

Abstract
Background
This is an updated version of the original Cochrane Review published in Issue 11, 2006 of the Cochrane Database of Systematic Reviews.
Epilepsy is a common neurological condition in which abnormal electrical discharges from the brain cause recurrent unprovoked seizures. It is believed that with effective drug treatment up to 70% of individuals with active epilepsy have the potential to become seizure‐free, and to go into long‐term remission shortly after starting drug therapy with a single antiepileptic drug (AED) in monotherapy.
The correct choice of first‐line AED for individuals with newly diagnosed seizures is of great importance. It is important that the choice of AEDs for an individual is made using the highest quality evidence regarding the potential benefits and harms of the various treatments.
Carbamazepine or lamotrigine are recommended as first‐line treatments for new onset focal seizures and as a first‐ or second‐line treatment for generalised tonic‐clonic seizures. Performing a synthesis of the evidence from existing trials will increase the precision of the results for outcomes relating to efficacy and tolerability and may assist in informing a choice between the two drugs.
Objectives
To review the time to treatment failure, remission and first seizure with lamotrigine compared to carbamazepine when used as monotherapy in people with focal onset seizures (simple or complex focal and secondarily generalised) or generalised onset tonic‐clonic seizures (with or without other generalised seizure types).
Search methods
We conducted the first searches for this review in 1997. For the most recent update, we searched the Cochrane Epilepsy Group Specialized Register, the Cochrane Central Register of Controlled Trials (CENTRAL) via the Cochrane Register of Studies Online (CRSO), MEDLINE, Clinical Trials.gov and the WHO International Clinical Trials Registry Platform on 26 February 2018, without language restrictions
Selection criteria
Randomised controlled trials comparing monotherapy with either carbamazepine or lamotrigine in children or adults with focal onset seizures or generalised onset tonic‐clonic seizures
Data collection and analysis
This was an individual participant data (IPD) review. Our primary outcome was time to treatment failure and our secondary outcomes were time to first seizure post randomisation, time to six‐month, 12‐month and 24‐month remission, and incidence of adverse events. We used Cox proportional hazards regression models to obtain trial‐specific estimates of hazard ratios (HRs) with 95% confidence intervals (CIs), using the generic inverse variance method to obtain the overall pooled HR and 95% CI.
Main results
We included 14 trials in this review. Individual participant data were available for 2572 participants out of 3787 eligible individuals from nine out of 14 trials: 68% of the potential data. For remission outcomes, a HR of less than one indicated an advantage for carbamazepine; and for first seizure and treatment failure outcomes, a HR of less than one indicated an advantage for lamotrigine.
The main overall results were: time to treatment failure for any reason related to treatment (pooled HR adjusted for seizure type: 0.71, 95% CI 0.62 to 0.82, moderate‐quality evidence), time to treatment failure due to adverse events (pooled HR adjusted for seizure type: 0.55 (95% CI 0.45 to 0.66, moderate‐quality evidence), time to treatment failure due to lack of efficacy (pooled HR for all participants: 1.03 (95% CI 0.75 to 1.41), moderate‐quality evidence) showing a significant advantage for lamotrigine compared to carbamazepine in terms of treatment failure for any reason related to treatment and treatment failure due to adverse events, but no different between drugs for treatment failure due to lack of efficacy.
Time to first seizure (pooled HR adjusted for seizure type: 1.26, 95% CI 1.12 to 1.41, high‐quality evidence) and time to six‐month remission (pooled HR adjusted for seizure type: 0.86, 95% CI 0.76 to 0.97, high‐quality evidence), showed a significant advantage for carbamazepine compared to lamotrigine for first seizure and six‐month remission. We found no difference between the drugs for time to 12‐month remission (pooled HR for all participants 0.91, 95% CI 0.77 to 1.07, high‐quality evidence) or time to 24‐month remission (HR for all participants 1.00, 95% CI 0.80 to 1.25, high‐quality evidence), however only two trials followed up participants for more than one year so evidence is limited.
The results of this review are applicable mainly to individuals with focal onset seizures; 88% of included individuals experienced seizures of this type at baseline. Up to 50% of the limited number of individuals classified as experiencing generalised onset seizures at baseline may have had their seizure type misclassified, therefore we recommend caution when interpreting the results of this review for individuals with generalised onset seizures.
The most commonly reported adverse events for both of the drugs across all of the included trials were dizziness, fatigue, gastrointestinal disturbances, headache and skin problems. The rate of adverse events was similar across the two drugs.
The methodological quality of the included trials was generally good, however there is some evidence that the design choice of masked or open‐label treatment may have influenced the treatment failure and withdrawal rates of the trials. Hence, we judged the quality of the evidence for the primary outcome of treatment failure to be moderate for individuals with focal onset seizures and low for individuals with generalised onset seizures. For efficacy outcomes (first seizure, remission), we judged the quality of evidence to be high for individuals with focal onset seizures and moderate for individuals with generalised onset seizures.
Authors' conclusions
Moderate quality evidence indicates that treatment failure for any reason related to treatment or due to adverse events occurs significantly earlier on carbamazepine than lamotrigine, but the results for time to first seizure suggested that carbamazepine may be superior in terms of seizure control. The choice between these first‐line treatments must be made with careful consideration. We recommend that future trials should be designed to the highest quality possible with consideration of masking, choice of population, classification of seizure type, duration of follow‐up, choice of outcomes and analysis, and presentation of results.
Plain language summary
Lamotrigine versus carbamazepine monotherapy (single medication treatment) for epilepsy
This is an updated version of the Cochrane Review previously published in Issue 11, 2016 of the Cochrane Database of Systematic Reviews.
Background
Epilepsy is a common neurological disorder in which abnormal electrical discharges from the brain cause recurrent seizures. We studied two types of epileptic seizures in this review: generalised onset seizures, in which electrical discharges begin in one part of the brain and move throughout the brain; and focal onset seizures, in which the seizure is generated in and affects one part of the brain (the whole hemisphere of the brain or part of a lobe of the brain). Focal seizures may become generalised (secondary generalisation) and move from one part of the brain throughout the brain. For around 70% of people with epilepsy, a single antiepileptic medication can control generalised onset or focal onset seizures.
This review applies to people with focal seizures (with or without secondary generalisation) and people with generalised tonic‐clonic seizures, a specific generalised seizure type. This review does not apply to people with other generalised seizure types such as absence seizures or myoclonic seizures, as the recommended treatments for these seizure types are different.
Objective
Carbamazepine and lamotrigine are first‐choice treatments for individuals with recently diagnosed epilepsy. The aim of this review was to compare how effective these drugs are at controlling seizures, to find out if they are associated with side effects that may result in individuals stopping the medication, and to inform a choice between these medications.
Methods
The last search for trials was in February 2018. We assessed the evidence from 14 randomised controlled trials comparing lamotrigine with carbamazepine. We were able to combine information for 2572 people from nine of the 14 trials; for the remaining 1215 people from five trials, information was not available to use in this review.
Results
The results of the review suggest that people are more likely to withdraw earlier from carbamazepine than lamotrigine treatment. The most common medicine‐related reason for withdrawal was side effects: 52% of total withdrawals in participants on carbamazepine and 36% of total withdrawals in participants on lamotrigine. The second most common medicine‐related cause for withdrawal was seizure recurrence: 58 of 719 total withdrawals (8%) on carbamazepine and 105 of 697 total withdrawals (15%) on lamotrigine.
The results suggest that recurrence of seizures after starting treatment with lamotrigine may happen earlier than treatment with carbamazepine. They also suggest that freedom from seizures for a period of six months may occur earlier on carbamazepine than lamotrigine. The majority of the people included in the 14 trials (88%) experienced focal seizures, so the results of this review apply mainly to people with this seizure type.
The most common side effects reported by participants during the trials were dizziness, fatigue, gastrointestinal problems, headaches and skin problems. These side effects were reported a similar number of times by people taking lamotrigine or carbamazepine.
Quality of the evidence
For people with focal onset seizures, we judged the quality of the evidence to be high for the outcomes of seizure recurrence and remission of seizures and we judged the quality of the evidence to be moderate for the outcome of treatment failure. The design of the trials (specifically, whether the people and treating clinicians knew which medication they were taking) may have influenced the rates of withdrawal from treatments. Up to 50% of people in the trials used in our results may have been wrongly classified as having generalised seizures; for people with generalised onset seizures, we judged the quality of the evidence to be moderate for the outcomes of seizure recurrence and remission of seizures and low quality for the outcome of treatment failure.
Conclusions
For people with focal onset seizures, lamotrigine and carbamazepine are effective treatments and a choice between these two treatments must be made carefully. More information is needed for people with generalised onset seizures. We recommend that all future trials comparing these medications, or any other antiepileptic medications, should be designed using high‐quality methods. Seizure types of people included in trials should also be classified very carefully to ensure that the results are also of high quality.
Summary of findings
Summary of findings for the main comparison. Summary of findings: lamotrigine compared with carbamazepine for epilepsy (primary outcomes).
| Lamotrigine compared with carbamazepine for epilepsy | ||||||
|
Patient or population: adults and children with focal onset or generalised onset seizures (generalised tonic‐clonic with or without other generalised seizure types) Settings: outpatients Intervention: lamotrigine Comparison: carbamazepine | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (trials) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Carbamazepine | Lamotrigine | |||||
|
Time to treatment failure (any reason related to treatment) All participants Range of follow‐up: 0 to 2420 days |
The median time to treatment failure was 1144 days in the carbamazepine group | The median time to treatment failure was 1813 days (669 days longer) in the lamotrigine group | HR 0.71 (0.62 to 0.82)a | 2481 (9 trials) | ⊕⊕⊕⊝ moderateb | HR of less than 1 indicates an advantage for lamotrigine. Treatment failure due to adverse events also occurred significantly earlier on carbamazepine compared to lamotrigine: HR 0.54 (95% CI 0.45 to 0.65, P<0.00001). There was no difference between lamotrigine and carbamazepine in terms of treatment failure due to lack of efficacy: HR 1.03 (95% CI 0.75 to 1.41, P=0.86) |
|
Time to treatment failure (any reason related to treatment) Subgroup: focal onset seizures Range of follow‐up: 0 to 2420 days |
The median time to treatment failure was 1149 days in the carbamazepine group | The median time to treatment failure was 1699 days (550 days longer) in the lamotrigine group | HR 0.74 (0.64 to 0.86) | 2182 (9 trials) | ⊕⊕⊕⊝ moderateb | HR of less than 1 indicates an advantage for lamotrigine. Treatment failure due to adverse events also occurred significantly earlier on carbamazepine compared to lamotrigine: HR 0.56 (95% CI 0.45 to 0.68, P<0.00001). Treatment failure due to lack of efficacy was not calculated for focal onset seizures subgroup due to small numbers of individuals withdrawing from treatment for lack of efficacy. |
|
Time to treatment failure (any reason related to treatment) Subgroup: generalised onset seizures Range of follow‐up: 0 to 1446 days |
The 25th percentile** of time to treatment failure was 57 days in the carbamazepine group | The 25th percentile** of time to treatment failure was 510 days (453 days longer) in the lamotrigine group | HR 0.51 (0.33 to 0.78) | 299 (6 trials) | ⊕⊕⊝⊝ lowc,d | HR of less than 1 indicates an advantage for lamotrigine Treatment failure due to adverse events also occurred significantly earlier on carbamazepine compared to lamotrigine: HR 0.49 (95% CI 0.27 to 0.88, P=0.02). Treatment failure due to lack of efficacy was not calculated for focal onset seizures subgroup due to small numbers of individuals withdrawing from treatment for lack of efficacy. |
| * Illustrative risks in the carbamazepine and lamotrigine groups are calculated at the median time to treatment failure (i.e. the time to 50% of participants failing or withdrawing from allocated treatment) within each group across all trials. The relative effect (pooled hazard ratio) shows the comparison of 'time to treatment failure' between the treatment groups. ** The 25th percentile of time to treatment failure (i.e. the time to 50% of participants failing or withdrawing from allocated treatment) is presented for the subgroup with generalised seizures as less than 50% of participants failed / withdrew from treatment, therefore the median time could not be calculated. Abbreviations: 95% CI: 95% confidence interval; HR: hazard ratio | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
a. Pooled hazard ratio for all participants adjusted for seizure type.
b. Downgraded once due to high risk of bias due to the open‐label design of five trials included in the analysis (Eun 2012; Lee 2011; Nieto‐Barrera 2001; Reunanen 1996; SANAD A 2007); the design of the trial may have influenced the withdrawal rates.
c. Downgraded once due to high risk of bias due to the open‐label design of three trials included in the analysis (Lee 2011; Reunanen 1996; SANAD A 2007); the design of the trial may have influenced the withdrawal rates.
d. Downgraded once due to potential misclassification of generalised onset seizures in up to 50% of participants in the trials.
Summary of findings 2. Summary of findings: lamotrigine compared with carbamazepine for epilepsy (secondary outcomes).
| Lamotrigine compared with carbamazepine for epilepsy | ||||||
|
Patient or population: adults and children with focal onset or generalised onset seizures (generalised tonic‐clonic with or without other generalised seizure types) Settings: outpatients Intervention: lamotrigine Comparison: carbamazepine | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI)1 | No of participants (trials) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Carbamazepine | Lamotrigine | |||||
|
Time to first seizure All participants Range of follow‐up: 0 to 2420 days |
The median time to first seizure was 232 days in the carbamazepine group | The median time to first seizure was 134 days (98 days shorter) in the lamotrigine group | HR 1.26 (1.12 to 1.41)a | 2476 (9 trials) | ⊕⊕⊕⊕ highb | HR of less than 1 indicates an advantage for lamotrigine |
|
Time to first seizure Subgroup: focal onset seizures Range of follow‐up: 0 to 2420 days |
The median time to first seizure was 208 days in the carbamazepine group | The median time to first seizure was 96 days (112 days shorter) in the lamotrigine group | HR 1.29 (1.14 to 1.45) | 2177 (9 trials) | ⊕⊕⊕⊕ highb | HR of less than 1 indicates an advantage for lamotrigine |
|
Time to first seizure Subgroup: generalised onset seizures Range of follow‐up: 0 to 853 days |
The median time to first seizure was 853 days in the carbamazepine group | The median time to first seizure was 337 days (516 days longer) in the lamotrigine group | HR 0.98 (0.65 to 1.48) | 277 (6 trials) | ⊕⊕⊝⊝ lowb,c | HR of less than 1 indicates an advantage for lamotrigine |
|
Time to 12‐month remission All participants Range of follow‐up: 0 to 2420 days |
The median time to 12‐month remission was 452 days in the carbamazepine group | The median time to 12‐month remission was 538 days (86 days longer) in the lamotrigine group | HR 0.91 (0.77 to 1.07) | 988 (2 trials) | ⊕⊕⊕⊕ highb | HR of less than 1 indicates an advantage for carbamazepine Time to 12‐month remission not presented by seizure type due to small numbers of participants with generalised onset seizures in the two trials |
| * Illustrative risks in the carbamazepine and lamotrigine groups are calculated at the median time to first seizure or time to 12‐month remission (i.e. the time to 50% of participants experiencing a first seizure or 12‐months of remission) within each group across all trials. The relative effect (pooled hazard ratio) shows the comparison of 'time to first seizure' or 'time to 12‐month remission' between the treatment groups. Abbreviations: 95% CI: 95% confidence interval; HR: hazard ratio | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
a. Pooled hazard ratio for all participants adjusted for seizure type.
b. High risk of bias due to the open‐label design in some of the included trials, however outcomes are objective and unlikely to be influenced by knowledge of drug allocation. No downgrade made.
c. Downgraded once due to potential misclassification of generalised onset seizures in up to 50% of participants in the trials.
Background
This review is an update of a previously published review in the Cochrane Database of Systematic Reviews (Issue 11, 2016) on 'Lamotrigine versus carbamazepine monotherapy for epilepsy: an individual participant data review' (Gamble 2006; Nolan 2016a)
Description of the condition
Epilepsy is a common neurological condition in which abnormal electrical discharges from the brain cause recurrent unprovoked seizures. Epilepsy is a disorder of many heterogenous seizure types, with an estimated incidence of 33 to 57 per 100,000 person‐years worldwide (Annegers 1999; Hirtz 2007; MacDonald 2000; Olafsson 2005; Sander 1996), accounting for approximately 1% of the global burden of disease (Murray 1994). The lifetime risk of epilepsy onset is estimated to be 1300 to 4000 per 100,000 person‐years (Hauser 1993; Juul‐Jenson 1983), and the lifetime prevalence could be as large as 70 million people worldwide (Ngugi 2010). It is believed that with effective drug treatment, up to 70% of individuals with active epilepsy have the potential to go into long‐term remission shortly after starting drug therapy (Cockerell 1995; Hauser 1993; Sander 2004), and around 70% of individuals can achieve seizure freedom using a single antiepileptic drug (AED) in monotherapy (Cockerell 1995). Current National Institute for Health and Care Excellence (NICE) guidelines recommend that both adults and children with epilepsy should be treated with monotherapy wherever possible (NICE 2012). The remaining 30% of individuals experience refractory or drug‐resistant seizures, which often require treatment with combinations of antiepileptic drugs (AEDs) or alternative treatments, such as epilepsy surgery (Kwan 2000).
We studied two seizure types in this review: generalised onset seizures, in which electrical discharges begin in one part of the brain and move throughout the brain; and focal onset seizures, in which the seizure is generated in and affects one part of the brain (the whole hemisphere of the brain or part of a lobe of the brain).
Description of the intervention
Carbamazepine was amongst the earliest 'traditional' medications licensed for the treatment of epileptic seizures and has been commonly used as monotherapy for focal onset and generalised onset seizures for over 30 years (Shakir 1980). Lamotrigine is among a 'second generation' of AEDs, licensed as monotherapy for epileptic seizures following demonstrations of efficacy compared to 'traditional' AEDs such as carbamazepine (Brodie 1995; Brodie 1999; Reunanen 1996).
Comparative trials have shown newer AEDs such as lamotrigine to be to be generally well tolerated as monotherapy in both adults and children, and related to fewer adverse events, fewer serious adverse events and fewer drug interactions with concomitant AEDs and other concomitant medications than 'traditional' first‐line AEDs such as carbamazepine (Brodie 1995; Brodie 1999; French 2007; Reunanen 1996).
Evidence regarding teratogenic effects (disturbances to foetal development) of carbamazepine and lamotrigine is conflicting and uncertain. It is thought that the risk of congenital malformation may be higher for women taking carbamazepine compared to the general population (Meador 2008; Morrow 2006), and carbamazepine has been shown to be associated with neural tube defects (Matlow 2012). The risk of malformations is thought to be lower for women taking lamotrigine than carbamazepine (Meador 2008), but the risk of malformation may increase with an increasing dose of lamotrigine (Morrow 2006). It is unclear whether taking carbamazepine or lamotrigine during pregnancy has any negative neurodevelopmental effects on the child (Bromley 2014).
Current NICE guidelines for adults and children recommend carbamazepine or lamotrigine as first‐line treatments for new onset focal seizures and as second‐line treatments for generalised tonic‐clonic seizures (NICE 2012). Lamotrigine is considered a suitable first‐line treatment for new onset generalised seizures if sodium valproate is considered unsuitable. Carbamazepine may be a suitable second‐line treatment for generalised onset seizures but may exacerbate myoclonic or absence seizures (Liporace 1994; Shields 1983; Snead 1985).
How the intervention might work
Antiepileptic medications suppress seizures by reducing neuronal excitability (disruption of the usual mechanisms of a neuron within the brain, which may lead to an epileptic seizure) (MacDonald 1995). Lamotrigine and carbamazepine are broad‐spectrum treatments suitable for many seizure types, and both have an anticonvulsant mechanism through blocking ion channels and binding with neurotransmitter receptors, or through inhibiting the metabolism or reuptake of neurotransmitters (Brodie 1996; Lees 1993; Ragsdale 1991).
Why it is important to do this review
With evidence that up to 70% of individuals with active epilepsy have the potential to go into long‐term remission of seizures shortly after starting drug therapy (Cockerell 1995; Hauser 1993; Sander 2004), the correct choice of first‐line antiepileptic therapy for individuals with newly diagnosed seizures is of great importance. It is important that the choice of AEDs for an individual is made using the highest quality evidence regarding the potential benefits and harms of various treatments. It is also important that the effectiveness and tolerability of AEDs appropriate to given seizure types are compared to one another.
Therefore the aim of this review is to summarise efficacy and tolerability from existing randomised controlled trials comparing lamotrigine and carbamazepine, two current first‐line recommended treatments for use in monotherapy for epileptic seizures. Performing a synthesis of the evidence from existing trials will increase the precision of the results for outcomes relating to efficacy and tolerability and may assist in informing a choice between the two drugs.
There are difficulties in undertaking a systematic review of epilepsy monotherapy trials as the important efficacy outcomes require analysis of time‐to‐event data (for example, time to first seizure after randomisation). Although methods have been developed to synthesise time‐to‐event data using summary information (Parmar 1998; Williamson 2002), the appropriate statistics are not commonly reported in published epilepsy trials (Nolan 2013a; Williamson 2000). Furthermore, although most epilepsy monotherapy trials collect seizure data, there has been no uniformity in the definition and reporting of outcomes. For example, trials may report time to 12‐month remission but not time to first seizure or vice versa, or some trials may define time to first seizure from the date of randomisation while others use the date of achieving maintenance dose. Trial investigators have also adopted differing approaches to the analysis, particularly with respect to the censoring of time‐to‐event data. For these reasons, we performed this review using individual participant data (IPD), which helps to overcome these problems. This review is one in a series of Cochrane IPD reviews investigating pair‐wise monotherapy comparisons (Marson 2000; Nevitt 2017b; Nolan 2013b; Nolan 2013c; Nolan 2016b; Nolan 2016c; Nolan 2016d). These data have also been included in IPD network meta‐analyses of anti‐epileptic drug monotherapy (Tudur Smith 2007; Nevitt 2017a)
Objectives
To review the time to treatment failure, remission and first seizure with lamotrigine compared to carbamazepine when used as monotherapy in people with focal onset seizures (simple or complex focal and secondarily generalised) or generalised onset tonic‐clonic seizures (with or without other generalised seizure types).
Methods
Criteria for considering studies for this review
Types of studies
Randomised controlled trials (RCTs) using either an adequate method of allocation concealment (e.g. sealed, opaque envelopes) or a 'quasi' method of randomisation (e.g. allocation by date of birth).
Trials may have been double‐blind, single‐blind or unblinded.
Trials must have included a comparison of lamotrigine monotherapy with carbamazepine monotherapy in individuals with epilepsy.
Types of participants
We included children or adults with focal onset seizures (simple focal, complex focal or secondarily generalised tonic‐clonic seizures) or generalised onset tonic‐clonic seizures, with or without other generalised seizure types (in other words, those who had only generalised tonic‐clonic seizures and those who had both generalised onset tonic‐clonic seizures and generalised seizures of other types (e.g. absence, myoclonic etc.)).
We excluded individuals with other generalised seizure types alone without generalised tonic‐clonic seizures (e.g. those who had only absence seizures without any generalised clonic tonic‐seizures) due to differences in first‐line treatment guidelines for other generalised seizure types (NICE 2012).
We included individuals with a new diagnosis of epilepsy, or who have had a relapse following antiepileptic monotherapy withdrawal.
Types of interventions
Carbamazepine or lamotrigine as monotherapy.
Types of outcome measures
Below is a list of outcomes investigated in this review. Reporting of these outcomes in the original trial report was not an eligibility requirement for this review.
Primary outcomes
Time to treatment failure (retention time). This was a combined outcome reflecting both efficacy and tolerability, as the following may have lead to failure of treatment: continued seizures, side effects, non‐compliance or the initiation of additional add‐on treatment. This is an outcome to which the participant makes a contribution and is the primary outcome measure recommended by the Commission on Antiepileptic Drugs of the International League Against Epilepsy (ILAE 1998; ILAE 2006).
Time to treatment failure is considered according to three definitions:
Time to treatment failure for any treatment related reason (continued seizures, side effects, non‐compliance or the initiation of additional add‐on treatment)
Time to treatment failure due to adverse events (i.e. side effects)
Time to treatment failure due to lack of efficacy (i.e. continued seizures)
Secondary outcomes
Time to first seizure post randomisation.
Time to achieve six‐month remission (seizure‐free period).
Time to achieve 12‐month remission (seizure‐free period).
Time to achieve 24‐month remission (seizure‐free period).
Incidence of adverse events (all reported whether related or unrelated to treatment) and adverse events leading to treatment failure.
Search methods for identification of studies
Electronic searches
The first searches for this review were run in 1997. Subsequent searches were run in July 2014, December 2015 and October 2016. For the most recent update we searched the following databases.
Cochrane Epilepsy Group Specialized Register (26 February 2018) using the search strategy set out in Appendix 1.
Cochrane Central Register of Controlled Trials (CENTRAL) via the Cochrane Register of Studies Online (CRSO, 26 February 2018) using the search strategy set out in Appendix 2.
MEDLINE (Ovid, 1946 to 26 February 2018) using the search strategy set out in Appendix 3.
ClinicalTrials.gov (26 February 2018) using the search terms 'Lamotrigine AND carbamazepine | Epilepsy'.
WHO International Clinical Trials Registry Platform (ICTRP, 26 February 2018) using the search terms ' Lamotrigine AND carbamazepine AND Epilepsy'.
We imposed no language restrictions.
Searching other resources
We reviewed the reference lists of retrieved studies to search for additional reports of relevant trials. We contacted Ciba Geigy (manufacturers of carbamazepine), GlaxoSmithKline (manufacturers of lamotrigine) and the original investigators of relevant trials identified by our search.
Data collection and analysis
Selection of studies
Two review authors (SJN and AGM) independently assessed trials for inclusion, resolving any disagreements by mutual discussion.
Data extraction and management
We requested the following individual participant data for all trials meeting our inclusion criteria.
Trial methods
Method of generation of random list
Method of concealment of randomisation
Stratification factors
Blinding methods
Participant covariates
Gender
Age
Seizure types
Time between first seizure and randomisation
Number of seizures prior to randomisation (with dates)
Presence of neurological signs
Electroencephalographic (EEG) results
Computerised tomography/magnetic resonance imaging (CT/MRI) results
Follow‐up data
Treatment allocation
Date of randomisation
Dates of follow‐up
Dates of seizures post randomisation or seizure frequency data between follow‐up visits
Dates of treatment withdrawal or treatment failure and reasons for treatment withdrawal or treatment failure
Dose
Dates of dose changes
For each trial for which we did not obtain individual participant data (IPD), we carried out an assessment to see whether any relevant aggregate level data had been reported or could be indirectly estimated using the methods of Parmar 1998, and Williamson 2002. Where graphical time‐to‐event data (e.g. Kaplan Meier curves) were published with or without corresponding effective numbers at risk, we used a Microsoft Excel spreadsheet (Excel 2010), to indirectly estimate hazard ratios (Tierney 2007).
Four trials including 1391 participants provided seizure data in terms of the number of seizures recorded between each follow‐up visit rather than specific dates of seizures (Eun 2012; Lee 2011; SANAD A 2007; Werhahn 2015). To enable the calculation of time‐to‐event outcomes, we applied linear interpolation to approximate dates of seizures between follow‐up visits. For example, if the trial recorded four seizures between two visits that occurred on 1 March 2010 and 1 May 2010 (interval of 61 days), then the date of first seizure would be approximately 13 March 2010. This allowed the computation of an estimate of the time to six‐month remission, 12‐month remission, 24‐month remission and first seizure.
We calculated time to six‐month, 12‐month and 24‐month remission from the date of randomisation to the date (or estimated date) that the individual had first been free of seizures for six, 12 or 24 months, respectively. If the person had one or more seizures during the trial, a six‐month, 12‐month or 24 month seizure‐free period could also occur between the estimated date of the last seizure during the trial and a period of six, 12 or 24 months of seizure freedom.
We calculated time to first seizure from the date of randomisation to the date that we estimated their first seizure to have occurred. If seizure data were missing for a particular visit, we censored these outcomes at the previous visit. We also censored these outcomes if the individual died or if follow‐up ceased prior to the occurrence of the event of interest. We used these methods in five trials including 1383 participants (Brodie 1995 A; Brodie 1995 B; Brodie 1999; Nieto‐Barrera 2001; Reunanen 1996), for which we directly received outcome data (dates of seizures after randomisation).
For all trials we received data for date and reason of withdrawal from the treatment or the date and reason for treatment failure. Time to treatment failure was calculated as date of randomisation to date of treatment failure. For the analysis of time‐to‐event, we defined an 'event' as treatment failure because of reasons related to the treatment (i.e. lack of efficacy, adverse events, or both lack of efficacy and adverse events, non‐compliance with the treatment regimen, withdrawal of consent from the trial, etc.). We censored the outcome if treatment failure or withdrawal of treatment was for reasons not related to the trial treatment: i.e. loss to follow‐up, death (not treatment or epilepsy‐related), treatment withdrawn due to remission, etc. We also censored individuals who were still on allocated treatment at the date of the end of follow‐up. We considered documented reasons for treatment failure or treatment withdrawal on a case‐by‐case basis for relation to treatment; two authors (SJN and AGM) independently classified reasons for treatment failure as events or censored and resolved any disagreements by discussion. If reasons for treatment failure were classified differently as events or censored in the included trials to our definitions, we conducted sensitivity analyses to account for differences in the definition of a treatment failure 'event' (see Sensitivity analysis).
For the analysis of 'Time to treatment failure due to adverse events,' only treatment failures which were documented to be due to adverse events (either as a sole reason or due to both a lack of efficacy and adverse events) were classed as an 'event' within time‐to‐event analyses and all other reasons for treatment failure were censored. Similarly, for the analysis of 'Time to treatment failure due to lack of efficacy' only treatment failures which were documented to be due to lack of efficacy (i.e. continued seizures, either as a sole reason or due to both a lack of efficacy and adverse events) were classed as an 'event' within time‐to‐event analyses and all other reasons for treatment failure were censored.
Assessment of risk of bias in included studies
Two review authors (SJN and JW) independently assessed the risk of bias for each trial using the Cochrane 'Risk of bias' tool, as described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We rated each of the following six domains as low, unclear or high risk of bias: method of generating random sequence, allocation concealment, blinding methods, incomplete outcome data, selective outcome reporting and other sources of bias. Any discrepancies in risk of bias judgements of the two review authors were resolved by discussion. In the event of the presence of high risk of bias in included trials (due to inadequate allocation concealment or lack of blinding), we planned sensitivity analyses excluding these trials.
Measures of treatment effect
We measured all outcomes in this review as time‐to‐event outcomes with the hazard ratio (HR) and 95% confidence interval (CI) used as the measure of treatment effect. We calculated outcomes from IPD provided, where possible, or extracted from published trials if possible.
Unit of analysis issues
The unit of allocation and analysis was the individual for all included trials; and no trials included in meta‐analyses were of a repeated measures (longitudinal) nature or of a cross‐over design.
One included trial allocated participants to three treatment arms, 100 mg/day lamotrigine, 200 mg/day lamotrigine or 600 mg/day carbamazepine (Reunanen 1996). In the primary analysis for all outcomes, we pooled the two lamotrigine arms and calculated a hazard ratio of lamotrigine compared to carbamazepine using the IPD provided. In sensitivity analysis, we calculated separate hazard ratios for 100 mg/day lamotrigine versus carbamazepine and 200 mg/day lamotrigine versus carbamazepine to examine any difference in the doses of lamotrigine compared to carbamazepine.
Dealing with missing data
For each trial that supplied IPD, we reproduced results from trial results where possible and performed the following consistency checks.
We cross‐checked trial details against any published report of the trial and contacted original trial authors if we found missing data, errors or inconsistencies. If trial authors could not resolve inconsistencies between the IPD and the published data, depending on the extent of the inconsistencies, we performed sensitivity analysis or excluded the data from the meta‐analysis.
We reviewed the chronological randomisation sequence and checked the balance of prognostic factors, taking account of factors stratified for in the randomisation procedure.
Assessment of heterogeneity
We assessed heterogeneity statistically using the Q test (P value less than 0.10 for significance) and the I² statistic (Higgins 2003) (greater than 50% indicating considerable heterogeneity), with output produced using the generic inverse variance approach outlined in Data and analyses, and visually by inspecting forest plots.
Assessment of reporting biases
Two review authors (SJN and JW) undertook all full quality and 'Risk of bias' assessments according to the methods outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). In theory, a review using IPD should overcome issues of reporting biases as unpublished data can be provided and unpublished outcomes calculated. We requested trial protocols with IPD for all trials. Any selective reporting bias detected could be assessed with the Outcome Reporting Bias In Trials (ORBIT) classification system (Kirkham 2010).
Data synthesis
We carried out our analysis on an intention‐to‐treat basis (that is, we analysed participants in the group to which they were randomised, irrespective of which treatment they actually received). Therefore, for the time‐to‐event outcomes, 'time to six‐month remission', 'time to 12‐month remission', 'time to 24 month remission' and 'time to first seizure post randomisation', we did not censor participants if treatment was withdrawn or if treatment failure occurred but follow‐up within the trial continued (e.g. if a participant continued to be followed up on a different treatment).
An intention‐to‐treat analysis tends toward finding no difference between treatments and we would have undertaken a secondary 'protocol correct' analysis as a sensitivity analysis if the primary analyses had suggested equivalence, in which case participants would have been censored at the time of treatment failure for seizure outcomes.
For all outcomes, we investigated the relationship between the time‐to‐event and treatment effect of the AEDs. We used Cox proportional hazards regression models to obtain trial‐specific estimates of log (hazard ratio) or treatment effect and associated standard errors in Stata Statistical Software, version 14 (Stata 2015). The model assumes that the ratio of hazards (risks) between the two treatment groups is constant over time (i.e. hazards are proportional). We tested this proportional hazards assumption of the Cox regression model for each outcome of each trial by testing the statistical significance of a time varying covariate in the model. We evaluated overall pooled estimates of hazard ratios (with 95% confidence intervals) using the generic inverse variance method. We expressed results as a hazard ratio (HR) and a 95% confidence interval (CI).
By convention, a HR greater than 1 indicates that an event is more likely to occur earlier on lamotrigine than on carbamazepine. Hence, for time to treatment failure or time to first seizure, a HR greater than 1 indicates a clinical advantage for carbamazepine (e.g. a HR of 1.2 would suggest a 20% increase in hazard of treatment failure from lamotrigine compared with carbamazepine), and for time to six‐month, 12‐month and 24‐month remission a HR greater than 1 indicates a clinical advantage for lamotrigine (i.e. the seizure‐free period occurs earlier on lamotrigine than carbamazepine).
Subgroup analysis and investigation of heterogeneity
There is a strong clinical belief that some AEDs are more effective in some seizure types than others (see Description of the intervention and How the intervention might work), therefore we stratified all analyses by seizure type (focal onset versus generalised onset), according to the classification of main seizure type at baseline. We classified focal seizures (simple or complex) and focal secondarily generalised seizures as focal epilepsy. We classified primarily generalised seizures as generalised epilepsy.
We conducted a Chi² test of interaction between treatment and epilepsy type. If we found significant statistical heterogeneity to be present, we performed meta‐analysis with a random‐effects model in addition to a fixed‐effect model, presenting the results of both models and performing sensitivity analyses to investigate differences in trial characteristics. If heterogeneity is found to be present in future updates and available data allow, we may investigate variables that may contribute to the variability (e.g. participant covariates, trial design) via regression models
Sensitivity analysis
We performed several sensitivity analyses to test the robustness of our results to characteristics of the included trials, as follows.
Definition of time to treatment failure: we classified reasons for treatment failure that were related to the trial treatment as 'events' and 'censored' reasons not related to treatment in the analysis of 'time to treatment failure'. If reasons for treatment failure were classified differently as events or censored in included trials to our definitions, we conducted sensitivity analyses to account for differences in the definition of a treatment failure 'event'.
One trial considered participants to have completed the trial and hence withdrew treatment if they experienced a seizure after week six (Reunanen 1996). This does not correspond with the treatment failure definition recommended by ILAE 1998, and used in this review. We included treatment failure data for the participants from this study, Reunanen 1996, in the primary analysis of time to treatment failure and excluded them in sensitivity analysis to examine any effect of the difference in definition of treatment failure.
Seizure dates: one trial did not include seizures that occurred during the first four weeks of the trial in efficacy analyses and dates of seizures before week four were not supplied to us (Nieto‐Barrera 2001). Therefore, we calculated seizure outcomes as the time to first seizure and time to six‐month remission after week four rather than after randomisation. We included seizure data from this study in the primary analysis of time to first seizure and time to six‐month remission and excluded them in sensitivity analysis to examine any effect of the difference in origin time of the seizure count.
Aggregate data: time to treatment failure was presented as summary statistics or graphically in four of the trials for which IPD were not available (Gilad 2007; Rowan 2005; Saetre 2007; Steinhoff 2005) and time to first seizure was presented graphically in three of the trials for which IPD were not available (Gilad 2007; Rowan 2005; Saetre 2007). In Saetre 2007, hazard ratios and 95% confidence intervals were published for both time‐to‐event outcomes. Due to the small number of events for the outcomes in two studies (Gilad 2007; Steinhoff 2005), it was possible to estimate individual treatment failure/seizure times from the graphs and therefore calculate an estimated hazard ratio. In Rowan 2005, we used indirect methods and approximate numbers at risk at a range of time points throughout the trials (described in Data extraction and management) to estimate the hazard ratios for the outcomes. These estimated hazard ratios are combined with the hazard ratios calculated from the trials providing IPD in sensitivity analysis.
Seizure freedom: all included trials were of at least 24 weeks' (around six months') duration. Those providing IPD that were over six months' duration contributed to the outcome 'time to six‐month remission of seizures' (Brodie 1995 A; Brodie 1995 B; Eun 2012; Lee 2011; Reunanen 1996; SANAD A 2007; Werhahn 2015). Two trials were of 24 weeks' duration (Brodie 1999; Nieto‐Barrera 2001).
We conducted sensitivity analysis calculating a pooled risk ratio of seizure freedom at six months and including the data from two studies (Brodie 1999; Nieto‐Barrera 2001), (assuming 24 weeks is approximately equal to six months) and the trials for which IPD were not available. We estimated seizure freedom at six months from the graph of time to first seizure published in Saetre 2007. We also conducted sensitivity analysis, calculating a pooled risk ratio of seizure freedom throughout the whole trial combining IPD and aggregated data from all included trials.
Misclassification of seizure type is a recognised problem in epilepsy, whereby some people with generalised seizures have been mistakenly classed as having focal onset seizures and vice versa. There is clinical evidence that individuals with generalised onset seizures are unlikely to have an 'age of onset' greater than 25 to 30 years (Malafosse 1994). Such misclassification impacted upon the results of three reviews in our series of pair‐wise reviews for monotherapy in epilepsy comparing carbamazepine, phenobarbitone, phenytoin and sodium valproate in which around 30% to 50% of participants analysed may have had their seizure type misclassified as generalised onset (Nolan 2016d; Nolan 2016b; Nevitt 2017b). Given the potential biases introduced into those reviews, we examined the distribution of age at onset for individuals with generalised seizures in the trials included in this review, to assess the potential impact of misclassification of seizure type on the outcomes.
Two trials recruited only individuals with focal onset seizures (Eun 2012; Werhahn 2015), therefore there were no participants with new onset generalised seizures over the age of 30 in these trials.
Two trials were designed to include participants with focal onset seizures only, however three participants in Nieto‐Barrera 2001, and nine participants in SANAD A 2007, were classified as having generalised onset seizures. Further, seizure type was missing for 85 participants in SANAD A 2007. We considered the individuals in these two trials to have a misclassification of seizure type. Overall:
in Brodie 1995 A, 20 out of the 54 participants (37%) classified as having generalised onset seizures were over the age of 30 at entry into the trial (and all over the age of 29 at seizure onset);
in Brodie 1995 B, 23 out of the 62 participants (37%) classified as having generalised onset seizures were over the age of 30 at entry into the trial (and all over the age of 29 at seizure onset);
in Brodie 1999, all 45 of the participants (100%) classified as having generalised onset seizures were over the age of 30 at entry into the trial (no age of onset data provided);
in Lee 2011, 9 out of the 15 participants (60%) classified as having generalised onset seizures were over the age of 30 at entry into the trial (no age of onset data provided);
in Reunanen 1996, 43 out of the 114 participants (38%) classified as having generalised onset seizures were over the age of 30 at entry into the trial (and all over the age of 23 at seizure onset).
In total, 152 out of 302 participants (50%) classified as having generalised onset seizures may have been wrongly classified as having new onset generalised seizures. To investigate misclassification for each outcome, we undertook two sensitivity analyses:
we reclassified the 152 individuals with generalised seizure types and age at onset greater than 30 as having focal onset seizures and repeated subgroup analysis;
for SANAD A 2007, we reclassified the 152 individuals with generalised seizure types and age at onset greater than 30, and the 85 individuals with missing seizure type, into an 'uncertain seizure type' group and repeated subgroup analysis with three groups.
Summary of findings and quality of the evidence (GRADE)
For the 2016 update, in a post hoc change from protocol, we have added two 'Summary of findings' tables to the review (outcomes in the tables decided before the update started based on clinical relevance).
Table 1 reports the primary outcome of 'time to treatment failure' in the subgroups of participants with focal onset seizures, generalised onset seizures and overall adjusted by epilepsy type.
Table 2 reports the secondary outcomes of 'time to first seizure' and 'time to 12‐month remission' in the subgroups of participants with focal onset seizures, generalised onset seizures and overall adjusted by epilepsy type. (Due to small numbers of participants with generalised seizures contributing to the outcome of time to 12‐month remission, an overall treatment effect for all participants is presented in Table 2.)
We determined the quality of the evidence using the GRADE approach, where we downgraded evidence in the presence of high risk of bias in at least one trial, indirectness of the evidence, unexplained heterogeneity or inconsistency, imprecision of results and high probability of publication bias. We downgraded evidence by one level if the limitation was considered serious and two levels if considered very serious, as judged by the review authors. Under the GRADE approach, evidence may also be upgraded if a large treatment effect is demonstrated with no obvious biases or if a dose‐response effect exists.
Results
Description of studies
Results of the search
We included five trials in previous versions of this review (Brodie 1995 A; Brodie 1995 B; Brodie 1999; Nieto‐Barrera 2001; Reunanen 1996).
For the 2016 update of this review, we identified 148 records from the databases and search strategies outlined in Electronic searches. We removed 40 duplicate records and screened the titles and abstracts of 108 records for inclusion in the review. We excluded 81 records based on the title and abstract and assessed 27 full‐text articles for inclusion in the review. We excluded 18 articles from the review (see Excluded studies below), classified one article as awaiting assessment (Korean Lamotrigine Study Group 2008) and included eight additional trials (Eun 2012; Gilad 2007; Lee 2011; Rowan 2005; Saetre 2007; SANAD A 2007; Steinhoff 2005; Werhahn 2015).
For the 2018 update of this review, we identified 52 records from the databases and search strategies outlined in Electronic searches. We removed 17 duplicate records and screened the titles and abstracts of 35 records for inclusion in the review. All 35 records were clearly irrelevant and were excluded. The one article which was classified as awaiting in assessment in the 2016 update of the review was now included (Korean Lamotrigine Study Group 2008). Therefore, we included a total of 14 studies were included in the review (see Included studies).
See Figure 1 for a Preferred Reporting Items for Systematic Reviews and Meta‐Analyses (PRISMA) study flow diagram.
1.

Study flow diagram.
Included studies
We identified 13 published reports that met the inclusion criteria for this review (Brodie 1995; Brodie 1999; Eun 2012; Gilad 2007; Korean Lamotrigine Study Group 2008; Lee 2011; Nieto‐Barrera 2001; Reunanen 1996; Rowan 2005; Saetre 2007; SANAD A 2007; Steinhoff 2005; Werhahn 2015). One of the published reports (Brodie 1995), contained results on two separate randomised controlled trials run on very similar protocols (Brodie 1995 A; Brodie 1995 B). Although the two trials were reported within the same publication we treated them as separate trials within this Cochrane Review; therefore we included a total of 14 trials in the review.
One trial recruited adults of all ages (Gilad 2007), and one trial recruited adults over the age of 16 (Lee 2011). One trial recruited children between the ages of 6 and 12 (Eun 2012). Two trials recruited individuals over the age of 12 (Reunanen 1996; Steinhoff 2005), and two recruited individuals over the age of 13 (Brodie 1995 A; Brodie 1995 B). One trial recruited individuals over the age of two (Nieto‐Barrera 2001), and one recruited individuals over the age of four (SANAD A 2007). Four trials recruited the elderly; two trials recruited individuals over the age of 60 (Rowan 2005; Werhahn 2015), and two recruited individuals over the age of 65 (Brodie 1999; Saetre 2007). The remaining trial did not state the eligible age ranges recruited (Korean Lamotrigine Study Group 2008).
Five trials were designed to recruit individuals with focal seizures only (Eun 2012; Lee 2011; Nieto‐Barrera 2001; SANAD A 2007; Werhahn 2015); however three of these trials did recruit some individuals with generalised onset seizures (Lee 2011; Nieto‐Barrera 2001; SANAD A 2007). We examine this seizure classification in sensitivity analysis. The remaining nine trials recruited individuals with focal or generalised tonic‐clonic seizures with or without other generalised seizure types.
Seven trials recruited individuals with new onset seizures (Brodie 1995 A; Brodie 1995 B; Brodie 1999; Eun 2012; Saetre 2007; Steinhoff 2005; Werhahn 2015). Four trials recruited individuals with new onset or untreated seizures (Korean Lamotrigine Study Group 2008; Lee 2011; Nieto‐Barrera 2001; Reunanen 1996), one trial recruited individuals with new onset, untreated or seizures treated to a “sub‐therapeutic” level (Rowan 2005), one trial recruited individuals with new onset, relapsed or recurrent seizures (failure of an AED not randomised in the trial) (SANAD A 2007), and one trial recruited individuals with new onset seizures following ischaemic stroke (Gilad 2007).
Four multicentre trials were conducted in the UK (Brodie 1995 A; Brodie 1995 B; Brodie 1999; SANAD A 2007). Two multicentre trials were conducted across Europe (Saetre 2007; Werhahn 2015), one multicentre trial across Europe and Mexico (Nieto‐Barrera 2001), and one multicentre trial across Europe and Australia (Reunanen 1996). One multicentre trial was conducted in Germany (Steinhoff 2005), one multicentre trial in the USA (Rowan 2005), three multicentre trials in the Republic of Korea (Eun 2012; Korean Lamotrigine Study Group 2008; Lee 2011), and one single‐centre trial was conducted in Israel (Gilad 2007).
We did not obtain individual participant data (IPD) for five trials including a total of 1215 participants. According to trial sponsor, GlaxoSmithKline, data could not be located for two trials (Korean Lamotrigine Study Group 2008; Saetre 2007), and data could not be provided due to restrictions over the anonymisation of datasets of trials conducted in Germany (Steinhoff 2005). For the other two trials, we made contact with the authors/sponsors who expressed interest in collaborating in this IPD meta‐analysis but at the time of writing, no data had been received (Gilad 2007; Rowan 2005). If IPD are received from these trials, we will include the data in future updates.
Individual participant data were available for the remaining nine trials, which recruited a total of 2572 participants, representing 68% of 3787 individuals from all 14 identified eligible trials. Data were available for the following participant characteristics (percentage of 2572 participants with data available): drug randomised (100%), sex (99%, data missing for 18 participants in SANAD A 2007), seizure type (97%, data missing for 85 participants in SANAD A 2007), age at randomisation (99%, data missing for 18 participants in SANAD A 2007, one participant in Nieto‐Barrera 2001, and two participants in Reunanen 1996), and number of seizures in the six months prior to randomisation (99%, missing for 18 participants in SANAD A 2007, one participant in Reunanen 1996, and six participants in Werhahn 2015). Time since first seizure to randomisation was provided for 691 participants out of 695 participants from four trials (Brodie 1995 A; Brodie 1995 B; Eun 2012 (data missing for one participant); Reunanen 1996 (data missing for three participants)).
Seven trials provided the results of neurological examinations for 1693 out of 1711 participants (99%) (Brodie 1995 A; Brodie 1995 B; Brodie 1999; Eun 2012; Lee 2011; Reunanen 1996; SANAD A 2007 (data for 18 participants missing)).
Six trials provided electroencephalographic (EEG) results for 710 out of 1044 participants (64%) (134 from Brodie 1995 A, 118 from Brodie 1995 B, 84 from Eun 2012, 110 from Lee 2011, 26 from Reunanen 1996, and 238 from Werhahn 2015).
Seven trials provided computerised tomography/magnetic resonance imaging (CT/MRI) results for 788 out of 1194 participants (66%) (94 from Brodie 1995 A, 92 from Brodie 1995 B, 149 from Brodie 1999, 84 from Eun 2012, 110 from Lee 2011, 21 from Reunanen 1996, and 238 from Werhahn 2015).
See the Characteristics of included studies table, Table 3 and Table 4 for further details.
1. Demographic characteristics of trial participants (trials providing individual participant data).
| Focal seizures: n (%) | Male gender: n (%) |
Age at entry (years): Mean (SD), range |
Aged > 30 and generalised seizures: n (%) |
Epilepsy duration (years): Mean (SD), range |
Number of seizures in prior 6 months: median (range) |
|||||||||||||
| LTG | CBZ | Missing | LTG | CBZ | Missing | LTG | CBZ | Missing | LTG | CBZ | Missing | LTG | CBZ | Missing | LTG | CBZ | Missing | |
| Brodie 1995 A | 44 (63%) | 38 (58%) | 0 | 28 (40%) | 28 (42%) | 0 | 35.3 (17.1), 15 to 71 | 32.5 (14.4), 13 to 69 | 0 | 11 | 9 | 0 | 2.2 (3.3), 0 to 17.9 | 1.8 (2.3), 0.3 to 11.0 | 0 | 4 (1 to 490) | 3 (1 to 960) | 0 |
| Brodie 1995 B | 27 (44%) | 35 (56%) | 0 | 26 (43%) | 30 (48%) | 0 | 30.9 (14.5), 14 to 86 | 29.1 (13.9), 14 to 81 | 0 | 12 | 11 | 0 | 1.4 (3.2), 0 to 19.4 | 1.2 (1.8), 0 to 7.1 | 0 | 3 (1 to 1020) | 3 (2 to 122) | 0 |
| Brodie 1999 | 72 (71%) | 33 (69%) | 0 | 55 (54%) | 28 (58%) | 0 | 77.3 (6.1), 65 to 94 | 76.2 (5.9), 66 to 88 | 0 | 30 | 15 | 0 | NA | NA | 150 | 3 (1 to 163) | 4.5 (1 to 108) | 0 |
| Eun 2012 | 43 (100%) | 41 (100%) | 0 | 24 (56%) | 24 (59%) | 0 | 9.2 (2.0), 6 to 13 | 8.3 (2.1), 5 to 12 | 0 | 0 | 0 | 0 | 0.6 (0.9), 0 to 4.5 | 0.5 (0.3), 0 to 1.4 | 1 | 3(2 to 11) | 3 (2 to 11) | 0 |
| Lee 2011 | 51 (89%) | 44 (83%) | 0 | 24 (42%) | 33 (62%) | 0 | 33.6 (12.6), 16 to 60 | 38.3 (11.5), 16 to 60 | 0 | 2 | 7 | 0 | NA | NA | 110 | 2(0 to 60) | 2 (0 to 200) | 0 |
| Nieto‐Barrera 2001 | 418 (99.5%) | 201 (99.5%) | 0 | 222 (53%) | 107 (53%) | 0 | 27.1 (21.7), 2 to 84 | 27.5 (21.0), 2 to 77 | 1 | 1 | 1 | 0 | NA | NA | 622 | 4 (1 to 9000) | 3 (1 to 3600) | 0 |
| Reunanen 1996 | 150 (65%) | 87 (72%) | 0 | 127 (55%) | 61 (50%) | 0 | 31.8 (14.0), 12 to 71 | 32.7 (14.6), 13 to 71 | 2 | 31 | 12 | 0 | 2.2 (3.2), 0 to 17.1 | 2.2 (3.7), 0.26 to 8 | 3 | 3(1 to 133) | 3 (1 to 145) | 1 |
| SANAD A 2007 | 329 (99%) | 333 (99%) | 85 | 205 (55%) | 204 (55%) | 18 | 36.8 (18.4), 6 to 83 | 39.3 (18.4), 5 to 82 | 18 | 46 | 42 | 0 | NA | NA | 727 | 2(0 to 1185) | 4 (0 to 466) | 19 |
| Werhahn 2015 | 118 (100%) | 121 (100%) | 0 | 69 (59%) | 65 (54%) | 0 | 70.8 (7.5), 60 to 88 | 71.8 (6.7), 60 to 89 | 0 | 0 | 0 | 0 | NA | NA | 239 | 2 (1 to 20) | 2 (1 to 90) | 6 |
CBZ = carbamazepine, LTG = lamotrigine; n = number of participants; NA = not applicable; SD = standard deviation
2. Baseline neurologic characteristics of participants (trials providing individual participant data).
| EEG normal: n (%) | CT scan normal: n (%) |
Neurological exam normal: n (%) |
|||||||
| LTG | CBZ | Missing | LTG | CBZ | Missing | LTG | CBZ | Missing | |
| Brodie 1995 A | 32 (46%) | 30 (46%) | 2 | 38 (84%) | 44 (90%) | 42 | 62 (89%) | 61 (92%) | 0 |
| Brodie 1995 B | 42 (73%) | 34 (56%) | 6 | 34 (77%) | 38 (79%) | 32 | 56 (92%) | 52 (83%) | 0 |
| Brodie 1999 | NA | NA | 150 | 39 (39%) | 23 (48%) | 1 | 59 (58%) | 31 (65%) | 0 |
| Eun 2012 | 3 (7%) | 3 (7%) | 0 | 38 (88%) | 37(90%) | 0 | 43 (100%) | 40 (98%) | 0 |
| Lee 2011 | 31 (54%) | 27 (51%) | 0 | 36 (63%) | 38 (72%) | 0 | 57 (100%) | 53 (100%) | 0 |
| Nieto‐Barrera 2001 | NA | NA | 622 | NA | NA | 622 | NA | NA | 622 |
| Reunanen 1996 | 9 (53%) | 4 (44%) | 325 | 11 (73%) | 5 (83%) | 330 | 202 (89%) | 103 (85%) | 0 |
| SANAD A 2007 | NA | NA | 756 | NA | NA | 756 | 277 (75%) | 281 (76%) | 18 |
| Werhahn 2015 | 45 (38%) | 37 (31%) | 1 | 26 (22%) | 26 (21%) | 1 | NA | NA | 239 |
CBZ = carbamazepine; CT = computerised tomography; EEG = electroencephalogram; LTG = lamotrigine; n = number of participants; NA = not applicable
Excluded studies
We excluded seven duplicate references (Eun 2008; Lee 2010; Ramsay 2003; Saetre 2006; Saetre 2009; Saetre 2010; Steinhoff 2004), and retained the most relevant primary reference for the trial in the review (Eun 2012; Lee 2011; Rowan 2005; Saetre 2007; Steinhoff 2005, respectively). We excluded five trials that did not compare lamotrigine and carbamazepine (Czapinski 1997; Gilliam 1998; Motte 1997; Steiner 1999; Stolarek 1994). We excluded three trials that were not of a monotherapy design (Carmant 2001; Fakhoury 2000; Jawad 1989). We excluded two trials that were not randomised (Martinez 2000; Zeng 2010). We excluded one trial that did not make a fully randomised comparison of lamotrigine and carbamazepine (Baxter 1998); lamotrigine was compared to the treating physician's choice of carbamazepine or sodium valproate. See Characteristics of excluded studies for further details.
Risk of bias in included studies
For further details, see the Characteristics of included studies table and Figure 2.
2.
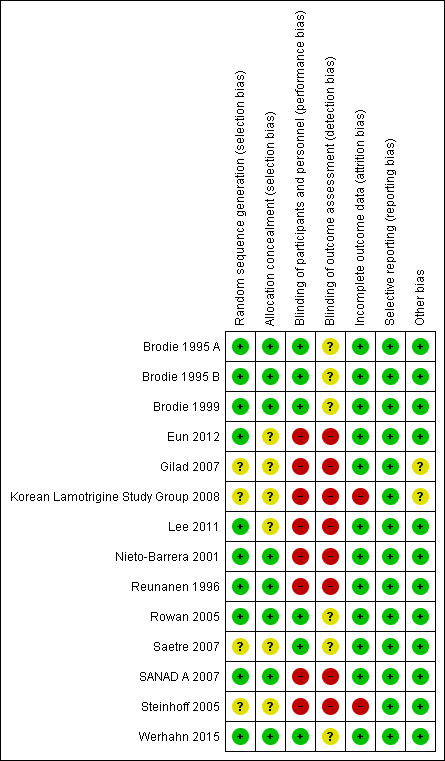
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
(1) Trials for which we received individual participant data (information reported in published papers or provided with IPD)
All nine trials used adequate methods of randomisation via computer‐generated random list and we judged them to be at low risk of bias (Brodie 1995 A; Brodie 1995 B; Brodie 1999; Eun 2012; Lee 2011; Nieto‐Barrera 2001; Reunanen 1996; SANAD A 2007; Werhahn 2015); two trials reported that block randomisation was used (Lee 2011; Werhahn 2015), and one trial reported that minimisation was used (SANAD A 2007).
Seven trials reported adequate methods of allocation concealment and we judged them to be at low risk of bias; five concealed treatment allocation with sealed, opaque envelopes (Brodie 1995 A; Brodie 1995 B; Brodie 1999; Nieto‐Barrera 2001; Reunanen 1996); one trial used telephone randomisation to a central allocation service (SANAD A 2007), and one trial used pharmacy allocation (Werhahn 2015). The remaining two trials did not report how allocation was concealed and we judged them to be at unclear risk of bias (Eun 2012; Lee 2011).
(2) Trials for which no individual participant data were available (information reported in published papers only)
One of the trials reported that blocked randomisation via a computer‐generated list and telephone randomisation to a central allocation service were used (Rowan 2005). We judged this trial to be at low risk of selection bias for random sequence generation and allocation concealment. The remaining four trials were described as "randomised" but did not provide information about the method of generation of the random list or allocation concealment so we judged them to be at unclear risk of bias (Gilad 2007; Korean Lamotrigine Study Group 2008; Saetre 2007; Steinhoff 2005).
Blinding
(1) Trials for which we received individual participant data (information reported in published papers or provided with IPD)
Four trials were double‐blind (participants and personnel) with the blinding achieved by using tablets of identical appearance; we judged these trials to be at low risk of performance bias (Brodie 1995 A; Brodie 1995 B; Brodie 1999; Werhahn 2015). In all four of these trials, the trial investigator was blinded but no information was provided as to whether other outcome assessors were blinded, therefore we judged all four trials to be at unclear risk of detection bias.
The remaining five trials were open‐label and we judged them to be at high risk of performance and detection bias (Eun 2012; Lee 2011; Nieto‐Barrera 2001; Reunanen 1996; SANAD A 2007).
(2) Trials for which no individual participant data were available (information reported in published papers only)
Two trials were double‐blind (participants and personnel) with the blinding achieved by using double dummy tablets; we judged these trials to be at low risk of performance bias (Rowan 2005; Saetre 2007). However, for these two trials no information was provided regarding blinding of outcome assessors therefore we judged the two trials to be at unclear risk of detection bias.
The remaining three trials were open‐label and we judged them to be at high risk of performance and detection bias (Gilad 2007; Korean Lamotrigine Study Group 2008; Steinhoff 2005).
Incomplete outcome data
(1) Trials for which we received individual participant data (information reported in published papers or provided with IPD)
In theory, a review using individual participant data should overcome issues of attrition bias as unpublished data can be provided, unpublished outcomes calculated and all randomised participants can be analysed by an intention‐to‐treat approach. All nine trials provided individual participant data for all randomised individuals and reported the extent of follow‐up for each individual (Brodie 1995 A; Brodie 1995 B; Brodie 1999; Eun 2012; Lee 2011; Nieto‐Barrera 2001; Reunanen 1996; SANAD A 2007; Werhahn 2015). We queried any missing data with the original trial authors. From the information provided by the authors, we deemed the small amount of missing data present (see Included studies) to be missing at random and not effecting our analysis.
(2) Trials for which no individual participant data were available (information reported in published papers only)
Three trials reported attrition rates and analysed all randomised participants using an intention‐to‐treat approach and we judged them to be at low risk of attrition bias (Gilad 2007; Rowan 2005; Saetre 2007). The remaining two trials did not analyse data for all randomised participants (Korean Lamotrigine Study Group 2008; Steinhoff 2005) and one of the trials did not state to which drug those excluded from analysis were randomised (Steinhoff 2005). This is not an intention‐to‐treat analysis therefore we judged these trials to be at high risk of attrition bias.
Selective reporting
(1) Trials for which we received individual participant data (information reported in published papers or provided with IPD)
In theory, a review using individual participant data should overcome issues of reporting biases as unpublished data can be provided and unpublished outcomes calculated. We sought trial protocols in all individual participant data requests and seven protocols were provided (Brodie 1995 A; Brodie 1995 B; Brodie 1999; Nieto‐Barrera 2001; Reunanen 1996; SANAD A 2007; Werhahn 2015). We received sufficient individual participant data to calculate all outcomes for all nine trials (depending on trial duration; e.g. time to 12‐month remission could not be calculated for a trial of 24 weeks etc.)
(2) Trials for which no individual participant data were available (information reported in published papers only)
Protocols were not available for any of the four trials, however a clinical summary report was provided for two trials from the sponsor (Saetre 2007; Steinhoff 2005), and case report forms of data collected were provided for one trial by the sponsor (Rowan 2005). All trials reported seizure and adverse event outcomes well, therefore we judged all five trials to be at low risk of selective reporting bias (Gilad 2007; Korean Lamotrigine Study Group 2008; Rowan 2005; Saetre 2007; Steinhoff 2005).
Other potential sources of bias
We identified no other sources of bias for 13 of the 14 included trials (Brodie 1995 A; Brodie 1995 B; Brodie 1999; Eun 2012; Korean Lamotrigine Study Group 2008; Lee 2011; Nieto‐Barrera 2001; Reunanen 1996; Rowan 2005; Saetre 2007; SANAD A 2007; Steinhoff 2005; Werhahn 2015). In one trial, it was unclear if all participants were receiving AED monotherapy treatment ('total number of AEDs' described in Table 1 of the publication), so we judged this trial to be at unclear risk of bias (Gilad 2007).
Effects of interventions
See Table 5 for details regarding the number of individuals (with individual participant data (IPD)) contributing to each analysis, Table 1 for a summary of the results for the primary outcome 'time to treatment failure' (stratified by epilepsy type), and Table 2 for a summary of results for the secondary outcomes 'time to first seizure' and 'time to 12‐month remission'.
3. Number of participants included in analyses (trials providing individual participant data).
| Number randomised | Time to treatment failure | Time to first seizure |
Time to 6‐ month remission1 |
Time to 12‐ month remission |
Time to 24‐ month remission |
|||||||||||||
| LTG | CBZ | Total | LTG | CBZ | Total | LTG | CBZ | Total | LTG | CBZ | Total | LTG | CBZ | Total | LTG | CBZ | Total | |
| Brodie 1995 A | 70 | 66 | 136 | 70 | 66 | 136 | 70 | 66 | 136 | 70 | 66 | 136 | NA | NA | NA | NA | NA | NA |
| Brodie 1995 B | 61 | 63 | 124 | 61 | 63 | 124 | 61 | 63 | 124 | 61 | 63 | 124 | NA | NA | NA | NA | NA | NA |
| Brodie 19991 | 102 | 48 | 150 | 102 | 48 | 150 | 102 | 48 | 150 | 102 | 48 | 150 | NA | NA | NA | NA | NA | NA |
| Eun 2012 | 43 | 41 | 84 | 43 | 41 | 84 | 43 | 41 | 84 | 43 | 41 | 84 | NA | NA | NA | NA | NA | NA |
| Lee 2011 | 57 | 53 | 110 | 57 | 53 | 110 | 57 | 53 | 110 | 57 | 53 | 110 | NA | NA | NA | NA | NA | NA |
| Nieto‐Barrera 20011,2 | 420 | 202 | 622 | 419 | 202 | 621 | 419 | 202 | 621 | 419 | 202 | 621 | NA | NA | NA | NA | NA | NA |
| Reunanen 1996 | 230 | 121 | 351 | 230 | 121 | 351 | 230 | 121 | 351 | 230 | 121 | 351 | NA | NA | NA | NA | NA | NA |
| SANAD A 20073 | 378 | 378 | 756 | 377 | 377 | 754 | 377 | 378 | 755 | 377 | 378 | 755 | 377 | 378 | 755 | 377 | 378 | 755 |
| Werhahn 20154 | 118 | 121 | 239 | 118 | 121 | 239 | 117 | 116 | 233 | 117 | 116 | 233 | 117 | 116 | 233 | NA | NA | NA |
| Total | 1479 | 1093 | 2572 | 1477 | 1092 | 2569 | 1476 | 1088 | 2564 | 1476 | 1088 | 2564 | 494 | 494 | 988 | 377 | 378 | 755 |
CBZ = carbamazepine; LTG = lamotrigine; NA: not applicable (trial duration not sufficient to measure the outcome).
1. Brodie 1999, and Nieto‐Barrera 2001, are of 24 weeks duration (approximately six months). The two trials are not included in the analyses of time to six‐month remission but are included in sensitivity analysis of seizure freedom at six months.
2. Follow‐up data are missing for one participant in Nieto‐Barrera 2001.
3. Treatment failure time missing for two participants and seizure data after follow‐up missing for one participant in SANAD A 2007.
4. Seizure data after follow‐up missing for six participants in Werhahn 2015.
For survival curve plots (cumulative incidence), see Figure 3; Figure 4; Figure 5; Figure 6; Figure 7; Figure 8; Figure 9; Figure 10; Figure 11; Figure 12 and Figure 13. We produced all cumulative incidence plots in Stata software version 14 (Stata 2015), using data from all trials providing IPD combined. We note that participants with event times of zero (i.e. those who withdrew from treatment or experienced seizure recurrence on the day of randomisation) are not included in the 'Numbers at risk' on the graphs and that data is not stratified by trial within these survival curve plots. All figures are intended to provide a visual representation of outcomes, extent of follow‐up and visual differences between seizure types. These graphs are not intended to show statistical significance and numerical values may vary compared to the text due to differences in methodology.
3.

Time to treatment failure for any reason related to treatment (CBZ: Carbamazepine; LTG: Lamotrigine)
4.

Time to treatment failure due to adverse effects (CBZ: Carbamazepine; LTG: Lamotrigine)
5.

Time to treatment failure due to lack of efficacy (CBZ: Carbamazepine; LTG: Lamotrigine)
6.
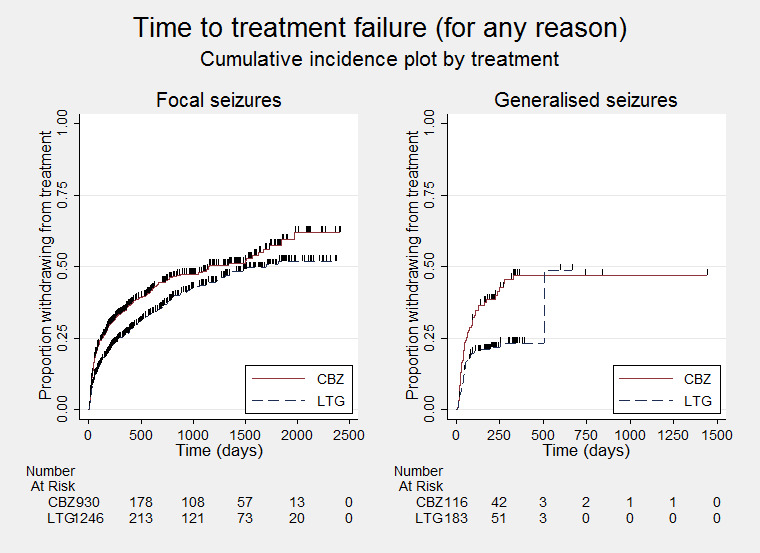
Time to treatment failure for any reason related to treatment ‐ by seizure type (CBZ: Carbamazepine; LTG: Lamotrigine)
7.

Time to treatment failure for due to adverse events ‐ by seizure type (CBZ: Carbamazepine; LTG: Lamotrigine)
8.
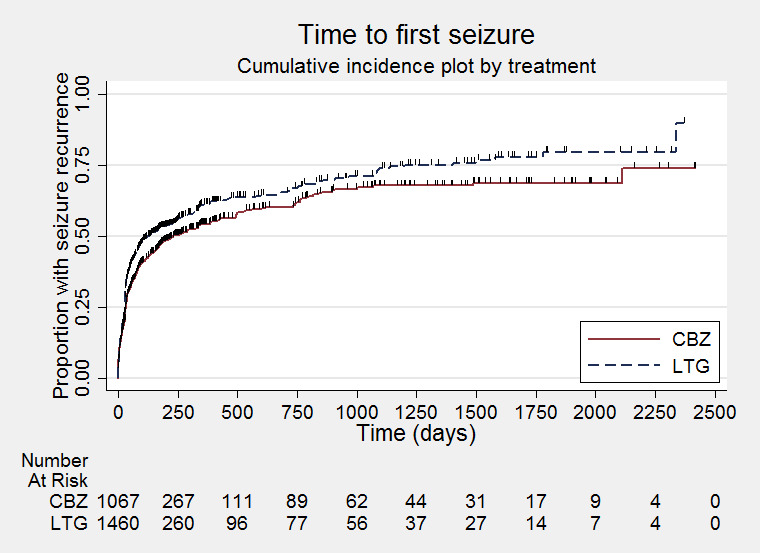
Time to first seizure (CBZ: Carbamazepine; LTG: Lamotrigine)
9.

Time to first seizure ‐ by seizure type (CBZ: Carbamazepine; LTG: Lamotrigine)
10.
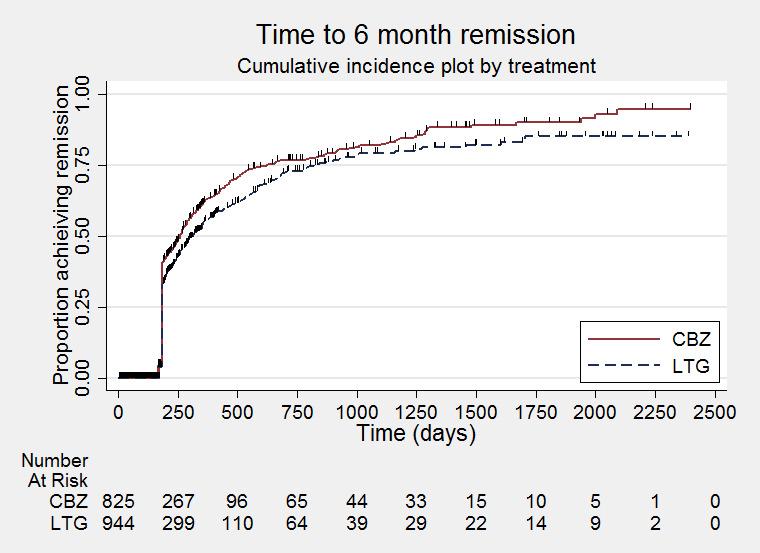
Time to six‐month remission (CBZ: Carbamazepine; LTG: Lamotrigine)
11.
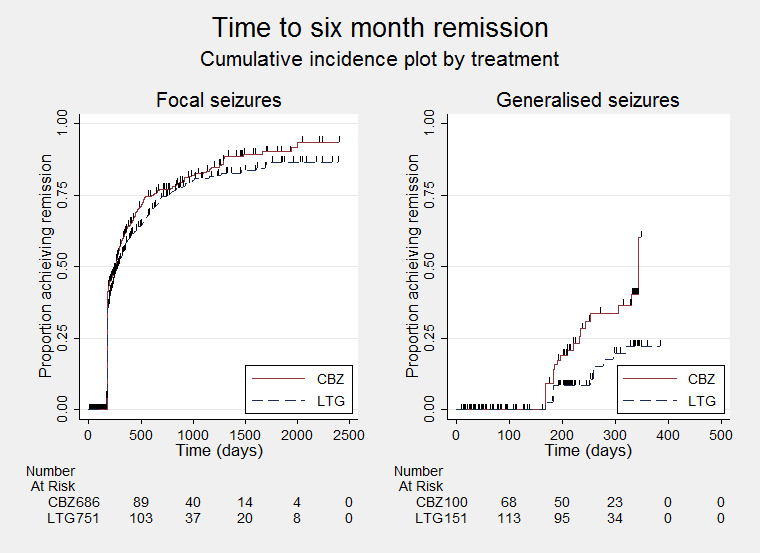
Time to six‐month remission ‐ by seizure type (CBZ: Carbamazepine; LTG: Lamotrigine)
12.
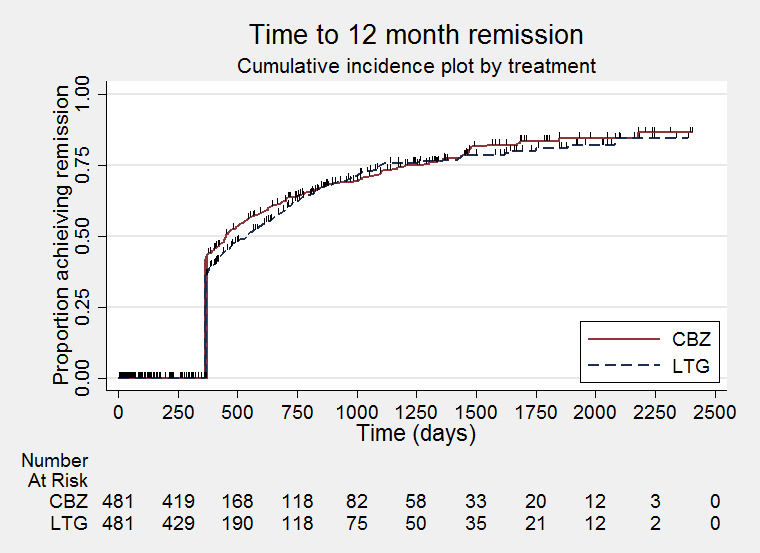
Time to 12‐month remission (CBZ: Carbamazepine; LTG: Lamotrigine)
13.
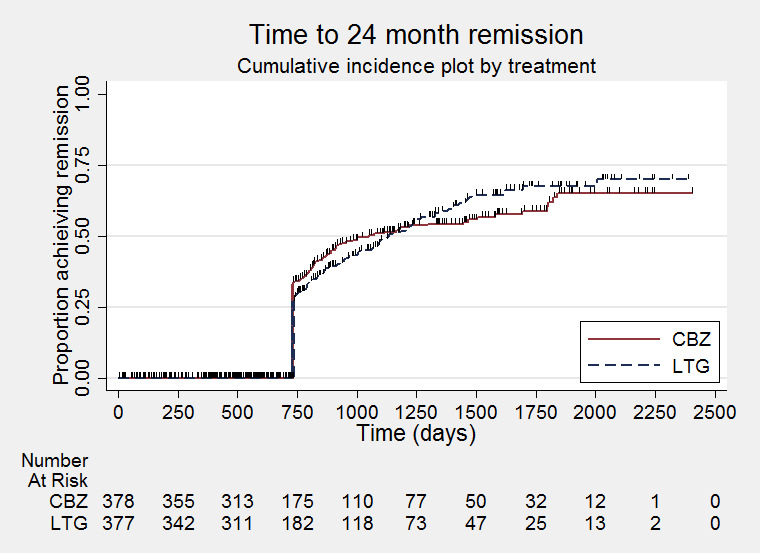
Time to 24‐month remission (CBZ: Carbamazepine; LTG: Lamotrigine)
We calculated all hazard ratios (HRs) presented below by generic inverse variance fixed‐effect meta‐analysis unless otherwise stated. All analyses met the assumption of proportional hazards (addition of time‐varying covariate into the model non‐significant) unless stated below.
Primary outcome
Time to treatment failure
For this outcome, a HR of less than 1 indicates a clinical advantage for lamotrigine. See Table 6 for reasons for premature termination for 3768 participants in all 14 included trials (missing data for 19 participants from one trial (Korean Lamotrigine Study Group 2008)), and how we classified these reasons in analysis of IPD. In one study, one participant randomised to lamotrigine had missing date and reason for treatment failure (Nieto‐Barrera 2001), and in another study two participants had missing dates of treatment failure (one withdrew from lamotrigine due to remission of seizures and one withdrew from carbamazepine due to 'other' reasons not related to the allocated drug) (SANAD A 2007).
4. Reasons for premature discontinuation (treatment failure).
|
Reason for early termination1 |
Classification in time‐to‐event analyses: Event | Classification in time‐to‐event analyses: Censored | Total | ||||||||||
| Adverse events | Inadequate response/seizure recurrence |
Both adverse events and inadequate response | Protocol violation/non‐compliance | Withdrew consent/participant choice3 | Other (treatment‐related)4 |
Illness or death (not treatment‐related) | Remission of seizures | Lost to follow‐up | Other (not treatment‐related)5 |
Completed trial | |||
| Brodie 1995 A | LTG | 18 | 3 | 0 | 3 | 1 | 0 | 1 | 0 | 1 | 0 | 43 | 70 |
| CBZ | 22 | 2 | 0 | 6 | 1 | 0 | 0 | 0 | 2 | 0 | 33 | 66 | |
| Brodie 1995 B | LTG | 7 | 3 | 0 | 5 | 2 | 0 | 0 | 0 | 1 | 0 | 43 | 61 |
| CBZ | 23 | 1 | 0 | 3 | 1 | 0 | 0 | 0 | 1 | 0 | 34 | 63 | |
| Brodie 1999 | LTG | 18 | 0 | 0 | 7 | 3 | 0 | 1 | 0 | 2 | 0 | 71 | 102 |
| CBZ | 20 | 0 | 0 | 3 | 2 | 2 | 0 | 0 | 1 | 0 | 20 | 48 | |
| Eun 2012 | LTG | 3 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 4 | 0 | 34 | 43 |
| CBZ | 3 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 2 | 0 | 35 | 41 | |
| Gilad 20072 | LTG | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 31 | 32 |
| CBZ | 10 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 21 | 32 | |
| Korean Lamotrigine Study Group 20082 | LTG | 24 | 11 | 0 | 0 | 0 | 52 | 0 | 0 | 0 | 0 | 165 | 252 |
| CBZ | 13 | 2 | 0 | 0 | 0 | 22 | 0 | 0 | 0 | 0 | 85 | 122 | |
| Lee 2011 | LTG | 4 | 3 | 0 | 2 | 7 | 0 | 0 | 0 | 2 | 0 | 39 | 57 |
| CBZ | 7 | 0 | 0 | 3 | 2 | 0 | 0 | 0 | 7 | 0 | 34 | 53 | |
| Nieto‐Barrera 20017 | LTG | 34 | 12 | 0 | 6 | 16 | 0 | 0 | 0 | 13 | 0 | 339 | 420 |
| CBZ | 26 | 4 | 0 | 11 | 2 | 0 | 0 | 0 | 3 | 0 | 156 | 202 | |
| Reunanen 1996 | LTG | 10 | 1 | 0 | 17 | 3 | 3 | 4 | 0 | 0 | 0 | 192 | 230 |
| CBZ | 12 | 0 | 0 | 11 | 6 | 0 | 2 | 0 | 0 | 0 | 90 | 121 | |
| Rowan 20052 | LTG | 20 | 7 | 0 | 15 | 24 | 0 | 7 | 0 | 10 | 5 | 112 | 200 |
| CBZ | 54 | 3 | 0 | 14 | 28 | 0 | 14 | 0 | 4 | 10 | 71 | 198 | |
| Saetre 20072 | LTG | 13 | 0 | 0 | 2 | 0 | 10 | 0 | 0 | 0 | 0 | 68 | 93 |
| CBZ | 23 | 0 | 0 | 1 | 0 | 6 | 0 | 0 | 0 | 0 | 61 | 91 | |
| SANAD A 20078 | LTG | 61 | 60 | 11 | 1 | 4 | 16 | 7 | 23 | 0 | 14 | 181 | 378 |
| CBZ | 104 | 43 | 20 | 2 | 1 | 7 | 10 | 25 | 0 | 15 | 151 | 378 | |
| Steinhoff 20052 | LTG | 7 | 1 | 0 | 0 | 13 | 3 | 0 | 0 | 0 | 0 | 64 | 88 |
| CBZ | 17 | 0 | 0 | 0 | 7 | 5 | 0 | 0 | 0 | 0 | 59 | 88 | |
| Werhahn 2015 | LTG | 31 | 2 | 0 | 6 | 13 | 1 | 0 | 0 | 0 | 0 | 65 | 118 |
| CBZ | 39 | 3 | 0 | 4 | 20 | 0 | 0 | 0 | 0 | 0 | 55 | 121 | |
| Total LTG | 251 | 105 | 11 | 64 | 86 | 85 | 20 | 23 | 33 | 19 | 1447 | 2144 | |
| Total CBZ | 373 | 58 | 20 | 58 | 71 | 42 | 27 | 25 | 20 | 25 | 905 | 1624 | |
| Total (all) | 624 | 163 | 31 | 122 | 157 | 127 | 47 | 48 | 53 | 44 | 2352 | 3768 | |
1. Primary reason for discontinuation specified ‐ participants may have withdrawn from allocated treatment for a combination of reasons.
2. Reasons for treatment failure extracted from trial publications for Gilad 2007, Korean Lamotrigine Study Group 2008; Rowan 2005, Saetre 2007, and Steinhoff 2005. Individual participant data for reasons for treatment failure provided for other trials.
3. Withdrawal of consent/participant choice classified as an event in this review but censored in included trial (SANAD A 2007). Sensitivity analysis classifying withdrawal of consent as a censored observation did not change the conclusions (results available on request).
4. Other treatment‐related reasons: investigator choice (Werhahn 2015), drug‐related death, pregnancy or perceived remission (SANAD A 2007). Specified only as 'other reason' for Brodie 1999; Reunanen 1996; Korean Lamotrigine Study Group 2008; Saetre 2007 and Steinhoff 2005.
5. Other reasons (not treatment‐related): epilepsy diagnosis changed (SANAD A 2007). Specified only as 'other reason' for Rowan 2005, and for seven participants in SANAD A 2007.
6. No information on whether participants withdrew from treatment or completed the study available for 19 participants
7. One participant (randomised to LTG) with date and reason for treatment failure missing.
8. Two participants with date of treatment failure missing so not included in analysis of time to treatment failure but with reasons for treatment failure provided (both censored: one withdrew from LTG due to remission of seizures, one withdrew from CBZ due to 'other' non‐treatment‐related reason).
Times to treatment failure and reasons for treatment withdrawal or treatment failure were available for 2569 participants from the nine trials providing IPD (99.9% of 2572 participants with IPD available included in this analysis, see Table 5) (Brodie 1995 A; Brodie 1995 B; Brodie 1999; Eun 2012; Lee 2011; Nieto‐Barrera 2001; Reunanen 1996; SANAD A 2007; Werhahn 2015).
Out of 3768 participants for whom we had reasons for treatment failure or withdrawal, 1416 participants prematurely withdrew from treatment (38%): 697 out of 2144 (33%) participants randomised to lamotrigine and 719 out of 1624 (44%) participants randomised to carbamazepine. We deemed 1224 participants (86% of total treatment failures) to have withdrawn for reasons related to the allocated drug: 602 (86% of treatment failures) on lamotrigine and 622 (87% of treatment failures) on carbamazepine and we classified these reasons as 'events' in the analysis. The most common treatment‐related reason for treatment failure was adverse events: 624 withdrawals (44% of total treatment failures), 251 (36% of total treatment failures) on lamotrigine and 373 (52% of total treatment failures) on carbamazepine.
We classed the other 192 reasons (95 on lamotrigine and 97 on carbamazepine) to be not related to the allocated drug and censored these participants in the analysis, in addition to the 2352 participants (1447 on lamotrigine and 905 on carbamazepine) who completed the trial without withdrawing or failing treatment.
Considering time to treatment failure for any reason related to the treatment, the overall pooled HR (for 2569 participants providing IPD from nine trials) was 0.73 (95% confidence interval (CI) 0.64 to 0.82, P < 0.00001, moderate‐quality evidence) indicating a statistically significant advantage with lamotrigine; in other words, treatment failure occurred significantly earlier on carbamazepine compared to lamotrigine in the nine included trials (Analysis 1.1). No important heterogeneity was present between trials (I2= 13%).
1.1. Analysis.

Comparison 1 Lamotrigine (LTG) versus carbamazepine (CBZ), Outcome 1 Time to treatment failure (any reason related to the treatment).
Considering time to treatment failure due to adverse events (all other reasons for treatment failure or treatment withdrawal censored in analysis), the overall pooled HR (for 2569 participants providing IPD from nine trials) was 0.54 (95% CI 0.45 to 0.65, P < 0.00001, moderate‐quality evidence) indicating a statistically significant advantage with lamotrigine; in other words, treatment failure due to adverse events occurred significantly earlier on carbamazepine compared to lamotrigine in the nine included trials (Analysis 1.2). No heterogeneity was present between trials (I2= 0%).
1.2. Analysis.
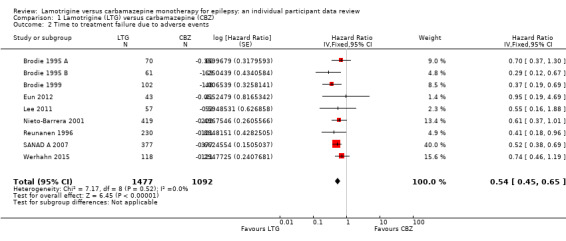
Comparison 1 Lamotrigine (LTG) versus carbamazepine (CBZ), Outcome 2 Time to treatment failure due to adverse events.
Considering time to treatment failure due to lack of efficacy (all other reasons for treatment failure or treatment withdrawal censored in analysis), 1874 participants provided IPD from five trials;, no participants withdrew from one or both of the drugs due to lack of efficacy in four trials (Brodie 1999; Eun 2012; Lee 2011; Reunanen 1996; see Table 6). The overall pooled HR was 1.03 (95% CI 0.75 to 1.41, P =0.86, moderate‐quality evidence) indicating no statistically significant difference between lamotrigine and carbamazepine (Analysis 1.3). No heterogeneity was present between trials (I2= 0%).
1.3. Analysis.
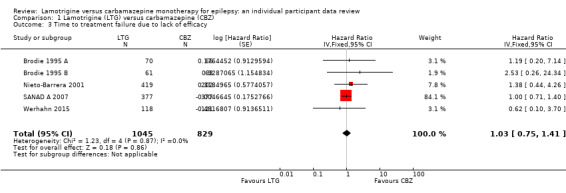
Comparison 1 Lamotrigine (LTG) versus carbamazepine (CBZ), Outcome 3 Time to treatment failure due to lack of efficacy.
Subgroup analyses: seizure type (focal versus generalised onset)
Seizure type was missing for 85 participants from SANAD A 2007, and nine participants were classified as having generalised onset seizures, even though the trial was designed to include only participants with focal onset seizures. Similarly, in Nieto‐Barrera 2001, including participants with focal onset seizures, three participants were classified as having generalised onset seizures. The latter three participants were excluded from the subgroup analyses (all completed the trial).
Considering time to treatment failure for any reason related to the treatment,for participants with generalised onset seizures (299 participants providing IPD from six trials), the pooled HR was 0.51 (95% CI 0.33 to 0.78, P = 0.002, I² = 18%, low‐quality evidence) and for participants with focal onset seizures (2182 participants providing IPD from nine trials) the pooled HR was 0.74 (95% CI 0.64 to 0.86, P = 0.0001, I² = 0%, moderate‐quality evidence) (Analysis 1.4), indicating a statistically significant advantage for lamotrigine over carbamazepine for both participants with focal onset and generalised onset seizures. Excluding the nine participants from SANAD A 2007 with generalised onset seizures produced similar results and did not change the conclusions.
1.4. Analysis.

Comparison 1 Lamotrigine (LTG) versus carbamazepine (CBZ), Outcome 4 Time to treatment failure (any reason related to the treatment) ‐ by seizure type.
The test for subgroup differences between focal and generalised onset seizures was not statistically significant (P = 0.10, I² = 63.4% for variability due to subgroup differences).
The overall pooled HR (adjusted for epilepsy type for 2481 participants from nine trials) was HR 0.71 (95% CI 0.62 to 0.82, P < 0.00001, moderate‐quality evidence). No important heterogeneity was present between trials overall (I2 =3%).
Considering time to treatment failure due to adverse events, for participants with generalised onset seizures (284 participants providing IPD from five trials), the pooled HR was 0.49 (95% CI 0.27 to 0.88, P = 0.002, I² = 52%, low‐quality evidence) and for participants with focal onset seizures (2182 participants providing IPD from nine trials) the pooled HR was 0.56 (95% CI 0.45 to 0.68, P<0.00001, I² = 0%, moderate‐quality evidence) (Analysis 1.5), indicating a statistically significant advantage for lamotrigine over carbamazepine for both participants with focal onset and generalised onset seizures. Excluding the nine participants from SANAD A 2007 with generalised onset seizures produced similar results and did not change the conclusions.
1.5. Analysis.
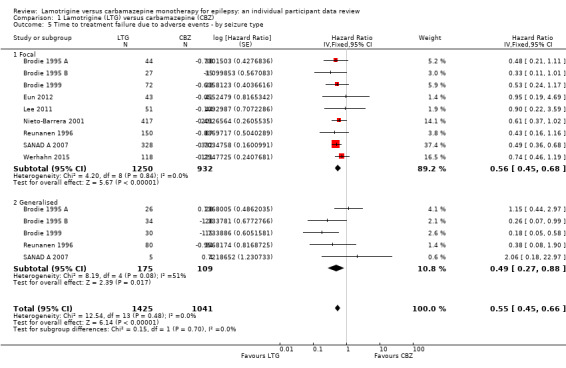
Comparison 1 Lamotrigine (LTG) versus carbamazepine (CBZ), Outcome 5 Time to treatment failure due to adverse events ‐ by seizure type.
The test for subgroup differences between focal and generalised onset seizures was not statistically significant (P = 0.70, I² =0% for variability due to subgroup differences).
The overall pooled HR (adjusted for epilepsy type for 2466 participants from nine trials) was HR 0.55 (95% CI 0.45 to 0.66, P < 0.00001, moderate‐quality evidence). No heterogeneity was present between trials overall (I² =0%).
Due to small numbers of participants withdrawing from treatment due to lack of efficacy (see Table 6), and even smaller numbers within each seizure type subgroup, subgroup analysis by seizure type was not conducted for time to treatment failure due to lack of efficacy was not conducted.
Sensitivity analyses
One study, Reunanen 1996, considered participants to have completed the trial and hence withdrew treatment if they experienced a seizure after week six. This does not correspond with the treatment failure definition recommended by the Commission on Antiepileptic Drugs of the International League Against Epilepsy (ILAE 1998); therefore we performed sensitivity analysis excluding this trial from Analysis 1.1 and Analysis 1.4. This sensitivity analysis produced similar results and did not change the conclusions.
One included trial allocated participants to three treatment arms: 100 mg/day lamotrigine (LTG100), 200 mg/day lamotrigine (LTG 200) or 600 mg/day carbamazepine (CBZ) (Reunanen 1996). See Table 7 for a sensitivity analysis comparing the primary analysis (LTG arms pooled versus CBZ), LTG 100 with the other arms in the trial, LTG 200 with the other arms in the trial, LTG 200 versus CBZ and LTG 100 versus CBZ. When including these alternative estimates in meta‐analysis, the pooled result is numerically similar and the conclusions unchanged.
5. Sensitivity analysis ‐ Reunanen 1996.
| Treatment | N | Comparator | N | Total | Time to treatment failure | Time to first seizure | Time to 6‐month remission | |||
| HR (95% CI) | P value | HR (95% CI) | P value | HR (95% CI) | P value | |||||
| Lamotrigine (both arms) | 230 | Carbamazepine | 121 | 351 | 0.59 (0.35 to 0.99) | 0.04 | 1.24 (0.86 to 1.79) | 0.25 | 0.84 (0.36 to 1.95) | 0.68 |
| Lamotrigine 200 mg | 115 | Lamotrigine 100 mg + carbamazepine | 236 | 351 | 0.47 (0.25 to 0.86) | 0.02 | 0.96 (0.67 to 1.36) | 0.8 | 0.62 (0.24 to 1.58) | 0.32 |
| Lamotrigine 100 mg | 115 | Lamotrigine 200 mg + carbamazepine | 236 | 351 | 1.05 (0.63 to 1.75) | 0.85 | 1.29 (0.91 to 1.83) | 0.15 | 1.33 (0.56 to 3.17) | 0.52 |
| Lamotrigine 200 mg | 115 | Carbamazepine | 121 | 236 | 0.41 (0.21 to 0.78) | 0.007 | 1.12 (0.73 to 1.72) | 0.59 | 0.63 (0.22 to 1.78) | 0.39 |
| Lamotrigine 100 mg | 115 | Carbamazepine | 121 | 236 | 0.73 (0.43 to 1.26) | 0.26 | 1.37 (0.90 to 2.07) | 0.14 | 1.10 (0.41 to 2.92) | 0.86 |
mg= milligrams per day; HR = hazard ratio; 95% CI = 95% confidence interval
From four out of the five trials without IPD available, we indirectly estimated aggregate hazard ratios of time to treatment failure due to adverse events (Gilad 2007), time to early termination (Rowan 2005), and retention time (Steinhoff 2005), and we extracted a published hazard ratio for time to all‐cause withdrawal (Saetre 2007). We were unable to estimate or extract an estimate from Korean Lamotrigine Study Group 2008. We note that these definitions do not directly correspond to our definition of treatment failure (i.e. all withdrawals and treatment failures are classed as events rather than only treatment‐related failures, see Table 6). Combining IPD and aggregate data, the pooled HR for 3391 participants from 13 included trials was 0.709 (95% CI 0.63 to 0.78, P < 0.00001, I² = 27%) (Analysis 1.6). This indicates that the advantage to lamotrigine over carbamazepine remains and is robust to the inclusion of treatment failure data of variable definitions.
1.6. Analysis.

Comparison 1 Lamotrigine (LTG) versus carbamazepine (CBZ), Outcome 6 Time to treatment failure (any reason related to the treatment, with aggregate data).
Given the subjective nature of the outcome of time to treatment failure, an outcome which can be influenced by the participant and personnel, we conducted a further subgroup analysis separating the trials that were of a double‐blind design (Brodie 1995 A; Brodie 1995 B; Brodie 1999; Rowan 2005; Saetre 2007; Werhahn 2015, 1231 participants included in analyses) and those which were an open‐label design (Eun 2012; Gilad 2007; Lee 2011; Nieto‐Barrera 2001; Reunanen 1996; Steinhoff 2005; SANAD A 2007, 2160 participants included in analyses). Including treatment failure data from 13 trials (aggregate data from four trials), we found the following results.
In the double‐blind trials, 242 out of 644 (38%) randomised participants withdrew from lamotrigine and 313 out of 587 (53%) withdrew from carbamazepine (in total 45% of participants withdrew from the randomised drug for any reason).
In the open‐label trials, 368 out of 1248 (29%) randomised participants withdrew from lamotrigine and 369 out of 915 (40%) withdrew from carbamazepine (in total 34% of participants withdrew from the randomised drug for any reason).
The advantage for lamotrigine over carbamazepine was slightly larger in the double‐blind trials (HR 0.65, 95% CI 0.56 to 0.75, P<0.00001, I² = 24%) than in the open‐label trials (HR 0.76, 95% CI 0.65 to 0.90, P=0.001, I² = 21%) (Analysis 1.7).
When including only the nine trials for which IPD were provided (double‐blind: Brodie 1995 A; Brodie 1995 B; Brodie 1999; Werhahn 2015; open‐label: Eun 2012; Lee 2011; Nieto‐Barrera 2001; Reunanen 1996; SANAD A 2007), the advantage for lamotrigine over carbamazepine was still slightly larger in the double‐blind trials (HR 0.68, 95% CI 0.57 to 0.81, P<0.0001, I² = 36%) than the open‐label trials (HR 0.77, 95% CI 0.65 to 0.92, P=0.003, I² = 0%)
1.7. Analysis.
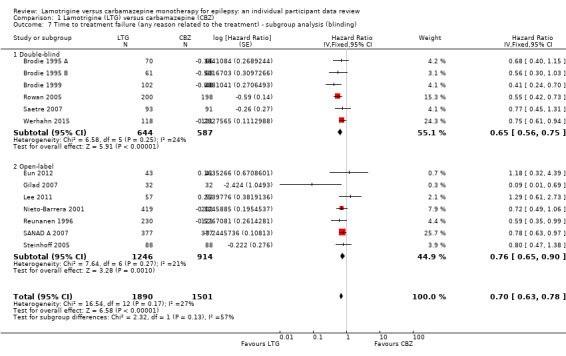
Comparison 1 Lamotrigine (LTG) versus carbamazepine (CBZ), Outcome 7 Time to treatment failure (any reason related to the treatment) ‐ subgroup analysis (blinding).
These results suggest that the design of a trial (i.e. whether or not a participant and their clinician is aware of the treatment a participant is taking) may influence the withdrawal rates of the trial, with participants significantly more likely to withdraw from a double‐blind trial than an open‐label trial (45% versus 34%: risk ratio (RR) 1.32 (95% CI 1.22 to 1.44, P < 0.00001)), and in turn this may influence the perceived effectiveness of the two drugs under comparison.
Following reclassification of 152 individuals from seven trials with potentially misclassified generalised onset seizures (Brodie 1995 A; Brodie 1995 B; Brodie 1999; Lee 2011; Nieto‐Barrera 2001; Reunanen 1996; SANAD A 2007), and 85 individuals with missing seizure type from one trial (SANAD A 2007), the results of the two sensitivity analyses are shown in Table 8. In summary, the results overall by seizure type and for individuals with focal onset seizures are very similar. Also, following reclassification, the advantage for lamotrigine over carbamazepine in those with generalised onset seizures is reduced and the test of difference between subgroups is no longer significant. There is a statistically significant advantage for lamotrigine over carbamazepine in those with uncertain seizure type.
6. Sensitivity analysis ‐ misclassification of seizure type.
| Time to treatment failure | Time to first seizure | Time to 6‐month remission | |
| Original analysis | F: HR 0.74, 95% CI (0.64 to 0.86) G: HR 0.51, 95% CI (0.33 to 0.78) O: HR 0.71, 95% CI (0.62 to 0.82) |
F: HR 1.29, 95% CI (1.14 to 1.45) G: HR 0.98, 95% CI (0.65 to 1.48) O: HR 1.26, 95% CI (1.12 to 1.41) |
F: HR 0.87, 95% CI (0.77 to 1.00) G: HR 0.78, 95% CI (0.55 to 1.11) O: HR 0.86, 95% CI (0.76 to 0.97) |
| Test of subgroup differences | Chi² = 2.73, df = 1 (P = 0.10), I² = 63.4% | Chi² = 1.53, df = 1 (P = 0.22), I² = 34.5% | Chi² = 0.37, df = 1 (P = 0.54), I² = 0% |
| Generalised onset and age at onset > 30 reclassified as focal onset | F: HR 0.72, 95% CI (0.62 to 0.83) G: HR 0.58, 95% CI (0.32 to 1.06) O: HR 0.71, 95% CI (0.62 to 0.82) |
F: HR 1.25, 95% CI (1.11 to 1.41) G: HR 1.17, 95% CI (0.67 to 2.04) O: HR 1.25, 95% CI (1.11 to 1.40) |
F: HR 0.85, 95% CI (0.75 to 0.97) G: HR 0.69, 95% CI (0.44 to 1.08) O: HR 0.84, 95% CI (0.74 to 0.95) |
| Test of subgroup differences | Chi² = 0.45, df = 1 (P = 0.50), I² = 0% | Chi² = 0.06, df = 1 (P = 0.81), I² = 0% | Chi² = 0.80, df = 1 (P = 0.37), I² = 0% |
| Generalised onset and age at onset > 30 reclassified as uncertain seizure type | F: HR 0.74, 95% CI (0.64 to 0.86) G: HR 0.58, 95% CI (0.32 to 1.06) U: HR 0.62, 95% CI (0.39 to 0.97) O: HR 0.72, 95% CI (0.63 to 0.83) |
F: HR 1.29, 95% CI (1.14 to 1.45) G: HR 1.17, 95% CI (0.67 to 2.04) U: HR 0.88, 95% CI (0.58 to 1.33) O: HR 1.24, 95% CI (1.11 to 1.39) |
F: HR 0.87, 95% CI (0.77 to 1.00) G: HR 0.69, 95% CI (0.44 to 1.08) U: HR 0.89, 95% CI (0.60 to 1.31) O: HR 0.86, 95% CI (0.76 to 0.97) |
| Test of subgroup differences | Chi² = 1.15, df = 2 (P = 0.56), I² = 0% | Chi² = 3.03, df = 2 (P = 0.22), I² = 33.9% | Chi² = 1.02, df = 2 (P = 0.60), I² = 0% |
CI = confidence interval; F = focal onset seizures; G = generalised onset seizures; HR = hazard ratio; O = overall pooled result adjusted by seizure type; U = uncertain seizure type.
Secondary outcomes
Time to first seizure post randomisation
For this outcome, a HR of less than 1 indicates a clinical advantage for lamotrigine.
Times to first seizure were available for 2564 participants from the nine trials providing IPD (99% of 2572 participants with IPD available included in this analysis, see Table 5) (Brodie 1995 A; Brodie 1995 B; Brodie 1999; Eun 2012; Lee 2011; Nieto‐Barrera 2001; Reunanen 1996; SANAD A 2007; Werhahn 2015).
Seizure recurrence was experienced by 1330 out of 2564 participants (52%), 805 out of 1476 (55%) on lamotrigine and 525 out of 1088 (48%) on carbamazepine. The overall pooled HR (for 2564 participants) was 1.22 (95% CI 1.09 to 1.37, P = 0.0004) indicating a statistically significant advantage to carbamazepine; in other words, participants experienced first seizure recurrence earlier on lamotrigine than carbamazepine in the nine included trials (Analysis 1.8). No heterogeneity was present between trials (I2= 0%).
1.8. Analysis.

Comparison 1 Lamotrigine (LTG) versus carbamazepine (CBZ), Outcome 8 Time to first seizure.
Subgroup analyses: seizure type (focal versus generalised onset)
Seizure type was missing for 85 participants from SANAD A 2007, and nine participants were classified as having generalised onset seizures, even though the trial was designed to include only participants with focal onset seizures. Similarly, in Nieto‐Barrera 2001, including participants with focal onset seizures, three participants were classified as having generalised onset seizures. The latter three participants were excluded from the subgroup analyses (all completed the trial).
For participants with generalised onset seizures (299 participants providing IPD from six trials), the pooled HR was 0.98 (95% CI 0.65 to 1.48, P = 0.94, moderate‐quality evidence), indicating no difference between the two drugs and for participants with focal onset seizures (2177 participants providing IPD from nine trials) the pooled HR was 1.29 (95% CI 1.14 to 1.45, P < 0.0001, high‐quality evidence) (Analysis 1.9), indicating a statistically significant advantage for carbamazepine. There was no evidence of a difference between the subgroups (test for subgroup differences P = 0.22). Excluding the nine participants from SANAD A 2007 with generalised onset seizures produced similar results and did not change the conclusions.
1.9. Analysis.
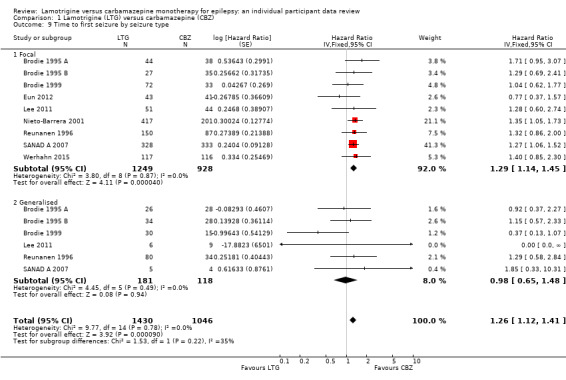
Comparison 1 Lamotrigine (LTG) versus carbamazepine (CBZ), Outcome 9 Time to first seizure by seizure type.
The overall pooled HR (adjusted for epilepsy type for 2476 participants from nine trials) was 1.26 (95% CI 1.12 to 1.41, P < 0.0001, high‐quality evidence). No heterogeneity was present between trials overall or by subgroups (I2 =0%).
Sensitivity analyses
Data from one trial could not be included for this outcome as the dates of seizures that occurred during the first four weeks of the trial were not supplied (Nieto‐Barrera 2001). A total of 216 participants (lamotrigine: 160, carbamazepine: 56) (35% of the number in the trial) experienced at least one seizure during the first four weeks, however dates of these seizures were not supplied. Therefore, for Nieto‐Barrera 2001, this outcome is calculated as 'time to first seizure after four weeks of treatment' rather than 'time to first seizure after randomisation'. Excluding this trial in sensitivity analysis produces very similar numerical results and the conclusions are unchanged.
One included trial allocated participants to three treatment arms: 100 mg/day lamotrigine (LTG100), 200 mg/day lamotrigine (LTG 200) or 600 mg/day carbamazepine (CBZ) (Reunanen 1996). See Table 7 for sensitivity analysis comparing the primary analysis (LTG arms pooled versus CBZ), LTG 100 with the other arms in the trial, LTG 200 with the other arms in the trial, LTG 200 versus CBZ and LTG 100 versus CBZ. When including these alternative estimates in meta‐analysis, the pooled result is numerically similar and the conclusions unchanged.
From three out of the five trials without IPD available, we indirectly estimated aggregate hazard ratios of time to first seizure from published graphs in two trials (Gilad 2007; Rowan 2005), and extracted a published hazard ratio of time to first seizure (Saetre 2007). We were unable to estimate or extract an estimate from Steinhoff 2005 or Korean Lamotrigine Study Group 2008. Combining IPD and aggregate data, the pooled HR for 3216 participants from the 12 included trials was 1.24 (95% CI 1.12 to 1.37, P < 0.00001, I2=15%) (Analysis 1.10). This again shows an advantage to carbamazepine over lamotrigine.
1.10. Analysis.
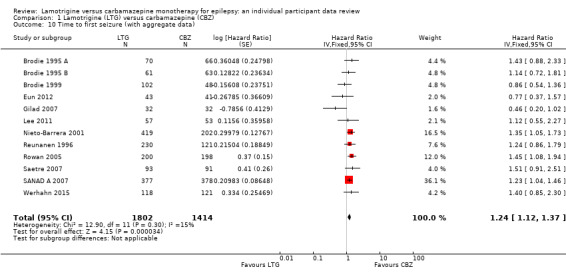
Comparison 1 Lamotrigine (LTG) versus carbamazepine (CBZ), Outcome 10 Time to first seizure (with aggregate data).
We were able to calculate or extract seizure freedom throughout the whole trial for all trials included in this review (in Rowan 2005, it was not stated whether any participants who withdrew experienced seizure recurrence, therefore we have conducted an intention‐to‐treat analysis of seizure freedom rather than seizure recurrence). For consistency with the primary analysis of the outcome, in Analysis 1.11 we have swapped event and non‐event so that a RR less than 1 indicates a clinical advantage for lamotrigine.
1.11. Analysis.
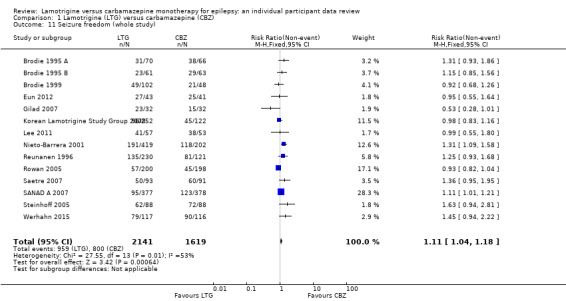
Comparison 1 Lamotrigine (LTG) versus carbamazepine (CBZ), Outcome 11 Seizure freedom (whole study).
The pooled RR of seizure freedom (for 3760 participants from 14 trials) is 1.11 (95% CI 1.04 to 1.18, P = 0.0006, I2=53%), indicating a statistically significant advantage to carbamazepine. Therefore, following the inclusion of seizure freedom/recurrence data from all included trials, the advantage to carbamazepine remains. There is now a large amount of heterogeneity present between the trials, which was not present in the IPD meta‐analysis (I2 =53%, Analysis 1.11). This may reflect the variable follow‐up lengths of the trials (from 24 weeks to over six years), or may reflect the way the aggregate data are presented in some of the trials; for example in Rowan 2005, seizure freedom rates are presented only for those who have completed the trial.
Following reclassification of 152 individuals from seven trials with potentially misclassified generalised onset seizures (Brodie 1995 A; Brodie 1995 B; Brodie 1999; Lee 2011; Nieto‐Barrera 2001; Reunanen 1996; SANAD A 2007), and 85 individuals with missing seizure type from one trial (SANAD A 2007), the results of the two sensitivity analyses are shown in Table 8. In summary, the results overall by seizure type and for individuals with focal onset seizures are very similar. Following reclassification, there is a slight advantage to carbamazepine, which is not statistically significant, for those with generalised onset seizures. For those with uncertain seizure type, there is a slight advantage to lamotrigine that is not statistically significant.
Time to achieve six‐month remission
For this outcome, a HR of less than 1 indicates a clinical advantage for carbamazepine.
Times to six‐month remission were available for 1793 participants from the seven trials providing IPD (70% of 2572 participants with IPD available included in this analysis, see Table 5) (Brodie 1995 A; Brodie 1995 B; Eun 2012; Lee 2011; Reunanen 1996; SANAD A 2007; Werhahn 2015). The remaining two trials were of 24 weeks duration so were not included in the analysis of time to six‐month remission but are included in the sensitivity analysis of seizure freedom (Brodie 1999; Nieto‐Barrera 2001).
Six‐month remission was achieved by 1113 out of 1793 participants (62%), 572 out of 955 (60%) on lamotrigine and 541 out of 838 (65%) on carbamazepine. The overall pooled HR (for 1793 participants) was 0.84 (95% CI 0.74 to 0.94, P = 0.003) indicating a statistically significant advantage to carbamazepine; in other words, participants experienced six‐month remission earlier on lamotrigine than carbamazepine in the seven included trials (Analysis 1.12). No heterogeneity was present between trials (I2= 0%).
1.12. Analysis.

Comparison 1 Lamotrigine (LTG) versus carbamazepine (CBZ), Outcome 12 Time to 6‐month remission.
Subgroup analyses: seizure type (focal versus generalised onset)
Seizure type was missing for 85 participants from SANAD A 2007, and nine participants were classified as having generalised onset seizures, even though the trial was designed to include only participants with focal onset seizures.
For participants with generalised onset seizures (254 participants providing IPD from five trials), the pooled HR was 0.78 (95% CI 0.55 to 1.11, P = 0.16, I2 =0%), indicating an advantage to carbamazepine that is not statistically significant, and for participants with focal onset seizures (1454 participants providing IPD from seven trials) the pooled HR was 0.87 (95% CI 0.77 to 1.00, P = 0.04, I2 =25%) (Analysis 1.13), indicating a statistically significant advantage for carbamazepine. There was no evidence of a difference between the subgroups (test for subgroup differences P = 0.54). Excluding the nine participants from SANAD A 2007 with generalised onset seizures produced similar results and did not change the conclusions.
1.13. Analysis.

Comparison 1 Lamotrigine (LTG) versus carbamazepine (CBZ), Outcome 13 Time to 6‐month remission by seizure type.
The overall pooled HR (adjusted for epilepsy type for 1708 participants from seven trials) was 0.86 (95% CI 0.76 to 0.97, P = 0.02) (Analysis 1.13) . No heterogeneity was present between trials overall (I2= 0%).
Sensitivity analyses
One included trial allocated participants to three treatment arms, 100 mg/day lamotrigine (LTG 100), 200 mg/day lamotrigine (LTG 200) or 600 mg/day carbamazepine (CBZ) (Reunanen 1996). See Table 7 for a sensitivity analysis comparing the primary analysis (LTG arms pooled versus CBZ), LTG 100 with the other arms in the trial, LTG 200 with the other arms in the trial, LTG 200 versus CBZ and LTG 100 versus CBZ. When including these alternative estimates in meta‐analysis, the pooled result is numerically similar and the conclusions unchanged.
We were able to calculate or extract seizure freedom at six months for all trials included in this review (estimated from the graph published in Saetre 2007). A RR less than 1 indicates a clinical advantage for carbamazepine.
The pooled RR of seizure freedom at six months (for 3760 participants from 14 trials) is RR 0.96 (95% CI 0.89 to 1.03, P = 0.25, I2 =21%) (Analysis 1.14), indicating no statistically significant advantage to either drug. As above, we note that the way the aggregate data are presented in some trials may influence the results; for example in Rowan 2005, seizure freedom rates are presented only for those who have completed the trial.
1.14. Analysis.

Comparison 1 Lamotrigine (LTG) versus carbamazepine (CBZ), Outcome 14 Seizure freedom at 6 months.
Following reclassification of 152 individuals from seven trials with potentially misclassified generalised onset seizures (Brodie 1995 A; Brodie 1995 B; Lee 2011; Reunanen 1996; SANAD A 2007), and 85 individuals with missing seizure type from one trial (SANAD A 2007), the results of the two sensitivity analyses are shown in Table 8. In summary, the results overall by seizure type are very similar to the subgroup analysis described above and the conclusions are unchanged. For those with uncertain seizure type, there is a slight advantage to carbamazepine that is not statistically significant.
In Lee 2011, analysis adjusted for epilepsy type, there was some evidence that the proportional hazards assumption of the Cox model may have been violated; the P value of the time‐varying covariate was 0.051. However, the time varying covariate is not significant in the analysis without adjustment for seizure type (P = 0.146).
Following visual inspection of a cumulative incidence plot (not shown but available from the authors), the curves appear to cross around 200 days, when less than 20% of randomised participants remain at risk in the trial. Therefore, we conclude that the crossing of the curves is likely to be due to small numbers of participants with generalised seizure types and small numbers remaining in the trial leading to changes and events being magnified at this time. The proportional hazards assumption of the Cox model was satisfied for all other trials included in analysis.
Time to achieve 12‐month (one‐year) remission
For this outcome, a HR of less than 1 indicates a clinical advantage for carbamazepine.
Times to 12‐month remission were available for 988 participants from the two trials providing IPD of sufficient duration (70% of 2572 participants with IPD available included in this analysis, see Table 5) (SANAD A 2007; Werhahn 2015).
Twelve‐month remission was achieved by 564 out of 998 participants (57%), 276 out of 474 (58%) on lamotrigine and 288 out of 474 (61%) on carbamazepine. The overall pooled HR (for 998 participants) was HR 0.91 (95% CI 0.77 to 1.07, P = 0.26, high‐quality evidence), indicating an advantage to carbamazepine that was not statistically significant (Analysis 1.15). No heterogeneity was present between trials (I2= 0%).
1.15. Analysis.

Comparison 1 Lamotrigine (LTG) versus carbamazepine (CBZ), Outcome 15 Time to 12‐month remission.
Subgroup analysis and sensitivity analysis
All participants had focal onset seizures in Werhahn 2015; and in SANAD A 2007, the design was to include only those with focal onset seizures. However, seizure type was missing for 85 participants, and nine participants were classified as having generalised onset seizures. Given the small numbers in the generalised onset group (which are likely to have been misclassified), we did not perform a sensitivity analysis by seizure type.
Instead we performed a subgroup analysis of those with focal seizures specified at baseline (all participants in Werhahn 2015, and 661 participants in SANAD A 2007) and those with uncertain seizure type (85 with missing seizure type and nine with generalised onset seizures in SANAD A 2007).
For those with uncertain seizure type (94 participants providing IPD from one trial), the HR was 0.81 (95% CI 0.47 to 1.37, P = 0.43) (Analysis 1.16), indicating an advantage to carbamazepine that is not statistically significant. For those with focal onset seizures (894 participants providing IPD from two trials), the pooled HR was 0.91 (95% CI 0.77 to 1.09, P = 0.31, I2= 0%), also indicating an advantage to carbamazepine that is not statistically significant (Analysis 1.16). There was no evidence of a difference between the subgroups (test for subgroup differences P = 0.66).
1.16. Analysis.
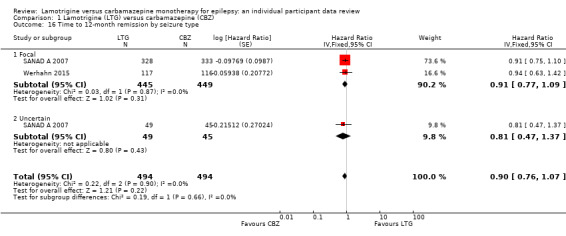
Comparison 1 Lamotrigine (LTG) versus carbamazepine (CBZ), Outcome 16 Time to 12‐month remission by seizure type.
Time to achieve 24‐month (two‐year) remission
For this outcome, a HR of less than 1 indicates a clinical advantage for carbamazepine.
Times to 24‐month remission were available for 755 participants from one trial providing IPD of sufficient duration (29% of 2572 participants with IPD available included in this analysis, see Table 5) (SANAD A 2007).
Twenty‐four month remission was achieved by 296 out of 755 participants (39%), 149 out of 377 (40%) on lamotrigine and 147 out of 378 (39%) on carbamazepine. The overall HR (for 755 participants from one trial) was 1.00 (95% CI 0.80 to 1.25, P = 0.99), indicating no statistically significant difference between the drugs (Analysis 1.17).
1.17. Analysis.

Comparison 1 Lamotrigine (LTG) versus carbamazepine (CBZ), Outcome 17 Time to 24‐month remission.
Subgroup analysis and sensitivity analysis
Seizure type was missing for 85 participants from SANAD A 2007, and nine participants were classified as having generalised onset seizures, even though the trial was designed to include only participants with focal onset seizures.
Given the small numbers in the generalised onset group (which are likely to have been misclassified), we did not perform a sensitivity analysis by seizure type.
Instead we performed a subgroup analysis of those with focal seizures specified at baseline (661 participants) and those with uncertain seizure type (85 with missing seizure type and nine with generalised onset seizures). For those with uncertain seizure type (94 participants providing IPD from one trial), the HR was 0.86 (95% CI 0.44 to 1.67, P = 0.65) (Analysis 1.18), indicating an advantage to carbamazepine that is not statistically significant. For those with focal onset seizures (661 participants providing IPD from two trials), the pooled HR was 1.06 (95% CI 0.83 to 1.35, P = 0.66), indicating an slight advantage to lamotrigine that is not statistically significant (Analysis 1.18). There was no evidence of a difference between the subgroups (test for subgroup differences P = 0.57).
1.18. Analysis.

Comparison 1 Lamotrigine (LTG) versus carbamazepine (CBZ), Outcome 18 Time to 24‐month remission by seizure type.
In SANAD A 2007 (analyses with and without adjustment for epilepsy type), there was evidence that the proportional hazards assumption of the Cox model may have been violated; the P value of the time‐varying covariate was 0.025.
Following visual inspection of a cumulative incidence plot (Figure 13), the curves appear to cross around 1200 days. Considering the distribution of events, all 24‐month remission events occurred before 1200 days; none of the 164 participants (82 in each treatment group) experienced remission after 1200 days. This observation would explain the apparent change in treatment effect over time (i.e. a difference in censoring times). The proportional hazards assumption of the Cox model was satisfied for all other trials included in analysis.
Incidence of adverse events
We were provided with individual participant data for adverse events experienced during the trial for nine trials (Brodie 1995 A; Brodie 1995 B; Brodie 1999; Eun 2012; Lee 2011; Nieto‐Barrera 2001; Reunanen 1996; SANAD A 2007; Werhahn 2015), and we extracted information relating to adverse events from the remaining five publications (Gilad 2007; Korean Lamotrigine Study Group 2008; Rowan 2005; Saetre 2007; Steinhoff 2005). Due to the wide range of events reported in the trials and the different methods of recording adverse events, we have not analysed adverse event data in meta‐analysis and provide a narrative report.
Seven trials provided very detailed information regarding all adverse events experienced by all participants during the trials (Brodie 1995 A; Brodie 1995 B; Brodie 1999; Nieto‐Barrera 2001; Reunanen 1996; SANAD A 2007; Werhahn 2015). This information is summarised in Table 9, Table 10 and Table 11.
7. Summary of adverse events experienced (seven trials providing detailed individual participant data).
| Trial |
Number experiencing adverse events |
Number of adverse events |
Number of adverse events per person (range) |
Number of drug‐related adverse events1 |
Number of adverse events requiring action/ treatment change |
Number of patients needing a treatment change/dose change |
|||||||||||
| LTG | CBZ | Total | LTG | CBZ | Total | LTG | CBZ | LTG | CBZ | Total | LTG | CBZ | Total | LTG | CBZ | Total | |
| Brodie 1995 A | 62 | 58 | 120 | 388 | 322 | 710 | 1 to 30 | 1 to 17 | 94 | 124 | 218 | 167 | 111 | 278 | 22 | 32 | 54 |
| Brodie 1995 B | 54 | 58 | 112 | 285 | 291 | 576 | 1 to 14 | 1 to 18 | 81 | 125 | 206 | 98 | 81 | 179 | 20 | 40 | 60 |
| Brodie 1999 | 91 | 41 | 132 | 338 | 173 | 511 | 1 to 12 | 1 to 10 | 109 | 73 | 182 | 92 | 66 | 158 | 39 | 27 | 66 |
| Eun 2012 | 3 | 6 | 9 | 5 | 8 | 13 | 1 to 30 | 1 to 2 | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| Lee 2011 | 4 | 6 | 10 | NA | NA | NA | NA | NA | 4 | 5 | 9 | NA | NA | NA | NA | NA | NA |
| Nieto‐Barrera 2001 | 218 | 120 | 338 | 524 | 277 | 801 | 1 to 10 | 1 to 11 | 238 | 152 | 390 | 116 | 82 | 198 | 70 | 54 | 124 |
| Reunanen 1996 | 124 | 77 | 201 | 451 | 243 | 694 | 1 to 14 | 1 to 8 | 138 | 169 | 307 | 156 | 52 | 208 | 23 | 36 | 59 |
| SANAD A 2007 | 229 | 260 | 489 | 1038 | 1339 | 2377 | 1 to 25 | 1 to 37 | NA | NA | NA | 447 | 665 | 1112 | 120 | 173 | 293 |
| Werhahn 2015 | 120 | 110 | 230 | 779 | 770 | 1549 | 1 to 53 | 1 to 30 | 291 | 382 | 673 | 147 | 159 | 306 | 64 | 65 | 129 |
CBZ = carbamazepine; LTG = lamotrigine; NA = information not available.
1. In Brodie 1995 A, Brodie 1995 B and Reunanen 1996 adverse events that are "definitely related", in Brodie 1999 and Nieto‐Barrera 2001 "a reasonable possibility" that adverse events are treatment‐related and in Werhahn 2015 adverse events are "related, probably related or possibility related".
8. Most commonly occurring adverse events (trials providing detailed individual participant data).
| Most commonly occurring adverse events | Brodie 1995 A | Brodie 1995 B | Brodie 1999 | Nieto‐Barrera 2001 | ||||||||||||
| LTG | CBZ | LTG | CBZ | LTG | CBZ | LTG | CBZ | |||||||||
| Events | Ppts | Events | Ppts | Events | Ppts | Events | Ppts | Events | Ppts | Events | Ppts | Events | Ppts | Events | Ppts | |
| Accidental injury/fracture | 2 | 2 | 1 | 1 | 3 | 3 | 2 | 2 | 19 | 12 | 4 | 3 | 7 | 7 | 1 | 1 |
| Aggression | 8 | 6 | 2 | 2 | 0 | 0 | 0 | 0 | 2 | 2 | 0 | 0 | 3 | 3 | 2 | 2 |
| Anorexia/weight loss | 2 | 2 | 0 | 0 | 6 | 4 | 0 | 0 | 2 | 2 | 1 | 1 | 6 | 5 | 0 | 0 |
| Anxiety/depression | 12 | 5 | 7 | 5 | 6 | 3 | 10 | 7 | 3 | 3 | 0 | 0 | 8 | 8 | 2 | 2 |
| Aphasia | 0 | 0 | 3 | 3 | 0 | 0 | 0 | 0 | 1 | 1 | 2 | 2 | 1 | 1 | 0 | 0 |
| Ataxia | 2 | 2 | 6 | 5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 2 | 3 | 3 |
| Chest infection/bronchitis | 11 | 6 | 12 | 8 | 3 | 3 | 1 | 1 | 16 | 12 | 4 | 4 | 18 | 15 | 8 | 8 |
| Cold/influenza | 17 | 15 | 4 | 4 | 8 | 8 | 10 | 9 | 7 | 7 | 1 | 1 | 25 | 19 | 11 | 11 |
| Concentration | 0 | 0 | 1 | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 4 | 4 | 1 | 1 |
| Confusion | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 2 | 1 | 1 | 0 | 0 | 0 | 0 |
| Cough/wheeze | 5 | 5 | 5 | 5 | 2 | 2 | 1 | 1 | 6 | 5 | 1 | 1 | 6 | 5 | 6 | 5 |
| Dental | 3 | 3 | 2 | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 6 | 6 | 3 | 3 |
| Dizzy/faint | 16 | 9 | 16 | 11 | 12 | 9 | 22 | 14 | 26 | 18 | 16 | 14 | 43 | 34 | 16 | 15 |
| Drowsy/fatigued | 32 | 21 | 52 | 31 | 34 | 20 | 49 | 36 | 25 | 17 | 21 | 15 | 36 | 34 | 45 | 40 |
| Gastrointestinal disturbances | 14 | 7 | 10 | 8 | 6 | 6 | 7 | 5 | 29 | 22 | 14 | 11 | 36 | 28 | 17 | 17 |
| Hair loss | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 |
| Headache/migraine | 77 | 27 | 31 | 17 | 48 | 24 | 52 | 22 | 14 | 10 | 8 | 8 | 56 | 46 | 16 | 14 |
| Impotence | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Increased/worsened seizures | 1 | 1 | 2 | 2 | 1 | 1 | 0 | 0 | 4 | 4 | 2 | 2 | 14 | 12 | 4 | 4 |
| Kidney/urinary problems | 3 | 2 | 2 | 2 | 4 | 4 | 1 | 1 | 12 | 10 | 4 | 4 | 4 | 4 | 1 | 1 |
| Memory problems | 7 | 5 | 2 | 2 | 5 | 3 | 3 | 2 | 4 | 4 | 0 | 0 | 2 | 2 | 1 | 1 |
| Menstrual problems | 3 | 3 | 16 | 12 | 0 | 0 | 4 | 4 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 |
| Mood/behavioural change | 9 | 5 | 6 | 5 | 1 | 1 | 6 | 6 | 5 | 4 | 0 | 0 | 7 | 7 | 4 | 4 |
| Nausea/vomiting | 17 | 13 | 15 | 11 | 26 | 18 | 21 | 9 | 21 | 17 | 8 | 6 | 26 | 23 | 13 | 11 |
| Pain | 19 | 13 | 9 | 6 | 23 | 13 | 7 | 5 | 20 | 17 | 7 | 7 | 13 | 8 | 4 | 2 |
| Pins and needles/tingling | 2 | 1 | 3 | 2 | 3 | 3 | 0 | 0 | 1 | 1 | 0 | 0 | 1 | 1 | 0 | 0 |
| Rash/skin problems | 25 | 21 | 20 | 13 | 32 | 15 | 32 | 23 | 31 | 19 | 30 | 14 | 49 | 46 | 32 | 30 |
| Sleep problems/dreams | 4 | 3 | 4 | 4 | 8 | 5 | 12 | 5 | 9 | 8 | 0 | 0 | 19 | 19 | 1 | 1 |
| Throat/tonsil infection | 11 | 7 | 7 | 6 | 6 | 5 | 3 | 3 | 1 | 1 | 0 | 0 | 15 | 14 | 7 | 7 |
| Tremor/twitch | 1 | 1 | 2 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 2 | 2 |
| Visual disturbance/nystagmus | 8 | 4 | 6 | 5 | 2 | 2 | 9 | 6 | 1 | 1 | 4 | 3 | 7 | 7 | 3 | 2 |
| Weight gain | 3 | 3 | 1 | 1 | 2 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 3 | 3 | 3 |
Table of most commonly occurring adverse events split into two for formatting reasons.
Events = number of adverse events reported; Ppts = number of participants reporting the adverse event (a participant could report the same type of adverse event multiple times).
LTG = lamotrigine; CBZ = carbamazepine
Most common adverse events are defined as events reported 10 or more times in at least one of the seven trials (Brodie 1995 A; Brodie 1995 B; Brodie 1999; Nieto‐Barrera 2001; Reunanen 1996; SANAD A 2007; Werhahn 2015). Less commonly reported adverse events are not summarised in this table but details are available on request from the review authors. General terminology for the type of adverse events was defined by the review authors based on the individual participant data provided.
9. Most commonly occurring adverse events continued (trials providing detailed individual participant data).
| Most commonly occurring adverse events | Reunanen 1996 | SANAD A 2007 | Werhahn 2015 | Total (across seven studies) | ||||||||||||
| LTG | CBZ | LTG | CBZ | LTG | CBZ | LTG | CBZ | |||||||||
| Events | Ppts | Events | Ppts | Events | Ppts | Events | Ppts | Events | Ppts | Events | Ppts | Events | Ppts | Events | Ppts | |
| Accidental injury/fracture | 2 | 2 | 0 | 0 | 29 | 19 | 10 | 10 | 16 | 15 | 14 | 7 | 78 | 60 | 32 | 24 |
| Aggression | 1 | 1 | 0 | 0 | 25 | 18 | 41 | 21 | 1 | 1 | 1 | 1 | 40 | 31 | 46 | 26 |
| Anorexia/weight loss | 3 | 2 | 0 | 0 | 12 | 11 | 16 | 13 | 1 | 1 | 0 | 0 | 32 | 27 | 17 | 14 |
| Anxiety/depression | 4 | 4 | 2 | 2 | 48 | 34 | 46 | 34 | 17 | 14 | 17 | 10 | 98 | 71 | 84 | 60 |
| Aphasia | 1 | 1 | 0 | 0 | 7 | 4 | 11 | 8 | 1 | 1 | 7 | 5 | 11 | 8 | 23 | 18 |
| Ataxia | 0 | 0 | 3 | 3 | 38 | 20 | 30 | 22 | 1 | 1 | 0 | 0 | 43 | 25 | 42 | 33 |
| Chest infection/bronchitis | 3 | 3 | 1 | 1 | 2 | 1 | 6 | 5 | 8 | 8 | 3 | 3 | 61 | 48 | 35 | 30 |
| Cold/influenza | 9 | 8 | 2 | 2 | 1 | 1 | 3 | 3 | 11 | 9 | 20 | 15 | 78 | 67 | 51 | 45 |
| Concentration | 3 | 3 | 4 | 3 | 8 | 7 | 11 | 11 | 5 | 5 | 3 | 3 | 20 | 19 | 21 | 20 |
| Confusion | 0 | 0 | 0 | 0 | 30 | 19 | 33 | 22 | 4 | 4 | 5 | 5 | 37 | 26 | 39 | 28 |
| Cough/wheeze | 3 | 3 | 0 | 0 | 4 | 4 | 1 | 1 | 14 | 11 | 13 | 11 | 40 | 35 | 27 | 24 |
| Dental | 6 | 5 | 0 | 0 | 7 | 7 | 16 | 11 | 3 | 2 | 2 | 2 | 27 | 25 | 25 | 20 |
| Dizzy/faint | 17 | 13 | 20 | 13 | 55 | 32 | 64 | 37 | 74 | 46 | 62 | 41 | 243 | 161 | 216 | 145 |
| Drowsy/fatigued | 56 | 40 | 77 | 47 | 125 | 72 | 267 | 123 | 30 | 24 | 51 | 46 | 338 | 228 | 562 | 338 |
| Gastrointestinal disturbances | 21 | 17 | 10 | 8 | 48 | 31 | 49 | 35 | 45 | 34 | 65 | 42 | 199 | 145 | 172 | 126 |
| Hair loss | 0 | 0 | 0 | 0 | 6 | 4 | 15 | 6 | 3 | 3 | 3 | 3 | 10 | 8 | 20 | 11 |
| Headache/migraine | 74 | 42 | 20 | 13 | 95 | 49 | 97 | 43 | 48 | 31 | 40 | 29 | 412 | 229 | 264 | 146 |
| Impotence | 1 | 1 | 0 | 0 | 5 | 4 | 17 | 5 | 0 | 0 | 0 | 0 | 6 | 5 | 17 | 5 |
| Increased/worsened seizures | 1 | 1 | 0 | 0 | 29 | 21 | 41 | 25 | 86 | 35 | 58 | 27 | 136 | 75 | 107 | 60 |
| Kidney/urinary problems | 4 | 3 | 2 | 2 | 4 | 3 | 10 | 8 | 16 | 16 | 18 | 17 | 47 | 42 | 38 | 35 |
| Memory problems | 4 | 4 | 3 | 3 | 38 | 23 | 71 | 34 | 7 | 6 | 7 | 7 | 67 | 47 | 87 | 49 |
| Menstrual problems | 15 | 9 | 13 | 7 | 4 | 4 | 3 | 2 | 0 | 0 | 0 | 0 | 22 | 16 | 37 | 26 |
| Mood/behavioural change | 5 | 5 | 4 | 1 | 32 | 22 | 56 | 34 | 2 | 2 | 6 | 5 | 61 | 46 | 82 | 55 |
| Nausea/vomiting | 21 | 15 | 15 | 11 | 38 | 23 | 54 | 35 | 30 | 23 | 37 | 24 | 179 | 132 | 163 | 107 |
| Pain | 18 | 15 | 1 | 1 | 14 | 9 | 15 | 12 | 55 | 28 | 28 | 20 | 162 | 103 | 71 | 53 |
| Pins and needles/tingling | 3 | 2 | 0 | 0 | 13 | 13 | 23 | 13 | 4 | 4 | 3 | 3 | 27 | 25 | 29 | 18 |
| Rash/skin problems | 33 | 26 | 17 | 14 | 65 | 36 | 99 | 65 | 23 | 20 | 39 | 32 | 258 | 183 | 269 | 191 |
| Sleep problems/dreams | 27 | 19 | 3 | 2 | 46 | 32 | 24 | 12 | 19 | 18 | 10 | 9 | 132 | 104 | 54 | 33 |
| Throat/tonsil infection | 13 | 10 | 1 | 1 | 2 | 2 | 1 | 1 | 6 | 4 | 4 | 3 | 54 | 43 | 23 | 21 |
| Tremor/twitch | 7 | 6 | 0 | 0 | 28 | 12 | 13 | 10 | 16 | 8 | 10 | 9 | 53 | 28 | 27 | 22 |
| Visual disturbance/nystagmus | 6 | 4 | 7 | 5 | 34 | 22 | 33 | 22 | 13 | 10 | 8 | 4 | 71 | 50 | 70 | 47 |
| Weight gain | 1 | 1 | 0 | 0 | 21 | 13 | 42 | 21 | 4 | 4 | 3 | 3 | 34 | 25 | 49 | 28 |
Table of most commonly occurring adverse events split into two for formatting reasons.
Events = number of adverse events reported; Ppts = number of participants reporting the adverse event (a participant could report the same type of adverse event multiple times).
LTG = lamotrigine; CBZ = carbamazepine
Most common adverse events are defined as events reported 10 or more times in at least one of the seven trials (Brodie 1995 A; Brodie 1995 B; Brodie 1999; Nieto‐Barrera 2001; Reunanen 1996; SANAD A 2007; Werhahn 2015). Less commonly reported adverse events are not summarised in this table but details are available on request from the review authors. General terminology for the type of adverse events was defined by the review authors based on the individual participant data provided.
The most common adverse events, reported 10 or more times in at least one of the seven trials are: accidental injury/fracture, aggression, anorexia/weight loss, anxiety/depression, aphasia, ataxia, chest infection/bronchitis, cold/influenza, concentration, confusion, cough/wheeze, dental, dizzy/faint, drowsy/fatigued, gastrointestinal disturbances, hair loss, headache/migraine, impotence, increased/worsened seizures, kidney/urinary problems, memory problems, menstrual problems, mood/behavioural change, nausea/vomiting, pain, pins and needles/tingling, rash/skin problems, sleep problems/dreams, throat/tonsil infection, tremor/twitch, visual disturbance/nystagmus, weight gain.
The five most commonly reported adverse events on both drugs were: dizzy/faint, drowsy/fatigued, gastrointestinal disturbances, headache/migraine, and rash/skin problems. Across all the trials, the rates of these common adverse events were similar for the two drugs. We did not statistically analyse adverse event data.
A summary for the other seven trials is as follows.
In Eun 2012, five events related to treatment were reported in three participants taking lamotrigine (all skin rashes). Eight events related to treatment were reported in six participants taking carbamazepine (six with tiredness/lethargy, two with skin rashes). All adverse events were described as non‐serious.
In Gilad 2007, two participants taking lamotrigine had adverse events: somnolence and dizziness, respectively. Twelve participants in the carbamazepine group had adverse events: three with nausea and vomiting, three with skin eruptions, two with confusion and one with overdose symptoms who was hospitalised
In Korean Lamotrigine Study Group 2008, adverse events reported were rash (12.8% of participants on lamotrigine and 8.6% of participants on carbamazepine), dizziness (6.8% of participants on lamotrigine and 10.1% of participants on carbamazepine), dyspnoea (0.4% of participants on lamotrigine and 7.0% of participants on carbamazepine), somnolence (0 of participants on lamotrigine and 4.7% of participants on carbamazepine), headache (5.3% of participants on lamotrigine and 0% of participants on carbamazepine), acne (1.1% of participants on lamotrigine and 0% of participants on carbamazepine), asthenia (0.8% of participants on lamotrigine and 1.6% of participants on carbamazepine), oesophageal ulceration (0.4% of participants on lamotrigine and 1.6% of participants on carbamazepine), abnormal liver function (0% of participants on lamotrigine and 1.6% of participants on carbamazepine). Non‐fatal serious adverse events were reported in 4 participants on lamotrigine; three with rash and one with post‐ictal psychosis. All serious adverse events were deemed to be related to treatment.
In Lee 2011, four participants taking lamotrigine reported skin rash (related to treatment), five participants taking carbamazepine reported skin rash (related to treatment) and one participant on carbamazepine reported mitral stenosis (unrelated to treatment).
In Rowan 2005, systemic and neurologic toxicities experienced were weight gain or weight loss (87.4% of participants on lamotrigine and 80.1% of carbamazepine), gastrointestinal problems (33.9% on lamotrigine and 32.2% on carbamazepine), hypersensitivity/severe hypersensitivity (3.3% on lamotrigine and 13.4% on carbamazepine), water retention (10.4% on lamotrigine and 8.8% on carbamazepine), hyponatraemia (6.6% on lamotrigine and 11.1% on carbamazepine), impotence (4.4% on lamotrigine and 7.6% on carbamazepine), and renal or liver disease (1.6% on lamotrigine and 4.1% on carbamazepine).
In Saetre 2007, for lamotrigine, 82 participants reported 378 events: 36 participants reported 53 gastrointestinal events, 20 participants reported 26 infections, 19 participants reported 36 musculoskeletal events, 44 participants reported 111 events of the nervous system, 13 participants reported 23 psychiatric events, 12 participants reported 20 skin problems, 11 participants reported 11 vascular disorders and two participants reported two events of the immune system and 24 participants reported 37 other events. For carbamazepine, 79 participants reported 310 events: 29 participants reported 46 gastrointestinal events, 13 participants reported 23 infections, 18 participants reported 29 musculoskeletal events, 45 participants reported 76 events of the nervous system, 12 participants reported 16 psychiatric events, 21 participants reported 25 skin problems, five participants reported six vascular disorders and 11 participants reported 14 events of the immune system and 27 participants reported 41 other events. For both treatments around half of the events were thought to be related to treatment, particularly dizziness, rash, headache, somnolence and gastrointestinal symptoms.
In Steinhoff 2005, the most frequent adverse events for lamotrigine were fatigue (14.8% of participants), rash, headache and nausea (5.7%), nervousness, sleep disorders, pruritus, alopecia and dizziness (4.5%). Three severe adverse events were reported, all possibly related to treatment (nausea and diarrhoea, leucopenia and fatigue). Most frequent adverse events for carbamazepine were fatigue (43.2%), amnesia and pruritus (10.2%), rash (9.1%), abnormal thoughts and abnormal gait (8%). Seven severe adverse events were reported – four were almost certainly related to treatment (rash, dermatitis, abnormal gait and fatigue), two were probably related to treatment (rash and hyponatraemia) and one was unrelated (astrocytoma).
Serious adverse events or adverse events requiring hospitalisation were reported in the seven trials providing detailed IPD (we note that some events were reported multiple times by participants), as follows.
In Brodie 1995 A, 16 serious adverse events were reported in seven participants. On lamotrigine, there were 13 events in five participants: suicidal ideation (unknown if related to treatment), knee arthroscopy (not related to treatment), recurrent seizures (related to treatment), aggression (unknown if related to treatment) and headache, diplopia, vertigo, photophobia and vomiting (in a single participant, all related to treatment) and on carbamazepine there were three events in two participants (meningioma not related to treatment and pain possibly related to treatment).
In Brodie 1995 B, seven serious adverse events were reported in six participants. On lamotrigine, there were five events in four participants: haematemesis and stomach ulcer (both related to treatment), appendicitis and tumour in two participants (none related to treatment). On carbamazepine, there were, two events in two participants: meningioma and tumour (neither related to treatment).
In Brodie 1999, 69 serious adverse events were reported in 36 participants. On lamotrigine, there were 36 events in 21 participants: six events in four participants were thought to be related to treatment (sickness, abdominal pain and vomiting in one participant, fracture, seizure recurrence and paranoia) and 30 events in 17 participants were not thought to be related to treatment (asthma, stroke in four participants, glaucoma and vomiting in one participant, urinary retention in one participant, chest infection and vomiting blood in one participant, fracture in two participants, seizure recurrence, vomiting, myalgia and chest pain in one participant, bronchospasm, atrial fibrillation and cardiac failure in one participant, tachycardia in one participant, myocardial infarction and pancreatitis in one participant). On carbamazepine, there were 33 events in 15 participants: eight events in four participants were thought to be related to treatment (rashes in three participants and diarrhoea and vomiting in one participant) and 25 events in 11 participants were not thought to be related to treatment (cerebrovascular accident and upper respiratory tract infection in two participants, leg cramps, myocardial infarction and collapse in one participant, stroke and bronchopneumonia in one participant, dizziness and syncope in one participant, high temperature, chest infection and vomiting in one participant, hyperglycaemic coma, pneumonia and septicaemia in one participant, angina and atrial fibrillation in one participant, falls and vagueness in one participant, ventricular failure and intestinal obstruction in one participant, and seizure recurrence in two participants).
In Nieto‐Barrera 2001, 40 serious adverse events were reported in 32 participants. On lamotrigine, there were 27 events in 23 participants: six events in five participants were thought to be related to treatment (rash, raised intracranial pressure, increased seizures, allergic reaction and vertigo) and 23 events in 18 participants were not thought to be related to treatment (change in seizure type, gastric infection, two participants with back pain, broken clavicle, febrile convulsions, tonic‐clonic seizures and related injury, intracranial bleeding, haematoma, low pressure of shunt system, complex seizure, traffic accident, pneumonia, gingivostomatitis, fractured elbow, infection, two sudden deaths and cerebral tumour). On carbamazepine, there were 13 events in nine participants: five events in three participants were thought to be related to treatment (diarrhoea and difficulty walking, allergic reaction and atrial fibrillation and hydrothorax) and eight events in six participants were not thought to be related to treatment (tumour, pneumonia, infection, febrile convulsions, embolisation, hepatitis B and cardiac arrest).
In Reunanen 1996, 12 serious adverse events were reported in seven participants (none related to treatment). On lamotrigine, there were 10 events in five participants: haematemesis and cerebral infarct, uterine bleeding and hysterectomy, cerebral and retinal emboli, pain and postoperative infection, and stroke. Three life‐threatening events were reported in three participants on lamotrigine (none related to treatment): carbon monoxide poisoning (fatal), myocardial infarction (fatal) and brain tumour. On carbamazepine, there were two events in two participants: pulmonary oedema and angioma.
In SANAD A 2007, 177 events resulting in hospitalisation were reported for 99 participants (it is not stated if the events were related to treatment). On lamotrigine, there were 86 events in 53 participants: worsening of seizures in 13 participants, seizure‐related injury in seven participants, cardiovascular events in five participants, stomach ulcer in two participants, infection in two participants, attempted suicide in one participant, rectal bleeding in one participant, pneumonia in one participant; swollen ear in one participant, enlarged prostate in one participant, bowel infection in one participant, malignancy in one participant, legionnaires disease in one participant, haemorrhage in one participant, stroke in one participant, meningioma in one participant, vomiting in one participant, hepatitis in one participant, constipation in one participant, allergic rash in one participant, aneurysm in one participant, vertigo in one participant, carcinoma in one participant, occipital arteriovenous malformations in one participant, Bell's palsy in one participant, allergic rash in one participant, lymphadenopathy in one participant, fractured clavicle in one participant, childbirth in one participant, miscarriage in one participant and toxicity in one participant. On carbamazepine, there were 91 events in 46 participants: worsening of seizures in 12 participants, cardiovascular events in five participants, attempted suicide in three participants, seizure‐related injury in three participants, allergic rash in two participants, antiphospholipid syndrome in one participant, arthritis in one participant, stomach cancer in one participant, urinary tract infection in one participant, disorientation in one participant, psychotic illness in one participant, exacerbation of chronic obstructive pulmonary disease in one participant, hysterectomy in one participant, torsion of testis in one participant, myringotomy in one participant, infection in one participant, worsening of seizures and visual disturbance in one participant, constipation in one participant, low serum in one participant, breast cancer in one participant, abdominal pain in one participant, ataxia in one participant, child birth in one participant, pneumonia in two participant and headache in one participant.
In Werhahn 2015, 120 serious adverse events were reported in 70 participants. On lamotrigine, there were 58 events in 34 participants: two events in two participants were thought to be related or possibly related to treatment (psychiatric disorder and hallucination) and 56 events in 33 participants were not thought to be related to treatment (worsening seizures in eight participants, gastroenteritis in one participant, transient ischaemic attack in two participants, myocardial infarction in two participants, alcohol poisoning in one participant, prostatic hyperplasia in one participant, sudden hearing loss in one participant, sudden death in one participant, cerebral infarction in one participant, astrocytoma in one participant, brain neoplasm in one participant, radius fracture in one participant, bursitis in one participant, head injury in one participant, osteoarthritis in one participant, pneumonia in one participant, urinary tract infection in one participant, herpes in one participant, angina in one participant, asthma in one participant, memory impairment in one participant, intestinal obstruction in one participant, vertebral fracture in one participant, suicidal ideation in one participant, meningioma in one participant and hernia in one participant). On carbamazepine, there were 62 events in 36 participants: four events in three participants were thought to be related to treatment (hepatic enzyme increased, liver disorder and allergic rash), 13 events in four participants were thought to be probably related to treatment (diarrhoea in one participant, headache and hyponatraemia in one participant, dizziness and nausea in two participants), eight events in five were thought to be possibly related to treatment (purpura in one participant, gastroenteritis in two participants, confusion in one participant, lupus erythematosus in one participant), and 28 events in 25 participants were not thought to be related to treatment (worsening seizures in six participants, pneumonia in two participants, sleep apnoea in one participant, cholecystitis in one participant, abdominal pain and nausea in one participant, pain in one participant, spine fusion surgery in one participant, acute coronary syndrome in one participant, gastrointestinal haemorrhage in one participant, death in one participant, hypertension in one participant, device occlusion in one participant, carcinoma in one participant, intestinal obstruction in one participant, melanoma in one participant, dementia in one participant, infectious peritonitis in one participant, pulmonary embolism in one participant, renal cancer in one participant and constipation in one participant).
Discussion
Summary of main results
The results of this review provide statistically significant, but moderate‐ to low‐quality evidence of an advantage for lamotrigine over carbamazepine for our primary global effectiveness outcome, time to treatment failure. Considering time to treatment failure for any reason related to treatment, for 2569 participants providing individual participant data (IPD) from nine trials, the pooled hazard ratio (HR) was 0.73 (95% confidence interval (CI) 0.64 to 0.82, P < 0.00001, moderate‐quality evidence). This advantage was also present in the 2182 participants with focal onset seizures (pooled HR 0.74, 95% CI 0.64 to 0.86, P = 0.0001, moderate‐quality evidence) and the 299 participants with generalised onset seizures (pooled HR 0.51, 95% CI 0.33 to 0.78, P = 0.002, low‐quality evidence) from the nine trials providing IPD.
The advantage also remained when incorporating aggregate data from four trials for which IPD were not available, allowing for alternative definitions of treatment failure from the definition used in this review (ILAE 1998), and allowing for blinded trial design.
There was also an advantage for lamotrigine over carbamazepine when considering time to treatment failure due to adverse events in all 2569 participants (pooled HR 0.54 (95% CI 0.45 to 0.65, P < 0.00001, moderate‐quality evidence)), the 2182 participants with focal onset seizures (pooled HR 0.56 (95% CI 0.45 to 0.68, P<0.00001, moderate‐quality evidence) and the 299 participants with generalised onset seizures (pooled HR 0.49 (95% CI 0.27 to 0.88, P = 0.002, low‐quality evidence) but no difference between lamotrigine and carbamazepine when considering time to treatment failure due to lack of efficacy in 1874 participants in five trials where at least one participant on each treatment failed treatment due to lack of efficacy (pooled HR 1.03 (95% CI 0.75 to 1.41, P =0.86), moderate‐quality evidence).
The results of this review provide statistically significant and high‐quality evidence of an advantage for carbamazepine over lamotrigine for our secondary efficacy outcomes, time to first seizure (pooled HR for 2564 participants: 1.22, 95% CI 1.09 to 1.37, P = 0.0004, high‐quality evidence) and time to six‐month remission (pooled HR for 1793 participants: 0.84, 95% CI 0.74 to 0.94, P = 0.003, high‐quality evidence). As above, this advantage was present in the subgroup of participants with focal onset seizures (high‐quality evidence), but in the smaller subgroup of participants with generalised onset seizures we found no significant difference between the drugs (moderate‐quality evidence).
We found no statistically significant difference between the drugs for the longer‐term outcomes of time to 12‐month remission and time to 24‐month remission (high‐quality evidence); however fewer data were available for inclusion in analyses at these time points due to the short duration of most included trials.
The most commonly reported adverse events for both of the drugs across all of the included trials were dizziness, fatigue, gastrointestinal disturbances, headache and skin problems. The rate of adverse events and serious adverse events was similar across the two drugs.
Overall completeness and applicability of evidence
We have gratefully received individual participant data (IPD) for 2572 individuals (68% of 3787 individuals from all eligible trials) from the authors or sponsors of nine trials (Brodie 1995 A; Brodie 1995 B; Brodie 1999; Eun 2012; Lee 2011; Nieto‐Barrera 2001; Reunanen 1996; SANAD A 2007; Werhahn 2015), which included a comparison of lamotrigine with carbamazepine for the treatment of epilepsy.
At the time of review, we have not been able to obtain IPD for the remaining five included trials with a total of 1215 participants. For three trials including 753 individuals (Korean Lamotrigine Study Group 2008; Saetre 2007; Steinhoff 2005), the trial sponsor confirmed that data could not be made available. For the other two trials, we made contact with the authors/sponsors who expressed interest in collaborating in this IPD meta‐analysis but at the time of writing, no data had been received (Gilad 2007; Rowan 2005).
If IPD are received from these trials, we will include the data in future updates. We were able to extract aggregate data from four out of the five trial publications to include in meta‐analysis for our primary outcome of 'time to treatment failure,' resulting in a similar pooled estimate to that from IPD only. Therefore, we do not believe that our failure to obtain IPD from 32% of eligible participants from the five trials has had a large impact on the applicability of the results of the review. We do, however, encourage caution when interpreting the numerical results of the review, particularly longer‐term remission outcomes for which only two trials were of sufficient duration. Given the results of this meta‐analysis it could be that, compared to carbamazepine, the initial doses of lamotrigine chosen were too low. Hence lamotrigine fared better for treatment failure as the dose chosen caused comparatively fewer side effects but was less effective at preventing seizures. This highlights the importance of measuring longer‐term seizure outcomes such as time to one‐ or two‐year remission from seizures, which would be much less affected by initial drug titration and initial target doses.
We have good evidence from previous reviews conducted by the Cochrane Epilepsy Group that misclassification of seizure type, particularly generalised seizure types, is an important issue in epilepsy trials (Nevitt 2017b; Nolan 2016b; Nolan 2016d). It is also likely in this review that a large proportion of individuals who were classified as experiencing generalised onset seizures at baseline had their seizure type wrongly classified, meaning that the results of the original trials and therefore the results of this review may have been confounded by classification bias. Following sensitivity analyses to account for this potential misclassification, the overall conclusions for our primary and secondary outcomes were not changed (see Summary of main results). However, due to the small proportion of total participants included experiencing generalised onset seizures (302 out of 2572, 12% of total participants) and up to 50% of those participants with potentially misclassified seizure type, the results of this review are primarily applicable to participants with focal onset seizures.
Quality of the evidence
The nine trials for which IPD were made available (as well as additional trial design information from trial authors/sponsors) were of generally good quality. Less information was available for trials without IPD available where risk of bias assessments were made only based on published information. Six trials were of a double‐blind design (Brodie 1995 A; Brodie 1995 B; Brodie 1999; Rowan 2005; Saetre 2007; Werhahn 2015), and eight trials were of an open‐label design (Eun 2012; Gilad 2007; Korean Lamotrigine Study Group 2008; Lee 2011; Nieto‐Barrera 2001; Reunanen 1996; Steinhoff 2005; SANAD A 2007). The results of this review suggest that the design of a trial (double‐blind versus open‐label) may influence the withdrawal and treatment failure rates of the trial, an outcome that is subjective and can be influenced by the participant or clinician. It is argued that an open‐label design is more pragmatic and reflective of 'real world' treatment for a trial of a chronic condition such as epilepsy where treatments are likely to be taken long‐term by participants (SANAD A 2007), and it is shown in this review that a significantly higher proportion of participants failed treatment in the trials with a double‐blind design compared to the open‐label trials (45% versus 34%, P < 0.0001, see Effects of interventions). However, in a trial of a 'new' compared to a 'standard' intervention, knowledge of the treatment allocation may influence the choice of the participant or clinician to continue taking the treatment, which may then influence the perceived effectiveness of the two drugs under comparison. Therefore, we have considered an open‐label design to potentially introduce bias into the results for the subjective outcomes of time to treatment failure, but not for the objective secondary outcomes of time to first seizure and remission.
Due to this potential risk of bias from an open‐label design, we have rated the evidence provided in this review according to Grading of Recommendations Assessment, Development and Evaluation (GRADE) criteria for our primary outcome of 'time to treatment failure' as 'moderate' for all participants and the subgroup of participants with focal onset seizures. Due to the limited number of participants with generalised onset seizures (and potential misclassification of seizure type), we have rated this evidence as low quality for the primary outcome (see Table 1).
For our secondary (objective) outcomes of time to first seizure and remission, we have rated the evidence as high‐quality (moderate‐quality in the subgroup of generalised onset seizures for the reasons stated above) (see Table 2).
Potential biases in the review process
We were able to include individual participant data (IPD) for 2572 out of 3787 eligible participants (68%) from nine out of 14 trials in this review and we were able to analyse all outcomes using IPD. Such an approach has many advantages, such as allowing the standardisation of definitions of outcomes across trials, and attrition and reporting biases are reduced as we can perform additional analyses and calculate additional outcomes from unpublished data. For the outcomes we used in this review that are of a time‐to‐event nature, an IPD approach is considered to be the 'gold standard' approach to analysis (Parmar 1998).
For reasons outside of our control, we were unable to obtain IPD for 1215 participants from five trials for inclusion in this review. However, following sensitivity analyses using aggregate data, we do not believe that the exclusion of 32% of eligible participants is likely to have impacted on the conclusions of this review (see Overall completeness and applicability of evidence).
Finally, we made some assumptions in the statistical methodology used in this review. Firstly, when we received only follow‐up dates and seizure frequencies, we used linear interpolation to estimate. We are aware that an individual's seizure patterns may be non‐linear; therefore, we recommend caution when interpreting the numerical results of the seizure‐related outcomes.
We also made an assumption that treatment effect for each outcome did not change over time (proportional hazards assumption, see Data synthesis). We are aware that in trials of long duration (e.g. SANAD A 2007, and Werhahn 2015, of over one year duration), the assumption of treatment effect remaining constant over time may not be appropriate; for example, there is likely to be a difference between participants who achieve immediate remission compared with participants who achieve later remission, and we encourage that results should be interpreted with this limitation in mind.
Agreements and disagreements with other studies or reviews
To our knowledge, together with previous versions of this review, this is the only systematic review and meta‐analysis that compares lamotrigine and carbamazepine monotherapy for focal onset seizures and generalised onset tonic‐clonic seizures. A network meta‐analysis has been published (Nevitt 2017a), comparing all direct and indirect evidence from lamotrigine, carbamazepine and other standard and new antiepileptic drugs licensed for monotherapy. The results of this review generally agree with the results of the network meta‐analysis.
Authors' conclusions
Implications for practice.
Current UK guidelines recommend carbamazepine or lamotrigine as first‐line treatment for adults and children with new onset focal seizures and sodium valproate for adults and children with new onset generalised seizures (NICE 2012).
For individuals with new onset focal seizures, the moderate‐quality evidence provided by this review suggests that lamotrigine is likely to be a more effective drug than carbamazepine in terms of treatment retention (treatment failure for any reason related to treatment or due to adverse events). However, high‐quality evidence provided by this review suggests that individuals are likely to achieve earlier remission and later seizure recurrence when taking carbamazepine compared to lamotrigine. Therefore a choice between these two first‐line treatments for individuals with new onset focal seizures must be carefully considered, taking the personal circumstances of an individual into account.
For individuals with new onset generalised seizures, the evidence in the review is limited and of moderate to low quality due to small numbers of participants with certain generalised seizure types recruited into the included trials. There is evidence that carbamazepine may exacerbate some generalised seizure types so should be used with caution in individuals with this seizure type (Liporace 1994; Shields 1983; Snead 1985). Lamotrigine may be an effective treatment option for new onset generalised seizures, but more evidence is required to confirm this.
Implications for research.
This review highlights the need for the design of future antiepileptic drug monotherapy trials that recruit individuals with specific epilepsy syndromes to be powered to detect a difference between particular antiepileptic drugs. An approach likely to reflect and inform clinical practice, as well as being statistically powerful, would be to recruit heterogeneous populations for whom epilepsy syndromes have been adequately defined, with testing for interaction between treatment and epilepsy syndrome. In view of potential problems of misclassification, syndromes will have to be well defined, with adequate checking mechanisms to ensure that classifications are accurate and a system to recognise uncertainty surrounding epilepsy syndromes in individuals within trials. It is also important that future trials are of a sufficient duration to measure long‐term effectiveness of antiepileptic drugs (treatments that will be life‐long for many individuals with epilepsy), as well as psychosocial, quality‐of‐life and health economic outcomes.
Consideration is also required in the design of a trial regarding whether to blind participants and outcome assessors to treatment allocation. While an open‐label design is a more pragmatic and practical approach for large, long‐term trials, when trials involve a new intervention compared to an established 'standard' intervention, masking of treatment may be important to avoid preconceptions over the relative effectiveness of the drugs.
The choice of outcomes at the design stage of a trial and the presentation of the results of outcomes, particularly of a time‐to‐event nature, require very careful consideration. While the majority of trials of a monotherapy design record an outcome measuring efficacy (seizure control) and an outcome measuring tolerability (adverse events), there is little uniformity between the definition of the outcomes and the reporting of the summary statistics related to the outcomes (Nolan 2013a), making an aggregate data approach to meta‐analysis in reviews of monotherapy trials impossible. Where trial authors cannot or will not make individual participant data available for analysis, we are left with no choice but to exclude a proportion of relevant evidence from the review, which may impact upon the interpretation of the results of the review and the applicability of the evidence and conclusions. The International League Against Epilepsy recommends that trials of a monotherapy design should adopt a primary effectiveness outcome of time to treatment failure (i.e. retention time) and should be of a duration of at least 48 weeks to allow for assessment of longer‐term outcomes, such as remission (ILAE 1998; ILAE 2006). If trials followed these recommendations, an aggregate data approach to meta‐analysis may be feasible, reducing the resources and time required from an individual participant data approach.
What's new
| Date | Event | Description |
|---|---|---|
| 26 February 2018 | New citation required but conclusions have not changed | Conclusions are unchanged. |
| 26 February 2018 | New search has been performed | Searches updated 26 February 2018; no new studies have been included. The term 'partial' has been replaced by 'focal', in accordance with the most recent classification of epilepsies of the International League Against Epilepsy (Scheffer 2017). |
History
Protocol first published: Issue 1, 1998 Review first published: Issue 1, 2006
| Date | Event | Description |
|---|---|---|
| 26 April 2017 | Amended | Declarations of interest updated. |
| 17 October 2016 | New search has been performed | Searches updated 17 October 2016; eight new studies have been included. |
| 17 October 2016 | New citation required but conclusions have not changed | Conclusions are unchanged. |
| 11 August 2009 | Amended | Contact details updated. |
| 27 August 2008 | Amended | Converted to new review format. |
Notes
Sarah J Nolan (lead author of the 2016 update) is now Sarah J Nevitt.
Acknowledgements
This review update was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Epilepsy Group. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS of the Department of Health.
We are greatly indebted to all of the original trialists that provided individual participant data and input into this review.
We are grateful to the Cochrane Epilepsy Group Information Specialist, Graham Chan, for performing all electronic searches.
We would like to thank Sarah White for her input into the original protocol, and to Carrol Gamble and Paula Williamson for contributions to the original review.
Appendices
Appendix 1. Epilepsy Specialized Register search strategy
The following was used for the latest update.
1. epilepax or lamictal or lamotrigin* AND INREGISTER
2. MeSH DESCRIPTOR Carbamazepine Explode All AND INREGISTER
3. biston or carbamazepin* or carbatrol or cbz or epitol or equetro or neurotop or tegretal or tegretol or teril or timonil AND INREGISTER
4. #2 OR #3 AND INREGISTER
5. #1 AND #4 AND INREGISTER
6. ((adjunct* or "add‐on" or "add on" or adjuvant* or combination* or polytherap*) not (monotherap* or alone or singl*)):TI AND INREGISTER
7. (#5 NOT #6) AND >31/07/2014:CRSCREATED AND INREGISTER
8. (epilepax or lamictal or lamotrigin*):AB,KW,MC,MH,TI AND CENTRAL:TARGET
9. MeSH DESCRIPTOR Carbamazepine Explode All AND CENTRAL:TARGET
10. (biston or carbamazepin* or carbatrol or cbz or epitol or equetro or neurotop or tegretal or tegretol or teril or timonil):AB,KW,MC,MH,TI AND CENTRAL:TARGET
11. #9 OR #10 AND CENTRAL:TARGET
12. #8 AND #11 AND CENTRAL:TARGET
13. ((adjunct* or "add‐on" or "add on" or adjuvant* or combination* or polytherap*) not (monotherap* or alone or singl*)):TI AND CENTRAL:TARGET
14. (#12 NOT #13) AND >31/07/2014:CRSINCENTRAL AND CENTRAL:TARGET
15. MESH DESCRIPTOR Epilepsy EXPLODE ALL AND CENTRAL:TARGET
16. MESH DESCRIPTOR Seizures EXPLODE ALL AND CENTRAL:TARGET
17. epilep* OR seizure* OR convuls* AND CENTRAL:TARGET
18. #15 OR #16 OR #17 AND CENTRAL:TARGET
19. #14 AND #18 AND CENTRAL:TARGET
20. #7 OR #19
Appendix 2. CENTRAL via CRSO search strategy
For the latest update, the following was used to search CENTRAL via the Cochrane Register of Studies Online (CRSO).
#1 epilepax OR lamictal OR lamotrigin*
#2 MESH DESCRIPTOR Carbamazepine EXPLODE ALL TREES
#3 biston OR carbamazepin* OR carbatrol OR cbz OR epitol OR equetro OR neurotop OR tegretol OR teril OR timonil
#4 #2 OR #3
#5 #1 AND #4
#6 (epilep* OR seizure* OR convuls*):TI,AB,KY
#7 MESH DESCRIPTOR Epilepsy EXPLODE ALL TREES
#8 MESH DESCRIPTOR Seizures EXPLODE ALL TREES
#9 #6 OR #7 OR #8
#10 eclampsia:TI
#11 #9 NOT #10
#12 #5 AND #11
#13 ((adjunct* OR "add‐on" OR "add on" OR adjuvant* OR combination* OR polytherap*) NOT (monotherap* or alone or singl*)):TI
#14 #12 NOT #13
#15 ("Conference Abstract"):PT AND INEMBASE
#16 #14 NOT #15
#17 * NOT INMEDLINE AND 03/12/2015 TO 17/10/2016:CD
#18 #16 AND #17
Earlier versions of this review used the following to search CENTRAL in the Cochrane Library.
#1 (lamotrigine OR lamictal)
#2 MeSH descriptor Carbamazepine explode all trees
#3 carbamazepine or tegretol
#4 (#1 AND ( #2 OR #3 ))
#5 MeSH descriptor Epilepsy explode all trees
#6 MeSH descriptor Seizures explode all trees
#7 epilep* or seizure* or convulsion*
#8 (#5 OR #6 OR #7)
#9 (#4 AND #8)
Appendix 3. MEDLINE search strategy
The following was used for the latest update. It is based on the Cochrane Highly Sensitive Search Strategy for identifying randomised trials in MEDLINE (Lefebvre 2011).
1. (lamotrigin$ or lamictal or epilepax).tw.
2. exp carbamazepine/ or (biston or carbamazepin$ or carbatrol or cbz or epitol or equetro or neurotop or tegret?l or teril or timonil).tw.
3. 1 and 2
4. exp Epilepsy/
5. exp Seizures/
6. (epilep$ or seizure$ or convuls$).tw.
7. 4 or 5 or 6
8. exp *Pre‐Eclampsia/ or exp *Eclampsia/
9. 7 not 8
10. (randomized controlled trial or controlled clinical trial or pragmatic clinical trial).pt. or (randomi?ed or placebo or randomly).ab.
11. clinical trials as topic.sh.
12. trial.ti.
13. 10 or 11 or 12
14. exp animals/ not humans.sh.
15. 13 not 14
16. 3 and 9 and 15
17. ((adjunct$ or "add‐on" or "add on" or adjuvant$ or combination$ or polytherap$) not (monotherap$ or alone or singl$)).ti.
18. 16 not 17
19. limit 18 to ed=20140731‐20180226
20. 18 not (1$ or 2$).ed.
21. 20 and (2014$ or 2015$ or 2016$ or 2017$ or 2018$).dt.
22. 19 or 21
23. remove duplicates from 22
Earlier versions of this review used the following search strategy, based on the previous Cochrane Highly Sensitive Search Strategy for MEDLINE as set out in Appendix 5b of the Cochrane Handbook for Systematic Reviews of Interventions (version 4.2.5, updated May 2005) (Higgins 2005).
1. randomized controlled trial.pt.
2. controlled clinical trial.pt.
3. exp Randomized Controlled Trials/
4. exp Random Allocation/
5. exp Double‐Blind Method/
6. exp Single‐Blind Method/
7. clinical trial.pt.
8. Clinical Trial/
9. (clin$ adj trial$).ab,ti.
10. ((singl$ or doubl$ or trebl$ or tripl$) adj (blind$ or mask$)).ab,ti.
11. exp PLACEBOS/
12. placebo$.ab,ti.
13. random$.ab,ti.
14. exp Research Design/
15. or/1‐14
16. (animals not humans).sh.
17. 15 not 16
18. lamotrigine.tw.
19. carbamazepine/ or carbamazepine.tw.
20. exp epilepsy/ or epilep$.tw.
21. exp seizures/ or seizure$.tw.
22. convulsion$.tw.
23. 18 and 19
24. 20 or 21 or 22
25. 23 and 24 and 17
Data and analyses
Comparison 1. Lamotrigine (LTG) versus carbamazepine (CBZ).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Time to treatment failure (any reason related to the treatment) | 9 | 2569 | Hazard Ratio (Fixed, 95% CI) | 0.73 [0.64, 0.82] |
| 2 Time to treatment failure due to adverse events | 9 | 2569 | Hazard Ratio (Fixed, 95% CI) | 0.54 [0.45, 0.65] |
| 3 Time to treatment failure due to lack of efficacy | 5 | 1874 | Hazard Ratio (Fixed, 95% CI) | 1.03 [0.75, 1.41] |
| 4 Time to treatment failure (any reason related to the treatment) ‐ by seizure type | 9 | 2481 | Hazard Ratio (Fixed, 95% CI) | 0.71 [0.62, 0.82] |
| 4.1 Focal | 9 | 2182 | Hazard Ratio (Fixed, 95% CI) | 0.74 [0.64, 0.86] |
| 4.2 Generalised | 6 | 299 | Hazard Ratio (Fixed, 95% CI) | 0.51 [0.33, 0.78] |
| 5 Time to treatment failure due to adverse events ‐ by seizure type | 9 | 2466 | Hazard Ratio (Fixed, 95% CI) | 0.55 [0.45, 0.66] |
| 5.1 Focal | 9 | 2182 | Hazard Ratio (Fixed, 95% CI) | 0.56 [0.45, 0.68] |
| 5.2 Generalised | 5 | 284 | Hazard Ratio (Fixed, 95% CI) | 0.49 [0.27, 0.88] |
| 6 Time to treatment failure (any reason related to the treatment, with aggregate data) | 13 | 3391 | Hazard Ratio (Fixed, 95% CI) | 0.70 [0.63, 0.78] |
| 7 Time to treatment failure (any reason related to the treatment) ‐ subgroup analysis (blinding) | 13 | 3391 | Hazard Ratio (Fixed, 95% CI) | 0.70 [0.63, 0.78] |
| 7.1 Double‐blind | 6 | 1231 | Hazard Ratio (Fixed, 95% CI) | 0.65 [0.56, 0.75] |
| 7.2 Open‐label | 7 | 2160 | Hazard Ratio (Fixed, 95% CI) | 0.76 [0.65, 0.90] |
| 8 Time to first seizure | 9 | 2564 | Hazard Ratio (Fixed, 95% CI) | 1.22 [1.09, 1.37] |
| 9 Time to first seizure by seizure type | 9 | 2476 | Hazard Ratio (Fixed, 95% CI) | 1.26 [1.12, 1.41] |
| 9.1 Focal | 9 | 2177 | Hazard Ratio (Fixed, 95% CI) | 1.29 [1.14, 1.45] |
| 9.2 Generalised | 6 | 299 | Hazard Ratio (Fixed, 95% CI) | 0.98 [0.65, 1.48] |
| 10 Time to first seizure (with aggregate data) | 12 | 3216 | Hazard Ratio (Fixed, 95% CI) | 1.24 [1.12, 1.37] |
| 11 Seizure freedom (whole study) | 14 | 3760 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.11 [1.04, 1.18] |
| 12 Time to 6‐month remission | 7 | 1793 | Hazard Ratio (Fixed, 95% CI) | 0.84 [0.74, 0.94] |
| 13 Time to 6‐month remission by seizure type | 7 | 1708 | Hazard Ratio (Fixed, 95% CI) | 0.86 [0.76, 0.97] |
| 13.1 Focal | 7 | 1454 | Hazard Ratio (Fixed, 95% CI) | 0.87 [0.77, 1.00] |
| 13.2 Generalised | 5 | 254 | Hazard Ratio (Fixed, 95% CI) | 0.78 [0.55, 1.11] |
| 14 Seizure freedom at 6 months | 14 | 3760 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.96 [0.89, 1.03] |
| 15 Time to 12‐month remission | 2 | 988 | Hazard Ratio (Fixed, 95% CI) | 0.91 [0.77, 1.07] |
| 16 Time to 12‐month remission by seizure type | 2 | 988 | Hazard Ratio (Fixed, 95% CI) | 0.90 [0.76, 1.07] |
| 16.1 Focal | 2 | 894 | Hazard Ratio (Fixed, 95% CI) | 0.91 [0.77, 1.09] |
| 16.2 Uncertain | 1 | 94 | Hazard Ratio (Fixed, 95% CI) | 0.81 [0.47, 1.37] |
| 17 Time to 24‐month remission | 1 | 755 | Hazard Ratio (Fixed, 95% CI) | 1.00 [0.80, 1.25] |
| 18 Time to 24‐month remission by seizure type | 1 | 755 | Hazard Ratio (Fixed, 95% CI) | 1.03 [0.82, 1.30] |
| 18.1 Focal | 1 | 661 | Hazard Ratio (Fixed, 95% CI) | 1.06 [0.83, 1.35] |
| 18.2 Uncertain | 1 | 94 | Hazard Ratio (Fixed, 95% CI) | 0.86 [0.44, 1.67] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Brodie 1995 A.
| Methods | Randomised, double‐blind, parallel‐group trial conducted in 8 centres in the UK 2 treatment arms: LTG and CBZ |
|
| Participants | Adults and children over the age of 13 with newly diagnosed epilepsy Number randomised: LTG = 70, CBZ = 66; 56 males (41%) 82 with focal seizures (60%) None had received previous AED treatment Mean age (range): 34 (13 to 71) years |
|
| Interventions | Monotherapy with LTG or CBZ for 48 weeks 4‐week escalation phase leading to LTG = 150 mg/day, CBZ = 600 mg/day Range of follow‐up: 0 to 398 days |
|
| Outcomes | Time to first seizure after 6 weeks of treatment Time to treatment withdrawal Proportion of randomised patients remaining seizure‐free during the last 40 and 24 weeks of trial Percentages of patients who reported adverse events |
|
| Notes | IPD provided by trial sponsor GlaxoSmithKline for time to treatment failure, time to first seizure and time to 6‐month remission | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer generated random sequence (information provided by drug manufacturer). Stratification by seizure type |
| Allocation concealment (selection bias) | Low risk | Allocation concealed by individual sealed opaque envelopes (information provided by drug manufacturer) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind achieved using LTG tablets formulated to be identical in appearance to CBZ tablets |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Trial investigator blinded, not stated if other outcome assessors were blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported; all randomised participants analysed from IPD provided (see footnote 2) |
| Selective reporting (reporting bias) | Low risk | All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | None identified |
Brodie 1995 B.
| Methods | Randomised, double‐blind, parallel‐group trial conducted in 8 centres in the UK 2 treatment arms: LTG and CBZ |
|
| Participants | Adults and children over the age of 13 with newly diagnosed epilepsy Number randomised: LTG = 61, CBZ = 63 56 males (45%) 62 with focal seizures (50%) None had received previous AED treatment Mean age (range): 30 (14 to 86) years |
|
| Interventions | Monotherapy with LTG or CBZ for 48 weeks 4‐week escalation phase leading to LTG = 150 mg/day, CBZ = 600 mg/day Range of follow‐up: 0 to 398 days |
|
| Outcomes | Time to first seizure after 6 weeks of treatment Time to treatment withdrawal Proportion of randomised patients remaining seizure‐free during the last 40 and 24 weeks of trial Percentages of patients who reported adverse events |
|
| Notes | IPD provided by trial sponsor GlaxoSmithKline for time to treatment failure, time to first seizure and time to 6‐month remission | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer generated random sequence (information provided by drug manufacturer). Stratification by seizure type |
| Allocation concealment (selection bias) | Low risk | Allocation concealed by individual sealed opaque envelopes (information provided by drug manufacturer) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind achieved using LTG tablets formulated to be identical in appearance to CBZ tablets |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Trial investigator blinded, not stated if other outcome assessors were blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported; all randomised participants analysed from IPD provided (see footnote 2) |
| Selective reporting (reporting bias) | Low risk | All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | None identified |
Brodie 1999.
| Methods | Randomised, multicentre, double‐blind, parallel‐group trial conducted in the UK 2 treatment arms: LTG and CBZ randomised in a 2:1 ratio |
|
| Participants | Adults over the age of 65 with newly diagnosed epilepsy with 2 or more seizures in the previous year with at least 1 seizure in the last 6 months Number randomised: LTG = 102, CBZ = 48 83 males (55%) 105 with focal seizures (70%) Not stated if any participants had received previous AED treatment Mean age (range): 77 (65 to 94) years |
|
| Interventions | Monotherapy with LTG or CBZ for 24 weeks 4‐week escalation phase leading to LTG = 100 mg/day, CBZ = 400 mg/day Range of follow‐up = 0 to 280 days |
|
| Outcomes | Time to first seizure after 6 weeks of treatment Time to treatment withdrawal Percentage of patients reporting an adverse event Proportion of patients who were both seizure‐free in the last 16 weeks of the trial and did not discontinue treatment |
|
| Notes | IPD provided by trial sponsor GlaxoSmithKline for time to treatment failure and time to first seizure (plus seizure freedom rates at 24 weeks) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random sequence (information provided by drug manufacturer). Participants randomised in a 2:1 ratio (LTG:CBZ) |
| Allocation concealment (selection bias) | Low risk | Allocation concealed with pharmacy‐dispensed treatment packs labelled with participant's trial number |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind achieved using LTG tablets formulated to be identical in appearance to CBZ tablets |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Trial investigator blinded, not stated if other outcome assessors were blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported; all randomised participants analysed from IPD provided (see footnote 2) |
| Selective reporting (reporting bias) | Low risk | All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | None identified |
Eun 2012.
| Methods | Randomised, multicentre, open‐label, parallel‐group trial conducted in 7 hospitals in the Republic of Korea 2 treatment arms: LTG and CBZ |
|
| Participants | Children between the ages of 6 and 12 with a new diagnosis of focal epilepsy and at least 2 seizures in the last 6 months Number randomised: LTG = 43, CBZ = 41 48 males (57%) 100% focal epilepsy Not stated if any participants had received previous AED treatment Mean age (range): 9 (5 to 13) years |
|
| Interventions | Monotherapy with LTG or CBZ for 32 weeks 8‐week escalation phase leading to LTG = 3 to 6 mg/kg/day, CBZ = 10 to 20 mg/kg/day Range of follow‐up: 12 to 788 days |
|
| Outcomes | Seizure‐free rate over 6 months (maintenance period) by treatment group Change in cognition (neuropsychological), behaviour and quality of life from screening to the end of the maintenance phase by treatment group Incidence of adverse events |
|
| Notes | IPD provided by trial author for time to treatment failure, time to first seizure and time to 6‐month remission No source of funding stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Each centre received a separate and independent computer‐generated random code list |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported; all randomised participants analysed from IPD provided (see footnote 2) |
| Selective reporting (reporting bias) | Low risk | All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | None identified |
Gilad 2007.
| Methods | Randomised single‐centre, open‐label, parallel‐group trial conducted at Tel Aviv University and Medical Centre, Israel 2 treatment arms: LTG and CBZ |
|
| Participants | Adults admitted to the neurological department with a first seizure event after an ischaemic stroke Number randomised: LTG = 32, CBZ = 32 46 males (72%) 100% focal seizures Unclear if any participants had received previous AED treatment Mean age (range): 67.5 (38 to 90) years |
|
| Interventions | Monotherapy with LTG or CBZ for 12 months Dose escalation phase (length not stated) leading to LTG 100 mg/day, CBZ 300 mg/day Range of follow‐up: not stated |
|
| Outcomes | The appearance of a second seizure under treatment or by finishing the 12‐month follow‐up without seizures Tolerability: incidence of adverse events Treatment withdrawals due to adverse events |
|
| Notes | Contact made with trial author who was willing to provide IPD but data never received. Aggregate data extracted from graphs in the publication. Stated in the title of the paper that LTG and CBZ were monotherapy treatments but Table 1 of the paper refers to total no. AED; unclear if all participants were receiving monotherapy treatment. No source of funding stated. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised in a 1:1 ratio, no further information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rate reported; all randomised participants included in analysis |
| Selective reporting (reporting bias) | Low risk | No protocol available. Seizure outcomes and adverse events well reported |
| Other bias | Unclear risk | Unclear if all participants were receiving monotherapy treatment |
Korean Lamotrigine Study Group 2008.
| Methods | Phase IV, open label, randomised, multicentre trial conducted in 21 Centres in Korea Two treatment arms: CBZ and LTG |
|
| Participants | Participants were untreated epileptics who had at least 2 unprovoked seizures (focal or generalised tonic clonic) during the last 24 weeks before the study start, more than 24 hours apart. Number randomised: CBZ=129, LTG=264 (ITT population) 154 male participants (39%); 288 participants (73%) with focal epilepsy Mean age (SD): CBZ=37.6 (15.8), LTG=34.2 (16.3) years |
|
| Interventions | Monotherapy with CBZ or LTG Permitted doses LTG: 100mg/day – 500mg/day for LTG , CBZ: 400mg/day – 1200mg/day. |
|
| Outcomes | Retention Rate at Study End Terminal 24 week seizure free rate and time interval from the end of dose titration phase to the first seizure |
|
| Notes | Full text of the trial published in Korean. Abstract and clinical trial summary available in English. IPD requested from trial sponsor Glaxo Smith Kline but data could not be located |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Trial described as randomised, no further information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Attrition rate reported, not all participants included in analysis, which is not an ITT approach |
| Selective reporting (reporting bias) | Low risk | Results for all outcomes summarised for all listed outcomes |
| Other bias | Unclear risk | None identified |
Lee 2011.
| Methods | Randomised, multicentre, open‐label, parallel‐group trial conducted in the Republic of Korea 2 treatment arms: LTG and CBZ |
|
| Participants | Adults over the age of 16 with newly diagnosed focal epilepsy or untreated focal epilepsy for at least 1 year Number randomised: LTG = 57, CBZ = 53 57 males (52%) 95 focal seizures (86%) Not stated how many participants had received previous AED treatment Mean age (range): 36 (16 to 60) years |
|
| Interventions | Monotherapy with LTG or CBZ for 48 weeks 8‐week escalation phase leading to LTG = 200 mg/day, CBZ = 600 mg/day Range of follow‐up: 14 to 337 days |
|
| Outcomes | Change of neuropsychological and cognitive scores from baseline: general intellectual ability, learning and memory, attention and executive function (group‐by‐time interaction) Frequency of psychological and health‐related quality of life symptoms Proportion with seizure freedom during the maintenance period |
|
| Notes | IPD provided by trial author for time to treatment failure, time to first seizure and time to 6‐month remission This trial was supported by a grant from GlaxoSmithKline Korea. No other funding sources stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Block randomisation (block size 4) via a computer randomisation program (information provided by trial author) |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported; all randomised participants analysed from IPD provided (see footnote 2) |
| Selective reporting (reporting bias) | Low risk | All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | None identified |
Nieto‐Barrera 2001.
| Methods | Randomised, multicentre, open‐label, parallel‐group trial conducted in Europe and Mexico 2 treatment arms: LTG and CBZ randomised in a 2:1 ratio |
|
| Participants | Adults and children over the age of 2 with newly diagnosed or currently untreated focal epilepsy with 2 or more seizures in the previous 6 months and with at least 1 seizure in the last 3 months Number randomised: LTG = 420, CBZ = 202 329 males (53%) 619 with focal seizures (99.5%) Not stated how many participants had received previous AED treatment Mean age (range): 27 (2 to 84) years |
|
| Interventions | Monotherapy with LTG or CBZ for 24 weeks 6‐week escalation phase leading to minimum of LTG 2 mg/kg/day age range 2 to 12 years, 200 mg/day age range 13 to 64 years and 100 mg/day age > 65 years. CBZ aged 2 to 12 years 5 to 40 mg/kg, age > 12 years 100 to 1500 mg/day Range of follow‐up: 0 to 245 days |
|
| Outcomes | Proportion of patients seizure‐free during the last 16 weeks of treatment Efficacy success: proportion of patients who did not withdraw before the end of week 18 and were seizure‐free in the last 16 weeks of the trial Time to withdrawal from the trial (proportion of patients completing the trial) Proportion of patients experiencing adverse events Treatment withdrawals due to adverse events |
|
| Notes | IPD provided by trial sponsor GlaxoSmithKline for time to treatment failure and time to first seizure (plus seizure freedom rates at 24 weeks) Dates of seizures during the first 4 weeks not provided with individual participant data |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random sequence. Participants randomised in a 2:1 ratio (LTG:CBZ), stratified by age group and country |
| Allocation concealment (selection bias) | Low risk | Allocation concealed by individual sealed, opaque envelopes |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported; all randomised participants analysed from IPD provided (see footnote 2) |
| Selective reporting (reporting bias) | Low risk | Protocol provided. All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | None identified |
Reunanen 1996.
| Methods | Randomised, double‐blind, parallel‐group trial conducted in 56 centres in Europe and Australia 3 treatment arms: LTG (200 mg/day), LTG (100 mg/day) and CBZ |
|
| Participants | Adults and children over the age of 12 with newly diagnosed, currently untreated or recurrent epilepsy with 2 or more seizures in the previous 6 months and with at least 1 seizure in the last 3 months. Participants must not have taken antiepileptic medication in the previous 6 months. Number randomised: LTG (200 mg) = 115, LTG (100 mg) = 116, CBZ = 121 188 males (54%) 237 with focal seizures (68%) Not stated how many participants had received previous AED treatment Mean age (range): 32 (12 to 71) years |
|
| Interventions | Monotherapy with LTG or CBZ for 30 weeks 4‐week escalation phase leading to LTG = 100 mg/day, LTG = 200 mg/day, CBZ = 600 mg/day Range of follow‐up: 0 to 378 days |
|
| Outcomes | Proportion seizure‐free after the first 6 weeks of treatment Time to first seizure Time to treatment withdrawal Frequency of adverse events with at least 5% incidence in any treatment group |
|
| Notes | IPD provided by trial sponsor GlaxoSmithKline for time to treatment failure, time to first seizure and time to 6‐month remission Participants considered to complete the trial if they experienced a seizure after the first 6 weeks |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random sequence (information provided by drug manufacturer) |
| Allocation concealment (selection bias) | Low risk | Allocation concealed by individual sealed, opaque envelopes (information provided by drug manufacturer) |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported; all randomised participants analysed from IPD provided (see footnote 2) |
| Selective reporting (reporting bias) | Low risk | All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | None identified |
Rowan 2005.
| Methods | Randomised, double‐blind, parallel‐group trial conducted in 18 Veterans Affairs Medical Centres in the United States 3 treatment arms: LTG, CBZ and gabapentin (GBP) |
|
| Participants | Adults over the age of 60 with newly diagnosed seizures, untreated or treated with sub‐therapeutic AED levels, with at least 1 seizure in the previous 3 months Number randomised: LTG = 200, CBZ = 198 378 males (95%) 299 with focal seizures (75%) Not stated how many participants had received previous AED treatment Mean age: 72 years, range not stated |
|
| Interventions | Monotherapy with LTG or CBZ for 12 months 6‐week escalation phase leading to LTG = 150 mg/day, CBZ = 600 mg/day Range of follow‐up: not stated |
|
| Outcomes | Retention in the trial for 12 months Seizure freedom at 12 months Time to 1st, 2nd, 5th and 10th seizure (time to seizures) Drug toxicity (incidence of systemic and neurologic toxicities) Serum drug levels and compliance Seizure‐free retention rates |
|
| Notes | IPD requested from trial sponsor, the Department of Veterans Affairs, USA. At the time of review, IPD have not been received. Aggregate data extracted from graphs in the publication | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Block randomisation (varying sizes) performed by site via a computer‐generated list |
| Allocation concealment (selection bias) | Low risk | Telephone randomisation used and pharmacy dispensed a prescription of the allocated drug (part of a blinded drug kit) to participants |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blinding achieved with double dummy tablets; doses of both increased and decreased simultaneously |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not specifically stated |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported. Most of the randomised participants included in analysis; 3 excluded due to site closure (not related to treatment) |
| Selective reporting (reporting bias) | Low risk | No protocol available but case report forms of data collected provided by the sponsor. Seizure outcomes and adverse events well reported |
| Other bias | Low risk | None identified |
Saetre 2007.
| Methods | Randomised, double‐blind, parallel‐group trial conducted in 29 centres across Croatia, Finland, France, Finland and Norway. 2 treatment arms: LTG, CBZ | |
| Participants | Adults over the age of 65 with newly diagnosed seizures, with a history of at least 2 seizures and at least 1 seizure in the previous 6 months. Participants must not have taken antiepileptic medication for more than 2 weeks in the previous 6 months and never taken CBZ or LTG. Number randomised: LTG = 94, CBZ = 92 102 males (54%) Proportion with focal seizures not stated Not stated how many participants had received previous AED treatment Mean age: 74 (65 to 91) years |
|
| Interventions | Monotherapy with LTG or CBZ for 40 weeks 4‐week escalation phase leading to LTG = 100 mg/day, CBZ = 400 mg/day Range of follow‐up: not stated |
|
| Outcomes | Retention in the trial (time to treatment withdrawal for any cause) Seizure freedom after week 4 Seizure freedom after week 20 Time to first seizure Adverse event reports Tolerability according to the Liverpool Adverse Event profile (AEP) |
|
| Notes | IPD requested from trial sponsor Glaxo Smith Kline but data could not be located Aggregate summary data extracted from the publication |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised, no other information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blinding achieved with double dummy tablets, packaged together |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not specifically stated |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported; all participants who received trial treatment were included in an intention‐to‐treat analysis |
| Selective reporting (reporting bias) | Low risk | No protocol available but clinical trial summary provided by the sponsor. Seizure outcomes and adverse events well reported |
| Other bias | Low risk | None identified |
SANAD A 2007.
| Methods | Randomised, multicentre, open‐label, parallel‐group trial conducted in the UK 5 treatment arms: LTG, CBZ, GBP, topiramate (TPM) and oxcarbazepine (OXC) |
|
| Participants | Adults and children over the age of 4 years with newly diagnosed focal epilepsy, relapsed focal epilepsy or failed treatment with a previous drug not used in this trial Number randomised: LTG = 378, CBZ = 378 409 males (54%) 662 focal epilepsy (88%) 139 had received previous AED treatment (18%) Mean age (range): 38 (5 to 83) years |
|
| Interventions | Monotherapy for LTG or CBZ (no fixed trial duration) Titration doses and maintenance doses decided by treating clinician Range of follow‐up: 17 to 2420 days |
|
| Outcomes | Time to treatment failure Time to 1‐year (12‐month) remission Time to 2‐year remission Time to first seizure Health‐related quality of life via the NEWQOL (Newly Diagnosed Epilepsy Quality of Life Battery) Health economic assessment and cost‐effectiveness of the drugs (cost per QALY gained and cost per seizure avoided) Frequency of clinically important adverse events |
|
| Notes | IPD provided for time to treatment failure, time to first seizure, time to 6‐month, time to 12‐month and time to 24‐month remission (trial conducted at our site and sponsored by the Health Technology Assessment programme of the National Institute of Health Research) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer minimisation program stratified by centre, sex and treatment history |
| Allocation concealment (selection bias) | Low risk | Telephone randomisation to a central randomisation allocation service |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported; all randomised participants analysed from IPD provided (see footnote 2) |
| Selective reporting (reporting bias) | Low risk | Protocol provided. All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | None identified |
Steinhoff 2005.
| Methods | Randomised, open‐label, parallel‐group trial conducted in 24 centres across Germany 4 treatment arms: LTG (2 arms), CBZ and sodium valproate (SV) Participants with focal and generalised epilepsy randomised separately to LTG or CBZ and LTG or SV respectively |
|
| Participants | Adults and children over the age of 12 with newly diagnosed epilepsy; at least 1 seizure and electroencephalographic imaging suggesting epilepsy Number randomised not stated; number included in analysis: LTG = 88, CBZ = 88 106 males (64%) 100% focal seizures Not stated how many participants had received previous AED treatment Mean age: 47.5 years, range not stated |
|
| Interventions | Monotherapy with LTG or CBZ for 22 to 26 weeks 4‐week escalation phase leading to LTG = 100 to 200 mg/day, CBZ = 600 to 1200 mg/day in adults and 600 to 1000 mg/day in children aged 11 to 15 Range of follow‐up: not stated |
|
| Outcomes | Number of seizure‐free patients during trial weeks 17 to 24 "Leaving the study" (retention rates) Adverse event rates |
|
| Notes | IPD requested from trial sponsor GlaxoSmithKline but data could not be provided due to restrictions over the de‐identification of datasets from trials conducted in Germany Aggregate data extracted from graphs in the publication Data from participants with focal seizures only included as this is the randomised comparison of LTG and CBZ |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised, no other information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Number of participants randomised to each group not reported (254 randomised and 239 analysed in the 4 arms of the trial). Reasons for exclusion stated but not to which drug these participants were randomised. |
| Selective reporting (reporting bias) | Low risk | No protocol available but clinical trial summary provided by the sponsor. Seizure outcomes and adverse events well reported |
| Other bias | Low risk | None identified |
Werhahn 2015.
| Methods | Randomised, double‐blind, parallel‐group trial conducted in 47 centres across Germany, Austria and Switzerland 3 treatment arms: LTG, CBZ and levetiracetam (LEV) |
|
| Participants | Adults over the age of 60 with newly diagnosed focal seizures, with a history of at least 2 seizures and at least 1 seizure in the previous 6 months. Participants must not have taken antiepileptic medication for more than 4 weeks. Number randomised: LTG = 118, CBZ = 121 135 males (56%) 100% focal epilepsy Not stated how many participants had received previous AED treatment Mean age (range): 71 (60 to 89) years |
|
| Interventions | Monotherapy with LTG or CBZ for 58 weeks 6‐week escalation phase leading to LTG = 100 mg/day, CBZ = 400 mg/day Range of follow‐up: 0 to 1508 days |
|
| Outcomes | Retention rate at week 58 Time to discontinuation from randomisation Seizure freedom rates at week 30 and week 58 Time to first seizure from randomisation Time to first drug‐related adverse event Adverse events (by severity) |
|
| Notes | IPD provided by trial author for time to treatment failure, time to first seizure, time to 6‐month and time to 12‐month remission Trial was sponsored by UCB |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A randomisation list for each centre (random permuted blocks) was prepared by the Interdisciplinary Centre for Clinical Trials (IZKS), Mainz, Germany |
| Allocation concealment (selection bias) | Low risk | The pharmacy of the University Hospital Mainz encapsulated the trial drugs and labelled the blinded medication including the randomisation number |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants and trial investigator blinded by the use of matching capsules |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Trial investigator blinded; not stated if other outcome assessors were blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rates reported; all randomised participants analysed from IPD provided (see footnote 2) |
| Selective reporting (reporting bias) | Low risk | Protocol provided. All outcomes reported or calculated with IPD provided (see footnote 2) |
| Other bias | Low risk | None identified |
1Abbreviations
AED: antiepileptic drug
CBZ: carbamazepine
IPD: individual participant data
ITT: intention‐to‐treat
LTG: lamotrigine
QALY: quality‐adjusted life year
2For trials for which IPD were provided attrition and reporting bias are reduced as attrition rates and unpublished outcome data are requested (Brodie 1995 A; Brodie 1995 B; Brodie 1999Nieto‐Barrera 2001; Reunanen 1996).
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Baxter 1998 | Participants randomised to lamotrigine and physician's choice of carbamazepine or valproate. No fully randomised comparison between lamotrigine and carbamazepine |
| Carmant 2001 | Not monotherapy |
| Czapinski 1997 | Wrong drug comparison |
| Eun 2008 | Conference abstract for full publication Eun 2012 |
| Fakhoury 2000 | Withdrawn to monotherapy. Design excluded. |
| Gilliam 1998 | Wrong drug comparison |
| Jawad 1989 | Not monotherapy |
| Lee 2010 | Conference abstract for full publication Lee 2011 |
| Martinez 2000 | Not randomised |
| Motte 1997 | Wrong drug comparison |
| Ramsay 2003 | Abstract of full publication Rowan 2005 |
| Saetre 2006 | Conference abstract for full publication Saetre 2007 |
| Saetre 2009 | Subset of Saetre 2007 |
| Saetre 2010 | Subset of Saetre 2010 |
| Steiner 1999 | Wrong drug comparison |
| Steinhoff 2004 | Abstract of full publication Steinhoff 2005 |
| Stolarek 1994 | Wrong drug comparison |
| Zeng 2010 | Not randomised |
Differences between protocol and review
For the 2018 update, 'Time to withdrawal of allocated treatment' was re‐defined as 'Time to treatment failure' due to feedback received from the Cochrane Editorial Unit regarding potential confusion regarding 'withdrawal' as a positive or negative outcome of anti‐epileptic monotherapy.
Additional analyses of 'Time to treatment failure' (due to lack of efficacy and due to adverse events) following feedback on published anti‐epileptic drug monotherapy reviews that these sub‐outcomes would be useful for clinical practice.
The term 'partial' has been replaced by 'focal', in accordance with the most recent classification of epilepsies of the International League Against Epilepsy (Scheffer 2017).
For the 2016 update, in a posthoc change, 'Summary of findings' tables were added to the review
December 2014: the title was changed to specify that the review uses individual participant data.
Contributions of authors
SJ Nevitt assessed studies for inclusion in the review update, obtained individual participant data from trial investigators for the review update, assessed risk of bias in all included studies, performed analyses in Stata version 14, added survival plots and a 'Summary of findings' table, and updated the text of the review.
C Tudur Smith provided statistical supervision and was involved with data analysis in the original review.
AG Marson independently assessed studies for inclusion, obtained individual participant data from trial investigators, provided guidance with the clinical interpretation of results, assessed eligibility and methodological quality of individual studies, and co‐wrote the original review.
J Weston independently assessed risk of bias in all included studies.
Sources of support
Internal sources
University of Liverpool, UK.
External sources
National Institute for Health Research (NIHR), UK.
Declarations of interest
SJ Nevitt: none known.
J Weston: none known.
AG Marson was Chief Investigator of SANAD A 2007. A consortium of pharmaceutical companies (GSK, EISAI, UCB Pharma) funded the National Audit of Seizure Management in Hospitals (NASH) through grants paid to the University of Liverpool. Professor Tony Marson is part funded by National Institute for Health Research Collaboration for Leadership in Applied Health Research and Care North West Coast (NIHR CLAHRC NWC).
C Tudur Smith was involved in the statistical analysis of SANAD A 2007.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Brodie 1995 A {published and unpublished data}
- Brodie MJ, Richens A, Yuen AWC, for UK Lamotrigine/Carbamazepine Monotherapy Trial Group. Double‐blind comparison of lamotrigine and carbamazepine in newly diagnosed epilepsy. Lancet 1995;345(8948):476‐9. [DOI] [PubMed] [Google Scholar]
- Gillham R. Use of SEALS, a quality of life instrument, in evaluating lamotrigine and carbamazepine monotherapy. Epilepsia 1995;36 (Suppl 3):S186‐7. [Google Scholar]
- Gillham R, Kane K, Bryant‐Comstock L, Brodie MJ. A double‐blind comparison of lamotrigine and carbamazepine in newly diagnosed epilepsy with health‐related quality of life as an outcome measure. Seizure 2000;9(6):375‐9. [DOI] [PubMed] [Google Scholar]
Brodie 1995 B {published and unpublished data}
- Brodie MJ, Richens A, Yuen AWC, for UK Lamotrigine/Carbamazepine Monotherapy Trial Group. Double‐blind comparison of lamotrigine and carbamazepine in newly diagnosed epilepsy. Lancet 1995;345(8948):476‐9. [DOI] [PubMed] [Google Scholar]
- Gillham R. Use of SEALS, a quality of life instrument, in evaluating lamotrigine and carbamazepine monotherapy. Epilepsia 1995;36 (Suppl 3):S186‐7. [Google Scholar]
- Gillham R, Kane K, Bryant‐Comstock L, Brodie MJ. A double‐blind comparison of lamotrigine and carbamazepine in newly diagnosed epilepsy with health‐related quality of life as an outcome measure. Seizure 2000;9(6):375‐9. [DOI] [PubMed] [Google Scholar]
Brodie 1999 {published and unpublished data}
- Brodie MJ, Giorgi L, and the Lamotrigine Elderly Study Group. A multicenter double‐blind randomised comparison between lamotrigine and carbamazepine in elderly patients with newly diagnosed epilepsy. Epilepsia 1998;39 (Suppl 6):72, Abstract no: D.08. [DOI] [PubMed] [Google Scholar]
- Brodie MJ, Overstall PW, Giorgi L. Multicentre, double‐blind, randomised comparison between lamotrigine and carbamazepine in elderly patients with newly diagnosed epilepsy. The UK Lamotrigine Elderly Study Group. Epilepsy Research 1999;37(1):81‐7. [DOI] [PubMed] [Google Scholar]
- Brodie MJ, Read CL, Gillham RA, Sweet RM, Kane K. Lack of neuropsychological effects of lamotrigine compared to carbamazepine as monotherapy. Epilepsia 1999;40 (Suppl 2):94. [Google Scholar]
Eun 2012 {published data only}
- Eun SH, Eun BL, Lee JS, Hwang YS, Kim KJ, Lee YM, et al. Effects of lamotrigine on cognition and behavior compared to carbamazepine as monotherapy for children with partial epilepsy. Brain and Development 2012;34(10):818‐23. [DOI] [PubMed] [Google Scholar]
Gilad 2007 {published data only (unpublished sought but not used)}
- Gilad R, Sadeh M, Rapoport A, Dabby R, Boaz M, Lampl Y. Monotherapy of lamotrigine versus carbamazepine in patients with post stroke seizure. Clinical Neuropharmacology 2007;30(4):189‐95. [DOI] [PubMed] [Google Scholar]
Korean Lamotrigine Study Group 2008 {published data only}
- Korean Lamotrigine Study Group. An Open, Randomized, Multicenter Comparative Clinical Trial of Lamotrigine and Carbamazepine as Initial Monotherapy in Previously Untreated Epilepsies. Journal of Korean Epilepsy Society 2008;12(1):27‐34. [Google Scholar]
Lee 2011 {published and unpublished data}
- Lee S‐A, Kim MJ, Lee H‐W, Heo K, Shin D‐J, Song H‐K, et al. The effect of recurrent seizures on cognitive, behavioral, and quality‐of‐life outcomes after 12 months of monotherapy in adults with newly diagnosed or previously untreated partial epilepsy. Epilepsy & Behavior 2015;53:202‐8. [DOI] [PubMed] [Google Scholar]
- Lee SA, Lee HW, Heo K, Shin DJ, Song HK, Kim OJ, et al. Cognitive and behavioral effects of lamotrigine and carbamazepine monotherapy in patients with newly diagnosed or untreated partial epilepsy. Seizure 2011;20(1):49‐54. [DOI] [PubMed] [Google Scholar]
Nieto‐Barrera 2001 {published and unpublished data}
- Nieto‐Barrera M, Brozmanova M, Capovilla G, Christie W, Pedersen B, Kane K, et al. A comparison of monotherapy with lamotrigine or carbamazepine in patients with newly diagnosed partial epilepsy. Epilepsy Research 2001;46(2):145‐55. [DOI] [PubMed] [Google Scholar]
Reunanen 1996 {published and unpublished data}
- Reunanen M, Dam M, Yuen AW. A randomised open multicentre comparative trial of lamotrigine and carbamazepine as monotherapy in patients with newly diagnosed or recurrent epilepsy. Epilepsy Research 1996;23(2):149‐55. [DOI] [PubMed] [Google Scholar]
- Severi S, Bianchi A, Zolo P, Muscas GC. Lamotrigine monotherapy in patients with partial epilepsy. Epilepsia 1995;36 (Suppl 3):S113. [Google Scholar]
- Severi S, Cantelmi T, Bianchi A, Zolo P. Efficacy and safety of lamotrigine in patients with partial epilepsy: preliminary data. Bollettino Lega Italiana Contro L'Epilessia 1993;82‐3:139‐43. [Google Scholar]
- Severi S, Muscas GC, Bianchi A, Zolo P. Efficacy and safety of lamotrigine monotherapy in partial epilepsy. Bollettino Lega Italiana Contro L'Epilessia 1994;86‐7:149‐51. [Google Scholar]
- Yuen AWC, Chapman A. Interim report on an open multicentre lamotrigine (Lamictal) versus carbamazepine monotherapy trial in patients with epilepsy. Canadian Journal of Neurological Sciences 1993;20 (Suppl 4):S150. [Google Scholar]
- Yuen AWC, Chapman A. Interim report on an open multicentre lamotrigine (Lamictal) versus carbamazepine monotherapy trial in patients with epilepsy. Epilepsia 1993;34 (Suppl 2):159. [Google Scholar]
Rowan 2005 {published data only (unpublished sought but not used)}
- Rowan AJ, Ramsay RE, Collins JF, Pryor F, Boardman KD, Uthman BM, et al and The V. A. Cooperative Study Group. New onset geriatric epilepsy: a randomised study of gabapentin, lamotrigine and carbamazepine. Neurology 2005;64(11):1868‐73. [DOI] [PubMed] [Google Scholar]
Saetre 2007 {published data only (unpublished sought but not used)}
- Saetre E, Perucca E, Isojarvi J, Gjerstad L and LAM 40089 Study Group. An international multicenter randomized double‐blind controlled trial of lamotrigine and sustained‐release carbamazepine in the treatment of newly diagnosed epilepsy in the elderly. Epilepsia 2007;48(7):1292‐302. [DOI] [PubMed] [Google Scholar]
SANAD A 2007 {published and unpublished data}
- Marson AG, Al‐Kharusi AM, Alwaidh M, Appleton R, Baker GA, Chadwick DW, et al and SANAD Study group. The SANAD study of effectiveness of carbamazepine, gabapentin, lamotrigine, oxcarbazepine, or topiramate for treatment of partial epilepsy: an unblinded randomised controlled trial. Lancet 2007;369(9566):1000‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Steinhoff 2005 {published data only (unpublished sought but not used)}
- Steinhoff BJ, Ueberall MA, Siemes H, Kurlemann G, Schmitz B, Bergmann L and the LAM‐SAFE Study Group. The LAM‐SAFE Study: Lamotrigine versus carbamazepine or valproic acid in newly diagnosed focal and generalised epilepsies in adolescents and adults. Seizure 2005;14(8):597‐605. [DOI] [PubMed] [Google Scholar]
Werhahn 2015 {published and unpublished data}
- Werhahn KJ, Trinka E, Dobesberger J, Unterberger I, Baum P, Deckert‐Schmitz M, et al. A randomized, double‐blind comparison of antiepileptic drug treatment in the elderly with new‐onset focal epilepsy. Epilepsia 2015;56(3):450‐9. [DOI: 10.1111/epi.12926] [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Baxter 1998 {unpublished data only}
- Baxter L, Cheesbrough A. An open randomised comparison of Lamictal (Lamotrigine) with physicians preferred choice of either valproate or carbamazepine as monotherapy in patients over 12 years of age with newly diagnosed epilepsy. Clinical Summary Report May 1998.
Carmant 2001 {published data only}
- Carmant L, Curtis P, Moorat AM. Switching paediatric patients to monotherapy: efficacy and tolerability for lamotrigine compared with carbamazepine. Epilepsia 2001;42 (Suppl 7):170. [Google Scholar]
Czapinski 1997 {published data only}
- Czapinski P, Terczynski A, Czapinska E. Open randomised comparative study of vigabatrin (VGB) and lamotrigine (LTG) efficacy in monotherapy of patients with drug‐resistant epilepsy with partial complex seizures resistant to carbamazepine. Epilepsia. 1997; Vol. 38 (Suppl 3):35.
- Czapinski P, Terczynski A, Czapinska E. Open randomized comparative study of vigabatrin (VGB) and lamotrigine (LTG) efficacy in monotherapy of patients with drug‐resistant epilepsy with partial complex seizures resistant to carbamazepine (CBZ). Journal of the Neurological Sciences 1997;150 (Suppl):S96. [Google Scholar]
- Czapinski P, Terczynski A, Czapiska E. Open randomized comparative study of vigabatrin (VGB) and lamotrigine (LTG) efficacy in monotherapy of patients with drug‐resistant epilepsy with partial complex seizures resistant to carbamazepine (CBZ). Journal of Neurology. 1997; Vol. 244 (Suppl 3):S34.
Eun 2008 {unpublished data only}
- Eun S, Eun B, Lee J, Lee Y, Hwang Y, Kim K, et al. The effects of cognition and behavior of lamotrigine compared to carbamazepine as monotherapy for children with partial epilepsy. Epilepsia 2008;49(Suppl 7):87, Abstract No: 1.200. [DOI] [PubMed] [Google Scholar]
Fakhoury 2000 {published data only}
- Fakhoury T, Gazda S, Nanry KP, Hammer AE, Barrett PS. Comparison of monotherapy with lamotrigine versus carbamazepine in patients with uncontrolled epilepsy with a broad spectrum of seizure types. Epilepsia 2000;41 (Suppl 7):107. [Google Scholar]
Gilliam 1998 {published data only}
- Gilliam F, Vazquez B, Sackellares JC, Chang GY, Messenheimer J, Nyberg J, et al. An active‐control trial of lamotrigine monotherapy for partial seizures. Neurology 1998;51(4):1018‐25. [DOI] [PubMed] [Google Scholar]
Jawad 1989 {published data only}
- Jawad S, Richens A, Goodwin G, Yuen WC. Controlled trial of lamotrigine (Lamictal) for refractory partial seizures. Epilepsia 1989;30(3):356‐63. [DOI] [PubMed] [Google Scholar]
Lee 2010 {published data only}
- Lee S‐A, Lee H‐W, Heo K, Song H‐K, Kim O‐J, Lee S‐M, et al. Cognitive and behavioral effects of lamotrigine and carbamazepine monotherapy in patients with newly diagnosed or untreated partial epilepsy. Epilepsia 2010;51(Suppl 4):116, Abstract no: p393. [DOI] [PubMed] [Google Scholar]
Martinez 2000 {published data only}
- Martinez W, Kaminow L, Nanry KP, Hammer AE, Barrett PS. Evaluation of lamotrigine versus carbamazepine, phenytoin, or divalproex sodium as monotherapy for epilepsy patients who failed or could not tolerate previous antiepileptic drug therapy. Epilepsia 2000;41 (Suppl 7):100, Abstract no: 2.044. [Google Scholar]
- Nanry KP, Martinez W, Li H, Hammer AE, Barrett PS. Epilepsy patients switched from older antiepileptic drugs to lamotrigine monotherapy show improvement in quality of life. Epilepsia 2000;41 (Suppl 7):176, Abstract no: 3.013. [Google Scholar]
Motte 1997 {published data only}
- Motte J, Trevathan E, Arvidsson JFV, Barrera MN, Mullens EL, Manasco P. Lamotrigine for generalized seizures associated with the Lennox‐Gastaut syndrome. New England Journal of Medicine 1997;337(25):1807‐12. [DOI] [PubMed] [Google Scholar]
Ramsay 2003 {published data only}
- Ramsay RE, Rowan AJ, Pryor FM, Collins JF, DVA Coop Study Group 428. Treatment of seizures in the elderly: final analysis from DVA Cooperative Study. Epilepsia 2003;44 (Suppl 9):170. [Google Scholar]
Saetre 2006 {published data only}
- Saetre E, Perucca E, Isojärvi J, Gjerstad L. An international multicenter double‐blind double‐dummy randomised trial comparing lamotrigine and slow‐release carbamazepine for treating newly diagnosed epilepsy in the elderly. Epilepsia. 7th European Congress on Epileptology, Helsinki, Finland. 2‐6 July 2006, 2006; Vol. 47:1.
Saetre 2009 {published data only}
- Saetre E, Abdelnoor M, Amlie JP, Tossebro M, Perucca E, Tauboll E, et al. Cardiac function and antiepileptic drug treatment in the elderly: a comparison between lamotrigine and sustained‐release carbamazepine. Epilepsia 2009;50(8):1841‐9. [DOI] [PubMed] [Google Scholar]
Saetre 2010 {published data only}
- Saetre E, Abdelnoor M, Perucca E, Tauboll E, Isojarvi J, Gjerstad L. Antiepileptic drugs and quality of life in the elderly: results from a randomized double‐blind trial of carbamazepine and lamotrigine in patients with onset of epilepsy in old age. Epilepsy and Behaviour 2010;17(3):395‐401. [DOI] [PubMed] [Google Scholar]
Steiner 1999 {published data only}
- Steiner TJ, Dellaportas CI, Findley LJ, Gross M, Gibberd FB, Perkin GD, et al. Lamotrigine monotherapy in newly diagnosed untreated epilepsy; a double‐blind comparison with phenytoin. Epilepsia 1999;40(5):601‐7. [DOI] [PubMed] [Google Scholar]
Steinhoff 2004 {published data only}
- Steinhoff BJ, Ueberall MA, Siemes H, Bergmann L. The lam‐safe study: lamotrigine versus carbamazepine and valproic acid in newly diagnosed focal and generalized epilepsies in adolescents and adults. Epilepsia 2004;45 (Suppl 3):68, Abstract no: 055. [DOI] [PubMed] [Google Scholar]
Stolarek 1994 {published data only}
- Stolarek I, Blacklaw J, Forrest G, Brodie MJ. Vigabatrin and lamotrigine in refractory epilepsy. Journal of Neurology, Neurosurgery, and Psychiatry 1994;57(8):921‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zeng 2010 {published data only}
- Zeng K, Wang X, Xi Z, Yan Y. Adverse effects of carbamazepine, phenytoin, valproate and lamotrigine monotherapy in epileptic adult Chinese patients. Clinical Neurology & Neurosurgery 2010;112(4):291‐5. [DOI] [PubMed] [Google Scholar]
Additional references
Annegers 1999
- Annegers JF, Dubinsky S, Coan SP, Newmark ME, Roht L. The incidence of epilepsy and unprovoked seizures in multiethnic, urban health maintenance organizations. Epilepsia 1999;40(4):502‐6. [DOI] [PubMed] [Google Scholar]
Brodie 1995
- Brodie MJ, Richens A, Yuen AWC, for UK Lamotrigine/Carbamazepine Monotherapy Trial Group. Double‐blind comparison of lamotrigine and carbamazepine in newly diagnosed epilepsy. Lancet 1995;345(8948):476‐9. [DOI] [PubMed] [Google Scholar]
Brodie 1996
- Brodie MJ, Dichter MA. Antiepileptic drugs. New England Journal of Medicine 1996;334(3):168‐75. [DOI] [PubMed] [Google Scholar]
Bromley 2014
- Bromley R, Weston J, Adab N, Greenhalgh J, Sanniti A, McKay AJ, et al. Treatment for epilepsy in pregnancy: neurodevelopmental outcomes in the child. Cochrane Database of Systematic Reviews 2014, Issue 10. [DOI: 10.1002/14651858.CD010236.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Cockerell 1995
- Cockerell OC, Johnson AL, Sander JW, Hart YM, Shorvon SD. Remission of epilepsy: results from the National General Practice Study of Epilepsy. Lancet 1995;346(8968):140‐4. [DOI] [PubMed] [Google Scholar]
Excel 2010 [Computer program]
- Microsoft. Microsoft Excel. Redmond, Washington: Microsoft, 2010.
French 2007
- French JA, Kanner AM, Bautista J, Abou‐Khalil B, Browne T, Harden CL, et al. Appendix C: Efficacy and tolerability of the new antiepileptic drugs I: Treatment of new onset epilepsy: Report of the Therapeutics and Technology Assessment Subcommittee and Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. CONTINUUM Lifelong Learning in Neurology 2007;13:203‐11. [DOI] [PubMed] [Google Scholar]
Hauser 1993
- Hauser WA, Annegers JF, Kurland LT. Incidence of epilepsy and unprovoked seizures in Rochester, Minnesota 1935 ‐ 1984. Epilepsia 1993;34:453‐68. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327:557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2005
- Higgins JPT, Green S. Cochrane Handbook for Systematic Reviews of Interventions 4.2.5 [updated May 2005]. Cochrane Database of Systematic Reviews. Chichester, UK: John Wiley & Sons Ltd, 2005, issue 3.
Higgins 2011
- Higgins JPT, Altman DG, Sterne JAC (editors). Chapter 8: Assessing risk of bias in included studies. Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). Available from handbook.cochrane.org. The Cochrane Collaboration, 2011. [Google Scholar]
Hirtz 2007
- Hirtz D, Thurman DJ, Gwinn‐Hardy K, Mohamed M, Chaudhuri AR, Zalutsky R. How common are the "common" neurologic disorders?. Neurology 2007;68:326‐37. [DOI] [PubMed] [Google Scholar]
ILAE 1998
- ILAE Commission on Antiepileptic Drugs. Considerations on designing clinical trials to evaluate the place of new antiepileptic drugs in the treatment of newly diagnosed and chronic patients with epilepsy. Epilepsia 1998;39(7):799‐803. [DOI] [PubMed] [Google Scholar]
ILAE 2006
- Glauser T, Ben‐Menachem E, Bourgeois B, Cnaan A, Chadwick D, Guerreiro C, et al. ILAE treatment guidelines: evidence based analysis of antiepileptic drug efficacy and effectiveness as initial monotherapy for epileptic seizures and syndromes. Epilepsia 2006;47(7):1094‐120. [DOI] [PubMed] [Google Scholar]
Juul‐Jenson 1983
- Juul‐Jenson P, Foldspang A. Natural history of epileptic seizures. Epilepsia 1983;24:297‐312. [DOI] [PubMed] [Google Scholar]
Kirkham 2010
- Kirkham JJ, Dwan KM, Altman DG, Gamble C, Dodd S, Smyth R, et al. The impact of outcome reporting bias in randomised controlled trials on a cohort of systematic reviews. BMJ 2010;340:c365. [DOI] [PubMed] [Google Scholar]
Kwan 2000
- Kwan P, Brodie MJ. Early identification of refractory epilepsy. New England Journal of Medicine 2000;342:314‐9. [DOI] [PubMed] [Google Scholar]
Lees 1993
- Lees G, Leach MJ. Studies on the mechanism of action of the novel anti‐convulsant lamotrigine (Lamictal) using primary neuroglial cultures from rat cortex. Brain Research 1993;612:190‐9. [DOI] [PubMed] [Google Scholar]
Lefebvre 2011
- Lefebvre C, Manheimer E, Glanville J. Chapter 6: Searching for studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from http://handbook.cochrane.org/.
Liporace 1994
- Liporace JD, Sperling MR, Dichter MA. Absence seizures and carbamazepine in adults. Epilepsia 1994;35(5):1026‐8. [DOI] [PubMed] [Google Scholar]
MacDonald 1995
- MacDonald RL, Kelly KM. Antiepileptic drug mechanisms of action. Epilepsia 1995;36(Suppl 2):S2‐12. [DOI] [PubMed] [Google Scholar]
MacDonald 2000
- MacDonald BK, Johnson AL, Goodridge DM, Cockerell OC, Sander JWA, Shorvon SD. Factors predicting prognosis of epilepsy after presentation with seizures. Annals of Neurology 2000;48:833‐41. [PubMed] [Google Scholar]
Malafosse 1994
- Malafosse A, Genton P, Hirsch E, Marescaux C, Broglin D, Bernasconi R. Idiopathic Generalised Epilepsies: Clinical, Experimental and Genetic. Eastleigh: John Libbey and Company, 1994. [Google Scholar]
Marson 2000
- Marson AG, Williamson PR, Hutton JL, Clough HE, Chadwick DW. Carbamazepine versus valproate monotherapy for epilepsy. Cochrane Database of Systematic Reviews 2000, Issue 3. [DOI: 10.1002/14651858.CD001030] [DOI] [PMC free article] [PubMed] [Google Scholar]
Matlow 2012
- Matlow J, Koren G. Is carbamazepine safe to take during pregnancy?. Canadian Family Physician 2012;58:163‐4. [PMC free article] [PubMed] [Google Scholar]
Meador 2008
- Meador K, Reynolds M, Crean S, Fahrbach K, Probst C. Pregnancy outcomes in women with epilepsy: a systematic reviews and meta‐analysis of published pregnancy registries and cohorts. Epilepsy Research 2008;81:1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Morrow 2006
- Morrow J, Russel A, Guthrie E, Parsons L, Robertson I, Waddell R, et al. Malformation risks of antiepileptic drugs in pregnancy: a prospective study from the UK Epilepsy and Pregnancy Register. Journal of Neurology, Neurosurgery, and Neuropsychiatry 2006;77(2):193‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Murray 1994
- Murray CJL, Lopez AD. Global comparative assessments in the health sector. World Health Organization 1994.
Nevitt 2017a
- Nevitt SJ, Sudell M, Weston J, Tudur Smith C, Marson A. Antiepileptic drug monotherapy for epilepsy: a network meta‐analysis of individual participant data. Cochrane Database of Systematic Reviews 2017, Issue 12. [DOI: 10.1002/14651858.CD011412.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Nevitt 2017b
- Nevitt SJ, Marson AG, Weston J, Tudur‐Smith C. Carbamazepine versus phenytoin monotherapy for epilepsy: an individual participant data review. Cochrane Database of Systematic Reviews 2017, Issue 2. [DOI: 10.1002/14651858.CD001911.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ngugi 2010
- Ngugi AK, Bottomley C, Kleinschmidt I, Sander JW, Newton CR. Estimation of the burden of active and life‐time epilepsy: a meta‐analytic approach. Epilepsia 2010;51:883‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
NICE 2012
- National Institute for Health and Care Excellence. Clinical Guidance 137: The epilepsies: the diagnosis and management of the epilepsies in adults and children in primary and secondary care. London: National Institute for Health and Care Excellence 2012.
Nolan 2013a
- Nolan SJ, Sutton L, Marson A, Tudur Smith C. Consistency of outcome and statistical reporting of time‐to‐event data: the impact on Cochrane Reviews and meta‐analyses in epilepsy. 21st Cochrane Colloquium: Better Knowledge for Better Health, Quebec City. 2013:114‐5.
Nolan 2013b
- Nolan SJ, Muller M, Tudur Smith C, Marson AG. Oxcarbazepine versus phenytoin monotherapy for epilepsy. Cochrane Database of Systematic Reviews 2013, Issue 5. [DOI: 10.1002/14651858.CD003615.pub3] [DOI] [PubMed] [Google Scholar]
Nolan 2013c
- Nolan SJ, Tudur Smith C, Pulman J, Marson AG. Phenobarbitone versus phenytoin monotherapy for partial onset seizures and generalised onset tonic‐clonic seizures. Cochrane Database of Systematic Reviews 2013, Issue 1. [DOI: 10.1002/14651858.CD002217.pub2] [DOI] [PubMed] [Google Scholar]
Nolan 2016b
- Nolan SJ, Marson AG, Weston J, Tudur‐Smith C. Carbamazepine versus phenobarbitone monotherapy for epilepsy: an individual participant data review. Cochrane Database of Systematic Reviews 2016, Issue 12. [DOI: 10.1002/14651858.CD001904.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Nolan 2016c
- Nolan SJ, Sudell M, Tudur Smith C, Marson A. Topiramate versus carbamazepine monotherapy for epilepsy: an individual participant data review. Cochrane Database of Systematic Reviews 2016, Issue 12. [DOI: 10.1002/14651858.CD012065.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Nolan 2016d
- Nolan SJ, Marson AG, Pulman J, Tudur Smith C. Phenytoin versus valproate monotherapy for partial onset seizures and generalised onset tonic‐clonic seizures: an individual participant data review. Cochrane Database of Systematic Reviews 2016, Issue 4. [DOI: 10.1002/14651858.CD001769.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Olafsson 2005
- Olafsson E, Ludvigsson P, Gudmundsson G, Hesdorfer D, Kjartansson O, Hauser WA. Incidence of unprovoked seizures and epilepsy in Iceland and assessment of the epilepsy syndrome classification: a prospective study. Lancet Neurology 2005;4:627‐34. [DOI] [PubMed] [Google Scholar]
Parmar 1998
- Parmar MK, Torri V, Stewart L. Extracting summary statistics to perform meta‐analyses of the published literature for survival endpoints. Statistics in Medicine 1998;17(24):2815‐34. [DOI] [PubMed] [Google Scholar]
Ragsdale 1991
- Ragsdale DS, Scheuer T, Catterall WA. Frequency and voltage‐dependent inhibition of type IIA Na+ channels,expressed in a mammalian cell line, by local anesthetic, antiarrhythmic, and anticonvulsant drugs. Molecular Pharmacology 1991;40(5):756‐65. [PubMed] [Google Scholar]
Sander 1996
- Sander JW, Shorvon SD. Epidemiology of the epilepsies. Journal of Neurology, Neurosurgery, and Psychiatry 1996;61(5):433‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sander 2004
- Sander JW. The use of anti‐epileptic drugs ‐ principles and practice. Epilepsia 2004;45(6):28‐34. [DOI] [PubMed] [Google Scholar]
Scheffer 2017
- Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017;58(4):512‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Shakir 1980
- Shakir RA. Sodium valproate, phenytoin and carbamazepine as sole anticonvulsants. The Place of Sodium Valproate in the Treatment of Epilepsy. London: Academic Press Inc (London) Ltd and the Royal Society of Medicine, 1980:7‐16. [Google Scholar]
Shields 1983
- Shields WD, Saslow E. Myoclonic, atonic, and absence seizures following institution of carbamazepine therapy in children. Neurology 1983;33:1487‐9. [DOI] [PubMed] [Google Scholar]
Snead 1985
- Snead OC, Hosey LC. Exacerbation of seizures in children by carbamazepine. New England Journal of Medicine 1985;313:916‐21. [DOI] [PubMed] [Google Scholar]
Stata 2015 [Computer program]
- StataCorp. Stata Statistical Software: Release 14. CollegeStation, TX: StataCorp LP, 2015.
Tierney 2007
- Tierney JF, Stewart LA, Ghersi D, Burdett S, Sydes MR. Practical methods for incorporating summary time‐to‐event data into meta‐analysis. Trials 2007;8:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tudur Smith 2007
- Tudur Smith C, Marson AG, Chadwick DW, Williamson PR. Multiple treatment comparisons in epilepsy monotherapy trials. Trials 2007;5(8):34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Williamson 2000
- Williamson PR, Marson AG, Tudur C, Hutton JL, Chadwick DW. Individual patient data meta‐analysis of randomized anti‐epileptic drug monotherapy trials. Journal of Evaluation in Clinical Practice 2000;6(2):205‐14. [DOI] [PubMed] [Google Scholar]
Williamson 2002
- Williamson PR, Tudur Smith C, Hutton JL, Marson AG. Aggregate data meta‐analysis with time‐to‐event outcomes. Statistics in Medicine 2002;21(11):3337‐51. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Gamble 2006
- Gamble C, Williamson PR, Marson AG. Lamotrigine versus carbamazepine monotherapy for epilepsy. Cochrane Database of Systematic Reviews 2006, Issue 1. [DOI: 10.1002/14651858.CD001031.pub2] [DOI] [PubMed] [Google Scholar]
Nolan 2016a
- Nolan SJ, Tudur Smith C, Weston J, Marson AG. Lamotrigine versus carbamazepine monotherapy for epilepsy: an individual participant data review. Cochrane Database of Systematic Reviews 2016, Issue 11. [DOI: 10.1002/14651858.CD001031.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Preston 1998
- Preston CL, Marson AG, Williamson PR. Lamotrigine versus carbamazepine monotherapy for epilepsy. Cochrane Database of Systematic Reviews 1998, Issue 1. [DOI: 10.1002/14651858.CD001031] [DOI] [PubMed] [Google Scholar]