

ABSTRACT
Targeting tau with immunotherapies is currently the most common approach taken in clinical trials of patients with Alzheimer’s disease. The most prominent pathological feature of tau is its hyperphosphorylation, which may cause the protein to aggregate into toxic assemblies that collectively lead to neurodegeneration. Of the phospho-epitopes, the region around Ser396/Ser404 has received particular attention for therapeutic targeting because of its prominence and stability in diseased tissue. Herein, we present the antigen-binding fragment (Fab)/epitope complex structures of three different monoclonal antibodies (mAbs) that target the pSer404 tau epitope region. Most notably, these structures reveal an antigen conformation similar to a previously described pathogenic tau epitope, pSer422, which was shown to have a β-strand structure that may be linked to the seeding core in tau oligomers. In addition, we have previously reported on the similarly ordered conformation observed in a pSer396 epitope, which is in tandem with pSer404. Our data are the first Fab structures of mAbs bound to this epitope region of the tau protein and support the existence of proteopathic tau conformations stabilized by specific phosphorylation events that are viable targets for immune modulation.
KEYWORDS: Monoclonal antibody, Alzheimer’s disease, tau protein, antibody-antigen complex, phospho-epitope
Introduction
Tau is a microtubule-associated protein required for cytoskeletal stability of neuronal axons throughout normal development. However, clinicopathological studies show a strong correlation between levels of modified tau protein and cognitive impairment.1,2 Although natively soluble, tau may undergo pathological modifications that cause it to aggregate into insoluble inclusions, which are the hallmark of neurodegenerative diseases collectively called tauopathies. In these diseases, tau becomes hyperphosphorylated (pTau), which may initiate its aggregation into soluble oligomers that eventually assemble into insoluble neurofibrillary tangles. Alzheimer’s disease (AD) is the most common tauopathy, but numerous less common diseases are characterized by these brain lesions, including progressive supranuclear palsy, corticobasal degeneration, Pick’s disease, and genetic variants linked to tau mutations such as familial frontotemporal dementia with Parkinsonism.3 Hence, pTau is a promising therapeutic target in all of the tauopathies. Currently, it is primarily being targeted by immunotherapy, with at least two active and seven passive tau immunotherapies in clinical trials as of the end of 2018.4,5
Tau is an intrinsically disordered protein with minimal stable secondary structures in its normal functional state.6 In addition, it coordinates microtubule attachment through frequent dynamic phosphorylation and dephosphorylation on multiple sites that also influence its conformation. Therefore, tau has many possible phosphorylation states and conformations, which makes identifying and targeting the appropriate aberrant protein a major hurdle for therapeutic discovery. Collectively, tau oligomers, larger aggregates, filaments and fibrils are thought to trigger microtubule disassembly, axon degeneration and dendritic spinal collapse7–9. It has been shown previously that hyperphosphorylation at amino acids Ser396 and Ser404 located in the C-terminal domain (numbered according to the 441 residue 2N4R tau isoform10) is a promising target for tau immunotherapy and related imaging diagnostics (Figure 1(a)).11–28 We recently described a Fab/epitope complex structure of a monoclonal antibody (mAb) specific for phosphorylated Ser396 (pSer396), and we present here antigen-binding fragment (Fab)/peptide crystal structures of three recently developed mAbs, 8B2, 6B2, and h4E6, targeting the Ser404 epitope region.11,12 Comparative structural analysis of our novel Ser404 mAbs, along with previously described pTau recognition, have revealed a common antigenic conformation that we believe is vital to the investigation of tau pathogenicity and development of antibody-based therapies to halt its progression or related diagnostic imaging probes.
Figure 1.
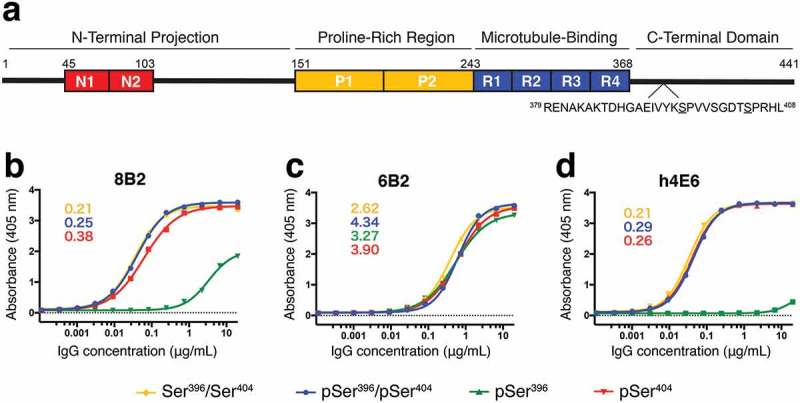
Antibody affinity to tau peptide measured by ELISA. (a) Schematic of tau 2N4R isoform labeled by known functional domains, including the two N-terminal acidic inserts (red, residues 45–103), two proline-rich regions (yellow, residues 151–243), microtubule-binding domains (blue, residues 244–368), and the C-terminal domain. The sequence of the region (residues 379–408) of interest is shown below with Ser396 and Ser404 underlined. (b) – (d) Binding curves from ELISA measurements for mAbs 8B2 (b), 6B2 (c), and h4E6 (d) comparing recognition of four differentially phosphorylated peptides (Table 1). Peptides pSer396/pSer404-tau (blue), pSer404-tau (red), and non-phosphorylated Ser396/Ser404-tau (yellow) were bound by all three mAbs while pSer396-tau (green) was only bound by 6B2 and, with a much lower affinity, by 8B2. Number insets are estimated Kd values (nM) from the best binding curves.
Results
MAb selectivity to Ser404-tau
To evaluate the selectivity of the panel of three mAbs, 8B2, 6B2, and h4E6, we used two types of enzyme-linked immunosorbent assays (ELISAs): 1) a standard solid phase assay in which binding of the antibodies to peptides coated onto the plate is examined (Figure 1), and 2) a competition ELISA in which solution phase peptide competes with binding of the antibody to the peptide coated on the plate (Figure 2).12 The competition ELISA clarifies how the antibodies recognize the peptides in their solid phase vs. solution, which can differ substantially. The four peptides used in the assay encompass the same tau sequence, but feature different combinatorial phosphorylation arrangements of Ser396 and Ser404 (Table 1) that may facilitate the identification of the epitope of each mAb.
Figure 2.
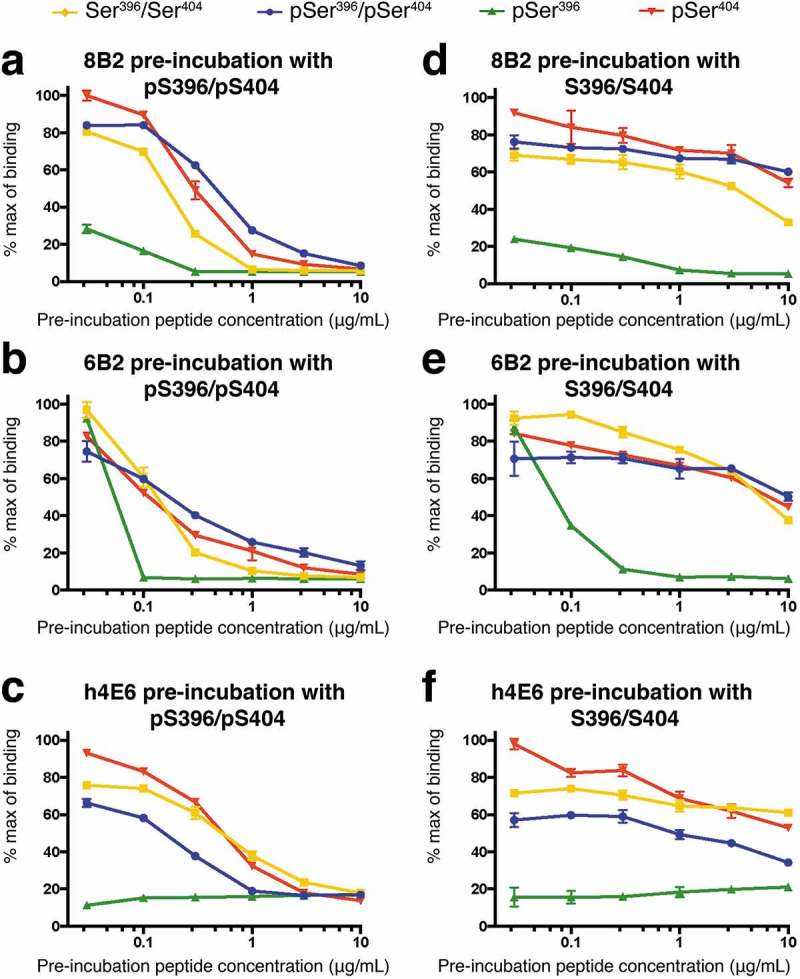
Phospho-selectivity confirmed by competition ELISA. Each mAb was pre-incubated with either the pSer396/pSer404 (a-c) or non-phosphorylated Ser396/Ser404 (d-f) peptide. The plate-bound peptide then competed for binding to the antibody in the pre-incubated solution. Note that distinct characteristics of the three mAbs, consistent with ELISA data in Figure 1, confirms their phospho-selectivity (see text for detailed discussions). Data are represented as mean values. See also Figure S1.
Table 1.
Peptides used for ELISA and crystallization experiments.
| ELISA and Crystallization | |
|---|---|
| pS396/pS404 | 386TDHGAEIVYKSPVVSGDTSPRHL408 |
| pS396 | 386TDHGAEIVYKSPVVSGDTSPRHL408 |
| pS404 | 386TDHGAEIVYKSPVVSGDTSPRHL408 |
| S396/S404 | 379RENAKAKTDHGAEIVYKSPVVSGDTSPRHL408 |
Binding curves from the solid phase assay were generated from ELISA data by maintaining plate-bound peptide concentrations and serially diluting IgG concentrations. This assay revealed that all three mAbs bind the non-phosphorylated Ser396/Ser404 peptide, the mono phosphorylated pSer404 peptide, and the di-phosphorylated pSer396/pSer404 peptide (Figure 1(b-d)). Interestingly, only 6B2 binds to mono-phosphorylated pSer396 peptide with similar affinity as to the other three peptides, while h4E6 does not recognize the pSer396 peptide, and 8B2 recognizes it only at high concentrations. In addition, the affinities of 6B2 to the three peptides that all the antibodies recognize well is ~10x lower than that of 8B2 and h4E6.
In the competition ELISA, we pre-incubated either the di-phosphorylated pSer396/pSer404 peptide or the non-phosphorylated Ser396/Ser404 peptide with each individual antibody, and then the pre-incubated complex was used to compete with the plate-bound peptides. Competition results from pre-incubation of the mAbs with the pSer396/pSer404 peptide (Figure 2(a-c)) were consistent with their binding profiles to the 4 peptides (Figure 1), i.e., the pSer396/pSer404 peptide in solution could compete with the three peptides that the mAbs bind well: the pSer396/pSer404 itself, the pSer404 peptide, and the Ser396/Ser404 peptide. In the case of 6B2, the required concentration of competition for the pSer396/pSer404 peptide was lower than that for mAbs 8B2 and h4E6 because 6B2 has lower affinity to those peptides (Figure 1(b)). The pSer396 peptide had little or no binding to 8B2 and h4E6, and the competition curves were very flat (Figure 2(a,c), respectively). Again, in the case of 6B2, the required competition concentration of the pSer396/pSer404 peptide for the pSer396 peptide was even lower than that for the other three peptides (Figure 2(b)).
Competition results from pre-incubation of the mAbs with the non-phosphorylated peptide (Figure 2(d-f)) were very different from that of the pSer396/pSer404 peptide. The non-phosphorylated peptide has little competition with the three peptides that bind the mAbs well (pSer396/pSer404 peptide, pSer404 peptide, as well as the non-phosphorylated peptide itself), but it could compete with the pSer396 peptide (Figure 2(e)). Competition ELISA of the original hybridoma-produced 4E6 recaptured the results of h4E6 (Figure S1). Taken together with the solid phase ELISA data in Figure 1, we concluded that phosphorylation at residue Ser396 compromises completely the recognition of h4E6 and partially of 8B2 unless the peptide is phosphorylated at residue Ser404. In order to elaborate on the ELISA findings, and to further characterize binding of the mAbs to their epitope, we used x-ray crystallography to resolve the atomic co-crystal structures of Fabs in complex with tau peptides.
Details of the 8B2 antibody-antigen interaction
We obtained two crystal structures of Fab 8B2: 1) an apo structure resolved to 1.9 Å resolution, and 2) a non-phosphorylated peptide-bound structure resolved to 1.8 Å resolution (Figure 3, S2, and S3). To crystallize the Fab/peptide complex, we screened all four peptides in Table 1, but only Fab 8B2 in complex with the non-phosphorylated peptide (379RENAKAKTDHGAEIVYKSPVVSGDTSPRHL408) was crystallized. We observed five C-terminal peptide residues (404SPRHL408) in the electron density (Figure 3), and interestingly, we found a tetrahedral density next to the side chain of Ser404 (Figure S2). Although there is no direct evidence that this is the density for a phosphate molecule, a phosphate molecule fit the density well, suggesting that the antigen-binding site can accommodate the side chain of pSer404, consistent with the fact that 8B2 can bind both phosphorylated and non-phosphorylated peptides (Figures 1 and 2). There is a binding pocket ~10 Å deep at the center of the 8B2 antigen-binding site that buries the C-terminal half of the epitope peptide (see below). The peptide has a straight linear conformation with a small bend between residues Arg406 and His407. Significant contacts between 8B2 and its epitope include a salt bridge, aromatic stacking, hydrophobic interactions, and a network of hydrogen bonds in addition to several contacts that coordinate the phosphate molecule (Figure 3(b,c)).
Figure 3.
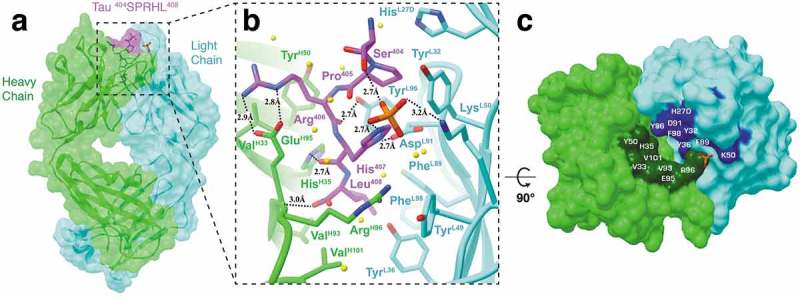
Fab structure of 8B2 in complex with a tau peptide. (a) Surface representation of the Fab 8B2/peptide complex with underlying ribbon display. Heavy and light chains are colored light green and cyan, respectively. The tau peptide is shown in magenta with the epitope sequence. Although the peptide used in crystallization is 23 residues in length, only five residues (404SPRHL408) are visible in the electron density map. (b) A front view ribbon representation of Fab 8B2 in complex with the non-phosphorylated tau peptide. Key residues involved in antigen-binding and a phosphate observed in the binding site next to the side chain of Ser404 are shown as sticks. Hydrogen bonds are represented by dashed black lines and labeled with bond distances. Water molecules are represented by yellow spheres. (c) Top-down view of the paratope. Surface areas of the contact residues from the heavy chain are shown in dark green and that from the light chain in blue. The phosphate observed at the binding site is shown as a reference. See also Figure S3.
At the bottom of the binding pocket, the Leu408 side chain is buried in a hydrophobic cove formed by residues PheL89 and PheL98 from light chain, and residues ValH93 and VH101 from the heavy chain (Figure 3(b)). The two peptide residues above Leu408, His407 and Arg406, are surrounded by side chains of several polar and charged residues and a water-mediated hydrogen bond network with the side chain of Arg406, which also forms a salt-bridge with the side chain of GluH95. The side chain of the next peptide residue, Pro405, is packed against the side chains of residues TyrL32 and HisL27D. At the very top of the epitope peptide, the OH group of Ser404 does not form any direct hydrogen bonds with the antibody itself, but does form a hydrogen bond with one of the oxygen atoms from the bound phosphate. The phosphate molecule is in turn coordinated by the side chains of LysL50, His407, a water molecule, and a glycerol molecule (not shown). Additional hydrogen bonds are coordinated by antibody side chain to peptide main chain interactions, including TyrL96 to the carboxyl group from Arg406, HisH35 to the carboxyl group from His407, ArgH96 main chain to the carboxyl group from Leu408, and the side chains of AspL91 and His407.
The apo Fab structure of mAb 8B2 shows unremarkable differences compared to the holo structure (Figure S3). However, comparison of the complementarity-determining region (CDR) loops reveal residues ArgH96 to TyrH99 in CDR-H3 in the peptide-bound structure may be positioned for the side chain of ArgH96 to contact the phosphate molecule and for several residues (AspH97, AsnH98, TyrH99) to face outwards from the binding interface. A second structural deviation comes from GluL34 in CDR-L1, whose side chain appears to rotate inwards towards the peptide.
Details of the 6B2 antibody-antigen interaction
The structure of Fab 6B2 in complex with the non-phosphorylated tau peptide (379RENAKAKTDHGAEIVYKSPVVSGDTSPRHL408) was resolved to 2.6 Å resolution (Figure 4). Like that for Fab 8B2, we screened all four peptides in Table 1, but only Fab 6B2 in complex with the non-phosphorylated peptide was crystallized. In the electron density, we observed six C-terminal peptide residues (403TSPRHL408) and, similar to that of Fab 8B2, a density near the side chain of Ser404 that could fit a phosphate molecule. This suggests that the antigen-binding site can accommodate the side chain of pSer404, consistent with the fact that 6B2 can bind both phosphorylated and non-phosphorylated peptides (Figures 1 and 2). Unlike the deep binding pocket of 8B2, the antigen-binding site of 6B2 is more of a shallow crevice between the heavy and light chains (Figure 4). Interestingly, the C-terminus of the epitope peptide is curved, making a half helical turn, deviating from the linear conformation seen in the peptide complexed with Fab 8B2. Significant contacts between 6B2 and its epitope include aromatic stacking, hydrophobic interactions, and a network of hydrogen bonds, in addition to several contacts that coordinate the phosphate molecule (Figure 4(b,c)).
Figure 4.
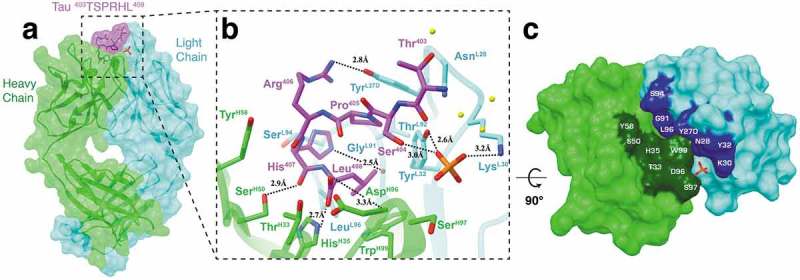
Fab structure of 6B2 in complex with a tau peptide. (a) Surface representation of the Fab 6B2/peptide complex with underlying ribbon display. Six residues (403TSPRHL408) are visible in the electron density map. (b) A detailed front view of Fab 6B2 in complex with a non-phosphorylated tau peptide. Again, a phosphate is observed next to the side chain of Ser404 at the antigen binding site. (c) Top-down view of the paratope surface.
The C-terminal residue Leu408 rests just on the side of CDR-H3 and is coordinated by hydrogen bonds between its carboxyl group and the side chain of HisH35 and the main chain of AspH96. In addition, residues LeuL96 and TrpH99 provide hydrophobic contacts that stabilize Leu408 within the antigen-binding crevice. His407 is located at the pitch of the half of a helical turn of the epitope, which allows strong interactions with both the heavy and light chains, including hydrogen bonds with the side chain of SerH50 and main chain of GlyL91 and contacts with the side chains of ThrH33 and SerL94. Arg406 is also located near the pitch of half of a helical turn of the epitope, but its side chain protrudes from the crevice and makes a hydrogen bond with the OH group of TyrL27D. Completing the half helical turn, Pro405 contacts ThrL92 while the side chain of Ser404 makes a hydrogen bond with the phosphate. At the very top of the epitope peptide, Thr403 makes a contact with the light chain residue AsnL28. In addition to the four aromatic residues previously mentioned (TyrL27D, TyrL32, HisH35, TrpH99), heavy chain residue TyrH58 contributes to the network of aromatic stacking. Finally, in addition to Ser404, the phosphate contacts SerH97 and Thr403 and forms hydrogen bonds with light chain residues LysL30 and TyrL32.
Details of the h4E6 antibody-antigen interaction and the similarity of its pSer404 epitope to the conformational tau pSer422 epitope
We obtained the crystal structure of Fab h4E6 in complex with a pSer404-tau peptide at a resolution of 3.0 Å (Figure 5(a-c)). In the electron density, we observed six C-terminal residues 403TpSPRHL408, including phosphorylated pSer404. Notably, the conformation of the peptide is similar to that bound to 8B2, but with a bigger backbone bend. We also detected an orphan phosphate between the peptide and the light chain. Similar to the other two structures, the C-terminus of the peptide is buried in the pocket at the center of the antigen-binding site. However, differing from 8B2 and 6B2, the pSer404 side chain is positioned towards the heavy chain, which results in an epitope peptide conformation with a slight helical twist in the opposite direction.
Figure 5.
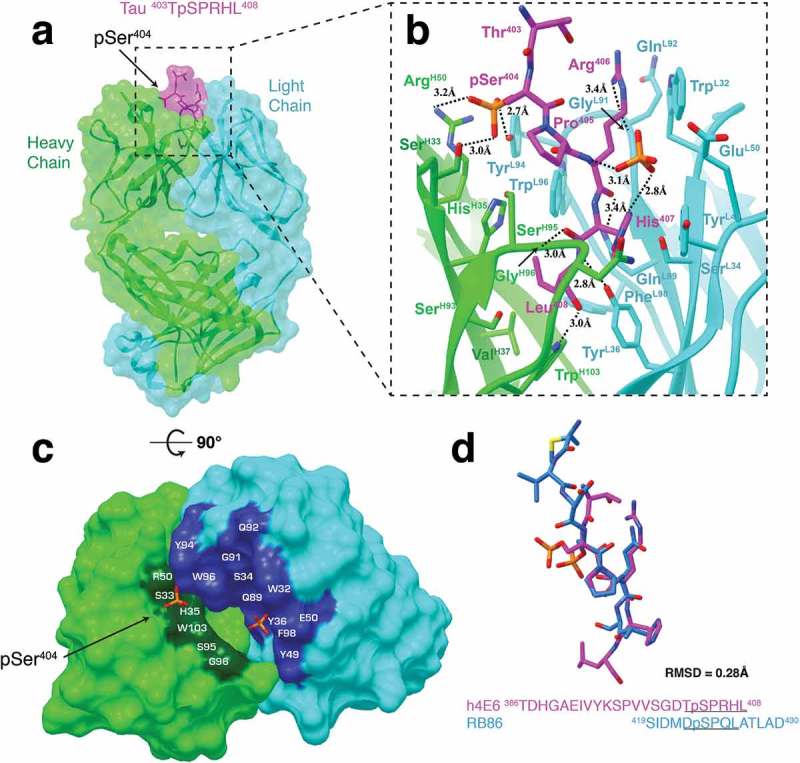
Fab structure of h4E6 in complex with pSer404-tau peptide. (a) Surface representation of Fab h4E6 in complex with a pSer404 tau peptide with underlying ribbon display. Six residues (403TpSPRHL408) are visible in the electron density map. (b) A detailed front view of the Fab h4E6 in complex. (c) Top-down view of the paratope surface. (d) Comparison of pSer404-tau peptide from our h4E6 complex (magenta) with a pSer422-tau peptide from the RB86 Fab complex structure (light blue, PDB: 5DMG). RMSD calculated using backbone atom pairs (403TpSPR406 and 421DpSPQ424). Note the similarity of the h4E6 backbone conformation around the phospho-serine residue with the RB86 epitope region, which has been described as another pathological phosphorylation site.
The h4E6-peptide complex exhibits the deepest antigen-binding pocket with the peptide residue Leu408 forming hydrogen bonds with PheL98 and TrpH103 and contacting residues HisH35, ValH37, SerH93, and TyrL36. Also making a significant number of contacts, peptide residue His407 forms a hydrogen bond between its carboxyl group and GlyH96, a hydrogen bond between its side chain and the phosphate, contacts residue SerL94, and stacks against the aromatic residue TyrL49. Arg406 is solely coordinated by the light chain and forms hydrogen bonds between its carboxyl group and residue GlnL89 and between its side chain and residue GlyL91. Arg406 also contacts residues GlnL92 and TrpL32. Pro405 provides a small kink in the peptide chain, which allows it to contact SerH95 in the heavy chain and TrpL96 in the light chain. Although Thr403 does not contact the antigen-binding surface, pSer404 is coordinated by hydrogen bonds between its phosphate and residues TyrL94, SerH33, and ArgH50. Notably, the frequency of SerH33 at residue position H33 (Kabat numbering) is <1% (analyzed by abYsis29). This position usually harbors a bulky aromatic residue, meaning this hypermutation likely facilitates the hydrogen bond in phospho-serine binding. Finally, the orphan phosphate is coordinated by hydrogen bonds to His407 and the amine group of Arg406, as well as contacts with Pro405, TyrL49 and GluL50.
In addition to pSer396 and pSer404, pSer422 is a known site of hyperphosphorylation30, and also an AD therapeutic target of interest (see Discussion). Interestingly, while Ser422 is not phosphorylated on tau in physiological conditions, like Ser396/Ser404 to some extent,31 it has been observed in a phosphorylated state in tauopathies and related animal models.32–35 Recently, the crystal Fab structure of mAb RB86 in complex with a pSer422-tau peptide (419SIDMVDpSPQLATLAD430) was elucidated to a resolution of 2.5 Å (PDB: 5DMG).36 The structure is composed of three Fab-peptide complexes in the asymmetric unit, with each of the peptides resolved to a different length around the pSer422 phospho-epitope. Nonetheless, all three peptides presented with high structural similarity to the pSer404 peptide from h4E6 (backbone RMSD = 0.28 Å) (Figure 5(d)). Most intriguingly, pSer422-tau has been described as having a β-sheet structure linked to properties of the seeding core in tau oligomers.37 Therefore, although the pSer404 and pSer422 epitopes differ in primary sequence, they share a similar secondary structure that may trigger pathogenic aggregation across numerous tauopathies.
Germline gene usage and overall features of the antigen-binding sites
It is important to identify antibody germline genes when analyzing the extent of diversity amongst a panel of antibodies. This is because parental antibodies may reveal conserved residue ‘hotspots’, which are important for subsequent humanization and optimization. mAbs 8B2 (IGHV1-14*01, IGKV1-117*01) and 6B2 (IGHV1-18*01, IGKV1-133*01) share similar murine V gene families for both heavy (IGHV1) and light (IGKV1) chains. Accordingly, both mAbs have 5 additional residues in CDR-L1 compared to h4E6 (IGHV14-3*02, IGKV15-103*01) (Figure 6(a)). However, despite differences in the primary sequence, all three mAbs share conserved residues that are important for antigen recognition. For example, H35 (Kabat numbering) in CDR-H1 is occupied by a histidine, which is used as an epitope contact in all three structures.
Figure 6.
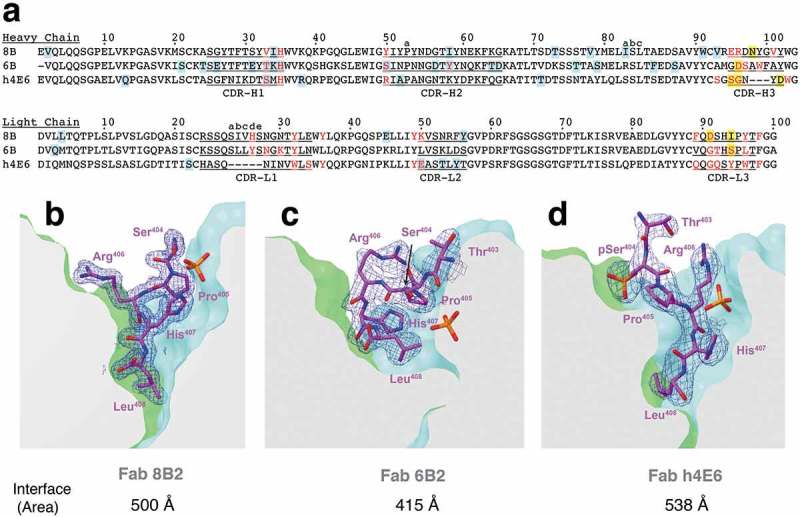
Sequence alignment and comparison of antigen-binding pockets. (a) Sequence alignment of the heavy and light chains from all three mAbs. CDR regions are underlined and labeled. Residues involved in antigen-binding are colored red. Blue highlighted residues signify deviations from germline V gene sequence and yellow highlighted residues signify deviations from VJ junction sequences (IMGT.org). (b)-(d) Antigen binding pockets of Fabs 8B2 (b), 6B2 (c), and h4E6 (d) were clipped in order to display the depth of antigen-binding. Peptides are shown as sticks in magenta with 2Fo-Fc map at contour level of 1σ shown in blue mesh. Antibody-peptide interface areas were calculated using PDBePISA.
Taken together, the ELISA binding profiles, atomic-level structural features, and sequence comparison provide a comprehensive analysis of the antigen-binding sites. Despite shared germline gene usage with Fab 8B2, and crystallization with the non-phosphorylated peptide, Fab 6B2 seems to be an outlier. 6B2 is the only mAb that binds pSer396 with similar affinity compared to the other three peptides (Figure 1(c)). However, competition ELISA data reveals that pSer396 cannot compete for binding with the pSer396/pSer404 or the non-phosphorylated peptide (Figure 2(b,e)). Furthermore, 6B2 binds all four peptides with an affinity ~10-fold less than both 8B2 and h4E6 (Figure 1). These observations are also supported by the Fab structure of 6B2, which reveals a shallower antigen-binding site (Figure 6(b-d)). The reduced surface area results in a half of a helical turn that can likely facilitate phosphorylation of residue Ser396, but does so with a binding affinity proportional to the antigen-binding area. Accordingly, this observation, along with shared germline gene usage, may explain why 8B2 is able to bind pSer396 at saturating concentrations (Figure 1(b)).
Discussion
The Ser396/Ser404 region of tau is a key epitope region for antibody-mediated immune modulation in development of an effective disease-modifying therapy for AD.4,38 Determining the crystal Fab structures of anti-Ser404 mAbs in complex with their epitopes not only can precisely map this epitope region, but also reveal its conformation and how it is targeted by Abs. We have obtained crystal Fab structures of three mAbs, known to bind to the Ser404 region,12 in complex with their epitope peptides. Our data showed that this epitope region can have an extended conformation, consistent with biological data that these regions in a hyperphosphorylated state can constitute a key seeding conformation for β-structured tau aggregation39–42. Each of these three mAbs has an antigen-binding pocket that can accommodate the epitope from its C-terminal end (Figure 6). This is an epitope-binding mode different from that of mAb C5.2, a pSer396-specific mAb, for which the epitope lies on top and along the antigen-binding surface.43 It is known that tau protein is often C-terminally truncated,44,45 and it is possible that residue 408 is a terminus of some of these truncated tau, allowing these mAbs to be reactive with tau paired helical filaments (PHF) isolated from human tissues (Figure S4).12
Our crystal structures provide an explanation of the binding data presented in Figures 1 and 2. While mAb h4E6 was crystallized with a phospho-peptide, we only obtained crystals of mAbs 8B2 and 6B2 with a non-phospho-peptide. However, in both 8B2 and 6B2 complex Fab structures, we observed a phosphate molecule right next to the side chain of Ser404, mimicking that of phosphorylated Ser404 and consistent with the fact that these mAbs can bind to both phospho- and non-phospho peptides (Figure 1). Interestingly, all three mAbs bind well the pSer404 peptide and the pSer396/pSer404 peptide, but they bind the pSer396 peptide differently (Figures 1 and 2). We observed in our structures that the very C-terminal end of the three epitopes are slightly different between the three mAbs: while the C-termini of 8B2 and h4E6 epitopes have a straight linear conformation, that of 6B2 has a curved, almost half of a helical turn (Figures 4 and 6). It is possible that when Ser396 alone is phosphorylated, the conformation of the C-terminus of the peptide becomes curved or less flexible, making it less accessible to 8B2 and h4E6 binding. This is consistent with data from the competition ELISA, which show that, in contrast to antibody binding to the pSer396/pSer404 peptide, binding to the pSer396 peptide is blocked by pre-incubation with both pSer396/pSer404 and non-phosphorylated peptides (Figure 2). These data indicate that the phosphorylation state of these two serines, Ser396 and Ser404, influences the conformation of this epitope region.
The Ser396/Ser404 region is a major target for antibody immune modulation, but the precise antibody binding sites and epitope conformations had not been well established. Together with our previously reported pSer396-specific mAb, C5.2, in complex with its pSer396 epitope,43 and the three structures reported here, we have provided a comprehensive structural view of this key epitope region. Our data revealed that the Ser396/Ser404 region can adopt an extended β-strand conformation, and the phosphorylation state of each serine may influence the overall conformation of this epitope region. In addition, mAb h4E6 has an epitope structure remarkably similar to the epitope of the pSer422-tau conformational rabbit mAb RB86 (Figure 5(d)), which was recently elucidated to 2.5 Å resolution (PDB: 5DMG). Interestingly, Ser422 is not usually phosphorylated on tau in physiological conditions,31 but it is in tauopathies and in related animal models.32–35 Furthermore, in pSer422 immunized animals, there is a decrease in aggregated tau and associated cognitive improvement,46,47 similar to immunotherapies targeting pSer396/pSer404.11–19,21,22,24,26–28 Recognition of a common conformation, which has been linked to phosphorylation-dependent tau aggregation, creates an opportunity for immunological recognition of precise pathological species, thus minimizing binding to normal tau protein.
Further comparison between Ser396, Ser404 and Ser422 reveals a shared serine/threonine-proline (S/T-P) motif, accounting for three of the 17 S/T-P motifs in 2N4R tau protein. This motif has for many years been linked to hyperphosphorylation by glycogen synthase kinase 3 (GSK3),48–50 a constitutively active, proline-directed serine/threonine kinase that plays a role in processes ranging from gene transcription to glycogen metabolism.51,52 Over-activity of GSK3 has been suggested to account for memory impairment, increased amyloid-β production, and local plaque-associated microglial-mediated inflammatory responses in AD.51 While a majority of the S/T-P sites are located in the proline-rich domain of tau protein, there are no S/T-P sites located in the microtubule-binding domain and Ser396, Ser404 and Ser422 are the only S/T-P sites in the C-terminal tail. There may be some significance in the structural similarities observed in antigen-recognition and in the location of these three hyperphosphorylated residues in a disordered region of tau known to contribute to tau aggregation in pathological conditions. Further investigation is needed to determine the mechanistic relationship between kinase activity, hyperphosphorylation and toxic aggregation. However, an agent inhibiting GSK3 recently failed in two Phase 2 trials.4 Perhaps targeting the individual S/T-P phospho-sites, as now is being pursued by immunotherapies, will be fruitful.4
Lastly, it is important to mention that mAbs 6B2 and 4E6 (original mouse mAb prototype of h4E6) have previously been functionally characterized.11,12,23–25 As shown by various techniques, both mAbs are able to enter neurons and detect pathological tau in mouse and culture models. However, these two antibodies differ in a number of ways. For example, although both are phospho-selective, as particularly seen in their binding to epitopes in solution, their affinities to various tau peptides and tau protein from normal and diseased brains differ.12 Likewise, their intracellular distribution following neuronal uptake is different. 4E6 is concentrated in cytosolic vesicles and co-localizes with PHF1 (a mAb known to target the pSer396/pSer404 epitope region53) -stained tau, while 6B2 has a more diffuse cytosolic distribution, and colocalizes better with MC1 (a conformational anti-tau mAb54) -stained tau. In addition, both mAbs were shown to be non-toxic and to reduce tau pathology in an ex vivo model.12 However, in an acute in vivo model, only 4E6 improved cognition and reduced soluble pTau, whereas 6B2 was ineffective. Furthermore, 4E6, but not 6B2, prevented toxicity, tau seeding, and reduced the spreading of pathological tau between neurons. Lastly, binding of 6B2 was found to have a higher affinity for aggregated PHF-enriched tau compared to 4E6, whereas 4E6 had a higher affinity for soluble PHF.24 This is consistent with our data, which show 6B2 has a shallower binding crevice and is more accommodating to the phosphorylation state of Ser396 and Ser404, which may be beneficial for recognition of a larger aggregated target. Overall, the data from these different studies suggest that binding to soluble PHF may be the key to identifying an efficacious therapy.
In conclusion, our findings provide valuable detailed insights into the pathologically important Ser396/Ser404-tau epitope region and how phosphorylation at these two sites may play a role in the generation of a common tau conformer found elsewhere in the protein. These insights support the rapidly growing practice of using tau antibodies as a potential disease-modifying treatment for AD and other tauopathies.
Materials and methods
Monoclonal antibody and synthetic peptides
MAbs 8B2, 6B2, and 4E6 were generated by GenScript as previously described.11,12 Briefly, wild type BALB/c mice were immunized with a peptide encompassing the pSer396/pSer404 region conjugated to keyhole limpet hemocyanin via a cysteine residue (cTDHGAEIVYK(pS)PVVSGDT(pS)PRHL). Hybridoma fusions were screened by ELISA and 8B2, 6B2, and 4E6 clones were selected based on binding to the phospho-tau immunogen. Although mAbs 4E6 and 6B2 were functionally characterized previously,11,12 mAb 8B2 has never been published before. Peptides used in ELISA and crystallization were synthesized by W.M. Keck Biotechnology Resource Center or GenScript (Table 1). The lyophilized peptides were solubilized in water to a stock concentration of 10 mg/mL before mixing with the Fabs for crystallization.
Fabs production and purification
The Fabs were prepared by papain digestion of IgG. Briefly, IgG and papain (Worthington, #LS003119) were mixed at a 1:15 molar ratio in a buffer (50 mM Tris pH 6.8 and 100 mM NaCl) containing 20 mM cysteine hydrochloride (Fisher Scientific, #BP376-100) and 0.1 M EDTA, pH 8.0. The reaction was incubated for 1 hour at 37°C and was stopped with the addition of 10 mM iodoacetamide (Bio-Rad, #163–2109). The Fabs were then isolated from the Fcs using a HiTrap Protein A affinity column (GE, # 17–0402-01). Finally, the Fabs were further purified using size-exclusion chromatography. The monodispersed peak containing the soluble Fabs was collected and concentrated to about 12 mg/mL for crystallization.
Enzyme-linked immunosorbent assay
Immulon 4 HBX 96-well microtiter plates (Thermo Fisher Scientific, # 3855) were coated with peptides diluted to 2 μg/mL in phosphate-buffered saline (PBS) and left overnight at 4°C (Table 1). Plates were then washed three times in PBS-T (PBS containing 0.05% Tween-20) and blocked in 5% non-fat milk and 3% bovine serum albumin in PBS for 2 hours at room temperature. Plates were subsequently washed 3 times in PBS-T before IgG was serially diluted (0.1 ng/mL – 20 µg/mL) in PBS and incubated for 1 hour at room temperature. Then, alkaline phosphatase-conjugated goat anti-mouse IgG and goat anti-human IgG (Southern Biotech, #2014–04) were diluted 1:2000 in PBS. Secondary antibodies were incubated for 30 minutes at room temperature. Finally, bound IgG was analyzed by addition of p-nitrophenyl phosphate substrate (Thermo Fisher Scientific, #34,047) and measurement at 405 nm on a VersaMax Microplate Reader.
For the competition ELISA, the same amount of peptides were coated in each well (10 µg/mL) and bound at 4°C overnight. MAbs (5 µg/mL) were pre-incubated with various concentrations of peptides (10– 0.04 µg/mL) for 30 min at room temperature in Superblock (Fisher Scientific, #37,515). The pre-incubated mixtures were then added to the plate and incubated for 2 hours at room temperature. Subsequently, horseradish peroxidase-conjugated anti-mouse IgG and anti-human IgG (1:3000) were added and incubated for 1 hour at room temperature. Three washes were performed between all steps with TBS-T (TBS containing 0.05% Tween-20). The plate was developed with TMB peroxidase EIA reagent (Thermo Fisher Scientific, #34,028) and stopped by 2 N sulfuric acid. Absorbance at 450 nm was read on a BioTek Synergy 2 plate reader.
Production of mAb h4E6
Mouse mAb 4E6 was originally developed through immunization and produced from hybridoma cells along with mAbs 6B2 and 8B2 (see above). For crystallization, recombinant human chimeric 4E6 (h4E6) was engineered and produced in order to overcome complications from the heterogeneity of Fab generated from the IgG produced by hybridoma cells. Briefly, mouse 4E6 was sequenced; its variable domains (Fvs) were synthesized (GenScript) and cloned into the XbaI/ApaI (heavy chain Fv) and XbaI/BsiWI (light chain Fv) sites of the modified expression vector pVRC8400 encoding a human IgG1 frame (PMID: 15,994,776). Equal amounts of heavy and light chain plasmid DNA were transiently transfected into HEK293S cells, cultured in Erlenmeyer flasks using 15 to 25% of the nominal volume, and rotated at 110 to 130 rpm under standard humidified conditions, 37°C and 5% CO2. Cells were allowed to secrete the recombinant protein for 5 days. Finally, mAb was isolated using a HiTrap Protein A affinity column and binding of the purified recombinant protein to tau peptides was confirmed with ELISA.
Crystallization, data collection, and structure determination
Concentrated Fab was mixed with the peptide at a 1:10 molar ratio. Crystallization conditions were screened and optimized using the vapor diffusion hanging drop method. Well-diffracted crystals of mAb h4E6 were obtained in a solution containing 25.5% polyethylene glycol 4000 and 0.17 M ammonium sulfate; crystals of mAb 6B2 were obtained in a solution of 2 M ammonium sulfate; crystals of mAb 8B2 were obtained in a solution of 23% polyethylene glycol 8000, 0.17 M ammonium acetate, and 0.085 M sodium cacodylate trihydrate pH 6.5; crystals of apo mAb 8B2 were obtained in a solution of 20% polyethylene glycol 6000, 1 M lithium chloride, and 0.1 M citric acid pH 4.0. X-ray diffraction data for h4E6 and 8B2 were collected at beamline 14–1 at the Stanford Synchrotron Radiation Lightsource (SSRL) and data for 6B2 was collected at beamline GM/CA-CAT at the Advanced Proton Source (APS), Argonne National Laboratory. The data sets were processed using the XDS software package55 and the structures determined by molecular replacement using initial models with high sequence similarity (Table 2). Multiple steps of refinement were carried out in COOT56 and PHENIX.57 The final structure analysis was performed in ICM58 and figures were generated with Chimera59 and PyMOL (http://pymol.org). The antigen-antibody interface areas were calculated by PDBePISA (EMBL-EBI).
Table 2.
Data collection and refinement statistics.
| 8B2 apo | 8B2 | 6B2 | h4E6 | |
|---|---|---|---|---|
| Data collection | ||||
| Space group | P22121 | P43212 | P1 | P1211 |
| Cell dimensions | ||||
| a, b, c (Å) | 53.4, 67.5, 240.2 | 68.8, 68.8, 198.8 | 54.0, 59.7, 143.8 | 79.2, 65.1, 97.4 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 89.4, 87.3, 82.6 | 90, 112, 90 |
| Resolution (Å) | 1.9 (1.96–1.9) | 1.79 (1.83–1.79) | 2.60 (2.69–2.60) | 2.99 (3.11–2.99) |
| Rmeas | 0.108 (0.559) | 0.078 (0.541) | 0.123 (0.585) | 0.143 (0.737) |
| I/σI | 35 (5.4) | 28.4 (4.3) | 5.92 (1.25) | 10.15 (2.16) |
| Completeness (%) | 94.8 (88.2) | 99.9 (99.78) | 97.60 (92.68) | 99.38 (98.93) |
| Redundancy | 10.7 (11.1) | 6.0 (6.8) | 1.8 (1.8) | 3.8 (3.8) |
| Refinement | ||||
| Resolution (Å) | 35.7–1.9 | 40.3–1.79 | 29.06–2.60 | 48.69– 2.99 |
| No. reflections | 65,961 (6038) | 45,390 (4437) | 53,304 (5074) | 18,575 (1841) |
| Rwork/Rfree | 18.3/23.5 | 17.8/21.8 | 19.1/23.3 | 20.2/25.9 |
| No. atoms | ||||
| Protein | 857 | 3397 | 13,316 | 6597 |
| Ligand/ion | 0 | 23 | 20 | 16 |
| Water | 1114 | 569 | 382 | 5 |
| B-factors | ||||
| Protein | 26.6 | 24.4 | 34.9 | 59.57 |
| Ligand/ion | 0 | 37.4 | 62.5 | 64.42 |
| Water | 32.3 | 33.9 | 33.4 | 38.35 |
| R.m.s. deviations | ||||
| Bond lengths (Å) | 0.007 | 0.006 | 0.009 | 0.004 |
| Bond angles (°) | 0.88 | 0.91 | 1.32 | 0.98 |
*Values in parentheses are for highest-resolution shell.
Data availability
The atomic coordinates and structure factors have been deposited in the RCSB Protein Data Bank under accession codes 6DC7 (8B2 apo), 6DC8 (8B2), 6DC9 (h4E6), and 6DCA (6B2).
Funding Statement
This work was supported in part by National Institute of Neurological Disorders and Stroke [NS077239] and [AG032611]; National Institute on Aging [AG032611].
Abbreviations
- AD
Alzheimer’s disease
- APS
The Advanced Photon Source
- CDR
complementarity-determining region
- ELISA
enzyme-linked immunosorbent assay
- Fab
antigen-binding fragment
- Fv
variable fragment domain
- GSK3
glycogen synthase kinase 3
- mAb
monoclonal antibody
- pTau
hyperphosphorylated Tau
- PHF
paired helical filament
- SSRL
The Stanford Synchrotron Radiation Lightsource
Acknowledgments
We thank Ruimin Pan for providing the plasmid for cloning the recombinant h4E6, and Senthil Kumar Krishnaswamy for help with sequencing of mAbs.
Author Contribution
Investigation, J.E.C., E.E.C.; Resources, E.E.C.; Formal Analysis, J.E.C., E.E.C., E.M.S., and X.P.K.; Validation, J.E.C. and X.P.K.; Writing - Original Draft, J.E.C.; Writing - Review & Editing, J.E.C., E.M.S., X.P.K.; Visualization, J.E.C.; Supervision, E.M.S. and X.P.K; Funding Acquisition, E.M.S. and X.P.K.
Disclosure of Potential Conflicts of Interest
E.M.S. is an inventor on various patents on immunotherapies and related diagnostics for neurodegenerative diseases that are assigned to New York University. Some of those focusing on the tau protein are licensed to and are being co-developed with H. Lundbeck A/S. J.E.C., E.E.C. and X.P.K. declare no competing interests.
Supplementary data
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Nelson PT, Alafuzoff I, Bigio EH, Bouras C, Braak H, Cairns NJ, Castellani RJ, Crain BJ, Davies P, Del Tredici K, et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol. 2012;71:362–81. doi: 10.1097/NEN.0b013e31825018f7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Braak H, Braak E.. Evolution of the neuropathology of Alzheimer’s disease. Acta Neurol Scand Suppl. 1996;165:3–12. [DOI] [PubMed] [Google Scholar]
- 3.Goedert M. Tau filaments in neurodegenerative diseases. FEBS Lett. 2018;592:2383–91. doi: 10.1002/1873-3468.13108. [DOI] [PubMed] [Google Scholar]
- 4.Congdon EE, Sigurdsson EM. Tau-targeting therapies for Alzheimer disease. Nat Rev Neurol. 2018;14:399–415. doi: 10.1038/s41582-018-0013-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sigurdsson EM. Tau immunotherapies for Alzheimer’s disease and related tauopathies: progress and potential pitfalls. J Alzheimers Dis. 2018;64:S555–S65. doi: 10.3233/JAD-179937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Skrabana R, Sevcik J, Novak M. Intrinsically disordered proteins in the neurodegenerative processes: formation of tau protein paired helical filaments and their analysis. Cell Mol Neurobiol. 2006;26:1083–95. doi: 10.1007/s10571-006-9083-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Butner KA, Kirschner MW. Tau-protein binds to microtubules through a flexible array of distributed weak sites. J Cell Biol. 1991;115:717–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wischik CM, Novak M, Edwards PC, Klug A, Tichelaar W, Crowther RA. Structural characterization of the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci U S A. 1988;85:4884–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A. 1986;83:4913–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goedert M, Jakes R. Expression of separate isoforms of human tau protein: correlation with the tau pattern in brain and effects on tubulin polymerization. Embo J. 1990;9:4225–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Congdon EE, Gu J, Sait HB, Sigurdsson EM. Antibody uptake into neurons occurs primarily via clathrin-dependent Fcgamma receptor endocytosis and is a prerequisite for acute tau protein clearance. J Biol Chem. 2013;288:35452–65. doi: 10.1074/jbc.M113.491001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gu J, Congdon EE, Sigurdsson EM. Two novel Tau antibodies targeting the 396/404 region are primarily taken up by neurons and reduce tau protein pathology. J Biol Chem. 2013;288:33081–95. doi: 10.1074/jbc.M113.494922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Asuni AA, Boutajangout A, Quartermain D, Sigurdsson EM. Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J Neurosci. 2007;27:9115–29. doi: 10.1523/JNEUROSCI.2361-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boutajangout A, Quartermain D, Sigurdsson EM. Immunotherapy targeting pathological tau prevents cognitive decline in a new tangle mouse model. J Neurosci. 2010;30:16559–66. doi: 10.1523/JNEUROSCI.4363-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boutajangout A, Ingadottir J, Davies P, Sigurdsson EM. Passive immunization targeting pathological phospho-tau protein in a mouse model reduces functional decline and clears tau aggregates from the brain. J Neurochem. 2011;118:658–67. doi: 10.1111/j.1471-4159.2011.07337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chai X, Wu S, Murray TK, Kinley R, Cella CV, Sims H, Buckner N, Hanmer J, Davies P, O’Neill MJ, et al. Passive immunization with anti-Tau antibodies in two transgenic models: reduction of tau pathology and delay of disease progression. J Biol Chem. 2011;286:34457–67. doi: 10.1074/jbc.M111.229633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bi M, Ittner A, Ke YD, Gotz J, Ittner LM. Tau-targeted immunization impedes progression of neurofibrillary histopathology in aged P301L tau transgenic mice. PLoS One. 2011;6:e26860. doi: 10.1371/journal.pone.0026860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Theunis C, Crespo-Biel N, Gafner V, Pihlgren M, Lopez-Deber MP, Reis P, Hickman DT, Adolfsson O, Chuard N, Ndao DM, et al. Efficacy and safety of a liposome-based vaccine against protein tau, assessed in in tau.P301L mice that model tauopathy. PLoS One. 2013;8:e72301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ittner A, Bertz J, Suh LS, Stevens CH, Gotz J, Ittner LM. Tau-targeting passive immunization modulates aspects of pathology in tau transgenic mice. J Neurochem. 2015;132:135–45. doi: 10.1111/jnc.12821. [DOI] [PubMed] [Google Scholar]
- 20.Umeda T, Eguchi H, Kunori Y, Matsumoto Y, Taniguchi T, Mori H, Tomiyama T. Passive immunotherapy of tauopathy targeting pSer413-tau: a pilot study in mice. Ann Clin Transl Neurol. 2015;2:241–55. doi: 10.1002/acn3.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sankaranarayanan S, Barten DM, Vana L, Devidze N, Yang L, Cadelina G, Hoque N, DeCarr L, Keenan S, Lin A, et al. Passive immunization with phospho-tau antibodies reduces tau pathology and functional deficits in two distinct mouse tauopathy models. PLoS One. 2015;10:e0125614. doi: 10.1371/journal.pone.0125614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krishnamurthy PK, Deng Y, Sigurdsson EM. Mechanistic studies of antibody-mediated clearance of tau aggregates using an ex vivo brain slice model. Front Psychiatry. 2011;2:59. doi: 10.3389/fpsyt.2011.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krishnaswamy S, Lin Y, Rajamohamedsait WJ, Rajamohamedsait HB, Krishnamurthy P, Sigurdsson EM. Antibody-derived in vivo imaging of tau pathology. J Neurosci. 2014;34:16835–50. doi: 10.1523/JNEUROSCI.2755-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Congdon EE, Lin Y, Rajamohamedsait HB, Shamir DB, Krishnaswamy S, Rajamohamedsait WJ, Verdier J-M, Robitzer M, Perrier V. Affinity of tau antibodies for solubilized pathological Tau species but not their immunogen or insoluble tau aggregates predicts in vivo and ex vivo efficacy. Mol Neurodegener. 2016;11:62. doi: 10.1186/s13024-016-0074-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shamir DB, Rosenqvist N, Rasool S, Pedersen JT, Sigurdsson EM. Internalization of tau antibody and pathological tau protein detected with a flow cytometry multiplexing approach. Alzheimers Dement. 2016;12:1098–107. doi: 10.1016/j.jalz.2016.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rajamohamedsait H, Rasool S, Rajamohamedsait W, Lin Y, Sigurdsson EM. Prophylactic active tau immunization leads to sustained reduction in both tau and amyloid-beta pathologies in 3xTg Mice. Sci Rep. 2017;7:17034. doi: 10.1038/s41598-017-17313-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nobuhara CK, DeVos SL, Commins C, Wegmann S, Moore BD, Roe AD, Costantino I, Frosch MP, Pitstick R, Carlson GA, et al. Tau antibody targeting pathological species blocks neuronal uptake and interneuron propagation of tau in vitro. Am J Pathol. 2017;187:1399–412. doi: 10.1016/j.ajpath.2017.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu W, Zhao L, Blackman B, Parmar M, Wong MY, Woo T, Yu F, Chiuchiolo MJ, Sondhi D, Kaminsky SM, et al. Vectored intracerebral immunization with the anti-tau monoclonal antibody PHF1 markedly reduces tau pathology in mutant tau transgenic mice. J Neurosci. 2016;36:12425–35. doi: 10.1523/JNEUROSCI.2016-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Swindells MB, Porter CT, Couch M, Hurst J, Abhinandan KR, Nielsen JH, Macindoe G, Hetherington J, Martin ACR. abYsis: integrated antibody sequence and structure-management, analysis, and prediction. J Mol Biol. 2017;429:356–64. doi: 10.1016/j.jmb.2016.08.019. [DOI] [PubMed] [Google Scholar]
- 30.Hasegawa M, Jakes R, Crowther RA, Lee VM, Ihara Y, Goedert M. Characterization of mAb AP422, a novel phosphorylation-dependent monoclonal antibody against tau protein. FEBS Lett. 1996;384:25–30. [DOI] [PubMed] [Google Scholar]
- 31.Bussière T, Hof PR, Mailliot C, Brown CD, Caillet-Boudin ML, Perl DP, Buée L, Delacourte A. Phosphorylated serine422 on tau proteins is a pathological epitope found in several diseases with neurofibrillary degeneration. Acta Neuropathol. 1999;97:221–30. [DOI] [PubMed] [Google Scholar]
- 32.Allen B, Ingram E, Takao M, Smith MJ, Jakes R, Virdee K, Yoshida H, Holzer M, Craxton M, Emson PC, et al. Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J Neurosci. 2002;22:9340–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deters N, Ittner LM, Gotz J. Divergent phosphorylation pattern of tau in P301L tau transgenic mice. Eur J Neurosci. 2008;28:137–47. doi: 10.1111/j.1460-9568.2008.06318.x. [DOI] [PubMed] [Google Scholar]
- 34.Ferrer I, Hernandez I, Boada M, Llorente A, Rey MJ, Cardozo A, Ezquerra M, Puig B. Primary progressive aphasia as the initial manifestation of corticobasal degeneration and unusual tauopathies. Acta Neuropathol. 2003;106:419–35. doi: 10.1007/s00401-003-0756-4. [DOI] [PubMed] [Google Scholar]
- 35.Augustinack JC, Schneider A, Mandelkow EM, Hyman BT. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer’s disease. Acta Neuropathol. 2002;103:26–35. [DOI] [PubMed] [Google Scholar]
- 36.Bujotzek A, Lipsmeier F, Harris SF, Benz J, Kuglstatter A, Georges G. VH-VL orientation prediction for antibody humanization candidate selection: A case study. MAbs. 2016;8:288–305. doi: 10.1080/19420862.2015.1117720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fitzpatrick AWP, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ, Crowther RA, Ghetti B, Goedert M, Scheres SHW. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature. 2017;547:185–90. doi: 10.1038/nature23002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pedersen JT, Sigurdsson EM. Tau immunotherapy for Alzheimer’s disease. Trends Mol Med. 2015;21:394–402. doi: 10.1016/j.molmed.2015.03.003. [DOI] [PubMed] [Google Scholar]
- 39.Barghorn S, Davies P, Mandelkow E. Tau paired helical filaments from Alzheimer’s disease brain and assembled in vitro are based on beta-structure in the core domain. Biochemistry. 2004;43:1694–703. doi: 10.1021/bi0357006. [DOI] [PubMed] [Google Scholar]
- 40.Berriman J, Serpell LC, Oberg KA, Fink AL, Goedert M, Crowther RA. Tau filaments from human brain and from in vitro assembly of recombinant protein show cross-β structure. Proc Natl Acad Sci USA. 2003; 100:9034–38. doi: 10.1073/pnas.1530287100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Margittai M, Langen R. Template-assisted filament growth by parallel stacking of tau. Proc Natl Acad Sci U S A. 2004;101:10278–83. doi: 10.1073/pnas.0401911101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Crowther RA. Straight and paired helical filaments in Alzheimer-disease have a common structural unit. Proc Natl Acad Sci U S A. 1991;88:2288–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chukwu JE, Pedersen JT, Pedersen LO, Volbracht C, Sigurdsson EM, Kong XP. Tau antibody structure reveals a molecular switch defining a pathological conformation of the tau protein. Sci Rep. 2018;8:6209. doi: 10.1038/s41598-018-24276-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kanmert D, Cantlon A, Muratore CR, Jin M, O’Malley TT, Lee G, Young-Pearse TL, Selkoe DJ, Walsh DM. C-terminally truncated forms of tau, but not full-length tau or its C-terminal fragments, are released from neurons independently of cell death. J Neurosci. 2015;35:10851–65. doi: 10.1523/JNEUROSCI.0387-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sato C, Barthelemy NR, Mawuenyega KG, Patterson BW, Gordon BA, Jockel-Balsarotti J, Sullivan M, Crisp MJ, Kasten T, Kirmess KM, et al. Tau kinetics in neurons and the human central nervous system. Neuron. 2018;97:1284–98 e7. doi: 10.1016/j.neuron.2018.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Troquier L, Caillierez R, Burnouf S, Fernandez-Gomez FJ, Grosjean ME, Zommer N, Sergeant N, Schraen-Maschke S, Blum D, Buee L. Targeting phospho-Ser422 by active tau immunotherapy in the THYTau22 mouse model: a suitable therapeutic approach. Curr Alzheimer Res. 2012;9:397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Collin L, Bohrmann B, Gopfert U, Oroszlan-Szovik K, Ozmen L, Gruninger F. Neuronal uptake of tau/pS422 antibody and reduced progression of tau pathology in a mouse model of Alzheimer’s disease. Brain. 2014;137:2834–46. doi: 10.1093/brain/awu213. [DOI] [PubMed] [Google Scholar]
- 48.Yang S-D, Song J-S, Yu J-S, Shiah S-G. Protein kinase FA/GSK-3 phosphorylates on Ser235-Pro and Ser404-Pro that are abnormally phosphorylated in Alzheimer’s disease brain. J Neurochem. 1993;61:1742–47. [DOI] [PubMed] [Google Scholar]
- 49.Hanger DP, Hughes K, Woodgett JR, Brion JP, Anderton BH. Glycogen synthase kinase-3 induces Alzheimer’s disease-like phosphorylation of tau: generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci Lett. 1992;147:58–62. [DOI] [PubMed] [Google Scholar]
- 50.Mandelkow EM, Drewes G, Biernat J, Gustke N, Van Lint J, Vandenheede JR, Mandelkow E. Glycogen synthase kinase-3 and the Alzheimer-like state of microtubule-associated protein tau. FEBS Lett. 1992;314:315–21. [DOI] [PubMed] [Google Scholar]
- 51.Hooper C, Killick R, Lovestone S. The GSK3 hypothesis of Alzheimer’s disease. J Neurochem. 2008;104:1433–39. doi: 10.1111/j.1471-4159.2007.05194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Beurel E, Grieco SF, Jope RS. Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol Ther. 2015;148:114–31. doi: 10.1016/j.pharmthera.2014.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Otvos L Jr., Feiner L, Lang E, Szendrei GI, Goedert M, Lee VM. Monoclonal antibody PHF-1 recognizes tau protein phosphorylated at serine residues 396 and 404. J Neurosci Res. 1994;39:669–73. doi: 10.1002/jnr.490390607. [DOI] [PubMed] [Google Scholar]
- 54.Jicha GA, Bowser R, Kazam IG, Davies P. Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J Neurosci Res. 1997;48:128–32. [DOI] [PubMed] [Google Scholar]
- 55.Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66:125–32. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. [DOI] [PubMed]
- 57.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–54. [DOI] [PubMed] [Google Scholar]
- 58.Abagyan R, Totrov M, Kuznetsov D. ICM - a new method for protein modeling and design - applications to docking and structure prediction from the distorted native conformation. J Comput Chem. 1994;15:488–506. doi: 10.1002/jcc.540150503. [DOI] [Google Scholar]
- 59.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The atomic coordinates and structure factors have been deposited in the RCSB Protein Data Bank under accession codes 6DC7 (8B2 apo), 6DC8 (8B2), 6DC9 (h4E6), and 6DCA (6B2).