

Abstract
Objective
To evaluate the safety and efficacy of low-dose naproxen for prevention of progression in presymptomatic Alzheimer disease (AD) among cognitively intact persons at risk.
Methods
Investigation of Naproxen Treatment Effects in Pre-symptomatic Alzheimer's Disease (INTREPAD), a 2-year double-masked pharmaco-prevention trial, enrolled 195 AD family history–positive elderly (mean age 63 years) participants screened carefully to exclude cognitive disorder (NCT-02702817). These were randomized 1:1 to naproxen sodium 220 mg twice daily or placebo. Multimodal imaging, neurosensory, cognitive, and (in ∼50%) CSF biomarker evaluations were performed at baseline, 3, 12, and 24 months. A modified intent-to-treat analysis considered 160 participants who remained on-treatment through their first follow-up examination. The primary outcome was rate of change in a multimodal composite presymptomatic Alzheimer Progression Score (APS).
Results
Naproxen-treated individuals showed a clear excess of adverse events. Among treatment groups combined, the APS increased by 0.102 points/year (SE 0.014; p < 10−12), but rate of change showed little difference by treatment assignment (0.019 points/year). The treatment-related rate ratio of 1.16 (95% confidence interval 0.64–1.96) suggested that naproxen does not reduce the rate of APS progression by more than 36%. Secondary analyses revealed no notable treatment effects on individual CSF, cognitive, or neurosensory biomarker indicators of progressive presymptomatic AD.
Conclusions
In cognitively intact individuals at risk, sustained treatment with naproxen sodium 220 mg twice daily increases frequency of adverse health effects but does not reduce apparent progression of presymptomatic AD.
Classification of evidence
This study provides Class I evidence that, for people who are cognitively intact, low-dose naproxen does not significantly reduce progression of a composite indicator of presymptomatic AD.
Alzheimer disease (AD) includes a decades-long period of presymptomatic biochemical, imaging, neurosensory, and subtle cognitive changes.1,2 Because cognitive changes emerge gradually, AD prevention trials using cognitive endpoints typically follow thousands of individuals for several years. Improved efficiency may result from use of multimodal composite indicators of early AD pathogenesis as well as subtle cognitive decline. One such indicator, the Alzheimer Progression Score (APS),3 assesses presymptomatic AD progression using Item Response Theory modeling. Predictive validity of the APS method has been demonstrated recently.3
Among potential preventive interventions in presymptomatic AD, nonsteroidal anti-inflammatory drugs (NSAIDs) have retained interest. Numerous observational studies have shown reduced incidence of AD in users of these drugs, at least in relatively young elderly.4 However, NSAID prevention trials have failed to show benefit, and instead have caused harm to older individuals,5 or those with imminent symptoms.6,7 These findings suggest that NSAID prevention trials must screen (preferably younger-old) participants carefully for cognitive or other prodromal AD symptoms.8 Such principles were applied when designing Investigation of Naproxen Treatment Effects in Pre-symptomatic Alzheimer's Disease (INTREPAD; NCT-02702817), a 2-year double-masked randomized trial of oral naproxen sodium 220 mg twice daily vs placebo for safety and efficacy against progression of AD-related change.
Methods
Primary research question
INTREPAD was designed to test the safety and efficacy of low-dose naproxen in reducing the rate of change of a composite indicator of presymptomatic AD. To that end, Class I evidence is provided here. An important additional objective of the work was to assess the practicality and utility of a more efficient approach to AD prevention trials using novel methods of recruitment, data collection, and analysis. A notable feature was the use of a composite primary efficacy outcome derived from a parallel observational program of cognitive, structural, and functional MRI, and neurosensory measures, along with CSF biomarkers.
Standard protocol approvals, registrations, and patient consents
INTREPAD (NCT-02702817) was approved by the institutional review board of McGill University. Recruitment occurred between November 2011 and March 2015. Data gathering ended March 28, 2017. All participants provided written informed consent for each trial procedure. Data were collected at the Douglas Mental Health University Institute, an affiliate of McGill University (Montréal). All research procedures complied with the ethical principles of the Declaration of Helsinki. A Data Monitoring Committee reviewed all safety and efficacy data prepared by a contract (unmasked) statistician on October 20, 2016, and upon completion (June 26, 2017).
Overview of participants and trial regimen
We recruited 462 healthy older individuals with a parental or multiple-sibling history of AD for participation in Pre-symptomatic Evaluation of Novel or Experimental Treatments for Alzheimer's Disease (PREVENT-AD), an observational cohort study of healthy persons aged 55+ without evidence of cognitive deficit.9 Among these, 195 eligible volunteers were randomized in INTREPAD to receive either low-dose naproxen sodium or placebo (figure 1). The primary efficacy analysis considered a modified intent-to-treat (mITT) group of 160 persons who remained on assigned treatments until their first follow-up evaluation 90 days after baseline. In all, 166 (85%) completed their participation per protocol (154 mITT and 12 others in the intent-to-treat (ITT) group with 2 years of follow-up and biomarker assessment). Participants who completed the trial on study treatments numbered 124.
Figure 1. Consort flow.

Fragile cognitive status indicates individuals with cognitive deficits suggestive of early mild cognitive impairment. GI = gastrointestinal; mITT = modified intent-to-treat; SAE = serious adverse event.
Initial integrity of participants' cognitive abilities was evaluated by telephone interview, followed by in-person assessment using the Montreal Cognitive Assessment (MoCA) and the Clinical Dementia Rating scale.10,11 In an early protocol change, we further verified intact cognitive status after baseline testing using the trial's principal cognitive assessment measure, the Repeatable Battery for Assessment of Neuropsychological Status (RBANS).12 As a consequence, 14 enrollees (3 originally assigned to naproxen and 11 to placebo) were considered unsuitable for further participation because of notable cognitive deficits that had escaped detection at baseline, suggesting early mild cognitive impairment (MCI). Final determination of cognitive eligibility relied for some on full neuropsychological evaluation.
Eligibility criteria
Other eligibility criteria included (1) at least 1 parent or 2 siblings with AD, (2) age ≥ 60 years, or ≥55 years if within 15 years of youngest-affected relative's onset, (3) health and social stability sufficient to enable participation for 5 years of longitudinal study, and (4) no contraindications to sustained treatment with naproxen sodium. Family history was ascertained from an expert's diagnosis of AD or, when necessary, via a brief, structured questionnaire (e-Methods; doi.org/10.5061/dryad.r58d342). Exclusion criteria included regular use (more than 4 doses per week) of corticosteroids, NSAIDs, other anti-inflammatory/immunosuppressant agents, or aspirin. A complete list of inclusion/exclusion criteria is available in the e-Methods.
Specification of primary efficacy outcome and initial power analysis
The original INTREPAD analysis plan called for a primary efficacy determination based on a composite of data that was not then specified. This composite was under development in data from the parallel (nontrial) parent PREVENT-AD Cohort, with near-identical characteristics.9 Initial power analyses considered a subset of data from the public use ADNI database. We simulated an attempt to demonstrate a true 25% reduction in the slope of the CSF concentration of total tau (t-tau) over 1 year, specifying 85% statistical power. This analysis suggested that the needed power could be provided by a sample of 228 aged cognitively normal participants assigned 1:1 to active drug vs placebo. We assumed that greater power would be provided by 2 (rather than 1) years of observation, and thus specified a target enrollment of 200 with an assumption that ∼80% would remain in an mITT primary analysis pool of persons who remained on study treatments through their first follow-up examination (but note below that results belied these assumptions).
When the primary efficacy outcome (APS)3 was fully specified, we performed a similar simulation in the parallel, nontrial PREVENT-AD Cohort. This simulation now relied on the Cohort's observed slope, random intercept variance, and error variance using a longitudinal random effects model. It suggested that 160 participants would afford 85% power to detect only a 50% difference in slope between 2 study arms, or 68% power to detect a 40% difference. Because the PREVENT-AD data did not include CSF biomarker measures, however, we expected that the trial's CSF assay results would provide a substantial increase in power, as had been the case in the observational BIOCARD study13 (see Discussion).
Randomization, masking, and provision of study drug
Using randomization.com, participants were randomized 1:1 in 34 permuted blocks of 6 to identical-appearing tablets of naproxen sodium 220 mg or placebo, both generously donated by Pharmascience (Montreal, Canada), for administration twice daily with meals. The Douglas Hospital pharmacy stored and prepared study drug in sealed dosage packets for each participant visit. Participants, the principal investigator, study staff, and all clinicians responsible for assessments or marker measures remained masked to treatment assignment until safety and efficacy analyses were complete. A protocol for interim unmasking was available but not needed. Only an external statistician (Daniel Morissette) had advance access to treatment assignment.
Assessment methods
Figure 2 shows the timeline for data gathering. Complete evaluations were performed at baseline and after 3, 12, and 24 months of treatment.
Figure 2. Trial timeline.

Full assessment (on site): baseline (BL), 3 months, 12 months, 24 months. Safety follow-up (on site): 1 month, 6 months, 18 months. Safety follow-up (telephone): 9 months, 15 months, 21 months. ** T1-weighted (EN, BL, 3 months, 12 months, 24 months), fluid-attenuated inversion recovery (EN, 24 months), diffusion-weighted imaging (EN, 24 months), arterial spin labeling (BL, 3 months, 12 months, 24 months), resting-state fMRI (BL, 3 months, 12 months, 24 months), gradient echo quantitative T2* task (BL, 3 months, 12 months, 24 months), task fMRI (BL, 3 months, 12 months, 24 months). EN = enrollment; LP = lumbar puncture; RBANS = repeatable battery for assessment of neuropsychological status; UPSIT = University of Pennsylvania Smell Identification Test.
Safety
Follow-up interviews (on-site or by telephone) for adverse events (AEs) were administered ad hoc or, at a minimum, every 3 months using structured medical history and review-of-system questionnaires. On-site safety evaluation included routine laboratory studies, ECG, and a brief physical and neurologic examination. Potentially important incidental MRI and other newly discovered health risks were referred for expert review. Research nurses rated AEs using the Cancer Therapy Evaluation Program Common Terminology Criteria, version 3.0. AEs were graded as mild, moderate, or severe after physician review, and each AE was also assigned a Preferred Term and a System Organ Class using the Medical Dictionary for Regulatory Activities classification system, version 19.1. Serious AEs (SAEs) that were life-threatening or required hospitalization were reported in real time to the McGill Research Ethics Board. Relationship of AEs and SAEs to treatment (assuming assignment to naproxen) was assessed by a study physician. Elective surgeries were not considered SAEs.
Compliance
Participants were asked to bring unused supplies of drug to trial visits. Research nurses evaluated compliance at each visit, inquiring when indicated into reasons for missed doses.
Cognitive and neurosensory performance
At baseline, 3, 12, and 24 months, neuropsychological performance was measured using the RBANS, which evaluates 5 cognitive domains (immediate memory, delayed memory, attention, language, and visuospatial abilities) and a total summary score. The RBANS is available in 4 equivalent versions (for longitudinal assessment) in both French and English. Version A was administered at baseline, and alternate forms were used in random order at follow-up visits. We developed correction factors to improve version equivalence and scored results without correction for age (often used clinically; see e-Methods, doi.org/10.5061/dryad.r58d342).
At each visit's fMRI session, participants were also given alternate versions of an episodic memory task (encoding and retrieval of objects).14 They were asked to identify whether items had been observed during the encoding period or were new at the retrieval session (details in e-Methods, doi.org/10.5061/dryad.r58d342). A correct response required a “hit” or correct rejection. Odor identification, a neurosensory faculty, was also tested using the 40-item “scratch and sniff” University of Pennsylvania Smell Identification Test (UPSIT).15,16 The latter, comprising 40 items administered in 4 randomly ordered booklets, was available in both French and English.
Neuroimaging markers
Brain structural and functional MRI were performed at baseline and 3-, 12-, and 24-month visits on a Siemens TIM Trio 3T MRI system (Siemens Medical Solutions, Erlangen, Germany) using the Siemens standard 12-channel head coil (figure 2 and e-Methods, doi.org/10.5061/dryad.r58d342). Using conventional pipelines, averages of gray matter density were calculated for 78 regions based on T1-weighted images using the SPM12 toolbox to define probabilistic gray matter maps. Cortical thickness was estimated from T1-weighted images using version 1.12 of the CIVET pipeline.17 Brain volumes were computed from the same images using a volumetric pipeline.18 Regional cerebral blood flow was evaluated using quantitative pipelines from single-echo pseudo-continuous arterial spin labeling acquisitions.19
CSF biomarkers
A subset of 93 participants in the mITT pool volunteered for up to 4 serial lumbar punctures (LPs) over the 2-year trial interval following their clinical and imaging evaluations. After an overnight fast, LPs were performed by P.R.-N. using an introducer and the Sprotte 24-gauge atraumatic needle. Samples of 20–30 mL were withdrawn by syringe and aliquoted (500 μL) into propylene cryotubes for storage at −80°C. We followed procedures specified by the BIOMARK-APD consortium of the EU Joint Program in Neurodegenerative Disease to measure CSF concentrations of the AD biomarkers Aβ1-42, t-tau, and phosphorylated tau (p-tau) with the Innotest ELISA assay kit (Fujirebio, Ghent, Belgium).
APOE genotype
APOE genotype was determined using RT-PCR amplified DNA and the PyroMark Q96 pyrosequencer (Qiagen, Toronto, Canada), as described previously.16
Primary efficacy outcome: The composite APS
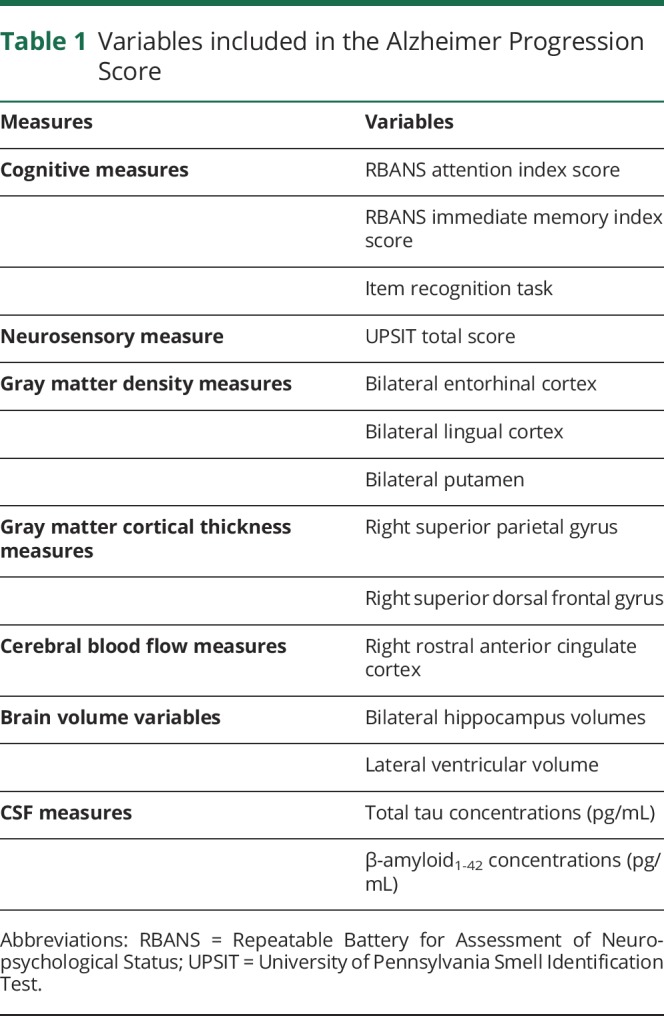
The primary efficacy outcome was annual rate of change in the APS using marker weights estimated beforehand in the nontrial PREVENT-AD Cohort. The APS is a composite that incorporates multimodal imaging, neurosensory, cognitive, and CSF markers, based on an assumption that change in each of these arises from a single underlying latent process (AD pathogenesis). Its scores are scaled as a standard normal distribution, with higher scores denoting increasing severity. Constituent measures are summarized in table 1. At each measurement, a uniform scheme of weightings for individual markers yielded a composite summary score. All available data were used to estimate individual scores, and missing data were accommodated in a process that essentially averaged over missing values. To verify robustness to missing data, Gross et al.20 had used iterative leave-one-out analyses in a similar outcome measure comprising 6 markers. Leoutsakos et al.3 also examined effects of missing CSF data on the APS, noting similarly satisfactory findings. The APS approach had been validated using data from the BIOCARD study,13 before being incorporated into INTREPAD efficacy analyses. In BIOCARD, the APS approach showed substantial abilities to predict subsequent conversion to MCI or AD dementia.3 Analyses there also showed temporal measurement invariance. Before applying the PREVENT-AD cohort-derived marker weights to the analysis of INTREPAD, we demonstrated that they provided excellent performance in the trial sample.3 However, the trial data (table 1) included 2 variables (CSF t-tau and Aβ1-42 levels) that were not available from nontrial participants. We incorporated these measures only after verifying that doing so did not materially alter the weights for the remaining variables. Full specification of variables and APS parameters preceded unmasking of treatment assignment.
Table 1.
Variables included in the Alzheimer Progression Score
Statistical analysis
We followed a statistical analysis plan finalized on June 8, 2017, by J.L. and the PREVENT-AD Research Group. To assure consideration to potentially important AEs occurring during the first 3 months of the trial, safety analyses were based on the full intention-to-treat population (n = 195). These analyses were based on summary listings of AEs, using a χ2 test for pairwise comparisons. The baseline characteristics of the treatment groups were compared pro forma using Fisher exact (for sex), χ2 (for number of APOE ε4 alleles), and 2-sample t tests (for age, education, parental age at AD onset, MoCA score, and APS).
The primary efficacy analysis was based on the mITT sample of participants who had remained on treatment through at least one follow-up (3-month) assessment (n = 160). Secondary outcomes were rate of change in cognition (RBANS total score), olfaction (UPSIT score), and CSF biomarkers of AD (Aβ1-42, t-tau, p-tau, t-tau/Aβ1-42, and p-tau/Aβ1-42 ratio) extending over the 24 months of treatment. For both primary and secondary efficacy analyses, we used longitudinal linear random effects models (random intercepts) to assess between-group differences in rates of change. We performed an additional post hoc analysis that included baseline APS score as a covariate. We also constructed additional exploratory models to test treatment effects on APS rate of change after first assigning participants into tertiles based on their baseline CSF Aβ1-42.
Version 9.4 of the SAS (SAS Institute, Cary, NC) statistical package and R (Core Team 2014) were used for analyses except for post hoc exploratory analyses, which were performed using MATLAB. APS scores were calculated using STAT, R, and MPLUS.21 All statistical tests were 2-sided. A p value ≤0.05 was considered to indicate statistical significance.
Data sharing
All de-identified data and related documentation from this trial are available upon request to qualified researchers without limit of time, subject to a standard data sharing agreement. The PREVENT-AD program is currently developing a less restrictive approach to data sharing through the Canadian Open Neuroscience Platform.
Results
Enrollment and study completion data
The naproxen- and placebo-assigned groups included 102 and 93 participants. The mITT analysis groups included 88 and 72 of these. The main reasons for exclusion of the remaining 35 were apparent ineligibility (discovered typically following review of baseline cognitive testing; n = 14), early appearance of intolerable adverse effects (n = 6), or voluntary withdrawal (many reasons given; n = 12). Compliance-to-completion rates (24 months on study drug) were 62% for participants assigned to naproxen and 66% for those given placebo (p = 0.579; figure 1). After the 3-month run-in, the most common cause for drug discontinuation was occurrence of new AEs (12 naproxen-assigned and 3 placebo-assigned individuals; figure 1).
Baseline characteristics
There were no important differences between naproxen and placebo-treated groups with respect to age, sex, or education. All participants were Caucasian, and there were no substantial imbalances across groups in APOE ε4 status, parental age at AD onset, or MoCA score. As noted below, however, we observed an unexpected difference by treatment assignment in baseline APS values in the mITT pool (table 2).
Table 2.
Baseline characteristics of Investigation of Naproxen Treatment Effects in Pre-symptomatic Alzheimer's Disease (INTREPAD) participants

Concomitant medications
Of the 102 naproxen-assigned persons in the ITT pool, 85 (83%) initiated use of concomitant medicines, regularly or for short intervals, over the interval of the trial. The comparable figure for those assigned to placebo was 66/93 (71%; p = 0.04; table e-3, doi.org/10.5061/dryad.r58d342). The apparent imbalance was attributable principally to initiation of lipid-lowering drugs during the trial but, notably, a similar imbalance was not evident in the mITT analysis pool. Baseline or anytime use of such drugs was also similar across the treatment arms in the mITT group and, accordingly, post hoc statistical adjustment for their use brought no appreciable change in the primary outcome results.
Safety outcomes
Table 3 shows the frequency of AEs occurring in ≥10% of either treatment arm. Gastrointestinal AEs prompted study drug discontinuation in 9 naproxen-assigned and 3 placebo-assigned participants. Constipation, dyspnea, hypertension, and petechiae were also substantially more common in persons receiving naproxen. However, as expected, this group reported pain less frequently. Overall, reports of any AE were more common in naproxen-assigned persons (98% vs 89%; p = 0.015). The principal contributors to this excess were vascular disorders, which were more common in the naproxen-assigned group (p = 0.023).
Table 3.
Adverse events (AEs)
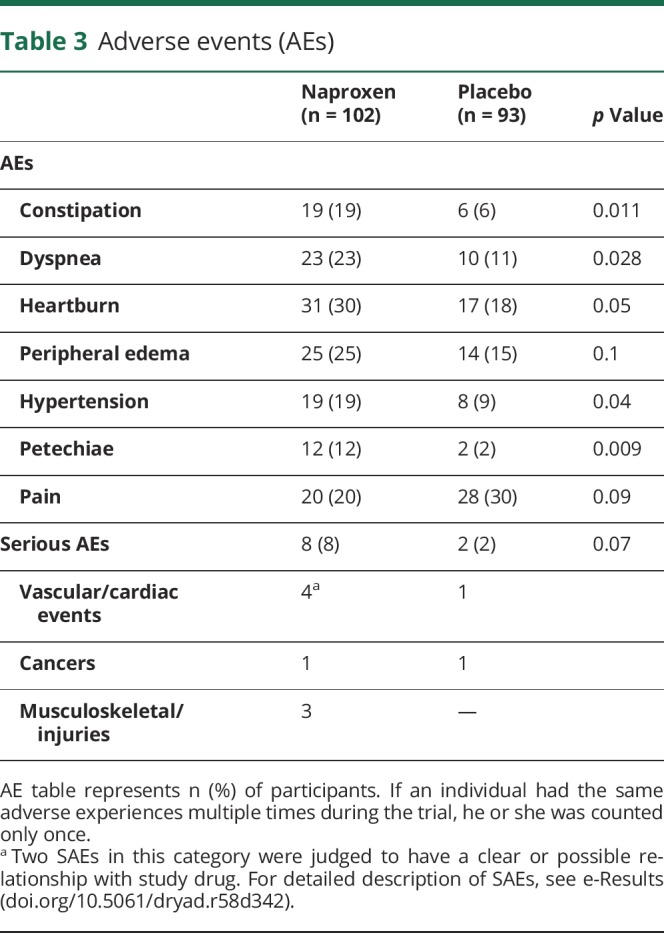
Of 10 participants reporting SAEs over 2 years (table e-4, doi.org/10.5061/dryad.r58d342), 8 had been assigned to naproxen. Three SAEs required study drug discontinuation, while 2 prompted treatment interruption. A further 3 participants were unable or unwilling (some upon physician's advice) to continue participation. No suspected unexpected serious adverse reaction occurred during the trial.
Among blood safety measures, both hemoglobin and hematocrit decreased in persons assigned to naproxen, but not in placebo-treated individuals. Between-group comparison of these measures showed strong differences at all time points except at 24 months (figures e-1 and e-2, doi.org/10.5061/dryad.r58d342). No differences were found in the 2 treatment groups with respect to other safety measurements (weight, pulse rate, systolic and diastolic blood pressure).
Primary efficacy outcome
Analyses of the primary outcome are summarized in figures 3 and 4. Among the combined treatment groups, the APS showed a clear increase over the 2-year trial period (β = 0.101 standard units/year; SE = 0.014; 95% confidence interval [CI] 0.074–0.130; p < 0.001). However, this change did not differ meaningfully between naproxen- and placebo-assigned participants after 3, 12, or 24 months of treatment. A longitudinal linear random effects model for APS showed a slight increase, if anything, in rate of change among naproxen-assigned persons, but this was well within chance expectation (time-by-treatment interaction β = +0.019 APS units/year for naproxen vs placebo; SE = 0.03; 95% CI –0.037 to +0.074; p = 0.51). The APS ratio for rate of change comparing naproxen- vs placebo-assigned mITT participants was 1.16 (bootstrapped 95% CI 0.64–1.96).
Figure 3. Treatment effects on Alzheimer Progression Score (APS).

Results for primary and exploratory secondary outcomes are represented. (A) There was no meaningful difference in APS rate of change between treatment groups. (B) Mean change from baseline (± standard error of the mean) in APS did not differ between the 2 treatment groups at any time during the treatment interval. However, APS for both groups increased after 12 and 24 months (*p < 0.05). Data are represented as point estimates (group means) with error bars (standard error of the mean). BL = Baseline; M = months.
Figure 4. Treatment effects on neurosensory and CSF biomarker measures.

Linear mixed effect models did not indicate any difference between naproxen- and placebo-assigned groups in rate of change of (A) cognitive or neurosensory or (B) biological markers of Alzheimer disease. University of Pennsylvania Smell Identification Test (UPSIT) scores decreased over the 2-year trial period for the whole group (*p < 0.05). Data are represented as point estimates (group means) with error bars (standard error of the mean). BL = baseline; p-tau = phosphorylated tau; RBANS = Repeatable Battery for the Assessment of Neuropsychological Status; t-tau = total tau.
Secondary efficacy analyses
We used similar linear random effects models in the mITT sample to evaluate treatment-related differences in rate of change of RBANS total index score, UPSIT score, and CSF AD biomarkers (Aβ1-42, t-tau, p-tau, t-tau/Aβ1-42, and p-tau/Aβ1-42). None of these comparisons suggested any benefit of naproxen treatment. Indeed, UPSIT results suggested a trend toward harm (β = −0.320, SE = 0.196; 95% CI –0.704 to +0.064; p = 0.102; figure 2B). Over the treatment interval, 4 INTREPAD enrollees (3 naproxen-assigned and 1 placebo-assigned) developed MCI22 or other suggestive cognitive deficiency sufficient to prompt discontinuation of study drug.
Exploratory efficacy analyses
Addition of covariates for APOE ε4 carrier status, age at baseline, years of education, and sex, as well as their interaction with time, did not materially affect the above findings.
As noted above, we observed that the naproxen-assigned group had higher baseline APS scores than placebo-assigned participants. This finding prompted us to carry out a post hoc addition of a term for baseline APS in the model used for the primary endpoint analysis. Addition of this term brought no consequential change from the primary endpoint results. We also analyzed whether higher baseline APS was associated with an increased rate of change in the outcome, regardless of treatment assignment. No such association was found. After partitioning participants into tertiles of Aβ1-42 concentration and testing for a triple interaction with time and treatment assignment, we also observed no effect of naproxen treatment on rate of change in APS among the different CSF Aβ1-42 tertiles.
Discussion
To assess the potential of the common NSAID naproxen sodium for prevention of AD, we tested this agent in INTREPAD, a 2-year randomized placebo-controlled trial among cognitively healthy persons at increased risk of dementia. We observed a clear increase over time in the trial's primary efficacy outcome (the APS composite representing several imaging metrics, a neurosensory modality, 3 cognitive measures, and, when available, CSF biomarkers). Assignment to naproxen produced an unambiguous increase in adverse events but no meaningful alteration in the rate of change for this primary efficacy outcome. Overall, our results suggest 2 points for discussion: (1) evaluation of a design intended to increase efficiency of AD prevention trials and (2) the trial's safety and efficacy results, and their ethical implications.
We employed several design features intended to improve efficiency for INTREPAD and, by implication, other AD prevention trials. First, we attempted to enrich participants' proportion of persons vulnerable to AD by requiring that enrollees have an affected parent or multiple siblings.23 Enrichment by requirement of a first-degree relative with AD is not novel, but the INTREPAD family history criteria were stronger. Especially in today's environment of greater longevity and increased awareness of AD-affected status (thus, ready identification), this more restrictive method is increasingly practical. It would seem easier to implement than a requirement of amyloid pathology demonstrable by PET24 or CSF analysis, or even homozygosity at the APOE ε4 risk allele.25 How this enhanced family history method of enrichment compares with the latter, more costly or invasive methods is unknown. However, we have recently shown that proximity to an index relative's age at symptom onset is related to increased AD biomarker load.26 This same metric was associated in a separate cohort with brain changes predictive of time to symptom onset.27 Given the method's reduced costs and subject burden, a cost/benefit comparison with other methods probably deserves consideration.
Second, we selected the composite APS as the trial's primary outcome. Relative to any single cognitive or biomarker indicator, composite outcomes of this sort should logically offer greater inference, especially in relatively early-stage presymptomatic AD. We chose the APS in preference to several similar composite indicators that had been less extensively validated.28,29 While additional efforts to validate the APS are warranted, it has shown demonstrable utility for evaluation of presymptomatic AD progress. Specifically, in BIOCARD, a 1 standard unit increase in APS predicted a 5-fold greater hazard of diagnostic progression over time.3 In the parallel PREVENT-AD cohort, we performed several simulations to compare the statistical power of the APS to that of its constituent markers. The APS provided more information than did any single endpoint, including all cognitive measures, and it also offered improved performance over a simple summing of z scores of its individual components (data not shown). We therefore suggest that the APS or similar multimodal composites show promise for prevention trials like INTREPAD.
Nonetheless, post hoc analyses suggested that our methods resulted in a trial with substantially less statistical power than had been originally projected. Our original power estimates were based in part on expectation that considerable information would be contributed by CSF biomarker data, available from more than half of the participants. That turned out not to be the case and, accordingly, the trial outcome's CI was sufficiently broad to suggest a notable possibility of type II error. Now having data on rate of change in the trial's outcomes, we can estimate that 2,250 person-years of observation (e.g., a sample of 1,125 followed over 2 years, or 563 followed for 4 years) would have been required for 80% power to detect a 30% reduction in the rate of APS change. Although much higher than originally estimated, this number still represents a considerable improvement over requirements of conventional prevention trial designs. For example, the ongoing A4 trial will follow ∼1,150 persons over ∼4.6 years (5,290 person-years) for its primary cognitive outcome.24,30 Similarly, the TOMMORROW trial had originally estimated a requirement of 5,800 persons over ∼4 years (29,200 person-years).31 By comparison, even ignoring costs of PET scans in the former or serial detailed psychometric assessments in the latter, and assuming costs proportional only to person-years of observation, the INTREPAD design may achieve cost savings of 57%–90% over traditional methods.
The INTREPAD safety results affirm prior data suggesting that, even in relatively low dosage among younger elderly persons, naproxen and other NSAIDs provoke harm in several health outcomes.32–34 Our findings that these risks can be held within bounds by careful monitoring does not obviate the ethical concern that NSAID treatments are potentially harmful and should be given for AD prevention only if they produce substantial reduction of AD risk. The INTREPAD results provide no evidence for such a reduction. This result is especially salient, inasmuch as the trial sample was relatively young for this sort of work, and chosen for a favorable risk/benefit balance in relation to NSAID treatment. Specifically, participants were on average ∼10 years younger than their affected parent or first-affected sibling, and were meticulously screened for incipient cognitive disorder.35 These attributes appear to weaken arguments that earlier NSAID trials failed to show benefit because their aging samples were too old or too near the cusp of symptom onset.36–38
The null efficacy outcomes of INTREPAD are reinforced by several observations. The CI around the trial's efficacy rate ratio suggests 95% certainty that the true treatment-related reduction of AD risk in this trial (or, presumably, in similar samples) does not exceed 36%. This conclusion is buttressed by a consistent absence of apparent benefit on any of the trial's secondary or exploratory outcomes. These results recall observations in ADAPT8 and prior NSAID treatment and prevention trials with null or negative results. While we cannot exclude possible benefits of NSAIDs in middle life, we can now suggest that such benefits would be nearly impossible to demonstrate in randomized prevention trials.
As regards our further objective of testing or demonstrating methods for improving efficiency in AD prevention trials, we offer several observations. Our enrichment strategy requiring parental or multiple-sibling family history of AD might have been improved by a further requirement specifying participants' age in relation to their index relatives' onsets. While evidently more informative than any single biomarker, or a composite of only a few such markers, our outcome could today probably be improved by incorporation of newer salient markers of preclinical disease. These might include threshold values for the traditional CSF biomarkers Aβ1-42, t-tau, or p-tau, or possibly neurofilament light chain as a precondition to enrollment. Power estimation and sample size requirement for such work should be clearer now than when we began this work. In this last sense, INTREPAD may be viewed as providing an approximate benchmark for work with samples having similar baseline characteristics.
In all events, this work has left us with extreme pessimism regarding any possible role of NSAIDs in AD prevention. Instead, our results may suggest reconsideration of inflammatory diseases (or a proinflammatory diathesis) as a possible explanation for the reduced AD incidence among NSAID users in observational studies.8
Acknowledgment
The authors thank the research participants and their study partners for their commitment to this work; members of the PREVENT-AD Research Group (preventad.loris.ca/acknowledgements/acknowledgements.php?date=2018-01-01); Daniel Morissette (StatExpert, Inc.) for executing the trial's data analysis plan; the Data Monitoring Committee, comprising Denis Evans (Chair), Bruce Lamb, James Neaton, Lon Schneider, and Doug Galasko; and Sharon Keays and Ruth Poole, Pharmascience, Inc., for assistance with drug supply and management.
Glossary
- AD
Alzheimer disease
- AE
adverse event
- APS
Alzheimer Progression Score
- CI
confidence interval
- INTREPAD
Investigation of Naproxen Treatment Effects in Pre-symptomatic Alzheimer's Disease
- ITT
intent-to-treat
- LP
lumbar puncture
- MCI
mild cognitive impairment
- mITT
modified intent-to-treat
- MoCA
Montreal Cognitive Assessment
- NSAID
nonsteroidal anti-inflammatory drug
- p-tau
phosphorylated tau
- PREVENT-AD
Pre-symptomatic Evaluation of Novel or Experimental Treatments for Alzheimer's Disease
- RBANS
Repeatable Battery for Assessment of Neuropsychological Status
- SAE
serious adverse event
- t-tau
total tau
- UPSIT
University of Pennsylvania Smell Identification Test
Appendix 1. Authors
Appendix 2. Coinvestigators
Footnotes
Study funding
This trial was supported by McGill University, by an unrestricted gift from Pfizer Canada, and by infrastructure support from the Canada Fund for Innovation. Dr. Breitner's efforts were supported by a Canada Research Chair award from the government of Canada. Genetic and laboratory work (J.P.) were supported by the Fonds de Recherche du Québec–Santé (FRQ-S) and the J.L. Levesque Foundation. P.-F.M. is supported by funding from the Canada First Research Excellence Fund, awarded to McGill University for the Healthy Brains for Healthy Lives initiative. M.-E.L.-M. and P.-F.M. received research support from the Lazlo & Etelka Kollar fellowship fund for this project. Additional support was provided by the Douglas Mental Health University Institute Foundation. The funding agencies had no role in study design, data analysis, or interpretation of results.
Disclosure
The authors report no disclosures relevant to the manuscript. Go to Neurology.org/N for full disclosures.
References
- 1.Jack CR Jr., Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol 2010;9:119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bateman RJ, Xiong C, Benzinger TL, et al. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med 2012;367:795–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leoutsakos JM, Gross AL, Jones RN, Albert MS, Breitner JCS. “Alzheimer's Progression Score”: development of a biomarker summary outcome for AD prevention trials. J Prev Alz Dis 2016;3:229–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.in t' Veld BA, Ruitenberg A, Hofman A, et al. Nonsteroidal antiinflammatory drugs and the risk of Alzheimer's disease. N Engl J Med 2001;345:1515–1521. [DOI] [PubMed] [Google Scholar]
- 5.Breitner JC, Haneuse SJ, Walker R, et al. Risk of dementia and AD with prior exposure to NSAIDs in an elderly community-based cohort. Neurology 2009;72:1899–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aisen PS, Davis KL, Berg JD, et al. A randomized controlled trial of prednisone in Alzheimer's disease: Alzheimer's Disease Cooperative Study. Neurology 2000;54:588–593. [DOI] [PubMed] [Google Scholar]
- 7.Thal LJ, Ferris SH, Kirby L, et al. A randomized, double-blind, study of rofecoxib in patients with mild cognitive impairment. Neuropsychopharmacology 2005;30:1204–1215. [DOI] [PubMed] [Google Scholar]
- 8.Adapt Research Group, Lyketsos CG, Breitner JC, Green RC, et al. Naproxen and celecoxib do not prevent AD in early results from a randomized controlled trial. Neurology 2007;68:1800–1808. [DOI] [PubMed] [Google Scholar]
- 9.Breitner JCS, Poirier J, Etienne PE, Leoutsakos JM; PREVENT-AD Research G. Rationale and structure for a new center for studies on prevention of Alzheimer's disease (StoP-AD). J Prev Alz Dis 2016;3:236–242. [DOI] [PubMed] [Google Scholar]
- 10.Hughes CP, Berg L, Danziger WL, Coben LA, Martin RL. A new clinical scale for the staging of dementia. Br J Psychiatry 1982;140:566–572. [DOI] [PubMed] [Google Scholar]
- 11.Nasreddine ZS, Phillips NA, Bedirian V, et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc 2005;53:695–699. [DOI] [PubMed] [Google Scholar]
- 12.Randolph C, Tierney MC, Mohr E, Chase TN. The repeatable battery for the assessment of neuropsychological status (RBANS): preliminary clinical validity. J Clin Exp Neuropsychol 1998;20:310–319. [DOI] [PubMed] [Google Scholar]
- 13.Albert M, Soldan A, Gottesman R, et al. Cognitive changes preceding clinical symptom onset of mild cognitive impairment and relationship to ApoE genotype. Curr Alzheimer Res 2014;11:773–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rajah MN, Languay R, Grady CL. Age-related changes in right middle frontal gyrus volume correlate with altered episodic retrieval activity. J Neurosci 2011;31:17941–17954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Doty RL, Shaman P, Dann M. Development of the University of Pennsylvania Smell Identification Test: a standardized microencapsulated test of olfactory function. Physiol Behav 1984;32:489–502. [DOI] [PubMed] [Google Scholar]
- 16.Lafaille-Magnan ME, Poirier J, Etienne P, et al. Odor identification as a biomarker of preclinical AD in older adults at risk. Neurology 2017;89:327–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zijdenbos AP, Forghani R, Evans AC. Automatic “pipeline” analysis of 3-D MRI data for clinical trials: application to multiple sclerosis. IEEE Trans Med Imaging 2002;21:1280–1291. [DOI] [PubMed] [Google Scholar]
- 18.Aubert-Broche B, Fonov VS, Garcia-Lorenzo D, et al. A new method for structural volume analysis of longitudinal brain MRI data and its application in studying the growth trajectories of anatomical brain structures in childhood. Neuroimage 2013;82:393–402. [DOI] [PubMed] [Google Scholar]
- 19.Wang J, Alsop DC, Song HK, et al. Arterial transit time imaging with flow encoding arterial spin tagging (FEAST). Magn Reson Med 2003;50:599–607. [DOI] [PubMed] [Google Scholar]
- 20.Gross AL, Mungas DM, Leoutsakos JS, Albert MS, Jones RN. Alzheimer's disease severity, objectively determined and measured. Alzheimers Dement 2016;4:159–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Muthén LK, Muthén BO. Mplus user's guide: Statistical analysis with latent variables: User's guide. Los Angeles: Muthén & Muthén; 2004. [Google Scholar]
- 22.Winblad B, Palmer K, Kivipelto M, et al. Mild cognitive impairment: beyond controversies, towards a consensus: report of the International Working Group on Mild Cognitive Impairment. J Intern Med 2004;256:240–246. [DOI] [PubMed] [Google Scholar]
- 23.Fratiglioni L, Ahlbom A, Viitanen M, Winblad B. Risk factors for late-onset Alzheimer's disease: a population-based, case-control study. Ann Neurol 1993;33:258–266. [DOI] [PubMed] [Google Scholar]
- 24.Sperling RA, Rentz DM, Johnson KA, et al. The A4 study: stopping AD before symptoms begin? Sci Transl Med 2014;6:228fs213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.A study of CAD106 and CNP520 versus placebo in participants at risk for the onset of clinical symptoms of Alzheimer's Disease [online]. Available at: clinicaltrials.gov/show/NCT02565511. Accessed May 2018.
- 26.Villeneuve S, Vogel JW, Gonneaud J, et al. Proximity to parental symptom onset and amyloid-beta burden in sporadic Alzheimer disease. JAMA Neurol 2018;75:608–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vogel JW, Vachon-Presseau E, Pichet Binette A, et al. Brain properties predict proximity to symptom onset in sporadic Alzheimer's disease. Brain 2018;141:1871–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jedynak BM, Lang A, Liu B, et al. A computational neurodegenerative disease progression score: method and results with the Alzheimer's Disease Neuroimaging Initiative cohort. Neuroimage 2012;63:1478–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xiong C, van Belle G, Chen K, et al. Combining multiple markers to improve the longitudinal rate of progression-application to clinical trials on the early stage of Alzheimer's disease. Stat Biopharm Res 2013;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Clinical trial of solanezumab for older individuals who may be at risk for memory loss [online]. Available at: clinicaltrials.gov/show/NCT02008357. Accessed May 2018.
- 31.Budur K, Welsh-Bohmer K, Burns D, et al. A pharmacogenetics-supported clinical trial to delay onset of mild cognitive impairment due to Alzheimer's disease using low-dose pioglitazone: an update on the TOMORROW study. Alzheimers Dement 2014;10:P809–P810. [Google Scholar]
- 32.Langman MJ, Weil J, Wainwright P, et al. Risks of bleeding peptic ulcer associated with individual non-steroidal anti-inflammatory drugs. Lancet 1994;343:1075–1078. [DOI] [PubMed] [Google Scholar]
- 33.Pirmohamed M, James S, Meakin S, et al. Adverse drug reactions as cause of admission to hospital: prospective analysis of 18 820 patients. BMJ 2004;329:15–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kearney PM, Baigent C, Godwin J, Halls H, Emberson JR, Patrono C. Do selective cyclo-oxygenase-2 inhibitors and traditional non-steroidal anti-inflammatory drugs increase the risk of atherothrombosis? Meta-analysis of randomised trials. BMJ 2006;332:1302–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hayden KM, Zandi PP, Khachaturian AS, et al. Does NSAID use modify cognitive trajectories in the elderly? The Cache County Study. Neurology 2007;69:275–282. [DOI] [PubMed] [Google Scholar]
- 36.Aisen PS, Schafer KA, Grundman M, et al. Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. JAMA 2003;289:2819–2826. [DOI] [PubMed] [Google Scholar]
- 37.Breitner JC, Baker LD, Montine TJ, et al. Extended results of the Alzheimer's disease anti-inflammatory prevention trial. Alzheimers Dement 2011;7:402–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leoutsakos JM, Muthen BO, Breitner JC, Lyketsos CG, ADAPT Research Team. Effects of non-steroidal anti-inflammatory drug treatments on cognitive decline vary by phase of pre-clinical Alzheimer disease: findings from the randomized controlled Alzheimer's Disease Anti-inflammatory Prevention Trial. Int J Geriatr Psychiatry 2012;27:364–374. [DOI] [PMC free article] [PubMed] [Google Scholar]