

ABSTRACT
We describe here the design, construction and validation of ALTHEA Gold Libraries™. These single-chain variable fragment (scFv), semisynthetic libraries are built on synthetic human well-known IGHV and IGKV germline genes combined with natural human complementarity-determining region (CDR)-H3/JH (H3J) fragments. One IGHV gene provided a universal VH scaffold and was paired with two IGKV scaffolds to furnish different topographies for binding distinct epitopes. The scaffolds were diversified at positions identified as in contact with antigens in the known antigen-antibody complex structures. The diversification regime consisted of high-usage amino acids found at those positions in human antibody sequences. Functionality, stability and diversity of the libraries were improved throughout a three-step construction process. In a first step, fully synthetic primary libraries were generated by combining the diversified scaffolds with a set of synthetic neutral H3J germline gene fragments. The second step consisted of selecting the primary libraries for enhanced thermostability based on the natural capacity of Protein A to bind the universal VH scaffold. In the third and final step, the resultant stable synthetic antibody fragments were combined with natural H3J fragments obtained from peripheral blood mononuclear cells of a large pool of 200 donors. Validation of ALTHEA Gold Libraries™ with seven targets yielded specific antibodies in all the cases. Further characterization of the isolated antibodies indicated KD values as human IgG1 molecules in the single-digit and sub-nM range. The thermal stability (Tm) of all the antigen-binding fragments was 75°C–80°C, demonstrating that ALTHEA Gold Libraries™ are a valuable source of specific, high affinity and highly stable antibodies.
KEYWORDS: Phage display, next-generation sequencing, Protein A filtration, semisynthetic libraries, lysozyme, human albumin, tumor necrosis factor
Introduction
Antibodies represent the fastest-growing segment of all the therapeutic proteins in the biotechnology industry.1 This remarkable progress is due in part to the exquisite specificity and high affinity of antibodies, compounded with their stability, solubility, clinical tolerability and relatively straightforward manufacturing process compared to other biologics. Yet, the overall process of bringing antibodies from the early discovery phase into the market is a long and expensive one, with the investment increasing exponentially as the potential antibody-based drug progresses from discovery to preclinical development to clinical trials and finally reaches the market.
Over decades, as therapeutic antibodies either moved through the process required for commercialization2 or failed to perform in preclinical development and clinical trials, it has become clear that many of the properties that account for the success of a therapeutic antibody in clinical trials, collectively called developability,3,4 are encoded in the primary amino acid sequence of the antibody. This implies that the developability profile of an antibody can be engineered in the early discovery phase, where the costs of development are still relatively modest. Hence, there is substantial interest in, and demand for, the design and implementation of more robust and efficient discovery platforms capable of generating antibody sequences with the optimal developability profile in the earliest stages of development.
Phage display has served as a key enabling technology for therapeutic antibody discovery.5 This technology platform, which was developed in the late 1980s and perfected during the past three decades,6–14 rests on the principle that more diverse, functional and larger libraries produce more diverse, specific and higher affinity antibodies. To generate larger and more diverse antibody libraries, three main approaches have been described in the literature, including: 1) naïve libraries composed of natural diversity;12 2) libraries based on rational design and synthesis of selected scaffolds;9,11,14 and 3) semisynthetic libraries, which combine synthetic and natural fragments.10
First-generation phage display antibody libraries were naïve.12 Although successful, these libraries included antibody genes toxic to Escherichia coli, and thus yielded low expression levels or no expression at all, which severely compromised the number of functional antibodies displayed in the library. Synthetic antibody libraries were developed to partially mitigate this limitation.11 In this alternative approach, the libraries were carefully designed by making assumptions on the number of scaffolds, positions to diversify, type of amino acids to include in the design and proportion of each amino acid per position to diversify. These assumptions did not always hold true, particularly at complementarity-determining region (CDR)-H3, which is a key element in defining the specificity and affinity of antibodies and by far the most diverse region of the antigen-binding site. Thus, while the structure of CDRs other than the CDR-H3 can be predicted with an accuracy of <1.0 Å,15 no method is currently available to reliably predict the CDR-H3 structure, limiting our ability to properly design the diversity at this region of the antigen-binding site
Obviously, the quality of the synthetic fragments comprising the library also affects its functionality. Nucleotide sequences with stop codons leading to truncated sequences do not produce functional antibody fragments fused to a virion particle. Insertions or deletions of one or two nucleotides change the reading frame of the gene sequence, generating stretches of amino acids that may impair folding or clones with hydrophobic amino acids that result in aggregation. These non-functional antibody variants lead to poorly performing antibody phage display libraries.
Further, antibodies selected from naïve and synthetic libraries, despite having the desired specificity and affinity, have often failed during the late development stages of formulation and manufacturing due to suboptimal developability profiles.16 For instance, some amino acids undergo posttranslational modifications such as deamidation of asparagine (N), oxidation of methionine (M), and isomerization of aspartic acid (D).17 Chemical modifications of these amino acids can result in heterogeneities in the antibody preparation or lack of potency if said amino acids are involved in the interaction with the antigen. In other instances, solvent-exposed tryptophan (W) residues can induce aggregation, leading to immunogenic reactions or lack of solubility at concentrations required for the therapeutic indication.16
Here, we describe the design, implementation and validation of highly functional semisynthetic Antibody Libraries for Therapeutic Antibody Discovery, called ALTHEA (from the Greek “to heal”) Gold Libraries™. These libraries combine highly stable and developable antibody variants with natural diversity at the CDR-H3/JH (H3J) region. To generate the libraries, we followed the strategy described in Figure 1. In the first step, fully synthetic primary antibody libraries (PLs) were designed, cloned, and displayed as single-chain variable fragments (scFvs) on the phage surface. Second, we performed a selection process in which the PLs were submitted to a heat shock and further selected with Protein A for in-frame and stable variants. We called the product of this step intermediate or filtered libraries (FLs). Third, highly functional and highly diverse secondary antibody libraries (SLs) - ALTHEA Gold LibrariesTM - were generated by combining FLs with natural H3J fragments obtained from a large pool of 200 donors.
Figure 1.
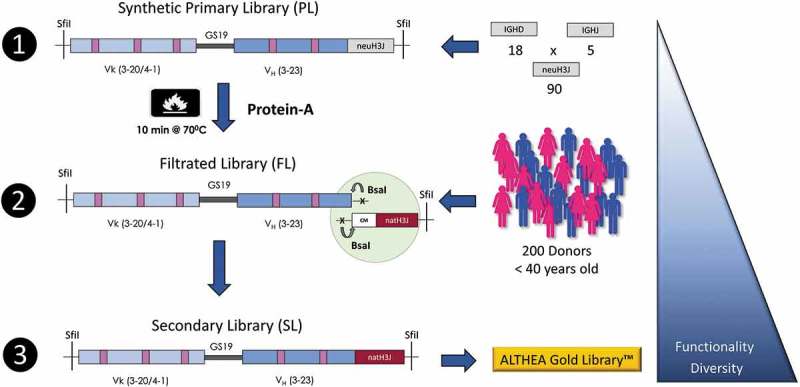
Construction of the ALTHEA Gold libraries™. Step 1: Primary synthetic scFv libraries (PLs) based on the two scFv scaffolds 3-20/3-23 and 4-01/3-23 combined with a set of 90 neutral H3J fragments were assembled and displayed on the phage. Step 2: PLs were heated at 70°C for 10 min and the surviving stable and well-folded scFv were rescued by Protein A to create the filtrated libraries (FL). Finally, in Step 3, the diversified scaffolds from the FLs were amplified by PCR using 5ʹ a universal primer matching CM in the FR-3 of VH and seamlessly assembled with a set of natural H3J fragments RT-PCR’ed from a large pool of 200 donors using the universal primer to build the secondary (SLs) ALTHEA Gold libraries™. The triangle on the right represents how the diversity and functionality of the libraries increased as the three-step process proceeded.
By using a Protein A binding assay and next-generation sequencing (NGS), we monitored how the functionality and diversity of the libraries evolved throughout the three-step construction process. The results indicated a progressive increase in functionality, stability and diversity. Moreover, by panning ALTHEA Gold Libraries™ with an array of seven diverse targets, specific scFvs were obtained in each case. Further characterization of selected antibodies indicated that their KD values were in the single-digit or sub-nM range, with thermal stabilities (Tm) above 75°C. These binding and biophysical parameters are consistent with the developability profile of therapeutic antibodies, thus demonstrating that ALTHEA Gold Libraries™ are a valuable source of specific and developable human antibodies. Importantly, by mining the information generated by NGS, we identified sequence patterns that explained the increase in stability after the filtration process. Such patterns should help in the design and implementation of even more robust and efficient antibody discovery platforms in the near future.
Results
ALTHEA VH and VL scaffolds
ALTHEA Gold Libraries™ consist of one VH scaffold and two distinct VL scaffolds. One of the VL scaffolds has a short CDR-L1 loop, whereas the other has a long one (Figure 2). By changing the length of CDR-L1 from a short to a long loop, antibodies alter the preference to bind protein or peptide targets, respectively.19,20 Therefore, by using the proper VL scaffold, antibodies against protein or peptide targets can be selected. When used in combination, this process would potentially generate antibodies that bind diverse epitopes on a given target.
Figure 2.
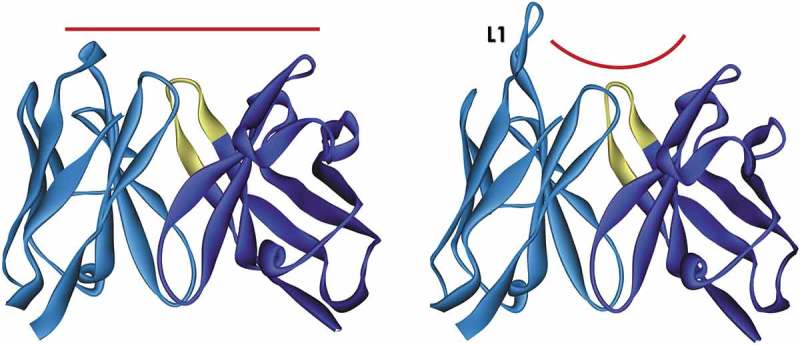
Ribbon representation of ALTHEA Gold Library™ scaffolds. The two scaffolds offer different topographies at the antigen-binding site. Left: scaffold 3-20/3-23 (PDB ID: 5I1c)18 has a short L1 loop creating a flat paratope favorable to protein epitopes. Right: scaffold 4-01/3-23 (PDB ID: 5I1d)18 has a long CDR-L1 loop (L1) creating a groove suitable for peptide binding.
The VL scaffold with a short L1 loop was assembled with the human IGKV3-20*01 germline gene combined with the human IGKJ4*01 joining region. The VL scaffold with the long L1 loop was built with the human IGKV4-01*01 germline gene also combined with the IGKJ4*01 germline gene. The universal VH scaffold was partially built with the human IGHV3-23*01 germline gene. The IGHV3-20*01 germline gene has been found9,21 to be dominant in the repertoire of functional human antibodies. It has also been used as a foundation to build numerous scFv and antigen-binding fragment (Fab) libraries for antibody discovery.9,10,14 Likewise, the two VL scaffolds have been observed to be prevalent in natural human antibodies, and have successfully been used to build antibody phage-displayed libraries.14,22–24 Thus, we expected that, by using these scaffolds, antibodies discovered via ALTHEA Gold Library™ would perform well both in vitro and in vivo settings and be amenable to therapeutic development and manufacturing. Further, antibodies encoded by these germline genes have recently been solved by X-ray crystallography in association with the Universal VH Scaffold.18 This knowledge certainly facilitated the design of ALTHEA Gold Library™.
In addition, the framework region 3 (FR-3) of the universal VH scaffold, being encoded by the IGHV3-23*01 germline gene, naturally binds Protein A of the bacterium Staphylococcus aureus.25,26 The Protein A binding site in the VH domain is formed by discontinuous amino acid stretches distant in the primary sequence and are brought together by folding. Therefore, Protein A offered a means to select for well-folded scFvs in the ALTHEA Gold Libraries™ construction process.
Thermostability profile of ALTHEA VH and VL scaffolds
To determine the optimal conditions for selection of functional and stable synthetic scFvs, we first studied the thermal stability and unfolding kinetics of the scaffolds combined with the CDR-H3 sequence of the CNTO-888 antibody.27 This configuration of the scaffolds, i.e., 3-20/3-23 and 4-01/3-23 combined with CNTO-888 CDR-H3, have been characterized recently,18 showing Tm values as Fabs of 75°C. We determined the thermal stability as scFvs in the phage context by displaying the scFvs as fusions to pIII and assessing Protein A binding after incubation for 10 min in a range of temperatures from 40°C to 80°C (Figure 3(a)). The 3-20/3-23 scFv started to unfold at 65°C, whereas, the 4-01/3-23 scFv did so at 55°C. The Tm, defined as 50% unfolding, of the 3-20/3-23 scFv was determined to be 75°C. The Tm of the 4-01/3-23 scFv was 65°C.
Figure 3.
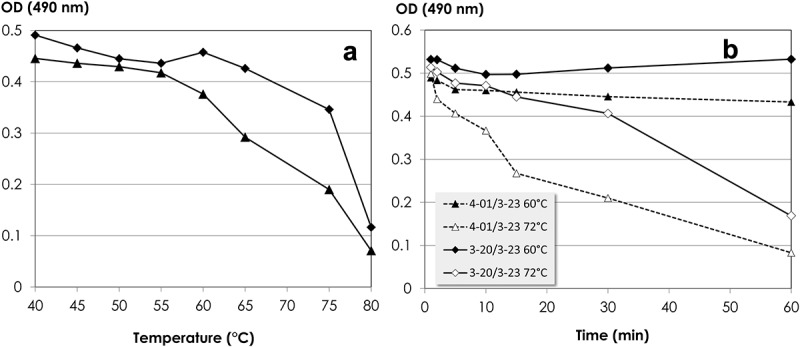
(a) Thermal unfolding kinetic of 3-20/3-23 (rhombus) and 4-01/3-23 (triangle) in the scFv configuration of ALTHEA Gold Library™. The 3-20/3-23 and 4-01/3-23 scFvs were built with the respective scaffolds and the CDR-H3 of CNTO 888 in the configuration of Figure 1. (b) Time-dependent thermal unfolding after incubation of 3-20/3-23 and 4-01/3-23 scFvs displayed on the phage surface at 60°C and 72°C.
The unfolding kinetics at 60°C and 72°C of the 3-20/3-23 and 4-01/3-23 scFvs are shown in Figure 3(b). At 60°C, both scFvs remained folded for up to one hour. At 72°C, 3-20/3-23 scFv-phage unfolded slowly with an ~20% decrease of the Protein A binding signal during the first 30 min. Afterwards, a quick drop of the ELISA signal was seen. The 4-01/3-23 scFv unfolded faster at 72°C, with a drop in the ELISA signal of ~30% in the first 10 min. The unfolding process accelerated afterwards. Therefore, we concluded that the 3-20/3-23 and 4-01/3-23 scFvs displayed in phage were stable at 60°C for at least one hour and could be incubated at 70°C for at least 10 min without significant unfolding and aggregation, i.e., <30%.
ALTHEA VH and VL scaffolds diversification
The universal VH scaffold and the two VL scaffolds were diversified at positions identified as in contact with antigens in known antigen-antibody complex structures (Supplementary Material; Library design). To this end, the structures of all the antibodies in complex with proteins and peptides deposited at the Protein Data Bank (PDB) were analyzed. The diversification regimes at the targeted positions for diversification were limited to amino acid distributions seen in natural antibodies and human germline sequences (Table 1). Amino acid residues associated with developability liabilities were avoided in the design. These residues included: 1) N followed by any amino acid but proline (XnoP) followed by serine (S) or threonine (S/T) [NXnoP(S/T)], which generate N-glycosylation sites; 2) D followed by glycine (G) [DG], which tends to isomerize; 3) N followed by G or serine [NG/S], which tends to deamidate; 4) solvent-exposed M, which tends to oxidize; and 5) solvent-exposed W, which leads to aggregation spots. The map of the diversity on the surface of scaffolds is depicted in Figure 4.
Table 1.
Scaffold diversification per CDR and per position.
| CDR | Positiona | Scaffold | ||
|---|---|---|---|---|
| VH 3-23 | VK 3-20 | VK 4-01 | ||
| H1 | 30 | ST | - | - |
| 31 | DNS | - | - | |
| 32 | YSTY | - | - | |
| 33 | AGQWY | - | - | |
| H2 | 50 | ADEGIRVWY | - | - |
| 52 | DQSY | - | - | |
| 53 | DEGRSY | - | - | |
| 55 | DGSY | - | - | |
| 56 | DGQSTY | - | - | |
| 58 | DKNRSY | - | - | |
| L1 | 30 | - | DQS | L |
| 30a | - | AHS | DGHRSY | |
| 30c | - | - | DGNSY | |
| 30f | - | - | EKY | |
| 31 | - | NST | N | |
| 32 | - | ANSY | DNRY | |
| L2 | 50 | ADGW | ADGW | |
| L3 | 91 | - | ADFGHRSY | GHQSWY |
| 92 | - | EGRSY | DQSTY | |
| 93 | - | ENQRSY | ENRSY | |
| 94 | - | AENRSWY | AENRSTY | |
| 96 | - | FILWY | FILWY | |
aAmino acid positions are defined using Chothia’s numbering (J Mol Biol. 196: 901–917, 1987).
Figure 4.
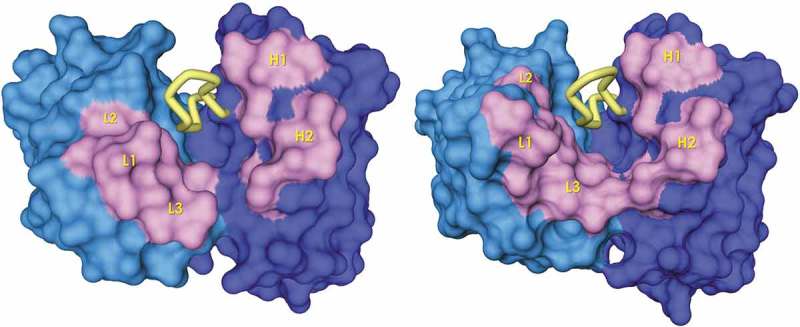
Solvent-exposed surface (antigen view) of ALTHEA Gold Library™ scaffolds. The CDR-H3 is represented as a yellow tube. The positions targeted for diversification are represented in pink. The figure was generated with a 1.7 Å-probe using Discovery Studio 2.5 (BIOVIA).
Neutral H3J fragments
We reasoned that, by using only one CDR-H3 sequence to generate the PLs, the diversity of amino acids in contact with, or nearby the CDR-H3, may be constrained to a few and specific residues to accommodate that unique loop under selection pressure for stable antibodies, thus restricting the diversity of the FLs. Therefore, a set of CDR-H3 sequences called “neutral H3J fragments” was designed by starting from the repertoire of human IGDH genes and combining them with human IGJH segments in the germline configuration (Supplementary Material; Neutral H3J fragments). We hypothesized that, since the repertoire of IGDH and IGJH segments co-evolved with the repertoire of IGHV and IGKV genes, the set of neutral H3J fragments had enough diversity to avoid biases in the amino acids at VH and VL residues in contact with or nearby the neutral H3J sequences during the selection process. We further thought that such an unbiased environment should be favorable to the selection for developable and diversified scaffolds, while supporting the cloning of natural H3J fragments in the third step of the library construction.
Construction of PLs
The designed and diversified scaffolds combined with the neutral H3J fragments were synthesized using trinucleotide phosphoramidites and assembled as scFvs in a VL-VH configuration with a glycine-rich linker (Figure 1). Trinucleotide phosphoramidites synthesis or trimer technology is based on synthetic codons instead of nucleotides, generating precise combinations of amino acids at targeted positions for diversifications while avoiding stop codons and unwanted amino acids that disrupt the folding of the scaffolds used to generate synthetic libraries.28 The quality of the synthetic fragments was assessed via Sanger sequencing of 96 fragments, indicating that 50% and 60% of sequences were in-frame and matched the designs of the 3-20/3-23 and 4-01/3-23 diversified scaffolds, respectively (Table 2).
Table 2.
ALTHEA Gold libraries™ construction process.
| PLs |
FLs |
SLs |
||||
|---|---|---|---|---|---|---|
| Librarya | PL1 | PL2 | FL1 | FL2 | SL1 | SL2 |
| Transformants (cfu) | 1.7 × 109 | 2.3 × 109 | 7.2 × 109 | 5.1 × 109 | 1.4 × 1010 | 1.1 × 1010 |
| In-frame Clones (%) | 61 | 84 | 95 | 97 | 85 | 85 |
| Protein-A Bindingb (%) | 59 | 70 | 90 | 89 | 83 | 85 |
| Heat-Shock Survivalc (%) | 49 | 56 | NDd | ND | 68 | 71 |
aData based on sample of 44 phage clones randomly picked in each library.
bDefined as more than 10% binding to Protein A relative to the binding of the control scaffolds.
cDefined as less than 10% binding to Protein A after 10 min at 70°C for phage clones having 25% or more binding to Protein A relative to the binding of the control scaffolds.
dND, not determined.
To generate the PLs, the synthetic scFv fragments were cloned in the phage display vector pADL-23c. PL1, based on 3-20/3-23 scaffolds, yielded 1.7 × 109 colony transforming units (cfu). PL2, based on 4-01/3-23 scaffolds, resulted in 2.3 × 109 cfu. Forty-four individual clones chosen at random from each PL were submitted to Sanger sequencing and showed a scFv insertion rate around 95%. All the scFv sequences were different, with 61% and 84% in-frame sequences matching the design of the 3-20/3-23 and 4-01/3-23 diversified scaffolds, respectively (Table 2). The difference in in-frame clones between the PLs (PL1 < PL2) was consistent with the difference observed in the quality of the synthetic fragments prior to cloning.
Selection for highly stable synthetic scFvs and generation of the FLs
The FLs were generated by incubating the PLs at 70°C for 10 min and rescuing the well-folded scFv variants with Protein A. This incubation condition was decided based on: 1) the unfolding kinetic of the scaffolds described above indicated that the ALTHEA VH and VL scaffolds did not significantly unfold at 70°C for 10 min; and 2) the least stable domain of the human IgG1 is the CH2,17 which unfolds at 68°C. Hence, incubation of the libraries at 70°C for at least 10 min could potentially yield a variety of stable scFvs suitable for therapeutic antibody development when expressed as IgGs.
After incubation of the PLs at 70°C for 10 min, the number of rescued clones was 7.2 × 109 and 5.1 × 109 cfu for FL1 and FL2, respectively. Considering that PL1 had 1.7 × 109 cfu and PL2 2.3 × 109 cfu, the number of rescued clones in the FLs assured a large coverage of the initial PLs diversity. In fact, analysis of 44 individual clones sequenced from each FL showed that all the sequences were different, with a proportion of in-frame scFvs of 95% and 97% for FL1 and FL2, respectively (Table 2). This indicated a significant improvement in the quality of the FLs with respect to PLs.
Natural H3J fragments and generation of the SLs
Natural H3J fragments were isolated from peripheral blood mononuclear cells (PBMCs) of a pool of 200 donors. Each donor provided 5 × 106 cells, with potentially 1 × 106 H3J fragments per donor. Hence, the pool of 200 donors contained up to 2 × 108 H3J sequences. The 200 donors included 100 females and 100 males under the age of 40 years. By limiting the age of donors to 40 years, we avoided CDR-H3 sequences from aged individuals. The repertoire of CDR-H3 sequences tend to be biased toward longer and more hydrophobic CDR-H3 loops with aging,29 in turn leading to more autoreactive antibodies than those of the repertoire of young individuals.
To simplify the RT-PCR process of the natural H3J fragments, we decided to focus on the most prevalent H3J fragments in the repertoire of circulating antibodies. To this end, we used a universal forward primer and three reverse primers. The universal forward primer was designed based on the finding that up to 95% of the circulating antibodies have a conserved motif (CM) at nucleotide level in the FR3 (Supplementary Material; Natural H3J fragments), which encodes the amino acid sequence ‘DTAVYYCA’ (residues H86 to H93), right before the CDR-H3. The reverse primers matched all the six human IGHJ germline genes (Supplementary Material; Natural H3J fragments).
The natural H3J fragments so amplified were combined with the FLs to generate the SLs, following the assembly strategy depicted on Figure 1. Briefly, the FL DNA and the natural H3J fragments were amplified by PCR with an overlap of four nucleotides GACA located at the 5ʹ-end of the CM sequence. During the amplification, a BsaI site was incorporated on each PCR product so that complementary overhangs over the GACA overlap could be generated by BsaI digestion. After purification, the FL and H3J fragments were mixed in equimolar amounts and concomitantly digested and ligated. The ligation products were further re-amplified as full scFvs before cloning in the phagemid vector to generate the two SLs independently. After electroporation, SL1 and SL2 reached 1.4 × 1010 cfu and 1.1 × 1010 cfu, respectively. The percentage of in-frame clones in a sample of 44 random clones as determined by Sanger sequencing was 85% in both SLs (Table 2), with all the clones having different sequences. This result indicated a 24% improvement of the in-frame clones in SL1 with respect to PL1.
Assessing the evolution of the functionality from PLs to SLs via Protein A
To determine whether the three-step process described above translated into a higher functionality of the libraries, the clones whose sequence was determined at each stage of the library-making (Table 2) were expressed as scFv-phage and assayed for Protein A binding at 37°C. The detailed results are provided in Supplementary Material; Protein A binding and summarized in Table 2. Most of the in-frame clones in the PLs bound Protein A. Only one clone in PL1 and four in PL2 did not bind Protein A. This resulted in 59% and 70% functional clones for PL1 and PL2, respectively. In the FLs, all the in-frame clones did bind Protein A, except two clones in FL1 and three in FL2. This yielded ~90% Protein A binders. In the SLs, only one in-frame clone in SL1 did not bind Protein A, whereas, all SL2 in-frame clones bound Protein A, for ~85% Protein A binders. It implied that the combination of natural H3J fragments with the synthetic fragments isolated from the FLs did not result in a significant loss of Protein A binding compared to the FLs, particularly for SL2.
We also assessed the gain in thermal stability of the SLs with respect to the PL by incubating the above-mentioned clones at 70°C for 10 min and measuring Protein A binding (Table 2 and Supplementary Material; Protein A binding). In PL1 and PL2, 49% and 56% of the clones survived the heat shock, respectively. The proportion of stable clones increased to 68% and 71% in SL1 and SL2, respectively. This corresponds to a significant 39% and 27% increase in thermostability for SL1 and SL2, respectively, with respect to the PLs.
Impact of the heat shock on sequence diversity as assessed by NGS
To study how incubation at 70°C for 10 min and rescuing well-folded scFvs with Protein A led to higher functionality and stability, two amplicons were prepared with plasmid DNA isolated from the PLs and FLs. Amplicon 1 covered the VL scaffolds (3-20 and 4-01), including the three VL CDRs. Amplicon 2 covered the CDR-H2 and the neutral H3J fragments. The NGS statistics are reported in Supplementary Material; NGS – PLs and FLs. In brief, over half a million sequences were obtained from the PL1 Amplicon 1 and close to 1.5 million for FL1. We observed a clear difference in the curated sequences from 76% up to 95% in PL1 with respect to FL1, suggesting that the filtration process enhanced the quality of the FL1 readable and productive sequences. For PL2 and FL2, the total number of sequences was higher than for PL1/FL1, with over 1.5 million for PL2 and over two million sequences for FL2. The number of curated sequences in both PL2 and FL2 was higher (96%) than for PL1 and similar (91%) to FL1. This observation again agreed with the suggestion that the quality of synthetic PL2 fragments was superior than that of PL1. The number of total sequences in Amplicon 2 for PLs and FLs was similar, with close to 1.5 million sequences for each library. The curated sequences were also similar, close to ~90%.
Analysis of the frequency of the neutral IGDH fragments contained in Amplicon 2 before and after the filtration process (Figure 5) indicated that all the designed fragments were represented in the PLs. The frequency of the designed fragments was within 2% of the expected value of 6% of an even distribution (1/18 neutral fragments) and similar in both PLs, suggesting no major bias due to the PLs preparation. The five IGHJ germline genes were also within 4% of their even distribution (20%) with a slight (5%) increase after filtration of IGHJ3 and IGHJ6 at the expense of IGHJ1 and IGHJ4 (data not shown). Again, the IGHJ frequencies were similar in both libraries, consistent with the suggestion that no major bias was introduced in the PLs preparation.
Figure 5.
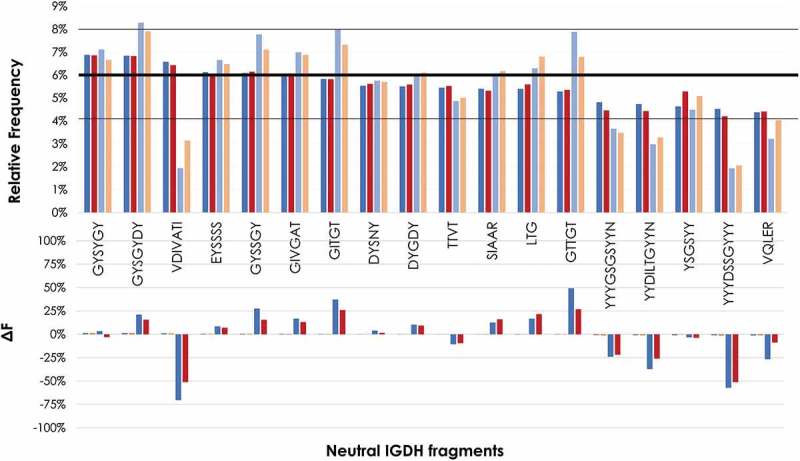
Frequency of eighteen neutral IGHD fragments before and after filtration as determined by NGS. Top panel: Frequency of PLs and FLs. The first two bars per IGHD correspond to the PLs (PL1: blue; PL2: dark red). The other two bars per IGHD correspond to the FLs (FL1: light blue; PL2: orange). A horizontal bold line at 6% represents the expected frequency of an even distribution of the 18 IGHD fragments. Two black lines at 4% and 8% represents the range of the frequency variation in the PLs. Bottom panel: Difference in frequency (ΔF) of FLs with respect to PLs. ΔF was calculated as: IGHD frequency of the FL minus IGHD frequency of PL and normalized by IGHD frequency of PL x 100; that is: [(IGHD of FL – IGHD of PL)/IGHD of PL] x100.
After the filtration process, the difference in frequency (ΔF) between PLs and FLs of some neutral IGDH fragments increased by more than 25% in both FLs with respect to the PLs, whereas other neutral IGDH fragments decreased by more than 50%. Although FL1 showed larger variations than FL2, both FL1 and FL2 showed a similar trend. Of note is the IGDH fragment “VDIVATI”, which decreased by close to 75% in PL1 and slightly over 50% in PL2. Interestingly, five of the seven amino acids of this IGDH fragment are hydrophobic. The longest IGDH fragments, which are rich in tyrosine (Y) residues, also showed a consistent decrease after the filtration process. In contrast, the IGDH fragments “GITGT” and “GTTGT”, which are mostly hydrophilic, showed a significant increase in their frequency after the filtration. Moreover, these two IGDH fragments differ by only one amino acid [isoleucine (I) to threonine (T) in position 2], with the fragment with the replacement to T being the one with a slightly higher frequency.
A similar analysis of the CDR-H2 contained in Amplicon 2 is shown in Figure 6. Overall, no significant ΔF change was seen between the two PLs, with ΔF below 10% when compared PL1 and PL2. This was in agreement with the previous observation that neither the cloning process nor the NGS data generation introduced a significant bias in the observed amino acid frequencies. After filtration, however, several similar changes were observed in both FL1 and FL2. The major change occurred at position H50, where the ΔF of isoleucine (I) between PLs and FLs decreased by ~50%, whereas, the ΔF of other hydrophobic amino acids such as valine (V) and W decreased by 20% or more, together with Y. The ΔF of G increased by more than 30% at H50. Y also decreased in the other diversified positions. Additional major adjustment after filtration occurred in the ΔF of glutamine (Q) in positions H52 and H56. Also, charged residues such as lysine (K) and arginine (R) in position H58, as well as serine S, increased their ΔF as a result of the filtration process. These increases, at the expense of a decreased Y, should have increased the polarity of the CDR-H2.
Figure 6.

Frequency of amino acids at the diversified positions in the CDR-H2 before and after filtration as determined by NGS. ΔF was calculated as in Figure 5 but for the amino acid frequency at a given position “i”, i.e., ΔFi = [(Amino acid Frequency at FLi – Amino acid Frequency at PLi)/Amino acid Frequency at PLi] x 100.
The CDR-L1 (Figure 7) and CDR-L3 (Figure 8) contained in Amplicon 1 also showed differences when comparing the ΔF of PLs and FLs. Overall, PL1:FL1 showed higher amino acid frequency adjustments than PL2:FL2 in both CDR-L1 and CDR-L3. This is consistent with the trend observed in Amplicon 2, where FL2 showed major adjustments compared to FL1. In the CDR-L1, the PL1:FL1 major ΔF changes occurred at position L32, with an increase of ~40% and 20% in the ΔF of asparagine (N) and S, respectively. In contrast, an ~35% ΔF decrease of Y occurred at that position. In PL2:FL2, relatively small but noticeable ΔF increases of around 10% were observed in several amino acids at positions L30a and L30c, with a significant ΔF decrease (~25%) of Y in position L30a and ~15% in position L30c. In the CDR-L3 of PL1:FL1, noticeable changes occurred in almost all positions (L92 to L94). In L92, a significant ΔF increase in glutamic acid (E) occurred upon filtration with a decrease in R. In position L93, an increase in E was observed, together with an increase in N and aspartic acid (D). In position L94 of FL2, a ΔF increase in E occurred upon filtration together with an increase in R and Y, in contrast to the ΔF decrease of S.
Figure 7.
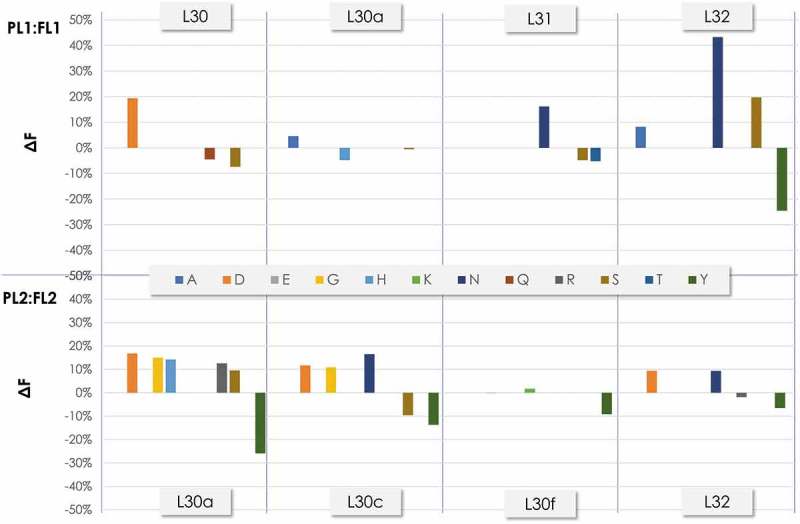
Frequency of the amino acids at the diversified positions in CDR-L1 before and after filtration as determined by NGS. ΔF was calculated as in Figure 6.
Figure 8.
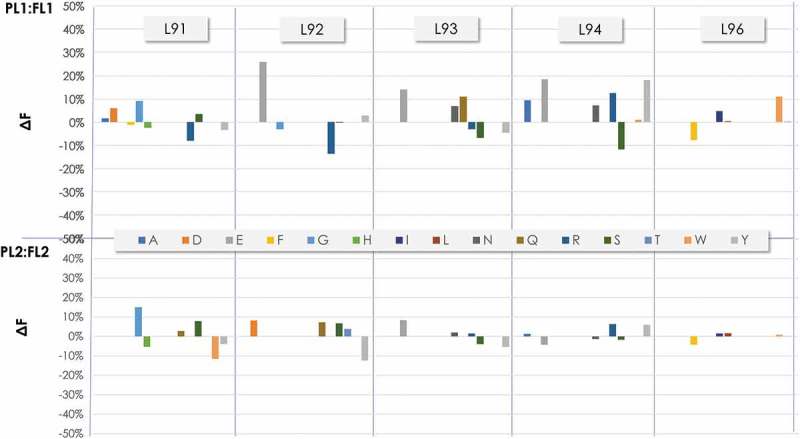
Frequency of the amino acids at the diversified positions in CDR-L3 before and after filtration as determined by NGS. ΔF was calculated as in Figure 6.
To understand whether the most significant ΔF changes observed upon filtration were corelated, we mapped the positions with the most significant ΔF variation onto the structure of the scaffolds (Figure 9(a,b)). In VH, major ΔF changes occurred in the side-chain of residues at positions H50 and H58, which are neighbor residues from parallel β-strands (Figure 9(c)). These residues point in the same direction, toward the interface with VL. The side-chain of H50 is mostly buried in the interface with VL and is in contact with side-chains of CDR-H3 residues; note that, in the structure, the methyl group of A H50 is at 3.4 Å of the hydroxyl group of Y H96. In the library design, given the buried nature of H50, it was diversified with hydrophobic residues. After the filtration process, however, hydrophobic residues (I and V) and bulky aromatic residues such as W and Y were selected negatively. In contrast, a positive selection of residues with small side-chains such as the germline alanine (A) and a high proportion of G occurred. Due to the proximity of H50 with the CDR-H3, this result was probably a consequence of the selection for hydrophilic IGDH fragments (see above). These changes, together with the negative selection of Y and positive selection of charged residues such as K and R at H58, should have generated a polar environment in this region of the antigen-binding site. If so, it could explain the increase in solubility and thermal stability of the scFv variants in the FLs.
Figure 9.
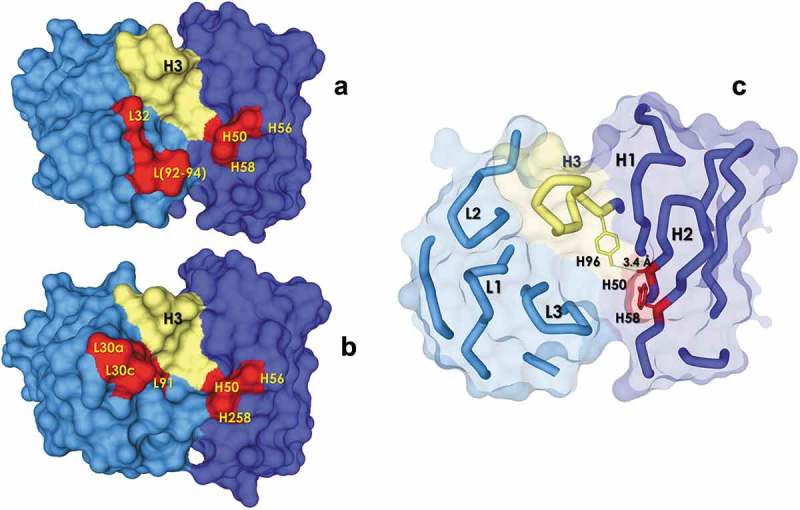
Map of the positions with a higher ΔF on the solvent exposed surface (antigen view) of the ALTHEA Gold Library™ primary scaffolds (a) SL1 and (b) SL2. The CDR-H3 is represented as a yellow tube. The positions a higher ΔF are represented in red. The Figure was generated with a 1.7 Å-probe using Discovery Studio 2.5 (BIOVIA). (c) H50 and H58 positions showing proximity between them and with the CDR-H3.
In VL, ΔF differences after filtration were more noticeable in PL1:FL1 with, in particular, a ΔF increase in N and S in L32 at the expense of a ΔF decrease of Y. In less proportion, D and N increased at L30 and L31, respectively. In the CDR-L3, a ΔF increase of charged residues (E) at positions L92 to L93 was seen, together with a ΔF increase of Y in position L94. Figure 9(a,b) show that, as in VH, these VL residues are in close proximity in the structure. Residues L30-32 are at the tip of the CDR-L1 with position L32 close to the CDR-H3, where the germline Y decreased, suggesting, together with a higher frequency of charged residues in the CDR-L3, an increase in polarity after filtration, like the observation made at VH. Therefore, it appears that the filtration process increased the stability of the FLs by: 1) favoring polar amino acids over hydrophobic ones, and 2) removing the excess Y in positions close to the CDR-H3 and the VH:VL interface.
Diversity of the natural H3J fragments
Analysis of the SLs was focused on the H3J diversity. We generated Amplicons 2 of the SL1 and SL2 following the same procedure as for PLs and FLs. The NGS statistics are reported in Supplementary Material; NGS – SLs. One million sequences were obtained for SL1 and over one million for SL2. The number of accepted sequences was around 80% in both SLs. The CDR-H3 length distribution (Figure 10(a)) followed a Gaussian curve typical of the human antibodies,7 with a peak at 12 amino acids in length. No difference was observed between SL1 and SL2, indicating that the pool of natural H3J fragments from the 200 donors was not biased towards a particular CDR-H3 length during cloning and/or the SLs preparation. As in the CDR-H3 length distribution, the IGHJ1-5 segments (Figure 10(b)) followed the expected usage seen in human antibody sequences except for IGHJ6. IGHJ6 had a lower frequency than expected in natural human antibodies31. The reported IGJH6 frequency is ~40%, followed by IGHJ4 with ~30%. In our case, IGHJ4 was the most prevalent IGHJ gene segment with ~40%, followed by the IGHJ6 with 20%. However, it was so designed during the RT-PCR amplification of the natural H3J fragments. The IGHJ6 encodes a stretch of five Y residues in the N-terminal region, which could lead to a destabilizing effect (see discussion section). Lowering the proportion of natural H3J fragments encoded by IGHJ6 was expected to allow selection of a higher number of developable antibodies. Of note, alleles “02” of the IGHJ3 and IGHJ5 genes were more frequent than alleles “01”. These alleles may represent predominant IGHJ3 and IGHJ5 genes in the sample of the 200 donors.
Figure 10.

(a) Natural H3J fragments CDR-H3 length distribution (Kabat’s definition)30 as determined by NGS. (b) Natural H3J fragments IGHJ distribution as determined by NGS. SL1: blue. SL2: red.
A more detailed analysis of the most prevalent 30 CDR-H3 sequences (Supplementary Material; NGS – SLs) indicated that the top sequences in SL1 and SL2 have around 1,000 copies, implying than these CDR-H3 sequences are in a 0.1% or less frequency in the sample of over a million sequences. In addition, the top 30 sequences of each library barely add up to 1%, meaning that 99% of all CDR-H3 sequences have only a few copies. Moreover, when comparing the most prevalent SL1 and SL2 sequences, only half are shared between the two SLs, pointing to the fact that the strategy of amplifying the natural H3J fragments with a universal forward primer and three reverse primers yielded a very diverse set of sequences.
Validation of ALTHEA gold libraries™ using diverse targets
Finally, to assess the potential of the SLs, ALTHEA Gold Libraries™, to produce specific, high affinity and highly stable antibodies, exploratory selections were performed with three well-known proteins: 1) human serum albumin HSA, a serum protein used to obtain antibodies that in conjunction with therapeutic antibodies has the potential to increase their half-life;32 2) tumor necrosis factor (TNF), a well-known target with several therapeutic antibodies in the market, including adalimumab, the top-selling drug worldwide;33 and 3) hen egg-white lysozyme (HEL), one of the protein models par excellence in studies of the antigen-antibody interaction and antibody engineering.22,34 A summary of the results of selections with these three targets is shown in Table 3.
Table 3.
Exploratory panning of ALTHEA Gold libraries™ with three disclosed targets.
| Unique clonesc |
KD (nM)f |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Target | Selectiona | Hit rateb | SL1 | SL2 | Yield (mg/L) d | Monomeric Content (%)e | Fab | IgG | Tm (°C)g |
| TNFα | IM, R4, EA | 20% | 1 | 1 | 50–60 | 92–97 | 590 | 0.094–8 | 77–80 |
| HSA | EP, R3, AE | 83% | - | 3 | 90 | 95 | 3.3 | 0.8 | 81 |
| HEL | MB, R3, AE | 6%-68% | 3 | 30 | - | - | - | - | - |
aIM: Immunotubes; R4: Four rounds of panning; EA: Elution with Adalimumab; EP: ELISA plates; R3: Three rounds of panning; AE: Acid Elution; MB: Magnetic beads.
bDefined as number of positive and specific clones (no BSA binding) divided by the number of assayed clones (44).
cDefined as unique sequences as determined by Sanger sequencing.
dEstimated concentration of IgG in the HEK293 culture.
ePercentage of protein in the peak of the proper size (~150 KDa) relative to the total mass as estimated by SEC-UPLC.
fKD estimated by BIAcore or ELISA; See Supplementary Material, Figure sF3.
gTm of the second thermal transition corresponding to Fab unfolding as determined by Thermal-Shift Analysis.
The HSA and TNF selections were performed in solid phase (Immunotubes or ELISA plates). For TNF selections, ALTHEA Gold Libraries™ (SL1 and SL2) were used in parallel. HSA selections were focused on SL2. In the case of HEL, the selections were also performed with both S1 and SL2 but in solution, with biotinylated HEL and streptavidin-coated magnetic beads to pull down the bound phages.
After the fourth round of selection with TNF, the specific phages were eluted by competition with adalimumab. The hit rate of SL1 was very low, with only one positive clone out of 44 (2%) assayed. In the case of SL2, the hit rate was higher 7/44 (16%). Sanger sequencing of the positive clones revealed two unique scFvs, one from each library. The HSA selections consisted of three rounds of panning, with 38 positive clones out of 44 (83%) assayed. Sanger sequencing of the positive clones indicated three unique scFvs. Solution panning for HEL after three rounds in solution yielded a hit rate of 6% (3/44) for SL1, with all three clones being unique. For SL2, the hit rate was 68% (30/44), with 30 unique sequences found once and one sequence showing up twice.
The HSA unique clone with a higher ELISA signal and the two positive TNF clones were converted to Fabs and IgGs for further characterization (see Table 3). The KD values of the three IgGs were in the single-digit or sub-nM range. The thermal stability (Tm) of the second transition, corresponding to the Fab, was above 75°C. These biophysical parameters, together with expression levels of 50–100 mg/L in transient expression, without optimization, and monomeric content of 90% or more after a single Protein A purification, are consistent with the developability profile of antibody candidates amenable to further development. The analysis of the HEL clones is still in progress.
Additional exploratory selections of ALTHEA Gold Libraries™ have been performed in our laboratory or in collaboration with other laboratories. These additional selections include four therapeutic targets: 1) a therapeutic enzyme of ~45 kDa, for which several attempts to obtain antibodies using other means, including immunization, failed; 2) a major histocompatibility molecule (MHC) in complex with a relevant peptide; 3) a therapeutically valuable ~17 kDa human protein extra-cellular domain (hECD); and 4) its mouse orthologue mECD, which shares only 50% identities with hECD. Highlights of the most relevant results are listed in Table 4.
Table 4.
Exploratory panning of ALTHEA Gold libraries™ with four undisclosed targets.
| Specific scFvs |
|||
|---|---|---|---|
| Target | SL1 | SL2 | Highlights |
| MHC/peptide | - | 1 | scFvs highly specific for the peptide as assessed by site directed mutagenesis. |
| Enzyme | 1 | 1 | No antibodies from other platforms were obtained. Best clone has a KD = 100 nM as scFv. |
| hECD | 2 | 3 | No-cross reactivity with mouse ortholog. Best clone has KD = 2 nM as scFv. |
| mECD | - | 4 | Two clones cross-reacted with the human ortholog. Best clone has KD = 60 nM as IgG1. |
The selections with the therapeutic enzyme yielded 15 positive clones out of 44 (34%) tested for binding to the target and BSA, with two unique specific scFvs as assessed by Sanger sequencing. The panning with the MHC/peptide was performed with subtraction using an irrelevant peptide in complex with the MHC molecule. One scFv specific for the relevant peptide was obtained. The scFv recognized specific solvent-exposed amino acids of the peptide as determined by site-directed mutagenesis of the peptide (data not shown). The hECD yielded 11 of 44 (25%) positive clones with five specific and unique scFvs. The best scFv showed a KD = 2.8 nM as assessed by BIAcore. Two other scFvs were converted to human IgGs and showed KD values of 1 nM and 10 nM. None of these three scFvs cross-reacted with the mouse ortholog, thus indicating that the scFvs were highly specific for the human target. The mECD yielded four specific antibodies; one of them was converted to IgG and the avidity assessed by ELISA yielding 20 nM.
In summary, ALTHEA Gold Libraries™ have been tested thus far with six non-related and diverse targets, plus two related, 50% identical, molecules. At least one specific antibody was obtained for each target, demonstrating the potential of ALTHEA Gold Libraries™ to generate specific antibodies. Three antibodies against two non-related targets (HSA and TNF) where further characterized, and shown to be high affinity and highly stable molecules.
Discussion
More the three decades ago, the seminal works by George Smith35,36 and Greg Winter6 gave birth to the phage display technology as a platform for peptide and antibody discovery. In the subsequent thirty plus years practicing phage display, it has been well established that the size and functionality of a library are critical parameters for success in isolating specific and high affinity antibodies from said library. Early selections from antibody phage-displayed libraries of 107 members generated ~90 nM antibody fragments to diverse proteins.12 Further studies using larger libraries, e.g., >1010 members,8,13 produced antibody fragments with single-digit nM or sub-nM affinities. Similarly, a synthetic library called the Griffiths’ library9 of >1010 members produced sub-nM binders, whereas antibody fragments of >100 nM were obtained when a small portion of the library containing only 107 clones was used in the selection process.
In parallel, as therapeutic antibody engineering and development evolved in the past decades, and dozens of antibodies were granted marketing approvals, it has been realized that reaching the desired specificity and affinity is not enough.4,16 Other biological and biophysical properties such as expression, stability and solubility should be considered during the early discovery phase, so that the risks of failure in the late therapeutic development phase are minimized. This is particularly true for antibodies isolated using phage display, where the discovery process is performed in vitro, without the filters imposed in vivo. In such cases, the selected antibodies must be extensively scrutinized to ensure they meet the proper threshold of stability, solubility and low aggregation at the concentrations needed for therapeutic development.17
We approached the design and construction of ALTHEA Gold Libraries™ keeping these concepts in mind, i.e., generation of large and functional libraries with enhanced stability and low aggregation. We followed a three-step construction process that led to a progressive increase in functionality and stability, culminating in a valuable source of antibodies, as showed by panning the libraries using different strategies and an array of seven non-related targets. In all the cases specific antibodies were obtained. The best anti-HSA antibody and the two anti-TNF ones had a biophysical profile that meet the success criteria of antibody candidates amenable to further development. In other instances, as with the MHC/peptide and hECD antigens, the antibodies were shown to be highly specific for the peptide or the human ortholog. Further characterization of the antibodies isolated with the hECD yielded single-digit nM antibodies after IgG conversion or as scFvs, the latter probably leading to sub-nM affinity after IgG conversion. These affinities are comparable with antibodies isolated from large libraries (see above). In our case, most of the selections were performed in solid phase and only a few clones (44) were screened and further characterized. Screening a larger number of clones (one thousand or more), as it is typically done in discovery campaigns, would likely yield more and even higher affinity antibodies. Other selection methods may lead to a higher discovery rate as well. In fact, the HEL panning was done with magnetic beads, which provides a higher apparent concentration of target. Sequencing of a limited number of positive clones pointed to a very high diversity of binders, indicating that the ALTHEA Gold Libraries™ have the potential to perhaps generate tens to hundreds of specific antibodies for each target.
To generate ALTHEA Gold Libraries™, our strategy departed from the classical linear library construction approach. We decoupled the selection for high stability from the diversity at the CDR-H3, which is a key element in defining the specificity and affinity of antibodies. First, we created fully synthetic libraries and selected for improved stability. Then, we combined the selected synthetic variants with natural H3J fragments derived from a large pool of donors. In so doing, we maximized the diversity of the natural H3J fragments while benefiting from the stability gained after submitting carefully designed synthetic libraries to harsh destabilizing conditions.
Selection for enhanced stability with Protein A combined with phage display has been used extensively.37–39 For instace, Jespers et al.38 submitted antibody libraries to incubations at 80°C and folded domains with enhaced thermal stability were selected with Protein A. Using the same library, Famm et al.37 extended the method to select for acid aggregation resitance. In both cases, however, the diversity of the initial pool of clones was reduced dramatically, i.e., the gain in stability was at the expense of the diversity. In our case, fresh diversity provided by the natural H3J fragments restored the diversity of the stable scaffold variants selected with Protein A after incubation at 70°C for 10 min.
It should be emphasized that, instead of generating fully synthetic final libraries by means of synthetic CDR-H3 fragments, we relied on natural H3J fragments to avoid making assumptions on the structure, length distribution and type of amino acids to be considered in the design. The natural H3J fragments were derived from a large pool of 200 donors to compensate for biases introduced by the antibody repertoire or immunological history of a few individuals. We also limited the age of donors to 40 years to avoid CDR-H3 sequences biased toward longer and more hydrophobic CDR-H3 loops. These decisions, together with the assumption that most of the natural H3J fragments were in-frame, led us to expect that most of the clones from the final semisynthetic libraries would be highly diverse and functional. In fact, the functionality of ALTHEA Gold Libraries™ (SLs) increased to ~85% of Protein A binders from ~65% Protein A binders in the PLs, implying a 20% improvement. In terms of number of functional clones in a library of 1010 variants, as the SLs are, this increase provided 2 × 109 additional antibody sequences to select from.
It was not obvious from prior knowledge that combining the stable variants comprising the FLs with diverse natural H3J fragments, which have not evolved to be stable at 70°C, would result in such an increase of 20%. Several factors may have led to this result. First, the selection of well-known and stable scaffolds to build the libraries. Second, the careful design of diversity based on positions often found in contact with antigens and, for the most part, solvent-exposed. Third, the combination of the diversified scaffolds with a set of neutral designed H3J fragments in the germline gene configuration. Fourth, the filtration process, which removed the excess of hydrophobic residues and tyrosine residues (see below) in certain diversified positions. Together, these factors led to a collection of highly stable variants in the FLs suited to accommodate, without a significant loss in stability, the repertoire of highly diverse and functional natural H3J fragments.
Actually, analyses of the ALTHEA Gold Libraries™ construction process by NGS revealed important lessons for future library designs. Over a million sequences obtained by NGS from the PLs and FLs indicated an increase in polar residues and a decrease in hydrophobic and Y residues in some designed positions after submitting the PLs for heating at 70°C for 10 min and rescuing the well-folded variants with Protein A. Mapping these positions in the structure of the scaffolds indicated that they are spatially close, and thus the changes observed upon filtration may be correlated. In a previous work,19 we noticed that polar amino acids, particularly R, N, D, S, T and G frequently occur at the antigen-binding site. Non-polar amino acids such as A, I, V, M, L and F are significantly underrepresented. Y was found in a high proportion in the antigen-binding site, consistent with previous studies. For instance, Lo Conte et al.40 observed that Y contributed to 17% of all amino acids in contact in the 19 antigen-antibody complexes available at that time. Similarly, an earlier work by Mian et al.41 reported the overuse of Y to contact antigens. Based in part on these early works and the observation that Y is a versatile amino acid, capable of generating diverse interactions with the antigens, Sidhu et al.42 elegantly showed that by creating antibody libraries with only Y and S or W and S, specific antibodies with affinity in the nM range can be obtained, similar to those obtained from more complex libraries.
Interestingly, Y was shown to be a destabilizing residue in certain positions of the antigen-binding site.43 Therefore, a phage display library for therapeutic antibody discovery needs to balance a high Y content, to generate versatile antigen-binding sites capable of producing highly specific and high affinity antibodies, but with not too high Y content, so that the stability and solubility of the potential therapeutic antibodies are not compromised. In our case, the filtration process seemed to have corrected the excess of Y and hydrophobic residues designed based on the available information on the structures, human antibody sequences and the repertoire of human germline genes. This “correction” led to a collection of highly stable diversified scaffolds capable of meshing well with the natural H3J fragments.
Finally, it should be noted that natural ligands other than Protein A bind folded variable regions of antibodies outside the antigen-binding site. For example, Protein L from the bacterial species Peptostreptococcus magnus and Protein M from mycoplasma strains, such as Mycoplasma pneumonia, Mycoplasma iowae, and Mycoplasma gallisepticum both bind the VL domain of antibodies.44 Protein M binds both kappa and lambda type antibodies.44 Protein L binds kappa antibodies encoded by the human IGKV-1, IGKV-2 and IGKV-4 gene families.45 More specifically, Protein L binds the IGKV4-01*01 germline gene, which is the VL scaffold of SL2. Although we did not use neither Protein L nor M to select well-folded antibodies, these ligands can be utilized alone or in conjunction with Protein A to prepare libraries with other scaffolds. For instance, the human VH domains of antibodies encoded by the gene families IGHV-1, IGHV-2, IGHV-4, IGHV-5, IGHV-6 and IGHV-7, which do not bind Protein A,25 could be diversified, combined with the neutral H3J fragments describe in the previous sections, paired with libraries of VL domains encoded by scaffolds built with members of the human IGKV-1, IGKV-2 and IGKV-4 gene families or scaffolds built with lambda-type antibodies, and submitted to diverse destabilizing conditions to select for well-folded antibodies with Protein L or M. The resultant libraries may serve as a substrate to build secondary libraries with natural H3J fragments to yield diverse antibody libraries for therapeutic antibody discovery, thus generalizing the ALTHEA Gold Library™ construction method here described to all the IGHV and IGLV genes of the human antibody repertoire.
Material and methods
PLs construction
The designs of the diversified VH and VL scaffolds were synthesized using trimer phosphoramidite technology in VL-linker-VH scFv configuration with IGKJ1*01 as joining region for the light chains, GS19 (GGGGSGGGGSGGGSGGGGS) as linker, and 90 neutral H3Js on the C-terminal side of VH 3-23. Two BglI/SfiI sites on each side were added for in-frame cloning in the pADL-23c vector (Antibody Design Lab, San Diego, CA) between the pelB leader peptide for periplasmic expression and the tags for detection. One microgram of each synthetic fragment 3-20/3-23 or 4-01/3-23 was digested with BglI restriction enzyme overnight at 37°C and ligated into the phagemid vector. After electroporation into electro-competent TG1 cells (Lucigen, Madison, WI), transformants were rescued in 2xYT medium supplemented with ampicillin at 37°C in the presence of glucose 1% w/v. Cells were harvested after overnight incubation, resuspended in fresh 2xYT medium supplemented with ampicillin and subsequently superinfected with M13KO7 helper phage (Antibody Design Labs, Cat No.: PH010L). No more than 55 min after transduction, kanamycin 50 µg/ml was added, the temperature was lowered to 30°C and the incubation was prolonged overnight. The day after, virions were purified by PEG precipitation following standard procedures.46
FLs generation
Twenty PCR tubes, each containing 100 µl of virions from PL1 at a concentration of 1.3 × 1013 virions/ml were, incubated at 70°C for 10 min in a thermal cycler and cooled down for 30 min on ice. In the meantime, 2 ml of magnetic beads (BioMag® Streptavidin, Cat No.: 84660-5 from Polysciences, Inc., Warrington, PA) were washed 2 times with tris-buffered saline (TBS) with 0.1% Tween 20 (TBST) and incubated with 500 µg of biotinylated Protein A (Life Technologies, Cat No.: 29989) for 30 min with agitation. After 3 washes with TBST, the beads were aliquoted in 10 microfuge tubes, resuspended in 400 µl TBST with 5% w/v nonfat dry milk and blocked for 30 min at room temperature. One-hundred µl of heated library were added to each tube and incubated for 2 h at room temperature on a rocker. After 5 washes with TBST and 5 washes with TBS, the bound phages were successively eluted with Trypsin-EDTA (Life Technologies; Cat No.: 25200056; 500 µl per tube) and 0.1 mM glycine pH 2.7 containing 1 mg/ml bovine serum albumin (BSA; 500 µl per tube) for 10 min at room temperature. After neutralization of the acid eluate with 1 M Tris pH 8.0, both eluates were combined and used to transduce XL10 Gold cells (Agilent Technologies, San Diego, CA). Bacteria were grown overnight at 37°C in 2xYT medium supplemented with ampicillin and 1% w/v glucose. The day after, plasmid DNA were prepared using a DNA MIDI kit (Macherey Nagel, Germany). For PL2, only 10 tubes containing each one 100 µl of virions at a concentration of 1.3 × 1013 virions/ml were processed in the same way for a total of half the number of initial virions.
Natural H3J fragments
The natural H3J fragments were obtained from the PBMCs of 200 healthy donors. Starting from the PBMCs, total RNA (tRNA) was individually isolated using Trizol (Invitrogen; Cat Nos.: 15596026 and 15596018). Pools of tRNAs from 10 donors were generated after determining the concentration by UV absorption and mixing the donors tRNAs in equal amounts to generate 20 tRNA pools. Each of the 20 pools were processed to isolate messenger RNA (mRNA) using the polyA Spin™ mRNA Isolation Kit (NEB, Cat No.: S1560S) following the manufacturer instructions. mRNA was used as template to generate cDNAs by reverse transcription using the OneTaq® RT-PCR Kit (NEB, Cat No.: E5310S) and a poly-T oligo. Double-stranded DNA containing the repertoire of natural H3J fragments was obtained by PCR using the forward primer ALhuVHFR3com_s 5ʹ- CACAGGTCTCGGACACGGCYGTGTATTACTGTGC containing a BsaI site (underlined) and annealing to CM and three reverse JH primers:
ALhujh1245_r: 5ʹ-TGTTGGCCTCCCGGGCCTGAGGAGACRGTGACCAGGGT,
ALhujh3_r: 5ʹ-TGTTGGCCTCCCGGGCCTGAAGAGACGGTGACCATTGTCC,
ALhujh6_r: 5ʹ-TGTTGGCCTCCCGGGCCTGAGGAGACGGTGACCGTGGTC,
all containing a BglI site (underlined). The quality of H3J fragments was assessed by cloning an aliquot of the final pool into a TOPO vector (Life Technologies). Sanger sequencing of 30 clones indicated that all H3J fragments were different, with length variation resembling the human CDR-H3 repertoire. The region introduced by the amplification primers for assembling the full scFvs and cloning into the vector 100% matched the expected nucleotide sequence.
SLs construction
The region corresponding to VL, the GS19 linker and VH just before the CM were amplified by PCR from the DNA isolated from the filtered primary libraries. The reverse primer was extended on its 5ʹend by the sequence 5ʹ-CACAGGTCTCG. For each library, natural H3J fragments (150 ng) were assembled with the primary filtered DNA fragments (600 ng) by simultaneous digestion with BsaI and ligation with T4 ligase for 4 h at 37°C. Sixty ng of the joined products (~7 × 1010 molecules) were further amplified by nested primers in separate PCR reactions. The two semisynthetic fragments so generated were ligated into the pADL-23c phagemid vector. A total of 5 µg of ligated products was electroporated into electro-competent TG1 for each library.
Protein A binding assay
COSTAR plates 3369 (Corning) were coated with Protein A (Sigma Aldrich, Cat No.: P6031) at 4 µg/ml in TBS overnight at 4°C. After blocking with TBST and 5% w/v nonfat dry milk for one hour, virions (~2.6 × 1012 virions/ml or a 1:4 v/v culture supernatant dilution) in TBST with 5% w/v nonfat dry milk were added to the wells in 2-fold serial dilutions and incubated for 2 h at 37°C. As a reference, virions derived from the parent scaffolds with the CDR-H3 of CNTO-888 cloned in the same vector were added and similarly diluted on the plate. Bound phages were detected with A4G1.6, a murine IgG1 monoclonal antibody conjugated to horseradish peroxidase (HRP) (Antibody Design Labs Cat No.: AS002). Binding of A4G1.6 to Protein A was inhibited by a large excess of polyclonal human IgG (100 µg/ml) added to the incubation buffer.
Amplicon preparation and NGS data generation
Plasmid DNA from PLs, FLs and SLs were purified using QIAGEN Plasmid Midi Kit (Cat No.: 12143) and used as templates to generate two amplicons of approximately 300 bps. The PCR reactions were performed as follows: 5 min start at 95°C followed by 10 cycles of 1 min at 95°C, 1 min at 67°C, 1 min at 72°C and terminated by a 10-min extension at 72°C. The PCR fragments were gel-purified using QIAquick PCR Purification Kit (Cat No.: 28104) and used as template to prepare the samples for the NGS following the manufacturer’s instructions. The NGS was performed in a Miseq platform from Illumina. FASTQ files were processed with the software AptaAnalyzer™ (AptaIT; Germany) using the BRC (B-cell receptors) functionality. The accepted output sequence files were further curated with in-house scripts to remove very short or long fragments not matching the sequence of the scaffolds. Truncated CDR-H3 sequences without the conserved cysteine H88 and tryptophan H102 were removed from the analyses.
Exploratory panning with diverse targets
Exploratory selections were performed in solid phase using either Immunotubes (Thermo Scientific; Cat No.: 444202) or ELISA plates (Thermo Scientific; Cat No.: 44-240421). The solid phase was coated with the target in a carbonate buffer at pH 9.0. Aliquots of 1011–1012 cfu from ALTHEA Gold Libraries™, covering 10 to 100 times the initial library diversity, were used in the first round of panning. Subsequent rounds were performed with the output of the previous round at 1012 cfu, after growing the eluted clones in agar plates overnight. After the third and fourth rounds of panning, direct phage ELISAs were performed to assay for the presence of specific clones. If a significant increase in OD between rounds was observed, screening for specific and soluble scFvs or phage clones followed in 96-well format with the target and BSA as negative control. As reporter reagent, Protein A/HRP (Thermo Fisher; Cat No.: 101023) was used for free scFv detection and specific binders were sent for Sanger sequencing and, after sequence clustering, unique clones were expressed as soluble scFvs and re-tested for binding to the target and BSA. The confirmed clones were expressed in 100-mL cultures and purified using HisTrap™ (GE Healthcare; Cat. No.: 29-0510-21). Affinity of the purified scFv was measured with a BIAcore.
Abbreviations
- A
Alanine
- ASA
Accessible Surface Area
- C
Cysteine
- CDR-H1
Complementarity-Determining Region 1 of VH
- CDR-H2
Complementarity-Determining Region 2 of VH
- CDR-H3
Complementarity-Determining Region 3 of VH
- CDR-L1
Complementarity-Determining Region 1 of VL
- CDR-L2
Complementarity-Determining Region 2 of VL
- CDR-L3
Complementarity-Determining Region 3 of VL
- CFU
colony transforming unit
- D
Aspartic acid
- E
Glutamic acid
- ECD
extra-cellular domain
- F
Phenylalanine
- FL
Filtrated Library
- FR
Framework region
- G
Glycine
- H
Histidine
- H3J
CDR-H3/JH fragments
- I
Isoleucine
- JH
Joining region of VH
- K
Lysine
- L
Leucine
- M
Methionine
- N
Asparagine
- NGS
Next-Generation Sequencing
- P
Proline
- PBMCs
Peripheral Blood Mononuclear Cells
- PL
Primary Library
- Q
Glutamine
- R
Arginine
- S
Serine
- scFv
single-chain variable fragment
- SL
Secondary Library
- T
Threonine
- Tm
Melting Temperature
- V
Valine
- VH and VL
Variable region of the heavy and light chain, respectively
- VL
variable light chain
- W
Tryptophan
- Y
Tyrosine
Acknowledgments
We would like to thank Lew Kotler and Jan Kieleczawa at Wyzer Biosciences for their assistance in designing the NGS amplicons and generating the NGS data. To Luis Vallejo and Said Vasquez for technical assistance with the expression, purification and biophysical characterization of the antibodies isolated from ALTHEA Gold Libraries™. This work was partially supported by the project “Laboratorio Nacional para Servicios Especializados de Investigación, Desarrollo e Innovación (I+D+i) para Farmoquímicos y Biotecnológicos, LANSEIDI-FarBiotec-CONACyT”.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Strohl WR. Current progress in innovative engineered antibodies. Protein Cell. 2018;9:86–120. doi: 10.1007/s13238-017-0457-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Almagro JC, Daniels-Wells TR, Perez-Tapia SM, Penichet ML.. Progress and challenges in the design and clinical development of antibodies for cancer therapy. Front Immunol. 2018;8:1751. doi: 10.3389/fimmu.2017.01751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chennamsetty N, Voynov V, Kayser V, Helk B, Trout BL. Prediction of aggregation prone regions of therapeutic proteins. J Phys Chem B. 2010;114:6614–6624. doi: 10.1021/jp911706q. [DOI] [PubMed] [Google Scholar]
- 4.Jain T, Sun T, Durand S, Hall A, Houston NR, Nett JH, Sharkey B, Bobrowicz B, Caffry I, Yu Y, et al. Biophysical properties of the clinical-stage antibody landscape. Proc Natl Acad Sci U S A. 2017;114:944–949. doi: 10.1073/pnas.1616408114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frenzel A, Schirrmann T, Hust M. Phage display-derived human antibodies in clinical development and therapy. MAbs. 2016;8:1177–1194. doi: 10.1080/19420862.2016.1212149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McCafferty J, Griffiths AD, Winter G, Chiswell DJ. Phage antibodies: filamentous phage displaying antibody variable domains. Nature. 1990;348:552–554. doi: 10.1038/348552a0. [DOI] [PubMed] [Google Scholar]
- 7.Finlay WJ, Almagro JC. Natural and man-made V-gene repertoires for antibody discovery. Front Immunol. 2012;3:342. doi: 10.3389/fimmu.2012.00198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Haard HJ, van Neer N, Reurs A, Hufton SE, Roovers RC, Henderikx P, de Bruïne AP, Arends JW, Hoogenboom HR. A large non-immunized human Fab fragment phage library that permits rapid isolation and kinetic analysis of high affinity antibodies. J Biol Chem. 1999;274:18218–18230. [DOI] [PubMed] [Google Scholar]
- 9.Griffiths AD, Williams SC, Hartley O, Tomlinson IM, Waterhouse P, Crosby WL, Kontermann RE, Jones PT, Low NM, Allison TJ. Isolation of high affinity human antibodies directly from large synthetic repertoires. Embo J. 1994;13:3245–3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoet RM, Cohen EH, Kent RB, Rookey K, Schoonbroodt S, Hogan S, Rem L, Frans N, Daukandt M, Pieters H, et al. Generation of high-affinity human antibodies by combining donor-derived and synthetic complementarity-determining-region diversity. Nat Biotechnol. 2005;23:344–348. doi: 10.1038/nbt1067. [DOI] [PubMed] [Google Scholar]
- 11.Knappik A, Ge L, Honegger A, Pack P, Fischer M, Wellnhofer G, Hoess A, Wölle J, Plückthun A, Virnekäs B. Fully synthetic human combinatorial antibody libraries (HuCAL) based on modular consensus frameworks and CDRs randomized with trinucleotides. J Mol Biol. 2000;296:57–86. doi: 10.1006/jmbi.1999.3444. [DOI] [PubMed] [Google Scholar]
- 12.Marks JD, Hoogenboom HR, Bonnert TP, McCafferty J, Griffiths AD, Winter G. By-passing immunization. Human antibodies from V-gene libraries displayed on phage. J Mol Biol. 1991;222:581–597. [DOI] [PubMed] [Google Scholar]
- 13.Vaughan TJ, Williams AJ, Pritchard K, Osbourn JK, Pope AR, Earnshaw JC, McCafferty J, Hodits RA, Wilton J, Johnson KS. Human antibodies with sub-nanomolar affinities isolated from a large non-immunized phage display library. Nat Biotechnol. 1996;14:309–314. doi: 10.1038/nbt0396-309. [DOI] [PubMed] [Google Scholar]
- 14.Shi L, Wheeler JC, Sweet RW, Lu J, Luo J, Tornetta M, Whitaker B, Reddy R, Brittingham R, Borozdina L, et al. De novo selection of high-affinity antibodies from synthetic fab libraries displayed on phage as pIX fusion proteins. J Mol Biol. 2010;397:385–396. doi: 10.1016/j.jmb.2010.01.034. [DOI] [PubMed] [Google Scholar]
- 15.Almagro JC, Teplyakov A, Luo J, Sweet RW, Kodangattil S, Hernandez-Guzman F, Gilliland GL. Second antibody modeling assessment (AMA-II). Proteins. 2014;82:1553–1562. doi: 10.1002/prot.24567. [DOI] [PubMed] [Google Scholar]
- 16.Bethea D, Wu S-J, Luo J, Hyun L, Lacy ER, Teplyakov A, Jacobs SA, O’Neil KT, Gilliland GL, Feng Y. Mechanisms of self-association of a human monoclonal antibody CNTO607. Protein Eng Des Sel. 2012;25:531–537. doi: 10.1093/protein/gzs047. [DOI] [PubMed] [Google Scholar]
- 17.Gilliland GL, Luo J, Vafa O, Almagro JC. Leveraging SBDD in protein therapeutic development: antibody engineering. Methods Mol Biol. 2012;841:321–349. doi: 10.1007/978-1-61779-520-6_14. [DOI] [PubMed] [Google Scholar]
- 18.Teplyakov A, Obmolova G, Malia TJ, Luo J, Muzammil S, Sweet R, Almagro JC, Gilliland GL. Structural diversity in a human antibody germline library. MAbs. 2016;8:1045–1063. doi: 10.1080/19420862.2016.1190060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raghunathan G, Smart J, Williams J, Almagro JC. Antigen-binding site anatomy and somatic mutations in antibodies that recognize different types of antigens. J Mol Recognit. 2012;25:103–113. doi: 10.1002/jmr.2158. [DOI] [PubMed] [Google Scholar]
- 20.Vargas-Madrazo E, Lara-Ochoa F, Almagro JC. Canonical structure repertoire of the antigen-binding site of immunoglobulins suggests strong geometrical restrictions associated to the mechanism of immune recognition. J Mol Biol. 1995;254:497–504. [DOI] [PubMed] [Google Scholar]
- 21.Glanville J, Zhai W, Berka J, Telman D, Huerta G, Mehta GR, Ni I, Mei L, Sundar PD, Day GMR, et al. Precise determination of the diversity of a combinatorial antibody library gives insight into the human immunoglobulin repertoire. Proc Natl Acad Sci U S A. 2009;106:20216–20221. doi: 10.1073/pnas.0909775106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Almagro JC, Quintero-Hernandez V, Ortiz-Leon M, Velandia A, Smith SL, Becerril B. Design and validation of a synthetic VH repertoire with tailored diversity for protein recognition. J Mol Recognit. 2006;19:413–422. doi: 10.1002/jmr.796. [DOI] [PubMed] [Google Scholar]
- 23.Cobaugh CW, Almagro JC, Pogson M, Iverson B, Georgiou G. Synthetic antibody libraries focused towards peptide ligands. J Mol Biol. 2008;378:622–633. doi: 10.1016/j.jmb.2008.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huovinen T, Syrjanpaa M, Sanmark H, Brockmann EC, Azhayev A, Wang Q, Vehniäinen M, Lamminmäki U. Two ScFv antibody libraries derived from identical VL-VH framework with different binding site designs display distinct binding profiles. Protein Eng Des Sel. 2013;26:683–693. doi: 10.1093/protein/gzt037. [DOI] [PubMed] [Google Scholar]
- 25.Hillson JL, Karr NS, Oppliger IR, Mannik M, Sasso EH. The structural basis of germline-encoded VH3 immunoglobulin binding to staphylococcal protein A. J Exp Med. 1993;178:331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Graille M, Stura EA, Corper AL, Sutton BJ, Taussig MJ, Charbonnier JB, Silverman GJ. Crystal structure of a Staphylococcus aureus protein A domain complexed with the Fab fragment of a human IgM antibody: structural basis for recognition of B-cell receptors and superantigen activity. Proc Natl Acad Sci U S A. 2000;97:5399–5404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Obmolova G, Teplyakov A, Malia TJ, Grygiel TL, Sweet R, Snyder LA, Gilliland GL. Structural basis for high selectivity of anti-CCL2 neutralizing antibody CNTO 888. Mol Immunol. 2012;51:227–233. doi: 10.1016/j.molimm.2012.03.022. [DOI] [PubMed] [Google Scholar]
- 28.Sondek J, Shortle D. A general strategy for random insertion and substitution mutagenesis: substoichiometric coupling of trinucleotide phosphoramidites. Proc Natl Acad Sci U S A. 1992;89:3581–3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dunn-Walters DK. The ageing human B cell repertoire: a failure of selection? Clin Exp Immunol. 2016;183:50–56. doi: 10.1111/cei.12700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kabat EA, Wu TT, Perry H, Gottesman K, Foeller C. Sequences of proteins of immunological interest. Fifth Edition NIH Publication No 91-3242 1991.
- 31.Arnaout R, Lee W, Cahill P, Honan T, Sparrow T, Weiand M, Nusbaum C, Rajewsky K, Koralov SB, Reindl M. High-resolution description of antibody heavy-chain repertoires in humans. PLoS One. 2011;6:e22365. doi: 10.1371/journal.pone.0022365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Adams R, Griffin L, Compson JE, Jairaj M, Baker T, Ceska T, West S, Zaccheo O, Davé E, Lawson AD, et al. Extending the half-life of a fab fragment through generation of a humanized anti-human serum albumin Fv domain: an investigation into the correlation between affinity and serum half-life. MAbs. 2016;8:1336–1346. doi: 10.1080/19420862.2016.1185581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao S, Chadwick L, Mysler E, Moots RJ. Review of biosimilar trials and data on adalimumab in rheumatoid arthritis. Curr Rheumatol Rep. 2018;20:57. doi: 10.1007/s11926-018-0769-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hwang WY, Almagro JC, Buss TN, Tan P, Foote J. Use of human germline genes in a CDR homology-based approach to antibody humanization. Methods. 2005;36:35–42. doi: 10.1016/j.ymeth.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 35.Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science. 1985;228:1315–1317. [DOI] [PubMed] [Google Scholar]
- 36.Scott JK, Smith GP. Searching for peptide ligands with an epitope library. Science. 1990;249:386–390. [DOI] [PubMed] [Google Scholar]
- 37.Famm K, Hansen L, Christ D, Winter G. Thermodynamically stable aggregation-resistant antibody domains through directed evolution. J Mol Biol. 2008;376:926–931. doi: 10.1016/j.jmb.2007.10.075. [DOI] [PubMed] [Google Scholar]
- 38.Jespers L, Schon O, Famm K, Winter G. Aggregation-resistant domain antibodies selected on phage by heat denaturation. Nat Biotechnol. 2004;22:1161–1165. doi: 10.1038/nbt1000. [DOI] [PubMed] [Google Scholar]
- 39.Rouet R, Lowe D, Christ D. Stability engineering of the human antibody repertoire. FEBS Lett. 2014;588:269–277. doi: 10.1016/j.febslet.2013.11.029. [DOI] [PubMed] [Google Scholar]
- 40.Lo Conte L, Chothia C, Janin J. The atomic structure of protein-protein recognition sites. J Mol Biol. 1999;285:2177–2198. [DOI] [PubMed] [Google Scholar]
- 41.Mian IS, Bradwell AR, Olson AJ. Structure, function and properties of antibody binding sites. J Mol Biol. 1991;217:133–151. [DOI] [PubMed] [Google Scholar]
- 42.Fellouse FA, Barthelemy PA, Kelley RF, Sidhu SS. Tyrosine plays a dominant functional role in the paratope of a synthetic antibody derived from a four amino acid code. J Mol Biol. 2006;357:100–114. doi: 10.1016/j.jmb.2005.11.092. [DOI] [PubMed] [Google Scholar]
- 43.Zhang K, Geddie ML, Kohli N, Kornaga T, Kirpotin DB, Jiao Y, Rennard R, Drummond DC, Nielsen UB, Xu L, et al. Comprehensive optimization of a single-chain variable domain antibody fragment as a targeting ligand for a cytotoxic nanoparticle. MAbs. 2015;7:42–52. doi: 10.4161/19420862.2014.985933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grover RK, Zhu X, Nieusma T, Jones T, Boreo I, MacLeod AS, Mark A, Niessen S, Kim HJ, Kong L, et al. A structurally distinct human mycoplasma protein that generically blocks antigen-antibody union. Science. 2014;343:656–661. doi: 10.1126/science.1246135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nilson BH, Solomon A, Bjorck L, Akerstrom B. Protein L from Peptostreptococcus magnus binds to the kappa light chain variable domain. J Biol Chem. 1992;267:2234–2239. [PubMed] [Google Scholar]
- 46.Marks JD, Bradbury A. Selection of human antibodies from phage display libraries. Methods Mol Biol. 2004;248:161–176. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.