

ABSTRACT
Loss of excision repair cross-complementation group 1 (ERCC1), frequently found in lung cancer, and mutations in breast cancer type 1/2 susceptibility genes (BRCA1/2), often found in ovarian, breast and prostate cancers, confer sensitivity to poly-(ADP-ribose) polymerase inhibitors (PARPi). Our work, and that of others, shows that PARPi selectively trigger tumor cell-autonomous immune phenotypes in ERCC1- or BRCA-defective contexts. This suggests that PARPi, used in appropriately selected populations, might mediate their therapeutic effects by potentiating anti-tumor immunity.
KEYWORDS: DNA damage response, DNA repair defects, PARP inhibitors, cGAS/STING, tumour cell-intrinsic immunity
DNA repair defects are a common hallmark of cancer that likely contributes to tumorigenesis by promoting genomic instability. This hallmark of cancer has been exploited therapeutically for over 50 years with the use of classical chemotherapies, which elicit cytotoxicity in tumor cells that are unable to effectively repair DNA damage. More recently, the identification of the synthetic lethal interaction between DNA damage induced by poly-(ADP-ribose) polymerase inhibitors (PARPi) and defects in the DNA repair tumor suppressor genes BRCA1 and BRCA2 (known as breast cancer type 1/2 susceptibility genes), has led to a more targeted approach to therapeutically exploiting specific DNA repair defects in biomarker-defined subsets of breast and ovarian cancers.1 For example, the PARPi olaparib, rucaparib, and niraparib have all been approved for the treatment of BRCA-gene mutant advanced ovarian cancer, and olaparib and talazoparib have been approved for the treatment of BRCA-gene mutant breast cancer. Pre-clinical work has also demonstrated that defects in other tumor suppressors that control DNA repair processes, such as excision repair cross-complementation group 1 (ERCC1) defects in models of non-small cell lung cancer (NSCLC), cause PARPi sensitivity.2 This has led to phase 2 clinical trials (e.g., NCT02679963) assessing the efficacy of PARPi in this setting.
Alongside this progress, immune-based therapies – notably antibodies targeting the programmed cell death-1/programmed death-ligand 1 (PD-1/PD-L1) immune checkpoint – have brought unprecedented survival benefits in many cancers including NSCLC. However, these sustained therapeutic responses are not seen in all patients; only 20% of patients with NSCLC currently benefit from these therapies and immunotherapy may even be deleterious in up to 15% of patients, who develop a hyper-progressive disease in response to treatment.3 As such, identifying evidence-based drug combinations that extend the proportion of patients who benefit from anti-PD-(L)1 therapy is an active area of study, as is understanding how to avoid hyper-progression. Recent discoveries that link the extent of tumor genomic instability and DNA repair defects to a better outcome on anti-PD-(L)1 therapies have led to the evaluation of anti-PD-(L)1 plus chemotherapy combination regimens. Such a strategy elicits impressive efficacy in advanced NSCLC; for example, the combined use of the anti-PD-1 antibody pembrolizumab with platinum-based doublet chemotherapy leads to a 12 months overall survival rate of 69.2%,4 an observation that has led to this combination becoming the new standard first-line treatment for advanced or metastatic NSCLC. Similarly, multiple clinical trials that evaluate anti-PD-(L)1 agents in combination with DNA repair-targeted therapies, such as PARPi, are being developed.
In many respects, some of these clinical advances have been made in the absence of strong pre-clinical data that supports the combined use of DNA repair-targeted therapies with immunotherapy. In particular, very little is known about the possible immunomodulatory properties of PARPi, and pre-clinical data supporting the combination of PARPi with anti-PD-(L)1 therapies are still scarce. Yet, a growing body of data has pointed towards a functional interplay between the DNA damage response (DDR) and anti-cancer immunity.5 The main areas of this interplay include genomic instability/tumor mutational load and the formation of tumor-specific neo-antigens, DNA damage-induced immunogenic cell death, and the activation of cytosolic nucleic acid sensing pathways in response to DNA insults. In this latter area, several lines of evidence now support that specific DNA repair defects in tumor cells can activate, in a cell-autonomous fashion, the innate immune system via the cyclic GMP-AMP synthase/stimulator of interferon genes (cGAS/STING) pathway.6,7 In normal cells, the cGAS/STING pathway detects foreign cytosolic DNA (for example, caused by viral infection) and activates the innate immune response by stimulating the expression of a series of pro-inflammatory genes, including interferon-family genes. But cGAS/STING signaling also likely plays a tumor suppressive role, by promoting anti-tumor immunity in response to cytosolic DNA caused by DNA damage and/or DNA repair defects in cancer cells. Prior to our work,7 exposing tumor cells to DNA-damaging agents such as irradiation or S-phase chemotherapies was known to trigger cGAS/STING signaling,8 but whether DNA damage caused by PARPi could elicit similar cell-autonomous immune responses was still unclear. We hypothesized that PARPi could selectively enhance tumor cell-autonomous immunity in tumor cells in which they also provoke synthetic lethality, such as ERCC1-deficient NSCLC cells or BRCA-gene mutant triple-negative breast cancer (TNBC) cells.
To test this hypothesis, we exploited in-house generated isogenic NSCLC models of ERCC1 deficiency (ERCC1-WT, -heterozygous, -knock out (KO) and -rescued clones) and recently described isogenic series of BRCA1-mutant TNBC SUM149 cells, that also have additional mutations that either reverse the homologous recombination defect via reversion of the pathogenic BRCA1 mutation (BRCA1-revertant cells) or cause PARPi resistance by mutation of the PARP1 gene itself (PARP1-null cells).7 Using RNA sequencing and gene set enrichment analysis in the ERCC1-isogenic NSCLC models, we found that when compared to ERCC1-WT cells, the transcriptomes of both ERCC1-KO and -heterozygous clones displayed an enrichment in the expression of genes related to immunity, notably genes involved in type I IFN signaling and cytokine signaling. Interestingly, this transcriptomic analysis – and further low-throughput validations at the protein level – also indicated a spontaneous re-expression of STING in ERCC1-KO clones (which was reversible upon re-expression of the functional ERCC1 isoform) and a profound upregulation of several chemotactic chemokines, including C-C motif chemokine ligand 5 (CCL5) and C-X-C motif chemokine ligand 10 (CXCL10), in cells harbouring a defect in ERCC1. In addition, using immunohistochemistry on a cohort of stage I-II resected lung adenocarcinoma, we found that NSCLC tumors with low ERCC1 expression had increased levels of tumor-infiltrating lymphocytes (TILs), suggesting that ERCC1 status may influence the nature of the immune infiltrate in these tumors.
We next sought to evaluate the effects of PARPi on a series of tumor cell-intrinsic immune phenotypes in ERCC1- or BRCA1-defective cells. Exposure of NSCLC or TNBC cells to several clinical PARPi led to a dose-dependent increase in the formation of cytosolic DNA. The extent of this increase was dependent upon the status of ERCC1 or BRCA1; indeed, whilst cytosolic DNA induction was limited in ERCC1-WT and BRCA1-revertant cells, we observed a much more substantial accumulation of cytosolic DNA in ERCC1-deficient and BRCA1-mutated cells, which displayed elevated levels of cytoplasmic chromatin fragments and micronuclei. Further, we found that cGAS, the critical DNA-binding factor of the cGAS/STING signaling cascade, localized to these micronuclei. We also noted that this increase in cytosolic DNA was associated with activation of STING signaling – as evidenced by the phosphorylation of TANK-binding kinase 1 (TBK1), interferon regulatory factors 3 and 7 (IFR3, IRF7), and nuclear factor-kappa B (NF-κB) –, a resultant type I IFN response and the release chemotactic chemokines, including CCL5 (Figure 1). Importantly, these effects were suppressed in ERCC1-rescued NSCLC cells and BRCA1-revertant TNBC cells, suggesting that extant DNA repair defects are required to induce profound cell-autonomous innate immune-related phenotypes in response to PARPi. We also used PARP1-null cells to assess whether these PARPi-induced effects were on-target effects caused by PARP1 inhibition, as opposed to the inhibition of other PARP-superfamily members; we found that genetic ablation of PARP1 completely reversed the activation of cGAS/STING signaling, supporting an on-target effect of PARPi.
Figure 1.
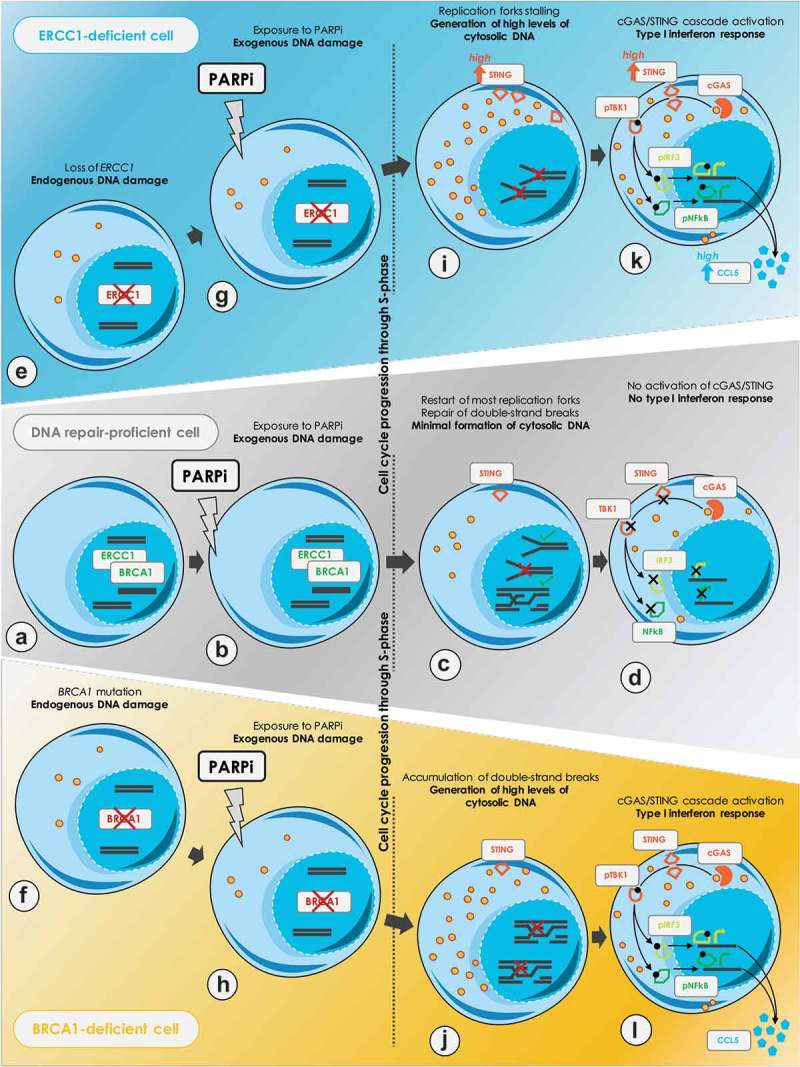
Proposed model of PARPi-induced cancer cell-autonomous immunity in DNA repair-deficient cancer cells.
A. DNA repair-proficient cells, such as wildtype excision repair cross-complementation group 1 (ERCC1) and breast cancer type 1 susceptibility gene (BRCA1) cells, adequately repair endogenous DNA lesions. B. Poly-(ADP-ribose) polymerase inhibitors (PARPi) exposure causes DNA damage, mostly initiated by PARP1 itself trapped onto the DNA at sites of spontaneous single-strand breaks (SSBs). Trapped-PARP1 generates stalled replication forks and subsequent double-strand breaks (DSBs) during DNA replication. C. In DNA repair-proficient cells, trapped-PARP1 lesions can be adequately excised, a process which is likely to be orchestrated by ERCC1. Following excision, BRCA1-mediated repair of DSBs occurs through homologous recombination, and replication restarts. D. Residual unrepaired lesions cause a minimal formation of cytoplasmic chromatin fragments, which are insufficient to trigger cyclic GMP-AMP synthase/stimulator of interferon genes (cGAS/STING) signaling cascade. By contrast, DNA repair-deficient cells, such as ERCC1-deficient and BRCA1-mutated cells are exposed to increase levels of endogenous DNA damage (E, F), which are further enhanced upon exposure to PARPi (G, H). I. In ERCC1-deficient cells, trapped-PARP1 lesions cannot be appropriately resolved, which prevents the subsequent processing of DSBs through homologous recombination. J. In BRCA1-mutated cells, several steps of homologous recombination are disabled and DSB repair cannot occur. K, L. In either case, this results in the accumulation of stalled replication forks, subsequent DSBs and unrepaired DNA lesions which eventually causes increased micronuclei formation and cytoplasmic chromatin fragments generation. These are detected by cGAS, which activates the cGAS/STING signaling cascade and results in a type I IFN response characterized by the secretion of C-C motif chemokine ligand 5 (CCL5) and other chemotactic cytokines. Abbreviations: pTBK1, phosphorylated TANK-binding kinase 1; pIRF3, phosphorylated interferon regulatory factor 3; pNF-κB, phosphorylated nuclear factor-kappa B.
Taken together, these experiments uncovered a novel and relatively unexpected role of PARPi, that is to modulate anti-tumor immunity through a tumor cell-intrinsic mechanism. Importantly, our study identified the genomic contexts (i.e., loss of ERCC1 or BRCA1 function) in which these immunomodulatory effects operate, thereby providing meaningful data for appropriately selecting patients in the clinic. Our findings are consistent with other recent publications;9,10 notably and very interestingly, Ding and colleagues reported that PARPi elicit potent anti-tumor immunity in BRCA1-deficient ovarian tumors following activation of STING signaling, not only in tumor cells but also in dendritic cells – thereby suggesting a link between innate immunity and systemic adaptive immune responses. In Ding et al.’s model, the addition of anti-PD-1 therapy enhanced the BRCA1-dependent and CD8+ T cell-dependent anti-tumor efficacy of PARPi – a finding that was also previously reported by Robillard et al..11
To conclude, our work,7 which uses a combination of rigorous and well-characterized isogenic systems, provides a scientific rationale for combining PARPi with anti-PD-(L)1 therapy in molecularly selected DNA repair-deficient populations. While several clinical trials are currently assessing this combination in BRCA1/2-mutant breast or ovarian cancers, other cancer types, such as NSCLC in which ERCC1 is frequently defective and anti-PD-(L)1 therapies have already shown impressive results, may also benefit from this therapeutic strategy. Such evaluation is underway.
Funding Statement
This work was funded by programme grants to SPV from Integrated Cancer Research Site (SIRIC) [SOCRATE-2 INCa-DGOS-INSERM_12551] and ATIP-Avenir, and programme grants to CJL from Cancer Research UK and Breast Cancer Now. RMC received funding from Fondation Philanthropia - Lombard Odier and Institut Servier.
Disclosure of Potential Conflicts of Interest
RMC has no conflicts of interest or financial interests to disclose. JCS is a full-time employee of Medimmune/AstraZeneca since September 2017. JCS has served in consulting or advisory roles for AstraZeneca, Astex, Clovis, GSK, GamaMabs, Lilly, MSD, Mission Therapeutics, Merus, Pfizer, PharmaMar, Pierre Fabre, Roche/Genentech, Sanofi, Servier, Symphogen, and Takeda. JCS has stock or other ownership in AstraZeneca and Gritstone. CJL has received research funding from: AstraZeneca, Merck KGaA, Artios. CJL has received consultancy, SAB membership or honoraria payments from Sun Pharma, GLG, Merck KGaA, Vertex, AstraZeneca, Tango, 3rd Rock, Ono Pharma, Artios. CJL has stock in Tango. CJL is also a named inventor on patents describing the use of DNA repair inhibitors and stands to gain from the development as part of the ICR “Rewards to Inventors” scheme. SPV has received research funding from Merck KGaA, Boehringer Ingelheim and Roche for unrelated research projects. As part of the Drug Development Department (DITEP), SPV is principal investigator or sub-investigator of clinical trials from Abbvie, Agios Pharmaceuticals, Amgen, Argen-X Bvba, Arno Therapeutics, Astex Pharmaceuticals, Astra Zeneca, Aveo, Bayer Healthcare Ag, Bbb Technologies Bv, Blueprint Medicines, Boehringer Ingelheim, Bristol Myers Squibb, Celgene Corporation, Chugai Pharmaceutical Co., Clovis Oncology, Daiichi Sankyo, Debiopharm S.A., Eisai, Eli Lilly, Exelixis, Forma, Gamamabs, Genentech, Inc., Glaxosmithkline, H3 Biomedicine, Inc, Hoffmann La Roche Ag, Innate Pharma, Iris Servier, Janssen Cilag, Kyowa Kirin Pharm. Dev., Inc., Loxo Oncology, Lytix Biopharma As, Medimmune, Menarini Ricerche, Merck Sharp & Dohme Chibret, Merrimack Pharmaceuticals, Merus, Millennium Pharmaceuticals, Nanobiotix, Nektar Therapeutics, Novartis Pharma, Octimet Oncology Nv, Oncoethix, Onyx Therapeutics, Orion Pharma, Oryzon Genomics, Pfizer, Pharma Mar, Pierre Fabre, Roche, Sanofi Aventis, Taiho Pharma, Tesaro Inc, and Xencor. SPV has participated to advisory boards for Merck KGaA and has benefited from reimbursement for attending symposia from AstraZeneca.
References
- 1.Lord CJ, Ashworth A.. PARP inhibitors: synthetic lethality in the clinic. Sci. 2017;355(6330):1152–1158. doi: 10.1126/science.aam7344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Postel-Vinay S, Bajrami I, Friboulet L, Elliott R, Fontebasso Y, Dorvault N, Olaussen KA, Andre F, Soria JC, Lord CJ, et al. A high-throughput screen identifies PARP1/2 inhibitors as a potential therapy for ERCC1-deficient non-small cell lung cancer. Oncogene. 2013;32(47):5377–5387. doi: 10.1038/onc.2013.311. [DOI] [PubMed] [Google Scholar]
- 3.Ferrara R, Mezquita L, Texier M, Lahmar J, Audigier-Valette C, Tessonnier L, Mazieres J, Zalcman G, Brosseau S, Le Moulec S, et al. Hyperprogressive disease in patients with advanced non-small cell lung cancer treated with PD-1/PD-L1 inhibitors or with single-agent chemotherapy. JAMA Oncol. 2018;4(11):1543–1552. doi: 10.1001/jamaoncol.2018.3676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gandhi L, Rodriguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, Domine M, Clingan P, Hochmair MJ, Powell SF, et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N Engl J Med. 2018;378(22):2078–2092. doi: 10.1056/NEJMoa1801005. [DOI] [PubMed] [Google Scholar]
- 5.Chabanon RM, Pedrero M, Lefebvre C, Marabelle A, Soria JC, Postel-Vinay S. Mutational landscape and sensitivity to immune checkpoint blockers. Clin Cancer Res. 2016;22(17):4309–4321. doi: 10.1158/1078-0432.CCR-16-0903. [DOI] [PubMed] [Google Scholar]
- 6.Parkes EE, Walker SM, Taggart LE, McCabe N, Knight LA, Wilkinson R, McCloskey KD, Buckley NE, Savage KI, Salto-Tellez M, et al. Activation of STING-dependent innate immune signaling by S-phase-specific DNA damage in breast cancer. J Natl Cancer Inst. 2017;109(1). doi: 10.1093/jnci/djx007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chabanon RM, Muirhead G, Krastev DB, Adam J, Morel D, Garrido M, Lamb A, Henon C, Dorvault N, Rouanne M, et al. PARP inhibition enhances tumor cell-intrinsic immunity in ERCC1-deficient non-small cell lung cancer. J Clin Invest. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature. 2017;548(7668):466–470. doi: 10.1038/nature23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ding L, Kim HJ, Wang Q, Kearns M, Jiang T, Ohlson CE, Li BB, Xie S, Liu JF, Stover EH, et al. PARP inhibition elicits STING-dependent antitumor immunity in Brca1-deficient ovarian cancer. Cell Rep. 2018;25(11):2972–80 e5. doi: 10.1016/j.celrep.2018.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shen J, Zhao W, Ju Z, Wang L, Peng Y, Labrie M, Yap TA, Mills GB, Peng G. PARPi triggers the STING-dependent immune response and enhances the therapeutic efficacy of immune checkpoint blockade independent of BRCAness. Cancer Res. 2019;79(2):311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robillard L, Nguyen M, Loehr A, Orsulic S, Kristeleit R, Lin K, Raponi M, Harding TC, Simmons AD. Proceedings of the AACR annual meeting 2017. Washington DC (USA); 2017. Abstract no. 3650. [Google Scholar]