

Abstract
Motivation
Synergistic drug combinations are a promising approach to achieve a desirable therapeutic effect in complex diseases through the multi-target mechanism. However, in vivo screening of all possible multi-drug combinations remains cost-prohibitive. An effective and robust computational model to predict drug synergy in silico will greatly facilitate this process.
Results
We developed DIGREM (Drug-Induced Genomic Response models for identification of Effective Multi-drug combinations), an online tool kit that can effectively predict drug synergy. DIGREM integrates DIGRE, IUPUI_CCBB, gene set-based and correlation-based models for users to predict synergistic drug combinations with dose–response information and drug-treated gene expression profiles.
Availability and implementation
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Over the past decade, many promising anticancer drug candidates have failed in clinical trials for various reasons, mostly related to a lack of efficacy and drug resistance (Holohan et al., 2013; Huang et al., 2014b; Printz, 2015). Because of these challenges, therapeutic discovery has been gradually shifting from a traditional ‘one gene, one drug, one disease’ paradigm to looking for synergistic multi-drug combinations (Al-Lazikani et al., 2012), which can potentially overcome the limitations of monotherapy by modulating multiple cellular processes simultaneously (Jia et al., 2009). Traditional high throughput screening is impractical for searching for effective multi-drug therapies because of the huge volume of possible combinations. It is much more efficient to predict drug synergism using an in silico approach and only validate the most promising combinations.
Ordinary differential equation-based methods simulate the dynamic effect of drug combinations by modeling the responses or activity levels of components in a pathway, and they have been successfully applied to certain pathways to explain kinase inhibitor cooperativity, e.g. the eGFR pathway (Araujo et al., 2005). However, an explicit description of the kinetics of a complex cellular network is usually unfeasible, which limits its broader application. Recently, drug synergism prediction has improved through the use of new computational models that incorporate massive next generation sequencing and protein–protein interaction data (Huang et al., 2014a; Zhao et al., 2011).
In order to facilitate the development of computational models for in silico drug screening, in 2012, the DREAM Challenge initiative in collaboration with the National Cancer Institute launched a community-based challenge for predicting compound pair synergism. The transcriptomic and dose–response data of 14 individual drugs were generated to predict the synergistic effect of all 91 possible drug pair combinations (Bansal et al., 2014). Prediction performances were evaluated by the experimentally derived synergistic effects and quantified using a probabilistic concordance index (PC-index). The PC-index of the submissions ranged from 0.61 to 0.42 (random guess has a PC-index of 0.5). The two best-performing methods, the Drug-Induced Genomic Residual Effect (DIGRE) model developed by our team and the IUPUI_CCBB model, have significantly higher PC-indexes (0.613 and 0.605, respectively) and much higher consistency compared to the other methods (Bansal et al., 2014). In this study, we further improved our DIGRE model by implementing a context-specific gene network.
Recently, several other new strategies have been proposed and applied to the DREAM dataset. Zhao et al. (2014) presented a correlation-based method using transcriptomic profiles; Sun et al. (2015) developed a new model named RACS that employed semi-supervised learning techniques. Hsu et al. (2016) proposed a gene set-based scoring algorithm. Nonetheless, open source implementation is still largely unavailable, which restricts the translation of computational predictions to real experimental discovery. To meet this growing demand, we developed the user-friendly online tool DIGREM (Drug-Induced Genomic REsponse Models), which integrates four models, DIGRE, IUPUI_CCBB and the abovementioned gene-set based method and correlation-based method (Fig. 1), for researchers to predict drug synergistic effects using their own data (details in Supplementary Note 1 and on website).
Fig. 1.
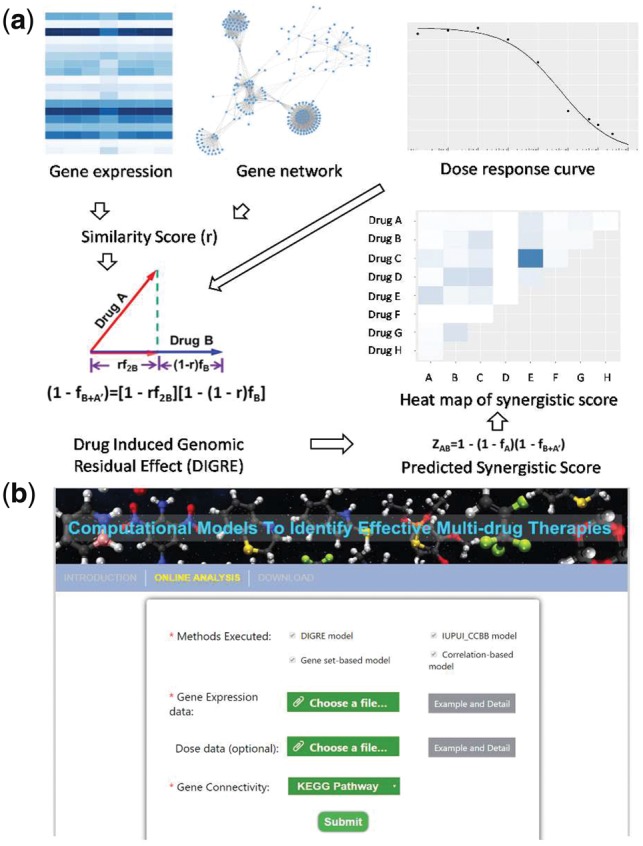
(a) DIGRE model workflow: (1) Drug-treated gene expression and gene–gene interaction network are used to generate similarity scores (r); (2) combine r and dose–response curve to compute the genomic residual effect; (3) then the synergistic score is calculated and visualized. (b) DIGREM website analysis page interface
2 Materials and methods
The DIGRE model is built on the assumption that when cells are treated by two drugs A and B sequentially, the transcriptomic change (genomic residual effect) induced by the first drug A will further contribute to the effect of the second drug B. The effect of drug combination can be simulated by the average effect of drug A followed by drug B, and vice versa. The implementation of DIGRE consists of three main steps: (i) Comparison of the differential gene expression induced by each individual drug in eight KEGG pathways related to cell growth (focused view, CGP), and neighboring genes in 32 cancer-relevant pathways (global view, GP), in order to generate the similarity score r. (ii) Estimation of the marginal cell survival rate reduction induced by drug B given the existence of drug A by integrating the similarity score r into the dose–response curves of both drug A and B. (iii) Estimation of the synergistic score by aggregating the effect of drug A and the marginal effect of drug B. For details about the DIGRE model, readers can refer to Supplementary Note 1 and Yang et al. (2015).
A more comprehensive and accurate gene network is a key component for the DIGRE model to better estimate r. DIGRE uses the KEGG pathway by default. User can also upload a self-constructed network if a context-specific gene network is available. We removed the dependency on dose–response data in the DIGRE model in order to make the model more generalizable to different scenarios. When dose–response data are not provided, the compound similarity scores are calculated and used as the synergistic scores to rank the drug pairs.
The IUPUI_CCBB model, gene-set-based model and correlation-based model are motivated by a similar idea as the DIGRE model: that a synergistic drug pair should perturb functionally similar genes, biological pathways or cellular processes. The IUPUI_CCBB model has a similar workflow as the DIGRE model except that it does not consider up-stream genes or the dose–response curve. A statistical test was applied to identify commonly disturbed genes in a core gene set by two drugs respectively, so replicates are required to ensure statistical power (Goswami et al., 2015). Rather than looking at individual genes or a defined gene set, the gene-set-based model calculates a gene-set enrichment score across all gene ontology and oncogenic signature gene-sets, and ranks drug pairs by an average percentage of commonly disturbed genes within the co-enriched gene sets (Hsu et al., 2016). The correlation-based method takes another approach. It calculates the correlation between the expression profiles of common differentially expressed genes induced by each drug in a drug pair.
3 Results
The original KEGG pathway DIGRE used in the challenge was first retrieved almost 4 years ago. It has now been updated with the most recent KEGG pathway information. As a result the accuracy of the DIGRE model in DREAM data has increased from 0.613 to 0.618. In addition, we have introduced a lymphoma-specific gene network constructed by a partial correlation based method (Peng et al., 2009), and the PC-index has further increased to 0.627 (Supplementary Table S1). When dose–response data are not provided, DIGRE model still maintains comparable predictive power, with a PC-index of 0.612. A more complete comparison of the different methods on the DREAM dataset can be found in Supplementary Figures S1–S3 and Supplementary Table S2.
4 Implementation
The DIGREM combo kit is implemented in R. By default, it will automatically run four models and report the predicted synergistic scores for each individual method. The drug pairs are ranked based on the synergistic score predicted by the DIGRE model, with the most synergistic pairs on the top. Replicates are required for IUPUI_CCBB, the gene-set-based and correlation-based models to run statistical tests and normalization (if replicates are not found, these methods will be skipped). A heat map and a bar plot will be displayed in the website to visualize the predicted drug synergy results by DIGRE model once the analysis is completed. The R package (DIGREsyn, https://github.com/Minzhe/DIGREsyn) and command line tool are also available on the website for analyzing large datasets on a local machine.
5 Conclusion
We introduced a user-friendly web-server, DIGREM, which is the first publicly accessible free software to prioritize synergistic drug pairs using transcriptomic and dose–response data. It will facilitate the discovery of synergistic drug combinations in large compound screening and lead to a better understanding of drug interaction.
Funding
This work was partially supported by the National Institutes of Health [5R01CA152301, 1R01GM115473, and 1R01CA172211], the Cancer Prevention Research Institute of Texas [RP120732-C2] and a subcontract from the National Cancer Institute [S15-82].
Conflict of Interest: none declared.
Supplementary Material
References
- Al-Lazikani B. et al. (2012) Combinatorial drug therapy for cancer in the post-genomic era. Nat. Biotechnol., 30, 679–692. [DOI] [PubMed] [Google Scholar]
- Araujo R.P. et al. (2005) A mathematical model of combination therapy using the EGFR signaling network. Biosystems, 80, 57–69. [DOI] [PubMed] [Google Scholar]
- Bansal M. et al. (2014) A community computational challenge to predict the activity of pairs of compounds. Nat. Biotechnol., 32, 1213–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami C.P. et al. (2015) A new drug combinatory effect prediction algorithm on the cancer cell based on gene expression and dose–response curve. CPT Pharmacometrics Syst. Pharmacol., 4, 80–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holohan C. et al. (2013) Cancer drug resistance: an evolving paradigm. Nat. Rev. Drug Discov., 13, 714–726. [DOI] [PubMed] [Google Scholar]
- Huang L. et al. (2014a) DrugComboRanker: drug combination discovery based on target network analysis. Bioinformatics, 30, i228–i236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M. et al. (2014b) Molecularly targeted cancer therapy: some lessons from the past decade. Trends Pharmacol. Sci., 35, 41–50. [DOI] [PubMed] [Google Scholar]
- Hsu Y. et al. (2016) A simple gene set-based method accurately predicts the synergy of drug pairs. BMC Syst. Biol., 10, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia J. et al. (2009) Mechanisms of drug combinations: interaction and network perspectives. Nat. Rev. Drug Discov., 8, 111–128. [DOI] [PubMed] [Google Scholar]
- Peng J. et al. (2009) Partial correlation estimation by joint sparse regression models. J. Am. Stat. Assoc., 104, 735–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Printz C. (2015) Failure rate: why many cancer drugs don't receive FDA approval, and what can be done about it. Cancer, 121, 1529–1530. [DOI] [PubMed] [Google Scholar]
- Sun Y. et al. (2015) Combining genomic and network characteristics for extended capability in predicting synergistic drugs for cancer. Nat. Commun., 6, 8481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J. et al. (2015) DIGRE: drug induced genomic residual effect model for successful prediction of multidrug effects. CPT Pharmacometrics Syst. Pharmacol., 4, 91–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J. et al. (2014) Predicting cooperative drug effects through the quantitative cellular profiling of response to individual drugs. CPT Pharmacometrics Syst. Pharmacol., 3, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X.M. et al. (2011) Prediction of drug combinations by integrating molecular and pharmacological data. PLoS Comput. Biol., 7, e1002323. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.