

Introductory Paragraph
Multiple sclerosis (MS) is a disease of the central nervous system, treated with disease-modifying therapies, including the biologic, interferon-beta (IFN-β). Up to 60% of IFN-β exposed MS patients develop abnormal biochemical liver test results1,2 and one in 50 experience drug-induced liver injury (DILI).3 Since genomic variation contributes to other forms of DILI,4,5 we aimed to identify biomarkers of IFN-β-induced liver injury using a two-stage genome-wide association study (GWAS). The rs2205986 variant, previously linked to differential expression of interferon regulatory factor (IRF)-6, surpassed genome-wide significance in the combined two-stage analysis (P=2.3×10−8, odds ratio=8.3, 95% CI=3.6–19.2). Analysis of an independent cohort of IFN-β-treated MS patients identified via electronic medical records (EMRs) revealed rs2205986 was also associated with increased peak levels of aspartate aminotransferase (AST, P=7.6×10−5) and alkaline phosphatase (ALP, P=4.9×10−4). We show that these findings may be applicable to predicting IFN-β-induced liver injury, offering insight into its safer use.
Main Letter
While the therapeutic options for MS are expanding, the IFN-βs remain the most widely used disease-modifying therapy. Liver injury secondary to IFN-β has potentially serious sequelae, yet, there are no means of predicting this adverse reaction. In the US, DILI is the leading cause of acute liver failure6 and the most common reason for drug withdrawal from the market.7 GWAS have successfully discovered variants of large effect sizes associated with DILI due to non-biologics using relatively small, but rigorously phenotyped cohorts.4,8 However, studies identifying variants associated with DILI from biologics, including IFN-β, have not been reported.
Patients who exhibited normal baseline biochemical liver test results prior to IFN-β exposure were included in this study (Online Methods). Cases met a published DILI definition9 and controls were exposed to IFN-β for ≥2 years, with all biochemical liver test results within the normal range. We recruited 170 patients from Canadian-based MS clinics for stage one analyses employing genome-wide genotyping. Upon exclusion of samples failing quality control (QC) or those of non-European genetic ancestry (Supplementary Fig. 1), 151 samples (38 cases, 113 controls) were subject to whole genome and HLA-allele imputation. Variants reaching P<1.0×10−6 in stage one were tested in stage two. The clinical and demographic characteristics of stage one participants were similar between cases and controls, apart from the controls being more likely to have a relapsing-remitting MS course (P=0.035, Supplementary Table 1).
Genome-wide analysis identified three regions associated with IFN-β-induced liver injury, after adjusting for disease course (Table 1, Supplementary Tables 2 and 3, Supplementary Fig. 2a). The strongest association was located on chromosome 1q32.2 (rs2205986 [G>A], P=1.9×10−7, OR=8.5, 95% CI 3.5–20.4, Table 1). This variant also surpassed our screening threshold unadjusted for covariates (P=3.1×10−7) and when adjusted for the first five principal components and MS disease course (P=2.6×10−7). HLA-region analyses did not identify any association with IFN-β-induced liver injury, including HLA-variants previously associated with DILI caused by other medications (Supplementary Table 4).
Table 1:
Pharmacogenomic association analyses for rs2205986 and IFN-β-induced liver injury in multiple sclerosis patients enrolled from Canada (stage one) and USA/Sweden (stage two)
| Genetic Variant Information | Population | Logistic Regression (Additive)a | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Variant | Annotation | Study stage | N cases | N controls | MAF cases | MAF controls | P-value | Odds Ratio | 95%CI |
|
rs2205986 1:210,116,112b |
IRF6 eQTL / SYT14 intronic |
Stage one | 38 | 113 | 0.24 | 0.04 | 1.9 × 10−7 | 8.5 | 3.5–20.4 |
| Stage two | 18 | 13 | 0.17 | 0.00 | 4.3 × 10−3 | - | - | ||
| Combined (Stage one + two) | 56 | 126 | 0.21 | 0.03 | 2.3 × 10−8 | 8.3 | 3.6–19.2 | ||
| BioVU ‘mild DILI’c | 9 | 78 | 0.22 | 0.05 | 0.048 | 4.3 | 1.02–17.8 | ||
Logistic regression was performed in stage one (adjusted for MS disease course) and two (adjusted for age)
GRCh37 assembly position (chromosome:base pair), HGVS notation: NC_000001.10:g.210116112G>A; and
defined as twice the upper limit of normal for either alanine or aspartate aminotransferase.
CI: confidence interval, eQTL: Expression quantitative trait locus, MAF: minor allele frequency, N: number.
The prioritized genomic regions were subsequently tested in stage two, using a separate cohort of MS subjects from the USA and Sweden (18 cases, 13 controls of European ancestry). Cases were significantly older (P=0.026), but were similar to controls across other characteristics (Supplementary Table 1). Of the variants tested in stage two, only the 1q32.2 region (rs2205986) was associated with IFN-β-induced liver injury (P=0.004, Table 1, Supplementary Table 3). This variant was only observed in cases (Supplementary Table 2) and the overall effect (combined stage one and two) surpassed genome-wide significance (P=2.3×10−8, OR=8.3).
Next, we evaluated array-genotyped MS patients receiving IFN-β identified via EMRs in the Vanderbilt University Medical Center repository (BioVU) to assess the influence of the rs2205986 DILI-risk variant on peak biochemical liver test results during IFN-β treatment. Of the four liver test results analyzed, rs2205986 was significantly associated with increased AST (P=7.6×10−5) and ALP (P=4.9×10−4) levels (Table 2, Supplementary Fig. 3). Each rs2205986 G-allele contributed, on average, to an increase of 52.3 units/L (ALP) and 29.4 units/L (AST).
Table 2:
Association analysis of rs2205986 genotype on peak biochemical liver tests results during IFN-β treatment in multiple sclerosis patients from the Vanderbilt University Medical Center (VUMC) repository, BioVU, USA
| Biochemical Liver Test | Measurement | aBeta coefficient | P-valuea | First recorded value (mean) | Highest recorded value (mean) | Mean highest value by genotype (GG / GA / AA) |
|---|---|---|---|---|---|---|
| ALP | Units/L | 52.28 | 4.9 × 10−4 | 79 | 101 | 255 / 140 / 94 |
| Upper limit of normal | 0.35 | 5.0 × 10−4 | NA | NA | NA | |
| AST | Units/L | 29.39 | 7.6 × 10−5 | 29 | 42 | 147 / 59 / 38 |
| Upper limit of normal | 0.73 | 7.6 × 10−5 | NA | NA | NA | |
| ALT | Units/L | 5.29 | 0.54 | 34 | 47 | 112 / 43 / 47 |
| Upper limit of normal | 0.10 | 0.54 | NA | NA | NA | |
| TBIL | mg/dL | 0.00 | 0.98 | 0.49 | 0.77 | 1.00 / 0.59 / 0.77 |
| Upper limit of normal | 0.00 | 0.98 | NA | NA | NA |
Linear regressions using additive genetic model for highest values, adjusted for age at biochemical liver test date, sex, and the first two principal components in n=87 MS patients exposed to interferon-beta. ALP, alkaline phosphatase; ALT; alanine transaminase; AST, aspartate aminotransferase; TBIL, total bilirubin, NA: Not applicable.
Of note, the one BioVU patient homozygous for the rs2205986 G-risk-allele presented with elevated peak alanine and aspartate aminotransferase (ALT, AST) and alkaline phosphatase (ALP) levels (Table 2), potentially indicating a marked increase in risk associated with this rare genotype. We performed a complementary analysis excluding this sample, which revealed a significant association with AST (P=0.017) but no association for the remaining liver test results. Further, since IFN-β-induced liver injury typically presents with a hepatocellular pattern, we also performed an exploratory case-control analysis of ‘mild DILI’ (ALT or AST >2x upper limit of normal) in the BioVU cohort. These analyses also detected evidence for an association with this phenotype (P=0.048; OR=4.3, 95% CI 1.02–17.8; Table 1), indicating the biomarker may also be useful in identifying milder forms of liver injury.
We also examined the frequency of the top stage one regions in a cohort of 1,319 disease-matched population controls that were unscreened for biochemical liver test abnormalities. This confirmed a higher frequency of rs2205986 in cases [minor allele frequency (MAF) 21.4%] compared to MS population controls (MAF 9.4%). Although these analyses were no longer genome-wide significant (P=3.0×10−4), the use of population controls are best suited to adverse drug reactions with prevalence rates of <1%.10 In contrast, 2% of IFN-β-treated MS patients develop DILI and up to 60% exhibit abnormal biochemical liver test results in a population-based cohort study.3 Since the BioVU biochemical liver test analyses indicated rs2205986 is associated with elevated peak liver test results, the depletion in MAF observed in drug-exposed/screened controls may have been caused by the removal of carriers with abnormal biochemical liver test results during the stringent selection process of controls.
Inspection of the 1q32.2 region revealed that only rs2205986 surpassed P<1.0×10−6, while 30 variants within a 266-kb linkage disequilibrium (LD) block displayed P<5.0×10−5 (Fig. 1). Upon adjusting for rs2205986, no variants were independently associated with DILI (P>0.05, Supplementary Table 5). Rs2205986 is an intronic synaptotagmin-14 (SYT14) variant, however this marker is approximately 4.5-kb from the nearest canonical exon and is not predicted to alter SYT14 transcription, splicing or expression. Notably, in silico annotation using the Genotype-Tissue Expression Project data revealed rs2205986 is an expression quantitative trait locus (eQTL) for the interferon regulatory factor 6 gene (IRF6, multi-tissueP=5.89×10−17),11 located 137-kb upstream from rs2205986 (Fig. 1).
Figure 1. Regional association plot of chromosome 1q32.2 demonstrating a pharmacogenomic association between rs2205986 and IFN-β-induced liver injury.
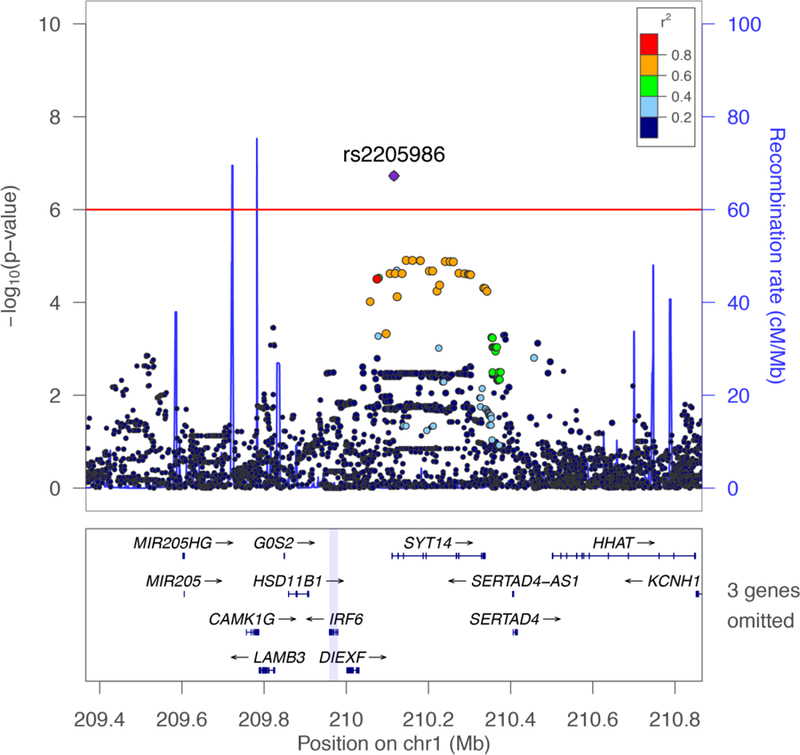
The interferon regulatory factor 6 (IRF6) gene is located ~132-kb upstream of synaptotagmin 14 (SYT14). Association results [primary y-axis, -log10(P-value)] are shown for genetic variants along with recombination rates (secondary y-axis, cM/Mb) for a 1.5 Mb region on chromosome 1. Each circle represents the -log10(P-value) from the logistic regression analysis, adjusted for MS disease course in the stage one case-control cohort (n=151). Genetic variants are coloured according to their pairwise correlation (r2) with rs2205986 (purple circle) using linkage disequilibrium data from the 1000 Genomes Project (European population). Three genes (MIR4260, TRAF3IP3, C1orf74) were omitted from the figure due to space requirements.
Interferon regulatory factors (IRFs) are a family of IFN transcription factors, which synchronize the type I IFN pathway.12 Many of the nine known IRFs are associated with promoting liver damage in another model of liver cell death: hepatic ischemia/reperfusion injury.13 Further, recent gene expression studies have identified IRF6 as an IFN-β drug response biomarker,14 while IRF3 has been implicated in tolvaptan-induced liver injury.15 IRF6 promotes apoptosis following brain injury,16 and previous case reports of MS patients experiencing IFN-β-induced liver injury have shown hepatocyte apoptosis,17 suggesting rs2205986-induced alterations in IRF6 expression may promote apoptosis in the presence of IFN-β. The rs2205986-IRF6 eQTL was not significant in liver tissue; however, the effect of this variant on gene expression may be amplified in the presence of IFN-β or the variant may exert its influence via the blood (Supplementary Fig. 4). Future studies should therefore investigate the influence of this eQTL on IFN-β-induced hepatic expression.
The HLA-region has been shown to confer risk to DILI caused by certain drugs;4,5 however, no HLA-alleles or variants outside the HLA-region previously been associated with DILI (e.g. glutathione S-transferase and ATP-binding cassette transporter genes)18,19 reached the screening threshold. As previous pharmacogenomic studies of DILI investigated non-biologics, these results might be expected given the differences in the metabolism of biologics compared to small molecule drugs,19 and the limited evidence surrounding IFN-β metabolism.20
We also incorporated rs2205986 into a predictive model for DILI: including rs2205986 significantly improved the prediction of liver injury, over clinical factors alone (P=0.0039, Supplementary Fig. 5). Rs2205986 had a specificity of 93.7% (95% CI=87.9–97.2) and sensitivity of 41.1% (95% CI=28.1–55.0). Notably, the only patient requiring a liver transplant was an rs2205986 carrier. The negative and positive predictive values of rs2205986 were 98.7% and 12.2% respectively and the number of individuals needed to screen for rs2205986 to prevent one case was 117. These metrics are similar to those reported for the testing of HLA-B*1502 and carbamazepine-induced Stevens-Johnson syndrome.21 Pharmacogenomic testing for HLA-B*1502 prior to carbamazepine use is recommended by the FDA for certain ancestries, highlighting the importance of the current findings. Future studies could consider incorporating additional variables; such as the absolute baseline liver biochemistry values into predictive models to further improve prediction.
To our knowledge, this is the first GWAS to investigate an adverse reaction due to a MS therapy and specifically DILI due to a biologic. These analyses were restricted to European genetic ancestry patients to minimize population stratification. However, since MS is known to be most prevalent in those with Northern European ancestry,22 these results are expected to be applicable to the majority of people with MS. Further, objectively defining DILI9 and applying stringent inclusion criteria for the controls in this study enhanced the statistical power.10 Nonetheless, sample size remains a limitation, and as a consequence, we were only able to identify one pharmacogenomic predictor of IFN-β-induced liver injury. Future studies of larger cohorts might improve the ability to detect additional variants of smaller effect.
In conclusion, we have identified an association between an IRF-related eQTL and IFN-β-induced liver injury. These findings have important implications for the development of strategies to reduce the occurrence of IFN-β-induced liver injury in MS patients. Pharmacogenomic testing for this variant prior to IFN-β therapy, rather than only monitoring liver enzymes during treatment, may prevent DILI in at-risk patients. Prevention of DILI in rs2205986-carriers could then be achieved by either considering alternative therapies or increased monitoring of liver injury. In addition, our findings set the stage for functional assessments of IRF6, rs2205986 and IFN-β treatment, to provide a mechanistic understanding of this pharmacogenomic association that can be specifically targeted to prevent DILI.
Online Methods
Study participants.
Subjects were eligible for inclusion in our study if they had either relapsing-remitting or secondary-progressive definite MS (based on Poser or McDonald criteria),23,24 documented exposure to an IFN-β product (IFN-β−1b subcutaneous, SC [250 mcg every other day], IFN-β−1a SC [22 mcg or 44 mcg 3x weekly] or IFN-β−1a intramuscular, IM [30 mcg weekly]) and had a normal baseline liver enzyme test. At least one alanine aminotransferase (ALT) test result was required for baseline assessment. Cases met the following definition, which includes at least one of the following criteria9: (1) ALT or aspartate aminotransferase (AST) ≥5x upper limit of normal (ULN); or (2) ALT ≥3x ULN with simultaneous elevation of bilirubin >2x ULN; or (3) alkaline phosphatase (ALP) >2x ULN. Controls were exposed to IFN-β for at least two years with all biochemical liver test results within normal limits based on the normal ranges for the site-specific laboratory.
As the first 15 months of IFN-β exposure is considered the greatest risk period for developing de novo ALT elevations,2 and all cases developed DILI within 700 days of beginning IFN-β,3 two years of IFN-β exposure was required to determine if a participant was truly a control. As expected due to the study design, stage one controls were exposed to IFN-β for a significantly longer duration (median: 82 months, IQR: 51–110.5 months) than cases (median: 4 months; IQR: 2.5–27.5 months, P=6.0 × 10−15). Moreover, all biochemical liver test results for controls had to be within the normal reporting range, which further limited the size of our control sample given that 30–60% of MS patients exposed to IFN-β will experience any de novo liver enzyme elevation.1,2 Although limiting our sample size for IFN-β-exposed controls, these stringent inclusion criteria increased the confidence in the clinical phenotype enhancing our power to detect genetic variants of clinical relevance.
Participants included in stage two were recruited from three sites: a USA-based clinic (Partners HealthCare MS Clinic, Boston, USA) and two national adverse drug reaction surveillance networks, situated in the USA (the Drug-Induced Liver Injury Network25,26) and Sweden (SWEDEGENE, http://www.swedegene.se/). Inclusion criteria for patients recruited from the Drug-Induced Liver Injury Network matched that from other centers except that two consecutive elevations of the same magnitude described above of ALT, AST or ALP were required.26 The relevant research ethics board of each participating institution approved the study and all participants provided written informed consent. This study complies with all relevant ethical regulations.
Clinical characterization.
Each patient’s medical record was reviewed prior to genotyping to capture demographic and clinical information and a comprehensive characterization of the adverse drug reaction, including drug exposure information and biochemical liver test results. The following information was collected from medical charts for all patients: demographics (sex, date of birth, self-reported ancestry), body mass index (BMI), MS disease characteristics (MS disease course at IFN-β initiation (relapsing-remitting (RR) or secondary-progressive (SP)), medications (IFN-β product (dose, route of administration, start and stop dates), concurrent medication usage (generic name, route of administration, dose, frequency, start and stop dates, where possible), and biochemical live test results [date of test, test result (value, if abnormal), reporting laboratory ULN, if abnormal].
Genotyping, quality control (QC) and imputation.
Genome-wide genotyping was performed for the stage one participants (n=170) using the Illumina MEGA array (1,705,969 genetic variants) followed by stringent sample and variant QC methods. The stage two cohort was either genotyped in the same manner (n=10) or as part of a genome-wide analysis of MS disease risk as described previously41 (n=24). For the participants genotyped as part of the MS disease risk study, access to imputed genotype data allowed for the extraction of the variants of interest where necessary.
The following combination of thresholds for QC metrics were implemented using either QCTOOL (version 2, http://www.well.ox.ac.uk/~gav/qctool_v2/), GTOOL (version 0.7.5, http://www.well.ox.ac.uk/~cfreeman/software/gwas/gtool.html) and PLINK (version 1.90).27 Genetic variants with a low call rate (<95%), a minor allele frequency <1% in both cases and controls and those deviating from Hardy-Weinberg equilibrium genotype distributions (P<1.0 × 10−6 in controls) were excluded. No samples were related (using an identity by descent estimation metric ≤ 0.15). Three patient samples in the stage one cohort were excluded due to low sample call rate. Non-autosomal markers were excluded from analyses.
Genotype Harmonizer (1.4.15)28 was used to ensure that variants were on the correct strand to facilitate imputation. Phasing was performed with SHAPEIT (version 2), followed by whole genome imputation using IMPUTE2 (version 2.3.2)28 and Phase 3 1000 Genomes Project reference panel.29 Markers with imputation info metrics ≥0.5 were included in the subsequent analyses. Imputation of classical HLA-alleles and HLA-region variants was performed with SNP2HLA (version 1.0.2),30 using the stage one cohort genotype data and the Type 1 Diabetes Genetics Consortium (T1DGC) reference panel. HLA-alleles and related variants with imputation scores R2≤0.5 and call rate<0.85 were excluded from the subsequent analyses.
Genotyping calls of genome-wide significant variants (rs2205986) were validated in the stage one cohort patients using TaqMan® genotyping assays (ThermoFisher Scientific, Waltham, USA and ThermoFisher Taqman® Genotyper Software) and exhibited 100% concordance with array genotype (Supplementary Fig. 6).
GWAS stage one and two statistical analyses.
Categorical variables [sex, MS disease course (relapsing-remitting or secondary-progressive), IFN-β product, liver injury pattern (hepatocellular, cholestatic or mixed), and concomitant hepatotoxic medication use] were summarized by frequency (percent), with age at IFN-β initiation and BMI (continuous variables) summarized using the median (interquartile range) or mean (standard deviation). Clinical and demographic factors were compared between cases and controls using the appropriate parametric (Pearson’s chi-square test, Student’s t-test) or non-parametric tests (Fisher’s exact test, Mann-Whitney U test), and associations with P<0.05 were considered significant (All P-values were 2-tailed).
Genetic ancestry was confirmed using principal components analysis (EIGENSTRAT method),35 which was subsequently compared to self-reported ethnicity, with patients excluded based on non-European ancestry. To minimize the potential confounding effects of population stratification, a total of 16 (stage one) and 3 (stage two) samples were removed from the analyses owing to non-European ancestry (Supplementary Fig. 1). The first 10 principal components were re-calculated within the individuals who were of European genetic ancestry in stage one (n=151), with no significant difference between cases and controls (Student’s t-test, P>0.1). Additionally, a genomic inflation factor of 1.06 indicates the stage one participants (n=151) utilized for genome-wide discovery, was not notably influenced by population stratification (Supplementary Fig. 2b).
The association for each genomic marker passing QC assessment with case/control status was tested using logistic regression in an additive model (adjusted for relevant clinical and demographic factors), with findings expressed as odds ratios with 95% confidence intervals. A screening threshold (P<1.0 × 10−6) was applied to the stage one cohort to prioritize variants for subsequent stage two analyses, where P<0.05 was considered significant. Associations reaching the standard genome-wide significance threshold (P<5.0 × 10−8) across the combined cohort (i.e. stage one and two) were considered to be statistically significant. For the HLA-region, an HLA-wide significance threshold of P<2.3 × 10−4 was set to account for Bonferroni correction for the 219 HLA-alleles present in the cohort. P<0.05 was considered significant for the replication of previously reported associations with HLA-alleles and DILI.
Genome-wide association analyses were performed with SNPTEST (version 2, https://mathgen.stats.ox.ac.uk/genetics_software/snptest/snptest.html), and other statistical analyses were performed using Golden Helix SVS (version 8.4, Bozeman, USA), IBM SPSS (version 22.0, Mississauga, Canada), or R for Statistical Computing (version 3.2.3). Plots (Manhattan plot, regional association plot and the ROC curve) were generated using LocusZoom,39 R for Statistical Computing or Golden Helix SVS.
BioVU electronic medical record analyses.
BioVU population. 279 MS patient samples that were previously genotyped at Vanderbilt University Medical Center (VUMC), USA were accessed. The samples are part of BioVU, a de-identified collection of DNA samples extracted from discarded blood and linked to de-identified electronic medical records (EMRs).31 All samples were identified as being from an individual with MS by previously published algorithms.32 The EMRs were evaluated manually to identify dates of IFN-β treatment; biochemical liver test results (ALT, AST, ALP and total bilirubin) were extracted from structured fields of the EMRs during IFN-β treatment. A total of 87 unique MS patients were exposed to IFN-β, had a sample and available biochemical liver test results during IFN-β treatment and were used in subsequent analyses. The highest value for each biochemical liver test result was identified independent of the other biochemical liver test results. For patients with more than one IFN-β treatment period, only the treatment period with the highest overall value was analyzed. Additionally, IFN-β-induced liver injury often presents with a hepatocellular pattern3, an exploratory analysis of ‘mild DILI’ was performed where cases were defined as either ALT or AST levels >2x ULN.
BioVU genotyping, quality control and statistical analyses.
Samples were genotyped on the Illumina MEGAEX array at VUMC. Quality control was performed by BioVU as previously described33 and array genotype data for rs2205986 was extracted. Relationship status was evaluated using PLINK27 and revealed no related individuals (identity by descent ≤0.15). Principal components were determined by multidimensional scaling in PLINK27. One patient-sample was excluded from the ALP analyses due to being an outlier (>3 standard deviations above the mean). BioVU association analyses were performed using PLINK.27 Rs2205986 genotype was analyzed using an additive genetic model by linear regression for association with the highest values for each of the four biochemical liver test results during IFN-β treatment, while logistic regression was employed in the case-control association analyses. BioVU linear regression analyses were adjusted for age at biochemical liver testing, sex, and the first two principal components.
Disease-matched population control analyses.
Genotype data (Illumina Human670-QuadCustom v1) for disease-matched population controls were obtained from the MS Wellcome Trust Case Control Consortium 2 cohort (EGAD00000000120)34 after approval by the relevant data access committees. We included MS patients recruited in North America and determined to be of Northern European genetic ancestry (Supplementary Fig. 7), leaving 1,319 patients for these analyses. QC, strand alignment and whole genome imputation was performed as described above.
Predictive test analyses.
Genomic markers of statistical significance were evaluated for specificity, sensitivity, negative predictive value (NPV), positive predictive value (PPV) and the number needed to screen within the combined patient cohort. NPV, PPV and number needed to screen, were calculated using sensitivity, specificity, and the population incidence of IFN-β-induced liver injury (2%).3 Post-test probabilities to estimate the proportion of patients testing positive for the variant who will develop DILI, were assessed using likelihood ratios and pre- and post-test odds (http://www.cebm.net/likelihood-ratios/). Receiver operating characteristic (ROC) curves, the corresponding area under the curve (AUC) estimates and 95% confidence intervals (95%CI) were generated for two predictive models of IFN-β-induced liver injury. The clinical model included age, IFN-β product, and sex (selected a priori based on previous DILI literature reporting significantly associated factors)36 and a separate model incorporated these same variables in addition to any significantly associated genomic variants. The ROC curves of these two prediction models were compared using the DeLong’s test.37,38
Supplementary Material
Acknowledgments
We gratefully acknowledge the participation of all patients and families who took part in this study across Canada, from Europe and from the USA. This work would not have been possible without their generous participation. We also acknowledge the contributions of the Canadian Pharmacogenomics Network for Drug Safety (CPNDS) Consortium (C. Hildebrand, A. Borrie, T. Wong, T. Bendyshe-Walton, N. Massah, F. Miao, M. Higginson, M. Staub, K. Shaw, B. Malkin and J. Stortz), the Winnipeg Health Sciences Centre MS Clinic (N. Hall, B. Stranger), London Health Sciences Centre MS Clinic (H. Rosehart), Dalhousie MS Clinic (K. Stadnyk, K. Sabourin, B. DeCoste) and Centre Hospitalier de L’Université de Montréal (É. Roger). We thank all the MS clinic neurologists who contributed to the study through patient examination and data collection. This study makes use of data generated by the Wellcome Trust Case-Control Consortium. A full list of the investigators who contributed to the generation of the data is available from www.wtccc.org.uk. Funding for the original Wellcome Trust Case-Control Consortium project was provided by the following Wellcome Trust awards: 076113, 085475 and 090355. This investigation was supported by a pilot grant from the British Columbia Clinical Genomics Network (PI: Tremlett). Doctoral Studentships provided additional funding for KK from the Canadian Institutes of Health Research (CIHR), the CIHR Drug Safety and Effectiveness Cross-Disciplinary Training Program (DSECT), the MS Society of Canada, the University of British Columbia and the Canadian Pharmacogenomics Network for Drug Safety. GEBW received fellowships from the CIHR and the CIHR DSECT programs. BID received stipends from the BC Children’s Hospital Research Institute, CIHR-DSECT, CIHR and the Michael Smith Foundation for Health Research during the period of this study. The Drug-Induced Liver Injury Network is supported by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) of the National Institutes of Health (NIH) as a Cooperative Agreements (U01s) under grants U01-DK065176 (Duke), U01-DK065201 (UNC), U01-DK065184 (Michigan), U01-DK065211 (Indiana), U01DK065193 (UConn), U01-DK065238 (UCSF/CPMC), U01-DK083023 (UTSW), U01-DK083027 (TJH/UPenn), U01-DK082992 (Mayo), U01-DK083020 (USC), U01-DK100928 (Icahn). Additional funding is provided by CTSA grants UL1 RR025761 (Indiana), UL1 RR025747 (UNC), and UL1 UL1 RR024986 (UMich). Additional funding is provided by CTSA grants UL1 RR025761 (Indiana), UL1 RR025747 (UNC), and UL1 UL1 RR024986 (UMich) and the Intramural Research Program of the National Cancer Institute. Vanderbilt University Medical Center BioVU is supported by institutional funding and by the Vanderbilt CTSA grant ULTR000445 from NCATS/NIH. Genome-wide genotyping was funded by P50GM115305 from NIGMS. HT and BC had full access to all of the data in the study and take full responsibility for the integrity of the data and the accuracy of the data analysis. The study funders had no role in the study design, data collection, data analysis, data interpretation, or writing of the report.
K.K. has a consulting agreement with Emerald Lake Safety. E.M.Y. is an investigator of clinical trials sponsored by Gilead Inc., Merck Inc., Vertex Inc., Hoffman LaRoche Inc., Abbvie Inc., Janssen Inc., Boeringher Ingelheim Inc., Intercept Inc., Genfit Inc., and has received honoraria for CME lectures from Merck Canada, Gilead Canada; has been a speaker at Advisory Board Meetings of Boeringher Ingelheim Canada, Hoffman LaRoche Canada, Abbvie Canada, Celgene Canada for which he received an honorarium and lastly, he is a member of the Gilead Canada compassionate release program adjudication committee for which he has received an honorarium. A.T. has received grant support from Hoffman la Roche, Sanofi Genzyme, Chugai, Novartis, and Biogen; consultancy for Biogen, EMD Serono, Hoffman la Roche, Sanofi Genzyme, and Teva Neuroscience. R.A.M. has conducted clinical trials for Sanofi-Aventis and receives research funding from the Canadian Institutes of Health Research (CIHR), the National MS Society, the MS Society of Canada, Research Manitoba, the Consortium of MS Centers, Crohn’s and Colitis Canada, and the Waugh Family Chair in Multiple Sclerosis. M.K. has no actual or potential conflict of interest in relation to this work, however, in the last two years M.K. or M.K.’s institution has received research support or grants from CIHR. T.L.C. was a principle investigator for a research study for Biogen Idec Canada; has received honoraria for lectures from EMD Merck Serono Canada, Biogen Idec Canada, Teva Canada Innovation, and Genzyme; has participated/spoken at Advisory Board Meetings of EMD Merck Serono, Teva Canada Innovation, Biogen Idec Canada, Genzyme, and Novartis for which an honorarium and travel support was received to attend the meeting. P.D. has received support as a consultant, membership on advisory councils, and grants from the pharmaceutical industry and is funded by the MS Society of Canada, and by the CIHR; but none represent a potential financial or non-financial conflict for this manuscript. N.C. has consulting agreements with Abbvie, Lilly, DS Biopharma (Afimmune), Tobira (Allergan), NuSirt, Celgene, Axovant, Shire, and Madrigal and research grants from Cumberland, Galectin and Intercept, but none represent a potential financial or non-financial conflict for this manuscript. M.W. and P.H. have no conflict of interest and they receive funding from the Swedish Research Council (Medicine 521–2011-2440 and 521–2014-3370), the Clinical Research Support (ALF) at Uppsala University, and MW also has grants from the Swedish Heart and Lung Foundation (20120557 and 20140291). Z.X. was a recipient of the Clinician Scientist Development Award from the National Multiple Sclerosis Society and the American Academy of Neurology and is supported by NINDS NIH K08-NS079493 and NINDS R01 NS098023. PDJ has research funding from Sanofi/Genzyme and Biogen and has consulted for Teva Pharmaceuticals and Sanofi/Genzyme. C.J.D.R. receives funding support from the CIHR, Canadian Foundation for Innovation, Canadian Hearing Foundation, BC Children’s Hospital Foundation, Child & Family Research Institute, Canadian Gene Cure Foundation, Teva Pharmaceutical Industries Inc., Genome BC, and the CIHR Drug Safety & Effectiveness Network. HT is the Canada Research Chair for Neuroepidemiology and Multiple Sclerosis and currently receives research support from the National MS Society, the CIHR, the MS Society of Canada and the MS Scientific Research Foundation. In the last five years, H.T. has received support from the Multiple Sclerosis Society of Canada (Don Paty Career Development Award); the Michael Smith Foundation for Health Research (Scholar Award) and the UK MS Trust. H.T. has received speaker honoraria and/or travel expenses to attend conferences from the Consortium of MS Centres (2013), the National MS Society (2014, 2016), ECTRIMS (2013–2018), Biogen Idec (2014), American Academy of Neurology (2013–2016). All speaker honoraria are either declined or donated to an MS charity or to an unrestricted grant for use by H.T.’s research group. B.C.C. currently receives research funding from the CIHR, Genome Canada, Genome British Columbia and British Columbia Children’s Hospital Research (Vancouver, Canada) and has previously held matching funds support for Genome Canada funding from Pfizer Canada (unrestricted).
Footnotes
Competing Financial Interests
The authors declare no competing financial interests.
Reporting summary. Further information on experimental design is available in the Nature Research Life Sciences Reporting Summary linked to this article.
Data availability. Data referenced in this study for the population control analysis are available from the Wellcome Trust Case Control Consortium 2 cohort under the following accession code: EGAD00000000120 (data access committee approval is required). All remaining data are not publicly available due to them containing information that could compromise research participant privacy or consent. Explicit consent to deposit data in public databases or to be shared outside the specific use of this research study was not obtained from the patients.
URLs. 1000 Genomes database, http://www.1000genomes.org; PLINK, https://www.cog-genomics.org/plink2; R for statistical computing 3.2.3, http://www.r-project.org/; Wellcome Trust Case Control Consortium, www.wtccc.org.uk; GTEx Portal, www.gtexportal.org.
References
- 1.Francis GS et al. Hepatic reactions during treatment of multiple sclerosis with interferon-beta-1a: incidence and clinical significance. Drug Saf 26, 815–27 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Chan S, Kingwell E, Oger J, Yoshida E & Tremlett H High-dose frequency beta-interferons increase the risk of liver test abnormalities in multiple sclerosis: a longitudinal study. Mult Scler 17, 361–7 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Kowalec K et al. Characteristics associated with drug-induced liver injury from interferon beta in multiple sclerosis patients. Expert Opin Drug Saf 13, 1305–17 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Daly AK et al. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat Genet 41, 816–9 (2009). [DOI] [PubMed] [Google Scholar]
- 5.Singer JB et al. A genome-wide study identifies HLA alleles associated with lumiracoxib-related liver injury. Nat Genet 42, 711–4 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Ostapowicz G et al. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med 137, 947–54 (2002). [DOI] [PubMed] [Google Scholar]
- 7.Navarro VJ & Senior JR Drug-related hepatotoxicity. N Engl J Med 354, 731–9 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Spraggs CF et al. HLA-DQA1*02:01 is a major risk factor for lapatinib-induced hepatotoxicity in women with advanced breast cancer. J Clin Oncol 29, 667–73 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Aithal GP et al. Case definition and phenotype standardization in drug-induced liver injury. Clin Pharmacol Ther 89, 806–15 (2011). [DOI] [PubMed] [Google Scholar]
- 10.Nelson MR et al. Genome-wide approaches to identify pharmacogenetic contributions to adverse drug reactions. Pharmacogenomics J 9, 23–33 (2009). [DOI] [PubMed] [Google Scholar]
- 11.GTEx Consortium. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–60 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Honda K & Taniguchi T IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol 6, 644–58 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Wang PX et al. Interferon regulatory factor 9 is a key mediator of hepatic ischemia/reperfusion injury. J Hepatol 62, 111–20 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Baranzini SE et al. Prognostic biomarkers of IFNb therapy in multiple sclerosis patients. Mult Scler 21, 894–904 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Mosedale M et al. Editor’s Highlight: Candidate Risk Factors and Mechanisms for Tolvaptan-Induced Liver Injury Are Identified Using a Collaborative Cross Approach. Toxicol Sci 156, 438–454 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin Y et al. Upregulation of Interferon Regulatory Factor 6 Promotes Neuronal Apoptosis After Traumatic Brain Injury in Adult Rats. Cell Mol Neurobiol 36, 27–36 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Byrnes V, Afdhal N, Challies T & Greenstein PE Drug induced liver injury secondary to interferon-beta (IFN-beta) in multiple sclerosis. Ann Hepatol 5, 56–9 (2006). [PubMed] [Google Scholar]
- 18.Lucena MI et al. Glutathione S-transferase m1 and t1 null genotypes increase susceptibility to idiosyncratic drug-induced liver injury. Hepatology 48, 588–96 (2008). [DOI] [PubMed] [Google Scholar]
- 19.Daly AK et al. Genetic susceptibility to diclofenac-induced hepatotoxicity: contribution of UGT2B7, CYP2C8, and ABCC2 genotypes. Gastroenterology 132, 272–81 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Kieseier BC The mechanism of action of interferon-beta in relapsing multiple sclerosis. CNS Drugs 25, 491–502 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Chen P et al. Carbamazepine-induced toxic effects and HLA-B*1502 screening in Taiwan. N Engl J Med 364, 1126–33 (2011). [DOI] [PubMed] [Google Scholar]
- 22.Milo R & Kahana E Multiple sclerosis: geoepidemiology, genetics and the environment. Autoimmun Rev 9, A387–94 (2010). [DOI] [PubMed] [Google Scholar]
- 23.Poser CM et al. New diagnostic criteria for multiple sclerosis: guidelines for research protocols. Ann Neurol 13, 227–31 (1983). [DOI] [PubMed] [Google Scholar]
- 24.Polman CH et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol 69, 292–302 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fontana RJ et al. Presentation and outcomes with clinically apparent interferon beta hepatotoxicity. Dig Dis Sci 58, 1766–75 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fontana RJ et al. Drug-Induced Liver Injury Network (DILIN) prospective study: rationale, design and conduct. Drug Saf 32, 55–68 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chang CC et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deelen P et al. Genotype harmonizer: automatic strand alignment and format conversion for genotype data integration. BMC Res Notes 7, 901 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.1000 Genomes Project Consortium et al. A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jia X et al. Imputing amino acid polymorphisms in human leukocyte antigens. PLoS One 8, e64683 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roden DM et al. Development of a large-scale de-identified DNA biobank to enable personalized medicine. Clin Pharmacol Ther 84, 362–9 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davis MF et al. Automated extraction of clinical traits of multiple sclerosis in electronic medical records. J Am Med Inform Assoc 20, e334–40 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guo Y et al. Illumina human exome genotyping array clustering and quality control. Nat Protoc 9, 2643–62 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.International Multiple Sclerosis Genetics Consortium et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 476, 214–9 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Price AL et al. Principal components analysis corrects for stratification in genome-wide association studies. Nature genetics 38, 904–909 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Lucena MI et al. Phenotypic characterization of idiosyncratic drug-induced liver injury: the influence of age and sex. Hepatology 49, 2001–9 (2009). [DOI] [PubMed] [Google Scholar]
- 37.Robin X et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics 12, 77 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.DeLong ER, DeLong DM & Clarke-Pearson DL Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics 44, 837–45 (1988). [PubMed] [Google Scholar]
- 39.Pruim RJ et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 26, 2336–7 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.