, adult-onset myopathy caused by mutation in GNE, a gene encoding 2 sequential enzymes critical to the biosynthesis of sialic acid (SA).1 GNE myopathy typically presents with distal muscle weakness, proceeding proximally with relative sparing of the quadriceps, compromising arm and leg muscle function. Disease progression is gradual and variable, with loss of ambulation ranging from 10 to 20 years after onset.2,3 Data from a GNE myopathy disease monitoring program (GNEM-DMP; NCT01784679) suggest that muscle strength can decline by roughly 10% in <2 years.4
Currently, there is no approved therapy and few clinical trials investigating treatments for GNE myopathy. Consistent with the hypothesis that disease pathology arises from decreased SA in skeletal muscle, preclinical models have shown efficacy by supplementing SA.5–9 Oral SA supplementation in a GNE myopathy mouse model (GNE D176V) increased serum SA in tissue and reduced pathology and loss of function in muscle.9 Since oral SA can be rapidly cleared from the body, an extended-release formulation of SA, aceneuramic acid extended-release (Ace-ER), was developed to promote muscle uptake from continuous, elevated serum SA.10
In a phase 2, dose-finding, placebo-crossover study, Ace-ER 6 g/d demonstrated stabilization in upper extremity (UE) strength in 47 patients with GNE myopathy over 48 weeks. Here, we report results of a phase 3, placebo-controlled study investigating the efficacy and safety of Ace-ER in 89 participants with GNE myopathy, the largest clinical trial to date in GNE myopathy.
Methods
Study population
Patients with GNE myopathy were enrolled at 13 sites from 7 countries: Bulgaria, Canada, France, Israel, Italy, United Kingdom, and United States. Some patients were recruited through the GNEM-DMP. Eligible participants met the following key inclusion criteria: 18–55 years of age; documented diagnosis due to previously demonstrated mutations in the GNE gene (genotyping was not conducted as part of the study); able to provide 2 dynamometry force values in elbow flexors with ≤15% variability in the dominant arm at screening; and able to walk ≥200 meters in the 6-minute walk test (6MWT) at screening, which indicates that patients are more likely to have sufficient muscle tissue to observe a treatment effect from a substrate replacement therapy. Patients were not eligible if they met the following key exclusion criteria: use of N-acetyl-d-mannosamine (ManNAc), SA, SA-related metabolites, IV immunoglobulin, or other substance that can be metabolized to produce SA in the body within 60 days of screening; >30 days treatment with SA (extended or immediate release) in prior clinical trial in the last year; hypersensitivity to SA or its excipients; and serum transaminase >3 times the upper limit of normal for age/sex, or serum creatinine >2 times upper limit at screening.
Study design and endpoints
UX001-CL301 (NCT02377921) was a phase 3, double-blind, placebo-controlled, multicenter, randomized, international study providing Class 1 evidence that Ace-ER does not improve muscle strength compared to placebo in patients with GNE myopathy (figure 1). Eligible patients were randomized 1:1, stratified by sex, to receive 6 g/d of Ace-ER or matching placebo orally 3 times a day for 48 weeks. Dose selection was based on the previous phase 2 trial and long-term extension, which demonstrated that 6 g/d of Ace-ER was efficacious while 3 g/d was not; 12 g/d was not more efficacious than 6 g/d, had a substantial pill burden, and was associated with a higher incidence of gastrointestinal adverse events.10–12 In addition, the placebo tablets in this phase 3 study were the same as those used in the phase 2 study.
Figure 1. UX001-CL301 study design.

6MWT = 6-minute walk test; Ace-ER = aceneuramic acid extended-release; GNEM = GNE myopathy.
The endpoints evaluated in UX001-CL301 were determined by analysis of phase 2 data (UX001-CL201; NCT01517880) assessing outcome measures capable of characterizing the pattern and extent of muscle weakness associated with GNE myopathy and functional limitations resulting from this weakness.13 All efficacy assessments were conducted on site at screening or baseline and every 8 weeks following baseline.
The primary endpoint was change from baseline in muscle strength based on the UE composite (UEC) score after 48 weeks of treatment and was used to compare treatment groups. Because coordinated engagement of multiple muscles is necessary for gross motor function and a composite muscle score can provide more clinically valuable information in a disease with variable decline across muscle groups, regulatory authorities agreed that the UEC was an appropriate primary efficacy endpoint. A hand-held dynamometer (HHD) was used to assess muscle strength. The UEC was calculated as the sum of the mean of bilateral HHD scores from grip, shoulder abductors, elbow flexors, and elbow extensors. Physical therapists at each site underwent extensive training to improve intrarater and inter-rater reliability on all HHD assessments.
For key secondary endpoints, the following change from baseline to week 48 was evaluated for both treatment groups: lower extremity (LE) muscle strength composite (LEC) score, muscle strength in knee extensors, and physical functioning as measured using the GNE myopathy–Functional Activity Scale (GNEM-FAS) mobility domain score.13,14 The GNEM-FAS is a validated instrument with qualitative and quantitative measures designed to assess functional impairment in GNE myopathy including mobility, UE function, and self-care; higher GNEM-FAS scores indicate a lower level of impairment. LEC was calculated as the sum of the following muscle groups using HHD: hip abductors and adductors, hip flexors and extensors, and knee flexors. Knee extensor muscle strength was also measured by HHD. The following additional secondary endpoints were also evaluated for both treatment groups at week 48: physical functioning as measured using the GNEM-FAS UE domain score13,14; UE function as measured by a timed weighted arm lift test15; LE function as measured by a timed Sit-to-Stand Test15,16; LE function as measured by distance walked in the 6MWT17; and the quality of life questionnaire for neuromuscular disease.18 Blood samples were collected at baseline and prior to drug administration every 8 weeks thereafter to assess change in free serum SA using liquid chromatography atmospheric pressure ionization mass spectrometry (response range 0.0400–20.0 μg/mL) performed by Intertek Pharmaceutical Services (San Diego, CA).
Safety assessments included the incidence, frequency, and severity of adverse events (AEs) as well as changes in vital signs and clinical laboratory tests. Changes in platelet level were also examined since a recent study reported on a potential link between mutations in GNE and thrombocytopenia.19 Safety assessments were collected at the same time points as efficacy assessments, as well as over the phone at treatment week 4 and approximately 28 days after the participant's last dose of study drug.
For additional details, the full protocol is provided in the supplementary materials.
Statistical analysis
The primary efficacy analysis set was based on participants with both baseline and postbaseline data. The 2 treatment groups were compared using the generalized estimating equation model with baseline, sex, and region (United States vs non–United States) as covariates. Data are summarized as mean, least squares mean (LSM), range, standard error, confidence interval (95% CI), and the number or percentage of participants. This phase 3 study aimed to enroll at least 80 participants to provide 90% power to detect a difference of about 5 kg in the change from baseline in UEC score between treatment groups, assuming an SD of 6 kg and a 2-sided α of 0.05. This calculation was based on the phase 2 study showing that patients able to walk more than 200 meters in the 6MWT at baseline experienced a greater effect of Ace-ER in the UEC; in addition, a change of 5 kg in UEC would be approximately a 9% decline in UE muscle strength in this phase 3 study population, and therefore suggested to be clinically meaningful.10 Safety analysis was based on all randomized patients who received at least 1 dose of study drug.
A regression analysis was performed to determine whether clinical outcomes were influenced by variables such as study site, baseline status, age, and time since diagnosis. Due to the heterogeneity of the population and lack of a full sample set, a genotype–phenotype analysis was not performed.
Standard protocol approvals, registrations, and patient consents
UX001-CL301 was designed, conducted, recorded, and reported in accordance with ethical principles, consistent with ICH GCP guidelines, and in accordance with applicable local, federal, and regulatory agency regulations. Institutional review boards at each site approved the protocol prior to site initiation. The study is registered with ClinicalTrials.gov (NCT02377921) and the EU Register (2014-005432-33). The principal investigator had access to the data and all authors have reviewed key findings and have access to study documents.
Data availability
Individual de-identified participant data will be available for 12 months from the time of publication upon request to researchers providing a methodologically sound proposal for analysis for research that contributes to the field of GNE myopathy. Proposals should be directed to clinicaldatarequest@ultragenyx.com. To gain access, data requestors will need to sign a data access and use agreement. Data will be shared via secured portal.
Results
Baseline demographics and characteristics
A total of 89 participants with GNE myopathy based on previous documentation of a pathogenic variant of the GNE/MNK enzyme were screened and randomized (figure e-1, doi.org/10.5061/dryad.h52h5t7). The safety analysis population included 44 participants treated with placebo and 45 participants treated with Ace-ER. Two participants, 1 from each treatment group, discontinued due to noncompliance; 1 of these 2 participants had no postbaseline assessments and was excluded from the placebo group for the primary efficacy data set, which included 43 participants treated with placebo and 45 participants treated with Ace-ER.
Baseline demographics and characteristics were generally well balanced between treatment groups (table 1). Both groups had similarly low free serum SA.20 Notably, despite the range in time from diagnosis to enrollment (0–20 years), both treatment groups showed substantial impairments in muscle strength at baseline compared with population norms, with the LE muscles more severely impaired than UE muscles21–24 (figure 2). The baseline percentage of normal predicted UE muscle strength, including grip, shoulder abductors, elbow flexors, and elbow extensors, ranged from 51% to 65% for placebo and 50% to 60% for the Ace-ER group. Baseline percentage of normal predicted LE muscle strength, including hip abductors, hip adductors, hip flexors, hip extensors, and knee flexors, ranged from 15% to 64% for the placebo group and 13% to 64% for the Ace-ER group; the percentage of normal muscle strength for hip flexors was the lowest compared to other muscles for both treatment groups. Knee extensor strength was similar for both treatment groups as well (percentage of normal predicted: placebo 62%, Ace-ER 58%) and demonstrated the relative sparing of the quadriceps characteristic of GNE myopathy.
Table 1.
Baseline characteristics
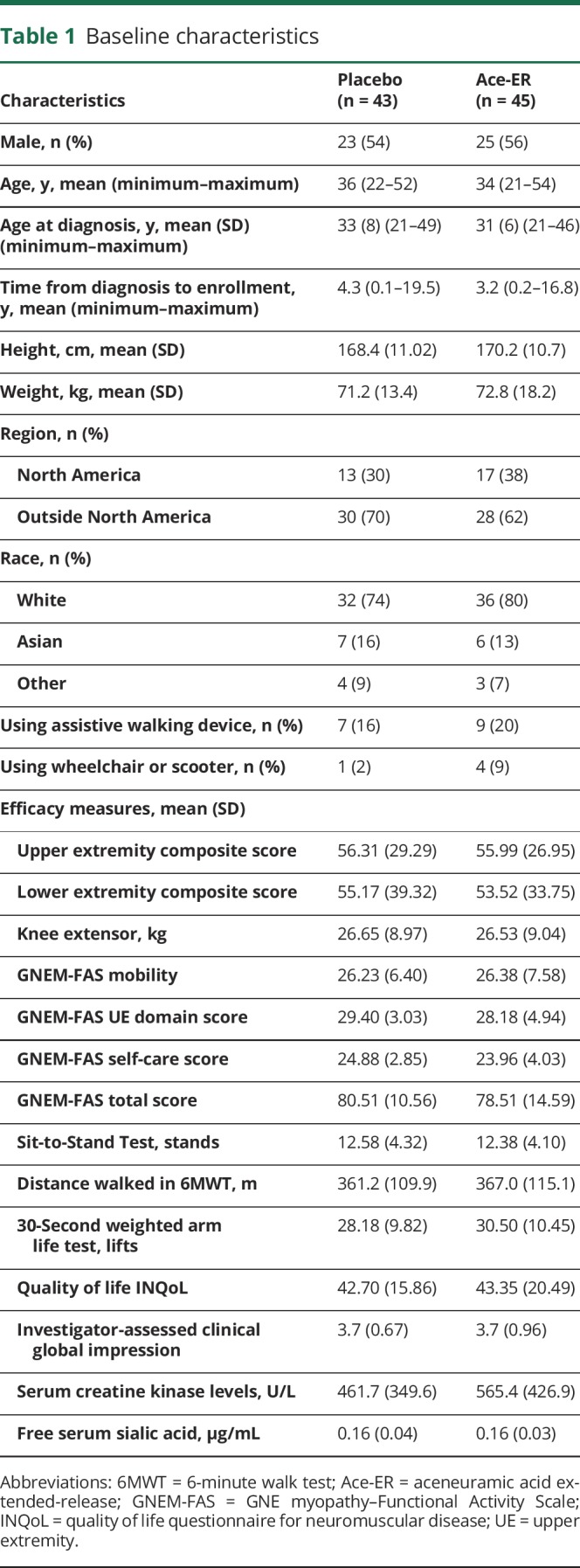
Figure 2. Percentage predicted muscle strength at baseline.

Ace-ER = aceneuramic acid extended-release.
Efficacy findings
Consistent with the phase 2 findings, serum SA increased from baseline (0.16 μg/mL) to week 48 (0.30 μg/mL) with Ace-ER treatment, but not with placebo treatment (0.16 μg/mL at both baseline and week 48; figure 3). The peak mean increase in serum SA, 2.3 times the mean baseline value, was observed at week 16. The change from baseline to week 48 in SA was significantly different between treatment groups (Ace-ER LSM change [95% CI] 0.14 [0.10 to 0.18]; placebo 0.00 [−0.01 to 0.02]; p < 0.0001).
Figure 3. Free serum sialic acid.

Data are expressed as mean ± standard error. Gray lines indicate placebo; blue lines indicate aceneuramic acid extended-release (Ace-ER).
UX001-CL301 did not meet the primary endpoint, as there was no significant difference between treatment groups in the change from baseline to week 48 for UEC score (LSM placebo −2.99 kg vs Ace-ER −2.25 kg; LSM difference 0.74, p = 0.54) (figure 4). The baseline UEC score was 56 kg for both placebo and Ace-ER. Week 48 UEC score was 53 kg for placebo and 54 kg for Ace-ER. The 2.99 kg decline in the placebo group was below the 5 kg target difference in the prevention of decline for which the study was designed. Similar results were obtained when analyzing the UEC as change in percentage predicted norm (LSM placebo −2.85% vs Ace-ER −2.05%; LSM difference 0.80, p = 0.55). The change from baseline for each individual muscle group within the UEC did not differ between treatment groups (table e-1, doi.org/10.5061/dryad.h52h5t7).
Figure 4. Change from baseline in primary and key secondary efficacy assessments.

(A–D) Data are expressed as mean change from baseline ± standard error. Gray lines indicate placebo; blue lines indicate aceneuramic acid extended-release (Ace-ER). GNEM-FAS = GNE myopathy–Functional Activity Scale; LEC = lower extremity composite; UEC = upper extremity composite.
After 48 weeks of treatment, there was also no significant difference between treatment groups for the 3 prespecified key secondary endpoints (figure 2). The baseline mean LEC score was 55 kg for placebo and 54 kg for Ace-ER. There was no significant difference between treatment groups in week 48 mean LEC score, with 53 kg for placebo and 51 kg for Ace-ER (LSM difference [95% CI] −1.49 [−5.83 to 2.86]; p = 0.50). The mean baseline knee extensor strength was 27 kg for both placebo and Ace-ER. Mean knee extensor strength at week 48 was similar between groups, with minimal to no increase (placebo 27 kg, Ace-ER 26 kg; LSM difference [95% CI] −0.40 [−2.38 to 1.58]; p = 0.69). The mean GNEM-FAS mobility score at baseline was 26 for both placebo and Ace-ER. There was no difference between treatment groups in mean GNEM-FAS mobility score at week 48, with 25 for placebo and 24 for Ace-ER (LSM difference [95% CI] −0.72 [−2.01 to 0.57]; p = 0.27). Finally, there was no significant difference in the change from baseline between treatments in any of the additional endpoints (table 2). Study site, baseline status, age, and time since diagnosis did not affect clinical outcome findings.
Table 2.
Change from baseline to week 48 in additional efficacy endpoints

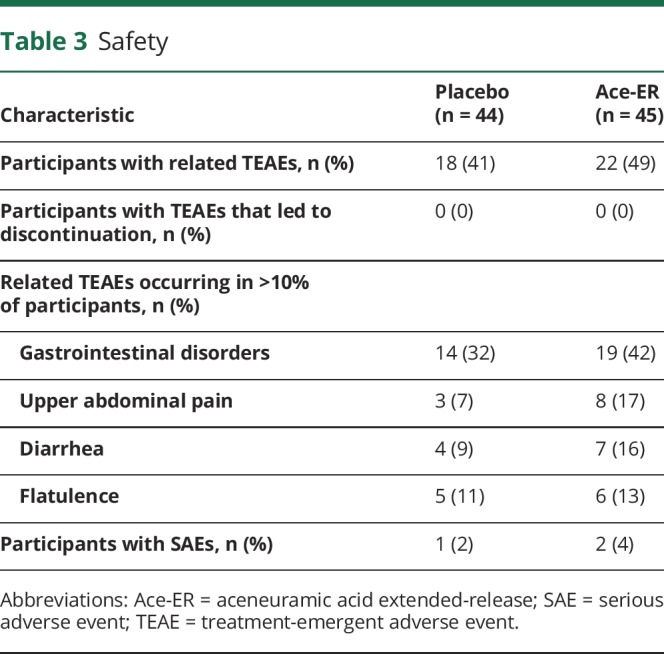
Safety
Overall, Ace-ER was well-tolerated and the most common AEs were mild to moderate gastrointestinal events (table 3). There were no deaths or grade 4 AEs. There were 2 serious AEs (SAEs) in the Ace-ER group (grade 3 acute myocardial infarction and grade 2 acute gastritis) and 1 SAE in the placebo group (abortion); all SAEs resolved and only the acute gastritis SAE was considered possibly treatment-related.
Table 3.
Safety
There were no noteworthy differences in vital signs and laboratory findings for either treatment group. Platelet counts at baseline were similar across treatment groups. At week 48, platelet count for 1 participant in Ace-ER shifted from normal to high but was not considered clinically significant. No noteworthy shift in platelet counts was observed in the placebo group.
Discussion
Very few clinical studies have been conducted for distal myopathies, and there have been no phase 3 clinical trials investigating treatments for any distal myopathy, including GNE myopathy.25 The rarity of distal myopathies results in a lack of confirmed diagnosis, patient cohorts, suitable and testable outcome measures, clinical trial expertise, and understanding disease course; this absence renders clinical research more challenging as compared to more common neurologic and neuromuscular diseases.
GNE myopathy is characterized by progressive loss of muscle strength due to impairment in the production of SA, which plays a role in muscle cellular health.26–29 The scientific rationale for the Ace-ER program was to replace the underlying deficiency in SA and thereby slow the progressive loss of muscle strength and function. UX001-CL301 investigated the efficacy and safety of Ace-ER compared to placebo in the first phase 3 study in GNE myopathy. The phase 3 study was unable to confirm the benefit observed in the prior phase 2 study. Compared to placebo, Ace-ER did not demonstrate a treatment benefit on primary or secondary measures of muscle strength and function; there was no significant treatment effect for Ace-ER compared to placebo in any exploratory endpoints. Overall, Ace-ER exhibited an acceptable safety profile.
Both the phase 2 study and this phase 3 study demonstrate that Ace-ER significantly increased serum SA.10 However, efficacy findings from the phase 3 study differed from findings in the phase 2 study. In the phase 2 study with 47 patients with GNE myopathy, Ace-ER 6 g/d resulted in maintenance of muscle strength in UEC score at 6 g/d compared with placebo at week 2410 and sustained responses at week 48. Variation in the population and size of these 2 studies may account for the differences in outcomes. The phase 3 study included 3 times the number of participants receiving Ace-ER 6/day for a full 48 weeks than the phase 2 study dose-finding placebo-crossover study. The phase 3 population was also more heterogeneous, with patients recruited from many more countries and with greater genetic heterogeneity (data not shown) than in the phase 2 (Israel and United States only). Further, participants in the phase 3 study were less impaired at baseline than in the phase 2 study. A greater percentage of participants reported using a walking device at baseline in the phase 2 study than the phase 3 study (33% vs 18%). At baseline, the GNEM-FAS total score in the phase 2 study was 69; participants in the Ace-ER and placebo group in the phase 3 study scored 79 and 81, respectively.
In addition to comparing the phase 2 and phase 3 study populations at baseline, we can contrast the magnitude of change in clinical outcomes through 24 weeks of treatment. Given that the expected treatment benefit was slowing of disease progression relative to placebo, the phase 3 study outcome was particularly sensitive to the extent of strength decline in placebo group. The decline in UEC score at week 24 in placebo groups was as follows: phase 2 study mean change (SD) −1.95 (3.10) kg; 5% decline vs phase 3 study change −1.06 (5.43) kg; 2%.4,10 Only after 48 weeks did the placebo group in the phase 3 study observe a 6% decline (−3.28 [5.83] kg); the placebo group did not continue beyond 24 weeks in the phase 2 study. Consistent with observations from this phase 3 study, recent findings from the GNEM-DMP, representing a larger natural history population of patients with GNE myopathy, also suggest a slower decline in UE muscle strength than observed in the phase 2 study; the mean decline in 74 patients was −2.02 kg and −3.31 kg after 12 and 24 months of monitoring without treatment, respectively.4 For treatment strategies that aim to slow disease progression, it is possible that a treatment duration longer than 48 weeks may be necessary to establish a treatment benefit.
Further biological hypotheses to be investigated include the lack of sufficient tissue levels (SA in muscle), compensatory downregulation of SA transport into the target tissue, altered cellular processing, or alternative, noncanonical functions of the GNE gene or protein itself. In the latter scenario, SA supplementation alone, however effective, would not restore muscle cell function.30 It is also possible that the underlying pathophysiology of the disease may extend beyond a deficiency in SA.
In spite of findings from this phase 3 study showing that Ace-ER was not superior to placebo, development of therapeutics that target the mechanism of disease of GNE myopathy, or the SA biosynthetic pathway, may still prove beneficial. SA does not penetrate muscles well. However, different formulations of extended-release SA may be able to further penetrate muscle cells, replenish the deficient pool of SA, and slow the rate of muscle decline. Oral treatment with ManNAc, a precursor in the SA biosynthetic pathway, prevented muscle weakness in a murine model of GNE myopathy and is now being studied in an open-label phase 2 study in patients with GNE myopathy (NCT02346461).31 Gene therapy is also being explored to treat GNE myopathy.1,32,33
UX001-CL301 was the largest clinical trial in patients with GNE myopathy and provided novel perspective on conducting trials in patients with distal myopathies. Due to the rarity of GNE myopathy, the phase 3 study included 13 clinical trial sites across 7 different countries. This geographic reach raises disease awareness and sets the stage for future studies in GNE myopathy. Findings from this phase 3 study also provide insight into the heterogeneity of GNE myopathy, especially with the rate of progression. In addition, in collaboration with TREAT-NMD, a network of clinicians and scientists performing neuromuscular research, data from this study alongside natural history data from the GNEM-DMP are available for interested parties in an effort to develop treatments for GNE myopathy. Understanding the differences between patients with GNE myopathy will continue to help develop outcome measures sensitive and specific enough for early detection of the therapy efficacy. One of the most promising highlights from the clinical trials investigating the efficacy and safety of Ace-ER was the development of the GNEM-FAS, another validated tool that will set the stage for future clinical studies in the GNE myopathy population.13,14
We investigated the efficacy and safety of an extended-release formulation of SA, Ace-ER, in the largest clinical trial in GNE myopathy, UX001-CL301. Overall, Ace-ER was well-tolerated, but did not demonstrate superiority over placebo in measures of muscle strength. The heterogeneity of patients participating in this study and the rate of disease progression provided valuable insight into how future studies can assess efficacy in clinical trials with distal myopathies.
Acknowledgment
The authors thank the patients, their parents/caregivers, and health care professionals who participated in this study, including Dean Miller; Ruth Wake; Dionne Moat; Robert Muni Lofra; Teresinha Evangelista, MD; Nikoletta Nikolenko, MD; Namita Goyal, MD; Sonia Nayyar, MD; Caroline Peyronnard, MD; Tim Lai, MD; Monica Lavian, DO; Brian Minton, BS; Patrick Tierney, RPT; Denise Davis, RPT; Cathy Chou, RPT; Nick Higgins; and Ed Connor, MD.
Glossary
- 6MWT
6-minute walk test
- Ace-ER
aceneuramic acid extended-release
- AE
adverse event
- CI
confidence interval
- GNEM-DMP
GNE myopathy disease monitoring program
- GNEM-FAS
GNE myopathy–Functional Activity Scale
- HHD
hand-held dynamometer
- LE
lower extremity
- LEC
lower extremity composite
- LSM
least squares mean
- ManNAc
N-acetyl-d-mannosamine
- SA
sialic acid
- SAE
serious adverse event
- UE
upper extremity
- UEC
upper extremity composite
Footnotes
Class of Evidence: NPub.org/coe
Author contributions
H.L., T.K., A.S., and E.K. contributed to the study design. H.L., A.B., Y.C., H.L., M.M., I.T., M.T., O.P., A.L., J.S., T.K., A.S., H.M., and E.K. contributed to the acquisition of data, analysis, and interpretation of study. All authors contributed to the development of this manuscript, and reviewed and approved the final version for submission.
Study funding
Supported by Ultragenyx Pharmaceutical Inc. The sponsor, Ultragenyx Pharmaceutical Inc., and investigators were responsible for collecting, analyzing, and interpreting data. The authors vouch for the completeness and accuracy of the data. This article was written with input from all authors and medical writing support provided by the sponsors. The lead author had access to the data and all authors decided to submit this manuscript. The project described was supported by the National Center for Research Resources and the National Center for Advancing Translational Sciences, NIH, through grant UL1 TR001414. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The Article Processing Charge was funded by Ultragenyx Pharmaceutical Inc.
Disclosure
H. Lochmüller has served as a clinical trial investigator for Ultragenyx Pharmaceutical Inc. Financial support to Newcastle University and the Newcastle NHS Trust for research projects and clinical trials was provided by AMO Pharma, Biogen, GW Pharma, Pfizer, PTC Therapeutics, Roche, and Ultragenyx. A. Behin has served as a clinical trial investigator for Ultragenyx Pharmaceutical Inc. Y. Caraco has served as a clinical trial investigator for Ultragenyx Pharmaceutical Inc. His institution, Hadassah University Hospital, has received financial support for research projects and clinical trials by EnteraBio, Bioblast, Boehringer Ingelheim, Sanofi Aventis, Synektik, LeukoDx, Novartis, BiolineRx, and Ultragenyx. H. Lau has served as a clinical trial investigator for Ultragenyx Pharmaceutical Inc. She has also conducted clinical research for Amicus, Biomarin, Genzyme/Sanofi, Pfizer, Mallinckrodt, Shire, and Ultragenyx. She has consulted for Actelion, Biomarin, Chiesi, Genzyme/Sanofi, Pfizer, Prevail Therapeutics, and Shire. She has served on an advisory board for Genzyme/Sanofi, Shire, and Ultragenyx. M. Mirabella has served as a clinical trial investigator for Ultragenyx Pharmaceutical Inc. I. Tournev has served as a clinical trial investigator for Ultragenyx Pharmaceutical Inc. M. Tarnopolsky has served as a clinical trial investigator for Ultragenyx Pharmaceutical Inc. He is the president and CEO of Exerkine Corporation. O. Pogoryelova has served as a clinical trial investigator for Ultragenyx Pharmaceutical Inc. A. Lai is employed by and holds stock options with Ultragenyx Pharmaceutical Inc. J. Shah is employed by and holds stock options with Ultragenyx Pharmaceutical Inc. T. Koutsoukos is employed and holds stock options with Ultragenyx Pharmaceutical Inc. A. Skrinar is employed and holds stock options with Ultragenyx Pharmaceutical Inc. H. Mansbach is employed and holds stock options with Ultragenyx Pharmaceutical Inc. E. Kakkis is employed and holds stock options with Ultragenyx Pharmaceutical Inc. T. Mozaffar has served as a clinical trial investigator for Ultragenyx Pharmaceutical Inc. Go to Neurology.org/N for full disclosures.
References
- 1.Nishino I, Carrillo-Carrasco N, Argov Z. GNE myopathy: current update and future therapy. J Neurol Neurosurg Psychiatry 2015;86:385–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nonaka I, Noguchi S, Nishino I. Distal myopathy with rimmed vacuoles and hereditary inclusion body myopathy. Curr Neurol Neurosci Rep 2005;5:61–65. [DOI] [PubMed] [Google Scholar]
- 3.Pogoryelova O, Cammish P, Mansbach H, et al. Phenotypic stratification and genotype-phenotype correlation in a heterogeneous, international cohort of GNE myopathy patients: first report from the GNE myopathy Disease Monitoring Program, registry portion. Neuromuscul Disord 2017;28:158–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Behin A, Mozaffar T, Tarnopolsky M, et al. GNE-myopathy (HIBM): upper and lower extremity muscle strength declines over time in a prospective study. Neuromuscul Disord 2017;27(suppl 2):S51–S270. [Google Scholar]
- 5.Varki NM, Varki A. Diversity in cell surface sialic acid presentations: implications for biology and disease. Lab Invest 2007;87:851–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chan YM, Lee P, Jungles S, et al. Substantial deficiency of free sialic acid in muscles of patients with GNE myopathy and in a mouse model. PLoS One 2017;12:e0173261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Malicdan MC, Noguchi S, Hayashi YK, Nonaka I, Nishino I. Prophylactic treatment with sialic acid metabolites precludes the development of the myopathic phenotype in the DMRV-hIBM mouse model. Nat Med 2009;15:690–695. [DOI] [PubMed] [Google Scholar]
- 8.Yonekawa T, Malicdan MC, Cho A, et al. Sialyllactose ameliorates myopathic phenotypes in symptomatic GNE myopathy model mice. Brain 2014;137:2670–2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Malicdan MC, Noguchi S, Tokutomi T, et al. Peracetylated N-acetylmannosamine, a synthetic sugar molecule, efficiently rescues muscle phenotype and biochemical defects in mouse model of sialic acid-deficient myopathy. J Biol Chem 2012;287:2689–2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Argov Z, Caraco Y, Lau H, et al. Aceneuramic acid extended release administration maintains upper limb muscle strength in a 48-week study of subjects with GNE myopathy: results from a phase 2, randomized, controlled study. J Neuromuscul Dis 2016;3:49–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Argov Z, Caraco Y, Lau H, et al. Oral sialic acid extended release (SA-ER) stabilizes upper extremity muscle strength in human GNE myopathy: a phase 2 study. Neurology 2014;83:e35. [Google Scholar]
- 12.Lau H, Argov Z, Caraco Y, et al. GO 27: randomized controlled phase 2 study of GNE myopathy subjects treated with placebo, 6 g or 3 g doses of extended-release sialic acid (SA-ER) and an extension study at a higher 12 g dose of combination extended and immediate release sialic acid (SA). Neuromuscul Disord 2014;24:923. [Google Scholar]
- 13.Argov Z, Bronstein F, Esposito A, et al. Characterization of strength and function in ambulatory adults with GNE myopathy. J Clin Neuromuscul Dis 2017;19:19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mayhew J, Bonner N, Arbuckle R, Turnbull A, Bowden A, Skrinar A. Development and preliminary evidence of the psychometric properties of the GNE myopathy functional activity scale. J Comp effectiveness Res 2017;7:381–395. [DOI] [PubMed] [Google Scholar]
- 15.Agarwal S, Kiely PD. Two simple, reliable and valid tests of proximal muscle function, and their application to the management of idiopathic inflammatory myositis. Rheumatology 2006;45:874–879. [DOI] [PubMed] [Google Scholar]
- 16.Ozalevli S, Ozden A, Itil O, Akkoclu A. Comparison of the Sit-to-Stand Test with 6 min walk test in patients with chronic obstructive pulmonary disease. Respir Med 2007;101:286–293. [DOI] [PubMed] [Google Scholar]
- 17.Chetta A, Zanini A, Pisi G, et al. Reference values for the 6-min walk test in healthy subjects 20–50 years old. Respir Med 2006;100:1573–1578. [DOI] [PubMed] [Google Scholar]
- 18.Vincent KA, Carr AJ, Walburn J, Scott DL, Rose MR. Construction and validation of a quality of life questionnaire for neuromuscular disease (INQoL). Neurology 2007;68:1051–1057. [DOI] [PubMed] [Google Scholar]
- 19.Izumi R, Niihori T, Suzuki N, et al. GNE myopathy associated with congenital thrombocytopenia: a report of two siblings. Neuromuscul Disord 2014;24:1068–1072. [DOI] [PubMed] [Google Scholar]
- 20.Sillanaukee P, Ponnio M, Jaaskelainen IP. Occurrence of sialic acids in healthy humans and different disorders. Eur J Clin Invest 1999;29:413–425. [DOI] [PubMed] [Google Scholar]
- 21.Bohannon RW. Reference values for extremity muscle strength obtained by hand-held dynamometry from adults aged 20 to 79 years. Arch Phys Med Rehabil 1997;78:26–32. [DOI] [PubMed] [Google Scholar]
- 22.NIMS. Muscular weakness assessment: use of normal isometric strength data: The National Isometric Muscle Strength (NIMS) Database Consortium. Arch Phys Med Rehabil 1996;77:1251–1255. [DOI] [PubMed] [Google Scholar]
- 23.Mathiowetz V, Kashman N, Volland G, Weber K, Dowe M, Rogers S. Grip and pinch strength: normative data for adults. Arch Phys Med Rehabil 1985;66:69–74. [PubMed] [Google Scholar]
- 24.Bohannon RW, Peolsson A, Massy-Westropp N, Desrosiers J, Bear-Lehman J. Reference values for adult grip strength measured with a Jamar dynamometer: a descriptive meta-analysis. Physiotherapy 2006;92:11–15. [Google Scholar]
- 25.Palmio J, Udd B. Myofibrillar and distal myopathies. Rev Neurol 2016;172:587–593. [DOI] [PubMed] [Google Scholar]
- 26.Noguchi S, Keira Y, Murayama K, et al. Reduction of UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase activity and sialylation in distal myopathy with rimmed vacuoles. J Biol Chem 2004;279:11402–11407. [DOI] [PubMed] [Google Scholar]
- 27.Saito F, Tomimitsu H, Arai K, et al. A Japanese patient with distal myopathy with rimmed vacuoles: missense mutations in the epimerase domain of the UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase (GNE) gene accompanied by hyposialylation of skeletal muscle glycoproteins. Neuromuscul Disord 2004;14:158–161. [DOI] [PubMed] [Google Scholar]
- 28.Andrew CG, Appel SH. Macromolecular characterization of muscle membranes I: proteins and sialic acid of normal and denervated muscle. J Biol Chem 1973;248:5156–5163. [PubMed] [Google Scholar]
- 29.Ervasti JM, Burwell AL, Geissler AL. Tissue-specific heterogeneity in alpha-dystroglycan sialoglycosylation: skeletal muscle alpha-dystroglycan is a latent receptor for Vicia villosa agglutinin b4 masked by sialic acid modification. J Biol Chem 1997;272:22315–22321. [DOI] [PubMed] [Google Scholar]
- 30.Harazi A, Becker-Cohen M, Zer H, Moshel O, Hinderlich S, Mitrani-Rosenbaum S. The interaction of UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase (GNE) and alpha-actinin 2 is altered in GNE myopathy M743T mutant. Mol Neurobiol 2017;54:2928–2938. [DOI] [PubMed] [Google Scholar]
- 31.Xu X, Wang AQ, Latham LL, et al. Safety, pharmacokinetics and sialic acid production after oral administration of N-acetylmannosamine (ManNAc) to subjects with GNE myopathy. Mol Genet Metab 2017;122:126–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nemunaitis G, Jay CM, Maples PB, et al. Hereditary inclusion body myopathy: single patient response to intravenous dosing of GNE gene lipoplex. Hum Gene Ther 2011;22:1331–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nemunaitis G, Maples PB, Jay C, et al. Hereditary inclusion body myopathy: single patient response to GNE gene Lipoplex therapy. J Gene Med 2010;12:403–412. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Individual de-identified participant data will be available for 12 months from the time of publication upon request to researchers providing a methodologically sound proposal for analysis for research that contributes to the field of GNE myopathy. Proposals should be directed to clinicaldatarequest@ultragenyx.com. To gain access, data requestors will need to sign a data access and use agreement. Data will be shared via secured portal.