

ABSTRACT
Cellular stress response is coordinated through the communication between mitochondria and the nucleus. However, whereas mitochondria are regulated by nuclear-encoded proteins, the nucleus was considered ungoverned by mitochondrial-encoded factors. We recently reported that a mitochondrial-encoded peptide directly regulates the nuclear genome upon cellular stress, indicating an integrated bi-genomic cross-communication mechanism.
KEYWORDS: Mitochondria, mitonuclear communication, mitochondrial-derived peptides, MDPs, MOTS-c, nuclear translocation, adaptive gene expression, stress response, stress resistance, homeostasis
Eukaryotic cells are multi-genomic systems, whereby each unique genome is compartmentalized in the nucleus, mitochondria and, in the case of plants, chloroplasts. The non-nuclear genomes evidently originate from endosymbiotic bacteria, which are thought to have provided several advantages, including bioenergetics, that allowed for eukaryotic evolution.1 For the past 1–2 billion years, the mitochondrial and nuclear genomes have co-evolved as the ancestral cells became functionally more complex.2,3 A lingering question remains on how the progress from an endosymbiotic relationship, which is not uncommon even today, to their establishment as a key organelle was coordinated. Of the many requisites, communication must have been key in fine-tuning the merger and consequent evolution of eukaryotic life. The ‘language’ that was used to mediate such communication presumably derived from mechanisms encoded in the original genomes of both parties, thus providing a foundation for eukaryotic cellular regulation. This notion is supported by the fact that more than 1,000 proteins encoded in the nuclear genome translocate to mitochondria to regulate various mitochondrial functions. On the contrary, mitochondrial DNA (mtDNA) has been traditionally known to host 13 protein coding genes, all of which are components of the electron transport chain (ETC) with no evident signaling roles. However, recent identification of short open reading frames (sORFs) encoded in mtDNA that yield bioactive peptides with regulatory roles, collectively categorized as mitochondrial-derived peptides (MDPs), provides another layer of mitochondrial communication. There are now eight published MDPs, including humanin,4–6 SHLP1–6 (small humanin-like peptide 1-6),7 and MOTS-c (mitochondrial open reading frame of the twelve S rRNA type-c),8 that have unique and overlapping biological significance.
In a recent report,9 we have shown that MOTS-c can dynamically translocate to the nucleus in response to cellular stress in an adenine monophosphate (AMP)-activated protein kinase (AMPK)-dependent manner. Within the nucleus, MOTS-c interacted with multiple stress-responsive transcription factors, including nuclear factor erythroid 2-related factor 2 (NFE2L2/NRF2), activating transcription factor 1 and 7 (ATF1/ATF7), and others (unpublished data from our lab). MOTS-c was associated with purified chromatin fractions, indicating interaction with DNA. Indeed, MOTS-c could bind to the promoter regions of NRF2-target genes that generally possess antioxidant response element (ARE) sequences, which required its hydrophobic core and cationic tail residues, and regulate downstream gene expression. A global profile of MOTS-c-dependent nuclear gene expression in response to glucose restriction, using RNA-seq, revealed that a broad range of genes, including bona fide NRF2-target genes, were involved. In addition, several transcription factor (TF)-binding motifs were enriched in the promoters of MOTS-c-regulated genes, such as ATF1/ATF7 and JUND (also known as JunD proto-oncogene, AP-1 transcription factor subunit), which are also related to NRF2. MOTS-c, but not nuclear-null mutants, significantly improved cellular survival under metabolic stress MOTS-c.
In summary, MOTS-c (i) is largely extra-nuclear and translocates to the nucleus upon cellular stress, (ii) requires the activation of AMPK, a master regulator of metabolic homeostasis, for nuclear transport, (iii) interacts with stress-responsive TFs and their target genes in the nucleus, and (iv) increases survival under metabolic deprivation. Together, these data demonstrate that MOTS-c is a mitochondrial-encoded regulator of the nuclear genome, the first of its kind to be reported, whose genetic message to the nucleus is to promote an adaptive stress response to maintain cellular homeostasis under deprived conditions. From a conceptual perspective, these findings indicate that the co-evolved mitonuclear genomes cross-regulate each other, providing evidence of the integrated genetic basis of bi-directional mitonuclear communication.
Our findings may also have considerable implications in cancer, which is a product of constant evolution and adaption with strong genetic and metabolic drivers (Figure 1). Cancer is a genetic disease, but the current approach in both basic research and translation development is largely focused on the nuclear genome, leaving the mitochondrial genome is the blind spot. Further, cancer is also considered a metabolic disease, in which mitochondria generally assume an altered metabolic role (i.e. Warburg effect) and exhibit altered communication to other cellular compartments10 to support the rampant growth. Notably, we previously reported that MOTS-c is an AMPK-dependent regulator of cellular metabolism and can not only retard the proliferation of the immortalized HEK293 cells, but also prevent diet-induced obesity and insulin-resistance and age-dependent insulin resistance in mice. Although there is much unveiled about the metabolic rewiring in cancer, how mitochondria communicate to other organelles in cancer is a critical question that remains largely enigmatic. This question is of special significance, again, for a major disease with strong metabolic and genetic bases. Thus, a large knowledge-gap currently exists on how the genetic network weaved by the two genomes are altered in cancers to provide evolutionary advantage for survival and growth.
Figure 1.
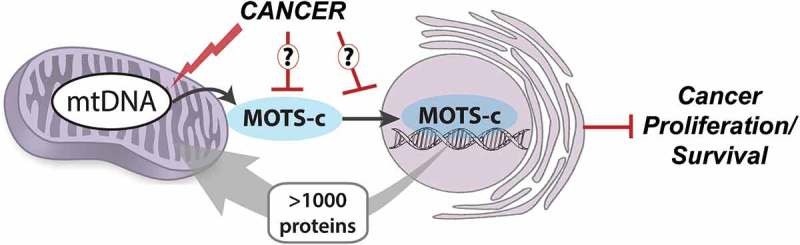
MOTS-c is a mitochondrial-encoded regulator of adaptive nuclear gene expression. There are over 1,000 nuclear-encoded proteins that reside in the mitochondria, but mitochondrial-encoded factors that actively regulate the nuclear genome have not been known. We recently reported that a peptide encoded in the mitochondrial DNA (mtDNA), MOTS-c (mitochondrial open reading frame of the twelve S rRNA type-c), dynamically translocates to the nucleus under metabolic stress to regulate nuclear transcription to promote cellular homeostasis. Because malignant cells are under constant evolutionary pressure to overcome hostile and stressful conditions for rampant growth, such mitochondrial-to-nuclear (mitonuclear) stress-responsive communication may be altered in tumors.
We believe we have just only begun to unravel an archaic communication system that existed throughout eukaryotic evolution. As with any unexplored territory, there are more questions remaining than answers provided. Some fundamental questions that will advance our knowledge base on MDPs include the regulatory mechanisms of their expression, mode of intra- and extra-cellular transport, and their impact on the nuclear gene regulation in aging and age-dependent chronic diseases, such as cancer.
Disclosure of Potential Conflicts of Interest
C.L. is a consultant for and a shareholder of CohBar, Inc.
References
- 1.Lane N, Martin WF.. Eukaryotes really are special, and mitochondria are why. Proc Natl Acad Sci USA. 2015;112(35):E4823–E4823. doi: 10.1073/pnas.1509237112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sunnucks P, Morales HE, Lamb AM, Pavlova A, & Greening C 2017. Integrative approaches for studying mitochondrial and nuclear genome co-evolution in oxidative phosphorylation. Front Genet. 8:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Quiros PM, Mottis A, Auwerx J. Mitonuclear communication in homeostasis and stress. Nat Rev Mol Cell Biol. 2016; doi: 10.1038/nrm.2016.23. [DOI] [PubMed] [Google Scholar]
- 4.Hashimoto Y, Niikura T, Tajima H, Yasukawa T, Sudo H, Ito Y, Kita Y, Kawasumi M, Kouyama K, Doyu M, et al. A rescue factor abolishing neuronal cell death by a wide spectrum of familial Alzheimer’s disease genes and Abeta. Proc Natl Acad Sci USA. 2001;98(11):6336–6341. doi: 10.1073/pnas.101133498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ikonen M, Liu B, Hashimoto Y, Ma L, Lee K-W, Niikura T, Nishimoto I, Cohen P. Interaction between the Alzheimer’s survival peptide humanin and insulin-like growth factor-binding protein 3 regulates cell survival and apoptosis. Proc Natl Acad Sci USA. 2003;100(22):13042–13047. doi: 10.1073/pnas.2135111100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guo B, Zhai D, Cabezas E, Welsh K, Nouraini S, Satterthwait AC, Reed JC. Humanin peptide suppresses apoptosis by interfering with Bax activation. Nature. 2003;423(6938):456–461. doi: 10.1038/nature01627. [DOI] [PubMed] [Google Scholar]
- 7.Cobb LJ, Lee C, Xiao J, Yen K, Wong RG, Nakamura HK, Mehta HH, Gao Q, Ashur C, Huffman DM, et al. Naturally occurring mitochondrial-derived peptides are age-dependent regulators of apoptosis, insulin sensitivity, and inflammatory markers. Aging (Albany NY). 2016;8(4):796–809. doi: 10.18632/aging.100943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee C, Zeng J, Drew BG, Sallam T, Martin-Montalvo A, Wan J, Kim S-J, Mehta H, Hevener AL, de Cabo R, et al. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metab. 2015;21(3):443–454. doi: 10.1016/j.cmet.2015.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim KH, Son JM, Benayoun BA, Lee C. The mitochondrial-encoded peptide MOTS-c translocates to the nucleus to regulate nuclear gene expression in response to metabolic stress. Cell Metab. 2018; doi: 10.1016/j.cmet.2018.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chattopadhyay E, Roy B. 2017. Altered mitochondrial signalling and metabolism in cancer. Front Oncol. 7:43. doi: 10.3389/fonc.2017.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]