

ABSTRACT
In antibody discovery, in-depth analysis of an antibody library and high-throughput retrieval of clones in the library are crucial to identifying and exploiting rare clones with different properties. However, existing methods have technical limitations, such as low process throughput from the laborious cloning process and waste of the phenotypic screening capacity from unnecessary repetitive tests on the dominant clones. To overcome the limitations, we developed a new high-throughput platform for the identification and retrieval of clones in the library, TrueRepertoire™. This new platform provides highly accurate sequences of the clones with linkage information between heavy and light chains of the antibody fragment. Additionally, the physical DNA of clones can be retrieved in high throughput based on the sequence information. We validated the high accuracy of the sequences and demonstrated that there is no platform-specific bias. Moreover, the applicability of TrueRepertoire™ was demonstrated by a phage-displayed single-chain variable fragment library targeting human hepatocyte growth factor protein.
KEYWORDS: Antibody discovery, monoclonal antibody, lead antibody, antibody library, phage display, antibody library sequencing, NGS, clone retrieval, rare clones
Introduction
In vitro display technologies enable fast selection of antibodies with desired properties (e.g., specific binding activity) from a highly diverse antibody library. The high diversity, which is derived from shuffling natural VH-VL pairs or mutagenesis, enhances the possibility of finding potent clones in the libraries. Additionall y, the association between the genotype and phenotype of clones in display systems allows for clone screening based on phenotype and, thereby, identification of the selected clones by genotype.1 Among the available in vitro display technologies, phage display is widely used due to the high transformation capacit y of phages and robustness of the process.2-4 For example, the first US Food and Drug Administration-approved human monoclonal antibody, adalimumab, was discovered t\ough phage display.5 Since the approval of adalimumab in 2002, many antibodies have been discovered and engineered through phage display, some of which have already been commercialized for clinical use.6
In phage display-based antibody discovery, a phage display library composed of diverse clones is constructed, and then clones with binding activity to a target antigen in the library are enriched through a binding activity-based enrichment method, i.e., biopanning. After several rounds of biopanning, the enriched clones are identified and retrieved for further characterization. In this identification and retrieval step, to increase the possibility to discover prominent lead antibodies, it is essential to retrieve many clones with distinct biochemical properties. These biochemical properties can be predicted by identifying the nucleotide sequence of the enriched clones. As a representative example, the number of binding epitopes targeted by the identified clones can be estimated by analyzing the distribution of the length and amino acid sequence of the heavy chain complementarity-determining region 3 (HCDR3).7,8
However, in most cases, few major clones are identified because dominant clones usually outnumber rare clones and make the selected pool of clones relatively homogeneous. Clones with prominent properties can exist in rare populations, but can be lost due to technical limitations. Also, clones with high binding activity to a target antigen can exist in a rare population in a library, due to the bias that results from the biopanning.9 This was shown in our previous research, where certain rare clones in a library were retrieved and confirmed to have binding activity to a target antigen.10 Thus, to retrieve these rare clones and exploit them, in-depth analysis of an antibody library is essential.
Improving throughput enables in-depth analysis of a library because sampling depth is increased. Conventionally, in vitro screening has been used to identify and retrieve clones in a phage display library.11 Next-generation sequencing (NGS) technology was introduced to improve the screening process in terms of sampling depth.10,12,13 However, the two existing methodologies, in vitro screening and the NGS-based process, still have several technical limitations in identifying and retrieving rare clones in the library in high throughput. For in vitro screening, colony picking used for clone isolation is throughput-limited due to its labor intensiveness, unless a picking machine is used. Even when a picking machine is used, an additional cost for its operation is incurred, and a risk of cross-contamination still exists. Additionally, Sanger sequencing used in the genotyping step has throughput limitations derived from its laborious preparation step and high cost. Moreover, the process of genotyping after phenotyping causes the phenotype test to overlap with the same clones, which dramatically reduces the screening efficiency. Due to the low throughput and the limitations associated with the phenotypic screening capacity, few clones are identified and retrieved in most cases.3,14,15
To overcome these limitations, methods using NGS were introduced. Through high-throughput sequencing of a phage display library by NGS, high-resolution information about the composition of phage display libraries could be obtained. Based on the high-resolution information, rare clones in the libraries can be detected and confirmed to have binding activity toward the target antigens. Additionally, the change in the order of the process, from genotyping after phenotyping to phenotyping after genotyping, eliminated the loss of screening capacity in the phenotyping step. However, NGS itself has technical limitations. First, bias on the composition of clones in a library can be introduced during PCR amplification.16 Second, NGS is vulnerable to sequencing and PCR errors.17 Third, due to the short read length, linkage information between heavy and light chains of antibody fragments is lost.18 Last, and most importantly, to retrieve clones in the library for phenotypic testing, PCR rescue or de novo gene synthesis must be used. Low throughput of the methods makes the retrieval step costly and time-consuming, thereby limiting the number of clones to be tested in the following experiments.12,19 Although unique molecular identifier (UMI) and single-cell sequencing techniques can be used to improve the NGS-based process, these techniques also rely on the low-throughput clone retrieval method.20-23 In this respect, existing methodologies are not adequate for in-depth analysis of a phage display library for detecting rare clones and high-throughput retrieval of their physical DNA in the library for further characterization.
We developed a new high-throughput cloning platform for antibody library screening, TrueRepertoire™, for in-depth analysis of phage display library and high-throughput retrieval of the clones’ physical DNA. Compared to conventional in vitro technologies, TrueRepertoire™ replaces the low-throughput colony picking system with a new high-throughput clone isolation system using a micro biochip and laser-based isolation method. Additionally, the low-throughput Sanger sequencing in the conventional process is replaced with a high-throughput clone identification system using multiplex PCR by barcoded primers, NGS, and barcode-based consensus sequence computation. Using TrueRepertoire™, highly accurate sequences of clones in an antibody display library can be obtained while preserving linkage information between heavy and light chains of the clones. Additionally, a substantial number of clones, including rare clones, in the library can be retrieved for use in subsequent experiments for further characterization (Figure 1). To validate the performance of TrueRepertoire™, the accuracy of consensus sequences from the clone identification system of TrueRepertoire™ was demonstrated through cross-validation by Sanger sequencing. Using an artificial library composed of clones with a pre-defined composition, we showed experimentally that TrueRepertoire™ has no platform-specific bias derived from the new clone isolation system. Finally, we demonstrated the applicability of TrueRepertoire™ by applying TrueRepertoire™ to a phage-displayed single-chain variable fragment (scFv) library.
Figure 1.
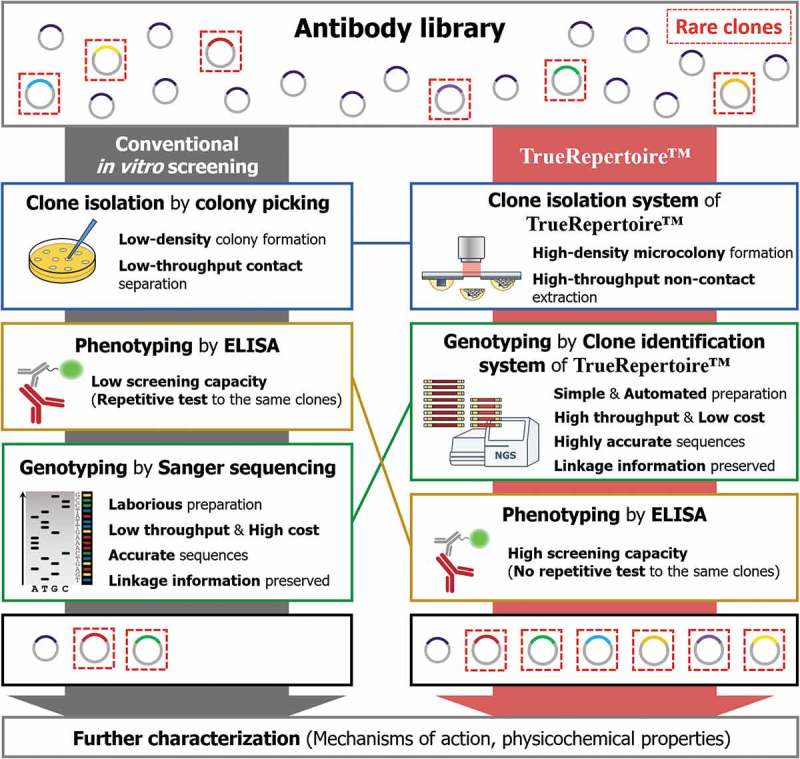
Comparison of the workflow between conventional in vitro screening and TrueRepertoire™. In the conventional in vitro screening process, clones in an antibody library are isolated by the colony picking method. The isolated clones are tested for their ability to bind to a target antigen by ELISA. Positive clones confirmed to have a binding activity are selected; then, the genotype of the clones is identified through Sanger sequencing. The low throughput and labor intensiveness of colony picking and Sanger sequencing limit the throughput. Moreover, the strategy of performing genotyping after phenotyping provokes the implementation of repetitive ELISA tests on the same clones, which reduces screening capacity. Overall, through the conventional in vitro screening process, in many cases, only a handful of clones are identified and retrieved for the following characterization. TrueRepertoire™ replaces colony picking with a new clone isolation system that enables the formation of microscale colonies in high density and extraction of the microcolonies in high throughput, without contact. The isolated clones are analyzed by the clone identification system of TrueRepertoire™. Through the clone identification system, highly accurate sequences of the clones are obtained based on the linkage information between heavy and light chains. Then, clones are selected based on the sequence information and tested for their binding activity by ELISA. Through the process, many clones, including rare clones, in the library can be identified and retrieved for further characterization.
Results
Micro biochip and laser-based isolation of TrueRepertoire™
Two critical technological compartments enable TrueRepertoire™: the micro biochip and laser-based isolation. Conventional contact-based colony picking was replaced with a new laser-based colony isolation system. In the isolation system, a micro biochip, termed Single-cell Separation and Incubation Chip capable of Light-mediated Extraction (SSICLE), is used to form intense amounts of microscale colonies on the chip. Following their formation, the microcolonies are extracted in high throughput from the chip using a pulsed infrared laser without contact.
SSICLE is composed of four main parts: 1) an indium tin oxide (ITO) coated layer, 2) a hydrophobic layer, 3) hydrophilic pillar structures, and 4) a cover glass. Microcolony-forming agarose droplets are formed around the pillar structures by self-assembly. Microorganisms are loaded into the agarose emulsion and grow to form microscale colonies. An aqueous solution consisting of the reagents for incubation, agarose for fixation, and microorganisms interacts with the hydrophilic pillar structures and the hydrophobic layer to form an agarose emulsion gathered around the pillar structures.24,25 The emulsions are sealed with oil to prevent evaporation, and each emulsion acts as an incubator for the loaded microorganisms. The whole structures, including the agarose emulsions, are patterned on an ITO-coated glass cover. The ITO-coated layer of the chip reacts to an infrared laser and vaporizes to produce pressure for extracting the agarose emulsions (Figure 2(a)).
Figure 2.
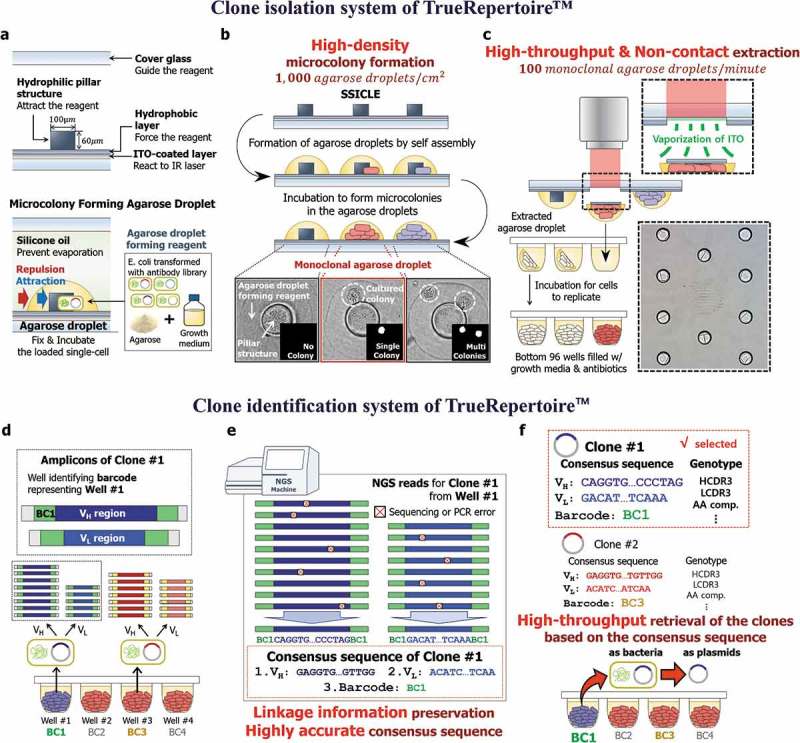
The clone isolation and clone identification systems of TrueRepertoire™. (a) The structure of SSICLE. In SSICLE, microcolony-forming agarose droplets can be patterned in high throughput by self-assembly. The reagent comprising the agarose droplet interacts with the hydrophilic structure and hydrophobic layer to form agarose droplets. Single cells of transformants of the antibody library can be loaded into the agarose droplets and grow to form microscale colonies. (b) Formation of the microcolonies on SSICLE. By using SSICLE, microcolonies can be formed on the chip in high density. The monoclonal agarose droplets, each of which contains a single colony, are detected by an automated imaging and processing system. (c) Extraction of the microcolonies by a laser. The ITO-coated layer of SSICLE reacts to an IR laser to extract the monoclonal agarose droplets. Through laser-based extraction, the microcolonies can be extracted in high throughput without contact. (d) Multiplex PCR with barcoded primers of the isolated clones. Plasmids of the clones isolated in the previous step are amplified by multiplex PCR with barcoded primers. Through the amplification, amplicons with a pre-defined set of barcodes for each variable region are produced. (e) Barcode-based sorting and computation of the consensus sequences. Based on the barcode information, NGS reads are sorted, and the consensus sequence corresponding to each clone is computed. In this step, sequencing and PCR errors can be excluded effectively. (f) Genotype-based selection of the clones. Using the consensus sequence, clones are analyzed and selected based on the genotype of the clones. Then, the clones are retrieved by referring to the barcode information representing the position of the well containing the selected clone.
The laser-based clone isolation system of TrueRepertoire™
As the first step of TrueRepertoire™, model organisms, especially bacteria, transformed with an antibody library are loaded onto SSICLE with adequate growth media for the model organisms and agarose for the fixation of cells. The chip loaded with the transformants is incubated to form microcolonies in the agarose emulsions. After incubation, the number of colonies in each incubator is counted by an automated imaging and processing system to confirm the monoclonality of the agarose emulsion (Figure 2(b)). Agarose emulsions with a single colony are detected in the previous step and are extracted from the chip into the bottom of 96 wells by a pulsed infrared laser (Figure 2(c)). To estimate the number of single colonies extractable from the chip, the expected number of monoclonal agarose droplets per chip according to lambda values of the Poisson distribution was calculated (Supplementary Figure 1). The non-contact-based extraction method using a pulsed laser eliminates the risk of cross-contamination. Additionally, the extraction step is fully automated, enabling high-throughput extraction of the clones (100 clones/minute, Supplementary Video. 1).
The clone identification system of TrueRepertoire™
After incubation of the bottom wells containing the isolated colonies in the clone isolation step, plasmids of the isolated colonies are amplified by multiplex PCR for the amplification of variable regions with barcoded primers. The barcodes of the primers contain plate and well information, which enables barcode-based sorting of NGS reads (Supplementary Figure 2). The PCR products are pooled and sequenced through the Illumina NGS platform (Figure 2(d)). Reads from the NGS run are analyzed based on the barcode information of the reads. The NGS reads are aligned according to the barcodes; then, a consensus sequence representing each well is computed. In this step, a conservative threshold value for the minimum number of reads for computing the consensus sequences is used to prevent the effect of errors induced during PCR and NGS (Supplementary Figure 3).26,27 For the filter-passed wells, 1 to 10 VH and 1 to 7 VL non-identical sequences were detected from a well when sequences with > 90% nucleotide sequence identity are considered as identical sequences. The mean read-counts ratio between the first major sequence and the second major sequence from a well was 55.1 and 60.0 for VH and VL, respectively (Supplementary Figure 4).28 The barcode-based amplification and following analysis provide several advantages over the conventional high-throughput sequencing of antibody libraries by NGS. First, the bias induced in the PCR amplification step is eliminated. Second, highly accurate sequences of clones are obtained. Third, linkage information between heavy and light chains of clones is preserved (Figure 2(e)).
The consensus sequences are used in the following genotypic analysis of the clones. Based on the results of the genotypic analysis, clones to be further characterized are selected and then retrieved by referring to the barcode information of the selected clones. The clones can be retrieved in the form of bacteria or plasmids (Figure 2(f)).
Validation of the clone identification system of TrueRepertoire™
The clone identification system of TrueRepertoire™ involves multiplex PCR with barcoded primers, NGS, and barcode-based reads analysis for computing the consensus sequence of clones. Through the clone identification system, consensus sequences for clones are computed and used for further genotypic analysis of the clones.
To demonstrate the accuracy of the consensus sequences, the sequences were cross-validated by Sanger sequencing. First, clones in phage-displayed scFv libraries were isolated. Then, the isolated clones were sequenced through the clone identification system of TrueRepertoire™ to obtain consensus sequences of the clones. Additionally, plasmids of the clones were extracted and sequenced through Sanger sequencing. The consensus sequences of TrueRepertoire™ were then compared with VH and VL regions of the sequences, derived from Sanger sequencing for the cross-validation (Supplementary Figure 5). A total 454 of consensus sequences of clones were cross-validated by Sanger sequencing via the abovementioned process. All 454 consensus sequences were perfectly matched to VH and VL regions of the corresponding Sanger results.
Validation of the clone isolation system of TrueRepertoire ™
TrueRepertoire™ uses a new method for the isolation of clones in an antibody library. The new method comprises SSICLE and non-contact extraction of colonies by a laser. Through the new clone isolation system, clones in the library can be isolated in high throughput.
To confirm that there is no platform-specific bias derived from the new clone isolation system, an artificial library with a pre-defined composition of clones was constructed. The library was composed of an equal concentration of plasmids, each containing a DNA sequence for the expression of a scFv molecule. Individual clones of the artificial library were selected randomly from a naïve phage-displayed human scFv library (Supplementary File. 1). To determine whether the clonal frequencies, which were intended to be uniform, of the library were reproduced, the artificial library was transformed into the ER2738 E. coli strain by electroporation. TrueRepertoire™ was then applied to the library. As a control, the conventional colony picking was also applied to the same library in parallel. Clones in the library were isolated by colony picking and then analyzed by the clone identification part of TrueRepertoire™ to achieve high throughput. Two technical replicate experiments were conducted for each condition: an experiment using TrueRepertoire™ and a control experiment using colony picking.
For the control, 1,103 and 1,415 clones for each technical replicate were isolated by colony picking following a general protocol described previously.29 Then, the isolated clones were analyzed by the clone identification system of TrueRepertoire™. The results indicated that the clonal frequencies of the clones were reproduced in the replicates (y = 0.9840x, p-value = 2.2ⅹ10−16, the linear regression for the replicate results, Figure 3(a)). However, the distribution slightly deviated from the intended uniform distribution (, KL-divergence between the distribution of clonal frequencies from colony picking and the uniform distribution of). Because the slight deviation was reproduced in both replicates, the deviation might have stemmed from factors inducing bias in the library composition other than a random effect, such as the variation in transformation efficiencies with plasmid form (Supplementary Figure 6).30
Figure 3.
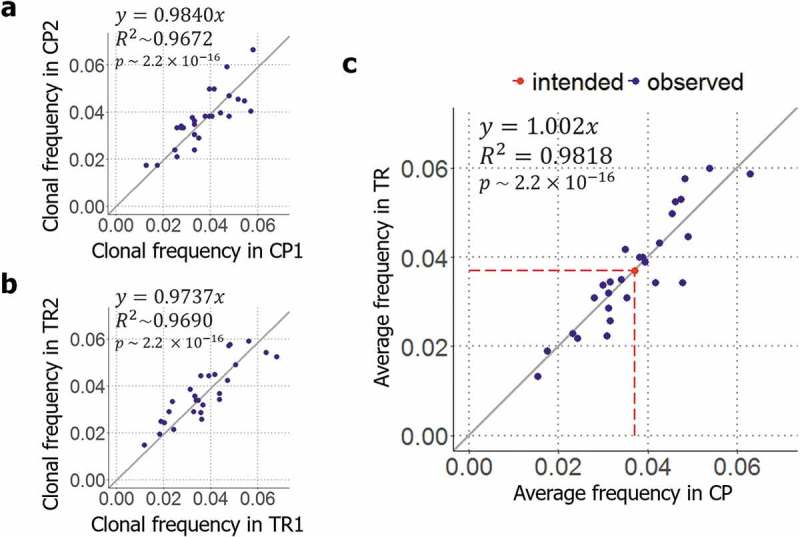
Validation of the clone isolation system of TrueRepertoire™. (a) Reproducibility of colony picking. To determine how the results are reproduced in colony picking, the distributions acquired from the two replicate control experiments were compared. (b) Reproducibility of TrueRepertoire™. To confirm the reproducibility of the platform, the results obtained from the two replicate experiments were compared. (c) Comparison between the results of TrueRepertoire™ and the control results. To determine whether a platform-specific bias exists in TrueRepertoire™ derived from the new clone isolation, the averaged clonal frequencies of two replicates of the platform were compared with the averaged clonal frequencies of two replicates of the control using colony picking.
For the experimental condition using TrueRepertoire™, 2,067 and 2,078 clones for each technical replicate were isolated and their genotypes were identified as described in the methods section. As a result, the clonal frequencies of the clones were reproduced in both replicates as in the control experiment (y = 0.9737x, p-value = 2.2ⅹ10−16, the linear regression for the replicate results, Figure 3(b)). The results obtained from the replicate experiment confirmed the reproducibility of TrueRepertoire™. A deviation from the uniform distribution was also observed (KL-divergence between the distribution of clonal frequencies from TrueRepertoire™ and the uniform distribution of).
To confirm the existence of platform-specific bias, the clonal frequencies from TrueRepertoire™ were compared with the clonal frequencies from the control experiment using colony picking. To make the comparison, averaged clonal frequency values of the two replicates for each condition were used. The averaged clonal frequencies of the clones identified through the two platforms, TrueRepertoire™ and colony picking, were confirmed to follow the same distribution (y = 1.002x, p-value = 2.2ⅹ10−16, the linear regression for the averaged clonal frequencies of two replicates from colony picking and TrueRepertoireTM, Figure 3(c)). In addition, clonal frequencies from the individual experiment for each condition, TrueRepertoire™ and colony picking, were also compared (Supplementary Figure 7).
All clones in the library were identified through TrueRepertoire™. A distortion of clonal frequencies from the intended frequencies, the uniform distribution of, was observed among the results yielded by TrueRepertoire™. However, the distortion was also observed in the results of the control experiment using colony picking, and the degrees of distortion for the platforms were similar (for colony picking and for TrueRepertoire™). For both platforms, the distortion is thought to have been introduced by the variation in transformation efficiencies with the forms of clonal plasmids. Thus, compared with conventional colony picking, TrueRepertoire™ has no platform-specific bias introduced by the new clone isolation system.
Application of TrueRepertoire™
To validate the applicability of TrueRepertoire™, TrueRepertoire™ was applied to a phage-displayed scFv library targeting human hepatocyte growth factor (hHGF) protein (Supplementary File. 2). The library was constructed from bone marrow of white leghorn chickens; the chickens were immunized with hHGF. Then, the library was enriched through five rounds of biopanning. The number of biopanning rounds was determined based on the output titer during the biopanning (Supplementary Table 1). Then, the library was analyzed by TrueRepertoire™. Additionally, for the comparison, the library was analyzed by NGS. Each variable region of the clones, the VH and VL regions, was amplified separately by PCR. The amplicons were then sequenced through the Illumina Miseq platform.
A total of 3,377 clones were isolated and analyzed by TrueRepertoire™. Among the analyzed clones, 955 clones were unique at the amino acid sequence level. Without considering the linkage information between heavy and light chains, the results feature 325 unique VH amino acid sequences and 905 unique VL amino acid sequences. To confirm that the results acquired from TrueRepertoire™ reflect the real distribution of clones in the library, the results of TrueRepertoire™ were compared with those of NGS. However, in NGS, linkage information between heavy and light chains is lost. Thus, in the TrueRepertoire™ results, frequencies of VH sequences were calculated by summing the clonal frequencies of the clones with the same VH sequence and then compared with the results of NGS for the VH region. The comparison confirmed that the clonal frequencies of clones from TrueRepertoire™ correspond with those from NGS (Figure 4(a)). Additionally, we analyzed whether linkage information acquired from TrueRepertoire™ was preserved in the NGS results. The linkages between heavy and light chains were analyzed for the 10 most frequent clones among the TrueRepertoire™ results. Among these 10, only two clones, the most frequent and fifth frequent clones, maintained their linkage information between heavy and light chains (Table 1).
Figure 4.
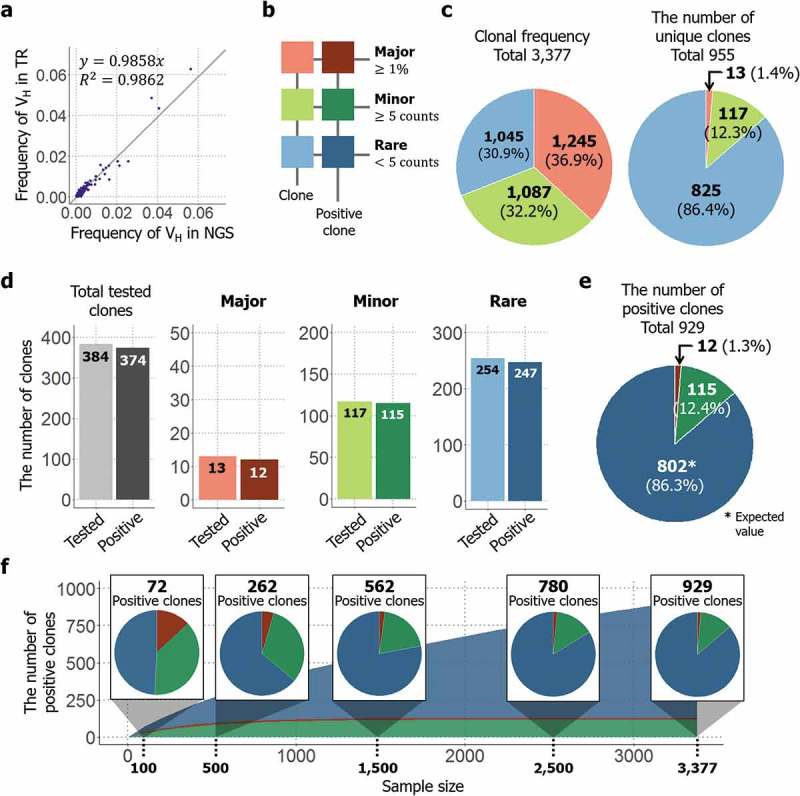
Application of TrueRepertoire™ to a phage-displayed scFv library targeting hHGF protein. (a) Comparison of the frequencies of a heavy-chain sequence of the identified clones between the NGS and TrueRepertoire™ results. To confirm that the clonal frequencies of the clones identified through TrueRepertoire™ reflect the real distribution of the library, the frequencies of the heavy-chain sequence were calculated and compared with those from NGS. (b) Groups of the identified clones. To clarify the distribution of the library, the clones were categorized into three groups, major, minor, and rare, according to the clonal frequency of the clones. The major group was denoted in red, the minor group in green, and the rare group in blue. Positive clones having binding activity to hHGF of each group were denoted in darker colors. (c) Composition of the clonal frequencies of the groups and the number of unique clones in each group. The clonal frequency of the groups refers to the sum of clonal frequencies of the clones in each group. (d) Binding activity test of the clones by ELISA. A total of 384 clones were retrieved and then tested for their binding activity to hHGF protein. All clones in the major and minor groups were retrieved and then tested for their binding activity. In the rare group, 254 clones were selected randomly from the total number of clones in the group and then tested for their binding activity. Among the tested 384 clones, 374 clones were confirmed to be positive clones with binding activity. (e) The expected composition of total positive clones in the library. The expected number of positive clones in the rare group was estimated based on the positive rate from the previous ELISA test. (f) The results of the sampling simulation. To determine how the number of positive clones and the composition of the positive clones changes as the throughput of TrueRepertoire™ increases, a sampling simulation was conducted.
Table 1.
Top 10 clones with respect to clonal frequency yielded by TrueRepertoire™ Rank of the clones was calculated based on clonal frequencies in the TrueRepertoire™ result. In the VH- VL pair column, “H” and “L” denote heavy and light chains each. The numbers following “H” and “L” denote the rank of the respective chains with the respect of the corresponding read frequencies in the NGS results. The result of the binding activity test using phage ELISA was also represented.
| TrueRepertoire™ |
NGS |
ELISA |
|||
|---|---|---|---|---|---|
| Rank | VH-VL pair | Clonal frequency (%) | VH frequency (%) | VL frequency (%) | Binding activity (O/X) |
| 1 | H1-L1 | 9.27 | 13.08 | 3.20 | O |
| 2 | H6-L8 | 4.83 | 3.56 | 0.96 | O |
| 3 | H2-L5 | 3.67 | 12.67 | 1.17 | O |
| 4 | H3-L13 | 2.37 | 7.62 | 0.65 | O |
| 5 | H9-L9 | 1.63 | 1.95 | 0.83 | O |
| 6 | H1-L40 | 1.54 | 13.08 | 0.31 | O |
| 7 | H14-L92 | 1.27 | 1.26 | 0.18 | O |
| 8 | H7-L47 | 1.18 | 2.45 | 0.28 | O |
| 9 | H2-L30 | 1.13 | 12.67 | 0.40 | O |
| 10 | H12-L31 | 1.10 | 1.39 | 0.37 | O |
To clarify the composition of the library, the identified clones were categorized into three groups – major, minor, and rare – according to the clonal frequency of the clones. Clones with a frequency greater than 1% of the total number of analyzed clones were categorized into the major group. For the remaining clones, if a clone had a clonal frequency of more than or equal to five, the clone was categorized into the minor group, and if the frequency was less than five, the clone was categorized into the rare group (Figure 4(b)).
As previously mentioned, 955 unique clones existed among the 3,377 analyzed clones. The clonal frequencies of the groups were calculated by summing the clonal frequencies of the clones in each group, and the values were confirmed to be similar to each other. Although the different groups showed approximately equal values with respect to clonal frequency, the number of unique clones in the rare group represented 86.4% of the total number of unique clones identified (Figure 4(c)). To calculate the number of positive clones with binding activity toward hHGF in each group, clones were selected from each group, then tested for their binding activity by phage ELISA. In total, 384 clones were tested as described in the methods section, among which 374 clones were confirmed to have binding activity toward hHGF (Supplementary Figure 8). All 13 clones in the major group and all 117 clones in the minor group were tested; 12 clones in the major group and 115 clones in the minor group were confirmed to have positive signals in the test. Regarding the rare group, 254 clones were selected randomly from among those in the rare group and then tested for their binding activity. Among the 254 clones, 247 clones were confirmed to have binding activity (Figure 4(d)). Using the positive rate from the test for the rare group, the expected number of positive clones in the rare group was estimated to be 825. Clones from not only the major and minor groups, but also from the rare group had a high positive rate in terms of binding activity. Thus, the rare group was also dominant in terms of the number of positive clones, like the case of the number of unique clones (Figure 4(e)). Additionally, to determine how the number of positive clones and the composition of the positive clones change as the throughput of TrueRepertoire™ increases, a sampling simulation was conducted. Data points were sampled randomly without replacement from the whole. Then, the expected number of positive clones in the sample was calculated based on the results of the phage ELISA test. Sampling was conducted in every 100 sample size from 100 to the whole sample size, 3,377. Sampling was performed 1,000 times at each sample size, and averaged values were then calculated and represented. As the throughput of TrueRepertoire™ increased, the number of positive clones increased following an approximately logarithmic scale. For the composition of the positive clones, at a sample size of 100, the number of positive clones from the major and minor groups represented more than 50% of the total number of positive clones. As the sample size increased, while the number of positive clones from the major and minor groups saturated early in the simulation, the number of positive clones from the rare group increased continuously. Eventually, clones from the rare group dominated among the positive clones in the latter phase of the simulation (Figure 4(f)). The high positivity of the result might be due to the selection of the chicken as host and the antigen from human. However, we confirmed that TrueRepertoireTM can be applied to other species as well (data not shown.).
To assess the functional diversity of the clones, HCDR3 of the clones was extracted and analyzed. A total of 115 unique HCDR3 clusters was identified, and a phylogenetic tree of the region was drawn following the analytical method described previously.31,32 The clusters had unique features in the use of amino acids and the length of the HCDR3 region (Figure 5(a)).
Figure 5.
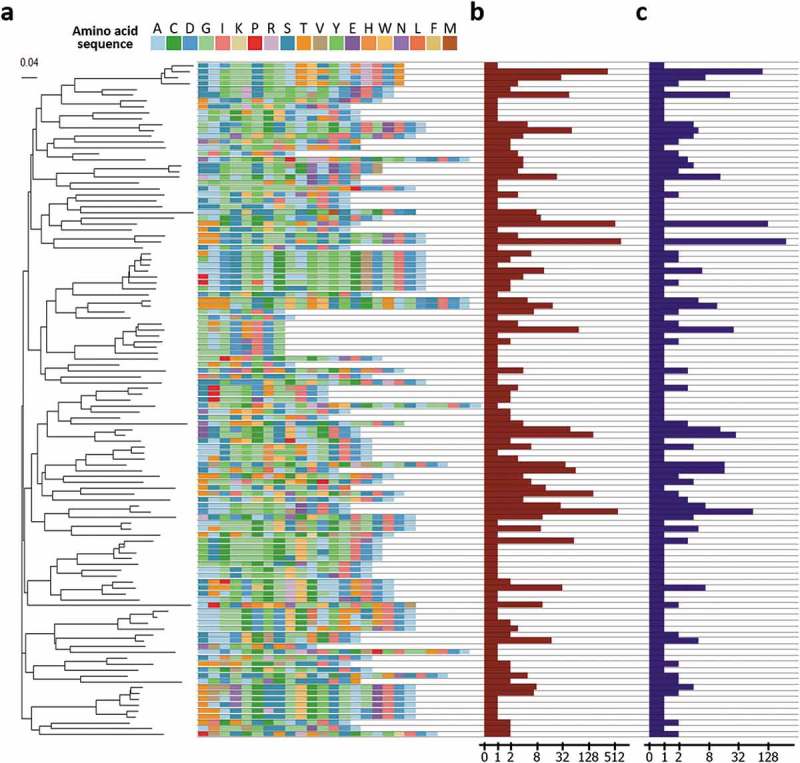
Analysis of HCDR3 region of the clones. (a) HCDR3 clusters of the clones and a phylogenetic tree of the clusters. To interrogate diversity at the functional level, the clones were clustered based on the sequences of the HCDR3 region. A total 115 unique clusters were identified and confirmed to have a diverse composition of amino acid sequences of the HCDR3 region and a heterogeneous length of the HCDR3 region. Then, the tree was built by ClustalW2 using the neighbor joining method. The scale bar indicates the probability of substitution. (b) Clonal frequencies of the clusters. Clonal frequencies of the cluster represent the sum of clonal frequencies of the clones having the HCDR3 sequence of the clusters. The clonal frequency can be interpreted as the importance of a cluster with respect to binding activity. In the following genotype-based screening step, this value can be used. (c) The number of unique clones in the cluster. This value represents the number of unique clones having the HCDR3 sequence of the cluster. Based on this value, the number of different clones with a specific HCDR3 sequence can be captured. The clones can be screened based on the genotype of the clones.
Additionally, the clonal frequency and number of unique clones corresponding to each HCDR3 cluster were represented together (Figure 5(b,c)). The clonal frequency of the clusters is the sum of clonal frequencies of clones having the HCDR3 amino acid sequence of the cluster. Considering the mechanism of biopanning, HCDR3 sequences of clusters with a high clonal frequency are expected to dominate through the biopanning process, possibly due to its superior affinity or higher stability. The clonal frequency can be used for prioritizing clones to be retrieved and further characterized. The number of unique clones in a cluster refers to the number of unique clones having the HCDR3 amino acid sequence of the cluster. This value can also be interpreted as the number of different retrievable clones having the HDCR3 sequence of the cluster. After capturing the possible number of retrievable clones of each cluster based on this value, the genotype of the clones in the group, rather than the HCDR3 sequence alone, can be analyzed. Then, the results of the genotype-based analysis can be used for prioritizing the clones based on the presence of post-translational modification motif, isoelectric point, or the expected immunogenicity.
Discussion
Here, we introduced a new robust and high-throughput platform, TrueRepertoire™, for the identification and retrieval of clones in a phage display library. TrueRepertoire™ is composed of two systems designed for clone isolation and clone identification. In the clone isolation system, microcolonies of clones in the library can be formed in high density using the micro biochip SSICLE, and the microcolonies can be extracted from the chip in high throughput without contact by laser. The isolated colonies can be analyzed by the clone identification system of the platform to compute highly accurate consensus sequence of the clones while preserving the linkage information between heavy and light chains of the clone. By using these novel systems, TrueRepertoire™ can identify and retrieve a massive number of clones, including rare clones, from a phage display library. We identified and retrieved 955 unique clones from the phage-displayed scFv library targeting hHGF by using TrueRepertoire™. In addition, as shown in the application of TrueRepertoire™ section, while the linkage information between heavy and light chains is preserved in TrueRepertoire™, the linkage information is lost in NGS. Thus, TrueRepertoire™ saves substantial effort and time by preserving the real linkage between heavy and light chains of clones. In the study discussed here, we validated that the consensus sequences have high accuracy and there is no platform specific bias in TrueRepertoire™. Also, we succeeded in identifying and retrieving many rare clones by applying the platform to the phage-displayed scFv library targeting hHGF. The retrieved clones were confirmed to have binding activity to the target antigen, hHGF. In addition, the functional diversity of the identified clones in the library was shown by analyzing the HCDR3 region of the clones. The clones had heterogeneous HCDR3 in the aspect of sequence length and amino acid composition. Considering the heterogeneity, the clones are expected to bind to different epitopes of hHGF, as reported previously.33
TrueRepertoireTM can identify and retrieve clones from antibody libraries at a much lower cost compared to the existing two methods, in vitro screening and NGS-based method. To elucidate the cost-effectiveness of the platform, a cost simulation was conducted on the hHGF case, as described in the Methods section and supplemental material. The simulation confirmed that the cost for TrueRepertoireTM was about 10-fold cheaper than the conventional in vitro screening and about 20-fold cheaper than the NGS-based method to retrieve 700 positive clones in the hHGF case. We assumed no labor cost for the convenience of the simulation. If labor cost is considered, the difference in cost between in vitro screening and TrueRepertoireTM will become higher because TrueRepertoireTM eliminates the labor-intensive clone isolation step and is an optimized process for the task. The cost of TrueRepertoireTM derived primarily from a single run of NGS; additional costs, which are incurred as the number of positive clones to be retrieved increases, were extremely low. On the other hand, the cost for in vitro screening and NGS-based method increases steeply as the number of positive clones to be retrieved increases. The cost was mainly from Sanger sequencing for in vitro screening and from gene synthesis for the NGS-based method (Supplementary Figure 9, Supplementary File. 3).
TrueRepertoire™ enables high-throughput genotype-based screening of the clones in a phage display library; thus, clones to be retrieved for further characterization can be prioritized, which reduces the cost for the subsequent characterization of the clones. In conventional antibody drug discovery, early screening and optimization steps focus on characteristics such as binding activity, potency, and stability for selection of lead constructs, while pharmacokinetic properties, which can influence both efficacy and toxicity, are typically characterized later in development and on a small number of lead monoclonal antibody constructs. Predicting pharmacokinetic properties of antibodies in the genotype-based screening can reduce the time needed for drug discovery and development by improving the lead monoclonal antibody selection process.34 TrueRepertoire™ provides a highly accurate consensus sequence of the clones. Additionally, the clonal frequency of the clones can be calculated based on the consensus sequences without a platform-specific bias. Using the consensus sequence and clonal frequency of the clones, characteristics of clones, including pharmacokinetics of antibodies, can be estimated. Therefore, clones can be selected based on genotype-based screening results, and the selected clones can be retrieved by referring to the barcode information of the consensus sequence.34-36 As a representative example of the genotype-based screening through TrueRepertoire™, charge distribution of antibodies can be analyzed by the consensus sequence of the clones, then clones expected to have poor pharmacokinetics can be excluded in the retrieval. There have been reports of antibodies with a high positive charge having poor pharmacokinetic profiles, and removing or repositioning the positive charges or counterbalancing with negative charges dramatically improved the pharmacokinetic profiles.37-39 The results of those reports can be introduced in the genotype-based screening to prevent clones with poor pharmacokinetics from being further characterized in the subsequent development. Thus, the efficiency of the development process can be enhanced.
Moreover, TrueRepertoire™ can enhance the screening efficiency of ELISA, which is followed for confirmation of binding activity of the retrieved clones. In the conventional in vitro screening, identification of the clones through Sanger sequencing is conducted after a phenotypic screening of the clones by ELISA, which provoke repetitive tests to the same clones. The waste of screening capacity increases the cost of the whole process and can limit the number of clones to be further characterized in the development. TrueRepertoire™ can provide refined sets of clones to be tested without overlap through sequence confirmation and genotype-based screening by using the consensus sequence of the clones, which can prevent repetitive tests and increase the screening capacity of the process. When the recombinant expression of a target antigen is not available, the high screening capacity of the platform acts as a critical advantage.
TrueRepertoire™ can also be applied to libraries other than a phage display library. The platform can be used to analyze such libraries and retrieve clones in those libraries. If a library is in a plasmid form or other forms that can be transformed into a model organism (i.e., bacteria), clones in the library can be identified and retrieved through TrueRepertoire™. For example, antibody libraries used in systems other than phage display or protein libraries used in the directed evolution of a protein can be processed with TrueRepertoire™. Additionally, TrueRepertoire™ can be used in microbiome research, particularly to identify and retrieve bacteria existing in the environment.
In summary, we developed a new robust platform, TrueRepertoire™, to identify and retrieve clones in high throughput in a phage display library. We showed that there is no platform-specific bias in TrueRepertoire™, and the applicability of the platform was demonstrated by finding many effective rare clones in a phage-displayed scFv library. TrueRepertoire™ provides a platform for high-throughput genotype-based screening of clones with advantages over previously developed processes. Additionally, TrueRepertoire™ is versatile. For example, it can be applied to antibody libraries other than phage display libraries, protein libraries used in the directed evolution of a protein, and microbiota native to a given environment.
Materials and methods
Fabrication of the single-cell separation & incubation chip capable of light-mediated extraction
To remove a contaminant on the surface, an ITO-deposited glass was washed thoroughly with absolute ethanol. To make a hydrophobic layer, 5% v/v solution of photo-initiator (2-hydroxy-2-methylpropiophenone, Sigma Aldrich) and PEGDA250 (polyethylene glycol diacrylate, 250, Sigma Aldrich) was spin-coated on the ITO-deposited glass (500 rpm – 5 s, 3000 rpm – 30 s, A/Del time – 5 s). After the coated hydrophobic layer was cured by ultraviolet light (365 nm, 85 mW, 10 s), residual uncured monomers of PEGDA250 were washed with absolute ethanol. To make 60 um of pillar structures, the same height of spacers was attached on both ends of the hydrophobic layer coated glass, then a photomask for the pillar structure was put on the spacers. Then, 5% v/v solution of the photoinitiator and PEGDA700 (Polyethylene glycol diacrylate, 700, Sigma Aldrich) was loaded to the gap between the photomask and the hydrophobic layer-coated glass. The pillar structures (60 um height) were patterned on the hydrophobic layer-coated glass by irradiating ultraviolet light on the photomask (365 nm, 85 mW, 5 s), then residual uncured monomers were washed with absolute ethanol. After making the spacers (180 um height) on both ends of the pillar structure patterned glass, a slide glass was put on the spacers and fixed to guide a solution loaded to the chip.
The clone isolation system of TrueRepertoire™
Transformation E. coli with an antibody library
To make transformants of an antibody library, E.coli (ER2738, Lucigen) was transformed with 1 ng of plasmid DNA for the library by electroporation according to the protocol from the manufacturer of the electroporation apparatus (MicroPulser™ Electroporator, Bio-rad).
Formation of the microcolony forming agarose droplets in SSICLE
The transformants were diluted with 1% m/m low gelling temperature agarose super optimal broth with catabolite repression (SOC) medium (2% w/v tryptone, 0.5% w/v yeast extract, 10 mM NaCl, 2.5 mM KCl, 10 mM MgCl2, 10 mM MgSO4, 20 mM glucose, and ddH2O) with antibiotics. The diluted transformants solution was loaded into SSICLE, then washed with silicone oil (Sigma Aldrich). Through this two-step manipulation, the microcolony forming agarose droplets were formed in high-throughput in SSICLE by self-assembly. After cooling the agarose droplets to fix the cells, the chip was incubated at 37 ℃ overnight in a cell incubator.
Image processing of the chip
After the incubation, images for the whole area of the chip were taken with an automated imaging system. The images were converted to gray-scale and contrast of the images was enhanced for the following image processing. Microcolonies in the agarose droplets were detected through several rounds of morphological operations, including erosion and dilation. After counting the number of microcolonies in each agarose droplet, agarose droplets with one microcolony were categorized as monoclonal agarose droplets, and the coordinates of the monoclonal agarose droplets were recorded for the following extraction step.
Extraction of the monoclonal agarose droplets
Using the coordinate information obtained in the image processing step, the monoclonal agarose droplets were extracted from the chip into 96-well filled with growth media and antibiotics by a fully automated in-house laser extraction system. The extraction system is comprised of stage controllers and a pulsed infra-red laser instrument. After the extraction, the bottom 96-well plates containing the extracted monoclonal agarose droplets were incubated overnight for E. coli in the agarose droplets to replicate sufficiently.
The clone identification system of TrueRepertoire™
Multiplex PCR with barcoded primers and NGS
After the overnight incubation, variable regions of the plasmids of E. coli in the 96-well plates were amplified by multiplex PCR with two sets of primers. The primers contain barcode regions containing plate and well information of the amplified E. coli. The multiplex PCR (95°C for 9 min 30 s, 26 cycles of 95°C for 25 s, 60°C for 25 s, and 72°C for 1 min, 72°C for 3 min) was performed with PCR reagent (10 µL of BioFACTTM 2× Taq polymerase Master Mix, 5 µL of nuclease free water, 1 µL of 10 µM forward barcoded primer, 1 µL of 10 µM reverse barcoded primer, and 1 µL of the growth media containing E. coli). After the multiplex PCR, the PCR products were pooled and purified using magnet beads. Then, the purified PCR products were prepared for NGS run (Miseq, 2300 bp, Illumina) following the protocol from the manufacturer.
Pre-processing and demultiplexing
Paired-end reads were merged using PEAR (Paired-End read merger, v0.9.6) and unmerged paired-end reads were eliminated.40 For quality control, merged reads with a low median quality PHRED score were additionally filtered out. Remaining reads were demultiplexed by the barcode region sequence. Then, the reads were tagged with the type of chains (heavy or light) and well location corresponding to the barcode sequence.
Sequence clustering
For each group of reads tagged with the same chain type and well location, the reads were clustered through the following steps. First, primer sequence was trimmed for each read, and sequence abundances (read count) were calculated based on the occurrence of the remaining immunoglobulin sequences. Sequence clusters were constructed using a greedy algorithm that sets the most abundant sequence as the representative sequence for each cluster and assigns new sequences to the closest cluster based on edit distance if the distance is smaller than a threshold and otherwise allocates a new cluster.
Well filtering
After the sequence clustering, some of the well data were discarded if either the VH or VL group was under one of the following conditions: 1) the relative abundance of the most abundant cluster is less than a threshold value, or 2) the read counts of the most abundant cluster of a group is less than a threshold value that is computed by a statistical analysis. (Supplementary Figure 3 and Supplementary Figure 4)
Extracting consensus sequence
Finally, a consensus sequence was constructed for each remaining group via the following steps. First, sequences in the most abundant cluster were aligned using Clustal Omega.41 Next, the most abundant nucleotide for each base position was calculated and assembled together to make a consensus sequence. Gaps were ignored after the construction.
Every bioinformatics process in this section not designating a software name in the text was processed using an in-house software of MSSIC platform.
Immunization of chicken for generation of anti-hHGF antibody
Six white leghorn chickens were immunized and boosted twice with 5 µg of hHGF (100-39H, Peprotech, Rocky Hill, NJ, USA). Blood samples were collected from the wing veins prior to immunization and during the immunization for ELISA to evaluate the immunization status.
Construction of combinatorial phage-displayed scFv library and biopanning
The chickens were sacrificed a week after the second boost. Bone marrow was harvested for total RNA isolation. Using the RNA, cDNA was synthesized to generate a phage-displayed scFv library as described previously.42 After the library construction, five rounds of bio-panning were performed against hHGF (PGA104, PanGen Biotech Inc., Suwon, Republic of Korea) conjugated magnetic beads as described previously.43
Phage ELISA
Clones obtained through TrueRepertoire™ were rescued by infection of helper phage and subjected to phage ELISA using hHGF coated microtiter plates (3690, Corning, NY, USA) to select binders as described previously.11 Briefly, the microtiter plates were coated overnight at 4℃ with 100 ng of hHGF (100-39H, Peprotech, Rocky Hill, NJ, USA) in coating buffer (0.1 M sodium bicarbonate, pH 8.6). The wells were blocked with 150 µL of 3% (w/v) bovine serum albumin (BSA) in phosphate-buffered saline (PBS) for 1 h at 37℃. The plates were then sequentially incubated with scFv-displaying phages in the culture supernatant and horseradish peroxidase-conjugated mouse anti-M13 monoclonal antibody (27–9421-01, GE Healthcare, Pittsburg, PA, USA) with intermittent washing using 0.05% (v/v) Tween 20 in PBS (PBST). Finally, the plates were washed again with 0.05% PBST followed by detection using 2,2ʹ-azino-bis-3-ethylbenzothiazoline-6-sulfonic acid solution (Pierce, Rockford, IL, USA). Absorbance was measured at 405 nm with a Multiscan Ascent microplate reader (Labsystems, Helsinki, Finland). As a control, binding activity to BSA was also tested according to the protocol described above. The expression of intact scFv molecules was confirmed by anti-human influenza hemagglutinin (clone 3F10, #11867423001, Roche, Indianapolis, IN, USA). Two replicate experiments were conducted for all the experimental conditions.
Cost simulation
The hHGF TrueRepertoireTM result was used for the construction of statistical population for the cost simulation (Supplementary File. 2). As the results of binding activity test of all of the clones were absent, we randomly assigned the positivity to the clones (See “POSITIVITY_ASSUMED” column in the file.). In in vitro screening and TrueRepertoireTM, clones were sampled randomly with replacement. In NGS-based method, unlike the in vitro screening and TrueRepertoireTM, there is no way to perfectly reconstruct VH-VL pairing, thus we restricted the simulation to VH sequences. In in vitro screening and TrueRepertoireTM, clones were randomly sampled with replacement. In each sampling point, sampling was conducted for 1,000 times, then the median cost value of the results was selected and displayed. In NGS-based method, clones were selected in order of the clonal frequency of the clones. Formulas used for the cost estimation and the cost for experiment instruments were listed in a separate datasheet (Supplementary File. 3).
Funding Statement
This research was supported by Global Research Development Center Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT(MSIT) (2015K1A4A3047345); the Brain Korea 21 Plus Project in 2018; grant of the Korea Health Technology R&D Project of the Korea Health Industry Development Institute (KHIDI) funded by the Ministry of Health & Welfare, Republic of Korea (HI18C2282); the Bio & Medical Technology Development Program of the National Research Foundation (NRF) funded by the Korean government (MSIT) (2018M3A9D7079488). This work was supported by the Brain Korea 21 Plus Project in 2018.
Abbreviations
- HCDR3
heavy-chain complementarity-determining region 3
- hHGF
human hepatocyte growth factor
- ITO
indium tin oxide
- NGS
next-generation sequencing
- scFv
single-chain variable fragment
- SSICLE
separation and incubation chip capable of light-mediated extraction
- UMI
unique molecular identifier
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed
Supplemental material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Bradbury ARM, Sidhu S, Dübel S, McCafferty J.. Beyond natural antibodies: the power of in vitro display technologies. Nat Biotechnol. 2011;29(3):245–54. doi: 10.1038/nbt.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee SY, Choi JH, Xu Z.. Microbial cell-surface display. Trends Biotechnol. 2003;21(1):45–52. doi: 10.1016/S0167-7799(02)00006-9. [DOI] [PubMed] [Google Scholar]
- 3.Hammers CM, Stanley JR.. Antibody phage display: technique and applications. J Invest Dermatol. 2014;134(2):e17. doi: 10.1038/jid.2013.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hust M, Dübel S. Mating antibody phage display with proteomics. Trends Biotechnol. 2004;22(1):8–14. doi: 10.1016/j.tibtech.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 5.Lorenz HM. Technology evaluation: adalimumab, Abbott laboratories. Curr Opin Mol Ther. 2002;4:185–90. [PubMed] [Google Scholar]
- 6.Nixon AE, Sexton DJ, Ladner RC. Drugs derived from phage display: from candidate identification to clinical practice. MAbs. 2014;6(1):73–85. doi: 10.4161/mabs.27240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peng H-P, Lee KH, Jian J-W, Yang A-S. Origins of specificity and affinity in antibody–protein interactions. Proc Natl Acad Sci USA. 2014;111(26):E2656–2665. doi: 10.1073/pnas.1401131111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu JL, Davis MM. Diversity in the CDR3 region of VH is sufficient for most antibody specificities. Immunity. 2000;13(1):37–45. doi: 10.1016/S1074-7613(00)00006-6. [DOI] [PubMed] [Google Scholar]
- 9.Derda R, Tang SKY, Li SC, Ng S, Matochko W, Jafari MR. Diversity of phage-displayed libraries of peptides during panning and amplification. Molecules. 2011;16(2):1776–803. doi: 10.3390/molecules16021776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang W, Yoon A, Lee S, Kim S, Han J, Chung J. Next-generation sequencing enables the discovery of more diverse positive clones from a phage-displayed antibody library. Exp Mol Med. 2017;49(3):e308. doi: 10.1038/emm.2017.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barbas CF III, Burton DR, Scott JK, Silverman GJ. Phage display: A laboratory manual. New York (NY): CSHL Press; 2001. [Google Scholar]
- 12.Ravn U, Gueneau F, Baerlocher L, Osteras M, Desmurs M, Malinge P, Magistrelli G, Farinelli L, Kosco-Vilbois MH, Fischer N. By-passing in vitro screening-next generation sequencing technologies applied to antibody display and in silico candidate selection. Nucleic Acids Res. 2010;38(21):e193. doi: 10.1093/nar/gkq789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ravn U, Didelot G, Venet S, Ng KT, Gueneau F, Rousseau F, Calloud S, Kosco-Vilbois M, Fischer N. Deep sequencing of phage display libraries to support antibody discovery. Methods. 2013;60(1):99–110. doi: 10.1016/j.ymeth.2013.03.001. [DOI] [PubMed] [Google Scholar]
- 14.Dias-Neto E, Nunes DN, Giordano RJ, Sun J, Botz GH, Yang K, Setubal JC, Pasqualini R, Arap W. Next-generation phage display: integrating and comparing available molecular tools to enable cost-effective high-throughput analysis. PLoS One. 2009;4(12):e8338. doi: 10.1371/journal.pone.0008338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rouet R, Jackson KJL, Langley DB, Christ D. Next-generation sequencing of antibody display repertoires. Front Immunol. 2018;9:118. doi: 10.3389/fimmu.2018.00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aird D, Ross MG, Chen WS, Danielsson M, Fennell T, Russ C, Jaffe DB, Nusbaum C, Gnirke A. Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol. 2011;12(2):R18. doi: 10.1186/gb-2011-12-2-r18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baum PD, Venturi V, Price DA. Wrestling with the repertoire: the promise and perils of next generation sequencing for antigen receptors. Eur J Immunol. 2012;42(11):2834–39. doi: 10.1002/eji.201242999. [DOI] [PubMed] [Google Scholar]
- 18.Wilson PC, Andrews SF. Tools to therapeutically harness the human antibody response. Nat Rev Immunol. 2012;12(10):709–19. doi: 10.1038/nri3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reddy ST, Ge X, Miklos AE, Hughes RA, Kang SH, Hoi KH, Chrysostomou C, Hunicke-Smith SP, Iverson BL, Tucker PW, et al. Monoclonal antibodies isolated without screening by analyzing the variable-gene repertoire of plasma cells. Nat Biotechnol. 2010;28(9):965–69. doi: 10.1038/nbt.1673. [DOI] [PubMed] [Google Scholar]
- 20.Chovanec P, Bolland DJ, Matheson LS, Wood AL, Krueger F, Andrews S, Corcoran AE. Unbiased quantification of immunoglobulin diversity at the DNA level with VDJ-seq. Nat Protoc. 2018;13(6):1232–52. doi: 10.1038/nprot.2018.021. [DOI] [PubMed] [Google Scholar]
- 21.Shugay M, Britanova OV, Merzlyak EM, Turchaninova MA, Mamedov IZ, Tuganbaev TR, Bolotin DA, Staroverov DB, Putintseva EV, Plevova K, et al. Towards error-free profiling of immune repertoires. Nat Methods. 2014;11(6):653–55. doi: 10.1038/nmeth.2960. [DOI] [PubMed] [Google Scholar]
- 22.DeKosky BJ, Kojima T, Rodin A, Charab W, Ippolito GC, Ellington AD, Georgiou G. In-depth determination and analysis of the human paired heavy- and light-chain antibody repertoire. Nat Med. 2015;21(1):86–91. doi: 10.1038/nm.3743. [DOI] [PubMed] [Google Scholar]
- 23.Gierahn TM, Wadsworth MH, Hughes TK, Bryson BD, Butler A, Satija R, Fortune S, Christopher Love J, Shalek AK. Seq-well: portable, low-cost RNA sequencing of single cells at high throughput. Nat Methods. 2017;14(4):395–98. doi: 10.1038/nmeth.4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McAvoy K, Jones D, Thakur RRS. Synthesis and characterisation of photocrosslinked poly(ethylene glycol) diacrylate implants for sustained ocular drug delivery. Pharm Res. 2018;35(2):36. doi: 10.1007/s11095-017-2298-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kang M, Park W, Na S, Paik S-M, Lee H, Park JW, Kim H-Y, Jeon NL. Capillarity guided patterning of microliquids. Small. 2015;11(23):2789–97. doi: 10.1002/smll.201403596. [DOI] [PubMed] [Google Scholar]
- 26.Kivioja T, Vähärautio A, Karlsson K, Bonke M, Enge M, Linnarsson S, Taipale J. Counting absolute numbers of molecules using unique molecular identifiers. Nat Methods. 2012;9(1):72–74. doi: 10.1038/nmeth.1778. [DOI] [PubMed] [Google Scholar]
- 27.Scott DW. Multivariate density estimation: theory, practice, and visualization. New York (NY): Wiley; 1992. [Google Scholar]
- 28.Chen Y, Kim SH, Shang Y, Guillory J, Stinson J, Zhang Q, Hötzel I, Hoi KH. Barcoded sequencing workflow for high throughput digitization of hybridoma antibody variable domain sequences. J Immunol Methods. 2018;455:88–94. doi: 10.1016/j.jim.2018.01.004. [DOI] [PubMed] [Google Scholar]
- 29.Lee CMY, Iorno N, Sierro F, Christ D. Selection of human antibody fragments by phage display. Nat Protoc. 2007;2(11):3001–08. doi: 10.1038/nprot.2007.448. [DOI] [PubMed] [Google Scholar]
- 30.Hanahan D. Studies on transformation of Escherichia coli with plasmids. J Mol Biol. 1983;166(4):557–80. doi: 10.1016/S0022-2836(83)80284-8. [DOI] [PubMed] [Google Scholar]
- 31.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al. ClustalW and ClustalX version 2. Bioinformatics. 2007;23(21):2947–48. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 32.Goujon M, McWilliam H, Li W, Valentin F, Squizzato S, Paern J, Lopez R. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res. 2010;38(Web Server issue):W695–9. doi: 10.1093/nar/gkq313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Umitsu M, Sakai K, Ogasawara S, Kaneko MK, Asaki R, Tamura-Kawakami K, Kato Y, Matsumoto K, Takagi J. Probing conformational and functional states of human hepatocyte growth factor by a panel of monoclonal antibodies. Sci Rep. 2016;6:33149. doi: 10.1038/srep33149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Avery LB, Wade J, Wang M, Tam A, King A, Piche-Nicholas N, Kavosi MS, Penn S, Cirelli D, Kurz JC, et al. Establishing in vitro in vivo correlations to screen monoclonal antibodies for physicochemical properties related to favorable human pharmacokinetics. MAbs. 2018;10(2):244–55. doi: 10.1080/19420862.2017.1417718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jain T, Sun T, Durand S, Hall A, Houston NR, Nett JH, Sharkey B, Bobrowicz B, Caffry I, Yu Y, et al. Biophysical properties of the clinical-stage antibody landscape. Proc Natl Acad Sci USA. 2017;114(5):944–49. doi: 10.1073/pnas.1616408114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sormanni P, Amery L, Ekizoglou S, Vendruscolo M, Popovic B. Rapid and accurate in silico solubility screening of a monoclonal antibody library. Sci Rep. 2017;7(1):8200. doi: 10.1038/s41598-017-07800-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Datta-Mannan A, Thangaraju A, Leung D, Tang Y, Witcher DR, Lu J, Wroblewski VJ. Balancing charge in the complementarity-determining regions of humanized mAbs without affecting pl reduces non-specific binding and improves the pharmacokinetics. MAbs. 2015;7(3):483–93. doi: 10.1080/19420862.2015.1016696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schoch A, Kettenberger H, Mundigl O, Winter G, Engert J, Heinrich J, Emrich T. Charge-mediated influence of the antibody variable domain on FcRn-dependent pharmacokinetics. Proc Natl Acad Sci USA. 2015;112(19):5997–6002. doi: 10.1073/pnas.1408766112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li B, Tesar D, Boswell CA, Cahaya HS, Wong A, Zhang J, Meng YG, Eigenbrot C, Pantua H, Diao J, et al. Framework selection can influence pharmacokinetics of a humanized therapeutic antibody through differences in molecule charge. MAbs. 2014;6(5):1255–64. doi: 10.4161/mabs.29809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang J, Kobert K, Flouri T, Stamatakis A. PEAR: A fast and accurate illumina paired-end reAd mergeR. Bioinformatics. 2014;30(5):614–20. doi: 10.1093/bioinformatics/btt593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using clustal omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee M-S, Lee JC, Choi CY, Chung J. Production and characterization of monoclonal antibody to botulinum neurotoxin type B light chain by phage display. Hybridoma. 2008;27(1):18–24. doi: 10.1089/hyb.2007.0532. [DOI] [PubMed] [Google Scholar]
- 43.Lee Y, Kim H, Chung J. An antibody reactive to the Gly63-Lys68 epitope of NT-proBNP exhibits O-glycosylation-independent binding. Exp Mol Med. 2014;46:e114. doi: 10.1038/emm.2014.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.