

Abstract
Background
Upper gastrointestinal (GI) bleeding due to stress ulcers contributes to increased morbidity and mortality in people admitted to intensive care units (ICUs). Stress ulceration refers to GI mucosal injury related to the stress of being critically ill. ICU patients with major bleeding as a result of stress ulceration might have mortality rates approaching 48.5% to 65%. However, the incidence of stress‐induced GI bleeding in ICUs has decreased, and not all critically ill patients need prophylaxis. Stress ulcer prophylaxis can result in adverse events such as ventilator‐associated pneumonia; therefore, it is necessary to evaluate strategies that safely decrease the incidence of GI bleeding.
Objectives
To assess the effect and risk‐benefit profile of interventions for preventing upper GI bleeding in people admitted to ICUs.
Search methods
We searched the following databases up to 23 August 2017, using relevant search terms: MEDLINE; Embase; the Cochrane Central Register of Controlled Trials; Latin American Caribbean Health Sciences Literature; and the Cochrane Upper Gastrointestinal and Pancreatic Disease Group Specialised Register, as published in the Cochrane Library (2017, Issue 8). We searched the reference lists of all included studies and those from relevant systematic reviews and meta‐analyses to identify additional studies. We also searched the World Health Organization International Clinical Trials Registry Platform search portal and contacted individual researchers working in this field, as well as organisations and pharmaceutical companies, to identify unpublished and ongoing studies.
Selection criteria
We included randomised controlled trials (RCTs) and quasi‐RCTs with participants of any age and gender admitted to ICUs for longer than 48 hours. We excluded studies in which participants were admitted to ICUs primarily for the management of GI bleeding and studies that compared different doses, routes, and regimens of one drug in the same class because we were not interested in intraclass effects of drugs.
Data collection and analysis
We used standard methodological procedures as recommended by Cochrane.
Main results
We identified 2292 unique records.We included 129 records reporting on 121 studies, including 12 ongoing studies and two studies awaiting classification.
We judged the overall risk of bias of two studies as low. Selection bias was the most relevant risk of bias domain across the included studies, with 78 studies not clearly reporting the method used for random sequence generation. Reporting bias was the domain with least risk of bias, with 12 studies not reporting all outcomes that researchers intended to investigate.
Any intervention versus placebo or no prophylaxis
In comparison with placebo, any intervention seems to have a beneficial effect on the occurrence of upper GI bleeding (risk ratio (RR) 0.47, 95% confidence interval (CI) 0.39 to 0.57; moderate certainty of evidence). The use of any intervention reduced the risk of upper GI bleeding by 10% (95% CI ‐12.0% to ‐7%). The effect estimate of any intervention versus placebo or no prophylaxis with respect to the occurrence of nosocomial pneumonia, all‐cause mortality in the ICU, duration of ICU stay, duration of intubation (all with low certainty of evidence), the number of participants requiring blood transfusions (moderate certainty of evidence), and the units of blood transfused was consistent with benefits and harms. None of the included studies explicitly reported on serious adverse events.
Individual interventions versus placebo or no prophylaxis
In comparison with placebo or no prophylaxis, antacids, H2 receptor antagonists, and sucralfate were effective in preventing upper GI bleeding in ICU patients. Researchers found that with H2 receptor antagonists compared with placebo or no prophylaxis, 11% less developed upper GI bleeding (95% CI ‐0.16 to ‐0.06; RR 0.50, 95% CI 0.36 to 0.70; 24 studies; 2149 participants; moderate certainty of evidence). Of ICU patients taking antacids versus placebo or no prophylaxis, 9% less developed upper GI bleeding (95% CI ‐0.17 to ‐0.00; RR 0.49, 95% CI 0.25 to 0.99; eight studies; 774 participants; low certainty of evidence). Among ICU patients taking sucralfate versus placebo or no prophylaxis, 5% less had upper GI bleeding (95% CI ‐0.10 to ‐0.01; RR 0.53, 95% CI 0.32 to 0.88; seven studies; 598 participants; moderate certainty of evidence). The remaining interventions including proton pump inhibitors did not show a significant effect in preventing upper GI bleeding in ICU patients when compared with placebo or no prophylaxis.
Regarding the occurrence of nosocomial pneumonia, the effects of H2 receptor antagonists (RR 1.12, 95% CI 0.85 to 1.48; eight studies; 945 participants; low certainty of evidence) and of sucralfate (RR 1.33, 95% CI 0.86 to 2.04; four studies; 450 participants; low certainty of evidence) were consistent with benefits and harms when compared with placebo or no prophylaxis. None of the studies comparing antacids versus placebo or no prophylaxis provided data regarding nosocomial pneumonia.
H2 receptor antagonists versus proton pump inhibitors
H2 receptor antagonists and proton pump inhibitors are most commonly used in practice to prevent upper GI bleeding in ICU patients. Proton pump inhibitors significantly more often prevented upper GI bleeding in ICU patients compared with H2 receptor antagonists (RR 2.90, 95% CI 1.83 to 4.58; 18 studies; 1636 participants; low certainty of evidence). When taking H2 receptor antagonists, 4.8% more patients might experience upper GI bleeding (95% CI 2.1% to 9%). Nosocomial pneumonia occurred in similar proportions of participants taking H2 receptor antagonists and participants taking proton pump inhibitors (RR 1.02, 95% CI 0.77 to 1.35; 10 studies; 1256 participants; low certainty of evidence).
Authors' conclusions
This review shows that antacids, sucralfate, and H2 receptor antagonists might be more effective in preventing upper GI bleeding in ICU patients compared with placebo or no prophylaxis. The effect estimates of any treatment versus no prophylaxis on nosocomial pneumonia were consistent with benefits and harms. Evidence of low certainty suggests that proton pump inhibitors might be more effective than H2 receptor antagonists. Therefore, patient‐relevant benefits and especially harms of H2 receptor antagonists compared with proton pump inhibitors need to be assessed by larger, high‐quality RCTs to confirm the results of previously conducted, smaller, and older studies.
Plain language summary
Interventions for preventing upper gastrointestinal bleeding in people on intensive care units
Review question
We reviewed the evidence about benefits and harms of interventions to prevent clinically important upper gastrointestinal (GI) bleeding in patients who were admitted to the intensive care unit (ICU).
Background
Stress ulcers are seen as superficial damage in the mucous lining of the stomach or intestines that can occur as the result of shock, sepsis, or trauma. Depending on the severity of the damage, afflicted areas may become sore and may start to bleed to varying degrees. Upper GI bleeding due to stress ulcers is a major contributor to increased severity of illness and death among people admitted to ICUs. However, standards of care have improved, and the incidence of upper GI bleeding in ICUs has decreased. Thus, not all critically ill patients need preventive treatment.
Stress ulcer prophylaxis can result in negative effects such as ventilator‐associated pneumonia (VAP). VAP is a lung infection caused by bacteria in people who are being mechanically ventilated. VAP usually manifests as fever, cough, and purulent sputum. The risk for VAP is increased in patients with severe illness, increased length of hospital stay, or use of stress ulcer prophylaxis. Hence, it is necessary to evaluate strategies that safely decrease the incidence of upper GI bleeding.
Study characteristics
The evidence is current to August 2017. We included 106 studies with a total of 15,027 critically ill participants of any age and any gender.
Key results
Relevant effects were found for the following drugs: H2 receptor antagonists, antacids, sucralfate, and proton pump inhibitors.
H2 receptor antagonists inhibit gastric acid secretion by blocking histamine receptors but can cause a small number of blood platelets (thrombocytopaenia), inflammation of the kidney (interstitial nephritis), and confusion. Antacids neutralise stomach acid but may cause diarrhoea or constipation. Proton pump inhibitors inhibit the final stage of gastric acid production, and it has been found that they may be associated with increased risk of Clostridium difficile diarrhoea. Ulcer protective agents, such as sucralfate, create a barrier between the gastric acid and the gastric mucosa by coating it. They may, however, cause constipation and interfere with the absorption of certain antibacterial agents.
In comparison with placebo or no preventive treatment, H2 receptor antagonists, antacids, and sucralfate might be effective in preventing clinically important upper GI bleeding in ICU patients. Hospital‐acquired pneumonia was most likely to occur in ICU patients taking either H2 receptor antagonists or sucralfate when compared with patients given placebo or no preventive treatment.
Evidence of low certainty suggests that proton pump inhibitors were more effective than H2 receptor antagonists in preventing upper GI bleeding in ICU patients. With proton pump inhibitors, 25 of 1000 people were likely to develop upper GI bleeding, and with H2 receptor antagonists, 73 of 1000 people (95% confidence interval 46 to 115 people) were likely to develop upper GI bleeding. The effect of H2 receptor antagonists versus proton pump inhibitors with respect to the risk for developing hospital‐acquired pneumonia was consistent with benefits and harms.
Quality of the evidence
Our certainty in the evidence ranged from low to moderate. For effects of different interventions compared with placebo or no prophylaxis, the certainty of evidence was moderate (H2 receptor antagonists) or low (antacids and sucralfate). For effects of H2 receptor antagonists compared with placebo or no preventive treatment on risk of hospital‐acquired pneumonia, the certainty of evidence was low. For effects of H2 receptor antagonists compared with proton pump inhibitors on hospital‐acquired pneumonia, the certainty of evidence was also low.
Summary of findings
Background
A glossary of the medical terms used in this review can be found in Appendix 1.
Description of the condition
Upper gastrointestinal (GI) bleeding due to stress ulcers is a major contributor to increased morbidity and mortality in people admitted to intensive care units (ICUs). Stress ulceration refers to GI mucosal injury related to the stress of being critically ill. The injury may vary from diffuse superficial ulcers to deep, bleeding lesions (Neligan 2006). The incidence of bleeding related to stress ulcers in critically ill people on mechanical ventilators ranges from less than 1% to 6% of patients admitted to ICU (Alhazzani 2013; Bardou 2015; Krag 2015). A study using the HEmorrhage MEasurement Tool (HEME) to assess GI bleeding in ICU patients observed that 5.2% of those with stress ulcers had major bleeds (Arnold 2007). ICU patients with major bleeding as a result of stress ulceration had mortality rates approaching 40% to 50% (Bardou 2015). Over the past few decades, however, with improved standards of care, the incidence of stress‐induced GI bleeding in ICUs and related mortality has decreased (Krag 2015). Hence, not all critically ill patients need prophylaxis (Penner 2005). Gastric bleeding can be diagnosed from a drop in blood pressure necessitating blood transfusion and a rapid heart rate (haemodynamic instability), 'coffee ground' aspiration, vomiting of blood (haematemesis), or passage of dark stools (melena) (Dorland 1995).
Bleeding from stress ulcers
Bleeding from stress ulcers may be trivial or profuse. Trivial bleeds may be detected only through laboratory tests, and profuse bleeding results in overt manifestations. Cook 1998a defined a clinically important bleed as overt bleeding with one of four features reflecting haemodynamic instability and blood loss. These include the following.
Drop in systolic or diastolic blood pressure (blood pressure during heart contractions or between two heartbeats) of 20 mmHg or more within 24 hours after upper GI bleeding.
Postural decrease in systolic blood pressure of 10 mmHg and accelerated pulse rate of 20 beats per minute; or evidence of significant blood loss (this may be difficult to demonstrate in intubated patients).
A drop in haemoglobin concentration of at least 2 g/dL needing two units of packed cell transfusion within 24 hours of the bleed.
Failure of a rise in haemoglobin concentration (in g/dL) of at least the number of units of blood transfused minus two.
Risk factors
Prophylaxis is often recommended for patients with major risk factors. These include:
patients who have a coagulopathy (a disorder of blood clotting); and
patients who require mechanical ventilation for longer than 48 hours.
In addition, prophylaxis is often recommended for patients who have two or more of the following risk factors for stress ulcers (AHSP 1999; Pfeffer 2007).
ICU stay longer than one week.
Sepsis or abnormally low blood pressure (hypotension).
Failure of the liver or kidneys.
History of peptic ulcer disease.
Use of high‐dose steroids (> 250 mg/d of hydrocortisone or equivalent).
Burns over > 35% of total body surface area.
Immediately after organ transplantation.
Head trauma with Glasgow Coma Scale score < 10.
Multiple trauma.
Occult bleeding for six days or longer.
Pathophysiology of stress ulcers
The aetiology ‐ cause or set of causes for a disease ‐ and pathophysiology of stress ulcers in ICU patients appear to be multi‐factorial. Normally, the splanchnic microcirculation and the gastric mucosa protect the mucosal epithelium ‐ the tissue lining the outer surface of organs ‐ from gastric acids, wherein bicarbonate ions secreted by the mucosa neutralise hydrogen ions. Reduced blood flow due to splanchnic hypoperfusion in critically ill patients, with resultant ischaemia and impaired cell oxygenation, releases nitric oxide (by increased levels of enzyme nitric oxide synthase) and oxygen free radicals and reduces prostaglandin synthesis. These mechanisms result in inflammation and cell death. Reperfusion hyperaemia due to elevated nitric oxide levels contributes to further cell death. Slowed upper gastric mobility and reduced secretion of bicarbonate by the gastric mucosa with resultant prolonged exposure of the damaged mucosa to gastric acids, as well as a decrease in mucosal repair mechanisms, also contribute to ulceration (Dorland 1995; Spirt 2006).
Consequences of upper GI bleeding
Major GI bleeding can be life‐threatening and can result in death. Minor bleeding can result in an increased need for blood transfusion and its attendant risks. It is therefore necessary to evaluate strategies that decrease the incidence of GI bleeding.
Complications of stress ulcer prophylaxis: ventilator‐associated pneumonia
Ventilator‐associated pneumonia is a common complication in mechanically ventilated patients. The reported incidence of ventilator‐associated pneumonia ranges from 1% to 28% (Chastre 2002; Rahbar 2006). A systematic review of observational studies and randomised studies revealed that the incidence of ventilator‐associated pneumonia ranged from 10% to 20% in people undergoing mechanical ventilation for longer than 48 hours (Safdar 2005).
Risk factors for ventilator‐associated pneumonia
The risk of ventilator‐associated pneumonia increases with the duration of mechanical ventilation continuing over 48 hours, but the risk is additionally elevated in people with contributory medical conditions. Additional sources of infection from tubes passing the trachea (endotracheal) or ventilator circuits and other feeding tubes, or from improper measures to prevent nosocomial infection in ICU staff, increase the risk of ventilator‐associated pneumonia (Augustyn 2007; CDC 2003; Masterton 2008).
The disadvantage of stress ulcer prophylaxis is that many of the interventions used for suppression of gastric acid raise the pH of gastric contents, alter gastric flora, and promote tracheobronchial colonisation and gastric colonisation of pathogenic bacteria ‐ the aspiration of which causes nosocomial pneumonia or ventilator‐associated pneumonia (Atherton 1978; Cook 1998b; Craven 1986). In addition to increasing mortality in critically ill patients, the afore mentioned effects can prolong length of hospital stay and increase costs (Safdar 2005). The elevated risk of developing ventilator‐associated pneumonia associated with stress ulcer prophylaxis may therefore offset the potential benefits of such therapy.
Diagnosis of ventilator‐associated pneumonia
The criteria used to establish the diagnosis of ventilator‐associated pneumonia vary. Traditionally, clinical features (fever, cough, and purulent sputum) combined with radiological evidence for pneumonia (new lung infiltrates or progression of infiltrates) and elevated white blood cell count in a patient on mechanical ventilation provide suggestive evidence. Confirmatory cultures from sputum or aspirates from the trachea or pleural fluid in such individuals increase the sensitivity of the diagnosis of bacterial causes of pneumonia. However, these traditional methods are not specific in ventilator‐associated pneumonia, and blood cultures are reported to have low sensitivity. Standardised techniques used in clinical research studies have included quantitative culture of specimens obtained from endotracheal aspirates and use of protected specimen brush (PSB) specimens after bronchoalveolar lavage ‐ a medical procedure to examine the lungs for lung disease ‐ or via blind catheterisation (non‐bronchoscopic protected bronchoalveolar lavage or protected specimen brush specimens). These techniques have improved the sensitivity and specificity of the diagnosis of ventilator‐associated pneumonia (CDC 2003; Masterton 2008).
Criteria commonly used to diagnose ventilator‐associated pneumonia include the clinical pulmonary infection score (CPIS), which has evolved from the original four criteria (fever, leucocytosis, positive sputum culture, and worsening chest X‐ray changes) to six criteria (plus increased oxygen requirement and semi‐quantitative cultures of tracheal aspirates with or without Gram stain). Criteria added to the CPIS help practitioners in selecting, modifying, and monitoring therapy, although their diagnostic accuracy is similar to that of traditional criteria (Masterton 2008).
Mortality from ventilator‐associated pneumonia
Individuals who contract ventilator‐associated pneumonia are already critically ill; therefore the death rate from ventilator‐associated pneumonia is high. Reported mortality rates range from 24% to 76%, with higher mortality attributed to specific situations (such as underlying disease and organ failure) or types of causative organisms (such as Pseudomonas or Acinetobacter) (CDC 2003; Chastre 2002). Mortality is doubled in critically ill patients with ventilator‐associated pneumonia when compared with those without ventilator‐associated pneumonia (Safdar 2005).
It is considered prognostically important to distinguish early‐onset from late‐onset ventilator‐associated pneumonia. The Working Party on Hospital Acquired Pneumonia of the British Society of Antimicrobial Chemotherapy defined the former as occurring during the first four days, and the latter as occurring five or more days after the start of mechanical ventilation. Early‐onset ventilator‐associated pneumonia is usually less severe and carries a better prognosis than late‐onset ventilator‐associated pneumonia (Chastre 2002; Masterton 2008).
Description of the intervention
Several pharmacological interventions are used for treatment and prevention of gastric ulcers, such as those that block histamine‐2 receptors (H2 receptor antagonists) (ranitidine, cimetidine, famotidine, etc.), proton pump inhibitors (esomeprazole, rabeprazole, omeprazole, lansoprazole, etc.), prostaglandin analogues (misoprostol), anticholinergics (pirenzepine, propantheline, etc.), antacids (sodium bicarbonate, magnesium hydroxide, etc.), and ulcer protectives (sucralfate, bismuth, etc.). Non‐pharmacological interventions such as enteral nutrition and early removal of tubes can also be used.
How the intervention might work
The interventions used in stress ulcer prophylaxis differ with respect to their mechanism of action, additional protective effects on the gastric mucosa, and effects on increasing gastric luminal pH and resultant potential to increase gastric bacterial colonisation, as well as their side effect profile (Mutlu 2001).
Antacids neutralise gastric acid in a dose‐dependent manner and raise the gastric pH. They have other beneficial cytoprotective effects but may cause an increase in magnesium and diarrhoea (magnesium‐based antacids) or may reduce phosphates and cause constipation (aluminium‐based antacids). Nursing costs are also increased owing to the need to administer antacids at frequent intervals (often hourly).
H2 receptor antagonists are believed to inhibit gastric acid secretion by blocking histamine receptors but have no additional cytoprotective effects and can cause thrombocytopaenia, interstitial nephritis, and confusion (especially in the elderly). Rapid intravenous infusions can cause bradycardia ‐ a slow heart rate ‐ and abnormally low blood pressure (hypotension), and many cytochrome P‐450‐mediated drug interactions and effects may occur (particularly with cimetidine).
Proton pump inhibitors act by inhibiting the final stage of gastric acid production but provide no additional cytoprotective effects on the gastric mucosa and do confer cytochrome P‐450‐mediated effects. However, it has been found that proton pump inhibitors may be associated with increased risk of Clostridium difficile diarrhoea (Arriola 2016; Cunningham 2003; Kwok 2012; Mutlu 2001). Yet, randomised controlled trials (RCTs) have reported very few data on this adverse event (Alhazzani 2017).
Prostaglandin analogues inhibit acid secretion and promote mucus and bicarbonate secretion that makes gastric contents alkaline, meaning that the pH is greater than 7, but they can cause diarrhoea and abdominal pain. However, they need to be administered four times daily.
Ulcer protective agents, such as sucralfate and colloidal bismuth subcitrate, create a barrier between gastric acid and the gastric mucosa by coating the mucosa. These agents have additional cytoprotective effects and do not effectively alter gastric pH, but they may cause constipation and may interfere with the absorption of certain antibacterial agents, such as tetracyclines and quinolones (Mutlu 2001).
Enteral nutrition may be delivered orally or through a feeding tube and helps to maintain gut integrity, modulate stress and the systemic immune response, and attenuate disease severity. Enteral nutrition might also reduce bacterial translocation and infective complications by maintaining the structural integrity of the gut. In addition, enteral nutrition is considered an effective means of providing stress ulcer prophylaxis, although it may raise gastric pH and theoretically increase rates of nosocomial infection or neutralise the effects of ulcer protective agents (Hinds 1999; McClave 2009).
Why it is important to do this review
ICU patients are at risk of developing stress ulcers. A proportion of these patients will develop clinically important bleeding, and the mortality rate in such patients is high (48.5% to 65%). Several RCTs, non‐randomised studies, and cohort studies have studied the role of different stress ulcer prophylactic drugs and strategies for prevention of stress ulcers and thereby upper GI bleeding. Stress ulcer prophylaxis is, however, a risk factor for the development of ventilator‐associated pneumonia. Thus the benefits of stress ulcer prophylaxis need to be balanced against this risk.
Previous systematic reviews and meta‐analyses were narrowed to comparisons of two interventions, were inconclusive, or generated conflicting results (Cook 1994b; Cook 1995b; Cook 1996; Lin 2010; Messori 2000). These systematic reviews did not include commonly used proton pump inhibitors. Several more recent systematic reviews investigated the risk‐benefit profile of bleeding prophylaxis in ICU patients (Alquraini 2017; Alshamsi 2016; Krag 2014; Pilkington 2012). They most often investigated the effects of a single drug class versus another drug class.
Guidelines for the management of hospital‐acquired pneumonia in the United Kingdom have been produced by the working party on hospital‐acquired pneumonia of the British Society for Antimicrobial Chemotherapy (Masterton 2008). This group considered the results of a systematic review ‐ Collard 2003 ‐ and a narrative synthesis of seven meta‐analyses (Cook 1991; Cook 1995; Cook 1996; Messori 2000; Tryba 1991; Tryba 1991b; Tryba 1995). Analyses yielded discordant results but overall suggested that risk of developing ventilator‐associated pneumonia was reduced when patients were given sucralfate compared with H2 receptor antagonists (although not when compared with placebo), and evidence from one study suggested that risk of clinically significant bleeding was increased when sucralfate was given to prevent upper GI bleeding compared with H2 receptor antagonists. Guidelines recommended that stress ulcer prophylaxis should be avoided in people on mechanical ventilation to preserve gastric function and reduce ventilator‐associated pneumonia. If stress ulcer prophylaxis is indicated, then the advantage of sucralfate in reducing the incidence of ventilator‐associated pneumonia needs to be balanced with its increased risk of clinically significant upper GI bleeding (Masterton 2008).
A guideline for stress ulcer prophylaxis in the intensive care unit provided by the Danish Society of Anesthesiology and Intensive Care Medicine recommended, first, that stress ulcer prophylaxis should not be used as a routine measure in all critically ill patients and, second, that proton pump inhibitors should be used over H2 receptor antagonists (Rorbaek Madsen 2014).
A guideline from 1999 released by the American Society of Health‐System Pharmacists recommended that antacids, H2 receptor antagonists, or sucralfate should be administered to prevent stress ulcers in adults (Armstrong 1999). An updated version of the guideline is expected in 2018. The Eastern Association for the Surgery of Trauma released a guideline in 2008 that recommended use of H2 receptor antagonists, cytoprotective agents, and some proton pump inhibitors as prophylactic treatment against stress ulcers and recommended against use of antacids (Guillamondegui 2008).
Still, a review of current evidence on different pharmacological and non‐pharmacological interventions for prophylaxis of upper GI bleeding comparing them versus no active treatment or other active treatments is needed to provide a comprehensive and systematic overview of the research evidence and the benefits and harms of bleeding prophylaxis in patients admitted to the ICU.
Objectives
To assess the effect and the risk‐benefit profile of interventions used to prevent upper GI bleeding in people admitted to level two and level three ICUs. These levels include all sick patients requiring support for organ failure/dysfunction in the ICU (Goldhil 2002).
Level two ICUs are for patients requiring detailed observation or intervention including support for a single failing organ system or postoperative care and patients stepping down from higher levels of care.
Level three ICUs are for patients requiring advanced respiratory support alone or basic respiratory support together with support for at least two organ systems.
Methods
Criteria for considering studies for this review
Types of studies
We followed methods as described in the review protocol (George 2010). We included randomised controlled trials (RCTs) and quasi‐randomised controlled studies.
Types of participants
People (any age and gender) admitted to level two or level three ICUs for longer than 48 hours. We excluded studies in which participants were admitted to the ICU primarily for management of upper GI bleeding.
Types of interventions
We compared the following interventions administered by any route and at any dose.
Drugs that reduce gastric acid secretion
H2 receptor antagonists: ranitidine, cimetidine, famotidine, roxatidine, nizatidine, loxatidine, etc.
Proton pump inhibitors: esomeprazole, rabeprazole, omeprazole, lansoprazole, pantoprazole, dexlansoprazole, etc.
Prostaglandin analogues: misoprostol, enprostil, rioprostil, etc.
Anticholinergics: pirenzepine, propantheline, oxyphenonium, doxepin, trimipramine, etc.
Potassium‐competitive acid blockers
Drugs that neutralise gastric acid (antacids)
Systemic: sodium bicarbonate, sodium citrate
Non‐systemic: magnesium hydroxide, magnesium trisilicate, aluminium hydroxide gel, magaldrate, calcium carbonate
Ulcer protectives
Sucralfate
Colloidal bismuth subcitrate
Ulcer healing drugs
Carbenoxolone sodium, deglycyrrhizinated liquorice
Others
Enteral and parenteral nutrition
Any other intervention used to reduce upper GI bleeding
Combinations of interventions (e.g. omeprazole‐bicarbonate combinations)
No prophylaxis
Placebo
We compared each class of drugs versus placebo or no prophylaxis (e.g. H2 receptor antagonists vs placebo or no prophylaxis), and we compared all classes of drugs against one another (e.g. H2 receptor antagonists vs proton pump inhibitors). We did not compare different drugs within a single class versus one another (e.g. ranitidine vs cimetidine) because this type of comparison did not fall within the focus of this review.
Types of outcome measures
Primary outcomes
Clinically important GI bleeding
For the purposes of this review, we used the definition used by study authors to define clinically important upper GI bleeding. We recorded details of the definition used
Secondary outcomes
Nosocomial pneumonia
Nosocomial pneumonia including ventilator‐associated pneumonia is defined as pneumonia contracted in a hospital (nosocomial) by a patient on mechanical ventilatory support (by endotracheal tube or tracheostomy) for longer than 48 hours (Masterton 2008; Mayhall 2001)
Criteria used in the review for the diagnosis of ventilator‐associated pneumonia will be those used by study authors. This outcome also includes incidence of nosocomial pneumonia because definitions used varied across study reports
Mortality
All‐cause mortality in ICU
All‐cause mortality in hospital
Duration of ICU stay
Duration of intubation
This also includes duration of mechanical ventilation because definitions varied across study reports.
Blood transfusion
Number of participants requiring transfusion
Number of units of blood transfused
Adverse events of interventions
Serious adverse events leading to discontinuation of treatment, prolongation of ICU stay, or disability
Any other adverse event (e.g. thrombocytopaenia related to H2 receptor antagonists, any other adverse event).
Search methods for identification of studies
We attempted to identify all relevant RCTs and quasi‐randomised studies (in which allocation to interventions was attempted but could be predicted) for inclusion, regardless of date or language of publication or publication status (published, unpublished, or in press) We also looked for ongoing studies.
We excluded non‐randomised studies.
Electronic searches
With the help of the Cochrane Information Specialist, we searched the following databases up to 23 August 2017, using the search terms listed in Appendix 2: MEDLINE; Embase; the Cochrane Central Register of Controlled Trials (CENTRAL), published in the Cochrane Library (2017, Issue 8); the Cochrane Upper Gastrointestinal and Pancreatic Disease Group Specialised Register; and Latin American Caribbean Health Sciences Literature (LILACS).
Searching other resources
We searched the reference lists of all included studies and of relevant systematic reviews and meta‐analyses to identify relevant studies. We also searched the World Health Organization (WHO) International Clinical Trials Registry Platform search portal (http://apps.who.int/trialsearch/) for ongoing clinical studies up to 23 August 2017. In addition, we searched all available conference proceedings until 2012 for relevant RCTs from the World Federation of Societies of Intensive and Critical Medicine (http://www.world‐critical‐care.org/) and the World Gastroenterology Organisation (http://www.worldgastroenterology.org/about‐wgo.html) websites. We contacted individual researchers working in this field, as well as organisations and pharmaceutical companies, to identify unpublished and ongoing studies.
Data collection and analysis
Selection of studies
Two review authors from a pool of four (ATG, IT, LEF, and RK) independently screened each citation and abstract yielded by the search strategy to identify potentially eligible studies. Two review authors from a pool of four (IT, LEF, PT, and JVP) independently checked the list of excluded studies to verify the appropriateness of reasons for their exclusion. We obtained and assessed full reports of potentially eligible studies for inclusion in the review based on the inclusion and exclusion criteria. If eligibility was unclear because information was inadequate or unclear, we attempted to contact study authors for clarification. Two review authors (IT, JJM) arranged for abstracts of articles written in non‐English languages to be translated for assessment against inclusion. We resolved disagreements through discussion and scrutinised each study report to ensure that RCTs with multiple publications were included only once by linking additional reports to the original study report included in the reference list of included studies. We documented reasons for exclusion of studies in the Characteristics of excluded studies tables.
Data extraction and management
Two review authors from a pool of four (IT, LEF, ATG, and RK) independently extracted data from studies using pre‐tested data extraction forms. We resolved disagreements related to data extraction by referring to the study report and by having discussions. When available, we extracted data on the following.
Population characteristics
Type of ICU care (level two or three); inclusion and exclusion criteria for participants, as well as their age and gender and the number of participants randomised to each group and included in the study overall.
Interventions
Details of interventions given (dose, route, duration); additional interventions used in each arm (e.g. enteral feeds, antibiotics).
Outcomes
Definitions or criteria used for the diagnosis of clinically significant upper GI bleeding (and the source of bleeding), pneumonia (and types of pathogens and sensitivity patterns, if available) and ventilator‐associated pneumonia; number of participants experiencing each outcome; and numbers of dropouts and withdrawals with reasons. When data were insufficient or missing, we attempted to contact the study authors.
For continuous outcomes, we extracted arithmetic mean values, standard deviations, and number of participants in each study arm for whom the outcome was assessed. We noted whether numbers assessed in the study were the numbers of participants who completed the study or the numbers randomised. If medians were reported, we attempted to extract ranges or interquartile ranges.
Assessment of risk of bias in included studies
Two review authors from a pool of six (IT, LEF, ATG, RK, PT, and JVP) independently assessed the risk of bias of each included study. We resolved disagreements by referring to the study report, by corresponding with the authors of the report, and by having discussions. We assessed each study on the domains of sequence generation; allocation concealment; blinding of participants, personnel, and outcome assessors; incomplete outcome data; selective reporting; and other sources of bias. We judged the risk of bias as 'high', 'low', or 'unclear', using guidelines (Higgins 2011) to make these judgements. We recorded this information for each included study in a 'Risk of bias' table in Review Manager (RevMan) 5.2 and summarised the risk of bias for each study in a summary figure and graph. We classified a study's overall risk of bias as high if the study had high risk of bias in any domain. Likewise, we classified the risk of bias as unclear if the study had no high risk of bias and had unclear risk of bias in any domain. Last, we classified a study's overall risk of bias as low if the study had low risk of bias in all domains.
We judged all included studies as being at low risk for detection bias for objectively determined outcomes of 'clinically important GI bleeding' and 'nosocomial pneumonia' if detected through a clear definition mentioned in the study, or if blinding of outcome assessors was clearly described. For other outcomes of interest, we judged the studies as having low risk of detection bias if these outcomes were mainly objective in nature. We judged studies as having unclear risk of detection bias on an outcome basis if they did not address upper GI bleeding or nosocomial pneumonia.
We classified studies that did not include a placebo arm and used different modes of administering study interventions as having high risk of performance bias, as it would not have been possible to blind study personnel and participants.
Measures of treatment effect
We used risk ratios for dichotomous outcomes and mean differences for continuous outcomes, with their respective 95% confidence intervals.
Unit of analysis issues
When outcomes were reported both at baseline and at follow‐up or at study endpoints, we used endpoint data preferentially over scores of the mean change from baseline because the standard deviation of this mean change for each treatment group often was not reported. Had only change scores been available from any study, we would have combined endpoint and change scores.
If studies had reported count data, we would have extracted the total number of events in each group, the total extent of person‐time at risk in each group, and the total number of participants in each group. If this information had not been available, we would have attempted to extract alternative summary statistics such as rate ratios and confidence intervals, if available. If count data had been presented as dichotomous outcomes, we would have extracted the number of participants in each intervention group and the number of participants in each intervention group who experienced at least one event. If count data were presented as continuous outcomes or as time‐to‐event outcomes, we would have attempted to extract the same information as outlined for continuous and time‐to‐event outcomes.
In case we would have identified any cluster‐randomised studies, and if their results had been adjusted for clustering, we would have combined the adjusted measures of effects. If results were not adjusted for clustering, we would have attempted to adjust the results by multiplying standard errors of the estimates by the square root of the design effect when the design effect was calculated as DEff = 1 + (M ‐ 1) ICC, where M is the average cluster size and ICC is the intracluster coefficient. If this was not possible, we would not have combined them in a meta‐analysis but would have presented these results in an additional table.
Dealing with missing data
We attempted to obtain missing data from study authors. When possible, we extracted data to allow an intention‐to‐treat analysis in which all randomised participants would be analysed in the groups to which they were originally assigned. Our primary analysis was a complete case analysis. If we noted a discrepancy in the numbers randomised and the numbers analysed for each treatment group, we calculated the percentage lost to follow‐up in each group and reported this information. If dropouts exceeded 10% for any study, we assigned the worst outcome to those lost to follow‐up for dichotomous outcomes and assessed the impact of this by performing sensitivity analyses.
For continuous data that were missing standard deviations, we calculated these from other available data such as standard errors, or we imputed them using the methods suggested in Deeks 2011. We did not make any assumptions about loss to follow‐up for continuous data, and we will analyse results for those who complete the study.
Assessment of heterogeneity
We assessed heterogeneity between studies by visually examining the forest plot to check for overlapping confidence intervals and by using the Chi² test for homogeneity with a 10% level of significance and the I² statistic. Although we acknowledge that this cutoff is arbitrary, we interpreted I² values from 0% to 40% as possibly not important, from 30% to 60% as moderate heterogeneity, from 50% to 90% as substantial heterogeneity, and from 75% to 100% as considerable heterogeneity, depending on whether inconsistency in results was due to differences in the direction of effect estimates between studies rather than to differences in the magnitude of effect estimates favouring an intervention, as well as the strength of evidence for heterogeneity seen in the P value for the Chi² test for heterogeneity (Deeks 2011).
Assessment of reporting biases
Apart from assessing the risk of selective outcome reporting considered under Assessment of risk of bias in included studies, we assessed the likelihood of potential publication bias by using funnel plots, provided that at least 10 studies were included in a meta‐analysis.
Data synthesis
We first compared interventions used to prevent upper GI bleeding versus placebo or no intervention in people admitted to intensive care units. We stratified analyses according to drug class of the active intervention used versus placebo or no treatment. We included three‐armed studies in these comparisons by splitting the comparison arm in two, as suggested in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a). We then compared specific interventions or combinations of interventions grouped under drug classes. For three‐armed studies, we considered only the two relevant arms in comparisons of drug classes.
We synthesised comparable data using the Mantel‐Haenszel method to derive pooled, weighted risk ratios in fixed‐effect meta‐analyses. We combined continuous data summarised by arithmetic means and standard deviations using the weighted mean difference. We used the random‐effects model for data synthesis when we identified heterogeneity as significant (see above) and found that it could not be explained by subgroup analyses (see below).
Had continuous data been summarised using geometric means, we would have combined them on the log scale using the generic inverse variance method and would have reported them on the natural scale. We would have compared count data using rate ratios when the total number of events in each group and the total extent of person‐time at risk in each group were available, or by using relative risks or weighted mean differences when data were presented in dichotomous or continuous form, respectively. We would have combined hazard ratios from survival data on the log scale by using the inverse variance method and would have presented them on the natural scale.
Subgroup analysis and investigation of heterogeneity
If data permitted, we intended to carry out the following subgroup analyses for each comparison.
Presence or absence of bleeding disorders (e.g. coagulopathies, defined as thrombocyte count < 50/nL, partial thromboplastin time (PTT) > 2 times the upper limit of the normal range, international normalised ratio (INR) > 1.5).
Pneumonia at the time of ICU admission.
Adults (≥ 18 years to 65 years) versus older adults (≥ 65 years) versus children and adolescents (< 18 years).
Use of co‐interventions that may affect outcomes (e.g. enteral feeds, prophylactic antibiotics, other measures used for selective decontamination of the digestive tract (SDD)).
For the outcome of nosocomial pneumonia, we planned to further subgroup data by development of early‐onset nosocomial pneumonia (within four days on ventilation) or late‐onset nosocomial pneumonia (five or more days on ventilation), or by rating data as unclear (if this subgrouping is not possible by using information in the report or by contacting study authors). We identified no studies within the same comparison that distinguished between early and late onset of pneumonia and determined that a subgroup analysis was not feasible.
Sensitivity analysis
We conducted sensitivity analyses to investigate robustness of results for the primary outcome by evaluating outcomes in studies with low risk of bias versus outcomes in studies with high or unclear risk of bias. We also undertook sensitivity analyses if studies reported dropout rates of 10% or greater, to ascertain differences in outcomes of intention‐to‐treat (ITT) analysis and analysis of completers. We assessed robustness of results using published and validated criteria to diagnose clinically important bleeding and nosocomial pneumonia.
Summarising results
We imported data for the following outcomes from Review Manager (RevMan) 5.2 into the Guideline Development Tool (GRADEpro 2015) and used this information to construct 'Summary of findings' (SoF) tables and to guide the conclusions of this review. We considered the following outcomes as critically important or important for clinical decisions for inclusion in these summary tables.
Clinically important upper GI bleeding.
Nosocomial pneumonia.
All‐cause mortality in ICU.
Duration of ICU stay.
Number of participants requiring blood transfusions.
Serious adverse events.
Results
Description of studies
See Characteristics of included studies,Characteristics of excluded studies,Characteristics of ongoing studies, and Characteristics of studies awaiting classification.
Results of the search
We retrieved 2802 records through database searching between July 2010 and August 2017. We identified 43 additional records from other sources, which included review of our personal files and records, correspondence with experts in the field, and review of bibliographies of review articles. After resolving duplicates, the final list included 2292 records. After discarding reports that clearly were not relevant, we identified 199 records as potentially eligible for inclusion. In the end, we included in the review 129 records reporting on 121 studies, of which 12 were ongoing studies and two were awaiting classification. We excluded 71 records. The process of study selection is described in Figure 1.
1.

PRISMA flow chart of included studies.
Included studies
We have described in the Characteristics of included studies tables the 107 competed studies that met the inclusion criteria for this review and have summarised them below. All studies took place in an ICU setting and included critically ill adults or children or both.
Participants, interventions, and comparisons
The 107 studies randomised a total of 15,057 participants to 27 comparisons involving 14 different treatment modalities. Most studies specifically mentioned that they randomised individuals who had no history of GI haemorrhage or peptic ulcer or gastritis or were not undergoing treatments for any of these conditions. The included studies included participants admitted to intensive care units, although the level of ICU into which participants were admitted was not clearly mentioned. Neither was a duration of ICU admission ≥ 48 hours ‐ a necessary inclusion criterion across studies. Among the included studies were five exclusively paediatric studies (Behrens 1994; Kuusela 1997; Lacroix 1986; Lopez‐Herce 1992; Yildizdas 2002); six quasi‐randomised studies (Borrero 1984; Borrero 1985; Borrero 1986; Brophy 2010; Martin 1980; Weigelt 1981); and seven studies reported as conference abstracts only (Fink 2003; Fogas 2013; Larson 1989; Luk 1982; Phillips 1998; Selvanderan 2016; Wee 2013).
Interventions versus placebo or no prophylaxis
We included a total of 32 studies involving 2151 individuals in any interventional arm (H2 receptor antagonists, antacids, sucralfate, proton pump inhibitors, prostaglandin analogues, or anticholinergics) (n = 1249) or in the placebo or no prophylaxis arms (n = 902).
H2 receptor antagonists versus placebo or no prophylaxis
We included a total of 26 studies involving 2210 individuals in the H2 receptor antagonist (ranitidine, cimetidine, or famotidine) arm (n = 1123) or in the placebo or no prophylaxis arm (n = 1087) (Apte 1992; Basso 1981; Ben‐Menachem 1994; Burgess 1995; Chan 1995; Darlong 2003; Friedman 1982; Groll 1986; Halloran 1980; Hanisch 1998; Kantorova 2004; Karlstadt 1990; Kaushal 2000; Lacroix 1986; Larson 1989; Lopez‐Herce 1992; Luk 1982; Martin 1993; Metz 1993; Peura 1985; Powell 1993; Reusser 1990; Rohde 1980; van den Berg 1985; Yildizdas 2002; Zinner 1981). Enteral feeding/nasogastric feeding/parenteral feeding was administered to many participants in the following studies: Apte 1992; Ben‐Menachem 1994; Darlong 2003; Halloran 1980; Kantorova 2004; Karlstadt 1990; Martin 1993; Peura 1985; and van den Berg 1985.
Proton pump inhibitors versus placebo or no prophylaxis
We included a total of five studies randomising 482 individuals to a proton pump inhibitor (omeprazole or pantoprazole) (n = 141) or to placebo or no prophylaxis (n = 133) (Ali 2016; Kantorova 2004; Powell 1993; Selvanderan 2016; Yildizdas 2002). Enteral feeds were administered alone in Kantorova 2004. Selvanderan 2016 did not report how many participants were randomised to each arm, so we were unable to add a total of 214 participants to the total numbers of participants in respective study arms.
Proton pump inhibitors plus sucralfate versus no prophylaxis
One study randomised 80 participants to receive proton pump inhibitors (omeprazole or lansoprazole) plus sucralfate (n = 40) or no prophylaxis (n = 40). Participants who did not receive prophylaxis did receive enteral nutrition (Fang 2014).
Prostaglandin analogues versus placebo or no prophylaxis
One study randomised 58 individuals to receive either a prostaglandin analogue (n = 29) or placebo (n = 29) (van Essen 1985).
Anticholinergics versus placebo or no prophylaxis
A total of two studies involving 131 individuals who received an anticholinergic (n = 59) or a placebo or no prophylaxis (n = 72) were included in the analysis (Hanisch 1998; Krakamp 1989). Ranitidine was administered to both arms in one study (Krakamp 1989).
Antacids versus placebo or no prophylaxis
A total of eight studies involving a total of 774 individuals in the antacid arm (n = 386) or placebo or no prophylaxis arm (n = 388) were included in the analysis (Basso 1981; Friedman 1982; Hastings 1978; Lopez‐Herce 1992; Luk 1982; Macdougall 1977; Pinilla 1985; Zinner 1981).
Sucralfate versus placebo or no prophylaxis
A total of seven studies involving a total of 598 individuals in the sucralfate arm (n = 302) and in the placebo or no prophylaxis arm (n = 296) were included in the analysis (Ben‐Menachem 1994; Darlong 2003; Eddleston 1994; Kantorova 2004; Kaushal 2000; Lopez‐Herce 1992; Yildizdas 2002). Enteral feeds/nasogastric feeding/parenteral feeds were administered to many participants in the following studies: Ben‐Menachem 1994 and Kantorova 2004.
Interventions compared with one another
H2 receptor antagonists versus proton pump inhibitors
We included a total of 20 studies involving 2370 individuals given H2 receptor antagonists (ranitidine, cimetidine, or famotidine) (n = 1037) or proton pump inhibitors (esomeprazole, omeprazole, rabeprazole, or pantoprazole) (n = 1333) (De Azevedo 2000; Bashar 2013; Brophy 2010; Conrad 2005; Fink 2003; Fogas 2013; Hata 2005; Kantorova 2004; Lee 2014; Levy 1997; Maasoumi 2016; Ng 2012; Phillips 1998; Powell 1993; Solouki 2009; Somberg 2008; Tabeefar 2012; Terzi 2009; Wee 2013; Yildizdas 2002). Enteral feeds/nasogastric feeding/parenteral feeds were administered to many participants in the following studies: Brophy 2010,Conrad 2005,Kantorova 2004,Solouki 2009, and Somberg 2008. Somberg 2008 randomised participants to four different intermittent dosing regimens of pantoprazole. These arms were combined to form a common interventional arm versus the H2 receptor antagonist, as the review did not aim to investigate efficacy of the same drug based on dose or mode of administration. Fogas 2013 did not report details on the H2 receptor antagonist and proton pump inhibitor used. One study did not report any outcomes of relevance for this review (Tabeefar 2012).
H2 receptor antagonists versus antacids
We included a total of 18 studies involving 1795 individuals in H2 receptor antagonist (ranitidine, cimetidine, or famotidine) (n = 957) or antacid interventional arms (n = 835) (Basso 1981; Cannon 1987; Friedman 1982; Kingsley 1985; Lamothe 1991; Lopez‐Herce 1992; Luk 1982; Martin 1980; Noseworthy 1987; Poleski 1986; Priebe 1980; Prod'hom 1994; Simms 1991; Stothert 1980; Thomason 1996; Tryba 1985; Weigelt 1981; Zinner 1981). Tryba 1985 administered 50 mg of pirenzipine daily to all randomised participants. Enteral feeds were administered to many participants (Cannon 1987; Simms 1991; Tryba 1985). Lamothe 1991 included four arms (ranitidine, cimetidine, famotidine, and antacids); H2 receptor antagonists were combined to form a common interventional arm versus antacids, as the review did not aim to investigate intraclass efficacy among included interventions.
H2 receptor antagonists versus sucralfate
We included a total of 26 studies involving 3352 individuals given H2 receptor antagonists (ranitidine, cimetidine, or famotidine) (n = 1772) or sucralfate (n = 1647) (Ben‐Menachem 1994; Cannon 1987; Cook 1998; Darlong 2003; De Azevedo 2000; Eddleston 1991; Fabian 1993; Kantorova 2004; Kappstein 1991; Kaushal 2000; Laggner 1988; Laggner 1989; Lopez‐Herce 1992; Maier 1994; Mustafa 1994; Ortiz 1998; Pickworth 1993; Prakash 2008; Prod'hom 1994; Ruiz‐Santana 1991; Ryan 1993; Simms 1991; Stoehr 2006; Thomason 1996; Tryba 1985; Yildizdas 2002). Tryba 1985 administered 50 mg of pirenzipine daily to all randomised participants. Enteral feeds/nasogastric feeding/parenteral feeds were administered to many participants in the following studies: Ben‐Menachem 1994,Cannon 1987,Cook 1998,Kantorova 2004,Mustafa 1994,Prod'hom 1994,Ruiz‐Santana 1991,Simms 1991, and Tryba 1985. Two studies randomised participants to more than one arm of H2 receptor antagonists (cimetidine bolus and continuous infusion) (Fabian 1993; Ortiz 1998). These arms were combined to form a common interventional arm versus sucralfate, as the review did not aim to investigate efficacy of the same drug based on dose or mode of administration.
H2 receptor antagonists versus anticholinergics
A total of four studies involved 599 individuals given H2 receptor antagonists (ranitidine or famotidine) (n = 307) or anticholinergics (n = 292) (Barandun 1985; Behrens 1994; Hanisch 1998; Tryba 1988). Parenteral nutrition was given at the time of endoscopy to participants in the second study alone.
H2 receptor antagonists versus prostaglandin analogues
One study randomised 127 individuals to receive H2 receptor antagonists (cimetidine) (n = 64) or a prostaglandin analogue (misoprostol) (n = 63) (Martin 1992).
H2 receptor antagonists versus teprenone
One study randomised 140 individuals to receive ranitidine (n = 70) or teprenone (n = 70) (Hata 2005).
H2 receptor antagonist + antacids versus sucralfate
A total of three studies involving 281 individuals in an interventional arm that combined H2 receptor antagonists with antacids (n = 144) or in the sucralfate arm (n = 137) were included in the analysis (Cioffi 1994; Driks 1987; Sirvent 1994). Enteral feeding/ parenteral nutrition was administered in Cioffi 1994 and Sirvent 1994. Conventional therapy with antacids, H2 receptor antagonists (cimetidine or ranitidine), or both was administered in Driks 1987; cimetidine was given in the first study and ranitidine in the third study.
Proton pump inhibitors versus teprenone
Hata 2005 randomised 140 individuals to receive rabeprazole (n = 70) or teprenone (n = 70).
Proton pump inhibitors plus naloxone versus naloxone
He 2017 randomised 120 participants to receive pantoprazole plus naloxone (n = 60) or naloxone alone (n = 60).
Proton pump inhibitors versus other medication
Lin 2016 randomised 120 individuals to receive lansoprazole (n = 60) or another medication not further specified (n = 60).
Antacids versus prostaglandin analogues
Skillman 1984 and Zinner 1989, which included 417 individuals in the antacid arm (n = 206) or the prostaglandin analogue arm (n = 211), were included in the analysis.
Antacids versus sucralfate
We included a total of 16 studies involving 1772 individuals given antacids (n = 884) or sucralfate (n = 888) (Bonten 1995; Borrero 1984; Borrero 1985; Borrero 1986; Bresalier 1987; Cannon 1987; Ephgrave 1998; Israsena 1987, Kitler 1990; Lopez‐Herce 1992; Mahul 1992; Prod'hom 1994; Simms 1991; Thomason 1996; Tryba 1987; Tryba 1985). Tryba 1985 administered 50 mg of pirenzipine daily to all randomised participants. Enteral feeds/nasogastric feeding/parenteral feeds were administered to many participants in the following studies: Bonten 1995,Cannon 1987,Ephgrave 1998,Mahul 1992,Prod'hom 1994,Simms 1991, and Tryba 1985.
Antacids versus bioflavonoids
Kitler 1990 randomised 198 individuals to receive an antacid (n = 113) or a bioflavonoid ('Maciadanol') (n = 85).
Sucralfate versus proton pump Inhibitors
We included a total of four studies involving a total of 424 individuals given sucralfate (n = 205) and a proton pump inhibitor (omeprazole, pantoprazole) (n = 219) (De Azevedo 2000; Kantorova 2004; Khorvash 2014; Yildizdas 2002). Enteral feeds/nasogastric feeding/parenteral feeds were administered to many participants in one study (Kantorova 2004).
Sucralfate versus bioflavonoids
Kitler 1990 randomised 198 individuals to receive sucralfate (n = 113) or a bioflavonoid ('Maciadanol') (n = 85).
Non‐pharmacological interventions
Total parenteral nutrition versus any other interventions plus total parenteral nutrition
Ruiz‐Santana 1991 randomised 73 individuals to receive total parenteral nutrition (n = 30), total parenteral nutrition plus ranitidine (n = 24), or total parenteral nutrition plus sucralfate (n = 19).
Bowel stimulation protocol versus no prophylaxis
Wang 2015 randomised 100 individuals to receive treatment through a bowel stimulation protocol including abdominal massage, rectal digital stimulation, and enema (n = 50) or no prophylaxis (n = 50).
Nasojejunal nutrition versus nasogastric nutrition
Davies 2012 randomised 180 individuals to nasojejunal (n = 91) or nasogastric nutrition (n = 89).
Enteral plus parenteral nutrition versus other nutrition regimens
Fan 2016 randomised 120 evenly to receive enteral plus parenteral nutrition or enteral nutrition alone or parenteral nutrition alone.
Funding sources
Most study reports that included information about funding sources mentioned that researchers had received funding from pharmaceutical companies that were involved in production of the tested interventions (23 studies). A total of 17 studies reported institutional funding from, for example, the hospitals involved in the study. Sixty studies provided no information about funding.
Excluded studies
We excluded 71 records from the review mainly for these reasons: 30 studies assessed the efficacy of drugs within the same class, 15 studies were not RCTs, participants in 15 studies were not admitted to the ICU or had an indication that did not fit our inclusion criteria, six studies assessed no health outcomes that were of relevance for the review, and five studies did not take place in an ICU setting. We have mentioned the reasons for exclusion more elaborately under Characteristics of excluded studies.
Ongoing studies
We retrieved 12 ongoing studies that might be relevant to this review and will be included in the next update.
Studies awaiting classification
We were unable to obtain the full text of two studies and classified them as studies awaiting classification (Labattut 1992; Morris 2001).
We retrieved from study registries 12 studies that were ongoing or finished with no publication identified (ACTRN12616000481471; EUCTR2015‐000318‐24‐DK; EudraCT 2007‐006102‐19; IRCT201104134578N2; ISRCTN12845429; Krag 2016; NCT00590928; NCT00702871; NCT02157376; NCT02290327; NCT02718261; NCT03098537).
Risk of bias in included studies
Assessments regarding risk of bias for all included studies are depicted in Figure 2 and Figure 3.
2.
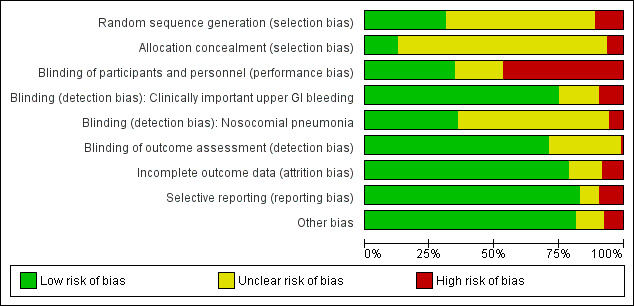
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
A total of 33 studies clearly mentioned the method employed for generating a random sequence (Bashar 2013; Ben‐Menachem 1994; Burgess 1995; Cannon 1987; Cook 1998; Davies 2012; Eddleston 1991; Eddleston 1994; Ephgrave 1998; Fabian 1993; Fang 2014; Hanisch 1998; Hastings 1978; Israsena 1987; Kantorova 2004; Kitler 1990; Kuusela 1997; Metz 1993; Ng 2012; Noseworthy 1987; Ortiz 1998; Prakash 2008; Priebe 1980; Prod'hom 1994; Rohde 1980; Ryan 1993; Skillman 1984; Solouki 2009; Somberg 2008; Stothert 1980; Tabeefar 2012; Thomason 1996; Yildizdas 2002). Thirteen studies mentioned the method employed to conceal the allocation, ensuring that they were clearly free from any selection bias (Ben‐Menachem 1994; Cook 1998; Davies 2012; Ephgrave 1998; Halloran 1980; Hanisch 1998; Kantorova 2004; Kuusela 1997; Martin 1993; Ng 2012; Noseworthy 1987; Prod'hom 1994; Somberg 2008).
Seven studies were quasi‐randomised studies and therefore were judged to have higher risk of selection bias compared with the other studies, which had unclear risk for the domains of 'sequence generation' and 'allocation concealment' (Borrero 1984; Borrero 1985; Borrero 1986; Brophy 2010; Martin 1980; Pickworth 1993; Weigelt 1981).
Blinding
Performance bias
Thirty‐seven studies clearly mentioned the methods employed for blinding participants and personnel across all outcomes, and so review authors judged their risk of performance bias as low (Barandun 1985; Bashar 2013; Bonten 1995; Burgess 1995; Chan 1995; Conrad 2005; Cook 1998; Ephgrave 1998; Fogas 2013; Friedman 1982; Groll 1986; Halloran 1980; Hanisch 1998; Kappstein 1991; Khorvash 2014; Krakamp 1989; Kuusela 1997; Lacroix 1986; Larson 1989; Lee 2014; Luk 1982; Martin 1992; Martin 1993; Metz 1993; Ng 2012; Pan 2004; Peura 1985; Pickworth 1993; Powell 1993; Selvanderan 2015; Selvanderan 2016; Stothert 1980; Tabeefar 2012; van den Berg 1985; van Essen 1985; Wee 2013; Zinner 1989).
Detection bias
Eighty studies reported that researchers used a clear definition for detection of upper GI bleeding, and review authors judged these studies as having low risk of detection bias for this outcome (Ali 2016; Apte 1992; Basso 1981; Ben‐Menachem 1994; Borrero 1984; Borrero 1985; Borrero 1986; Bresalier 1987; Brophy 2010; Burgess 1995; Cannon 1987; Chan 1995; Cioffi 1994; Conrad 2005; Cook 1998; Darlong 2003; Davies 2012; Driks 1987; Eddleston 1991; Eddleston 1994; Ephgrave 1998; Fabian 1993; Fink 2003; Friedman 1982; Groll 1986; Halloran 1980; Hanisch 1998; Kantorova 2004; Kappstein 1991; Karlstadt 1990; Kaushal 2000; Kingsley 1985; Kitler 1990; Krakamp 1989; Kuusela 1997; Lacroix 1986; Laggner 1988; Lamothe 1991; Larson 1989; Lee 2014; Levy 1997; Lin 2016; Lopez‐Herce 1992; Luk 1982; Macdougall 1977; Maier 1994; Martin 1980; Martin 1992; Martin 1993; Metz 1993; Ng 2012; Ortiz 1998; Peura 1985; Pickworth 1993; Pinilla 1985; Poleski 1986; Prakash 2008; Priebe 1980; Prod'hom 1994; Reusser 1990; Rohde 1980; Ruiz‐Santana 1991; Ryan 1993; Selvanderan 2015; Selvanderan 2016; Skillman 1984; Solouki 2009; Somberg 2008; Stothert 1980; Terzi 2009; Thomason 1996; Tryba 1985; Tryba 1987; Tryba 1988; van den Berg 1985; van Essen 1985; Wang 2015; Weigelt 1981; Zinner 1981; Zinner 1989). Thirty‐eight studies reported that researchers used a clear definition for detection of nosocomial pneumonia, and review authors judged their risk of detection bias as low (Apte 1992; Bashar 2013; Ben‐Menachem 1994; Bonten 1995; Cioffi 1994; Conrad 2005; Cook 1998; Davies 2012; Driks 1987; Eddleston 1991; Eddleston 1994; Ephgrave 1998; Fabian 1993; Fogas 2013; Hanisch 1998; Israsena 1987; Kantorova 2004; Kappstein 1991; Khorvash 2014; Laggner 1989; Lee 2014; Lin 2016; Mahul 1992; Maier 1994; Martin 1993; Metz 1993; Mustafa 1994; Pickworth 1993; Prakash 2008; Prod'hom 1994; Ryan 1993; Sirvent 1994; Solouki 2009; Somberg 2008; Thomason 1996; Tryba 1987; Tryba 1988; Yildizdas 2002).
Incomplete outcome data
High risk of attrition bias was suspected in eight studies (Barandun 1985; Fabian 1993; Hanisch 1998; Israsena 1987; Khorvash 2014; Rohde 1980; Ruiz‐Santana 1991; Terzi 2009). Although two studies conducted per‐protocol analyses for outcomes of interest and excluded participants, reports show an imbalance between interventional arms with respect to final numbers of participants (Fabian 1993; Ruiz‐Santana 1991). Therefore, review authors judged the likelihood of attrition bias as high. Only one strata of the study was available for analysis (Rohde 1980), and 16% of participants were not accounted for in two studies, respectively (Barandun 1985; Terzi 2009).
Fourteen studies, which reported that researchers conducted a per‐protocol analysis for outcomes of interest and excluded participants, showed no imbalance between interventional arms with respect to final numbers of participants (Bresalier 1987; Hanisch 1998; Kantorova 2004; Lopez‐Herce 1992; Poleski 1986; Prod'hom 1994; Reusser 1990; Skillman 1984; Stothert 1980; Thomason 1996; van den Berg 1985; van Essen 1985; Yildizdas 2002; Zinner 1989). Therefore review authors judged the likelihood of bias due to attrition as low.
The following studies were unclear on the numbers of participants initially randomised to the interventional arms: Fink 2003,Groll 1986,Kitler 1990,Phillips 1998, and Prakash 2008. Therefore review authors determined that they had unclear risk of attrition bias.
Selective reporting
All studies analysed and reported intended outcomes except the following.
Barandun 1985 and Laggner 1988 were unclear on reporting of adverse events of interventions.
Ortiz 1998 and Chan 1995 showed selective or unclear reporting on secondary outcomes of interest.
Rohde 1980 reported outcomes for only one strata of the study (participants with polytrauma); all other strata were excluded from analysis.
Thomason 1996 conducted an intention‐to‐treat analysis for the outcomes of pneumonia and all‐cause mortality only.
Sirvent 1994, van den Berg 1985, Lopez‐Herce 1992, and Macdougall 1977 did not report all‐cause mortality separately for each intervention arm.
Zinner 1989 was unclear on the total number of participants in each arm when reporting adverse events due to interventions.
Chan 1995 reported more outcomes than initially proposed in the Methods section.
We evaluated risk of reporting bias for clinical upper GI bleeding for the following comparisons: any intervention versus no prophylaxis or placebo, H2 receptor antagonists versus no prophylaxis or placebo, H2 receptor antagonists versus proton pump inhibitors, H2 receptor antagonists versus antacids, H2 receptor antagonists versus sucralfate, and antacids versus sucralfate. These were the only comparisons that involved at least 10 studies. Visual inspection of funnel plots revealed no evidence of reporting bias (Figure 4; Figure 5; Figure 6; Figure 7; Figure 8; Figure 9).
4.

Funnel plot of comparison: 1 Interventions versus placebo or no prophylaxis, outcome: 1.1 Clinically important upper GI bleeding.
5.

Funnel plot of comparison: 2 H2 receptor antagonists versus placebo or no prophylaxis, outcome: 2.1 Clinically important upper GI bleeding.
6.

Funnel plot of comparison: 9 H2 receptor antagonists versus proton pump inhibitors, outcome: 9.1 Clinically important upper GI bleeding.
7.

Funnel plot of comparison: 10 H2 receptor antagonists versus antacids, outcome: 10.1 Clinically important upper GI bleeding.
8.
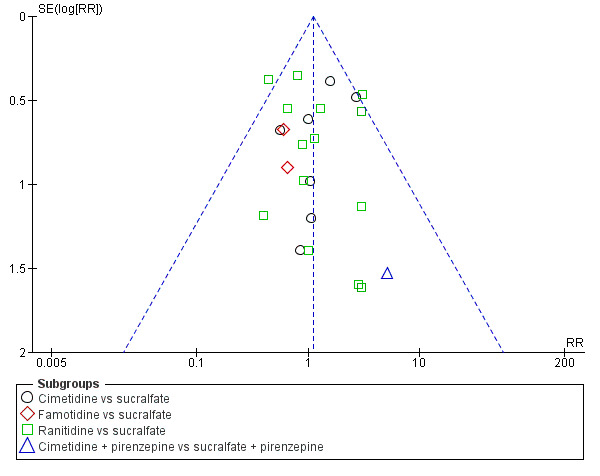
Funnel plot of comparison: 11 H2 receptor antagonists versus sucralfate, outcome: 11.1 Clinically important upper GI bleeding.
9.

Funnel plot of comparison: 19 Antacids versus sucralfate, outcome: 19.1 Clinically important upper GI bleeding.
Other potential sources of bias
Review authors classified 20 studies as having unclear or high risk of bias, as reports were unclear on the baseline characteristics of randomised participants (Ali 2016; Basso 1981; Behrens 1994; Bresalier 1987; Conrad 2005; Darlong 2003; Fogas 2013; He 2017; Lacroix 1986; Larson 1989; Levy 1997; Maasoumi 2016; Macdougall 1977; Mahul 1992; Ortiz 1998; Ruiz‐Santana 1991; Selvanderan 2015; Selvanderan 2016; Wang 2015; Wee 2013). Three studies were published only as conference abstracts and did not report enough data for assessment of other biases (Fogas 2013; Selvanderan 2016; Wee 2013). These reasons are elaborately described in the risk of bias tables for these respective studies.
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7; Table 8; Table 9
Summary of findings for the main comparison. Interventions compared with placebo or no prophylaxis for preventing upper gastrointestinal bleeding in people admitted to intensive care units.
| Any intervention compared with placebo or no prophylaxis for preventing upper gastrointestinal bleeding in people admitted to intensive care units | ||||||
| Patient or population: people admitted to intensive care units Setting: ICU Intervention: any intervention Comparison: placebo or no prophylaxis | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo or no prophylaxis | Risk with Interventions | |||||
|
Clinically important upper GI bleeding Follow‐up: 15 days† |
Study population | RR 0.47 (0.39 to 0.57) | 3207 (30 RCTs) | ⊕⊕⊕⊝ MODERATEa | ||
| 188 per 1000 | 88 per 1000 (73 to 107) | |||||
|
Nosocomial pneumonia Follow‐up: 48 hours after extubation‡ |
Study population | RR 1.15 (0.90 to 1.48) | 1331 (9 RCTs) | ⊕⊕⊝⊝ LOWb,c | ||
| 143 per 1000 | 164 per 1000 (129 to 211) | |||||
|
All‐cause mortality in ICU Follow‐up: 4 weeks§ |
Study population | RR 1.10 (0.90 to 1.34) | 2159 (19 RCTs) | ⊕⊕⊝⊝ LOWb,d | ||
| 152 per 1000 | 168 per 1000 (137 to 204) | |||||
|
Duration of ICU stay Follow‐up: not reported |
Mean duration of ICU stay ranged from 8.6 to 11.1 days | MD 0.24 days higher (1.13 days lower to 1.61 higher days) | ‐ | 447 (2 RCTs) | ⊕⊕⊝⊝ LOWb,e | |
|
Number of participants requiring blood transfusion Follow‐up: 48 hours after discharge‡ |
Study population | RR 0.63 (0.41 to 0.97) | 981 (9 RCTs) | ⊕⊕⊕⊝ MODERATEf | ||
| 96 per 1000 | 60 per 1000 (39 to 93) | |||||
| Serious adverse events | Not reported | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI).
‡Duration of follow‐up reported in one study. †Duration of follow‐up reported in four studies. §Duration of follow‐up reported in five studies. CI: confidence interval; GI: gastrointestinal; ICU: intensive care unit; MD: mean difference; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence. High certainty: We are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect. Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded by one level for risk of bias because of high risk of selection bias in one study, high risk of performance bias in nine studies, high risk of detection bias in five studies, high risk of attrition bias in four studies, high risk of reporting bias in five studies, and high risk of other biases in four studies.
bDowngraded by one level for imprecision because effect estimate and 95% CI were compatible with benefit and harm.
cDowngraded by one level for risk of bias because of high risk of performance bias in three studies, high risk of detection bias in one study, and high risk of attrition bias in two studies.
dDowngraded by one level for risk of bias because of high risk of performance bias in seven studies and high risk of attrition bias in two studies.
eDowngraded by one level for risk of bias because of high risk of performance bias in one study.
fDowngraded by one level for risk of bias because of high risk of selection bias in one study, high risk of performance bias in two studies, high risk of attrition bias in one study, high risk of reporting bias in one study, and high risk of other biases in one study.
Summary of findings 2. H2 receptor antagonists compared with placebo or no prophylaxis for preventing upper gastrointestinal bleeding in people admitted to intensive care units.
| H2 receptor antagonists compared with placebo or no prophylaxis for preventing upper gastrointestinal bleeding in people admitted to intensive care units | ||||||
| Patient or population: people admitted to intensive care units Setting: ICU Intervention: H2 receptor antagonists Comparison: placebo or no prophylaxis | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo or no prophylaxis | Risk with H2 receptor antagonists | |||||
|
Clinically important upper GI bleeding Follow‐up: 15 days/weeks† |
Study population | RR 0.50 (0.36 to 0.70) | 2149 (24 RCTs) | ⊕⊕⊕⊝ MODERATEa | ||
| 182 per 1000 | 91 per 1000 (65 to 127) | |||||
|
Nosocomial pneumonia Follow‐up: 48 hours after extubation‡ |
Study population | RR 1.12 (0.85 to 1.48) | 945 (8 RCTs) | ⊕⊕⊝⊝ LOWb,c | ||
| 146 per 1000 | 164 per 1000 (124 to 216) | |||||
|
All‐cause mortality in ICU Follow‐up: 4 weeks§ |
Study population | RR 1.12 (0.88 to 1.42) | 1428 (14 RCTs) | ⊕⊕⊝⊝ LOWb,d | ||
| 145 per 1000 | 162 per 1000 (127 to 205) | |||||
|
Duration of ICU stay Follow‐up: not reported |
Mean duration of ICU stay ranged from 8.6 to 11.1 days | MD 0.73 days higher (0.92 days lower to 2.38 days higher) | ‐ | 230 (2 RCTs) | ⊕⊕⊝⊝ LOWb,e | |
|
Number of participants requiring blood transfusions Follow‐up: 48 hours after extubationǁ |
Study population | RR 0.58 (0.36 to 0.95) | 655 (7 RCTs) | ⊕⊕⊕⊝ MODERATEf | ||
| 112 per 1000 | 65 per 1000 (40 to 107) | |||||
| Serious adverse events | Not reported | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI).
†Duration of follow‐up reported in four studies. ‡Duration of follow‐up reported in two studies. §Duration of follow‐up reported in five studies. ǁDuration of follow‐up reported in one study. CI: confidence interval; GI: gastrointestinal; ICU: intensive care unit; MD: mean difference; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence. High certainty: We are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect. Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded by one level for risk of bias because of high risk of selection bias in one study, high risk of performance bias in eight studies, high risk of attrition bias in two studies, high risk of reporting bias in four studies, and high risk of other biases in three studies.
bDowngraded by one level for imprecision because 95% CI was compatible with benefit and harm.
cDowngraded by one level for risk of bias because of high risk performance bias in three studies and high risk of attrition bias in one study.
dDowngraded by one level for risk of bias because of high risk of performance bias in three studies and high risk of attrition bias in one study.
eDowngraded by one level for risk of bias because of high risk of performance bias in one study.
fDowngraded by one level for risk of bias because of high risk of selection bias in one study, high risk of performance bias in two studies, high risk of attrition bias in one study, and high risk of other biases in one study.
Summary of findings 3. Proton pump inhibitors compared with placebo or no prophylaxis for preventing upper gastrointestinal bleeding in people admitted to intensive care units.
| Proton pump inhibitors compared with placebo or no prophylaxis for preventing upper gastrointestinal bleeding in people admitted to intensive care units | ||||||
| Patient or population: people admitted to intensive care units Setting: ICU Intervention: proton pump inhibitors Comparison: placebo or no prophylaxis | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo or no prophylaxis | Risk with proton pump inhibitors | |||||
|
Clinically important upper GI bleeding Follow‐up: not reported |
Study population | RR 0.63 (0.18 to 2.22) | 237 (3 RCTs) | ⊕⊕⊝⊝ LOWa,b | ||
| 49 per 1000 | 31 per 1000 (9 to 108) | |||||
|
Nosocomial pneumonia Follow‐up: not reported |
Study population | RR 1.24 (0.77 to 1.98) | 227 (2 RCTs) | ⊕⊕⊝⊝ LOWa,c | ||
| 188 per 1000 | 233 per 1000 (145 to 372) | |||||
|
All‐cause mortality in ICU Follow‐up: not reported |
Study population | RR 1.09 (0.60 to 1.99) | 258 (3 RCTs) | ⊕⊕⊝⊝ LOWa,c | ||
| 134 per 1000 | 146 per 1000 (80 to 266) | |||||
|
Duration of ICU stay Follow‐up: not reported |
Mean duration of ICU stay ranged from 8.6 to 11.1 days | MD 0.03 days lower (1.63 days lower to 1.58 days higher) | ‐ | 227 (2 RCTs) | ⊕⊕⊝⊝ LOWa,c | |
| Number of participants requiring blood transfusion | Not reported | |||||
| Serious adverse events | Not reported | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; GI: gastrointestinal; ICU: intensive care unit; MD: mean difference; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence. High certainty: We are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect. Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded by one level for imprecision because 95% CI was compatible with benefit and harm.
bDowngraded by one level for risk of bias because of high risk of performance bias in one study and high risk of attrition bias in one study.
cDowngraded by one level for risk of bias because of high risk of performance bias in one study.
Summary of findings 4. Antacids compared with placebo or no prophylaxis for preventing upper gastrointestinal bleeding in people admitted to intensive care units.
| Antacids compared with placebo or no prophylaxis for preventing upper gastrointestinal bleeding in people admitted to intensive care units | ||||||
| Patient or population: people admitted to intensive care units Setting: ICU Intervention: antacids Comparison: placebo or no prophylaxis | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo or no prophylaxis | Risk with antacids | |||||
| Clinically important upper GI bleeding Follow‐up: not reported |
Study population | RR 0.49 (0.25 to 0.99) | 774 (8 RCTs) | ⊕⊕⊝⊝ LOWa,b | ||
| 170 per 1000 | 83 per 1000 (43 to 168) | |||||
| Nosocomial pneumonia | Not reported | |||||
| All‐cause mortality in ICU Follow‐up: not reported |
Study population | RR 1.01 (0.53 to 1.96) | 300 (2 RCTs) | ⊕⊕⊝⊝ LOWc,d | ||
| 161 per 1000 | 163 per 1000 (85 to 316) | |||||
| Duration of ICU stay | Not reported | |||||
| Number of participants requiring blood transfusions Follow‐up: not reported |
Study population | RR 0.94 (0.30 to 2.96) | 226 (2 RCTs) | ⊕⊕⊝⊝ LOWc,e | ||
| 45 per 1000 | 43 per 1000 (14 to 135) | |||||
| Serious adverse events | Not reported | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; GI: gastrointestinal; ICU: intensive care unit; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence. High certainty: We are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect. Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded by one level for inconsistency because of moderate heterogeneity; I² = 56%.
bDowngraded by one level for risk of bias because of high risk of performance bias in five studies, high risk of detection bias in one study, high risk of reporting bias in two studies, and high risk of other biases in one study.
cDowngraded by one level for imprecision because 95% CI was compatible with benefit and harm.
dDowngraded by one level for risk of bias because of high risk of performance bias in two studies.
eDowngraded by one level for risk of bias because of high risk of performance bias in one study.
Summary of findings 5. Sucralfate compared with placebo or no prophylaxis for preventing upper gastrointestinal bleeding in people admitted to intensive care units.
| Sucralfate compared with placebo or no prophylaxis for preventing upper gastrointestinal bleeding in people admitted to intensive care units | ||||||
| Patient or population: people admitted to intensive care units Setting: ICU Intervention: sucralfate Comparison: placebo or no prophylaxis | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo or no prophylaxis | Risk with sucralfate | |||||
| Clinically important upper GI bleeding Follow‐up: 15 days† |
Study population | RR 0.53 (0.32 to 0.88) | 598 (7 RCTs) | ⊕⊕⊕⊝ MODERATEa | ||
| 108 per 1000 | 57 per 1000 (35 to 95) | |||||
| Nosocomial pneumonia Follow‐up: not reported |
Study population | RR 1.33 (0.86 to 2.04) | 450 (4 RCTs) | ⊕⊕⊝⊝ LOWb,c | ||
| 122 per 1000 | 163 per 1000 (105 to 249) | |||||
| All‐cause mortality in ICU Follow‐up: 15 days† |
Study population | RR 0.97 (0.66 to 1.43) | 500 (5 RCTs) | ⊕⊕⊝⊝ LOWb,d | ||
| 165 per 1000 | 160 per 1000 (109 to 236) | |||||
| Duration of ICU stay Follow‐up: not reported |
Mean duration of ICU stay ranged from 8.6 to 11.1 days | MD 0.02 days lower (1.70 days lower to 1.65 days higher) | ‐ | 224 (2 RCTs) | ⊕⊕⊝⊝ LOWb,e | |
| Number of participants requiring blood transfusion Follow‐up: not reported |
Study population | RR 0.60 (0.15 to 2.44) | 200 (1 RCT) | ⊕⊕⊝⊝ LOWb,e | ||
| 50 per 1000 | 30 per 1000 (8 to 122) | |||||
| Serious adverse events | Not reported | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). †Duration of follow‐up reported in only one study. CI: confidence interval; GI: gastrointestinal; ICU: intensive care unit; MD: mean difference; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence. High certainty: We are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect. Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded by one level for risk of bias because of high risk of performance bias in five studies, high risk of reporting bias in one study, and high risk of other biases in one study.
bDowngraded by one level for imprecision because 95% CI was compatible with benefit and harm.
cDowngraded by one level for risk of bias because of high risk of performance bias in two studies.
dDowngraded by one level for risk of bias because of high risk of performance bias in three studies.
eDowngraded by one level for risk of bias because of high risk of performance bias in one study.
Summary of findings 6. H2 receptor antagonists compared with proton pump inhibitors for preventing upper gastrointestinal bleeding in people admitted to intensive care units.
| H2 receptor antagonists compared with proton pump inhibitors for preventing upper gastrointestinal bleeding in people admitted to intensive care units | ||||||
| Patient or population: people admitted to intensive care units Setting: ICU Intervention: H2 receptor antagonists Comparison: proton pump inhibitors | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with proton pump inhibitors | Risk with H2 receptor antagonists | |||||
| Clinically important upper GI bleeding Follow‐up: not reported |
Study population | RR 2.90 (1.83 to 4.58) | 1636 (13 RCTs) | ⊕⊕⊝⊝ LOWa,b | ||
| 25 per 1000 | 73 per 1000 (46 to 115) | |||||
| Nosocomial pneumonia Follow‐up: 30 days† |
Study population | RR 1.02 (0.77 to 1.35) | 1256 (10 RCTs) | ⊕⊕⊝⊝ LOWc,d | ||
| 123 per 1000 | 126 per 1000 (95 to 166) | |||||
| All‐cause mortality in ICU Follow‐up: 30 days† |
Study population | RR 0.96 (0.78 to 1.19) | 1564 (12 RCTs) | ⊕⊕⊝⊝ LOWc,e | ||
| 158 per 1000 | 152 per 1000 (124 to 189) | |||||
| Duration of ICU stay Follow‐up: not reported |
Mean duration of ICU stay ranged from 7.7 to 23.6 days | MD 0.14 days higher (1.14 days lower to 1.41 days higher) | ‐ | 482 (5 RCTs) | ⊕⊕⊝⊝ LOWc,f | |
| Number of participants requiring blood transfusion Follow‐up: not reported |
Study population | RR 1.98 (0.75 to 5.21) | 575 (3 RCTs) | ⊕⊕⊕⊝ MODERATEc | ||
| 17 per 1000 | 35 per 1000 (13 to 91) | |||||
| Serious adverse events | Not reported | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). †Duration of follow‐up reported in only one study. CI: confidence interval; GI: gastrointestinal; ICU: intensive care unit; MD: mean difference; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence. High certainty: We are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect. Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded by one level for inconsistency because of substantial heterogeneity; I² = 59%.
bDowngraded by one level for risk of bias because of high risk of selection bias in one study, high risk of performance bias in five studies, high risk of detection bias in two studies, and high risk of other biases in one study.
cDowngraded by one level for imprecision because 95% CI was compatible with benefit and harm.
dDowngraded by one level for risk of bias because of high risk of performance bias in four studies, high risk of detection bias in two studies, and high risk of other biases in one study.
eDowngraded by one level for risk of bias because of high risk of performance bias in five studies, high risk of attrition bias in one study, and high risk of other biases in one study.
fDowngraded by one level for risk of bias because of high risk of performance bias in three studies, high risk of attrition bias in one study, and high risk of other biases in one study.
Summary of findings 7. H2 receptor antagonists compared with antacids for preventing upper gastrointestinal bleeding in people admitted to intensive care units.
| H2 receptor antagonists compared with antacids for preventing upper gastrointestinal bleeding in people admitted to intensive care units | ||||||
| Patient or population: people admitted to intensive care units Setting: ICU Intervention: H2 receptor antagonists Comparison: antacids | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with antacids | Risk with H2 receptor antagonists | |||||
| Clinically important upper GI bleeding Follow‐up: 25 days† |
Study population | RR 0.96 (0.67 to 1.36) | 1700 (16 RCTs) | ⊕⊕⊝⊝ LOWa,b | ||
| 86 per 1000 | 82 per 1000 (57 to 117) | |||||
| Nosocomial pneumonia Follow‐up: 25 days‡ |
Study population | RR 1.05 (0.81 to 1.36) | 581 (4 RCTs) | ⊕⊕⊝⊝ LOWa,c | ||
| 280 per 1000 | 294 per 1000 (227 to 381) | |||||
| All‐cause mortality in ICU Follow‐up: 25 days§ |
Study population | RR 1.01 (0.66 to 1.55) | 1321 (11 RCTs) | ⊕⊝⊝⊝ VERY LOWa,d,e | ||
| 163 per 1000 | 165 per 1000 (108 to 253) | |||||
| Duration of ICU stay | Not reported | |||||
| Number of participants requiring blood transfusion Follow‐up: not reported |
Study population | RR 2.49 (1.35 to 4.62) | 744 (6 RCTs) | ⊕⊕⊕⊝ MODERATEf | ||
| 30 per 1000 | 75 per 1000 (41 to 139) | |||||
| Serious adverse events | Not reported | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI).
†Duration of follow‐up reported in two studies. ‡Duration of follow‐up reported in one study. §Duration of follow‐up reported in three studies. CI: confidence interval; GI: gastrointestinal; ICU: intensive care unit; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence. High certainty: We are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect. Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded by one level for imprecision because 95% CI was compatible with benefit and harm.
bDowngraded by one level for risk of bias because of high risk of selection bias in two studies, high risk of performance bias in 12 studies, high risk of detection bias in two studies, and high risk of reporting bias in two studies.
cDowngraded by one level for risk of bias because of high risk of selection bias in one study, high risk of performance bias in four studies, high risk of detection bias in one study, and high risk of reporting bias in one study.
dDowngraded by one level for inconsistency because of moderate heterogeneity; I² = 53%.
eDowngraded by one level for risk of bias because of high risk of selection bias in two studies, high risk of performance bias in nine studies, and high risk of reporting bias in one study.
fDowngraded by one level for risk of bias because of high risk of selection bias in one study and high risk of performance bias in four studies.
Summary of findings 8. H2 receptor antagonists compared with sucralfate for preventing upper gastrointestinal bleeding in people admitted to intensive care units.
| H2 receptor antagonists compared with sucralfate for preventing upper gastrointestinal bleeding in people admitted to intensive care units | ||||||
| Patient or population: people admitted to intensive care units Setting: ICU Intervention: H2 receptor antagonists Comparison: sucralfate | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with sucralfate | Risk with H2 receptor antagonists | |||||
| Clinically important upper GI bleeding Follow‐up: 15 days† |
Study population | RR 1.10 (0.87 to 1.41) | 3316 (24 RCTs) | ⊕⊕⊝⊝ LOWa,b | ||
| 66 per 1000 | 73 per 1000 (58 to 93) | |||||
| Nosocomial pneumonia Follow‐up: 25 days‡ |
Study population | RR 1.22 (1.07 to 1.40) | 3041 (17 RCTs) | ⊕⊕⊕⊝ MODERATEc | ||
| 189 per 1000 | 230 per 1000 (202 to 264) | |||||
| All‐cause mortality in ICU Follow‐up: 25 days§ |
Study population | RR 1.09 (0.95 to 1.24) | 3178 (21 RCTs) | ⊕⊕⊝⊝ LOWa,d | ||
| 204 per 1000 | 222 per 1000 (194 to 253) | |||||
| Duration of ICU stay Follow‐up: 2 weeks |
Mean duration of ICU stay ranged from 7.9 to 13.7 days | MD 0.01 days higher (1.92 days lower to 1.95 days higher) | ‐ | 1791 (6 RCTs) | ⊕⊝⊝⊝ VERY LOWa,e,f | |
| Number of participants requiring blood transfusion Follow‐up: until death or dischargeǁ |
Study population | RR 1.25 (0.70 to 2.23) | 1095 (9 RCTs) | ⊕⊕⊝⊝ LOWa,g | ||
| 35 per 1000 | 43 per 1000 (24 to 77) | |||||
| Serious adverse events | Not reported | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). †Duration of follow‐up reported in five studies. ‡Duration of follow‐up reported in three studies. §Duration of follow‐up reported in six studies. ǁDuration of follow‐up reported in one study. CI: confidence interval; GI: gastrointestinal; ICU: intensive care unit; MD: mean difference; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence. High certainty: We are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect. Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded by one level for imprecision because 95% CI was compatible with benefit and harm.
bDowngraded by one level for risk of bias because of high risk of selection bias in one study, high risk of performance bias in 20 studies, high risk of detection bias in two studies, high risk of attrition bias in two studies, high risk of reporting bias in four studies, and high risk of other biases in two studies.
cDowngraded by one level for risk of bias because of high risk of selection bias in two studies, high risk of performance bias in 12 studies, high risk of detection bias in one study, high risk of attrition bias in one study, and high risk of reporting bias in two studies.
dDowngraded by one level for risk of bias because of high risk of selection bias in two studies, high risk of performance bias in 16 studies, high risk of detection bias in one study, high risk of attrition bias in two studies, high risk of reporting bias in three studies, and high risk of other biases in one study.
eDowngraded by one level for inconsistency because of considerable heterogeneity; I² = 82%.
fDowngraded by one level for risk of bias because of high risk of performance bias in four studies and high risk of attrition bias in one study.
gDowngraded by one level for risk of bias because of high risk of performance bias in eight studies, high risk of attrition bias in one study, high risk of reporting bias in one study, and high risk of other biases in one study.
Summary of findings 9. Antacids compared with sucralfate for preventing upper gastrointestinal bleeding in people admitted to intensive care units.
| Antacids compared with sucralfate for preventing upper gastrointestinal bleeding in people admitted to intensive care units | ||||||
| Patient or population: people admitted to intensive care units Setting: ICU Intervention: antacids Comparison: sucralfate | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with sucralfate | Risk with antacids | |||||
| Clinically important upper GI bleeding Follow‐up: 21 days† |
Study population | RR 1.00 (0.72 to 1.39) | 1772 (16 RCTs) | ⊕⊕⊝⊝ LOWa,b | ||
| 66 per 1000 | 66 per 1000 (47 to 91) | |||||
| Nosocomial pneumonia Follow‐up: 25 days‡ |
Study population | RR 1.04 (0.84 to 1.30) | 996 (7 RCTs) | ⊕⊕⊝⊝ LOWa,c | ||
| 232 per 1000 | 242 per 1000 (195 to 302) | |||||
| All‐cause mortality in ICU Follow‐up: 25 days† |
Study population | RR 1.15 (0.93 to 1.40) | 1249 (11 RCTs) | ⊕⊕⊝⊝ LOWa,d | ||
| 206 per 1000 | 237 per 1000 (192 to 289) | |||||
| Duration of ICU stay Follow‐up: not reported |
Mean duration of ICU stay ranged from 10.4 to 16.8 days | MD 2.5 days lower (6.61 days lower to 1.61 days higher) | ‐ | 227 (2 RCTs) | ⊕⊕⊝⊝ LOWa,e | |
| Number of participants requiring blood transfusion Follow‐up: until discharge or onset of GI bleeding§ |
Study population | RR 0.73 (0.40 to 1.34) | 667 (6 RCTs) | ⊕⊕⊝⊝ LOWa,f | ||
| 52 per 1000 | 38 per 1000 (21 to 69) | |||||
| Serious adverse events | Not reported | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI).
†Duration of follow‐up reported in four studies. ‡Duration of follow‐up reported in two studies. §Duration of follow‐up reported in one study. CI: confidence interval; GI: gastrointestinal; ICU: intensive care unit; MD: mean difference; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence. High certainty: We are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect. Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded by one level for imprecision because 95% CI was compatible with benefit and harm.
bDowngraded by one level for risk of bias because of high risk of selection bias in three studies, high risk of performance bias in 12 studies, high risk of detection bias in one study, high risk of attrition bias in one study, high risk of reporting bias in two studies, and high risk of other biases in two studies.
cDowngraded by one level for risk of bias because of high risk of performance bias in four studies, high risk of detection bias in one study, high risk of reporting bias in one study, and high risk of other biases in one study.
dDowngraded by one level for risk of bias because of high risk of selection bias in three studies, high risk of performance bias in eight studies, high risk of attrition bias in one study, high risk of reporting bias in one study, and high risk of other biases in one study.
eDowngraded by one level for risk of bias because of high risk of attrition bias in one study.
fDowngraded by one level for risk of bias because of high risk of selection bias in one study, high risk of performance bias in six studies, and high risk of other biases in one study.
Interventions versus placebo or no prophylaxis
For this comparison, data on eight of our pre‐defined outcomes were available.
Clinically important upper GI bleeding
Clinically important upper GI bleeding was analysed (Analysis 1.1) for the comparison of any intervention (H2 receptor antagonists, proton pump inhibitors, prostaglandin analogues, anticholinergics, and sucralfate) versus placebo or no prophylaxis. We identified 31 studies that contributed data to this outcome, which we categorised according to drug class. There seems to be a beneficial effect of any intervention versus placebo or no prophylaxis on the occurrence of clinically important upper GI bleeding (risk ratio (RR) 0.47, 95% confidence interval (CI) 0.39 to 0.57; moderate certainty of evidence). We have elaborately mentioned further results below under each respective comparison.
1.1. Analysis.

Comparison 1 Interventions versus placebo or no prophylaxis, Outcome 1 Clinically important upper GI bleeding.
Nosocomial pneumonia
Nosocomial pneumonia (which included ventilator‐associated pneumonia) was analysed (Analysis 1.2) for the comparison of any interventions (H2 receptor antagonists, proton pump inhibitors, anticholinergics, and sucralfate vs placebo or no prophylaxis). We identified nine studies that contributed data to this outcome and categorised these data according to drug class. With respect to the occurrence of differences in nosocomial pneumonia between study groups (RR 1.15, 95% CI 0.90 to 1.48; low certainty of evidence), the use of any intervention versus no intervention or prophylaxis was consistent with benefits and harms. We have elaborately mentioned these results below under each respective comparison.
1.2. Analysis.

Comparison 1 Interventions versus placebo or no prophylaxis, Outcome 2 Nosocomial pneumonia.
All‐cause mortality in ICU
All‐cause mortality in the ICU was analysed (Analysis 1.3) for the comparison of any interventions (H2 receptor antagonists, proton pump inhibitors, prostaglandin analogues, anticholinergics, antacids, and sucralfate vs placebo or no prophylaxis). We identified 18 studies that contributed data to this outcome and organised the data from these studies according to drug class. With respect to all‐cause mortality in the ICU, the effect estimate of any intervention versus no intervention or prophylaxis was consistent with benefit and harm (RR 1.10, 95% CI 0.90 to 1.34; low certainty of evidence). We have elaborately mentioned these results below under each respective comparison.
1.3. Analysis.

Comparison 1 Interventions versus placebo or no prophylaxis, Outcome 3 All‐cause mortality in ICU.
All‐cause mortality in hospital
All‐cause mortality in the hospital was analysed (Analysis 1.4) for the comparison of any interventions (H2 receptor antagonists, proton pump inhibitors, antacids, and sucralfate vs placebo or no prophylaxis). We identified five studies that contributed data to this outcome and categorised the data from these studies according to drug class. The effect estimate of any intervention versus no intervention or prophylaxis was consistent with benefits and harms with respect to all‐cause mortality in the hospital (RR 1.15, 95% CI 0.85 to 1.55). We have elaborately mentioned these results below under each respective comparison.
1.4. Analysis.

Comparison 1 Interventions versus placebo or no prophylaxis, Outcome 4 All‐cause mortality in hospital.
Duration of ICU stay
Duration of ICU stay was analysed (Analysis 1.5) for the comparison of any interventions (H2 receptor antagonists, proton pump inhibitors, and sucralfate vs placebo or no prophylaxis). We identified two studies that contributed data to this outcome and categorised the data from these studies according to drug class. The effect estimate of any intervention versus no intervention or prophylaxis was consistent with benefits and harms with respect to duration of ICU stay (mean difference (MD) 0.24 days, 95% CI ‐1.13 to 1.61; low certainty of evidence). We have elaborately mentioned these results below under each respective comparison.
1.5. Analysis.
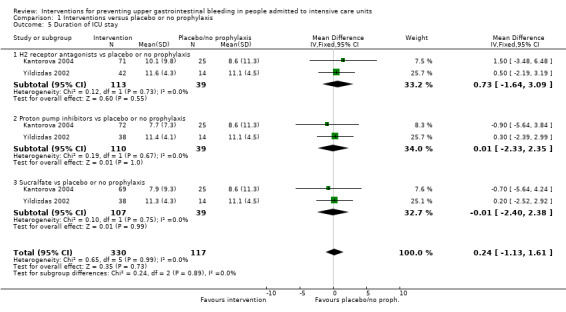
Comparison 1 Interventions versus placebo or no prophylaxis, Outcome 5 Duration of ICU stay.
Duration of intubation
Duration of intubation was analysed (Analysis 1.6) for the comparison of any interventions (H2 receptor antagonists, proton pump inhibitors, and sucralfate vs placebo or no prophylaxis). We identified two studies that contributed data to this outcome and categorised the data from these studies according to drug class. The effect estimate of any intervention versus no intervention or prophylaxis was consistent with benefits and harms with respect to duration of intubation (MD 0.87 days, 95% CI ‐0.58 to 2.31; low certainty of evidence). We have elaborately mentioned these results below under each respective comparison.
1.6. Analysis.
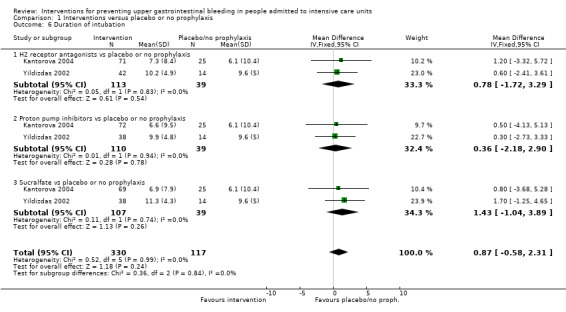
Comparison 1 Interventions versus placebo or no prophylaxis, Outcome 6 Duration of intubation.
Number of participants requiring blood transfusions
The number of participants requiring blood transfusions was analysed (Analysis 1.7) for the comparison of any interventions (H2 receptor antagonists, antacids, and sucralfate vs placebo or no prophylaxis). We identified nine studies that contributed data to this outcome and categorised the data from these studies according to drug class. Data seem to show a reduction in the number of participants in the intervention group requiring blood transfusion (RR 0.63, 95% CI 0.41 to 0.97; moderate certainty of evidence). We have elaborately mentioned these results below under each respective comparison.
1.7. Analysis.

Comparison 1 Interventions versus placebo or no prophylaxis, Outcome 7 Number of participants requiring blood transfusions.
Units of blood transfused
The number of units of blood transfused was analysed (Analysis 1.8) for the comparison of any interventions (H2 receptor antagonists and sucralfate vs placebo or no prophylaxis). We identified two studies that contributed data to this outcome and categorised the data from these studies according to drug class. The effect estimate of any intervention versus no intervention or prophylaxis was consistent with benefits and harms with respect to the number of units of blood transfused (MD 0.09 units, 95% CI ‐0.99 to 1.17). We have elaborately mentioned these results below under each respective comparison.
1.8. Analysis.

Comparison 1 Interventions versus placebo or no prophylaxis, Outcome 8 Units of blood transfused.
H2 receptor antagonists versus placebo or no prophylaxis
For this comparison, data on nine of our pre‐defined outcomes were available.
Clinically important upper GI bleeding
We identified 24 studies relevant to this outcome (N = 2149) and stratified data into six groups according to drug class members of H2 receptor antagonists. For this outcome, we found evidence suggesting that H2 receptor antagonists reduced the risk of clinically important upper GI bleeding compared with placebo or no prophylaxis (8.3% vs 17.7%; RR 0.50, 95% CI 0.36 to 0.70; Analysis 2.1). We judged the certainty of the evidence as moderate. This outcome had moderate levels of heterogeneity (Chi² = 38.78, df = 23.0 (P = 0.02), I² = 40%).
2.1. Analysis.

Comparison 2 H2 receptor antagonists versus placebo or no prophylaxis, Outcome 1 Clinically important upper GI bleeding.
Cimetidine versus placebo
We found 10 studies that were relevant to this comparison (N = 772). For this comparison, the effect was consistent with benefits and harms (RR 0.53, 95% CI 0.28 to 1.02). For this outcome, heterogeneity was substantial (Chi² = 22.25, df = 9.0 (P = 0.008), I² = 60%; Analysis 2.1).
Famotidine versus placebo
For this comparison, we identified a single relevant study (N = 146). The effect was consistent with benefits and harms (RR 2.11, 95% CI 0.20 to 22.79; Analysis 2.1).
Ranitidine versus placebo
We found five relevant studies for this comparison (N = 446). Evidence suggested a beneficial effect of H2 receptor antagonists compared with placebo within this comparison (RR 0.36, 95% CI 0.17 to 0.77; Analysis 2.1).
Cimetidine versus no prophylaxis
For this comparison, we identified three relevant studies (N = 516). In this subgroup, the effect was consistent with benefits and harms (RR 0.59, 95% CI 0.23 to 1.48). For this comparison, heterogeneity was moderate (Chi² = 3.53, df = 2.0 (P = 0.17), I² = 43%) (Analysis 2.1).
Famotidine versus no prophylaxis
We identified a single study for this comparison (N = 50). We found evidence of benefit of H2 receptor antagonists compared with no prophylaxis (RR 0.30, 95% CI 0.09 to 0.96; Analysis 2.1).
Ranitidine versus no prophylaxis
We found four studies that were relevant to this comparison (N = 219). The effect of H2 receptor antagonists versus no prophylaxis was consistent with benefits and harms (RR 0.51, 95% CI 0.26 to 1.00; Analysis 2.1). One study with 19 participants in the ranitidine group and 21 participants in the no prophylaxis group reported no events of clinically important upper GI bleeding (Reusser 1990).
Nosocomial pneumonia
We identified eight studies that were relevant to this outcome (N = 945) and divided the data into five subcategories in accordance with our protocol. The effect of H2 receptor antagonists versus no prophylaxis on nosocomial pneumonia was consistent with benefits and harms (15.8% vs 14.6%; RR 1.12, 95% CI 0.85 to 1.48; Analysis 2.2). We judged the certainty of this evidence as low.
2.2. Analysis.

Comparison 2 H2 receptor antagonists versus placebo or no prophylaxis, Outcome 2 Nosocomial pneumonia.
Cimetidine versus placebo
We identified two relevant studies for this comparison (N = 204). The effect of cimetidine versus placebo on nosocomial pneumonia was consistent with benefits and harms (RR 0.34, 95% CI 0.06 to 2.00). This comparison showed moderate heterogeneity (Chi² = 1.59, df = 1.0 (P = 0.21), I² = 37%; Analysis 2.2).
Famotidine versus placebo
We included a single study for this comparison (N = 146). The effect of famotidine versus placebo was consistent with benefits and harms (RR 1.48, 95% CI 0.49 to 4.45; Analysis 2.2).
Ranitidine versus placebo
We found two studies that were relevant to this comparison (N = 277). The effect was consistent with benefits and harms (RR 0.79, 95% CI 0.47 to 1.31; Analysis 2.2).
Cimetidine versus no prophylaxis
We included a single study in this comparison (N = 200). The effect was consistent with benefits and harms (RR 2.17, 95% CI 0.86 to 5.47; Analysis 2.2).
Ranitidine versus no prophylaxis
We found two studies that were relevant to this comparison (N = 118). Within this comparison, the effect was consistent with benefits and harms (RR 1.33, 95% CI 0.93 to 1.90; Analysis 2.2).
All‐cause mortality in ICU
We identified 14 studies that contributed data to this outcome (N = 1428). With respect to all‐cause mortality in the ICU, the effect of H2 receptor antagonists versus no prophylaxis on nosocomial pneumonia was consistent with benefits and harms (15.8% vs 14.3%; RR 1.12, 95% CI 0.88 to 1.42; Analysis 2.3). We judged the certainty of this evidence as low.
2.3. Analysis.

Comparison 2 H2 receptor antagonists versus placebo or no prophylaxis, Outcome 3 All‐cause mortality in ICU.
Cimetidine versus placebo
We found four studies that were relevant to this comparison (N = 478). Data showed no clear difference between H2 receptor antagonists and placebo (RR 1.05, 95% CI 0.66 to 1.68; Analysis 2.3).
Famotidine versus placebo
We identified a single study for this comparison (N = 146). Data show no clear difference between H2 receptor antagonists and placebo (RR 1.32, 95% CI 0.55 to 3.16; Analysis 2.3).
Ranitidine versus placebo
We found two relevant studies for this comparison (N = 148). We found no evidence showing a clear difference between the two groups (RR 0.69, 95% CI 0.31 to 1.54; Analysis 2.3). One study (N = 21) reported no events of all‐cause mortality in both groups on the ICU (Powell 1993).
Cimetidine versus no prophylaxis
We found two relevant studies for this comparison (N = 400). Data show no clear difference between H2 receptor antagonists and no prophylaxis within this comparison (RR 1.00, 95% CI 0.61 to 1.63). For this outcome, heterogeneity was considerable (Chi² = 5.12, df = 1.0 (P = 0.02), I² = 80%; Analysis 2.3).
Famotidine versus no prophylaxis
We found one study that was relevant to this comparison (N = 50). For this comparison, we found no evidence showing a clear difference between the two groups (RR 1.25, 95% CI 0.59 to 2.64; Analysis 2.3).
Ranitidine versus no prophylaxis
We found four studies that were relevant to this comparison (N = 206). Data show no clear difference between H2 receptor antagonists and no prophylaxis (RR 1.58, 95% CI 0.97 to 2.58; Analysis 2.3).
All‐cause mortality in hospital
For this outcome, we found four relevant studies (N = 487). Data seemed to show no difference between H2 receptor antagonists and placebo or no prophylaxis with respect to all‐cause mortality in the hospital (18.4% vs 15.7%; RR 1.16, 95% CI 0.79 to 1.70; Analysis 2.4).
2.4. Analysis.

Comparison 2 H2 receptor antagonists versus placebo or no prophylaxis, Outcome 4 All‐cause mortality in hospital.
Famotidine versus placebo
We found one study that was relevant to this comparison (N = 146). For this comparison, we found no evidence of a clear difference between the two groups (RR 0.89, 95% CI 0.43 to 1.86; Analysis 2.4).
Ranitidine versus placebo
We found one study that was relevant to this comparison (N = 101). For this comparison, we did not find evidence of a clear difference between the two groups (RR 0.35, 95% CI 0.01 to 8.47; Analysis 2.4).
Cimetidine versus no prophylaxis
We identified a single study for this comparison (N = 200). We found no evidence of a clear difference between the two groups (RR 1.47, 95% CI 0.88 to 2.46; Analysis 2.4).
Ranitidine versus no prophylaxis
We found one study that was relevant to this comparison (N = 40). Data show no clear difference between H2 receptor antagonists and no prophylaxis (RR 0.92, 95% CI 0.33 to 2.53; Analysis 2.4).
Duration of ICU stay
We identified two studies relevant to this outcome (N = 230). Data seemed to show no difference between H2 receptor antagonists and placebo or no prophylaxis with respect to duration of ICU stay (MD 0.73 days, 95% CI ‐0.92 to 2.38; Analysis 2.5). We judged the certainty of this evidence as low.
2.5. Analysis.

Comparison 2 H2 receptor antagonists versus placebo or no prophylaxis, Outcome 5 Duration of ICU stay.
Famotidine versus placebo
We identified a single study for this comparison (N = 146). We found no evidence of a clear difference between the two groups (MD 1.50 days, 95% CI ‐1.93 to 4.93; Analysis 2.5).
Ranitidine versus no prophylaxis
We found one study that was relevant to this comparison (N = 84). We found no evidence of a clear difference between the two groups (MD 0.50 days, 95% CI ‐1.38 to 2.38; Analysis 2.5). An additional study reported insufficient data for inclusion in a formal meta‐analysis (Reusser 1990). Researchers randomised 19 participants to the ranitidine arm and 21 to the control arm. The mean duration of ICU stay was 13 days (range 6 to 40 days) in the ranitidine group and 17 days (range 5 to 30 days) in the control group.
Ranitidine versus placebo
We identified two studies that looked at this outcome but did not report enough data to be included in formal meta‐analyses (N = 154) (Hanisch 1998; Reusser 1990). In the included studies, 76 participants received ranitidine and 78 received placebo. The mean duration of ICU stay was 9.7 days (2 to 95) and 12.6 days (2 to 58) in Hanisch 1998, and 13 days (16 to 40) and 17 days (5 to 30) in Reusser 1990.
Cimetidine versus no prophylaxis
We identified one study that did not report sufficient information for inclusion in the meta‐analysis (N = 200) (Ben‐Menachem 1994). The duration of ICU stay was 4 days (2 to 9 days) in the cimetidine group (n = 100) and 3 days (2 to 6 days) in the group that received no prophylaxis (n = 100).
Duration of intubation
For this outcome, we found two relevant studies and categorised data into two comparisons (N = 230). Data seemed to show no difference between H2 receptor antagonists and placebo or no prophylaxis with respect to duration of intubation (MD 0.79 days, 95% CI ‐0.95 to 2.54; Analysis 2.6). Apte 1992 assessed the duration of intubation but did not report enough data for inclusion in meta‐analyses.
2.6. Analysis.

Comparison 2 H2 receptor antagonists versus placebo or no prophylaxis, Outcome 6 Duration of intubation.
Famotidine versus placebo
We found one study that was relevant to this comparison (N = 146). We found no evidence of a clear difference between the two groups (MD 1.20 days, 95% CI ‐1.86 to 4.26; Analysis 2.6).
Ranitidine versus no prophylaxis
We identified a single study for this comparison (N = 84). Data show no clear difference between H2 receptor antagonists and no prophylaxis (MD 0.60 days, 95% CI ‐1.52 to 2.72; Analysis 2.6).
Ranitidine versus placebo
We identified two studies that looked at this outcome but did not report enough data for inclusion in formal meta‐analyses (N = 148) (Apte 1992; Hanisch 1998). In the included studies, 73 participants received ranitidine and 75 received placebo. The mean duration of intubation was 8.2 days (2 to 93) and 10.2 days (2 to 55) in Hanisch 1998, and 7.5 days (3 to 28) and 12.5 days (3 to 63) in Apte 1992.
Number of participants requiring blood transfusions
Data seemed to show a beneficial effect of H2 receptor antagonists compared with placebo or no prophylaxis with respect to the number of participants requiring blood transfusions (6.4% vs 11.2%; RR 0.58, 95% CI 0.36 to 0.95; seven studies; 655 participants; Analysis 2.7). We judged the certainty of this evidence as moderate.
2.7. Analysis.

Comparison 2 H2 receptor antagonists versus placebo or no prophylaxis, Outcome 7 Number of participants requiring blood transfusions.
Cimetidine versus placebo
We found three studies that were relevant to this comparison (N = 107). For this outcome, within this comparison, we found evidence suggesting that H2 receptor antagonists were different in their effect compared with placebo (RR 0.39, 95% CI 0.19 to 0.79; Analysis 2.7). Results favoured cimetidine over placebo.
Ranitidine versus placebo
We identified two relevant studies for this comparison (N = 148). We found no evidence of a clear difference between the two groups (RR 1.05, 95% CI 0.24 to 4.60). This comparison had substantial heterogeneity (Chi² = 1.89, df = 1.0 (P = 0.17), I² = 47%; Analysis 2.7).
Cimetidine versus no prophylaxis
We found two studies that were relevant to this comparison (N = 400). Data show no clear difference between H2 receptor antagonists and no prophylaxis (RR 0.77, 95% CI 0.35 to 1.71; Analysis 2.7).
Ranitidine versus no prophylaxis
We found two studies that were relevant to this comparison (N = 74) (Apte 1992; Reusser 1990). For this outcome, within this comparison, we found no evidence suggesting that H2 receptor antagonists were different in their effects compared with no prophylaxis. Researchers reported no events in either treatment group, and we could not include these studies in the meta‐analysis.
Units of blood transfused
For this outcome, we found two relevant studies and categorised data into two comparisons (N = 209). Data seemed to show no difference between H2 receptor antagonists and placebo or no prophylaxis with respect to the number of units of blood transfused (MD 0.33 units, 95% CI ‐0.04 to 0.70; Analysis 2.8). This outcome had considerable heterogeneity (Chi² = 9.49, df = 1.0 (P = 0.0), I² = 89%).
2.8. Analysis.

Comparison 2 H2 receptor antagonists versus placebo or no prophylaxis, Outcome 8 Units of blood transfused.
Cimetidine versus placebo
We found one study that was relevant to this comparison (N = 9). Evidence suggested a difference between H2 receptor antagonists and placebo (MD ‐4.35 units, 95% CI ‐7.35 to ‐1.35; Analysis 2.8). One study reported 11 blood transfusions in the cimetidine group (n = 26) and 41 in the placebo group (n = 24) and reported no standard deviation (Halloran 1980). Another study reported that the mean volume of blood transfused was 600 mL (0 to 900 mL) in the cimetidine group (n = 114) and 550 mL (0 to 1200 mL) in the placebo group (n = 107) (Groll 1986).
Cimetidine versus no prophylaxis
We included a single study in this comparison (N = 200). We found evidence suggesting a difference between H2 receptor antagonists and no prophylaxis favouring no prophylaxis (MD 0.40 units, 95% CI 0.03 to 0.77; Analysis 2.8).
Ranitidine vs placebo
We identified one study that looked at this outcome but did not report enough data for inclusion in formal meta‐analyses (Hanisch 1998). In the included study, 57 participants received ranitidine and 57 received placebo. A mean of two units of blood was transfused in each of the three participants with GI bleeding in the ranitidine group, and 12 units was transfused in the single surviving participant with GI bleeding in the placebo group.
Adverse events of interventions
For this outcome we found eight relevant studies and divided the data into 12 comparisons according to type of adverse event.
Diarrhoea
We found two studies that were relevant to this comparison (N = 225). We found no evidence of a clear difference between the two groups (RR 1.30, 95% CI 0.57 to 2.96; Analysis 2.9).
2.9. Analysis.

Comparison 2 H2 receptor antagonists versus placebo or no prophylaxis, Outcome 9 Adverse events of interventions.
Thrombocytopaenia
We found one study that was relevant to this comparison (N = 200). We found no evidence of a clear difference between the two groups (RR 3.0, 95% CI 0.12 to 72.77; Analysis 2.9).
Hypophosphataemia
We identified a single study in this comparison (N = 25). We found no evidence of a clear difference between the two groups (RR 3.75, 95% CI 0.17 to 84.02; Analysis 2.9).
Mental confusion
We identified five relevant studies for this comparison (N = 657). We found evidence that H2 receptor antagonists increased the risk for mental confusion compared with placebo or no prophylaxis (RR 2.01, 95% CI 1.10 to 3.65; Analysis 2.9).
Nausea and vomiting
We found two studies that were relevant to this comparison (N = 287). We found no evidence of a clear difference between the two groups (RR 0.46, 95% CI 0.09 to 2.35; Analysis 2.9).
Increased creatinine levels
We included a single study in this comparison (N = 2000. Evidence showed no clear difference between H2 receptor antagonists and placebo or no prophylaxis (RR 1.04, 95% CI 0.64 to 1.69; Analysis 2.9).
Erythema (superficial reddening of the skin)
We found one study that was relevant to this comparison (N = 70). Data showed no clear difference between H2 receptor antagonists and placebo or no prophylaxis (RR 3.00, 95% CI 0.13 to 71.22; Analysis 2.9).
Pancreatitis
We found one study that was relevant to this comparison (N = 39). We found no evidence of a clear difference between the two groups (RR 0.29, 95% CI 0.01 to 6.66; Analysis 2.9). .
Chest infection
We found one study that was relevant to this comparison (N = 101). Data showed no clear difference between H2 receptor antagonists and placebo or no prophylaxis (RR 1.74, 95% CI 0.92 to 3.30; Analysis 2.9).
Delirium
We found one study that was relevant to this comparison (N = 39. Data showed no clear difference between H2 receptor antagonists and placebo or no prophylaxis (RR 2.59, 95% CI 0.11 to 59.93; Analysis 2.9).
Hallucinations
We included a single study in this comparison (N = 39). We found no evidence of a clear difference between the two groups (RR 2.59, 95% CI 0.11 to 59.93; Analysis 2.9).
Severe bleeding
We included a single study in this comparison (N = 101). Data showed no clear difference between H2 receptor antagonists and placebo or no prophylaxis (RR 0.35, 95% CI 0.01 to 8.47; Analysis 2.9).
Proton pump inhibitors versus placebo or no prophylaxis
For this comparison, data on six of our pre‐defined outcomes were available. Omeprazole was administered in all three studies providing data for this comparison. Placebo was administered in two studies as comparator, and in one study no prophylaxis was given as comparator.
Clinically important upper GI bleeding
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 3.1). In the three included studies that contributed data to the analyses, 114 participants received proton pump inhibitors and 123 participants received either placebo or no prophylaxis. The event occurred in 2.6% and 4.9% of participants in the two groups. Data seemed to show no difference between proton pump inhibitors and placebo or no prophylaxis with respect to the occurrence of clinically important upper GI bleeding (RR 0.63, 95% CI 0.18 to 2.22). We judged the certainty of the evidence as low.
3.1. Analysis.

Comparison 3 Proton pump inhibitors versus placebo or no prophylaxis, Outcome 1 Clinically important upper GI bleeding.
We identified two additional studies. Selvanderan 2016 included 214 participants and did not report how many participants were randomised to the two interventions. Powell 1993 included 21 participants in the proton pump inhibitor arm and 10 in the placebo arm. Both trials reported no events of clinically important upper GI bleeding in either group.
Omeprazole versus placebo
We found one relevant study for this comparison (N = 147. We found no evidence of a clear difference between the two groups (RR 1.04, 95% CI 0.07 to 16.34; Analysis 3.1).
Omeprazole versus no prophylaxis
We identified a single study for this comparison (N = 80). We found no evidence of a clear difference between the two groups (RR 0.74, 95% CI 0.13 to 4.18; Analysis 3.1).
Pantoprazole versus placebo
We included a single study for this comparison (N = 10). We found no evidence of a clear difference between the two groups (RR 0.28, 95% CI 0.02 to 4.66; Analysis 3.1).
Nosocomial pneumonia
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 3.2). In the two included studies, 110 participants received proton pump inhibitors and 117 received placebo. Nosocomial pneumonia occurred in 22.8% of the group that received proton pump inhibitors and 18.7% of the group that received placebo or no prophylaxis. Data seemed to show no difference between proton pump inhibitors and placebo or no prophylaxis with respect to the occurrence of nosocomial pneumonia (RR 1.24, 95% CI 0.77 to 1.98). We judged the certainty of the evidence as low.
3.2. Analysis.

Comparison 3 Proton pump inhibitors versus placebo or no prophylaxis, Outcome 2 Nosocomial pneumonia.
We identified an additional study that was published as a conference abstract and did not report enough data for formal meta‐analysis (Selvanderan 2016). Researchers randomised a total of 214 participants to receive pantoprazole or placebo. One participant in the placebo group and two in the pantoprazole group developed ventilator‐associated complications including pneumonia. Data showed eight and 12 cases of clinician‐adjudicated nosocomial pneumonia in pantoprazole and control groups, respectively.
Omeprazole versus placebo
We identified a single study for this comparison (N = 147). Data show no clear difference between proton pump inhibitors and placebo within this comparison (RR 1.67, 95% CI 0.57 to 4.86; Analysis 3.2).
Omeprazole versus no prophylaxis
We included a single study in this comparison (N = 80). We found no evidence of a clear difference between the two groups. Nosocomial pneumonia occurred in 44.7% and 40.5% of participants in the groups that received omeprazole or no prophylaxis, respectively (RR 1.11, 95% CI 0.66 to 1.84; Analysis 3.2).
All‐cause mortality in ICU
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 3.3). In the three included studies, 131 participants received proton pump inhibitors and 127 received either placebo or no prophylaxis. The event occurred in 13.3% and 13.4% of participants in the two groups. Data seemed to show no difference between proton pump inhibitors and placebo or no prophylaxis with respect to all‐cause mortality in the ICU (RR 1.09, 95% CI 0.60 to 1.99). We judged the certainty of this evidence as low.
3.3. Analysis.
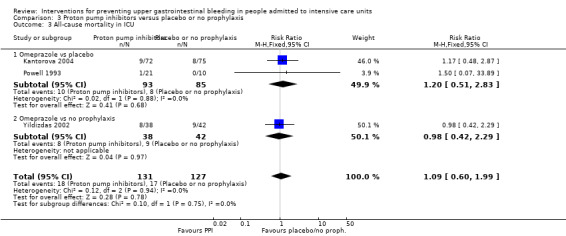
Comparison 3 Proton pump inhibitors versus placebo or no prophylaxis, Outcome 3 All‐cause mortality in ICU.
We identified an additional study that was published as a conference abstract and did not provide enough data for formal meta‐analysis (Selvanderan 2016). Researchers randomised a total of 214 participants to receive pantoprazole or placebo. The study reported an adjusted hazard ratio for mortality with pantoprazole of 1.68 (95% CI 0.97 to 2.90).
Omeprazole versus placebo
We found two relevant studies for this comparison (N = 178). We found no evidence of a clear difference between the two groups (RR 1.20, 95% CI 0.51 to 2.83; Analysis 3.3).
Omeprazole versus no prophylaxis
We included a single study in this comparison (N = 80). Data show no clear difference between proton pump inhibitors and no prophylaxis within this comparison with respect to all‐cause mortality in the ICU (RR 0.98, 95% CI 0.42 to 2.29; Analysis 3.3).
All‐cause mortality in hospital
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 3.4). In the included study, 75 participants received proton pump inhibitors and 75 received placebo. All‐cause mortality occurred in 18.7% of the group that received proton pump inhibitors and 17.3% of the group that received placebo. Data seemed to show no difference between proton pump inhibitors and placebo or no prophylaxis with respect to all‐cause mortality in hospital (RR 1.08, 95% CI 0.54 to 2.13).
3.4. Analysis.

Comparison 3 Proton pump inhibitors versus placebo or no prophylaxis, Outcome 4 All‐cause mortality in hospital.
Duration of ICU stay
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 3.5). In the two included studies, 110 participants received proton pump inhibitors and 117 received either placebo or no prophylaxis. Data seemed to show no difference between proton pump inhibitors and placebo or no prophylaxis with respect to length of ICU stay (MD ‐0.03 days, 95% CI ‐1.63 to 1.58; two studies; 227 participants; Analysis 3.5). We judged the certainty of this evidence as low.
3.5. Analysis.

Comparison 3 Proton pump inhibitors versus placebo or no prophylaxis, Outcome 5 Duration of ICU stay.
Omeprazole versus placebo
We included a single study in this comparison (N = 147). We found no evidence of a clear difference between the two groups (MD ‐0.90 days, 95% CI ‐3.96 to 2.16; Analysis 3.5).
Omeprazole versus no prophylaxis
We included a single study in this comparison (N = 80). Data show no clear difference between proton pump inhibitors and no prophylaxis (MD 0.30 days, 95% CI ‐1.58 to 2.18; Analysis 3.5).
Duration of intubation
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 3.6). In the two included studies, 110 participants received sucralfate and 107 received either placebo or no prophylaxis. Data seemed to show no difference between proton pump inhibitors and placebo or no prophylaxis with respect to duration of intubation (MD 0.36 days, 95% CI ‐1.43 to 2.15).
3.6. Analysis.
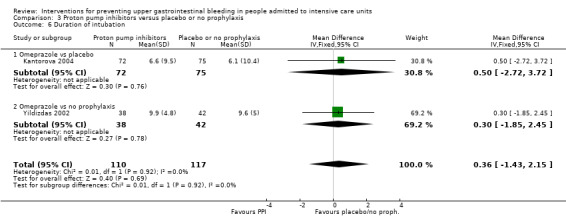
Comparison 3 Proton pump inhibitors versus placebo or no prophylaxis, Outcome 6 Duration of intubation.
Omeprazole versus placebo
We included a single study in this comparison (N = 147). Data show no difference between proton pump inhibitors and placebo with respect to duration of intubation (MD 0.50 days, 95% CI ‐2.72 to 3.72; Analysis 3.6).
Omeprazole versus no prophylaxis
We found one study that was relevant to this comparison (N = 800). We found no evidence of a clear difference between the two groups (MD 0.30 days, 95% CI ‐1.85 to 2.45; Analysis 3.6).
Pirenzepine versus placebo
We identified one study that looked at this outcome but did not report enough data for inclusion in formal meta‐analyses (Hanisch 1998). In the included study, 44 participants received pirenzepine and 57 received placebo. The mean duration of intubation was 8.2 days (2 to 32) and 10.2 days (2 to 55) in the two groups, respectively.
Adverse events
One study including 214 participants compared pantoprazole versus placebo (Selvanderan 2015). Researchers tested 30 participants assigned to pantoprazole and 40 assigned to placebo for Clostridium difficile infection. One participant in the pantoprazole group was infected with C difficile.
Proton pump inhibitors plus sucralfate versus placebo or no prophylaxis
For this comparison, data on one of our pre‐defined outcomes were available. One study compared early and late administration of omeprazole plus lansoprazole plus sucralfate versus no prophylaxis. For this comparison, we included only participants with late start of therapy (48 to 72 hours) to fit the inclusion criteria of this review. We did not include in the analysis the group with early start of therapy (12 to 24 hours). The only outcome measured that was of relevance to this review was the occurrence of clinically important upper GI bleeding.
Clinically important upper GI bleeding
Data seemed to show no difference between proton pump inhibitors plus sucralfate and placebo or no prophylaxis with respect to the occurrence of clinically important upper GI bleeding (20.0% vs 37.5%; RR 0.53, 95% CI 0.26 to 1.12; one study; 80 participants who contributed data to this comparison (Analysis 4.1). We judged the certainty of this evidence as moderate.
4.1. Analysis.
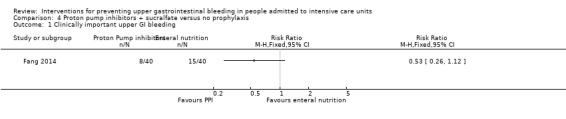
Comparison 4 Proton pump inhibitors + sucralfate versus no prophylaxis, Outcome 1 Clinically important upper GI bleeding.
Prostaglandin analogues versus placebo or no prophylaxis
For this comparison, data on two of our pre‐defined outcomes were available. Only one study provided data for this comparison. It included 58 people, of whom 29 received prostaglandin analogues and 29 received placebo.
Clinically important upper GI bleeding
Data seemed to show no difference between prostaglandin analogues and placebo with regard to the occurrence of clinically important upper GI bleeding (10.3% in both groups; RR 1.00, 95% CI 0.22 to 4.55; one study; 58 participants; Analysis 5.1). We judged the certainty of this evidence as moderate.
5.1. Analysis.
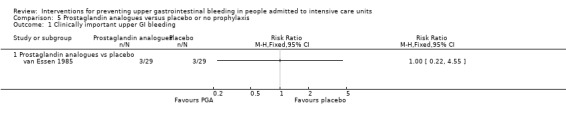
Comparison 5 Prostaglandin analogues versus placebo or no prophylaxis, Outcome 1 Clinically important upper GI bleeding.
All‐cause mortality in ICU
Data seemed to show no difference between prostaglandin analogues and placebo with regard to all‐cause mortality in the ICU (27.6% and 24.1%; RR 1.14, 95% CI 0.48 to 2.74; one study; 58 participants; Analysis 5.2). We judged the certainty of this evidence as moderate.
5.2. Analysis.

Comparison 5 Prostaglandin analogues versus placebo or no prophylaxis, Outcome 2 All‐cause mortality in ICU.
Anticholinergics versus placebo or no prophylaxis
For this comparison, data on three of our pre‐defined outcomes were available. Two studies provided data for this comparison. Both studies administered pirenzepine to a total of 59 participants and placebo or placebo plus ranitidine to 72 participants. In Krakamp 1989, ranitidine was the co‐intervention given to both arms.
Clinically important upper GI bleeding
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 6.1). In the two studies that contributed data towards this outcome, the event occurred in 10.2% and 9.7% of participants. Data seemed to show no difference between anticholinergics and placebo or no prophylaxis with respect to the occurrence of clinically important upper GI bleeding (RR 0.95, 95% CI 0.36 to 2.51). We judged the certainty of this evidence as low.
6.1. Analysis.

Comparison 6 Anticholinergics versus placebo or no prophylaxis, Outcome 1 Clinically important upper GI bleeding.
Pirenzepine versus placebo
We found one study that was relevant to this comparison (N = 101). We found no evidence of a clear difference between the two groups (RR 1.94, 95% CI 0.34 to 11.13; Analysis 6.1).that
Pirenzepin plus ranitidine versus placebo plus ranitidine
We found one study that was relevant to this comparison (N = 30). Data show no clear difference between the two groups (RR 0.60, 95% CI 0.17 to 2.07; Analysis 6.1).
Nosocomial pneumonia
We classified the study on the basis of its intervention and comparison arms (as shown in Analysis 6.2). In the single included study, 44 participants received anticholinergics and 57 received placebo. The event occurred in 22.7% and 21.1% of participants in both groups. Data seemed to show no difference between anticholinergics and placebo with respect to the occurrence of nosocomial pneumonia (RR 1.08, 95% CI 0.51 to 2.27; one study; 101 participants). We judged the certainty of this evidence as low.
6.2. Analysis.

Comparison 6 Anticholinergics versus placebo or no prophylaxis, Outcome 2 Nosocomial pneumonia.
All‐cause mortality in ICU
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 6.3). In the two included studies, 59 participants received anticholinergics and 72 received placebo alone or placebo with ranitidine. The event occurred in 25.4% and 20.8% of participants in both groups. Data seemed to show no difference between anticholinergics and placebo with respect to all‐cause mortality in the ICU (RR 1.23, 95% CI 0.66 to 2.30). We judged the certainty of this evidence as low.
6.3. Analysis.

Comparison 6 Anticholinergics versus placebo or no prophylaxis, Outcome 3 All‐cause mortality in ICU.
Pirenzepine versus placebo
We included a single study in this comparison (N = 101). We found no evidence of a clear difference between the two groups (RR 1.30, 95% CI 0.65 to 2.60; Analysis 6.3).
Pirenzepin plus ranitidine versus placebo plus ranitidine
We found one study that was relevant to this comparison (N = 30. We found no evidence of a clear difference between the two groups (RR 1.0, 95% CI 0.24 to 4.18; Analysis 6.3).
Duration of ICU stay
We identified one study that looked at this outcome but did not report enough data for inclusion in formal meta‐analyses (Hanisch 1998). In the included study, 44 participants received pirenzepine and 57 received placebo. The mean duration of ICU stay was 9.9 days (2 to 39) and 12.6 days (2 to 58), respectively.
Duration of intubation
We identified one study that looked at this outcome but did not report enough data for inclusion in formal meta‐analyses (Hanisch 1998). In the included study, 44 participants received pirenzepine and 57 received placebo. The mean duration of intubation was 8.0 days (2 to 32) and 10.2 days (2 to 55), respectively.
Units of blood transfused
We identified one study that looked at this outcome but did not report enough data for inclusion in formal meta‐analyses (Hanisch 1998). In the included study, 44 participants received pirenzepine and 57 received placebo. The mean number of units of blood transfused was four units in each of the three patients with GI bleeding in the pirenzepine group and 12 units in the single surviving participant with GI bleeding in the placebo group.
Antacids versus placebo or no prophylaxis
For this comparison, data on five of our pre‐defined outcomes were available. Among the eight studies that provided data for this comparison, two studies administered placebo and five studies provided no prophylaxis.
Clinically important upper GI bleeding
Data seemed to show a beneficial effect of antacids compared with placebo or no prophylaxis with respect to the occurrence of clinically important upper GI bleeding. The outcome occurred in 7.2% of the antacids group and in 17.7% of the placebo group (RR 0.49, 95% CI 0.25 to 0.99; eight studies; 774 participants; Analysis 7.1). We judged the certainty of this evidence as low. This outcome had substantial heterogeneity (Chi² = 15.92, df = 7.0 (P = 0.03), I² = 56%).
7.1. Analysis.
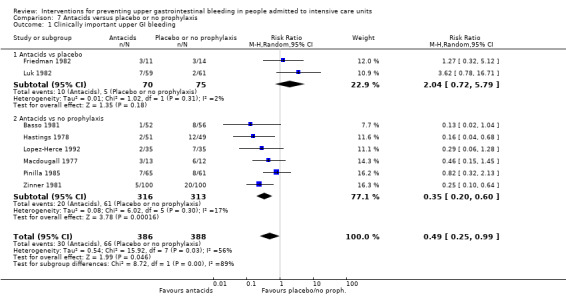
Comparison 7 Antacids versus placebo or no prophylaxis, Outcome 1 Clinically important upper GI bleeding.
Antacids versus placebo
We found two studies that were relevant to this comparison (N = 145). Data show no clear difference between antacids and placebo (RR 2.04, 95% CI 0.72 to 5.79; Analysis 7.1).
Antacids versus no prophylaxis
We found six studies that were relevant to this comparison (N = 629). For this outcome, within this comparison, we found evidence suggesting that antacids had a beneficial effect compared with no prophylaxis (RR 0.35, 95% CI 0.20 to 0.60; Analysis 7.1).
All‐cause mortality in ICU
Data seemed to show no difference between anticholinergics and no prophylaxis with respect to all‐cause mortality in the ICU. The event occurred in 15.9% of the antacids group and in 16.1% of the group that received no prophylaxis (RR 1.01, 95% CI 0.53 to 1.96; two studies; 300 participants; Analysis 7.2). We judged the certainty of the evidence as low.
7.2. Analysis.

Comparison 7 Antacids versus placebo or no prophylaxis, Outcome 2 All‐cause mortality in ICU.
All‐cause mortality in the hospital
Data seemed to show no difference between anticholinergics and no prophylaxis with respect to all‐cause mortality in the hospital. The event occurred in 30.7% of the intervention group and in 21.3% of the control group (RR 1.44, 95% CI 0.79 to 2.64; one study; 126 participants; Analysis 7.3).
7.3. Analysis.

Comparison 7 Antacids versus placebo or no prophylaxis, Outcome 3 All‐cause mortality in hospital.
Duration of intubation
We identified one study that did not report enough data for inclusion in formal meta‐analysis (Pinilla 1985). Study participants received either antacids (n = 65) or no specific prophylaxis (n = 61). The mean duration of ventilation in the antacids group was 136.4 hours (range 0 to 360) and in the control group 173.6 hours (range 12 to 552).
Number of participants requiring blood transfusions
Data seemed to show no difference between antacids and no prophylaxis with respect to the number of participants requiring blood transfusion. In the intervention group, 4.3% of participants required blood transfusion, and in the control group, 4.5% required blood transfusion (RR 0.94, 95% CI 0.30 to 2.96; two studies; 226 participants; Analysis 7.4). We judged the certainty of the evidence as low.
7.4. Analysis.

Comparison 7 Antacids versus placebo or no prophylaxis, Outcome 4 Number of participants requiring blood transfusions.
Adverse events of interventions
We identified four studies relevant to this outcome and categorised data according to seven adverse events.
Diarrhoea
We included four relevant studies in this comparison (N = 395). For this outcome, within this comparison, we found evidence suggesting that antacids increased the risk of diarrhoea compared with placebo or no prophylaxis. Diarrhoea occurred in 16.4% of the intervention group and 4.5% of the control group (RR 3.56, 95% CI 1.83 to 6.94; Analysis 7.5). For this comparison, heterogeneity was moderate (Chi² = 4.99, df = 3.0 (P = 0.17), I² = 39%).
7.5. Analysis.

Comparison 7 Antacids versus placebo or no prophylaxis, Outcome 5 Adverse events of interventions.
Hypomagnesaemia
We included a single study in this comparison (N = 25). We found no evidence of a clear difference between the two groups (RR 3.75, 95% CI 0.17 to 84.02; Analysis 7.5). Researchers reported one event in the intervention group and none in the control group.
Hypophosphataemia
We found two studies that were relevant to this comparison (N = 225). For this outcome, we found evidence suggesting that antacids increased the risk of hypophosphataemia compared with placebo or no prophylaxis (16.2% and 2.6%; RR 5.48, 95% CI 1.81 to 16.61; Analysis 7.5). Data showed 18 events in the intervention group and three in the control group.
Hypermagnesaemia
We included a single study in this comparison (N = 100). Data show no clear difference between antacids and placebo or no prophylaxis (RR 6.73, 95% CI 0.36 to 127.02; Analysis 7.5). Researchers reported three events in the intervention group and none in the control group.
Nausea and vomiting
We found three studies that were relevant to this subgroup (N = 370). Data show no clear difference between antacids and placebo or no prophylaxis (RR 2.39, 95% CI 0.86 to 6.64; Analysis 7.5). Researchers reported 11 events in the intervention group and four in the control group.
Mental confusion
We included a single study in this comparison (N = 200). Data show no clear difference between antacids and placebo or no prophylaxis (RR 1.27, 95% CI 0.61 to 2.67; Analysis 7.5). Fourteen events occurred in the intervention group and 11 in the control group.
Creatinine increase
We found one relevant study (N = 200). We found no evidence of a clear difference between the two groups (RR 1.17, 95% CI 0.73 to 1.87; Analysis 7.5). The event occurred in 28 and 24 participants in the two groups, respectively.
Sucralfate versus placebo or no prophylaxis
For this comparison, data on nine of our pre‐defined outcomes were available. Among the seven studies that provided data for this comparison, placebo was administered in two studies and no prophylaxis was given in five studies.
Clinically important upper GI bleeding
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 8.1). Data seemed to show a beneficial effect of sucralfate compared with placebo or no prophylaxis with respect to the occurrence of clinically important upper GI bleeding. The event occurred in 6.3% of participants in the intervention group and in 10.8% of participants of the control group (RR 0.53, 95% CI 0.32 to 0.88; seven studies; 598 participants; Analysis 8.1). We judged the certainty of this evidence as moderate.
8.1. Analysis.

Comparison 8 Sucralfate versus placebo or no prophylaxis, Outcome 1 Clinically important upper GI bleeding.
Sucralfate versus placebo
We identified two relevant studies (N = 170). Data show no clear difference between sucralfate and placebo (RR 1.40, 95% CI 0.30 to 6.62; Analysis 8.1). For this comparison, heterogeneity was moderate (Chi² = 1.53, df = 1.0 (P = 0.22), I² = 35%).
Sucralfate versus no prophylaxis
We identified five relevant studies for this comparison (N = 428). We found evidence for a beneficial effect of sucralfate compared with no prophylaxis (RR 0.46, 95% CI 0.26 to 0.80; Analysis 8.1).
Nosocomial pneumonia
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 8.2). We identified four studies relevant to this outcome (N = 450). Data seemed to show no difference between sucralfate and placebo or no prophylaxis with respect to the occurrence of ventilator‐associated pneumonia. The event occurred in 15.9% and 12.2% of the two groups, respectively (RR 1.33, 95% CI 0.86 to 2.04; Analysis 8.2). We judged the certainty of this evidence as low.
8.2. Analysis.

Comparison 8 Sucralfate versus placebo or no prophylaxis, Outcome 2 Nosocomial pneumonia.
Sucralfate versus placebo
We identified two relevant studies (N = 170). Data show no clear difference between sucralfate and placebo (RR 1.43, 95% CI 0.49 to 4.16; Analysis 8.2).
Sucralfate versus no prophylaxis
We found two relevant studies (N = 280). We found no evidence of a clear difference between the two groups (RR 1.30, 95% CI 0.82 to 2.07; Analysis 8.2). This comparison had moderate heterogeneity (Chi² = 1.51, df = 1.0 (P = 0.22), I² = 34%).
All‐cause mortality in ICU
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 8.3). Data seemed to show no difference between sucralfate and placebo or no prophylaxis with respect to all‐cause mortality in the ICU. The event occurred in 16.3% and 16.5% in the two groups, respectively (RR 0.97, 95% CI 0.66 to 1.43; five studies; 500 participants). We judged the certainty of this evidence as low.
8.3. Analysis.

Comparison 8 Sucralfate versus placebo or no prophylaxis, Outcome 3 All‐cause mortality in ICU.
Sucralfate versus placebo
We identified two relevant studies (N = 170). Data show no clear difference between sucralfate and placebo (RR 0.92, 95% CI 0.48 to 1.80; Analysis 8.3).
Sucralfate versus no prophylaxis
We found three studies that were relevant to this review (N = 330). We found no evidence of a clear difference between the two groups (RR 0.99, 95% CI 0.62 to 1.60; Analysis 8.3).
All‐cause mortality in hospital
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 8.4). Data seemed to who no difference between sucralfate and placebo or no prophylaxis with respect to all‐cause mortality in the hospital. The event occurred in 17.8% and 18.3% in the two groups, respectively (RR 0.97, 95% CI 0.62 to 1.52; two studies; 344 participants).
8.4. Analysis.

Comparison 8 Sucralfate versus placebo or no prophylaxis, Outcome 4 All‐cause mortality in hospital.
Sucralfate versus placebo
We included a single study in this comparison (N = 144). Data show no clear difference between sucralfate and placebo (RR 1.09, 95% CI 0.54 to 2.18; Analysis 8.4).
Sucralfate versus no prophylaxis
We included a single study in this comparison (N = 200). We found no evidence of a clear difference between the two groups (RR 0.89, 95% CI 0.49 to 1.62; Analysis 8.4).
Duration of ICU stay
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 8.5). Data seemed to show no difference between sucralfate and placebo or no prophylaxis with respect to duration of ICU stay (MD ‐0.02 days, 95% CI ‐1.70 to 1.65; two studies; 224 participants). We judged the certainty of this evidence as low.
8.5. Analysis.

Comparison 8 Sucralfate versus placebo or no prophylaxis, Outcome 5 Duration of ICU stay.
Sucralfate versus placebo
We found one study that was relevant to this comparison (N = 144). We found no evidence of a clear difference between the two groups (MD ‐0.70 days, 95% CI ‐4.07 to 2.67; Analysis 8.5).
Sucralfate versus no prophylaxis
We found one study that was relevant to this comparison (N = 80). We found no evidence of a clear difference between the two groups (MD 0.20 days, 95% CI ‐1.73 to 2.13; Analysis 8.5). We identified one study that did not report sufficient information for inclusion in the meta‐analysis (Ben‐Menachem 1994). In this study, the duration of ICU stay was 3 days (2 to 8.5 days) in the sucralfate group (n = 100) and 3 days (2 to 6 days) in the group that received no prophylaxis (n = 100).
Duration of intubation
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 8.6). Data seemed to show no difference between sucralfate and placebo or no prophylaxis with respect to duration of intubation (MD 1.42, 95% CI ‐0.27 to 3.10; two studies; 224 participants).
8.6. Analysis.

Comparison 8 Sucralfate versus placebo or no prophylaxis, Outcome 6 Duration of intubation.
Sucralfate versus placebo
We included a single study in this comparison (N = 144). Data show no clear difference between sucralfate and placebo (MD 0.80 days, 95% CI ‐2.20 to 3.80; Analysis 8.6).
Sucralfate versus no prophylaxis
We found one study that was relevant to this comparison (N = 80). Data show no clear difference between sucralfate and no prophylaxis (MD 1.70 days, 95% CI ‐0.34 to 3.74; Analysis 8.6).
Number of participants requiring blood transfusion
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 8.7). Data seemed to show no difference between sucralfate and no prophylaxis with respect to the number of participants requiring blood transfusion. Blood transfusion was required by 3% and 5% of the two groups, respectively (RR 0.60, 95% CI 0.15 to 2.44; one study; 200 participants). We judged the certainty of this evidence as low.
8.7. Analysis.

Comparison 8 Sucralfate versus placebo or no prophylaxis, Outcome 7 Number of participants requiring blood transfusions.
Units of blood transfused
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 8.8). Data seemed to show an unfavourable effect of sucralfate with respect to the number of units of blood transfused (MD 0.80, 95% CI 0.32 to 1.28; one study; 200 participants).
8.8. Analysis.

Comparison 8 Sucralfate versus placebo or no prophylaxis, Outcome 8 Units of blood transfused.
Adverse events of interventions
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 8.9). For this outcome, we found a single study that measured nausea and vomiting as an adverse event. Data seemed to show no difference between sucralfate and placebo or no prophylaxis with respect to the occurrence of adverse events. The sucralfate group included four participants with nausea and vomiting and the placebo group included none (RR 9.00, 95% CI 0.50 to 161.13; one study; 70 participants).
8.9. Analysis.
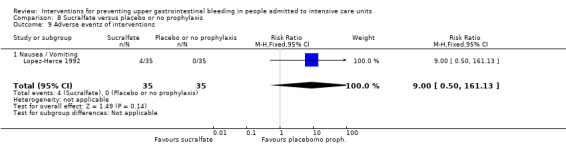
Comparison 8 Sucralfate versus placebo or no prophylaxis, Outcome 9 Adverse events of interventions.
H2 receptor antagonists versus proton pump inhibitors
For this comparison, data on eight of our pre‐defined outcomes were available. Among the 18 studies that provided data for this comparison, ranitidine was administered in nine studies, cimetidine in two, and famotidine in six. Two studies combined the pantoprazole arms (Fink 2003; Somberg 2008), and one study combined the omeprazole arms to form a common interventional arm for comparison with H2 receptor antagonists (Powell 1993), as the review did not aim to investigate intraclass efficacy among included interventions. Tabeefar 2012 compared pantoprazole (n = 11) and ranitidine (n = 8), but reported no outcomes that were of relevance for this review.
Clinically important upper GI bleeding
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 9.1). Data seemed to show increased risk of clinically important upper GI bleeding under H2 receptor antagonists when compared with proton pump inhibitors. The event occurred in 6.9% and 1.7% of participants in the two groups, respectively (RR 2.90, 95% CI 1.83 to 4.58; 13 studies; 1636 participants). Five studies with 525 participants reported that no events occurred in either group. We judged the certainty of this evidence as low.
9.1. Analysis.

Comparison 9 H2 receptor antagonists versus proton pump inhibitors, Outcome 1 Clinically important upper GI bleeding.
Cimetidine versus omeprazole
We found one study that was relevant to this comparison (N = 359). We found no evidence of a clear difference between the two groups (RR 1.4, 95% CI 0.55 to 3.61; Analysis 9.1).
Cimetidine versus pantoprazole
We found one study that was relevant to this comparison (N = 202) (Somberg 2008). We found no evidence of a difference between the two groups, but no events were reported in either group.
Famotidine versus lansoprazole
We included a single study in this comparison (N = 51). We found no evidence of a clear difference between the two groups (RR 3.63, 95% CI 0.15 to 84.98; Analysis 9.1).
Famotidine versus omeprazole
We included a single study in this comparison (N = 143). Data show no clear difference between famotidine and omeprazole (RR 2.03, 95% CI 0.19 to 21.87; Analysis 9.1).
Famotidine versus pantoprazole
We found two studies that were relevant to this comparison (N = 159). Data show no clear difference between famotidine and pantoprazole (RR 0.73, 95% CI 0.18 to 3.04). This comparison had substantial heterogeneity (Chi² = 2.44, df = 1.0 (P = 0.12), I² = 59%; Analysis 9.1).
Famotidine versus esomeprazole
We identified two relevant studies for this comparison (N = 371). For this outcome, we found evidence suggesting that famotidine seemed to increase the risk for upper GI bleeding compared with esomeprazole (RR 7.53, 95% CI 1.39 to 40.85; Analysis 9.1).
Ranitidine versus omeprazole
Five studies contributed data to this comparison (N = 413). We found evidence of increased risk of GI bleeding with ranitidine compared with omeprazole (RR 4.08, 95% CI 1.99 to 8.36; Analysis 9.1). Powell 1993 (N = 31) reported no events in either of the two study arms.
Ranitidine versus pantoprazole
We found two studies that were relevant to this comparison (N = 213). We found no evidence of a clear difference between ranitidine and pantoprazole, and no events were reported in either treatment group (Analysis 9.1).
Ranitidine versus rabeprazole
We found one study that was relevant to this comparison (N = 140). Data show no clear difference between ranitidine and rabeprazole (RR 9.00, 95% CI 0.49 to 164.09; Analysis 9.1).
H2 receptor antagonists (not defined) versus proton pump inhibitors (not defined)
We included a single study in this comparison (N = 79) (Fogas 2013). We found no evidence of a clear difference between the two groups, and no events were reported in either treatment group (Analysis 9.1).
Nosocomial pneumonia
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 9.2). Data seemed to show no difference between H2 receptor antagonists and proton pump inhibitors with respect to the occurrence of nosocomial pneumonia. The event occurred in 13.3% and 12.1% of participants in the two groups, respectively (RR 1.02, 95% CI 0.77 to 1.35; eight studies; 1256 participants). We judged the certainty of this evidence as low.
9.2. Analysis.

Comparison 9 H2 receptor antagonists versus proton pump inhibitors, Outcome 2 Nosocomial pneumonia.
Cimetidine versus omeprazole
We found one study that was relevant to this comparison (N = 359). Data show no clear difference between cimetidine and omeprazole (RR 0.84, 95% CI 0.45 to 1.54; Analysis 9.2).
Cimetidine versus pantoprazole
We identified a single study for this comparison (N = 202). Data show no clear difference between cimetidine and pantoprazole (RR 0.89, 95% CI 0.28 to 2.91; Analysis 9.2).
Famotidine versus esomeprazole
We included a single study in this comparison (N = 60). Data show no clear difference between famotidine and esomeprazole (RR 1.00, 95% CI 0.07 to 15.26; Analysis 9.2).
Famotidine versus omeprazole
We included a single study in this comparison (N = 143). Data show no clear difference between famotidine and omeprazole (RR 0.89, 95% CI 0.34 to 2.32; Analysis 9.2).
Ranitidine versus omeprazole
We identified five relevant studies for this comparison (N = 413). Data show no clear difference between ranitidine and omeprazole (RR 1.19, 95% CI 0.80 to 1.75; Analysis 9.2). This comparison had moderate heterogeneity (Chi² = 6.07, df = 4.0 (P = 0.19), I² = 34%).
H2 receptor antagonists (not defined) versus proton pump inhibitors (not defined)
We found one study that was relevant to this comparison (N = 79). Data show no clear difference between H2 receptor antagonists and proton pump inhibitors (RR 1.03, 95% CI 0.47 to 2.26; Analysis 9.2).
All‐cause mortality in ICU
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 9.3). Data seemed to show no difference between H2 receptor antagonists and proton pump inhibitors with respect to all‐cause mortality in the ICU. The event occurred in 19.3% and 16.3% of participants in the two groups, respectively (RR 0.96, 95% CI 0.78 to 1.19; 12 studies; 1564 participants). We judged the certainty of this evidence as low.
9.3. Analysis.
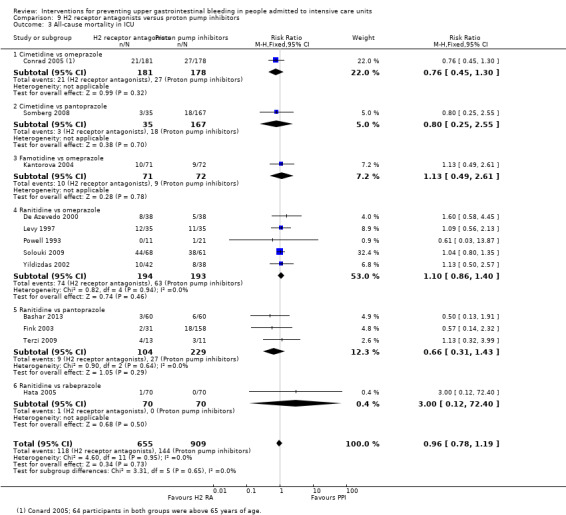
Comparison 9 H2 receptor antagonists versus proton pump inhibitors, Outcome 3 All‐cause mortality in ICU.
Cimetidine versus omeprazole
We found one study that was relevant to this comparison (N = 359). We found no evidence of a clear difference between the two groups (RR 0.76, 95% CI 0.45 to 1.30; Analysis 9.3).
Cimetidine versus pantoprazole
We found one study that was relevant to this comparison (N = 202). We found no evidence of a clear difference between the two groups (RR 0.80, 95% CI 0.25 to 2.55; Analysis 9.3).
Famotidine versus omeprazole
We found one study that was relevant to this comparison (N = 143). We found no evidence of a clear difference between the two groups (RR 1.13, 95% CI 0.49 to 2.61; Analysis 9.3).
Ranitidine versus omeprazole
We identified five relevant studies for this comparison (N = 387). We found no evidence of a clear difference between the two groups (RR 1.10, 95% CI 0.86 to 1.40; Analysis 9.3).
Ranitidine versus pantoprazole
We found three studies that were relevant to this comparison (N = 333). Data show no clear difference between the two groups (RR 0.66, 95% CI 0.31 to 1.43; Analysis 9.3).
Ranitidine versus rabeprazole
We found one study that was relevant to this comparison (N = 140). We found no evidence of a clear difference between the two groups (RR 3.00, 95% CI 0.12 to 72.40; Analysis 9.3).
All‐cause mortality in the hospital
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 9.4). Data seemed to show no difference between H2 receptor antagonists and proton pump inhibitors with respect to all‐cause mortality in the hospital. The event occurred in 5.5% and 7.2% of participants, respectively (RR 0.72, 95% CI 0.37 to 1.43; two studies; 454 participants).
9.4. Analysis.

Comparison 9 H2 receptor antagonists versus proton pump inhibitors, Outcome 4 All‐cause mortality in hospital.
Famotidine versus esomeprazole
We found one study that was relevant to this comparison (N = 311). We found no evidence of a clear difference between the two groups (RR 0.37, 95% CI 0.04 to 3.49; Analysis 9.4).
Famotidine versus omeprazole
We included a single study in this comparison (N = 143). Data show no clear difference between famotidine and omeprazole (RR 0.80, 95% CI 0.39 to 1.63; Analysis 9.4).
Duration of ICU stay
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 9.5). Data seemed to show no difference between H2 receptor antagonists and proton pump inhibitors with respect to duration of ICU stay (MD 0.14 days, 95% CI ‐1.14 to 1.41; five studies; 482 participants). We judged the certainty of this evidence as low.
9.5. Analysis.
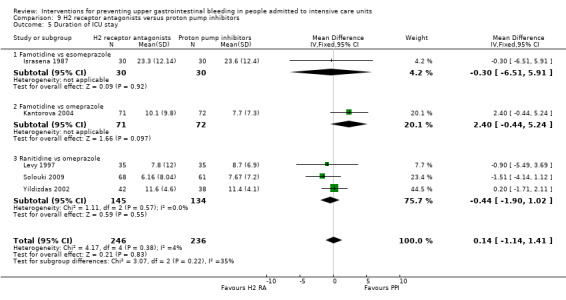
Comparison 9 H2 receptor antagonists versus proton pump inhibitors, Outcome 5 Duration of ICU stay.
Famotidine versus esomeprazole
We included a single study in this comparison (N = 60). We found no evidence of a clear difference between the two groups (MD ‐0.30 days, 95% CI ‐6.51 to 5.91; Analysis 9.5).
Famotidine versus omeprazole
We found one study that was relevant to this comparison (N = 143). Data show no clear difference between the two groups (MD 2.40 days, 95% CI ‐0.44 to 5.24; Analysis 9.5).
Famotidine versus pantoprazole
We included a single study in this comparison (N = 129). We found no evidence that famotidine was clearly different in its effects compared with pantoprazole, for no standard deviation was reported in the study (Wee 2013).
Ranitidine versus omeprazole
We found three studies that were relevant to this comparison (N = 279). We found no evidence of a clear difference between the two groups (MD ‐0.44 days, 95% CI ‐1.90 to 1.02; Analysis 9.5).
Ranitidine versus pirenzepine
We identified one study that looked at this outcome but did not report enough data for inclusion in formal meta‐analyses (Hanisch 1998). In the included study, 44 participants received pirenzepine and 57 received ranitidine. The mean duration of ICU stay was 9.9 (2 to 39) days and 9.7 (2 to 95) days in the two groups, respectively.
Duration of intubation
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 9.6). We identified six studies relevant to this outcome (N = 671). However, only five studies contributed data to our meta‐analysis. We found no difference between H2 receptor antagonists and proton pump inhibitors with respect to duration of intubation (MD ‐0.35 days, 95% CI ‐1.48 to 0.78; five studies; 542 participants).
9.6. Analysis.

Comparison 9 H2 receptor antagonists versus proton pump inhibitors, Outcome 6 Duration of intubation.
Famotidine versus omeprazole
We included a single study in this comparison (N = 143). We found no evidence of a clear difference between the two groups (MD 0.70 days, 95% CI ‐2.24 to 3.64; Analysis 9.6)
Famotidine versus pantoprazole
We found one study that was relevant to this comparison (N = 129). For this outcome, data seemed to show no difference in effect between famotidine (6.3 days; n = 61) and pantoprazole (6.5 days; n = 68). However, data (no SD/SE) reported were insufficient for inclusion in study meta‐analyses (Analysis 9.6). We identified an additional study that was published as a conference abstract and did not report enough data for formal meta‐analysis (Wee 2013). Researchers included 61 participants in the famotidine group and 68 in the pantoprazole group. The mean duration of intubation was 6.3 days in the famotidine group and 6.5 days in the pantoprazole group.
Ranitidine versus omeprazole
We identified three relevant studies for this comparison (N = 279). We found no evidence of a clear difference between the two groups (MD ‐0.78 days, 95% CI ‐2.24 to 0.67; Analysis 9.6).
Ranitidine versus pantoprazole
We found one study that was relevant for this comparison (N = 120). Data show no clear difference between ranitidine and pantoprazole (MD 0.07 days, 95% CI ‐2.18 to 2.32; Analysis 9.6).
Ranitidine versus pirenzepine
We identified one study that looked at this outcome but did not report enough data for inclusion in formal meta‐analyses (Hanisch 1998). In the included study, 44 participants received pirenzepine and 57 received ranitidine. The mean duration of intubation was 8.2 (2 to 32) days and 8.2 (2 to 93) days in the two groups, respectively.
Number of participants requiring blood transfusion
Data seemed to show no difference between H2 receptor antagonists and proton pump inhibitors with respect to the number of participants requiring blood transfusion (3.8% and 1.7%; RR 1.98, 95% CI 0.75 to 5.21; three studies; 575 participants; Analysis 9.7). We judged the certainty of this evidence as low.
9.7. Analysis.
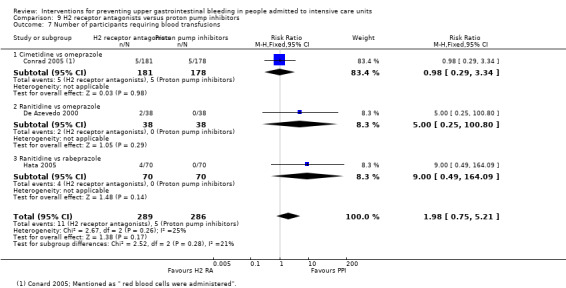
Comparison 9 H2 receptor antagonists versus proton pump inhibitors, Outcome 7 Number of participants requiring blood transfusions.
Cimetidine versus omeprazole
We included a single study in this comparison (N = 359). Data show no clear difference between cimetidine and omeprazole (RR 0.98, 95% CI 0.29 to 3.34; Analysis 9.7).
Ranitidine versus omeprazole
We found one study that was relevant to this comparison (N = 76). We found no evidence of a clear difference between the two groups (RR 5.00, 95% CI 0.25 to 100.80; Analysis 9.7).
Ranitidine versus rabeprazole
We included a single study in this comparison (N = 140). Data show no clear difference between ranitidine and rabeprazole (RR 9.00, 95% CI 0.49 to 164.09; Analysis 9.7).
Adverse events of interventions
For this outcome, we found five relevant studies and categorised data according to 12 adverse events.
Pyrexia
We identified a single study that reported on this adverse event (N = 202). Data show no clear difference between H2 receptor antagonists and proton pump inhibitors (RR 0.93, 95% CI 0.05 to 19.03; Analysis 9.8).
9.8. Analysis.
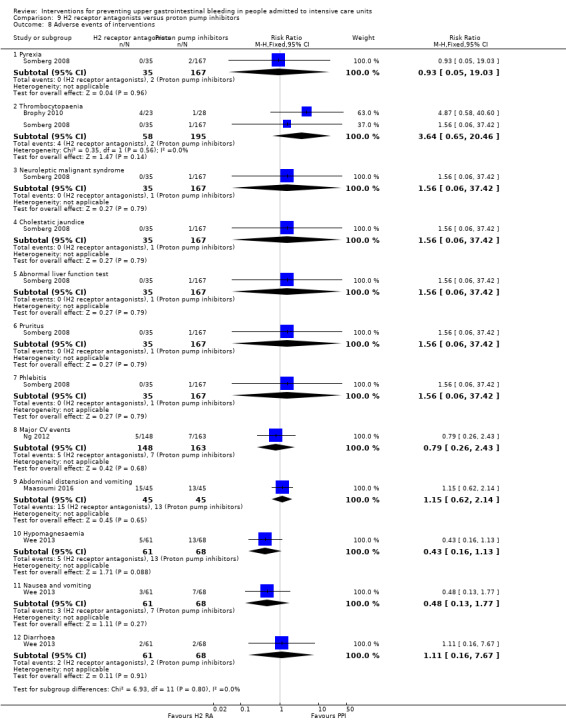
Comparison 9 H2 receptor antagonists versus proton pump inhibitors, Outcome 8 Adverse events of interventions.
Thrombocytopaenia
Two relevant studies reported on this adverse event (N = 253). Data show no clear difference between H2 receptor antagonists and proton pump inhibitors for thrombocytopaenia (RR 3.64, 95% CI 0.65 to 20.46; Analysis 9.8).
Neuroleptic malignant syndrome
We identified a single study that reported on this adverse event (N = 202). We found no evidence of a clear difference between the two groups (RR 1.56, 95% CI 0.06 to 37.42; Analysis 9.8).
Cholestatic jaundice
We found one study that reported on this adverse event (N = 202). We found no evidence of a clear difference between the two groups (RR 1.56, 95% CI 0.06 to 37.42; Analysis 9.8).
Abnormal liver function test
A single study reported on this adverse event (N = 202). Data show no clear difference between H2 receptor antagonists and proton pump inhibitors (RR 1.56, 95% CI 0.06 to 37.42; Analysis 9.8).
Pruritus
We found one study that reported on this adverse event (N = 202). Data show no clear difference between H2 receptor antagonists and proton pump inhibitors (RR 1.56, 95% CI 0.06 to 37.42; Analysis 9.8).
Phlebitis
A single study reported on this adverse event (N = 202). Data show no clear difference between H2 receptor antagonists and proton pump inhibitors (RR 1.56, 95% CI 0.06 to 37.42; Analysis 9.8).
Major cardiovascular events
We found a single study that reported on this adverse event (N = 311). We found no evidence of a clear difference between the two groups (RR 0.79, 95% CI 0.26 to 2.43; Analysis 9.8).
Abdominal distension and vomiting
We included a single study in this comparison (N = 90). We found no evidence of a clear difference between the two groups (RR 1.15, 95% CI 0.62 to 2.14; Analysis 9.8).
Hypomagnesaemia
We included a single study in this comparison (N = 129). We found no evidence of a clear difference between the two groups (RR 0.43, 95% CI 0.16 to 1.13; Analysis 9.8).
Nausea and vomiting
We included a single study in this comparison (N = 129). We found no evidence of a clear difference between the two groups (RR 0.48, 95% CI 0.13 to 1.77; Analysis 9.8).
C difficile‐related diarrhoea
We included a single study in this comparison (N = 129). We found no evidence of a clear difference between the two groups (RR 1.11, 95% CI 0.16 to 7.67; Analysis 9.8).
H2 receptor antagonists versusantacids
For this comparison, data on seven of our pre‐defined outcomes were available. Among the 18 studies that provided data for this comparison, ranitidine was administered in four studies and cimetidine in 13; one study included four arms (ranitidine, cimetidine, famotidine, and antacids) (Lamothe 1991). H2 receptor antagonists were combined to form a common interventional arm versus antacids, as the review did not aim to investigate intraclass efficacy among included interventions. Tryba 1985 administered pirenzepine as a concomitant medication to both cimetidine and antacid arms.
Clinically important upper GI bleeding
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 10.1). In the 16 included studies, 884 participants received ranitidine, cimetidine, or famotidine, and 816 received antacids. Clinically important upper GI bleeding occurred in 8.4% and 8.6% of participants in both groups. Data seemed to show no difference between the two groups with respect to the occurrence of clinically important upper GI bleeding (RR 0.96, 95% CI 0.67 to 1.36; 16 studies; 1700 participants). We judged the certainty of this evidence as low.
10.1. Analysis.

Comparison 10 H2 receptor antagonists versus antacids, Outcome 1 Clinically important upper GI bleeding.
Nosocomial pneumonia
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 10.2). In the four included studies, 288 participants received ranitidine or cimetidine, and 293 received antacids. Nosocomial pneumonia occurred in 29.2% and 28.0% of participants in the two groups, respectively. Data seemed to show no difference between the two groups with respect to the occurrence of the outcome of interest (RR 1.05, 95% CI 0.81 to 1.36). We judged the certainty of this evidence as low.
10.2. Analysis.
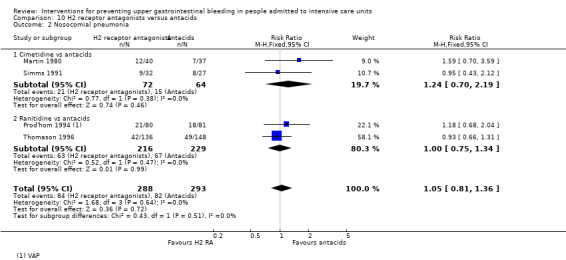
Comparison 10 H2 receptor antagonists versus antacids, Outcome 2 Nosocomial pneumonia.
Cimetidine versus antacids
We identified two relevant studies for this comparison (N = 136). Data show no clear difference between cimetidine and antacids (RR 1.24, 95% CI 0.70 to 2.19; Analysis 10.2).
Ranitidine versus antacids
We included two relevant studies in this comparison (N = 445). Data show no clear difference between ranitidine and antacids (RR 1.00, 95% CI 0.75 to 1.34; Analysis 10.2).
All‐cause mortality in ICU
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 10.3). In the 11 included studies, 472 participants received cimetidine, 33 received cimetidine plus pirenzepine, and 178 received ranitidine. Overall, 638 participants received antacids. Tryba 1985 administered pirenzepine as a concomitant medication to both cimetidine and antacid arms. All‐cause mortality occurred in 16.1% and 16.3% of the participants in both groups. Data seemed to show no difference between the two groups with respect to all‐cause mortality (RR 1.01, 95% CI 0.66 to 1.55). We judged the certainty of this evidence as very low.
10.3. Analysis.

Comparison 10 H2 receptor antagonists versus antacids, Outcome 3 All‐cause mortality in ICU.
Cimetidine versus antacids
We found eight studies that were relevant to this comparison (N = 885). We found no evidence of a clear difference between the two groups (RR 1.05, 95% CI 0.69 to 1.59). Heterogeneity was moderate (Chi² = 10.02, df = 7.0 (P = 0.19), I² = 30%; Analysis 10.3).
Cimetidine plus pirenzepine versus antacid plus pirenzepine
We included a single study in this comparison (N = 66). We found no evidence of a clear difference between the two groups (RR 1.25, 95% CI 0.37 to 4.25; Analysis 10.3).
Ranitidine versus antacids
We found two studies that were relevant to this comparison (N = 370). We found no evidence of a clear difference between the two groups (RR 1.13, 95% CI 0.14 to 8.97). This comparison had considerable heterogeneity (Chi² = 6.71, df = 1.0 (P = 0.01), I² = 85%; Analysis 10.3).
All‐cause mortality in hospital
We classified the study on the basis of its intervention and comparison arms (as shown in Analysis 10.4). In the single included study, 80 participants received ranitidine and 81 received antacids. The event occurred in 33.8% and 39.5% of participants in the two groups. Data seemed to show no difference between the two groups with respect to all‐cause mortality in the hospital (RR 0.85, 95% CI 0.57 to 1.29).
10.4. Analysis.

Comparison 10 H2 receptor antagonists versus antacids, Outcome 4 All‐cause mortality in hospital.
Duration of intubation
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 10.5). In the three included studies, 64 participants received cimetidine and 57 received antacids. Data seemed to show no difference between the two groups with respect to duration of intubation (MD ‐0.81 days, 95% CI ‐3.85 to 2.23).
10.5. Analysis.

Comparison 10 H2 receptor antagonists versus antacids, Outcome 5 Duration of intubation.
Number of participants requiring blood transfusions
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 10.6). In the six included studies, 379 participants received cimetidine or ranitidine and 365 received antacids. The event occurred in 8.2% and 3.0% of participants in both groups. Data seemed to show increased risk of blood transfusion in the H2 receptor antagonists arm compared with the antacids arm (RR 2.49, 95% CI 1.35 to 4.62). We judged the certainty of this evidence as moderate.
10.6. Analysis.
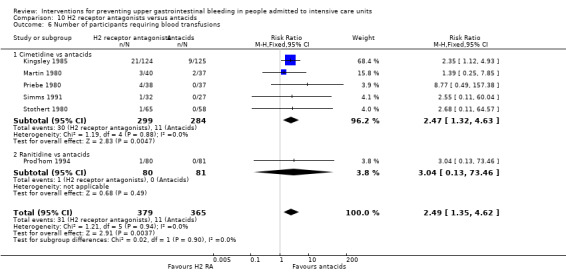
Comparison 10 H2 receptor antagonists versus antacids, Outcome 6 Number of participants requiring blood transfusions.
Cimetidine versus antacids
We found five studies that were relevant to this comparison (N = 583). We found evidence of a difference between cimetidine and antacids (RR 2.47, 95% CI 1.32 to 4.63; Analysis 10.6). Results favour the antacids arm.
Ranitidine versus antacids
We included a single study in this comparison (N = 161). We found no evidence of a clear difference between the two groups (RR 3.04, 95% CI 0.13 to 73.46; Analysis 10.6).
Number of units of blood transfused
We identified one study that addressed this outcome but did not report enough data for inclusion in a formal meta‐analysis (Priebe 1980). The study included 38 participants who received cimetidine and 37 who received antacids. Blood transfusions were required only for participants with GI bleeding, all of whom were included in the cimetidine group. It was reported that one participant received two units of blood and three participants received a total of four units of blood.
Adverse events of interventions
We identified 12 studies relevant to this outcome and divided the data into 12 groups according to adverse events.
Diarrhoea
We found seven studies that were relevant to this comparison (N = 863). We found evidence of a difference between H2 receptor antagonists and antacids. Darrhoea occurred in 2.1% and 10.4% of participants in the two groups, respectively (RR 0.23, 95% CI 0.13 to 0.43; six studies; 777 participants; Analysis 10.7). Results favour the H2 receptor antagonist arm. This comparison had moderate heterogeneity (Chi² = 11.3, df = 5.0 (P = 0.05), I² = 56%).
10.7. Analysis.

Comparison 10 H2 receptor antagonists versus antacids, Outcome 7 Adverse events of interventions.
Thrombocytopaenia
We found four studies that were relevant to this comparison (N = 452). We found no evidence of a clear difference between the two groups (RR 1.40, 95% CI 0.93 to 2.09; Analysis 10.7). This comparison had moderate heterogeneity (Chi² = 5.88, df = 3.0 (P = 0.12), I² = 49%).
Nausea and vomiting
We included four relevant studies in this comparison (N = 380). We found no evidence of a clear difference between the two groups (RR 0.46, 95% CI 0.19 to 1.1; Analysis 10.7).
Hypophosphataemia
We included two relevant studies in this comparison (N = 108). Data show no clear difference between H2 receptor antagonists and antacids (RR 0.24, 95% CI 0.04 to 1.3; Analysis 10.7).
Hypomagnesaemia
We included a single study in this comparison (N = 22). We found no evidence of a clear difference between the two groups (RR 0.33, 95% CI 0.02 to 7.39; Analysis 10.7).
Increase in creatinine
We found two studies that were relevant to this comparison (N = 286). Data show no clear difference between H2 receptor antagonists and antacids (RR 0.85, 95% CI 0.56 to 1.28; Analysis 10.7).
Mental confusion
We found four studies that were relevant to this comparison (N = 476). We found no evidence of a clear difference between the two groups (RR 1.26, 95% CI 0.77 to 2.07; Analysis 10.7).
Hypermagnesaemia
We included two relevant studies in this comparison (N = 115). Data show no clear difference between H2 receptor antagonists and antacids (RR 0.58, 95% CI 0.17 to 2.03; Analysis 10.7).
Rash/Erythema
We found two studies that were relevant to this comparison (N = 231). We found no evidence of a clear difference between the two groups (RR 3.02, 95% CI 0.32 to 28.53; Analysis 10.7).
Alkalosis
Alkalosis is a primary rise in the plasma bicarbonate concentration. We included a single study in this comparison (N = 75). We found no evidence of a clear difference between the two groups (RR 0.32, 95% CI 0.01 to 7.73; Analysis 10.7).
Dryness of mouth
We included a single study in this comparison (N = 67). We found no evidence of a clear difference between the two groups (RR 5.15, 95% CI 0.26 to 103.33; Analysis 10.7).
Leucopaenia
We included a single study in this comparison (N = 161). We found no evidence of a clear difference between the two groups (RR 3.04, 95% CI 0.13 to 73.46; Analysis 10.7).
H2 receptor antagonists versus sucralfate
For this comparison, data on nine of our pre‐defined outcomes were available. Among the 25 studies that provided data for this comparison, ranitidine was administered in 15 studies, cimetidine in seven, and famotidine in two. Tryba 1985 administered pirenzepine as a concomitant medication to both cimetidine and sucralfate arms. Two studies randomised participants to more than one arm of H2 receptor antagonists (cimetidine bolus and continuous infusion) (Fabian 1993; Ortiz 1998). These arms were combined to form a common interventional arm versus sucralfate, as the review did not aim to investigate efficacy based on dose or mode of administration of the same drug. We included one study that compared ranitidine (n = 15) with sucralfate (n = 15) but did not provide data on any of the outcomes relevant to this review.
Clinically important upper GI bleeding
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 11.1). In the 24 included studies, 1761 participants received ranitidine, cimetidine, or famotidine, and 1638 received sucralfate. Clinically important upper GI bleeding occurred in 7.6% and 6.6% of participants in the two groups, respectively. Data seemed to show no difference between the two groups with respect to the occurrence of clinically important upper GI bleeding (RR 1.10, 95% CI 0.87 to 1.41; 24 studies; 3316 participants). An additional study with 83 participants reported no events of clinically important upper GI bleeding in either treatment group (Pickworth 1993). We judged the certainty of this evidence as low.
11.1. Analysis.

Comparison 11 H2 receptor antagonists versus sucralfate, Outcome 1 Clinically important upper GI bleeding.
Cimetidine versus sucralfate
We included seven relevant studies in this comparison (N = 873). We found no evidence of a clear difference between the two groups (RR 1.37, 95% CI 0.87 to 2.14; Analysis 11.1).
Famotidine versus sucralfate
We included two relevant studies in this comparison (N = 190). Data show no clear difference between H2 receptor antagonists and sucralfate (RR 0.62, 95% CI 0.21 to 1.78; Analysis 11.1).
Ranitidine versus sucralfate
We found 14 studies that were relevant to this comparison (N = 2186). Data show no clear difference between H2 receptor antagonists and sucralfate (RR 1.03, 95% CI 0.76 to 1.39; Analysis 11.1). An additional study with 83 participants reported no events of clinically important upper GI bleeding in either treatment group (Pickworth 1993).
Cimetidine plus pirenzepine versus sucralfate plus pirenzepine
We included a single study in this comparison (N = 67). We found no evidence of a clear difference between the two groups (RR 5.15, 95% CI 0.26 to 103.33; Analysis 11.1).
Nosocomial pneumonia
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 11.2). In the 17 included studies, 1547 participants received ranitidine or cimetidine or famotidine, and 1460 received sucralfate. Nosocomial pneumonia occurred in 23.5% and 18.9% of participants in both groups. Data seemed to show increased risk of nosocomial pneumonia in the H2 receptor antagonist group compared with the sucralfate group (RR 1.22, 95% CI 1.07 to 1.40; 17 studies; 3041 participants). We judged the certainty of the evidence as moderate.
11.2. Analysis.
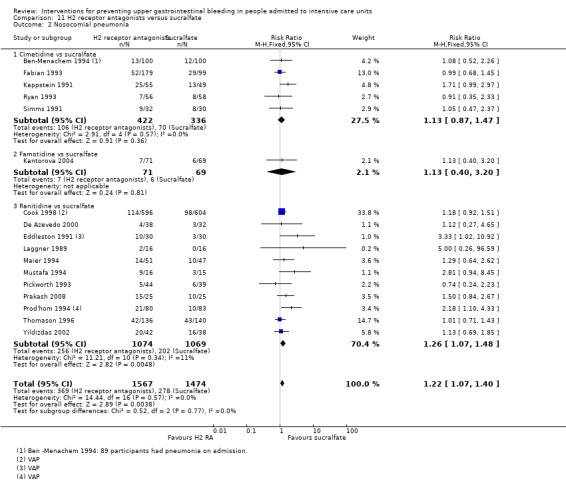
Comparison 11 H2 receptor antagonists versus sucralfate, Outcome 2 Nosocomial pneumonia.
Cimetidine versus sucralfate
We included five relevant studies in this comparison (N = 758). Data show no clear difference between cimetidine and sucralfate (RR 1.13, 95% CI 0.87 to 1.47; Analysis 11.2).
Famotidine versus sucralfate
We found one study that was relevant to this comparison (N = 140). We found no evidence of a clear difference between the two groups (RR 1.13, 95% CI 0.40 to 3.20; Analysis 11.2).
Ranitidine versus sucralfate
We found 11 studies that contributed data to this comparison (N = 2143). For this outcome, within this comparison, we found evidence suggesting that ranitidine was clearly different in its effects compared with sucralfate (RR 1.26, 95% CI 1.07 to 1.48; Analysis 11.2). One study with 70 participants reported no events in either treatment arm (Lopez‐Herce 1992).
All‐cause mortality in ICU
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 11.3). In the 21 included studies (N = 3178), 1652 participants received cimetidine, ranitidine, or famotidine, and 1526 received sucralfate. All‐cause mortality occurred in 22.0% and 20.4% of participants in both groups. We found no difference between the two groups with respect to all‐cause mortality in the ICU (RR 1.09, 95% CI 0.95 to 1.24). We judged the certainty of this evidence as low.
11.3. Analysis.

Comparison 11 H2 receptor antagonists versus sucralfate, Outcome 3 All‐cause mortality in ICU.
Cimetidine versus sucralfate
We found six studies that were relevant to this comparison (N = 814). Data show no clear difference between H2 receptor antagonists and sucralfate (RR 1.18, 95% CI 0.91 to 1.54; Analysis 11.3).
Famotidine versus sucralfate
We found two studies that were relevant to this comparison (N = 190). We found no evidence of a clear difference between the two groups (RR 1.23, 95% CI 0.69 to 2.19; Analysis 11.3).
Ranitidine versus sucralfate
We included 12 relevant studies in this comparison (N = 2107). We found no evidence of a clear difference between the two groups (RR 1.04, 95% CI 0.88 to 1.22; Analysis 11.3).
Cimetidine plus pirenzepine versus sucralfate plus pirenzepine
We included a single study in this comparison (N = 67). We found no evidence of a clear difference between the two groups (RR 1.29, 95% CI 0.38 to 4.38; Analysis 11.3).
All‐cause mortality in hospital
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 11.4). In the four included studies (N = 717), 366 participants received cimetidine, ranitidine, or famotidine, and 351 received sucralfate. All‐cause mortality in the hospital occurred in 22.7% and 20.2% of participants in both groups. Data showed no significant differences between the two groups (RR 1.14, 95% CI 0.86 to 1.50).
11.4. Analysis.

Comparison 11 H2 receptor antagonists versus sucralfate, Outcome 4 All‐cause mortality in hospital.
Cimetidine versus sucralfate
We found two studies that were relevant to this comparison (N = 413). Data show no clear difference between H2 receptor antagonists and sucralfate (RR 1.28, 95% CI 0.86 to 1.92; Analysis 11.4). For this comparison, heterogeneity was moderate (Chi² = 1.9, df = 1.0 (P = 0.17), I² = 47%).
Ranitidine versus sucralfate
We included a single study in this comparison (N = 164). We found no evidence of a clear difference between the two groups (RR 1.11, 95% CI 0.71 to 1.74; Analysis 11.4).
Famotidine versus sucralfate
We included a single study in this comparison (N = 140). Data show no clear difference between H2 receptor antagonists and sucralfate (RR 0.82, 95% CI 0.40 to 1.71; Analysis 11.4).
Duration of intubation
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 11.5). In the 10 included studies (N = 1751), 864 participants received cimetidine, ranitidine, or famotidine, and 887 received sucralfate. Data seemed to show no difference between the two groups with respect to duration of intubation (MD 0.22 days, 95% CI ‐1.55 to 2.00). This outcome had considerable heterogeneity (Chi² = 50.79, df = 9.0 (P = 0.0), I² = 82%).
11.5. Analysis.
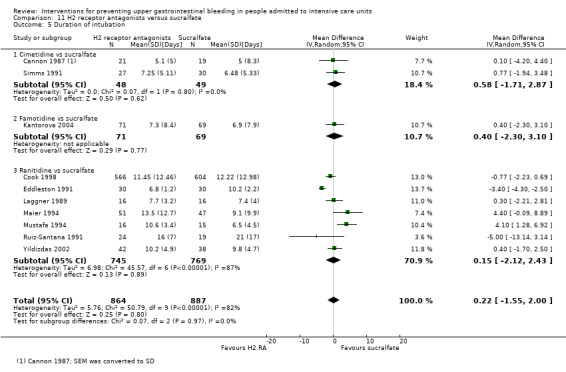
Comparison 11 H2 receptor antagonists versus sucralfate, Outcome 5 Duration of intubation.
Cimetidine versus sucralfate
We identified two relevant studies for this comparison (N = 97). Data show no clear difference between H2 receptor antagonists and sucralfate (MD 0.58 days, 95% CI ‐1.71 to 2.87; Analysis 11.5). We identified an additional study that did not provide enough data for formal meta‐analysis (Kappstein 1991). Of the included participants, 55 received cimetidine and 49 received sucralfate. The mean duration of mechanical ventilation was 5.36 days in the cimetidine group and 5.02 days in the sucralfate group. We identified an additional study that did not report enough data on the duration of intubation for inclusion in a formal meta‐analysis (Ryan 1993). This study included 56 participants in the cimetidine group and 58 in the sucralfate group. The mean duration of intubation was 5.1 days in the cimetidine group and 5.6 days in the sucralfate group.
Famotidine versus sucralfate
We included a single study in this comparison (N = 140). Data show no clear difference between H2 receptor antagonists and sucralfate (MD 0.40 days, 95% CI ‐2.30 to 3.10; Analysis 11.5).
Ranitidine versus sucralfate
We included seven relevant studies in this comparison (N = 1514). Data show no clear difference between H2 receptor antagonists and sucralfate (MD 0.15 days, 95% CI ‐2.12 to 2.43; Analysis 11.5). For this outcome, heterogeneity was considerable (Chi² = 45.57, df = 6.0 (P = 0.0), I² = 86%). Cook 1998 reported insufficient data for inclusion in our meta‐analyses. The median duration of intubation was seven days and eight days, respectively, in the ranitidine group (n = 596) and the sucralfate group (n = 604).
Duration of ICU stay
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 11.6). In the six included studies (N = 1791), 904 participants received ranitidine or famotidine and 887 received sucralfate. Data seemed to show no difference between the two groups with respect to duration of ICU stay (MD 0.01 days, 95% CI ‐1.92 to 1.95). For this outcome, heterogeneity was considerable (Chi² = 28.48, df = 5.0 (P = 0.0), I² = 82%) and heterogeneity could not be explained by differences among the different subcomparisons. We judged the certainty of this evidence as very low.
11.6. Analysis.

Comparison 11 H2 receptor antagonists versus sucralfate, Outcome 6 Duration of ICU stay.
Cimetidine versus sucralfate
We found one study that was relevant to this comparison (N = 213). We found no evidence of a clear difference between the two groups (MD 0.0 days, 95% CI ‐3.05 to 3.05; Analysis 11.6). Another study with 300 participants did not provide sufficient data for inclusion in our meta‐analyses, but data seemed to show no difference between cimetidine (median number of days = 4; n = 100) and sucralfate (median number of days = 3; n = 100) or control (median number of days = 3; n = 100) with respect to duration of ICU stay (Ben‐Menachem 1994).
Famotidine versus sucralfate
We found one study that was relevant to this comparison (N = 140). We found no evidence of a clear difference between the two groups (MD 2.2 days, 95% CI ‐0.96 to 5.36; Analysis 11.6).
Ranitidine versus sucralfate
We found four studies that were relevant to this comparison (N = 1438). We found no evidence of a clear difference between the two groups (MD ‐0.43 days, 95% CI ‐2.70 to 1.84; Analysis 11.6). This comparison had considerable heterogeneity (Chi² = 21.37, df = 3.0 (P = 0.0), I² = 85%). Cook 1998 reported insufficient data for inclusion in meta‐analyses. The median length of ICU stay in both groups was nine days (n = 596 and 604, respectively).
Number of participants requiring blood transfusion
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 11.7). In the nine included studies (N = 1095), 603 participants received cimetidine or ranitidine and 492 received sucralfate. Blood transfusions were required by 4.5% and 3.5% of participants in the two groups. Data seemed to show no difference between study arms with respect to the number of participants requiring blood transfusion (RR 1.25, 95% CI 0.70 to 2.23). We judged the certainty of this evidence as low.
11.7. Analysis.

Comparison 11 H2 receptor antagonists versus sucralfate, Outcome 7 Number of participants requiring blood transfusion.
Cimetidine versus sucralfate
We included five relevant studies in this comparison (N = 732). We found no evidence of a clear difference between the two groups (RR 1.00, 95% CI 0.47 to 2.16; Analysis 11.7).
Ranitidine versus sucralfate
We found four studies that were relevant to this comparison (N = 363). We found no evidence of a clear difference between the two groups (RR 1.77, 95% CI 0.71 to 4.39; Analysis 11.7).
Units of blood transfused
We classified the single study that contributed data for this outcome from its intervention and comparison arms (as shown in Analysis 11.8) (Ben‐Menachem 1994). In the included study (N = 200), 100 participants received cimetidine and 100 received sucralfate. The mean number of units of blood transfused was 1.6 (1.3) and 2.0 (2.0). Data seemed to show no difference between the two groups with respect to the mean number of units of blood transfused (MD ‐0.40, 95% CI ‐0.87 to 0.07). An additional study did not provide enough data (no SE or SD reported) for inclusion in formal meta‐analysis (Fabian 1993). This study reported that the mean number of required blood transfusions was 9.0 in the sucralfate group (n = 206) and 10.3 in the cimetidine group (n = 410).
11.8. Analysis.
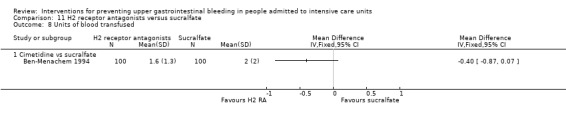
Comparison 11 H2 receptor antagonists versus sucralfate, Outcome 8 Units of blood transfused.
Adverse events of interventions
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 11.9). We identified six studies relevant to this outcome and divided the data from these studies according to eight adverse events.
11.9. Analysis.
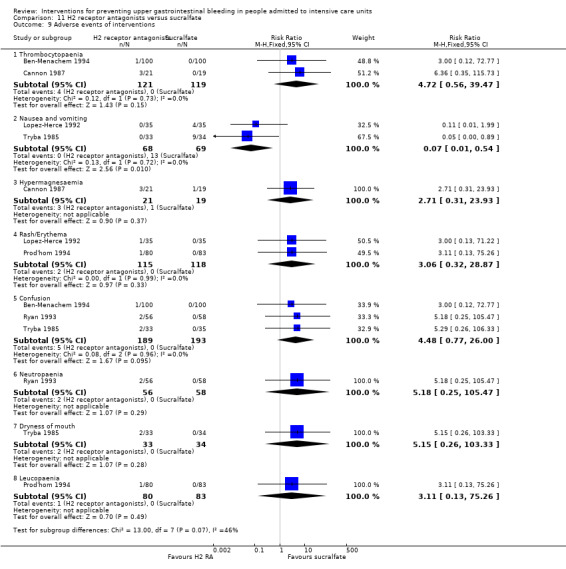
Comparison 11 H2 receptor antagonists versus sucralfate, Outcome 9 Adverse events of interventions.
Thrombocytopaenia
We found two studies that were relevant to this adverse event (N = 240). We found no evidence of a clear difference between the two groups (RR 4.72, 95% CI 0.56 to 39.47; Analysis 11.9).
Nausea and vomiting
We found two studies that were relevant to this comparison (N = 137). Nausea and vomiting seemed to be reduced in the H2 receptor antagonists arm (RR 0.07, 95% CI 0.01 to 0.54; two studies; 137 participants; Analysis 11.9).
Hypermagnesaemia
We found one study that was relevant to this adverse event (N = 40). Data show no clear difference between H2 receptor antagonists and sucralfate (RR 2.71, 95% CI 0.31 to 23.93; Analysis 11.9).
Rash/Erythema
We found two relevant studies for this adverse event (N = 233). We found no evidence of a clear difference between the two groups (RR 3.06, 95% CI 0.32 to 28.87; Analysis 11.9).
Confusion
We identified three relevant studies for this adverse event (N = 382). We found no evidence of a clear difference between the two groups (RR 4.48, 95% CI 0.77 to 26.0; Analysis 11.9).
Neutropaenia
We found one study that was relevant to this adverse event (N = 114). We found no evidence of a clear difference between the two groups (RR 5.18, 95% CI 0.25 to 105.47; Analysis 11.9).
Dryness of mouth
We identified a single study for this adverse event (N = 67). Data show no clear difference between H2 receptor antagonists and sucralfate for this adverse event (RR 5.15, 95% CI 0.26 to 103.33; Analysis 11.9).
Leucopaenia
We found one study that was relevant to this adverse event (N = 163). We found no evidence of a clear difference between the two groups (RR 3.11, 95% CI 0.13 to 75.26; Analysis 11.9).
H2 receptor antagonists versus anticholinergics
For this comparison, data on five of our pre‐defined outcomes were available. Among the four studies that provided data for this comparison, ranitidine was administered in two studies, cimetidine in one, and famotidine in one.
Clinically important upper GI bleeding
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 12.1). In the three included studies (N = 556), 285 participants received either ranitidine or cimetidine, and 271 received pirenzepine. Clinically important upper GI bleeding occurred in 4.2% and 3.0% of participants in the two groups. Data seemed to show no difference between the two groups with respect to the occurrence of clinically important upper GI bleeding (RR 1.37, 95% CI 0.58 to 3.26). We rated the certainty of this evidence as low.
12.1. Analysis.

Comparison 12 H2 receptor antagonists versus anticholinergics, Outcome 1 Clinically important upper GI bleeding.
Cimetidine versus pirenzepine
We included a single study in this comparison (N = 55). Data show no clear difference between H2 receptor antagonists and anticholinergics (RR 1.45, 95% CI 0.26 to 7.99; Analysis 12.1).
Ranitidine versus pirenzepine
We included two relevant studies in this comparison (N = 501). For this comparison, we found no evidence of a clear difference between the two groups (RR 1.35, 95% CI 0.50 to 3.67; Analysis 12.1).
Nosocomial pneumonia
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 12.2). In the three included studies (N = 544), 279 participants received ranitidine or famotidine, and 265 received pirenzepine. Nosocomial pneumonia occurred in 6.5% and 5.3% of participants in both groups. Data seemed to show no difference between the two groups with respect to occurrence of the outcome of interest (RR 0.96, 95% CI 0.50 to 1.84). We judged the certainty of this evidence as low.
12.2. Analysis.

Comparison 12 H2 receptor antagonists versus anticholinergics, Outcome 2 Nosocomial pneumonia.
Famotidine versus pirenzepine
We found one study that was relevant to this comparison (N = 43). Data show no clear difference between H2 receptor antagonists and anticholinergics (RR 0.32, 95% CI 0.01 to 7.42; Analysis 12.2).
Ranitidine versus pirenzepine
We found two studies that were relevant to this comparison (N = 501). Data show no clear difference between H2 receptor antagonists and anticholinergics (RR 1.03, 95% CI 0.53 to 2.01). This comparison had moderate heterogeneity (Chi² = 1.42, df = 0.23 (P = 0.11), I² = 30%; Analysis 12.2).
All‐cause mortality in ICU
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 12.3). In the two included studies (N = 501), 257 participants received ranitidine and 244 received pirenzepine. All‐cause mortality occurred in 5.8% and 6.6% of participants in the two groups. Data seemed to show no difference between the two groups with respect to occurrence of the outcome of interest (RR 0.89, 95% CI 0.21 to 3.87). This outcome had substantial heterogeneity (Chi² = 4.08, df = 1.0 (P = 0.04), I² = 75%). We judged the certainty of this evidence as very low.
12.3. Analysis.

Comparison 12 H2 receptor antagonists versus anticholinergics, Outcome 3 All‐cause mortality in ICU.
Duration of ICU stay
We identified one study that looked at this outcome but did not report sufficient data for inclusion in formal meta‐analyses (Hanisch 1998). In the included study, 57 participants received ranitidine and 44 received pirenzepine. The mean duration of ICU stay was 9.7 days (2 to 95) and 9.9 days (2 to 39), respectively.
Duration of intubation
We identified one study that looked at this outcome but did not report sufficient data for inclusion in formal meta‐analyses (Hanisch 1998). In the included study, 57 participants received ranitidine and 44 received pirenzepine. The mean duration of mechanical ventilation was 8.2 days (2 to 93) and 8.0 days (2 to 32), respectively.
Number of participants requiring blood transfusions
We classified the study on the basis of its intervention and comparison arms (as shown in Analysis 12.4). In the included study, 57 participants received ranitidine and 44 received pirenzepine. The event occurred in 5.3% and 6.8% of participants in the two groups. Data seemed to show no difference between the two groups with respect to the number of participants requiring blood transfusion (RR 0.77, 95% CI 0.16 to 3.64). We judged the certainty of this evidence as low.
12.4. Analysis.

Comparison 12 H2 receptor antagonists versus anticholinergics, Outcome 4 Number of participants requiring blood transfusion.
Number of units of blood transfused
We identified one study that looked at this outcome but did not report sufficient data for inclusion in formal meta‐analyses (Hanisch 1998). In the included study, 57 participants received ranitidine and 44 received pirenzepine. The mean number of units of blood transfused was two units for each of the three participants with GI bleeding in the ranitidine group, and four units for each of the four participants with GI bleeding in the pirenzepine group.
Adverse events of interventions
We classified studies on the basis of their intervention and comparison arms and adverse events of interventions (as shown in Analysis 12.5). Researchers reported no adverse events other than tachycardia and high temperature.
12.5. Analysis.

Comparison 12 H2 receptor antagonists versus anticholinergics, Outcome 5 Adverse events of interventions.
Tachycardia
A single study looked at this adverse event (N = 55). Data show no clear difference between H2 receptor antagonists and anticholinergics for tachycardia (RR 0.11, 95% CI 0.01 to 1.90; Analysis 12.5).
High temperature
We found one study that looked at this adverse event (N = 43). For this adverse event, we found no evidence of a clear difference between the two groups (RR 0.53, 95% CI 0.21 to 1.32; Analysis 12.5).
H2 receptor antagonists versus prostaglandin analogues
For this comparison, data on two of our pre‐defined outcomes were available. One study provided data for this comparison. Study groups received ranitidine (n = 64) and misoprostol (n = 63), respectively. The only outcomes measured that were relevant for this review were clinically important upper GI bleeding and all‐cause mortality in the ICU.
Clinically important upper GI bleeding
We classified this study on the basis of intervention and comparison arms (as shown in Analysis 13.1). In the single included study, 64 participants received cimetidine and 63 received misoprostol. Clinically important upper GI bleeding occurred in 4.7% and 11.1% of participants in the two groups. Data seemed to show no difference between the two groups with respect to the occurrence of clinically important upper GI bleeding (RR 0.42, 95% CI 0.11 to 1.56). We judged the certainty of this evidence as low.
13.1. Analysis.

Comparison 13 H2 receptor antagonists versus prostaglandin analogues, Outcome 1 Clinically important upper GI bleeding.
All‐cause mortality in ICU
We classified this study on the basis of intervention and comparison arms (as shown in Analysis 13.2). In the included study, 64 participants received cimetidine and 63 received misoprostol. All‐cause mortality occurred in 39.1% and 30.2% of participants in the two groups. Data seemed to show no difference between the two groups with respect to all‐cause mortality (RR 1.30, 95% CI 0.80 to 2.10). We judged the certainty of this evidence as moderate.
13.2. Analysis.
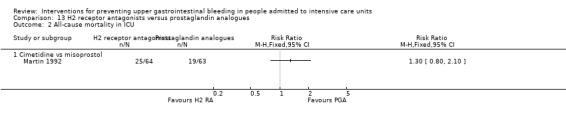
Comparison 13 H2 receptor antagonists versus prostaglandin analogues, Outcome 2 All‐cause mortality in ICU.
H2 receptor antagonists versus teprenone
For this comparison, data on three of our pre‐defined outcomes were available. One study provided data for this comparison. Study groups received ranitidine or teprenone, respectively. The only outcomes measured that were relevant for this review were clinically important upper GI bleeding, all‐cause mortality in the ICU, and number of participants requiring blood transfusion.
Clinically important upper GI bleeding
We classified this study on the basis of intervention and comparison arms (as shown in Analysis 14.1). In the included study, 70 participants received ranitidine and 70 received teprenone. Clinically important upper GI bleeding occurred in 5.7% and 5.7% of participants in the two groups. Data seemed to show no difference between the two groups with respect to the occurrence of clinically important upper GI bleeding (RR 1.00, 95% CI 0.26 to 3.84). We judged the certainty of this evidence as low.
14.1. Analysis.
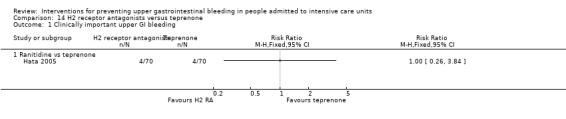
Comparison 14 H2 receptor antagonists versus teprenone, Outcome 1 Clinically important upper GI bleeding.
All‐cause mortality in ICU
We classified this study on the basis of intervention and comparison arms (as shown in Analysis 14.2). In the included study, 70 participants received ranitidine and 70 received teprenone. All‐cause mortality in the ICU occurred in 1.4% and 1.4% of participants in the two groups. Data seemed to show no difference between the two groups with respect to all‐cause mortality (RR 1.00, 95% CI 0.06 to 15.67). We judged the certainty of this evidence as moderate.
14.2. Analysis.

Comparison 14 H2 receptor antagonists versus teprenone, Outcome 2 All‐cause mortality in ICU.
Number of participants requiring blood transfusions
We classified this study on the basis of intervention and comparison arms (as shown in Analysis 14.3). In the included study, 70 participants received ranitidine and 70 received teprenone. Blood transfusions were required by 5.7% and 5.7% of participants in both groups. Data seemed to show no difference between the two groups with respect to the number that needed blood transfusion (RR 1.00, 95% CI 0.26 to 3.84). We judged the certainty of this evidence as moderate.
14.3. Analysis.

Comparison 14 H2 receptor antagonists versus teprenone, Outcome 3 Number of participants requiring blood transfusion.
H2 receptor antagonists plus antacids versus sucralfate
For this comparison, data on six of our pre‐defined outcomes were available. Among the three studies that provided data for this comparison, cimetidine was administered in Cioffi 1994, ranitidine was administered in Sirvent 1994, and either ranitidine or cimetidine was administered in Driks 1987, along with antacids, and outcomes were compared with those following administration of sucralfate.
Clinically important upper GI bleeding
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 15.1). In the two included studies that contributed data to this outcome, 119 participants received ranitidine or cimetidine plus antacids, and 111 received sucralfate. Clinically important upper GI bleeding occurred in 1.7% and 8.1% of participants in the two groups. Data seemed to show a beneficial effect of H2 receptor antagonists plus antacids compared with sucralfate with respect to the occurrence of upper GI bleeding (RR 0.24, 95% CI 0.06 to 0.95). We judged the certainty of this evidence as moderate.
15.1. Analysis.

Comparison 15 H2 receptor antagonist + antacids versus sucralfate, Outcome 1 Clinically important upper GI bleeding.
Cimetidine plus antacids versus sucralfate
We included a single study in this comparison (N = 100). We found no evidence of a clear difference between the two groups (RR 0.14, 95% CI 0.01 to 2.70).
Ranitidine plus antacids versus sucralfate
We included a single study in this comparison (N = 51) (Sirvent 1994). We found no evidence of a clear difference between the two groups, for no events were reported in either treatment group.
Cimetidine or ranitidine plus antacids versus sucralfate
We included a single study in this comparison (N = 130). We found no evidence of a clear difference between the two groups (RR 0.29, 95% CI 0.06 to 1.41).
Nosocomial pneumonia
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 15.2). In the three included studies, 144 participants received cimetidine or ranitidine plus antacids, and 137 received sucralfate. Nosocomial pneumonia occurred in 25.0% and 24.1% of participants in the two groups. For this outcome, heterogeneity was substantial (Chi² = 6.36, df = 2.0 (P = 0.04), I² = 69%), and data seemed to show no difference between the two groups with respect to the occurrence of nosocomial pneumonia (RR 1.09, 95% CI 0.51 to 2.32). We judged the certainty of this evidence as very low.
15.2. Analysis.

Comparison 15 H2 receptor antagonist + antacids versus sucralfate, Outcome 2 Nosocomial pneumonia.
Cimetidine plus antacids versus sucralfate
We included a single study in this comparison (N = 100). We found no evidence of a clear difference between the two groups (RR 0.53, 95% CI 0.26 to 1.07).
Ranitidine plus antacids versus sucralfate
We included a single study in this comparison (N = 51). We found no evidence of a clear difference between the two groups (RR 1.27, 95% CI 0.64 to 2.53).
Cimetidine or ranitidine plus antacids versus sucralfate
We included a single study in this comparison (N = 130). We found no evidence of a clear difference between the two groups (RR 2.02, 95% CI 0.89 to 4.58).
All‐cause mortality in ICU
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 15.3). In the two included studies, 119 participants received cimetidine or ranitidine plus antacids, and 111 received sucralfate. All‐cause mortality occurred in 35.3% and 25.2% of participants in the two groups. Data seemed to show no difference between the two groups with respect to all‐cause mortality in the ICU (RR 1.38, 95% CI 0.92 to 2.05).
15.3. Analysis.

Comparison 15 H2 receptor antagonist + antacids versus sucralfate, Outcome 3 All‐cause mortality in ICU.
Cimetidine plus antacids versus sucralfate
We included a single study in this comparison (N = 100). We found no evidence of a clear difference between the two groups (RR 1.00, 95% CI 0.46 to 2.19).
Cimetidine or ranitidine plus antacids versus sucralfate
We included a single study in this comparison (N = 130). We found no evidence of a clear difference between the two groups (RR 1.57, 95% CI 0.99 to 2.50).
Duration of ICU stay
We classified this study on the basis of its intervention and comparison arms (as shown in Analysis 15.4). In the included study, 69 participants received cimetidine or ranitidine plus antacids, and 61 received sucralfate. Data seemed to show no difference between the two groups with respect to duration of ICU stay (MD 3.60 days, 95% CI ‐1.11 to 8.31). We judged the certainty of this evidence as low.
15.4. Analysis.

Comparison 15 H2 receptor antagonist + antacids versus sucralfate, Outcome 4 Duration of ICU stay.
Duration of intubation
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 15.5). In the two included studies, 119 participants received cimetidine or ranitidine plus antacids, and 111 received sucralfate. Data seemed to show no difference in duration of intubation for both study groups (MD ‐1.24 days, 95% CI ‐13.82 to 11.33). Heterogeneity was substantial for this outcome (Chi² = 4.32, df = 1.0 (P = 0.04), I² = 77%).
15.5. Analysis.
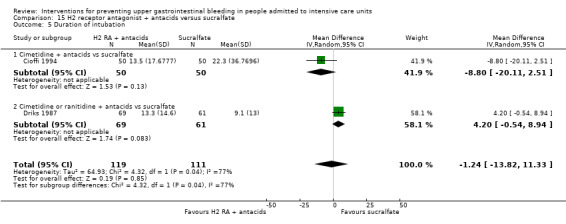
Comparison 15 H2 receptor antagonist + antacids versus sucralfate, Outcome 5 Duration of intubation.
Cimetidine plus antacids versus sucralfate
We included a single study in this comparison (N = 100). We found evidence showing a clear difference between the two groups (MD ‐8.80 days, 95% CI ‐20.11 to ‐2.51). Results favour the cimetidine plus antacids arm.
Cimetidine or ranitidine plus antacids versus sucralfate
We included a single study in this comparison (N = 130). We found no evidence of a clear difference between the two groups (MD 4.20 days, 95% CI ‐0.54 to 8.94).
Number of participants requiring blood transfusions
We classified this study on the basis of its intervention and comparison arms (as shown in Analysis 15.6). In the included study, 69 participants received cimetidine or ranitidine plus antacids, and 61 received sucralfate. Blood transfusions were required by 0% and 1.6% of participants in both groups. Data seemed to show no difference between the two groups with respect to the number of participants requiring blood transfusion (RR 0.30, 95% CI 0.01 to 7.12). We judged the certainty of this evidence as low.
15.6. Analysis.

Comparison 15 H2 receptor antagonist + antacids versus sucralfate, Outcome 6 Number of participants requiring blood transfusion.
Proton pump inhibitors versus teprenone
For this comparison, data on two of our pre‐defined outcomes were available, and only one study contributed data.
Clinically important upper GI bleeding
We classified this study on the basis of its intervention and comparison arms (as shown in Analysis 16.1). In the single included study, 70 participants received proton pump inhibitors and 70 received teprenone. Clinically important upper GI bleeding occurred in 0% and 5.7% of participants in both groups. We found no evidence of a clear difference between the two groups in this comparison (RR 0.11, 95% CI 0.01 to 2.03; Analysis 16.1). We judged the certainty of this evidence as low.
16.1. Analysis.

Comparison 16 Proton pump inhibitors versus teprenone, Outcome 1 Clinically important upper GI bleeding.
All‐cause mortality in ICU
We classified this study on the basis of its intervention and comparison arms (as shown in Analysis 16.2). In the single included study, 70 participants received proton pump inhibitors and 70 received teprenone. All‐cause mortality occurred in 0% and 0.7% of participants in both groups. Data show no clear difference between proton pump inhibitors and teprenone (RR 0.33, 95% CI 0.01 to 8.04). We judged the certainty of this evidence as moderate.
16.2. Analysis.

Comparison 16 Proton pump inhibitors versus teprenone, Outcome 2 All‐cause mortality in ICU.
Number of participants requiring blood transfusion
We classified this study on the basis of its intervention and comparison arms (as shown in Analysis 16.3). In the single included study, 70 participants received proton pump inhibitors and 70 received teprenone. Data show no clear difference between proton pump inhibitors and teprenone with respect to the number of participants requiring blood transfusion (RR 0.11, 95% CI 0.01 to 2.03). We judged the certainty of this evidence as moderate.
16.3. Analysis.

Comparison 16 Proton pump inhibitors versus teprenone, Outcome 3 Number of participants requiring blood transfusion.
Proton pump inhibitors plus naloxone versus naloxone
For this comparison, data on three of our pre‐defined outcomes were available. One study contributed data towards this comparison (He 2017). In the single included study, 60 participants received proton pump inhibitors (pantoprazole) plus naloxone, and 60 received naloxone alone.
Clinically important upper GI bleeding
We classified this study on the basis of its intervention and comparison arms (as shown in Analysis 17.1). Clinically important upper GI bleeding occurred in 1.6% and 8.33% of participants of the two groups, respectively. We found no evidence of a clear difference between the two groups in this comparison (RR 0.19, 95% CI 0.02 to 1.65; one study; 120 participants).
17.1. Analysis.

Comparison 17 Proton pump inhibitor plus naloxone versus naloxone, Outcome 1 Clinically important upper GI bleeding.
All‐cause mortality in the hospital
We classified this study on the basis of its intervention and comparison arms (as shown in Analysis 17.2). All‐cause mortality occurred in 5.0% and 21.7% of participants in the two groups, respectively. Data seemed to show a beneficial effect of pantoprazole plus naloxone on all‐cause mortality in the hospital (RR 0.23, 95% CI 0.06 to 0.89; one study; 120 participants).
17.2. Analysis.
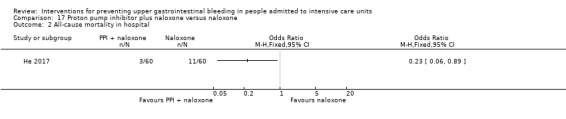
Comparison 17 Proton pump inhibitor plus naloxone versus naloxone, Outcome 2 All‐cause mortality in hospital.
Adverse events
We classified this study on the basis of its intervention and comparison arms (as shown in Analysis 17.3). Gastrointestinal discomfort, the only adverse event reported, occurred in 10.0% and 18.33% of participants in the two groups, respectively. We found no evidence of a clear difference between the two groups for this comparison (RR 0.40, 95% CI 0.14 to 1.14; one study; 120 participants).
17.3. Analysis.

Comparison 17 Proton pump inhibitor plus naloxone versus naloxone, Outcome 3 Adverse events ‐ gastrointestinal discomfort.
Proton pump inhibitors versus other medication (not defined)
For this comparison, data on three of our pre‐defined outcomes were available. One study contributed data towards this comparison. In the single included study, 60 participants received proton pump inhibitors (lansoprazole), and 60 received other medication not further specified.
Clinically important upper GI bleeding
We classified this study on the basis of its intervention and comparison arms (as shown in Analysis 18.1). Clinically important upper GI bleeding occurred in 0% and 10.0% of participants in the two groups, respectively. We found no evidence of a clear difference between the two groups for this comparison (RR 0.08, 95% CI 0.00 to 1.34).
18.1. Analysis.

Comparison 18 Proton pump inhibitors versus other medication (not defined), Outcome 1 Clinically important upper GI bleeding.
Nosocomial pneumonia
We classified this study on the basis of its intervention and comparison arms (as shown in Analysis 18.2). Nosocomial pneumonia occurred in 6.7% and 10% of participants in the two groups, respectively. We found no evidence of a clear difference between the two groups in this comparison (RR 0.67, 95% CI 0.20 to 2.24).
18.2. Analysis.
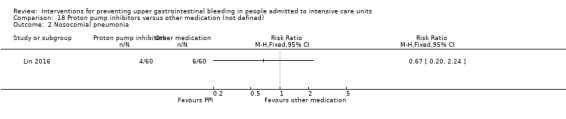
Comparison 18 Proton pump inhibitors versus other medication (not defined), Outcome 2 Nosocomial pneumonia.
All‐cause mortality in hospital
For this outcome, we found a single study (N = 120). All‐cause mortality in the hospital occurred in 10% and 0% of participants in the two groups, respectively. Data seemed to show no clear difference between proton pump inhibitors and other medication (not defined) (RR 5.00, 95% CI 0.25 to 102.00).
Antacids versus sucralfate
For this comparison, data on eight of our pre‐defined outcomes were available. Among the 16 studies that provided data for this comparison, one study administered pirenzepine as a co‐intervention in both arms (Tryba 1985).
Clinically important upper GI bleeding
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 19.1). In the 16 included studies (N = 1772), 890 participants received antacids and 882 received sucralfate. The event occurred in 6.6% of participants in both groups. Data seemed to show no clear differences between the two groups with respect to the occurrence of clinically important upper GI bleeding (RR 1.00, 95% CI 0.72 to 1.39). We judged the certainty of this evidence as low.
19.1. Analysis.

Comparison 19 Antacids versus sucralfate, Outcome 1 Clinically important upper GI bleeding.
Antacids versus sucralfate
We found 15 studies that were relevant to this comparison (N = 1705). We found no evidence of a clear difference between the two groups (RR 0.96, 95% CI 0.69 to 1.35; Analysis 19.1).
Antacid plus pirenzepine versus sucralfate plus pirenzepine
We found one study that was relevant to this comparison (N = 67). Data seemed to show no clear difference between antacids and sucralfate (RR 5.15, 95% CI 0.26 to 103.33; Analysis 19.1).
Nosocomial pneumonia
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 19.2). In the seven studies that contributed to this outcome, 501 participants received antacids and 495 received sucralfate. The event occurred in 24.4% and 23.2% of participants in the two groups. Data seemed to show no clear difference between the two groups with respect to the occurrence of nosocomial pneumonia (RR 1.04, 95% CI 0.84 to 1.30). We judged the certainty of this evidence as low.
19.2. Analysis.

Comparison 19 Antacids versus sucralfate, Outcome 2 Nosocomial pneumonia.
All‐cause mortality in ICU
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 19.3). In the 11 studies that contributed to this outcome (N = 1249), 628 participants received antacids and 621 received sucralfate. The event occurred in 23.9% and 20.6% of participants in the two groups. Data seemed to show no clear difference between the two groups with respect to all‐cause mortality in the ICU (RR 1.15, 95% CI 0.93 to 1.40). We judged the certainty of this evidence as low.
19.3. Analysis.
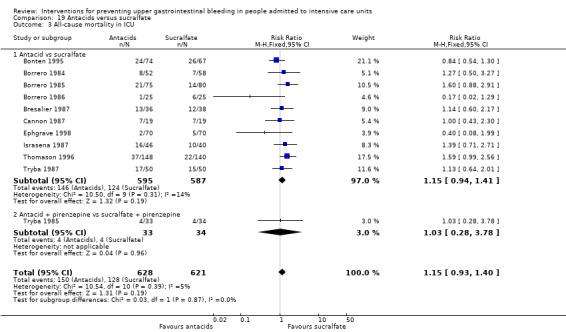
Comparison 19 Antacids versus sucralfate, Outcome 3 All‐cause mortality in ICU.
Antacid versus sucralfate
We found 10 studies that were relevant to this comparison (N = 1182). Data seemed to show no clear difference between antacids and sucralfate (RR 1.15, 95% CI 0.94 to 1.41; Analysis 19.3).
Antacid plus pirenzepine versus sucralfate plus pirenzepine
We found one study that was relevant to this comparison (N = 67). Data seemed to show no clear difference between antacids plus pirenzepine and sucralfate plus pirenzepine (RR 1.03, 95% CI 0.28 to 3.78; Analysis 19.3).
All‐cause mortality in hospital
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 19.4). In the three studies that contributed to this outcome (N = 450), 227 participants received antacids and 223 received sucralfate. The event occurred in 33.0% and 33.2% of participants in both groups. Data seemed to show no clear difference between the two groups with respect to all‐cause mortality in the hospital (RR 0.98, 95% CI 0.69 to 1.39). This outcome had moderate heterogeneity (Chi² = 3.38, df = 2.0 (P = 0.18), I² = 41%).
19.4. Analysis.

Comparison 19 Antacids versus sucralfate, Outcome 4 All‐cause mortality in hospital.
Duration of ICU stay
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 19.5). In the two studies that contributed to this outcome, 120 participants received antacids and 107 received sucralfate. Data seemed to show no clear difference between the two groups with respect to duration of ICU stay (MD ‐2.50 days, 95% CI ‐6.61 to 1.61). We rated the certainty of this evidence as low.
19.5. Analysis.

Comparison 19 Antacids versus sucralfate, Outcome 5 Duration of ICU stay.
Duration of intubation
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 19.6). In the four studies that contributed to this outcome (N = 281), 142 participants received antacids and 139 received sucralfate. Data seemed to show no clear difference between the two groups with respect to duration of intubation (standardised mean difference (SMD) ‐0.18, 95% CI ‐0.41 to 0.06).
19.6. Analysis.
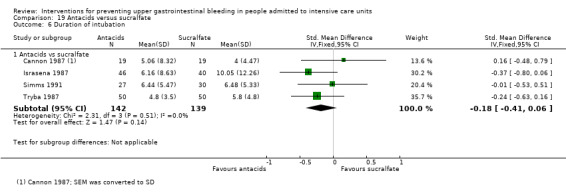
Comparison 19 Antacids versus sucralfate, Outcome 6 Duration of intubation.
Number of participants requiring blood transfusion
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 19.7). In the six studies that contributed to this outcome (N = 667), 338 participants received antacids and 329 received sucralfate. The event occurred in 3.6% and 5.2% of participants in the two groups. Data seemed to show no clear difference between the two groups with respect to the number of participants requiring blood transfusion (RR 0.73, 95% CI 0.40 to 1.34). We judged the certainty of this evidence as low.
19.7. Analysis.
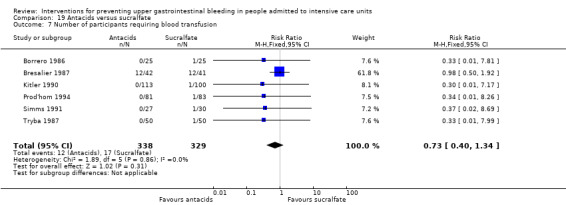
Comparison 19 Antacids versus sucralfate, Outcome 7 Number of participants requiring blood transfusion.
Adverse events of interventions
We classified studies on the basis of their intervention and comparison arms and adverse events of the intervention (as shown in Analysis 19.8). We identified nine studies that were relevant to this outcome and categorised data into six adverse events as reported in these studies.
19.8. Analysis.

Comparison 19 Antacids versus sucralfate, Outcome 8 Adverse events of interventions.
Diarrhoea
We identified six relevant studies for this outcome (N = 599). For this outcome, we found evidence suggesting that antacids were clearly different in their effects compared with sucralfate. This event occurred in 11.4% and 0% of the participants in both groups (RR 12.4, 95% CI 3.88 to 39.64; Analysis 19.8). Results favour the sucralfate arm.
Hypermagnesaemia
We found four studies that were relevant to this outcome (N = 317). For this outcome, we found evidence suggesting that antacids were clearly different in their effects compared with sucralfate (RR 4.72, 95% CI 1.24 to 17.95; Analysis 19.8). This event occurred in 6.5% and 0.6% of participants in the two groups. Results favour the sucralfate arm.
Nausea and vomiting
We included three relevant studies for this outcome (N = 223). For the occurrence of nausea and vomiting, we found no evidence of a clear difference between the two groups (RR 0.63, 95% CI 0.28 to 1.41; Analysis 19.8). This event occurred in 7.0% and 11.9% of participants in the two groups.
Thrombocytopaenia
We included a single study that looked at this outcome (N = 38). Data show no clear difference between antacids and sucralfate with respect to the occurrence of thrombocytopaenia (RR 5.00, 95% CI 0.26 to 97.70; Analysis 19.8). This event occurred in 10.5% and 0% of the participants in both groups.
Severe alkalosis
We found one study that was relevant to this outcome (N = 100). Data show no clear difference between antacids and sucralfate (RR 3.00, 95% CI 0.13 to 71.92; Analysis 19.8). This event occurred in 2.0% and 0% of the participants in both groups.
Allergic reactions
We found one study that was relevant to this outcome (N = 100). For this comparison, we found no evidence of a clear difference between the two groups (RR 0.20, 95% CI 0.01 to 4.06; Analysis 19.8). Allergic reactions occurred in 0% and 4.0% of participants in the two groups.
Antacids versus prostaglandin analogues
For this comparison, data on three of our pre‐defined outcomes were available. Two studies contributed data for this comparison.
Clinically important upper GI bleeding
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 20.1). In the two included studies (N = 329), 162 participants received antacids and 185 participants received prostaglandin analogues. This event occurred in 2.4% and 7.8% of participants in the two groups. Data seemed to show a beneficial effect of antacids compared with prostaglandin analogues on the occurrence of clinically important upper GI bleeding (RR 0.33, 95% CI 0.12 to 0.91). Results favour antacids. We judged the certainty of this evidence as moderate.
20.1. Analysis.

Comparison 20 Antacids versus prostaglandin analogues, Outcome 1 Clinically important upper GI bleeding.
All‐cause mortality in ICU
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 20.2). In the two studies that contributed to this outcome (N = 417), 206 participants received antacids and 211 received prostaglandin analogues. This event occurred in 6.3% and 7.6% of participants in both groups. Data show no difference between the two groups with respect to all‐cause mortality in the ICU (RR 0.84, 95% CI 0.42 to 1.67). We judged the certainty of this evidence as low.
20.2. Analysis.
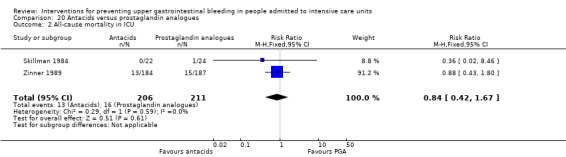
Comparison 20 Antacids versus prostaglandin analogues, Outcome 2 All‐cause mortality in ICU.
Adverse events of interventions
We classified this study on the basis of its intervention and comparison arms and adverse events of the intervention (as shown in Analysis 20.3).
20.3. Analysis.

Comparison 20 Antacids versus prostaglandin analogues, Outcome 3 Adverse events of interventions.
Diarrhoea
We found only one study that was relevant to this comparison (N = 368). We found no evidence of a clear difference between the two groups (RR 0.92, 95% CI 0.64 to 1.33; Analysis 20.3).
Elevated serum bicarbonate
We found one study that was relevant to this comparison (N = 338). We found evidence of a clear difference between antacids and prostaglandin analogues with respect to the occurrence of elevated serum bicarbonate (RR 2.21, 95% CI 1.27 to 3.87; Analysis 20.3).
Phospate levels below 2.5 mg/dL
We found one study that was relevant to this outcome (N = 276). We found evidence of a clear difference between antacids and prostaglandin analogues for the occurrence of phosphate levels below 2.5 mg/dL (RR 1.66, 95% CI 1.01 to 2.73; Analysis 20.3). Results favour prostaglandin analogues.
Antacids versus bioflavonoids
For this comparison, data on two of our pre‐defined outcomes were available. Only one study contributed data towards this comparison. In the single included study, 113 participants received antacids and 85 received bioflavonoids.
Clinically important upper GI bleeding
We classified this study on the basis of its intervention and comparison arms (as shown in Analysis 21.1). Clinically important upper GI bleeding occurred in 5.3% of participants in the antacid arm and in 8.2% of participants in the bioflavonoid arm. Data show no difference between the two groups with respect to the occurrence of clinically important upper GI bleeding (RR 0.64, 95% CI 0.22 to 1.85; 198 participants; one study). We judged the certainty of this evidence as low.
21.1. Analysis.
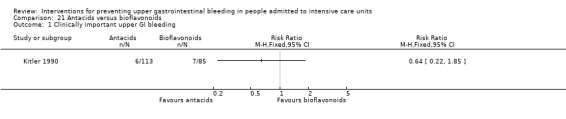
Comparison 21 Antacids versus bioflavonoids, Outcome 1 Clinically important upper GI bleeding.
Number of participants requiring blood transfusion
We classified this study on the basis of its intervention and comparison arms (as shown in Analysis 21.2). In the single included study, 113 participants received antacids and 85 received bioflavonoids. This event occurred in 2.4% of participants in the bioflavonoid arm and in no participants in the antacids arm. Data show no difference between the two groups with respect to the number of participants requiring blood transfusion (RR 0.15, 95% CI 0.01 to 3.10; 198 participants; one study). We judged the certainty of this evidence as low.
21.2. Analysis.

Comparison 21 Antacids versus bioflavonoids, Outcome 2 Number of participants requiring blood transfusion.
Sucralfate versus proton pump inhibitors
For this comparison, data on eight of our pre‐defined outcomes were available. Three studies contributed data for this comparison.
Clinically important upper GI bleeding
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 22.1). In the three included studies (N = 287), 139 participants received sucralfate and 148 received proton pump inhibitors (omeprazole). Clinically important upper GI bleeding occurred in 5.8% and 2% of participants in both groups. Data showed no difference between the two groups with respect to the occurrence of clinically important upper GI bleeding (RR 2.58, 95% CI 0.77 to 8.63). We judged the certainty of this evidence as low.
22.1. Analysis.

Comparison 22 Sucralfate versus proton pump inhibitors, Outcome 1 Clinically important upper GI bleeding.
Ventilator‐associated pneumonia
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 22.2). In the four studies that contributed to this outcome (N = 424), 210 participants received sucralfate and 214 received proton pump inhibitors. Nosocomial pneumonia occurred in 16.6% and 25.2% of participants in the two groups. Data showed no difference between the two groups with respect to the occurrence of nosocomial pneumonia (RR 0.67, 95% CI 0.41 to 1.09). We judged the certainty of this evidence as low.
22.2. Analysis.

Comparison 22 Sucralfate versus proton pump inhibitors, Outcome 2 Nosocomial pneumonia.
Sucralfate versus omeprazole
We found three studies that were relevant to this comparison (N = 287; n = 139 in the sucralfate arm and n = 148 in the proton pump inhibitor arm). We found no evidence of a difference between the two interventions (RR 0.88, 95% CI 0.57 to 1.36).
Sucralfate versus pantoprazole
We found one study that was relevant to this comparison (N = 137). We found evidence of a clear difference between sucralfate and proton pump inhibitors (RR 0.39, 95% CI 0.20 to 0.75). Results favour the sucralfate arm.
All‐cause mortality in ICU
We classified studies n the basis of their intervention and comparison arms (as shown in Analysis 22.3). In the four studies that contributed to this outcome (N = 424), 205 participants received sucralfate and 219 received proton pump inhibitors (omeprazole). All‐cause mortality in the ICU occurred in 15.6% and 14.6% of participants in both groups. Data show no significant differences between the two groups with respect to all‐cause mortality in the ICU (RR 1.07, 95% CI 0.68 to 1.68). We judged the certainty of this evidence as low.
22.3. Analysis.

Comparison 22 Sucralfate versus proton pump inhibitors, Outcome 3 All‐cause mortality in ICU.
Sucralfate versus omeprazole
We included three relevant studies in this comparison (N = 287). Data showed no clear difference between sucralfate and proton pump inhibitors (RR 1.26, 95% CI 0.75 to 2.11; Analysis 22.3).
Sucralfate versus pantoprazole
We included a single study in this comparison (N = 137). We found no evidence of a clear difference between the two groups (RR 0.65, 95% CI 0.25 to 1.68; Analysis 22.3).
All‐cause mortality in hospital
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 22.4). In the two studies that contributed to this outcome (N = 278), 140 participants received sucralfate and 138 received proton pump inhibitors. This event occurred in 13.5% and 17.4% of participants in the two groups. Data showed no significant difference between the two groups with respect to all‐cause mortality in the hospital (RR 0.79, 95% CI 0.46 to 1.37).
22.4. Analysis.

Comparison 22 Sucralfate versus proton pump inhibitors, Outcome 4 All‐cause mortality in hospital.
Sucralfate versus omeprazole
We included a single study in this comparison (N = 141). Data show no clear difference between sucralfate and proton pump inhibitors (RR 0.97, 95% CI 0.49 to 1.91; Analysis 22.4).
Sucralfate versus pantoprazole
We included a single study in this comparison (N = 137). Data show no clear difference between sucralfate and proton pump inhibitors (RR 0.56, 95% CI 0.21 to 1.45; Analysis 22.4).
Duration of ICU stay
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 22.5). In the two studies that contributed to this outcome (N = 217), 107 participants received sucralfate and 110 received proton pump inhibitors. Data seemed to show no difference between the two groups with respect to duration of ICU stay (MD 0.01 days, 95% CI ‐1.68 to 1.70). We judged the certainty of this evidence as low.
22.5. Analysis.

Comparison 22 Sucralfate versus proton pump inhibitors, Outcome 5 Duration of ICU stay.
Duration of intubation
We classified studies on the basis of their intervention and comparison arms (as shown in Analysis 22.6). In the three studies that contributed to this outcome (N = 354), 173 participants received sucralfate and 181 received proton pump inhibitors. Data seemed to show no significant difference between the two groups with respect to duration of intubation (MD ‐0.16 days, 95% CI ‐1.61 to 1.28).
22.6. Analysis.

Comparison 22 Sucralfate versus proton pump inhibitors, Outcome 6 Duration of intubation.
Number of participants requiring blood transfusion
We classified this studies on the basis of their intervention and comparison arms (as shown in Analysis 22.7). In the single study that contributed to this outcome (N = 70), 32 participants received sucralfate and 38 received proton pump inhibitors. Blood transfusions were required by 6.25% and 0% of participants in the two groups, respectively. Data seemed to show differences between the two groups with respect to the number of participants requiring blood transfusion (RR 5.91, 95% CI 0.29 to 118.78). We judged the certainty of this evidence as moderate.
22.7. Analysis.

Comparison 22 Sucralfate versus proton pump inhibitors, Outcome 7 Number of participants requiring blood transfusion.
Adverse events of interventions
For this outcome, we found a single study and categorised data according to eight adverse events (N = 1096).
Fever
For this outcome, we found evidence suggesting that sucralfate was clearly different in its effects compared with proton pump inhibitors (RR 0.81, 95% CI 0.70 to 0.94; Analysis 22.8). Researchers reported 54 and 62 events in both treatment arms, respectively. Results favour the sucralfate arm.
22.8. Analysis.

Comparison 22 Sucralfate versus proton pump inhibitors, Outcome 8 Adverse events of interventions.
Leucocytosis
For this outcome, we found evidence suggesting that sucralfate was clearly different in its effects compared with proton pump inhibitors (RR 0.66, 95% CI 0.55 to 0.80; Analysis 22.8). Researchers reported 45 and 63 events in both treatment arms, respectively. Results favour the sucralfate arm.
Sudden purulent sputum
Data show no clear difference between sucralfate and proton pump inhibitors for this outcome (RR 0.85, 95% CI 0.38 to 1.86; Analysis 22.8). Researchers reported 10 and 11 events in the two treatment arms, respectively.
Sudden cough or aggravation of coughing
We found evidence of a clear difference between sucralfate and proton pump inhibitors within this outcome (RR 0.23, 95% CI 0.07 to 0.79; Analysis 22.8). Researchers reported 3 and 12 events in the two treatment arms, respectively. Results favour the sucralfate arm.
Dyspnoea (breathing discomfort)
We found evidence of a clear difference between sucralfate and proton pump inhibitors within this outcome (RR 0.68, 95% CI 0.54 to 0.87; Analysis 22.8). Researchers reported 39 and 53 events in the two treatment arms, respectively. Results favour the sucralfate arm.
Rales or bronchial sounds
We found evidence of a clear difference between sucralfate and proton pump inhibitors for this outcome (RR 0.31, 95% CI 0.19 to 0.51; Analysis 22.8). Researchers reported 14 and 42 events in the two treatment arms, respectively. Results favour the sucralfate arm.
Aggravation of blood gas exchange
For this comparison, we found no evidence of a clear difference between the two groups (RR 0.76, 95% CI 0.49 to 1.18; Analysis 22.8). Researchers reported 23 and 28 events in the two treatment arms, respectively.
Change in sputum quality
For this outcome, we found evidence suggesting that sucralfate was clearly different in its effects compared with proton pump inhibitors (RR 0.23, 95% CI 0.13 to 0.40; Analysis 22.8). Researchers reported 11 and 45 events in the two treatment arms, respectively. Results favour the sucralfate arm.
Sucralfate versus bioflavonoids
For this comparison, data on two of our pre‐defined outcomes were available. Only one study contributed data towards this comparison.
Clinically important upper GI bleeding
We classified this study on the basis of its intervention and comparison arms (as shown in Analysis 23.1). In the single included study (N = 185), 100 participants received sucralfate and 85 received bioflavonoids. Clinically important upper GI bleeding occurred in 9% of participants in the antacid arm and in 8.2% of participants in the bioflavonoid arm. Data showed no clear difference between the two groups with respect to the occurrence of clinically important upper GI bleeding (RR 1.09, 95% CI 0.43 to 2.81). We judged the certainty of this evidence as low.
23.1. Analysis.
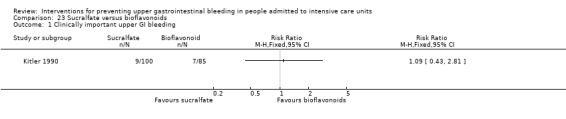
Comparison 23 Sucralfate versus bioflavonoids, Outcome 1 Clinically important upper GI bleeding.
Number of participants requiring blood transfusions
We classified this study on the basis of its intervention and comparison arms (as shown in Analysis 23.2). In the single included study (N = 185), 100 participants received antacids and 85 received bioflavonoids. Blood transfusions were required by 1% of participants in the sucralfate arm and by 2.4% of participants in the bioflavonoid arm. Data showed no clear difference between the two groups with respect to the number of participants requiring blood transfusion (RR 0.42, 95% CI 0.04 to 4.61). Results did not favour any intervention. We judged the certainty of this evidence as low.
23.2. Analysis.
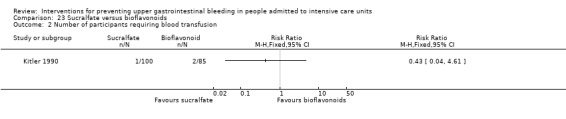
Comparison 23 Sucralfate versus bioflavonoids, Outcome 2 Number of participants requiring blood transfusion.
Total parenteral nutrition versus any other intervention plus total parenteral nutrition
For this comparison, data on three of our pre‐defined outcomes were available. One study contributed data towards this comparison. In the single included study, 30 participants received total parenteral nutrition, 24 received H2 receptor antagonist plus total parenteral nutrition, and 19 received sucralfate plus total parenteral nutrition. We compared the effects of total parenteral nutrition versus H2 receptor antagonists plus total parenteral nutrition and total parenteral nutrition versus sucralfate plus total parenteral nutrition separately.
Clinically important upper GI bleeding
For this outcome, we found a single study and categorised data by two comparisons. We judged the certainty of this evidence as low.
Total parenteral nutrition versus H2 receptor antagonists plus total parenteral nutrition alone
We classified this study on the basis of its intervention and comparison arms (as shown in Analysis 24.1). This event occurred in 3.3% of participants in the total parenteral nutrition arm and in 4.2% of participants in the H2 receptor antagonist (ranitidine) plus total parenteral nutrition arm. Data showed no significant differences between the two groups with respect to the occurrence of clinically important upper GI bleeding (RR 0.80, 95% CI 0.05 to 12.14; 54 participants; one study).
24.1. Analysis.
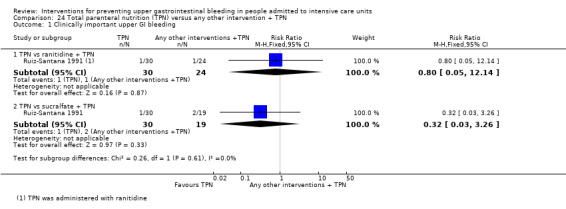
Comparison 24 Total parenteral nutrition (TPN) versus any other intervention + TPN, Outcome 1 Clinically important upper GI bleeding.
Total parenteral nutrition versus sucralfate plus total parenteral nutrition
We classified this study on the basis of its intervention and comparison arms (as shown in Analysis 24.1). Clinically important upper GI bleeding occurred in 3.3% of participants in the total parenteral nutrition arm and in 10.5% of participants in the sucralfate plus total parenteral nutrition arm. Data seemed to show no difference between the two groups with respect to the occurrence of clinically important upper GI bleeding (RR 0.32, 95% CI 0.03 to 3.26; 49 participants; one study). Results did not favour any intervention.
All‐cause mortality in ICU
For this outcome, we found a single study and categorised data by two comparisons. We judged the certainty of this evidence as low.
Total parenteral nutrition versus H2 receptor antagonists plus total parenteral nutrition
We classified this study on the basis of its intervention and comparison arms (as shown in Analysis 24.2). All‐cause mortality in the ICU occurred in 2.3% of participants in the total parenteral nutrition arm and in 2.1% of participants in the H2 receptor antagonist plus total parenteral nutrition arm. Data seemed to show no difference between the two groups with respect to all‐cause mortality in the ICU (RR 1.12, 95% CI 0.41 to 3.09; 54 participants; one study). Results did not favour any intervention.
24.2. Analysis.
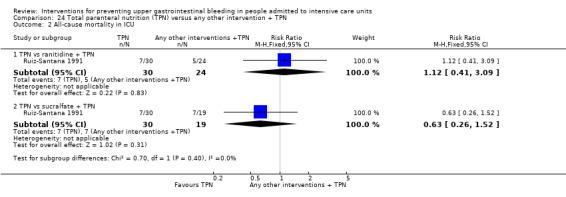
Comparison 24 Total parenteral nutrition (TPN) versus any other intervention + TPN, Outcome 2 All‐cause mortality in ICU.
Total parenteral nutrition versus sucralfate plus total parenteral nutrition
We classified this study on the basis of its intervention and comparison arms (as shown in Analysis 24.2). All‐cause mortality in the ICU occurred in 23.3% of participants in the total parenteral nutrition arm and in 36.8% of participants in the sucralfate plus total parenteral nutrition arm. Data seemed to show no difference between the two groups with respect to all‐cause mortality in the ICU (RR 0.63, 95% CI 0.26 to 1.52; 49 participants; one study). Results did not favour any intervention.
Duration of intubation
For this outcome, we found a single study and categorised data by two comparisons. We judged the certainty of this evidence as low.
Total parenteral nutrition versus H2 receptor antagonists plus total parenteral nutrition
We classified this study on the basis of its intervention and comparison arms (as shown in Analysis 24.3). Data seemed to show no difference between the two groups with respect to duration of intubation (MD ‐2.00 days, 95% CI ‐9.53 to 5.53; 54 participants; one study).
24.3. Analysis.

Comparison 24 Total parenteral nutrition (TPN) versus any other intervention + TPN, Outcome 3 Duration of intubation.
Total parenteral nutrition versus sucralfate plus total parenteral nutrition
We classified this study on the basis of its intervention and comparison arms (as shown in Analysis 24.3). Data seemed to show no difference between the two groups with respect to duration of intubation (MD 3.00, 95% CI ‐1.50 to 7.50; 49 participants; one study).
Bowel stimulation versus no prophylaxis
One study contributed data towards this comparison. In the single included study, 50 participants received a bowel stimulation protocol and 50 participants received no prophylaxis. This comparison had a single outcome.
Clinically important upper GI bleeding
We classified this study on the basis of its intervention and comparison arms (as shown in Analysis 25.1). Clinically important upper GI bleeding occurred in 14.0% and 36.0% of participants in the two groups, respectively. We found evidence of a clear difference between bowel stimulation and no prophylaxis (RR 0.39, 95% CI 0.18 to 0.85). Results favour the bowel stimulation protocol arm.
25.1. Analysis.

Comparison 25 Bowel stimulation versus no prophylaxis, Outcome 1 Clinically important upper GI bleeding.
Nasojejunal nutrition versus nasogastric nutrition
For this comparison, data on four of our pre‐defined outcomes were available. One study contributed data towards this comparison. In the single included study, 91 participants received nasojejunal nutrition and 89 received nasogastric nutrition.
Clinically important upper GI bleeding
We classified this study on the basis of its intervention and comparison arms (as shown in Analysis 26.1). Clinically important upper GI bleeding occurred in 15.4% and 5.6% of participants in the two groups, respectively. For this outcome, we found evidence suggesting that nasojejunal nutrition was clearly different in its effect compared with nasogastric nutrition (RR 2.74, 95% CI 1.03 to 7.28). Results favour nasogastric nutrition. We judged the certainty of this evidence as moderate.
26.1. Analysis.

Comparison 26 Nasojejunal nutrition versus nasogastric nutrition, Outcome 1 Clinically important upper GI bleeding.
Nosocomial pneumonia
We classified this study on the basis of its intervention and comparison arms (as shown in Analysis 26.2). Nosocomial pneumonia occurred in 19.8% and 21.3% of participants in the two groups. Data seemed to show no difference between nasojejunal nutrition and nasogastric nutrition (RR 0.93, 95% CI 0.52 to 1.65). We judged the certainty of this evidence as low.
26.2. Analysis.

Comparison 26 Nasojejunal nutrition versus nasogastric nutrition, Outcome 2 Nosocomial pneumonia.
All‐cause mortality in hospital
We classified this study on the basis of its intervention and comparison arms (as shown in Analysis 26.3). All‐cause mortality occurred in 14.3% and 13.5% of participants in the two groups. Data seemed to show no clear difference between nasojejunal nutrition and nasogastric nutrition (RR 1.06, 95% CI 0.51 to 2.19).
26.3. Analysis.

Comparison 26 Nasojejunal nutrition versus nasogastric nutrition, Outcome 3 All‐cause mortality in hospital.
Duration of ICU stay
This study did not report enough data for inclusion in formal meta‐analyses. The median duration of the ICU stay was 11 days and 10 days in the nasogastric group (n = 89) and the nasojejunal group (n = 91), respectively.
Duration of mechanical ventilation
This study did not report enough data for inclusion in formal meta‐analyses. The median duration of mechanical ventilation was eight days in both the nasogastric group (n = 89) and the nasojejunal group (n = 91).
Adverse events of interventions
We classified this study on the basis of its intervention and comparison arms (as shown in Analysis 26.4). Researchers reported 77 and 76 adverse events, including vomiting, witnessed aspiration, abdominal distension, and diarrhoea, in the two groups, respectively. We found no evidence of a clear difference between the two groups in this comparison (RR 0.99, 95% CI 0.88 to 1.12).
26.4. Analysis.

Comparison 26 Nasojejunal nutrition versus nasogastric nutrition, Outcome 4 Adverse events of interventions.
Enteral plus parenteral nutrition versus other nutrition regimens
For this comparison, data on five of our pre‐defined outcomes were available. One study contributed data towards this comparison (Fan 2016). In the single included study with three treatment arms, 40 participants received enteral plus parenteral nutrition and 80 received other nutrition regimens, 40 received enteral nutrition, and 40 received parenteral nutrition.
Nosocomial pneumonia
We classified this study on the basis of its intervention and comparison arms (as shown in Analysis 27.1). Nosocomial pneumonia occurred in 27.5% and 35.0% of participants in the two groups, respectively. Data seemed to show no clear difference between enteral plus parenteral nutrition and interventions in the other study groups (RR 0.79, 95% CI 0.44 to 1.40; one study; 120 participants).
27.1. Analysis.

Comparison 27 Enteral plus parenteral nutrition versus other nutrition regimens, Outcome 1 Nosocomial pneumonia.
All‐cause mortality in the hospital
We classified this study on the basis of its intervention and comparison arms (as shown in Analysis 27.2). All‐cause mortality in the hospital occurred in 10.0% and 36.2% of participants in the two groups, respectively. All‐cause mortality seemed to be lower in the group receiving enteral plus parenteral nutrition than in the groups receiving other nutrition regimens (RR 0.20, 95% CI 0.06 to 0.60; one study; 120 participants).
27.2. Analysis.

Comparison 27 Enteral plus parenteral nutrition versus other nutrition regimens, Outcome 2 All‐cause mortality in hospital.
Duration of ICU stay
We classified this study on the basis of its intervention and comparison arms (as shown in Analysis 27.3). Duration of ICU stay seemed to be shorter in the study group receiving enteral plus parenteral nutrition compared with groups receiving other nutrition regimens (MD ‐5.98 days, 95% CI ‐8.81 to ‐3.16; one study; 120 participants).
27.3. Analysis.

Comparison 27 Enteral plus parenteral nutrition versus other nutrition regimens, Outcome 3 Duration of ICU stay.
Duration of intubation
We classified this study on the basis of its intervention and comparison arms (as shown in Analysis 27.4). Duration of intubation seemed to be shorter in the study group receiving enteral plus parenteral nutrition compared with the groups receiving other nutrition regimens (MD ‐7.37 days, 95% CI ‐9.29 to ‐5.45; one study; 120 participants).
27.4. Analysis.
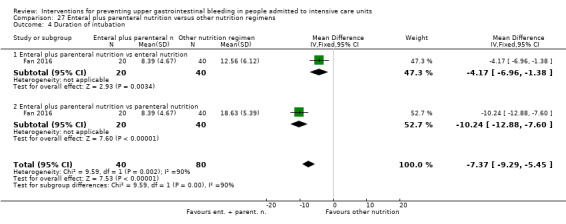
Comparison 27 Enteral plus parenteral nutrition versus other nutrition regimens, Outcome 4 Duration of intubation.
Adverse events of interventions
We classified this study on the basis of its intervention and comparison arms (as shown in Analysis 27.5 and following).
27.5. Analysis.
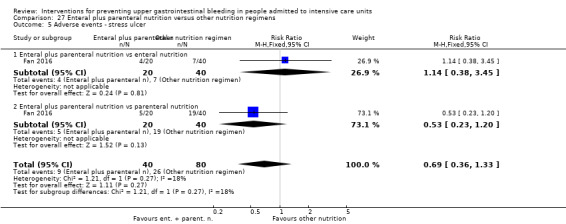
Comparison 27 Enteral plus parenteral nutrition versus other nutrition regimens, Outcome 5 Adverse events ‐ stress ulcer.
Occurence of stress ulcers
Data seemed to show no differences between study groups with respect to the occurrence of stress ulcers (RR 0.69, 95% CI 0.36 to 1.33; one study; 120 participants). Researchers reported nine events in the intervention group and 26 in the control groups.
Diarrhoea
Data seemed to show no difference between study groups with respect to the occurrence of diarrhoea (RR 0.42, 95% CI 0.17 to 1.02; one study; 120 participants). Researchers reported that the event occurred in eight participants in the intervention group and in 30 participants in control groups.
Pyaemia
Data seemed to show no difference between study groups with respect to the occurrence of pyaemia (RR 0.88, 95% CI 0.37 to 2.09; one study; 120 participants). Researchers reported 10 events in the intervention group and 22 events in the control groups.
Intracranial infection
Data seemed to show no difference between study groups with respect to the occurrence of intracranial infection (RR 0.43, 95% CI 0.15 to 1.25; one study; 120 participants). Researchers reported five events in the intervention group and 20 events in the control groups.
Hypoproteinaemia
Data seemed to show a beneficial effect of enteral plus parenteral nutrition on the occurrence of hypoproteinaemia (RR 0.11, 95% CI 0.04 to 0.27; one study; 120 participants). Researchers reported seven events in the intervention group and 54 events in the control groups.
Subgroup analyses
Presence of bleeding disorders
We identified three studies that explicitly reported including patients with bleeding disorders (Conrad 2005; Kantorova 2004; Martin 1992). Only two studies compared a common intervention and a comparator (H2 receptor antagonists vs proton pump inhibitors). Subgroup analyses provided no evidence of a difference between studies that explicitly included patients with bleeding disorders and those that did not include such patients (test for subgroup differences P value = 0.09). Further, there seemed to be no difference between groups with respect to the occurrence of nosocomial pneumonia (test for subgroup differences P value = 0.37) and all‐cause mortality in the ICU (test for subgroup differences P value = 0.52). Researchers provided no data on other outcomes and comparisons for this subgroup that could be formally included in meta‐analysis.
Pneumonia at time of admission
We identified four studies that explicitly reported including participants with pneumonia at the time of ICU admission (Ben‐Menachem 1994; Karlstadt 1990; Martin 1993; Metz 1993). The proportion of participants with pneumonia ranged from 2.33% to 21% in these studies, respectively. All studies compared the effects of H2 receptor antagonists (cimetidine or ranitidine) versus placebo or no prophylaxis. Subgroup analyses provided no evidence of a difference between studies that explicitly included patients with pneumonia at the time of admission and those that did not include such patients (test for subgroup differences P value = 0.28). Data showed no difference between groups with respect to the occurrence of nosocomial pneumonia (test for subgroup differences P value = 0.54) and all‐cause mortality in the ICU (test for subgroup differences P value = 0.20). Researchers provided no data on other outcomes and comparisons for this subgroup that could be formally analysed in meta‐analysis.
Infants and children
We identified five studies that explicitly reported including only children and adolescents (Behrens 1994; Kuusela 1997; Lacroix 1986; Lopez‐Herce 1992; Yildizdas 2002). All participants in these studies were aged from 0 to 20 years. Two studies compared effects of H2 receptor antagonists versus placebo, and three studies had multiple study arms including H2 receptor antagonists, anticholinergics (Behrens 1994), antacids (Lopez‐Herce 1992), sucralfate (Lopez‐Herce 1992;Yildizdas 2002), and proton pump inhibitors (Yildizdas 2002), as well as placebo or no prophylaxis. In infants and adolescents, data showed no difference between H2 receptor antagonists and placebo or no prophylaxis with respect to the occurrence of clinically important upper GI bleeding (test for subgroup differences P value = 0.10), nosocomial pneumonia (test for subgroup differences P value = 0.84), or all‐cause mortality in the ICU (test for subgroup differences P value = 0.52); duration of ICU stay (test for subgroup differences P value = 0.62); duration of intubation (test for subgroup differences P value = 0.75); or number of participants requiring blood transfusion (test for subgroup differences P value = 0.37). Researchers provided no data on other outcomes of this comparison for this subgroup that could be formally included in meta‐analysis.
We detected no differences between H2 receptor antagonists and sucralfate with respect to the occurrence of clinically important upper GI bleeding in infants (test for subgroup differences P value = 0.61), nosocomial pneumonia (test for subgroup differences P value = 0.75), all‐cause mortality in the ICU (test for subgroup differences P value = 0.92), duration of ICU stay (test for subgroup differences P value = 0.84), or duration of intubation (test for subgroup differences P value = 0.90). We found no data on other outcomes in this comparison for this subgroup that could be formally analysed in meta‐analysis. Moreover, researchers provided no data on other outcomes and comparisons for this subgroup that could be formally analysed in meta‐analysis.
The reporting quality of the included studies did not permit other subgroup analyses. No studies explicitly included only adults over the age of 65 years. Thus, insufficient data were available for analysis of the effect of bleeding prophylaxis in older adults only. Furthermore, use of co‐interventions and the level of detail of reporting about co‐interventions varied strongly among studies, so that meaningful subgroup analyses were not possible with respect to differences in effect due to co‐interventions.
Sensitivity analyses
Studies with low risk of bias versus studies with unclear or high risk of bias
Only two studies had an overall low risk of bias (Cook 1998; Ephgrave 1998), and one study had low risk of bias, when risk of performance and detection bias was assessed for upper GI bleeding only (Ng 2012).
Cook 1998 compared H2 receptor antagonists (ranitidine) (n = 596) versus sucralfate (n = 604). This study found lower risk of upper GI bleeding with H2 receptor antagonists (RR 0.44, 95% CI 0.21 to 0.92; one study; 1200 participants) compared with studies with unclear and high risk of bias (RR 1.28, 95% CI 0.98 to 1.66; 23 studies; 2199 participants). Data seemed to show no difference between study groups with respect to the occurrence of nosocomial pneumonia in the study with low risk of bias (RR 1.18, 95% CI 0.92 to 1.51; one study; 1200 participants), and studies with unclear or high risk of bias found increased risk of nosocomial pneumonia (RR 1.21, 95% CI 1.02 to 1.43; 14 studies; 1807 participants). The latter result favoured the sucralfate arm. Data show no differences between studies with low risk of bias and studies with unclear or high risk of bias in the comparison of H2 receptor antagonists versus sucralfate with respect to the outcome all‐cause mortality in the ICU, duration of intubation, and duration of ICU stay. No studies reported data on other outcomes pre‐defined in this review, so that no further sensitivity analyses were possible.
Ephgrave 1998 compared antacids (n = 70) versus sucralfate (n = 70) and reported no differences in effect between the study with low risk of bias and studies with unclear or high risk of bias with respect to the outcomes clinically important upper GI bleeding, nosocomial pneumonia, and all‐cause mortality in the ICU. No other outcomes of this comparison were available for sensitivity analysis.
Ng 2012 compared an H2 receptor antagonist (famotidine) (n = 149) versus a proton pump inhibitor (esomeprazole) (n = 164) and reported no differences in effect between the study with low risk of bias and studies with unclear or high risk of bias with respect to the outcomes clinically important upper GI bleeding and all‐cause mortality in the hospital. No studies reported data on other outcome pre‐defined in this review, so that no further sensitivity analyses were possible.
Studies with attrition greater than 10%
Six studies had an attrition rate greater than 10% (Barandun 1985;Fabian 1993;Hanisch 1998;Israsena 1987;Ruiz‐Santana 1991;Terzi 2009).
Barandun 1985 compared H2 receptor antagonists (n = 28) versus anticholinergics (n = 27) and noted no apparent differences in this comparison with respect to the occurrence of clinically important upper GI bleeding when excluding from analysis the study with an attrition rate of 10% or higher. No other outcomes of this comparison were available for sensitivity analysis.
Fabian 1993 compared H2 receptor antagonists (n = 410) versus sucralfate (n = 206) and noted no apparent differences in this comparison with respect to the occurrence of clinically important upper GI bleeding, nosocomial pneumonia, all‐cause mortality in the ICU or the hospital, duration of ICU stay, and the number of participants requiring blood transfusions when excluding from analysis the study with an attrition rate of 10% or higher. No other outcomes of this comparison were available for sensitivity analysis.
Hanisch 1998 compared an H2 receptor antagonist (ranitidine) (n = 57) versus an anticholinergic (pirenzepine) (n = 44) or placebo (n = 57) and noted no apparent difference in effect size and direction when excluding the study with an attrition rate of 10% from the comparison of H2 receptor antagonists versus anticholinergics or placebo. Neither did direction or strength of effect change when the study with an attrition rate of 10% was removed from the comparison of anticholinergics and placebo.
Israsena 1987 compared antacids (n = 47) versus sucralfate (n = 40) and noted no apparent difference in effect size and direction when excluding from analysis the study with an attrition rate of 10%.
Ruiz‐Santana 1991 was the only study comparing total parenteral nutrition with total parenteral nutrition plus other medications. So, no sensitivity analysis could be performed.
Terzi 2009 compared an H2 receptor antagonist (ranitidine) (n = 10) verus a proton pump inhibitor (pantoprazole) (n = 10) and noted no apparent difference in effect size and direction when excluding from analysis the study with an attrition rate of 10%.
Studies with published and validated criteria to diagnose clinically important upper GI bleeding and nosocomial pneumonia
Lack of detail regarding criteria used to diagnose the primary endpoints of the review did not allow sensitivity analysis.
Discussion
This review includes 107 randomised controlled trials that randomised 15,057 participants admitted to the intensive care unit (ICU) to receive interventions to prevent clinically important upper gastrointestinal (GI) bleeding. The various interventions yielded 27 sets of comparisons.
Summary of main results
Clinically important upper GI bleeding due to stress ulcers is a major contributor to increased morbidity and mortality among patients admitted to ICU. The main objective of this review was to assess whether interventions that are commonly used to prevent upper GI bleeding in patients admitted to the ICU are effective in preventing such bleeding, when used alone or in combination. Interventions most commonly used were H2 receptor antagonists, proton pump inhibitors, anticholinergics, antacids, sucralfate, and prostaglandin analogues. However, one of the most common adverse events associated with using these drugs, as discussed before, consists of raised pH of gastric contents, altered gastric flora, and promotion of tracheobronchial and gastric colonisation with pathogenic bacteria ‐ aspiration of which can cause nosocomial pneumonia or ventilator‐associated pneumonia. Some of the included studies investigated ventilator‐associated pneumonia as one of the outcomes, whereas other studies looked into this outcome in a more generic way and addressed it as 'nosocomial pneumonia'. Because ventilator‐associated pneumonia is considered a type of nosocomial pneumonia, we included results of studies that looked into either of these outcomes under the common term 'nosocomial pneumonia'.
Effect of any intervention versus placebo or no prophylaxis
In comparison with placebo, research findings suggest a beneficial effect of any prophylactic intervention on the occurrence of clinically important upper GI bleeding (risk ratio (RR) 0.47, 95% confidence interval (CI) 0.39 to 0.57; moderate certainty of evidence). Use of any intervention reduced the risk of clinically important upper GI bleeding by 9% (95% CI ‐11% to ‐7%). Data seemed to show no difference between any intervention and placebo or no prophylaxis with respect to the occurrence of any adverse events, including nosocomial pneumonia (low certainty of evidence), all‐cause mortality in the ICU (low certainty of evidence) or the hospital, duration of ICU stay (low certainty of evidence), duration of intubation (low certainty of evidence), number of participants requiring blood transfusions (moderate certainty of evidence), and units of blood transfused.
Effects of single drug classes versus placebo or no prophylaxis
Among the different interventions that were compared with placebo or no prophylaxis, researchers found that antacids, H2 receptor antagonists, antacids, and sucralfate seemed to be effective in preventing clinically important upper GI bleeding in ICU patients and produced significant results. Investigators found that H2 receptor antagonists reduced the risk of clinically important upper GI bleeding by 11% (95% CI ‐0.16 to ‐0.06) compared with placebo or no prophylaxis (RR 0.50, 95% CI 0.36 to 0.70; moderate certainty of evidence). Antacids reduced the risk of bleeding by 9% (95% CI ‐0.17 to ‐0.00) compared with placebo or no prophylaxis. Among ICU patients, sucralfate reduced the risk by 5% (95% CI ‐0.10 to ‐0.01) compared with placebo or no prophylaxis (RR 0.53, 95% CI 0.32 to 0.88; low certainty of evidence). Remaining interventions were not found to be significantly associated with lower rates of occurrence of upper GI bleeding in ICU patients when compared with placebo or no prophylaxis. This might be due to lack of sufficiently large randomised controlled trials (RCTs) in these comparisons.
Results of the review indicate that nosocomial pneumonia might occur more often in ICU patients taking H2 receptor antagonists or sucralfate when compared with ICU patients given placebo or no prophylaxis. Data seem to show no difference between H2 receptor antagonists and placebo or no prophylaxis in the occurrence of nosocomial pneumonia (RR 1.12, 95% CI 0.85 to 1.48; low certainty of evidence). Similarly, there seems to be no difference between sucralfate and placebo or no prophylaxis in the occurrence of nosocomial pneumonia (RR 1.33, 95% CI 0.86 to 2.04; low certainty of evidence).
Effects of one drug class versus another drug class
When H2 receptor antagonists were compared with proton pump inhibitors, evidence of low certainty suggested that proton pump inhibitors were significantly more effective in preventing upper GI bleeding in ICU patients (RR 2.90, 95% CI 1.83 to 4.58). Low‐certainty evidence also suggested no apparent difference in the incidence of nosocomial pneumonia with H2 receptor antagonists compared with proton pump inhibitors (RR 1.02, 95% CI 0.77 to 1.35). Results indicated that the usefulness of proton pump inhibitors over H2 receptor antagonists to prevent upper GI bleeding in ICU patients likely outweighs the possible adverse event of nosocomial pneumonia.
Evidence of low certainty suggested that H2 receptor antagonists compared with sucralfate were not significantly different in preventing upper GI bleeding in ICU patients (RR 1.10, 95% CI 0.87 to 1.41). However, evidence of moderate certainty shows that under H2 receptor antagonists, participants had increased risk of nosocomial pneumonia (RR 1.22, 95% CI 1.07 to 1.40). Evidence of low quality suggests that H2 receptor antagonists and antacids were comparable with respect to the outcomes of upper GI bleeding (RR 0.96, 95% CI 0.67 to 1.36) and nosocomial pneumonia in ICU patients (RR 1.05, 95% CI 0.81 to 1.36).
Evidence of moderate certainty showed that when H2 receptor antagonists plus antacids were compared with sucralfate, GI bleeding occurred in fewer participants taking H2 receptor antagonists plus antacid than those taking sucralfate (RR 0.24, 95% CI 0.06 to 0.95). However, use of H2 receptor antagonists plus antacids was comparable with use of sucralfate in the incidence of nosocomial pneumonia (RR 1.09, 95% CI 0.51 to 2.32; very low certainty of evidence).
This review found no other relevant differences between interventions and comparator interventions with regard to the occurrence of clinically important upper GI bleeding and nosocomial pneumonia.
Overall completeness and applicability of evidence
Completeness
We searched multiple sources for relevant randomised controlled studies and included and excluded studies on the basis of inclusion and exclusion criteria. We believe that we have identified all randomised controlled studies relevant to the objectives of this review. Still evidence gaps indicate that further research would be helpful in guiding decision‐making. One of these gaps reflects lack of evidence about the effects of bleeding prophylaxis on different population subgroups, including people with pneumonia at the time of ICU admission, as well as the effects of co‐interventions such as different feeding regimens, antibiotics, etc., on the effectiveness of bleeding prophylaxis. Furthermore, evidence from RCTs is needed on the effects of proton pump inhibitors on Clostridium difficile infection‐related diarrhoea. This adverse event has been observed in non‐randomised studies but rarely in RCTs. C difficile infection‐related diarrhoea was not included as an outcome of this review at the stage of formulating the review protocol. We identified only two studies that reported on this adverse event (Selvanderan 2015; Wee 2013).
Applicability
Upper GI bleeding occurs in ICU patients as the result of stress ulcers, and excessive blood loss further increases mortality and morbidity among ICU patients. The various interventions that are compared in this review are available worldwide and can prevent bleeding from stress ulcers in ICU patients. Furthermore, no compelling evidence suggests that any interventions clearly increase the risk of ventilator‐associated pneumonia or nosocomial pneumonia, which might further warrant their usage in ICU patients. Evidence from this review shows that among the different interventions, antacids, H2 receptor antagonists, sucralfate, and proton pump inhibitors confer the greatest benefit with respect to the occurrence of clinically important upper GI bleeding. We included studies that investigated the effects of bleeding prophylaxis in a broad spectrum of participants. Populations in these studies included different age groups, both genders, and different reasons for admission, such as burns, trauma, etc. In addition, we included studies that had been conducted in a variety of countries and ICU settings.
Quality of the evidence
We assessed the overall certainty of evidence using the GRADE approach (Schunemann 2008). This approach integrates evaluations regarding study limitations with judgements regarding inconsistency of results, indirectness, or deviations from accepted practice in the way interventions and comparisons were given, as well as populations studied, choice of outcomes, and methods of ascertainment used; imprecision in the estimates in terms of statistical and clinical importance; and the likelihood that publication bias affected the estimates. Overall, the main reasons for downgrading the certainty of evidence were risk of bias in the included studies and imprecision of the overall effect estimate.
Any intervention compared with placebo or no prophylaxis
Certainty of the evidence for any intervention versus placebo or no prophylaxis was moderate for the primary outcome of preventing clinically important upper GI bleeding in ICU patients, meaning that we are moderately confident in the effect estimate. The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different (Table 1) (Balshem 2011). Certainty of the evidence was judged as low for the outcomes of nosocomial pneumonia, all‐cause mortality in the ICU, duration of ICU stay, and duration of intubation. This means that our confidence in the effect estimate is limited. The true effect may be substantially different from the estimate of the effect. Certainty of evidence for the number of participants requiring blood transfusion was judged as moderate. However, usefulness of the interventions in preventing upper GI bleeding in ICU patients seems to outweigh their adverse effects, such as nosocomial pneumonia, when compared with placebo or no prophylaxis.
H2 receptor antagonists compared with placebo or no prophylaxis
Certainty of the evidence for H2 receptor antagonists versus placebo or no prophylaxis was moderate for the primary outcome of preventing clinically important upper GI bleeding in ICU patients (Table 2). Overall certainty of the evidence was judged as low for the outcomes of nosocomial pneumonia, all‐cause mortality in the ICU, duration of ICU stay, and duration of intubation. Certainty for the number of participants requiring blood transfusions was judged as moderate. However, usefulness of the interventions in preventing upper GI bleeding in ICU patients seems to outweigh their adverse effects such as nosocomial pneumonia, when compared with placebo or no prophylaxis.
Antacids compared with placebo or no prophylaxis
Overall certainty of the evidence for antacids versus placebo or no prophylaxis was low for the primary outcome of preventing clinically important upper GI bleeding in ICU patients, as well as for the outcomes all‐cause mortality in the ICU and numbers of participants requiring blood transfusion. None of the included studies reported on nosocomial pneumonia. Given the fact that antacids are no longer the first choice for preventing clinically important upper GI bleeding in ICU patients, further research most probably is not needed for this comparison (Table 4).
Sucralfate compared with placebo or no prophylaxis
Although evidence suggests that sucralfate might have a protective effect when compared with placebo or no prophylaxis, overall certainty of the evidence was moderate for the outcome of clinically important upper GI bleeding in ICU patients. Overall certainty of the evidence was judged as low for the outcomes of nosocomial pneumonia, all‐cause mortality in the ICU, duration of ICU stay, and numbers of participants requiring blood transfusion (Table 5).
H2 receptor antagonists compared with proton pump inhibitors
Overall certainty of the evidence for H2 receptor antagonists versus proton pump inhibitors was low for the primary outcome of preventing clinically important upper GI bleeding in ICU patients, as well as for the outcomes of nosocomial pneumonia, all‐cause mortality in the ICU, and duration of ICU stay. Certainty of the evidence for the outcome of number of participants requiring blood transfusion was judged as moderate (Table 6).
H2 receptor antagonists compared with antacids
Overall certainty of the evidence for H2 receptor antagonists versus antacids was low for clinically important upper GI bleeding and nosocomial pneumonia. Certainty was judged as very low for all‐cause mortality in the ICU and as moderate for the number of participants requiring blood transfusion. However, antacids are no longer the first line of choice for preventing GI bleeding in ICU patients, and further evidence probably is not needed for this comparison (Table 7).
H2 receptor antagonists compared with sucralfate
Overall certainty of the evidence was low for H2 receptor antagonists versus sucralfate for the outcome clinically important upper GI bleeding in ICU patients. Overall certainty of the evidence was judged as moderate for the outcome of nosocomial pneumonia. Moreover, certainty of the evidence for the outcomes of all‐cause mortality in the hospital and number of participants requiring blood transfusion was low, and certainty was very low for the outcome duration of ICU stay (Table 8). This means that we have very little confidence in the effect estimate. The true effect is likely to be substantially different from the estimate of effect.
Antacids compared with sucralfate
Overall certainty of the evidence for antacids versus sucralfate was low for clinically important upper GI bleeding, nosocomial pneumonia, all‐cause mortality in the ICU, duration of ICU stay, and number of participants requiring blood transfusion (Table 9).
Potential biases in the review process
We used standard methods as described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We chose critical and important outcomes for the 'Summary of findings' tables before extracting data from the included studies by using the GRADE approach through discussion.
Given the large numbers of comparisons and outcomes in this review, the review might be prone to false‐positive results. Therefore, all results must be interpreted with caution.
Temporal changes in clinical care and response to ICU patients at increased risk for upper GI bleeding could have contributed to heterogeneity and increased the uncertainty of findings. Exploratory sensitivity analyses by year of publication not reported in further detail did not alter the consistency of study results nor the direction of effect. Included trials included participants with different indications at baseline, which is very important to consider when one is interpreting the findings of this review. Furthermore, treatment regimens varied across the included studies.
Because of the small number of included studies, we did not perform the following subgroup analyses: studies that explicitly included only adults over the age of 65 years, and studies that used any co‐interventions. Additionally, we could not perform the following sensitivity analyses owing to lack of available data from the included trials: studies with published and validated criteria for diagnosis of clinically important upper GI bleeding and nosocomial pneumonia. Results from subgroup analyses must be interpreted with caution because the number of included studies is relatively small and the power to detect any statistical differences in effect is limited. Information from such subgroup and sensitivity analyses would be relevant for guiding decision‐making in practice.
Agreements and disagreements with other studies or reviews
In recent years, several non‐Cochrane reviews have investigated the risk‐benefit profile of bleeding prophylaxis in ICU patients and their findings have been published (Alhazzani 2017; Alquraini 2017; Alshamsi 2016; Barkun 2012; Krag 2014; Pilkington 2012). The most recent reviews confirmed the beneficial effect of stress ulcer prophylaxis on clinically important upper GI bleeding, as is supported by this Cochrane review.
Alhazzani 2017 conducted a network meta‐analysis of randomised controlled studies and found that proton pump inhibitors might be more effective than H2 receptor antagonists but might also increase the risk of ventilator‐associated pneumonia. Similar results were yielded when Barkun 2012 conducted a meta‐analysis. Review authors concluded that routine stress ulcer prophylaxis is not warranted, and that benefits and harms should be carefully considered in practice.
Alquraini 2017 compared sucralfate versus H2 receptor antagonists in ICU patients for prevention of upper GI bleeding and the incidence of pneumonia. This review found no difference between the two treatments in the occurrence of upper GI bleeding but found a lower incidence of nosocomial pneumonia in the sucralfate arm, which confirms the findings of this review.
Alshamsi 2016 compared proton pump inhibitors and H2 receptor antagonists and, among others, their effects on prevention of upper GI bleeding and nosocomial pneumonia. Results of their review confirm our results that proton pump inhibitors were more effective in preventing upper GI bleeding in critically ill patients when compared with H2 receptor antagonists, and that effects of both interventions on the incidence of pneumonia were similar.
Krag 2014 conducted a systematic review using Cochrane methods and compared the effects of H2 receptor antagonists or proton pump inhibitors versus placebo or no treatment. They reported lower risk of upper GI bleeding in the pooled effect of all studies with treatment versus placebo or no treatment. However, they emphasised that this result was not maintained in an analysis of low risk of bias studies only. Similar to our findings, risk of pneumonia was not significantly different between treatment and no prophylaxis or placebo.
Last, Pilkington 2012 compared H2 receptor antagonists versus proton pump inhibitors and found no difference between the two treatments with respect to the occurrence of upper GI bleeding and pneumonia. These findings cannot be confirmed by our review, possibly because we included newer studies.
Overall, existing and recent systematic reviews confirm the results of this review. However, their scope is limited to reporting the effects of two interventions compared with each other or of one or two interventions compared with placebo or no prophylaxis. Published reviews are of reasonable quality as assessed by Cochrane methods (Alshamsi 2016; Krag 2014). and two reviews used the GRADE approach to assess the certainty of evidence (Alhazzani 2017; Alquraini 2017). Therefore, our review constitutes the most comprehensive and most up‐to‐date piece of work on this important medical topic.
Authors' conclusions
Implications for practice.
This review found evidence of moderate certainty to show that tested interventions might be effective in preventing upper GI bleeding in ICU patients compared with placebo or no prophylaxis. Further, sucralfate or H2 receptor antagonists might be effective in preventing upper gastrointestinal bleeding in ICU patients compared with placebo or no prophylaxis. Further, the effect estimate of proton pump inhibitors versus placebo or no prophylaxis is not significant, but it does not preclude potential benefit in preventing upper GI bleeding. Nosocomial pneumonia was slightly more frequent among patients receiving prophylaxis, but this adverse event might be outweighed by the benefits of the interventions. Results regarding the effectiveness of preventing upper GI bleeding must be interpreted with caution because most studies included in the comparison H2 receptor antagonists versus placebo or no prophylaxis were published in the 1980s and 1990s, and only evidence of low certainty (due to inconsistency and risk of bias) suggests that proton pump inhibitors are more effective than H2 receptor antagonists.
Decisions in practice should be based on individual assessments of patient needs and underlying conditions. Recent evidence indicates that prevention of upper GI bleeding with H2 receptor antagonists results in reduced cost and increased survival compared with use of proton pump inhibitors (Hammond 2017;MacLaren 2014). Still, local context must be considered in selection of treatment.
Implications for research.
Our findings reveal low‐certainty evidence on assessment of nosocomial pneumonia in ICU patients undergoing prophylaxis for upper GI bleeding. Given that antacids are almost no longer used in practice, further good quality research is needed to assess the risk of nosocomial pneumonia in patients receiving sucralfate, H2 receptor antagonists, or protons pump inhibitors. Provided H2 receptor antagonists and proton pump inhibitors are the drugs used most often, and proton pump inhibitors seem to be more effective in preventing clinically important upper GI bleeding, the effect of proton pump inhibitors on any adverse event including nosocomial pneumonia and mortality must be investigated in meta‐analyses or explored by larger, high‐quality RCTs conducted to confirm the positive risk‐benefit balance. In addition, more research is needed on the effectiveness of different treatments planned on admission to the ICU.
Acknowledgements
The first draft of this document was the output of a workshop organised by the South Asian Cochrane Network at Christian Medical College, in Vellore, India. Prathap Tharyan also contributed considerably to the first draft of this review.
Hsiu‐Han Chan co‐ordinated data extraction and quality assessment for three included studies that are published in the Chinese language. Yang‐Sheng Lin, MD, Division of Gastroenterology, Department of Internal Medicine, MacKay Memorial Hospital, Taipei Campus, Taiwan; and Sheng‐Wei Cheng, Division of Gastroenterology, Department of Internal Medicine, Wan Fang Hospital, Taipei Medical University, Taipei, Taiwan extracted data from three studies using the Chinese language.
Cathy Yuan conducted literature searches for the review update in June 2016 and August 2017.
Appendices
Appendix 1. Glossary of medical terms
Antimicrobial – active against microbes.
Aspiration – action or process of drawing breath.
Bicarbonate ions ‐ bicarbonate serves a crucial biochemical role in the physiological pHbuffering system.
Bronchial ‐ related to the bronchi or bronchioles.
Creatinine – compound that is produced by metabolism of creatine and is excreted in the urine.
Cytoprotective – providing protection to cells against harmful agents.
Enteral nutrition – nutrition passing through the intestine, either naturally via the mouth and oesophagus, or through an artificial opening.
Flora ‐ population of microbes inhabiting the outside or inside surfaces of the body.
Gastric – concerning the stomach.
Gram stain – method used to distinguish different types of bacteria by staining them.
Haemoglobin ‐ red protein responsible for transporting oxygen into the blood of vertebrates.
Hyperaemia ‐ excess of blood in the vessels supplying an organ or another part of the body.
Hypermagnesaemia ‐ high level of magnesium in the blood.
Hypomagnesaemia ‐ low level of magnesium in the blood.
Hypoperfusion ‐ decreased flow of blood through an organ.
Hypophosphataemia ‐ abnormally low level of phosphate in the blood.
Infiltrate ‐ substance denser than air such as pus, blood, or protein that lingers within the parenchyma of the lungs.
Interstitial nephritis – type of bacterial infection of the kidneys.
Ischaemia ‐ inadequate blood supply to an organ or part of the body.
Lesion ‐ region in an organ or tissue that has suffered damage through injury or disease.
Leucocytosis ‐ increase in the number of white cells in the blood.
Leucopaenia ‐ reduction in the number of white cells in the blood.
Luminal ‐ concerning the interior of the gastrointestinal tract.
Microcirculation ‐ circulation of the blood in the smallest blood vessels.
Morbidity ‐ the condition of being diseased.
Mortality ‐ the number of deaths in a given area or period, or from a particular cause.
Mucosal ‐ concerning the mucosa.
Neutropaenia ‐ presence of abnormally few neutrophils in the blood.
Nosocomial – originating in a hospital.
Occult bleeding – bleeding that is detectable only chemically or microscopically.
Cytochrome P‐450 mediated drug interaction ‐ a drug interaction mediated via the cytochrome P‐450, a family of isozymes responsible for biotransformation of many drugs via oxidation.
Pathogenic – causing disease.
Pathophysiology ‐ disordered physiological processes associated with disease or injury.
Pancreatitis ‐ inflammation of the pancreas.
Peptic ‐ related to digestion.
pH ‐ figure expressing the acidity or alkalinity of a solution.
Pharmacological ‐ concerned with uses, effects, and modes of action of drugs.
Pleural – concerning the serous membranes lining the thorax and enveloping the lungs.
Pneumonia ‐ lung inflammation caused by bacterial or viral infection.
Postural – concerning the position in which someone holds his or her body when standing or sitting.
Prophylaxis ‐ preventive treatment.
Prostaglandin ‐ group of compounds with varying hormone‐like effects.
Pulmonary – related to the lungs.
Purulent ‐ consisting of, containing, or discharging pus.
Pyaemia ‐ blood poisoning caused by spread in the bloodstream of pus‐forming bacteria.
Quinolones ‐ antibiotic derived from quinoline.
Rales ‐ abnormal rattling sounds heard when unhealthy lungs are examined with a stethoscope.
Renal – related to the kidneys.
Reperfusion ‐ action of restoring the flow of blood to an organ or tissue.
Sepsis ‐ presence in tissues of harmful bacteria and their toxins.
Splanchnic ‐ related to the viscera or internal organs.
Tetracyclines ‐ any of a large group of antibiotics with a molecular structure containing four rings.
Thrombocytopaenia ‐ condition characterised by abnormally low levels of thrombocytes in the blood.
Tracheo‐bronchial colonisation ‐ colonisation of the lower airways by bacteria.
Ulceration – development of or affected by an ulcer.
Ventilator ‐ appliance for artificial respiration.
Appendix 2. CENTRAL search strategy
(stomach or antrum or antral or pyloric or pylorus or gastri$ or epigastr$ or duodenal or duodenum or gastro‐duodenal or gastroduodenal or oeso*ag* or esp*ag* or "upper GI" or UGI).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name]
(h*emorrhag$ or bleed$ or re‐bleed$ or rebleed$).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name]
1 and 2
exp Gastrointestinal Hemorrhage/
(gastr$ adj3 mucosal adj3 injur$).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name]
exp injury/ and exp gastric mucosa/
or/3‐6
(stress adj3 ulcer$).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name]
(h*emorrhag$ or bleed$ or re‐bleed$ or rebleed$).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name]
(ulcer$ or lesion$).mp.
exp Peptic Ulcer/
9 and (10 or 11)
8 or 12
h*ematemesis.mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name]
(melena or melaena).mp.
(coffee adj1 ground).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name]
or/14‐16
7 or 13 or 17
exp Intensive Care/ or exp Critical Illness/ or exp Critical care/ or exp Intensive care units/ or exp Monitoring, Physiologic/
exp Enteral nutrition/
(Nasogastric adj2 (intubat* or tube*)).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name]
exp Intubation, Gastrointestinal/
exp Feeding Apparatus/ or exp Nutritional Support/ or exp Enteric Feeding/ or exp Tube Feeding/
nasoduodenal tube*.mp.
force feeding$.mp.
or/19‐25
exp Histamine H2 Antagonists/
H2 receptor Antagonist*.mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name]
H2 blocker*.mp.
(ranitidine or azanplus or biotidin or pylorid or raciran or raniberl or ranisen or ranitidin or rantec or sostril or taladine or tritec or wal‐zan or zantac).mp.
(cimetidine or acitak or altramet or biomet or dyspamet or eureceptor or galenamet or histodil or peptimax or phimetin or tagamet or ultec or zita).mp.
(famotidine or fluxid or leader acid reducer or mylanta or pepcid).mp.
(roxatidine or Roxit).mp.
(Nizatidine or axid or zinga).mp.
exp proton pump inhibitors/
(omeprazole or losec or nexium or prilosec or rapinex or zegerid).mp.
(Esomeprazole or Sompraz or Zoleri or Nexium or Lucen or Esopral or Axagon or Nexiam).mp.
(Rabeprazole or aciphex or dexrabeprazole or pariet or rablet).mp.
(pantoprazole or protium or protonix).mp.
(lansoprazole or agopton or bamalite or lanzoprazole or lanzor or monolitum or ogast or ogastro or opiren or prevacid or prezal or pro ulco or promeco or takepron or ulpax or zoton).mp.
prostaglandins e, synthetic/
(misoprostol or cytotec or glefos).mp.
enprostil.mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name]
rioprostil.mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name]
Cholinergic Antagonists/
(Pirenzepine or Gastrozepin or Bisvanil or Gasteril or Leblon or Pirenzepin or Pirenzepina or Pirenzepine Gastrozepin or Pirenzepinum or Tabe or Ulcosan).mp.
(Propantheline or Pro‐Banthine).mp.
(Oxyphenonium or Antrenyl or Argicillin or Methacin or Methocidin or Methocidine or Methocidinum or Metocidina or Hydroxymethylgramicidin or Oxiphenonum).mp.
(Doxepin or adapin or apodoxepin or aponal or deptran or desidox or doneurin or doxepia or doxepinbiomo or espadox or mareen or prudoxin or quitaxon or silenor or sinequan or xepin or zonalon).mp.
(Trimipramine or rhotrimine or stangyl or surmontil).mp.
exp antacids/
(sodium bicarbonate or baking soda or bicarb or bicarbonate of soda or carbonic acid monosodium salt or citrocarbonate or monosodium carbonate or neut or neutra caine or soda bic or sodium acid carbonate or sodium hydrogen carbonate).mp.
(sodium citrate or 2‐hydroxypropanetricarboxylic acid or albright's solution or bicitra or citra or citrate or citric acid or cystemme or cystoleve or cytra‐2 or liqui‐dualcitra or oracit or shohl's solution or uralyt).mp.
(magnesium hydroxide or antacids magnesium oh or brucite or magnesium hydrate or mil‐par or milk of magnesia).mp.
(aluminium hydroxide gel or aldrox or algeldrate or alhydrogel or aloh‐gel or alternagel or alu‐cap or alu‐tab or alugel or aluminium hydroxide mixture bp or aluminium hydroxide oral suspension bp or aluminum hydroxide or aluminum hydroxide gel or amphojel or andursil or basalgel or brasivil or brimos or dialume or hydrated alumina or pepsamer or rocgel).mp.
(magnesium trisilicate or sepiolite or Alenic Alka or Gaviscon or Genaton).mp.
(Magaldrate or Gadral or Magaltop or Magralibi or Riopan).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name]
(calcium carbonate or alka‐mints or alkets or alkums or amilac or amitone or antacid extra strength or aragonite or cacit or caco or cal oys or cal‐gest or calcarb or calceos or calci mix or calcichew or calcidrink or calcitab or calcite or calcium antacid or calcium concentrate or calcium liquid softgel or calcium milk or calcium oyster or calcium‐based antacid or calmicid or caltrate or caltro or chalk or chewable calcium or chooz or dicarbosil or equilet or extra strength mylanta calci tabs or limestone or maalox antacid barrier or maalox children or maalox quick dissolve or marble or milk of calcium or mor‐cal or mylanta child or nephro calci or o‐calcium or os‐cal or oscal or oysco or oyst cal or oyster calcium or oyster shell or pepto‐bismol children or rolaids calcium rich or rolaids sodium free or rolaids softchews or super calcium or surpass or titralac or tums or ultra mylanta calci tabs or uni‐cal or uni‐mint or vaterite).mp.
(sucralfate or aluminum sucrose sulfate or antepsin or carafate or ulcerban or ulcogant or ulsanic or xactdose).mp.
(colloidal bismuth subcitrate or bi‐citrate or biselic or bismofarma or bismuth citrate or bismuth subcitrate or bismuth tripotassium dicitrate or bissubcit or de‐nol or de‐noltab or gastrodenol or sucrato or tripotassium dicitratobismuthate or ventrisol‐polfa).mp.
(carbenoxolone sodium or bioplex or bioral or carbenoxalone or carbosan or disodium enoxolone or duogastrone or pharmaxolon or sanodin).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name]
Deglycyrrhizinised Liquorice.mp.
or/27‐62
18 and 26 and 63
Update search on 15 June 2016 and 22 August 2017:
exp Upper Gastrointestinal Tract/
(stomach or antrum or antral or pyloric or pylorus or gastri* or gastro* or duoden* or epigastr* or gastritis or duodenitis).tw,kw.
(esophag* or oesophag* or gastroduoden* or gastrointestinal).tw,kw.
exp Peptic Ulcer/
(peptic or ulcer* or upper GI or UGI or PUD).tw,kw.
or/1‐5
(hemorrhag* or haemorrhag* or bleed* or rebleed* or re‐bleed* or bled or rebled).tw,kw.
(mucosa* adj2 (injur* or damage* or lesion*)).tw,kw.
exp Hemorrhage/
or/7‐9
6 and 10
exp Gastrointestinal Hemorrhage/
exp melena/
(melena or melaena).tw,kw.
exp hematemesis/
(haematemesis or hematemesis).tw,kw.
((coffee adj ground) or "UGIB").tw,kw.
or/11‐17
exp Intensive Care Units/
exp Critical Illness/
exp Critical care/
(intensive care or critical care or respiratory care or recovery room or close attention unit* or special care unit* or coronary care or burn unit* or ICU or RCU or CCU).tw,kw.
((critical or critically or severe or serious*) adj2 (ill or illness)).tw,kw.
((head or brain or severe or multiple) adj2 (injur* or trauma*)).tw,kw.
exp Respiration, Artificial/
(((mechanical* or artificial or controlled) adj2 (ventilat* or respiration)) or on ventilator or ventilated patients).tw,kw.
((prophylaxis or prophylactic or prevent*) and (((after or post) adj2 (surgery or operat*)) or postoperat* or sepsis or hepatic failure* or respiratory failure* or renal failure* or burns or transplantation or stroke or incubated or acute pancreatitis or intracerebral h*morrhag* or intracerebral bleed*)).ti,ab.
or/19‐27
18 and 28
(stress adj2 (ulcer* or gastritis or bleed* or hemorrhage* or haemorrhage*)).tw,kw.
29 or 30
limit 31 to yr="2012 ‐Current"
remove duplicates from 32
Appendix 3. MEDLINE search strategy
randomized controlled trial.pt.
controlled clinical trial.pt.
randomized.ab.
placebo.ab.
drug therapy.fs.
randomly.ab.
trial.ab.
groups.ab.
or/1‐8
exp animals/ not humans.sh.
9 not 10
(stomach or antrum or antral or pyloric or pylorus or gastri$ or epigastr$ or duodenal or duodenum or gastro‐duodenal or gastroduodenal or oeso*ag* or esp*ag* or "upper GI" or UGI).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name]
(h*emorrhag$ or bleed$ or re‐bleed$ or rebleed$).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name]
12 and 13
exp Gastrointestinal Hemorrhage/
(gastr$ adj3 mucosal adj3 injur$).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name]
exp injury/ and exp gastric mucosa/
or/14‐17
(stress adj3 ulcer$).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name]
(h*emorrhag$ or bleed$ or re‐bleed$ or rebleed$).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name]
(ulcer$ or lesion$).mp.
exp Peptic Ulcer/
20 and (21 or 22)
19 or 23
h*ematemesis.mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name]
(melena or melaena).mp.
(coffee adj1 ground).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name]
or/25‐27
18 or 24 or 28
exp Intensive Care/ or exp Critical Illness/ or exp Critical care/ or exp Intensive care units/ or exp Monitoring, Physiologic/
exp Enteral nutrition/
(Nasogastric adj2 (intubat* or tube*)).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name]
exp Intubation, Gastrointestinal/
exp Feeding Apparatus/ or exp Nutritional Support/ or exp Enteric Feeding/ or exp Tube Feeding/
nasoduodenal tube*.mp.
force feeding$.mp.
or/30‐36
exp Histamine H2 Antagonists/
H2 receptor Antagonist*.mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name]
H2 blocker*.mp.
(ranitidine or azanplus or biotidin or pylorid or raciran or raniberl or ranisen or ranitidin or rantec or sostril or taladine or tritec or wal‐zan or zantac).mp.
(cimetidine or acitak or altramet or biomet or dyspamet or eureceptor or galenamet or histodil or peptimax or phimetin or tagamet or ultec or zita).mp.
(famotidine or fluxid or leader acid reducer or mylanta or pepcid).mp.
(roxatidine or Roxit).mp.
(Nizatidine or axid or zinga).mp.
exp proton pump inhibitors/
(omeprazole or losec or nexium or prilosec or rapinex or zegerid).mp.
(Esomeprazole or Sompraz or Zoleri or Nexium or Lucen or Esopral or Axagon or Nexiam).mp.
(Rabeprazole or aciphex or dexrabeprazole or pariet or rablet).mp.
(pantoprazole or protium or protonix).mp.
(lansoprazole or agopton or bamalite or lanzoprazole or lanzor or monolitum or ogast or ogastro or opiren or prevacid or prezal or pro ulco or promeco or takepron or ulpax or zoton).mp.
prostaglandins e, synthetic/
(misoprostol or cytotec or glefos).mp.
enprostil.mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name]
rioprostil.mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name]
Cholinergic Antagonists/
(Pirenzepine or Gastrozepin or Bisvanil or Gasteril or Leblon or Pirenzepin or Pirenzepina or Pirenzepine Gastrozepin or Pirenzepinum or Tabe or Ulcosan).mp.
(Propantheline or Pro‐Banthine).mp.
(Oxyphenonium or Antrenyl or Argicillin or Methacin or Methocidin or Methocidine or Methocidinum or Metocidina or Hydroxymethylgramicidin or Oxiphenonum).mp.
(Doxepin or adapin or apodoxepin or aponal or deptran or desidox or doneurin or doxepia or doxepinbiomo or espadox or mareen or prudoxin or quitaxon or silenor or sinequan or xepin or zonalon).mp.
(Trimipramine or rhotrimine or stangyl or surmontil).mp.
exp antacids/
(sodium bicarbonate or baking soda or bicarb or bicarbonate of soda or carbonic acid monosodium salt or citrocarbonate or monosodium carbonate or neut or neutra caine or soda bic or sodium acid carbonate or sodium hydrogen carbonate).mp.
(sodium citrate or 2‐hydroxypropanetricarboxylic acid or albright's solution or bicitra or citra or citrate or citric acid or cystemme or cystoleve or cytra‐2 or liqui‐dualcitra or oracit or shohl's solution or uralyt).mp.
(magnesium hydroxide or antacids magnesium oh or brucite or magnesium hydrate or mil‐par or milk of magnesia).mp.
(aluminium hydroxide gel or aldrox or algeldrate or alhydrogel or aloh‐gel or alternagel or alu‐cap or alu‐tab or alugel or aluminium hydroxide mixture bp or aluminium hydroxide oral suspension bp or aluminum hydroxide or aluminum hydroxide gel or amphojel or andursil or basalgel or brasivil or brimos or dialume or hydrated alumina or pepsamer or rocgel).mp.
(magnesium trisilicate or sepiolite or Alenic Alka or Gaviscon or Genaton).mp.
(Magaldrate or Gadral or Magaltop or Magralibi or Riopan).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name]
(calcium carbonate or alka‐mints or alkets or alkums or amilac or amitone or antacid extra strength or aragonite or cacit or caco or cal oys or cal‐gest or calcarb or calceos or calci mix or calcichew or calcidrink or calcitab or calcite or calcium antacid or calcium concentrate or calcium liquid softgel or calcium milk or calcium oyster or calcium‐based antacid or calmicid or caltrate or caltro or chalk or chewable calcium or chooz or dicarbosil or equilet or extra strength mylanta calci tabs or limestone or maalox antacid barrier or maalox children or maalox quick dissolve or marble or milk of calcium or mor‐cal or mylanta child or nephro calci or o‐calcium or os‐cal or oscal or oysco or oyst cal or oyster calcium or oyster shell or pepto‐bismol children or rolaids calcium rich or rolaids sodium free or rolaids softchews or super calcium or surpass or titralac or tums or ultra mylanta calci tabs or uni‐cal or uni‐mint or vaterite).mp.
(sucralfate or aluminum sucrose sulfate or antepsin or carafate or ulcerban or ulcogant or ulsanic or xactdose).mp.
(colloidal bismuth subcitrate or bi‐citrate or biselic or bismofarma or bismuth citrate or bismuth subcitrate or bismuth tripotassium dicitrate or bissubcit or de‐nol or de‐noltab or gastrodenol or sucrato or tripotassium dicitratobismuthate or ventrisol‐polfa).mp.
(carbenoxolone sodium or bioplex or bioral or carbenoxalone or carbosan or disodium enoxolone or duogastrone or pharmaxolon or sanodin).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name]
Deglycyrrhizinised Liquorice.mp.
or/38‐73
29 and 37 and 74
11 and 75
Update search on 15 June 2016 and 22 August 2017:
exp Upper Gastrointestinal Tract/
(stomach or antrum or antral or pyloric or pylorus or gastri* or gastro* or duoden* or epigastr* or gastritis or duodenitis).tw,kw.
(esophag* or oesophag* or gastroduoden* or gastrointestinal).tw,kw.
exp Peptic Ulcer/
(peptic or ulcer* or upper GI or UGI or PUD).tw,kw.
or/1‐5
(hemorrhag* or haemorrhag* or bleed* or rebleed* or re‐bleed* or bled or rebled).tw,kw.
(mucosa* adj2 (injur* or damage* or lesion*)).tw,kw.
exp Hemorrhage/
or/7‐9
6 and 10
exp Gastrointestinal Hemorrhage/
exp melena/
(melena or melaena).tw,kw.
exp hematemesis/
(haematemesis or hematemesis).tw,kw.
((coffee adj ground) or "UGIB").tw,kw.
or/11‐17
exp Intensive Care Units/
exp Critical Illness/
exp Critical care/
(intensive care or critical care or respiratory care or recovery room or close attention unit* or special care unit* or coronary care or burn unit* or ICU or RCU or CCU).tw,kw.
((critical or critically or severe or serious*) adj2 (ill or illness)).tw,kw.
((head or brain or severe or multiple) adj2 (injur* or trauma*)).tw,kw.
exp Respiration, Artificial/
(((mechanical* or artificial or controlled) adj2 (ventilat* or respiration)) or on ventilator or ventilated patients).tw,kw.
((prophylaxis or prophylactic or prevent*) and (((after or post) adj2 (surgery or operat*)) or postoperat* or sepsis or hepatic failure* or respiratory failure* or renal failure* or burns or transplantation or stroke or incubated or acute pancreatitis or intracerebral h*morrhag* or intracerebral bleed*)).ti,ab.
or/19‐27
18 and 28
(stress adj2 (ulcer* or gastritis or bleed* or hemorrhage* or haemorrhage*)).tw,kw.
29 or 30
randomized controlled trial.pt.
controlled clinical trial.pt.
random*.ab.
placebo.ab.
drug therapy.fs.
trial.ab.
groups.ab.
or/32‐38
exp animals/ not humans.sh.
39 not 40
31 and 41
limit 42 to yr="2012 ‐Current"
Appendix 4. Embase search strategy
Clinical trial/
Randomized controlled trial/
Randomization/
Single‐Blind Method/
Double‐Blind Method/
Cross‐Over Studies/
Random Allocation/
Placebo/
Randomi?ed controlled trial$.tw.
Rct.tw.
Random allocation.tw.
Randomly allocated.tw.
Allocated randomly.tw.
(allocated adj2 random).tw.
Single blind$.tw.
Double blind$.tw.
((treble or triple) adj blind$).tw.
Placebo$.tw.
Prospective study/
or/1‐19
Case study/
Case report.tw.
Abstract report/ or letter/
or/21‐23
20 not 24
(stomach or antrum or antral or pyloric or pylorus or gastri$ or epigastr$ or duodenal or duodenum or gastro‐duodenal or gastroduodenal or oeso*ag* or esp*ag* or "upper GI" or UGI).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name]
(h*emorrhag$ or bleed$ or re‐bleed$ or rebleed$).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name]
26 and 27
gastrointestinal hemorrhage/ or duodenum bleeding/ or peptic ulcer bleeding/ or stomach hemorrhage/ or upper gastrointestinal bleeding/
(gastr$ adj3 mucosal adj3 injur$).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name]
or/28‐30
(stress adj3 ulcer$).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name]
(h*emorrhag$ or bleed$ or re‐bleed$ or rebleed$).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name]
(ulcer$ or lesion$).mp.
exp Peptic Ulcer/
33 and (34 or 35)
32 or 36
h*ematemesis.mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name]
(melena or melaena).mp.
(coffee adj1 ground).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name]
or/38‐40
31 or 37 or 41
intensive care/ or exp artificial feeding/ or artificial ventilation/ or exp Critical Illness/ or exp Intensive care units/ or patient monitoring/
(Nasogastric adj2 (intubat* or tube*)).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name]
exp digestive tract intubation/
exp Feeding Apparatus/ or exp Nutritional Support/
nasoduodenal tube*.mp.
force feeding$.mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name]
or/43‐48
exp histamine H2 receptor antagonist/
H2 receptor Antagonist*.mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name]
H2 blocker*.mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name]
(ranitidine or azanplus or biotidin or pylorid or raciran or raniberl or ranisen or ranitidin or rantec or sostril or taladine or tritec or wal‐zan or zantac).mp.
(cimetidine or acitak or altramet or biomet or dyspamet or eureceptor or galenamet or histodil or peptimax or phimetin or tagamet or ultec or zita).mp.
(famotidine or fluxid or leader acid reducer or mylanta or pepcid).mp.
(roxatidine or Roxit).mp.
(Nizatidine or axid or zinga).mp.
exp proton pump inhibitors/
(omeprazole or losec or nexium or prilosec or rapinex or zegerid).mp.
(Esomeprazole or Sompraz or Zoleri or Nexium or Lucen or Esopral or Axagon or Nexiam).mp.
(Rabeprazole or aciphex or dexrabeprazole or pariet or rablet).mp.
(pantoprazole or protium or protonix).mp.
(lansoprazole or agopton or bamalite or lanzoprazole or lanzor or monolitum or ogast or ogastro or opiren or prevacid or prezal or pro ulco or promeco or takepron or ulpax or zoton).mp.
prostaglandin E/
(misoprostol or cytotec or glefos).mp.
enprostil.mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name]
rioprostil.mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name]
cholinergic receptor blocking agent/
(Pirenzepine or Gastrozepin or Bisvanil or Gasteril or Leblon or Pirenzepin or Pirenzepina or Pirenzepine Gastrozepin or Pirenzepinum or Tabe or Ulcosan).mp.
(Propantheline or Pro‐Banthine).mp.
(Oxyphenonium or Antrenyl or Argicillin or Methacin or Methocidin or Methocidine or Methocidinum or Metocidina or Hydroxymethylgramicidin or Oxiphenonum).mp.
(Doxepin or adapin or apodoxepin or aponal or deptran or desidox or doneurin or doxepia or doxepinbiomo or espadox or mareen or prudoxin or quitaxon or silenor or sinequan or xepin or zonalon).mp.
(Trimipramine or rhotrimine or stangyl or surmontil).mp.
antacid agent/
(sodium bicarbonate or baking soda or bicarb or bicarbonate of soda or carbonic acid monosodium salt or citrocarbonate or monosodium carbonate or neut or neutra caine or soda bic or sodium acid carbonate or sodium hydrogen carbonate).mp.
(sodium citrate or 2‐hydroxypropanetricarboxylic acid or albright's solution or bicitra or citra or citrate or citric acid or cystemme or cystoleve or cytra‐2 or liqui‐dualcitra or oracit or shohl's solution or uralyt).mp.
(magnesium hydroxide or antacids magnesium oh or brucite or magnesium hydrate or mil‐par or milk of magnesia).mp.
(aluminium hydroxide gel or aldrox or algeldrate or alhydrogel or aloh‐gel or alternagel or alu‐cap or alu‐tab or alugel or aluminium hydroxide mixture bp or aluminium hydroxide oral suspension bp or aluminum hydroxide or aluminum hydroxide gel or amphojel or andursil or basalgel or brasivil or brimos or dialume or hydrated alumina or pepsamer or rocgel).mp.
(magnesium trisilicate or sepiolite or Alenic Alka or Gaviscon or Genaton).mp.
(Magaldrate or Gadral or Magaltop or Magralibi or Riopan).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name]
(calcium carbonate or alka‐mints or alkets or alkums or amilac or amitone or antacid extra strength or aragonite or cacit or caco or cal oys or cal‐gest or calcarb or calceos or calci mix or calcichew or calcidrink or calcitab or calcite or calcium antacid or calcium concentrate or calcium liquid softgel or calcium milk or calcium oyster or calcium‐based antacid or calmicid or caltrate or caltro or chalk or chewable calcium or chooz or dicarbosil or equilet or extra strength mylanta calci tabs or limestone or maalox antacid barrier or maalox children or maalox quick dissolve or marble or milk of calcium or mor‐cal or mylanta child or nephro calci or o‐calcium or os‐cal or oscal or oysco or oyst cal or oyster calcium or oyster shell or pepto‐bismol children or rolaids calcium rich or rolaids sodium free or rolaids softchews or super calcium or surpass or titralac or tums or ultra mylanta calci tabs or uni‐cal or uni‐mint or vaterite).mp.
(sucralfate or aluminum sucrose sulfate or antepsin or carafate or ulcerban or ulcogant or ulsanic or xactdose).mp.
(colloidal bismuth subcitrate or bi‐citrate or biselic or bismofarma or bismuth citrate or bismuth subcitrate or bismuth tripotassium dicitrate or bissubcit or de‐nol or de‐noltab or gastrodenol or sucrato or tripotassium dicitratobismuthate or ventrisol‐polfa).mp.
(carbenoxolone sodium or bioplex or bioral or carbenoxalone or carbosan or disodium enoxolone or duogastrone or pharmaxolon or sanodin).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name]
Deglycyrrhizinised Liquorice.mp.
or/50‐85
42 and 49 and 86
25 and 87
Update search on 15 June 2016 and 22 August 2017:
exp upper gastrointestinal tract/
(stomach or antrum or antral or pyloric or pylorus or gastri* or gastro* or duoden* or epigastr* or gastritis or duodenitis).tw,kw.
(esophag* or oesophag* or gastroduoden* or gastrointestinal).tw,kw.
exp peptic ulcer/
(peptic or ulcer* or upper GI or UGI or PUD).tw,kw.
or/1‐5
(hemorrhag* or haemorrhag* or bleed* or rebleed* or re‐bleed* or bled or rebled).tw,kw.
(mucosa* adj2 (injur* or damage* or lesion*)).tw,kw.
exp bleeding/
or/7‐9
6 and 10
exp gastrointestinal hemorrhage/
exp melena/
(melena or melaena).tw,kw.
exp hematemesis/
(haematemesis or hematemesis).tw,kw.
((coffee adj ground) or "UGIB").tw,kw.
or/11‐17
exp intensive care unit/
exp critical illness/
exp intensive care/
(intensive care or critical care or respiratory care or recovery room or close attention unit* or special care unit* or coronary care or burn unit* or ICU or RCU or CCU).tw,kw.
((critical or critically or severe or serious*) adj2 (ill or illness)).tw,kw.
((head or brain or severe or multiple) adj2 (injur* or trauma*)).tw,kw.
exp artificial ventilation/
(((mechanical* or artificial or controlled) adj2 (ventilat* or respiration)) or on ventilator or ventilated patients).tw,kw.
((prophylaxis or prophylactic or prevent*) and (((after or post) adj2 (surgery or operat*)) or postoperat* or sepsis or hepatic failure* or respiratory failure* or renal failure* or burns or transplantation or stroke or incubated or acute pancreatitis or intracerebral h*morrhag* or intracerebral bleed*)).ti,ab.
or/19‐27
18 and 28
(stress adj2 (ulcer* or gastritis or bleed* or hemorrhage* or haemorrhage*)).tw,kw.
29 or 30
random:.tw.
placebo:.mp.
double‐blind:.tw.
32 or 33 or 34
31 and 35
limit 36 to yr="2012 ‐Current"
Appendix 5. LILACS search strategy
Descriptors in Health Sciences (DeCS) were used to account for Portuguese, Spanish and English terms in LILACS.
(E02.760.190.400 or N02.278.388.493 or E02.760.190) [DeCS Category] and (C06.405.227 or C06.405.227.400 or A12.459.764 or C06.405.227.700) [DeCS Category]
Update search on 15 June 2016 and 22 August 2017:
(((tw:(ulcer OR stomach OR antrum OR antral OR gastri* OR gastro* OR duoden* OR epigastr* OR gastritis OR duodenitis OR esophageal* OR oesophageal*)) AND (tw:(hemorrhag* OR haemorrhag* OR bleed* OR rebleed* OR re‐bleed* OR bled OR rebled))) OR (tw: (stress ulcer*)) OR (mh: (c06.405.227)) ) AND ( (mh: (e02.760.190)) OR (tw: (intensive care OR critical care OR critically ill OR mechanical* ventilat*))) AND (instance:"regional") AND ( db:("LILACS") AND type_of_study:("clinical_trials"))
Data and analyses
Comparison 1. Interventions versus placebo or no prophylaxis.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinically important upper GI bleeding | 30 | 3132 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.47 [0.39, 0.57] |
| 1.1 H2 receptor antagonists vs placebo or no prophylaxis | 24 | 1844 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.46 [0.37, 0.59] |
| 1.2 Proton pump inhibitors vs placebo or no prophylaxis | 3 | 159 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.57 [0.13, 2.59] |
| 1.3 Prostagladin analogues vs placebo or no prophylaxis | 1 | 58 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.22, 4.55] |
| 1.4 Anticholinergics vs placebo or no prophylaxis | 2 | 103 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.87 [0.30, 2.49] |
| 1.5 Antacids vs placebo or no prophylaxis | 7 | 515 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.39 [0.23, 0.63] |
| 1.6 Sucralfate vs placebo or no prophylaxis | 7 | 453 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.47 [0.25, 0.87] |
| 2 Nosocomial pneumonia | 9 | 1331 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.15 [0.90, 1.48] |
| 2.1 H2 receptor antagonists vs placebo or no prophylaxis | 8 | 788 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.06 [0.77, 1.46] |
| 2.2 Proton pump inhibitors vs placebo or no prophylaxis | 2 | 149 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.13 [0.59, 2.17] |
| 2.3 Anticholinergics vs placebo or no prophylaxis | 1 | 72 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.06 [0.43, 2.59] |
| 2.4 Sucralfate vs placebo or no prophylaxis | 4 | 322 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.59 [0.84, 3.01] |
| 3 All‐cause mortality in ICU | 19 | 2159 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.10 [0.90, 1.34] |
| 3.1 H2 receptor antagonists vs placebo or no prophylaxis | 14 | 1209 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.16 [0.89, 1.53] |
| 3.2 Proton pump inhibitors vs placebo or no prophylaxis | 3 | 180 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.05 [0.46, 2.38] |
| 3.3 Prostagladin analogues vs placebo or no prophylaxis | 1 | 58 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.14 [0.48, 2.74] |
| 3.4 Anticholinergics vs placebo or no prophylaxis | 2 | 103 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.22 [0.59, 2.56] |
| 3.5 Antacids vs placebo or no prophylaxis | 2 | 250 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.02 [0.58, 1.79] |
| 3.6 Sucralfate vs placebo or no prophylaxis | 5 | 359 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.58, 1.51] |
| 4 All‐cause mortality in hospital | 5 | 857 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.15 [0.85, 1.55] |
| 4.1 H2 receptor antagonists vs placebo or no prophylaxis | 4 | 387 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.14 [0.71, 1.83] |
| 4.2 Proton pump inhibitors vs placebo or no prophylaxis | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.17 [0.42, 3.22] |
| 4.3 Antacids vs placebo or no prophylaxis | 1 | 126 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.44 [0.79, 2.64] |
| 4.4 Sucralfate vs placebo or no prophylaxis | 2 | 244 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.53, 1.68] |
| 5 Duration of ICU stay | 2 | 447 | Mean Difference (IV, Fixed, 95% CI) | 0.24 [‐1.13, 1.61] |
| 5.1 H2 receptor antagonists vs placebo or no prophylaxis | 2 | 152 | Mean Difference (IV, Fixed, 95% CI) | 0.73 [‐1.64, 3.09] |
| 5.2 Proton pump inhibitors vs placebo or no prophylaxis | 2 | 149 | Mean Difference (IV, Fixed, 95% CI) | 0.01 [‐2.33, 2.35] |
| 5.3 Sucralfate vs placebo or no prophylaxis | 2 | 146 | Mean Difference (IV, Fixed, 95% CI) | ‐0.01 [‐2.40, 2.38] |
| 6 Duration of intubation | 2 | 447 | Mean Difference (IV, Fixed, 95% CI) | 0.87 [‐0.58, 2.31] |
| 6.1 H2 receptor antagonists vs placebo or no prophylaxis | 2 | 152 | Mean Difference (IV, Fixed, 95% CI) | 0.78 [‐1.72, 3.29] |
| 6.2 Proton pump inhibitors vs placebo or no prophylaxis | 2 | 149 | Mean Difference (IV, Fixed, 95% CI) | 0.36 [‐2.18, 2.90] |
| 6.3 Sucralfate vs placebo or no prophylaxis | 2 | 146 | Mean Difference (IV, Fixed, 95% CI) | 1.43 [‐1.04, 3.89] |
| 7 Number of participants requiring blood transfusions | 9 | 981 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.63 [0.41, 0.97] |
| 7.1 H2 receptor antagonists vs placebo or no prophylaxis | 7 | 605 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.57 [0.35, 0.94] |
| 7.2 Antacids vs placebo or no prophylaxis | 2 | 226 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.30, 2.96] |
| 7.3 Sucralfate vs placebo or no prophylaxis | 1 | 150 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.75 [0.13, 4.34] |
| 8 Units of blood transfused | 2 | 309 | Mean Difference (IV, Random, 95% CI) | 0.09 [‐0.99, 1.17] |
| 8.1 H2 receptor antagonists vs placebo or no prophylaxis | 2 | 159 | Mean Difference (IV, Random, 95% CI) | ‐1.73 [‐6.37, 2.90] |
| 8.2 Sucralfate vs placebo or no prophylaxis | 1 | 150 | Mean Difference (IV, Random, 95% CI) | 0.80 [0.25, 1.35] |
Comparison 2. H2 receptor antagonists versus placebo or no prophylaxis.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinically important upper GI bleeding | 24 | 2149 | Risk Ratio (M‐H, Random, 95% CI) | 0.50 [0.36, 0.70] |
| 1.1 Cimetidine vs placebo | 10 | 772 | Risk Ratio (M‐H, Random, 95% CI) | 0.53 [0.28, 1.02] |
| 1.2 Famotidine vs placebo | 1 | 146 | Risk Ratio (M‐H, Random, 95% CI) | 2.11 [0.20, 22.79] |
| 1.3 Ranitidine vs placebo | 5 | 446 | Risk Ratio (M‐H, Random, 95% CI) | 0.36 [0.17, 0.77] |
| 1.4 Cimetidine vs no prophylaxis | 3 | 516 | Risk Ratio (M‐H, Random, 95% CI) | 0.59 [0.23, 1.48] |
| 1.5 Famotidine vs no prophylaxis | 1 | 50 | Risk Ratio (M‐H, Random, 95% CI) | 0.3 [0.09, 0.96] |
| 1.6 Ranitidine vs no prophylaxis | 4 | 219 | Risk Ratio (M‐H, Random, 95% CI) | 0.51 [0.26, 1.00] |
| 2 Nosocomial pneumonia | 8 | 945 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.12 [0.85, 1.48] |
| 2.1 Cimetidine vs placebo | 2 | 204 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.34 [0.06, 2.00] |
| 2.2 Famotidine vs placebo | 1 | 146 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.48 [0.49, 4.45] |
| 2.3 Ranitidine vs placebo | 2 | 277 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.79 [0.47, 1.31] |
| 2.4 Cimetidine vs no prophylaxis | 1 | 200 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.17 [0.86, 5.47] |
| 2.5 Ranitidine vs no prophylaxis | 2 | 118 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.33 [0.93, 1.90] |
| 3 All‐cause mortality in ICU | 14 | 1428 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.12 [0.88, 1.42] |
| 3.1 Cimetidine vs placebo | 4 | 478 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.05 [0.66, 1.68] |
| 3.2 Famotidine vs placebo | 1 | 146 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.32 [0.55, 3.16] |
| 3.3 Ranitidine vs placebo | 2 | 148 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.69 [0.31, 1.54] |
| 3.4 Cimetidine vs no prophylaxis | 2 | 400 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.61, 1.63] |
| 3.5 Famotidine vs no prophylaxis | 1 | 50 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.25 [0.59, 2.64] |
| 3.6 Ranitidine vs no prophylaxis | 4 | 206 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.58 [0.97, 2.58] |
| 4 All‐cause mortality in hospital | 4 | 487 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.16 [0.79, 1.70] |
| 4.1 Famotidine vs placebo | 1 | 146 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.89 [0.43, 1.86] |
| 4.2 Ranitidine vs placebo | 1 | 101 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.35 [0.01, 8.47] |
| 4.3 Cimetidine vs no prophylaxis | 1 | 200 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.47 [0.88, 2.46] |
| 4.4 Ranitidine vs no prophylaxis | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.92 [0.33, 2.53] |
| 5 Duration of ICU stay | 2 | 230 | Mean Difference (IV, Fixed, 95% CI) | 0.73 [‐0.92, 2.38] |
| 5.1 Famotidine vs placebo | 1 | 146 | Mean Difference (IV, Fixed, 95% CI) | 1.5 [‐1.93, 4.93] |
| 5.2 Ranitidine vs no prophylaxis | 1 | 84 | Mean Difference (IV, Fixed, 95% CI) | 0.5 [‐1.38, 2.38] |
| 6 Duration of intubation | 2 | 230 | Mean Difference (IV, Fixed, 95% CI) | 0.79 [‐0.95, 2.54] |
| 6.1 Famotidine vs placebo | 1 | 146 | Mean Difference (IV, Fixed, 95% CI) | 1.20 [‐1.86, 4.26] |
| 6.2 Ranitidine vs no prophylaxis | 1 | 84 | Mean Difference (IV, Fixed, 95% CI) | 0.60 [‐1.52, 2.72] |
| 7 Number of participants requiring blood transfusions | 7 | 655 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.58 [0.36, 0.95] |
| 7.1 Cimetidine vs placebo | 3 | 107 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.39 [0.19, 0.79] |
| 7.2 Ranitidine vs placebo | 2 | 148 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.05 [0.24, 4.60] |
| 7.3 Cimetidine vs no prophylaxis | 2 | 400 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.77 [0.35, 1.71] |
| 8 Units of blood transfused | 2 | 209 | Mean Difference (IV, Fixed, 95% CI) | 0.33 [‐0.04, 0.70] |
| 8.1 Cimetidine vs placebo | 1 | 9 | Mean Difference (IV, Fixed, 95% CI) | ‐4.35 [‐7.35, ‐1.35] |
| 8.2 Cimetidine vs no prophylaxis | 1 | 200 | Mean Difference (IV, Fixed, 95% CI) | 0.40 [0.03, 0.77] |
| 9 Adverse events of interventions | 8 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 9.1 Diarrhoea | 2 | 225 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.30 [0.57, 2.96] |
| 9.2 Thrombocytopenia | 1 | 200 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.12, 72.77] |
| 9.3 Hypophosphatemia | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.75 [0.17, 84.02] |
| 9.4 Mental confusion | 5 | 657 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.01 [1.10, 3.65] |
| 9.5 Nausea and vomiting | 2 | 287 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.46 [0.09, 2.35] |
| 9.6 Increased creatinine levels | 1 | 200 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.04 [0.64, 1.69] |
| 9.7 Erythema | 1 | 70 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.13, 71.22] |
| 9.8 Pancreatitis | 1 | 39 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.29 [0.01, 6.66] |
| 9.9 Chest infection | 1 | 101 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.74 [0.92, 3.30] |
| 9.10 Delirium | 1 | 39 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.59 [0.11, 59.93] |
| 9.11 Hallucinations | 1 | 39 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.59 [0.11, 59.93] |
| 9.12 Severe bleeding | 1 | 101 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.35 [0.01, 8.47] |
Comparison 3. Proton pump inhibitors versus placebo or no prophylaxis.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinically important upper GI bleeding | 3 | 237 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.63 [0.18, 2.22] |
| 1.1 Omeprazole vs placebo | 1 | 147 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.04 [0.07, 16.34] |
| 1.2 Omeprazole vs no prophylaxis | 1 | 80 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.74 [0.13, 4.18] |
| 1.3 Pantoprazole vs placebo | 1 | 10 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.28 [0.02, 4.66] |
| 2 Nosocomial pneumonia | 2 | 227 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.24 [0.77, 1.98] |
| 2.1 Omeprazole vs placebo | 1 | 147 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.67 [0.57, 4.86] |
| 2.2 Omeprazole vs no prophylaxis | 1 | 80 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.11 [0.66, 1.84] |
| 3 All‐cause mortality in ICU | 3 | 258 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.09 [0.60, 1.99] |
| 3.1 Omeprazole vs placebo | 2 | 178 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.20 [0.51, 2.83] |
| 3.2 Omeprazole vs no prophylaxis | 1 | 80 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.98 [0.42, 2.29] |
| 4 All‐cause mortality in hospital | 1 | 150 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.08 [0.54, 2.13] |
| 4.1 Omeprazole vs placebo | 1 | 150 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.08 [0.54, 2.13] |
| 5 Duration of ICU stay | 2 | 227 | Mean Difference (IV, Fixed, 95% CI) | ‐0.03 [‐1.63, 1.58] |
| 5.1 Omeprazole vs placebo | 1 | 147 | Mean Difference (IV, Fixed, 95% CI) | ‐0.90 [‐3.96, 2.16] |
| 5.2 Omeprazole vs no prophylaxis | 1 | 80 | Mean Difference (IV, Fixed, 95% CI) | 0.30 [‐1.58, 2.18] |
| 6 Duration of intubation | 2 | 227 | Mean Difference (IV, Fixed, 95% CI) | 0.36 [‐1.43, 2.15] |
| 6.1 Omeprazole vs placebo | 1 | 147 | Mean Difference (IV, Fixed, 95% CI) | 0.5 [‐2.72, 3.72] |
| 6.2 Omeprazole vs no prophylaxis | 1 | 80 | Mean Difference (IV, Fixed, 95% CI) | 0.30 [‐1.85, 2.45] |
Comparison 4. Proton pump inhibitors + sucralfate versus no prophylaxis.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinically important upper GI bleeding | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
Comparison 5. Prostaglandin analogues versus placebo or no prophylaxis.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinically important upper GI bleeding | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.1 Prostaglandin analogues vs placebo | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 All‐cause mortality in ICU | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
Comparison 6. Anticholinergics versus placebo or no prophylaxis.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinically important upper GI bleeding | 2 | 131 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.95 [0.36, 2.51] |
| 1.1 Pirenzepine vs placebo | 1 | 101 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.94 [0.34, 11.13] |
| 1.2 Pirenzepin + ranitidine vs placebo + ranitidine | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.6 [0.17, 2.07] |
| 2 Nosocomial pneumonia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2.1 Pirenzepine vs placebo | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 All‐cause mortality in ICU | 2 | 131 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.23 [0.66, 2.30] |
| 3.1 Pirenzepine vs placebo | 1 | 101 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.30 [0.65, 2.60] |
| 3.2 Pirenzepine + ranitidine vs placebo + ranitidine | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.24, 4.18] |
Comparison 7. Antacids versus placebo or no prophylaxis.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinically important upper GI bleeding | 8 | 774 | Risk Ratio (M‐H, Random, 95% CI) | 0.49 [0.25, 0.99] |
| 1.1 Antacids vs placebo | 2 | 145 | Risk Ratio (M‐H, Random, 95% CI) | 2.04 [0.72, 5.79] |
| 1.2 Antacids vs no prophylaxis | 6 | 629 | Risk Ratio (M‐H, Random, 95% CI) | 0.35 [0.20, 0.60] |
| 2 All‐cause mortality in ICU | 2 | 300 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.53, 1.96] |
| 2.1 Antacids vs no prophylaxis | 2 | 300 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.53, 1.96] |
| 3 All‐cause mortality in hospital | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3.1 Antacids vs no prophylaxis | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Number of participants requiring blood transfusions | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4.1 Antacids vs no prophylaxis | 2 | 226 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.30, 2.96] |
| 5 Adverse events of interventions | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.1 Diarrhoea | 4 | 395 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.56 [1.83, 6.94] |
| 5.2 Hypomagnesaemia | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.75 [0.17, 84.02] |
| 5.3 Hypophosphataemia | 2 | 225 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.48 [1.81, 16.61] |
| 5.4 Hypermagnesaemia | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 6.73 [0.36, 127.02] |
| 5.5 Nausea and vomiting | 3 | 370 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.39 [0.86, 6.64] |
| 5.6 Mental confusion | 1 | 200 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.27 [0.61, 2.67] |
| 5.7 Creatinine increase | 1 | 200 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.17 [0.73, 1.87] |
Comparison 8. Sucralfate versus placebo or no prophylaxis.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinically important upper GI bleeding | 7 | 598 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.53 [0.32, 0.88] |
| 1.1 Sucralfate vs placebo | 2 | 170 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.40 [0.30, 6.62] |
| 1.2 Sucralfate vs no prophylaxis | 5 | 428 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.46 [0.26, 0.80] |
| 2 Nosocomial pneumonia | 4 | 450 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.33 [0.86, 2.04] |
| 2.1 Sucralfate vs placebo | 2 | 170 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.43 [0.49, 4.16] |
| 2.2 Sucralfate vs no prophylaxis | 2 | 280 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.30 [0.82, 2.07] |
| 3 All‐cause mortality in ICU | 5 | 500 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.66, 1.43] |
| 3.1 Sucralfate vs placebo | 2 | 170 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.92 [0.48, 1.80] |
| 3.2 Sucralfate vs no prophylaxis | 3 | 330 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.99 [0.62, 1.60] |
| 4 All‐cause mortality in hospital | 2 | 344 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.62, 1.52] |
| 4.1 Sucralfate vs placebo | 1 | 144 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.09 [0.54, 2.18] |
| 4.2 Sucralfate vs no prophylaxis | 1 | 200 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.89 [0.49, 1.62] |
| 5 Duration of ICU stay | 2 | 224 | Mean Difference (IV, Fixed, 95% CI) | ‐0.02 [‐1.70, 1.65] |
| 5.1 Sucralfate vs placebo | 1 | 144 | Mean Difference (IV, Fixed, 95% CI) | ‐0.70 [‐4.07, 2.67] |
| 5.2 Sucralfate vs no prophylaxis | 1 | 80 | Mean Difference (IV, Fixed, 95% CI) | 0.20 [‐1.73, 2.13] |
| 6 Duration of intubation | 2 | 224 | Mean Difference (IV, Fixed, 95% CI) | 1.42 [‐0.27, 3.10] |
| 6.1 Sucralfate vs placebo | 1 | 144 | Mean Difference (IV, Fixed, 95% CI) | 0.80 [‐2.20, 3.80] |
| 6.2 Sucralfate vs no prophylaxis | 1 | 80 | Mean Difference (IV, Fixed, 95% CI) | 1.70 [‐0.34, 3.74] |
| 7 Number of participants requiring blood transfusions | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 7.1 Sucralfate vs no prophylaxis | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Units of blood transfused | 1 | 200 | Mean Difference (IV, Fixed, 95% CI) | 0.8 [0.32, 1.28] |
| 8.1 Sucralfate vs no prophylaxis | 1 | 200 | Mean Difference (IV, Fixed, 95% CI) | 0.8 [0.32, 1.28] |
| 9 Adverse events of interventions | 1 | 70 | Risk Ratio (M‐H, Fixed, 95% CI) | 9.0 [0.50, 161.13] |
| 9.1 Nausea / Vomiting | 1 | 70 | Risk Ratio (M‐H, Fixed, 95% CI) | 9.0 [0.50, 161.13] |
Comparison 9. H2 receptor antagonists versus proton pump inhibitors.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinically important upper GI bleeding | 13 | 1636 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.90 [1.83, 4.58] |
| 1.1 Cimetidine vs omeprazole | 1 | 359 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.40 [0.55, 3.61] |
| 1.2 Famotidine vs lansoprazole | 1 | 51 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.63 [0.15, 84.98] |
| 1.3 Famotidine vs omeprazole | 1 | 143 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.03 [0.19, 21.87] |
| 1.4 Famotidine vs pantoprazole | 2 | 159 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.73 [0.18, 3.04] |
| 1.5 Famotidine vs esomeprazole | 2 | 371 | Risk Ratio (M‐H, Fixed, 95% CI) | 7.53 [1.39, 40.85] |
| 1.6 Ranitidine vs omeprazole | 5 | 413 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.08 [1.99, 8.36] |
| 1.7 Ranitidine vs rabeprazole | 1 | 140 | Risk Ratio (M‐H, Fixed, 95% CI) | 9.0 [0.49, 164.09] |
| 2 Nosocomial pneumonia | 10 | 1256 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.02 [0.77, 1.35] |
| 2.1 Cimetidine vs omeprazole | 1 | 359 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.84 [0.45, 1.54] |
| 2.2 Cimetidine vs pantoprazole | 1 | 202 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.89 [0.28, 2.91] |
| 2.3 Famotidine vs esomeprazole | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.07, 15.26] |
| 2.4 Famotidine vs omeprazole | 1 | 143 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.89 [0.34, 2.32] |
| 2.5 Ranitidine vs omeprazole | 5 | 413 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.19 [0.80, 1.75] |
| 2.6 H2 receptor antagonists (not defined) vs proton pump inhibitors (not defined) | 1 | 79 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.03 [0.47, 2.26] |
| 3 All‐cause mortality in ICU | 12 | 1564 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.96 [0.78, 1.19] |
| 3.1 Cimetidine vs omeprazole | 1 | 359 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.76 [0.45, 1.30] |
| 3.2 Cimetidine vs pantoprazole | 1 | 202 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.80 [0.25, 2.55] |
| 3.3 Famotidine vs omeprazole | 1 | 143 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.13 [0.49, 2.61] |
| 3.4 Ranitidine vs omeprazole | 5 | 387 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.10 [0.86, 1.40] |
| 3.5 Ranitidine vs pantoprazole | 3 | 333 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.66 [0.31, 1.43] |
| 3.6 Ranitidine vs rabeprazole | 1 | 140 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.12, 72.40] |
| 4 All‐cause mortality in hospital | 2 | 454 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.72 [0.37, 1.43] |
| 4.1 Famotidine vs esomeprazole | 1 | 311 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.37 [0.04, 3.49] |
| 4.2 Famotidine vs omeprazole | 1 | 143 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.80 [0.39, 1.63] |
| 5 Duration of ICU stay | 5 | 482 | Mean Difference (IV, Fixed, 95% CI) | 0.14 [‐1.14, 1.41] |
| 5.1 Famotidine vs esomeprazole | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐0.30 [‐6.51, 5.91] |
| 5.2 Famotidine vs omeprazole | 1 | 143 | Mean Difference (IV, Fixed, 95% CI) | 2.40 [‐0.44, 5.24] |
| 5.3 Ranitidine vs omeprazole | 3 | 279 | Mean Difference (IV, Fixed, 95% CI) | ‐0.44 [‐1.90, 1.02] |
| 6 Duration of intubation | 5 | 542 | Mean Difference (IV, Fixed, 95% CI) | ‐0.35 [‐1.48, 0.78] |
| 6.1 Famotidine vs omeprazole | 1 | 143 | Mean Difference (IV, Fixed, 95% CI) | 0.70 [‐2.24, 3.64] |
| 6.2 Ranitidine vs omeprazole | 3 | 279 | Mean Difference (IV, Fixed, 95% CI) | ‐0.78 [‐2.24, 0.67] |
| 6.3 Ranitidine vs pantoprazole | 1 | 120 | Mean Difference (IV, Fixed, 95% CI) | 0.07 [‐2.18, 2.32] |
| 7 Number of participants requiring blood transfusions | 3 | 575 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.98 [0.75, 5.21] |
| 7.1 Cimetidine vs omeprazole | 1 | 359 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.98 [0.29, 3.34] |
| 7.2 Ranitidine vs omeprazole | 1 | 76 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.0 [0.25, 100.80] |
| 7.3 Ranitidine vs rabeprazole | 1 | 140 | Risk Ratio (M‐H, Fixed, 95% CI) | 9.0 [0.49, 164.09] |
| 8 Adverse events of interventions | 5 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 8.1 Pyrexia | 1 | 202 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.05, 19.03] |
| 8.2 Thrombocytopaenia | 2 | 253 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.64 [0.65, 20.46] |
| 8.3 Neuroleptic malignant syndrome | 1 | 202 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.56 [0.06, 37.42] |
| 8.4 Cholestatic jaundice | 1 | 202 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.56 [0.06, 37.42] |
| 8.5 Abnormal liver function test | 1 | 202 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.56 [0.06, 37.42] |
| 8.6 Pruritus | 1 | 202 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.56 [0.06, 37.42] |
| 8.7 Phlebitis | 1 | 202 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.56 [0.06, 37.42] |
| 8.8 Major CV events | 1 | 311 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.79 [0.26, 2.43] |
| 8.9 Abdominal distension and vomiting | 1 | 90 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.15 [0.62, 2.14] |
| 8.10 Hypomagnesaemia | 1 | 129 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.43 [0.16, 1.13] |
| 8.11 Nausea and vomiting | 1 | 129 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.48 [0.13, 1.77] |
| 8.12 Diarrhoea | 1 | 129 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.11 [0.16, 7.67] |
Comparison 10. H2 receptor antagonists versus antacids.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinically important upper GI bleeding | 16 | 1700 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.67, 1.36] |
| 1.1 Cimetidine vs antacids | 11 | 1155 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.65, 1.78] |
| 1.2 Cimetidine + pirenzepine vs antacid + pirenzepine | 1 | 66 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.15, 6.68] |
| 1.3 Ranitidine vs antacids | 4 | 479 | Risk Ratio (M‐H, Random, 95% CI) | 0.72 [0.42, 1.23] |
| 2 Nosocomial pneumonia | 4 | 581 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.05 [0.81, 1.36] |
| 2.1 Cimetidine vs antacids | 2 | 136 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.24 [0.70, 2.19] |
| 2.2 Ranitidine vs antacids | 2 | 445 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.00 [0.75, 1.34] |
| 3 All‐cause mortality in ICU | 11 | 1321 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.66, 1.55] |
| 3.1 Cimetidine vs antacids | 8 | 885 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.69, 1.59] |
| 3.2 Cimetidine + pirenzepine vs antacid + pirenzepine | 1 | 66 | Risk Ratio (M‐H, Random, 95% CI) | 1.25 [0.37, 4.25] |
| 3.3 Ranitidine vs antacids | 2 | 370 | Risk Ratio (M‐H, Random, 95% CI) | 1.13 [0.14, 8.97] |
| 4 All‐cause mortality in hospital | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1 Ranitidine vs antacids | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Duration of intubation | 3 | 121 | Mean Difference (IV, Random, 95% CI) | ‐0.81 [‐3.85, 2.23] |
| 5.1 Cimetidine vs antacids | 3 | 121 | Mean Difference (IV, Random, 95% CI) | ‐0.81 [‐3.85, 2.23] |
| 6 Number of participants requiring blood transfusions | 6 | 744 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.49 [1.35, 4.62] |
| 6.1 Cimetidine vs antacids | 5 | 583 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.47 [1.32, 4.63] |
| 6.2 Ranitidine vs antacids | 1 | 161 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.04 [0.13, 73.46] |
| 7 Adverse events of interventions | 12 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 7.1 Diarrhoea | 6 | 777 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.23 [0.13, 0.43] |
| 7.2 Thrombocytopaenia | 4 | 452 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.40 [0.93, 2.09] |
| 7.3 Nausea and vomiting | 4 | 380 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.46 [0.19, 1.10] |
| 7.4 Hypophosphataemia | 2 | 108 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.24 [0.04, 1.30] |
| 7.5 Hypomagnesaemia | 1 | 22 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.02, 7.39] |
| 7.6 Increase in creatinine | 2 | 286 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.85 [0.56, 1.28] |
| 7.7 Mental confusion | 4 | 476 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.26 [0.77, 2.07] |
| 7.8 Hypermagnesaemia | 2 | 115 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.58 [0.17, 2.03] |
| 7.9 Rash/Erythema | 2 | 231 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.02 [0.32, 28.53] |
| 7.10 Alkalosis | 1 | 75 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.32 [0.01, 7.73] |
| 7.11 Dryness of mouth | 1 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.15 [0.26, 103.33] |
| 7.12 Leucopaenia | 1 | 161 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.04 [0.13, 73.46] |
Comparison 11. H2 receptor antagonists versus sucralfate.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinically important upper GI bleeding | 24 | 3316 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.10 [0.87, 1.41] |
| 1.1 Cimetidine vs sucralfate | 7 | 873 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.37 [0.87, 2.14] |
| 1.2 Famotidine vs sucralfate | 2 | 190 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.62 [0.21, 1.78] |
| 1.3 Ranitidine vs sucralfate | 14 | 2186 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.03 [0.76, 1.39] |
| 1.4 Cimetidine + pirenzepine vs sucralfate + pirenzepine | 1 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.15 [0.26, 103.33] |
| 2 Nosocomial pneumonia | 17 | 3041 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.22 [1.07, 1.40] |
| 2.1 Cimetidine vs sucralfate | 5 | 758 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.13 [0.87, 1.47] |
| 2.2 Famotidine vs sucralfate | 1 | 140 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.13 [0.40, 3.20] |
| 2.3 Ranitidine vs sucralfate | 11 | 2143 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.26 [1.07, 1.48] |
| 3 All‐cause mortality in ICU | 21 | 3178 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.09 [0.95, 1.24] |
| 3.1 Cimetidine vs sucralfate | 6 | 814 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.18 [0.91, 1.54] |
| 3.2 Famotidine vs sucralfate | 2 | 190 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.23 [0.69, 2.19] |
| 3.3 Ranitidine vs sucralfate | 12 | 2107 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.04 [0.88, 1.22] |
| 3.4 Cimetidine + pirenzepine vs sucralfate + pirenzepine | 1 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.29 [0.38, 4.38] |
| 4 All‐cause mortality in hospital | 4 | 717 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.14 [0.86, 1.50] |
| 4.1 Cimetidine vs sucralfate | 2 | 413 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.28 [0.86, 1.92] |
| 4.2 Ranitidine vs sucralfate | 1 | 164 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.11 [0.71, 1.74] |
| 4.3 Famotidine vs sucralfate | 1 | 140 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.82 [0.40, 1.71] |
| 5 Duration of intubation | 10 | 1751 | Mean Difference (IV, Random, 95% CI) | 0.22 [‐1.55, 2.00] |
| 5.1 Cimetidine vs sucralfate | 2 | 97 | Mean Difference (IV, Random, 95% CI) | 0.58 [‐1.71, 2.87] |
| 5.2 Famotidine vs sucralfate | 1 | 140 | Mean Difference (IV, Random, 95% CI) | 0.40 [‐2.30, 3.10] |
| 5.3 Ranitidine vs sucralfate | 7 | 1514 | Mean Difference (IV, Random, 95% CI) | 0.15 [‐2.12, 2.43] |
| 6 Duration of ICU stay | 6 | 1791 | Mean Difference (IV, Random, 95% CI) | 0.01 [‐1.92, 1.95] |
| 6.1 Cimetidine vs sucralfate | 1 | 213 | Mean Difference (IV, Random, 95% CI) | 0.0 [‐3.05, 3.05] |
| 6.2 Famotidine vs sucralfate | 1 | 140 | Mean Difference (IV, Random, 95% CI) | 2.20 [‐0.96, 5.36] |
| 6.3 Ranitidine vs sucralfate | 4 | 1438 | Mean Difference (IV, Random, 95% CI) | ‐0.43 [‐2.70, 1.84] |
| 7 Number of participants requiring blood transfusion | 9 | 1095 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.25 [0.70, 2.23] |
| 7.1 Cimetidine vs sucralfate | 5 | 732 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.00 [0.47, 2.16] |
| 7.2 Ranitidine vs sucralfate | 4 | 363 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.77 [0.71, 4.39] |
| 8 Units of blood transfused | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 8.1 Cimetidine vs sucralfate | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 9 Adverse events of interventions | 6 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 9.1 Thrombocytopaenia | 2 | 240 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.72 [0.56, 39.47] |
| 9.2 Nausea and vomiting | 2 | 137 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.07 [0.01, 0.54] |
| 9.3 Hypermagnesaemia | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.71 [0.31, 23.93] |
| 9.4 Rash/Erythema | 2 | 233 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.06 [0.32, 28.87] |
| 9.5 Confusion | 3 | 382 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.48 [0.77, 26.00] |
| 9.6 Neutropaenia | 1 | 114 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.18 [0.25, 105.47] |
| 9.7 Dryness of mouth | 1 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.15 [0.26, 103.33] |
| 9.8 Leucopaenia | 1 | 163 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.11 [0.13, 75.26] |
Comparison 12. H2 receptor antagonists versus anticholinergics.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinically important upper GI bleeding | 3 | 556 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.37 [0.58, 3.26] |
| 1.1 Cimetidine vs pirenzepine | 1 | 55 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.45 [0.26, 7.99] |
| 1.2 Ranitidine vs pirenzepine | 2 | 501 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.35 [0.50, 3.67] |
| 2 Nosocomial pneumonia | 3 | 544 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.96 [0.50, 1.84] |
| 2.1 Famotidine vs pirenzepine | 1 | 43 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.32 [0.01, 7.42] |
| 2.2 Ranitidine vs pirenzepine | 2 | 501 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.03 [0.53, 2.01] |
| 3 All‐cause mortality in ICU | 2 | 501 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.21, 3.87] |
| 3.1 Ranitidine vs pirenzepine | 2 | 501 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.21, 3.87] |
| 4 Number of participants requiring blood transfusion | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1 Ranitidine vs pirenzepine | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Adverse events of interventions | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.1 Tachycardia | 1 | 55 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.11 [0.01, 1.90] |
| 5.2 High temperature | 1 | 43 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.53 [0.21, 1.32] |
Comparison 13. H2 receptor antagonists versus prostaglandin analogues.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinically important upper GI bleeding | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.1 Cimetidine vs misoprostol | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 All‐cause mortality in ICU | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2.1 Cimetidine vs misoprostol | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 14. H2 receptor antagonists versus teprenone.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinically important upper GI bleeding | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.1 Ranitidine vs teprenone | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 All‐cause mortality in ICU | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2.1 Ranitidine vs teprenone | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Number of participants requiring blood transfusion | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3.1 Ranitidine vs teprenone | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 15. H2 receptor antagonist + antacids versus sucralfate.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinically important upper GI bleeding | 2 | 230 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.24 [0.06, 0.95] |
| 1.1 Cimetidine + antacids vs sucralfate | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.14 [0.01, 2.70] |
| 1.2 Cimetidine or ranitidine + antacids vs sucralfate | 1 | 130 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.29 [0.06, 1.41] |
| 2 Nosocomial pneumonia | 3 | 281 | Risk Ratio (M‐H, Random, 95% CI) | 1.09 [0.51, 2.32] |
| 2.1 Cimetidine + antacids vs sucralfate | 1 | 100 | Risk Ratio (M‐H, Random, 95% CI) | 0.53 [0.26, 1.07] |
| 2.2 Ranitidine + antacids vs sucralfate | 1 | 51 | Risk Ratio (M‐H, Random, 95% CI) | 1.27 [0.64, 2.53] |
| 2.3 Cimetidine or ranitidine + antacids vs sucralfate | 1 | 130 | Risk Ratio (M‐H, Random, 95% CI) | 2.02 [0.89, 4.58] |
| 3 All‐cause mortality in ICU | 2 | 230 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.38 [0.92, 2.05] |
| 3.1 Cimetidine + antacids vs sucralfate | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.46, 2.19] |
| 3.2 Cimetidine or ranitidine + antacids vs sucralfate | 1 | 130 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.57 [0.99, 2.50] |
| 4 Duration of ICU stay | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 5 Duration of intubation | 2 | 230 | Mean Difference (IV, Random, 95% CI) | ‐1.24 [‐13.82, 11.33] |
| 5.1 Cimetidine + antacids vs sucralfate | 1 | 100 | Mean Difference (IV, Random, 95% CI) | ‐8.8 [‐20.11, 2.51] |
| 5.2 Cimetidine or ranitidine + antacids vs sucralfate | 1 | 130 | Mean Difference (IV, Random, 95% CI) | 4.20 [‐0.54, 8.94] |
| 6 Number of participants requiring blood transfusion | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
Comparison 16. Proton pump inhibitors versus teprenone.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinically important upper GI bleeding | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.1 Proton pump inhibitors vs teprenone | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 All‐cause mortality in ICU | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2.1 Proton pump inhibitors vs teprenone | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Number of participants requiring blood transfusion | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3.1 Proton pump inhibitors vs teprenone | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 17. Proton pump inhibitor plus naloxone versus naloxone.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinically important upper GI bleeding | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2 All‐cause mortality in hospital | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3 Adverse events ‐ gastrointestinal discomfort | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
Comparison 18. Proton pump inhibitors versus other medication (not defined).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinically important upper GI bleeding | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2 Nosocomial pneumonia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3 All‐cause mortality in hospital | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
18.3. Analysis.

Comparison 18 Proton pump inhibitors versus other medication (not defined), Outcome 3 All‐cause mortality in hospital.
Comparison 19. Antacids versus sucralfate.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinically important upper GI bleeding | 16 | 1772 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.00 [0.72, 1.39] |
| 1.1 Antacids vs sucralfate | 15 | 1705 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.96 [0.69, 1.35] |
| 1.2 Antacid + pirenzepine vs sucralfate + pirenzepine | 1 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.15 [0.26, 103.33] |
| 2 Nosocomial pneumonia | 7 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 Antacids vs sucralfate | 7 | 996 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.04 [0.84, 1.30] |
| 3 All‐cause mortality in ICU | 11 | 1249 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.15 [0.93, 1.40] |
| 3.1 Antacid vs sucralfate | 10 | 1182 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.15 [0.94, 1.41] |
| 3.2 Antacid + pirenzepine vs sucralfate + pirenzepine | 1 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.03 [0.28, 3.78] |
| 4 All‐cause mortality in hospital | 3 | 450 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.69, 1.39] |
| 5 Duration of ICU stay | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5.1 Antacids vs sucralfate | 2 | 227 | Mean Difference (IV, Fixed, 95% CI) | ‐2.50 [‐6.61, 1.61] |
| 6 Duration of intubation | 4 | Std. Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 6.1 Antacids vs sucralfate | 4 | 281 | Std. Mean Difference (IV, Fixed, 95% CI) | ‐0.18 [‐0.41, 0.06] |
| 7 Number of participants requiring blood transfusion | 6 | 667 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.73 [0.40, 1.34] |
| 8 Adverse events of interventions | 9 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 8.1 Diarrhoea | 6 | 599 | Risk Ratio (M‐H, Fixed, 95% CI) | 12.40 [3.88, 39.64] |
| 8.2 Hypermagnesaemia | 4 | 317 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.72 [1.24, 17.95] |
| 8.3 Nausea and vomiting | 3 | 223 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.63 [0.28, 1.41] |
| 8.4 Thrombocytopaenia | 1 | 38 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.0 [0.26, 97.70] |
| 8.5 Severe alkalosis | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.13, 71.92] |
| 8.6 Allergic reactions | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.2 [0.01, 4.06] |
Comparison 20. Antacids versus prostaglandin analogues.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinically important upper GI bleeding | 2 | 329 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.12, 0.91] |
| 2 All‐cause mortality in ICU | 2 | 417 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.84 [0.42, 1.67] |
| 3 Adverse events of interventions | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 Diarrhoea | 1 | 368 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.92 [0.64, 1.33] |
| 3.2 Elevated serum bicarbonate | 1 | 338 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.21 [1.27, 3.87] |
| 3.3 Phospate levels < 2.5 mg/dL | 1 | 276 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.66 [1.01, 2.73] |
Comparison 21. Antacids versus bioflavonoids.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinically important upper GI bleeding | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2 Number of participants requiring blood transfusion | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
Comparison 22. Sucralfate versus proton pump inhibitors.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinically important upper GI bleeding | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 Sucralfate vs omeprazole | 3 | 287 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.58 [0.77, 8.63] |
| 2 Nosocomial pneumonia | 4 | 424 | Risk Ratio (M‐H, Random, 95% CI) | 0.67 [0.41, 1.09] |
| 2.1 Sucralfate vs omeprazole | 3 | 287 | Risk Ratio (M‐H, Random, 95% CI) | 0.88 [0.57, 1.36] |
| 2.2 Sucralfate vs pantoprazole | 1 | 137 | Risk Ratio (M‐H, Random, 95% CI) | 0.39 [0.20, 0.75] |
| 3 All‐cause mortality in ICU | 4 | 424 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.07 [0.68, 1.68] |
| 3.1 Sucralfate vs omeprazole | 3 | 287 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.26 [0.75, 2.11] |
| 3.2 Sucralfate vs pantoprazole | 1 | 137 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.65 [0.25, 1.68] |
| 4 All‐cause mortality in hospital | 2 | 278 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.79 [0.46, 1.37] |
| 4.1 Sucralfate vs omeprazole | 1 | 141 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.49, 1.91] |
| 4.2 Sucralfate vs pantoprazole | 1 | 137 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.56 [0.21, 1.45] |
| 5 Duration of ICU stay | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5.1 Sucralfate vs omeprazole | 2 | 217 | Mean Difference (IV, Fixed, 95% CI) | 0.01 [‐1.68, 1.70] |
| 6 Duration of intubation | 3 | 354 | Mean Difference (IV, Fixed, 95% CI) | ‐0.16 [‐1.61, 1.28] |
| 6.1 Sucralfate vs omeprazole | 2 | 217 | Mean Difference (IV, Fixed, 95% CI) | 0.02 [‐1.56, 1.60] |
| 6.2 Sucralfate vs pantoprazole | 1 | 137 | Mean Difference (IV, Fixed, 95% CI) | ‐1.10 [‐4.69, 2.49] |
| 7 Number of participants requiring blood transfusion | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 7.1 Sucralfate vs omeprazole | 1 | 70 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.91 [0.29, 118.78] |
| 8 Adverse events of interventions | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 Fever | 1 | 137 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.70, 0.94] |
| 8.2 Leucocytosis | 1 | 137 | Risk Ratio (M‐H, Random, 95% CI) | 0.66 [0.55, 0.80] |
| 8.3 Sudden purulent sputum | 1 | 137 | Risk Ratio (M‐H, Random, 95% CI) | 0.85 [0.38, 1.86] |
| 8.4 Sudden cough or aggravation of coughing | 1 | 137 | Risk Ratio (M‐H, Random, 95% CI) | 0.23 [0.07, 0.79] |
| 8.5 Dyspnoea | 1 | 137 | Risk Ratio (M‐H, Random, 95% CI) | 0.68 [0.54, 0.87] |
| 8.6 Rales or bronchial sounds | 1 | 137 | Risk Ratio (M‐H, Random, 95% CI) | 0.31 [0.19, 0.51] |
| 8.7 Aggravation of blood gas exchange | 1 | 137 | Risk Ratio (M‐H, Random, 95% CI) | 0.76 [0.49, 1.18] |
| 8.8 Change in sputum quality | 1 | 137 | Risk Ratio (M‐H, Random, 95% CI) | 0.23 [0.13, 0.40] |
Comparison 23. Sucralfate versus bioflavonoids.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinically important upper GI bleeding | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2 Number of participants requiring blood transfusion | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
Comparison 24. Total parenteral nutrition (TPN) versus any other intervention + TPN.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinically important upper GI bleeding | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 TPN vs ranitidine + TPN | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.8 [0.05, 12.14] |
| 1.2 TPN vs sucralfate + TPN | 1 | 49 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.32 [0.03, 3.26] |
| 2 All‐cause mortality in ICU | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 TPN vs ranitidine + TPN | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.12 [0.41, 3.09] |
| 2.2 TPN vs sucralfate + TPN | 1 | 49 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.63 [0.26, 1.52] |
| 3 Duration of intubation | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 3.1 TPN vs ranitidine + TPN | 1 | 54 | Mean Difference (IV, Fixed, 95% CI) | ‐2.0 [‐9.53, 5.53] |
| 3.2 TPN vs sucralfate + TPN | 1 | 49 | Mean Difference (IV, Fixed, 95% CI) | 3.0 [‐1.50, 7.50] |
Comparison 25. Bowel stimulation versus no prophylaxis.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinically important upper GI bleeding | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
Comparison 26. Nasojejunal nutrition versus nasogastric nutrition.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinically important upper GI bleeding | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2 Nosocomial pneumonia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3 All‐cause mortality in hospital | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4 Adverse events of interventions | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
Comparison 27. Enteral plus parenteral nutrition versus other nutrition regimens.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Nosocomial pneumonia | 1 | 120 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.79 [0.44, 1.40] |
| 1.1 Enteral plus parenteral nutrition vs enteral nutrition | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.6 [0.29, 1.25] |
| 1.2 Enteral plus parenteral nutrition vs parenteral nutrition | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.25 [0.47, 3.33] |
| 2 All‐cause mortality in hospital | 1 | 120 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.20 [0.06, 0.60] |
| 2.1 Enteral plus parenteral nutrition vs enteral nutrition | 1 | 60 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.26 [0.05, 1.30] |
| 2.2 Enteral plus parenteral nutrition vs parenteral nutrition | 1 | 60 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.15 [0.03, 0.74] |
| 3 Duration of ICU stay | 1 | 120 | Mean Difference (IV, Fixed, 95% CI) | ‐5.98 [‐8.81, ‐3.16] |
| 3.1 Enteral plus parenteral nutrition vs enteral nutrition | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐3.81 [‐7.59, ‐0.03] |
| 3.2 Enteral plus parenteral nutrition vs parenteral nutrition | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐8.72 [‐12.97, ‐4.47] |
| 4 Duration of intubation | 1 | 120 | Mean Difference (IV, Fixed, 95% CI) | ‐7.37 [‐9.29, ‐5.45] |
| 4.1 Enteral plus parenteral nutrition vs enteral nutrition | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐4.17 [‐6.96, ‐1.38] |
| 4.2 Enteral plus parenteral nutrition vs parenteral nutrition | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐10.24 [‐12.88, ‐7.60] |
| 5 Adverse events ‐ stress ulcer | 1 | 120 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.69 [0.36, 1.33] |
| 5.1 Enteral plus parenteral nutrition vs enteral nutrition | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.14 [0.38, 3.45] |
| 5.2 Enteral plus parenteral nutrition vs parenteral nutrition | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.53 [0.23, 1.20] |
| 6 Adverse events ‐ diarrhoea | 1 | 120 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.42 [0.17, 1.02] |
| 6.1 Enteral plus parenteral nutrition vs enteral nutrition | 1 | 60 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.17 [0.05, 0.59] |
| 6.2 Enteral plus parenteral nutrition vs parenteral nutrition | 1 | 60 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.42 [0.35, 5.73] |
| 7 Adverse events ‐ pyaemia | 1 | 120 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.88 [0.37, 2.09] |
| 7.1 Enteral plus parenteral nutrition vs enteral nutrition | 1 | 60 | Odds Ratio (M‐H, Fixed, 95% CI) | 4.11 [0.87, 19.41] |
| 7.2 Enteral plus parenteral nutrition vs parenteral nutrition | 1 | 60 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.37 [0.11, 1.21] |
| 8 Adverse events ‐ intracranial infection | 1 | 120 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.43 [0.15, 1.25] |
| 8.1 Enteral plus parenteral nutrition vs enteral nutrition | 1 | 60 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.83 [0.19, 3.63] |
| 8.2 Enteral plus parenteral nutrition vs parenteral nutrition | 1 | 60 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.23 [0.05, 1.15] |
| 9 Adverse events ‐ hypoproteinaemia | 1 | 120 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.11 [0.04, 0.27] |
| 9.1 Enteral plus parenteral nutrition vs enteral nutrition | 1 | 60 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.20 [0.06, 0.72] |
| 9.2 Enteral plus parenteral nutrition vs parenteral nutrition | 1 | 60 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.04 [0.01, 0.19] |
27.6. Analysis.

Comparison 27 Enteral plus parenteral nutrition versus other nutrition regimens, Outcome 6 Adverse events ‐ diarrhoea.
27.7. Analysis.

Comparison 27 Enteral plus parenteral nutrition versus other nutrition regimens, Outcome 7 Adverse events ‐ pyaemia.
27.8. Analysis.

Comparison 27 Enteral plus parenteral nutrition versus other nutrition regimens, Outcome 8 Adverse events ‐ intracranial infection.
27.9. Analysis.

Comparison 27 Enteral plus parenteral nutrition versus other nutrition regimens, Outcome 9 Adverse events ‐ hypoproteinaemia.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Ali 2016.
| Methods | Parallel‐group randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 10 participants Number analysed: 10 participants Pantoprazole
Placebo
Inclusion criteria
Exclusion criteria: ‐ Baseline imbalances: ‐ |
|
| Interventions |
Pantoprazole
Placebo
Adherence to regimen: ‐ Duration of trial: ‐ Duration of follow‐up: ‐ |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
|
|
| Notes |
Setting: ICU Source of funding: ‐ Conflicts of interest: ‐
Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: ‐ |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: mentioned only that participants were randomised to treatment. Not enough information on method of randomisation |
| Allocation concealment (selection bias) | Unclear risk | Comment: no information about allocation concealment reported |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: mentioned only that the trial was double‐blind. No information about the method of blinding |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: mentioned that outcome assessment was done blinded to intervention. Presence of 1 or more macroscopic abnormalities (erythema or oedema, erosions, ulcerations, and nasogastric tube lesions) and GI bleeding was assessed, blinded to intervention, at endoscopy by a single experienced gastroenterologist Quote: "assessed, blinded to intervention, at endoscopy by a single experienced gastroenterologist" |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: The outcome was not addressed in this trial |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: mentioned that outcome assessment was done blinded to intervention |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: Conference abstract reports about 10 participants from a larger prospective trial. Those who received > 5 doses of pantoprazole or placebo (n = 84) were eligible for the endoscopy substudy, but unclear why data from only 10 participants are reported |
| Selective reporting (reporting bias) | Low risk | Comment: Outcomes reported in Methods section are also reported in Results section |
| Other bias | Unclear risk | Comment: not enough information reported in conference abstract to assess other biases |
Apte 1992.
| Methods | Open‐label parallel‐group randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 34 participants Number analysed: 34 participants Ranitidine
No prophylaxis
Inclusion criteria
Exclusion criteria
Baseline imbalances: Participants were tracheotomised patients with tetanus. Maximum tetanus severity score was 11 (4 to 16) in the ranitidine group and 10 (6 to 16) in the control group. Groups were similar with respect to age and gender distribution |
|
| Interventions |
Ranitidine
No prophylaxis
Adherence to regimen: 34 tracheotomised participants who were admitted to medical ICU with tetanus were randomly assigned to ranitidine or control group within 24 hours of tracheal intubation. Six participants who had pneumonia before tracheostomy or had ranitidine before randomisation were excluded. All remaining participants were studied until 48 hours after extubation Duration of trial: ‐ Duration of follow‐up: studied daily until 48 hours after tracheal extubation |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
|
|
| Notes |
Setting: Medical ICU in Department of Medicine, Department of Microbiology, King Edward Memorial Hospital, Parel, Bombay, India Source of funding: Quote: "Study supported, in part, by a grant from Seth GS medical College and KEM Hospital research Society”; Torrent Pharmaceuticals provided ranitidine Conflicts of interest: ‐ Ethics approval: Quote: "The study was approved by the institutional ethics committee" Informed consent: Quote: "The study was approved by the institutional ethics committee [with] waived informed consent" Comment: not obtained, as mentioned in the trial report Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Trial included patients with tetanus. Trial reports that gram‐negative bacilli were the predominant organisms that caused pneumonia. Participants treated with ranitidine developed pneumonia significantly earlier (median 3 days, range 1 to 5) than participants given control (median 5 days, range 3 to 14 days) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not enough information on method of randomisation reported |
| Allocation concealment (selection bias) | Unclear risk | Comment: not enough information on method of allocation concealment reported |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: This trial compared an intervention vs no prophylaxis (blinding was not possible) |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: GI bleeding was an objective outcome that was detected as per the definition in the trial protocol |
| Blinding (detection bias) Nosocomial pneumonia | Low risk | Comment: Pneumonia was an objective outcome that was detected as per the definition in the trial protocol |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: All other outcomes of interest were objective in nature |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All participants who were randomised were included in analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: This trial was supported in part by a grant from Seth GS Medical College and KEM Hospital Research Society. The role of the sponsor in the conduct and reporting of the trial is unclear. No other form of bias is suspected |
Barandun 1985.
| Methods | Double‐blind parallel‐group randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 66 participants Number analysed: 55 participants Pirenzepine
Cimetidine
Inclusion criteria
Exclusion criteria
Baseline imbalances: baseline imbalances comparable, also in terms of severity of injury/trauma |
|
| Interventions |
Pirenzepine
Cimetidine
Adherence to regimen: 11 participants withdrew from the trial. However, no reasons are mentioned in the trial report. Also no mention of which interventional group these 11 participants belonged to Duration of trial: ‐ Duration of follow‐up: not clearly mentioned, probably until discharge |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
|
|
| Notes |
Setting: Surgical ICU, Chur, Switzerland Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not enough information reported on method of sequence generation |
| Allocation concealment (selection bias) | Unclear risk | Comment: not enough information reported on method of allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: This appears to be a double‐dummy placebo‐controlled trial, in which participants from group 1 got a placebo that looks like the intervention in group 2 and vice versa. Thus personnel would have been blinded and likelihood of performance bias is low |
| Blinding (detection bias) Clinically important upper GI bleeding | Unclear risk | Comment: not enough information reported on the criteria for diagnosis of upper GI bleeding |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: The trial did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: This appears to be a double‐dummy placebo‐controlled trial, in which participants from group 1 got a placebo that looks like the intervention in group 2 and vice versa. Thus the likelihood of detection bias seems low |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: 11 participants withdrew from the trial, but reasons for withdrawal and the group to which they were randomised are not clearly mentioned in the trial report |
| Selective reporting (reporting bias) | Unclear risk | Comment: All intended outcomes were reported but no clear mention of the number of participants in the cimetidine group who had confusion and high K levels |
| Other bias | Low risk | Comment: The trial report is unclear on the source of funding. No other sources of bias detected |
Bashar 2013.
| Methods | Double‐blind parallel‐group randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 146 participants Number analysed: 120 participants Ranitidine
Pantoprazole
Inclusion criteria
Exclusion criteria
Baseline imbalances: We found no statistically significant differences between the 2 groups regarding baseline characteristics, such as age, sex, or APACHE II |
|
| Interventions |
Ranitidine
Pantoprazole
Adherence to regimen: ‐ Duration of trial: July 2011 to July 2012 Duration of follow‐up: ‐ |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought in review but not reported in trial
Outcomes reported, but not used in the review
|
|
| Notes |
Setting: ICU, Iran Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Trial was approved by the Ethics Committee of Hamedan University of Medical Sciences. Informed consent: Written informed consent was obtained from legal guardians of participants Clinical trials registration: ‐ Sample size calculation: ‐ Conflicts of interest: ‐ |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "The patients were randomised using online random allocation software (www.allocationsoftware.com)" |
| Allocation concealment (selection bias) | Unclear risk | Comment: not enough information reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The patients and the attending intensivists responsible for data collection were blinded to the assigned groups" |
| Blinding (detection bias) Clinically important upper GI bleeding | Unclear risk | Comment: Trial did not address this outcome |
| Blinding (detection bias) Nosocomial pneumonia | Low risk | Quote: "Patients underwent chest radiography which was repeated at least twice a week" Comment: Objective outcome measurement unlikely to introduce bias |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Quote: "The patients and intensivists responsible for data collection were blinded to the assigned groups" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All participants were followed up until discharge |
| Selective reporting (reporting bias) | Low risk | Comment: All outcomes listed in the Methods section were reported in the Results section |
| Other bias | Low risk | Comment: no other sources of bias suspected |
Basso 1981.
| Methods | Single‐blind randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 168 participants Number analysed: 168 participants Cimetidine
Antacids (Maalox)
No prophylaxis
Inclusion criteria
Exclusion criteria
Baseline imbalances: Risk categories and risk factors in the 3 groups were comparable |
|
| Interventions |
Cimetidine
Antacids (Maalox)
No prophylaxis
Adherence to regimen: 16 participants died, 6 did not comply with therapy, and 9 were transferred to other institutions. Therefore, 31 participants did not complete 10 days of the trial Duration of trial: March 1978 to April 1979 Duration of follow‐up: not clearly mentioned in trial report |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported but not used in review
|
|
| Notes |
Setting: ICU, University of Rome Source of funding: ‐ Ethics approval: ‐ Informed consent: Quote: "Informed consent was obtained from either the participant or their closest relative" Clinical trials registration: not provided Sample size calculation: ‐ |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “The study was done in a single blind manner, assigning the treatment according to a list of randomised values” Comment: not enough information reported on method of sequence generation |
| Allocation concealment (selection bias) | Unclear risk | Comment: not enough information reported on method of allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: This was not a placebo‐controlled trial and personnel were not blinded |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Quote: "The observer assessing the occurrence of gastrointestinal bleeding did not know the type of prophylactic measures the patient was receiving" |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: The trial did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Unclear risk | Comment: Unclear whether outcome assessors were blinded for other outcomes |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: All randomised participants were part of the final analysis |
| Selective reporting (reporting bias) | Unclear risk | Comment: All‐cause mortality in ICU and units of blood transfused were not mentioned separately for each interventional arm. Unclear whether this contributed to reporting bias |
| Other bias | Unclear risk | Comment: source of funding and baseline characteristics unclear |
Behrens 1994.
| Methods | Open‐label randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 43 participants Number analysed: 43 participants Pirenzepine
Famotidine
No prophylaxis
Inclusion criteria
Exclusion criteria
Baseline imbalances: There were no differences between groups that did not receive any prophylaxis (not randomised) and the group that did receive prophylaxis (randomised to pirenzepine and famotidine) |
|
| Interventions |
Pirenzepine
Famotidine
No prophylaxis
Adherence to regimen: ‐ Duration of trial: October 1988 to November 1991 Duration of follow‐up: ‐ |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported in report but not used in review
|
|
| Notes |
Setting: University Children's Hospital, Department of Pediatric Gastroenterology, Erlangen, Germany Source of funding: ‐ Conficts of interest: ‐ Ethics approval: Quote: "The study was approved by the committee on human research of the University of Erlangen‐Nurnberg" Informed consent: ‐ Clinical trial registration: ‐ Sample size calculation: ‐ Additional notes: Gastric cultures were positive in 95% of participants with mean gastric pH > 4 and in 80% of participants with mean gastric pH < 4. Five and 4 participants in the 2 groups required mechanical ventilation. Candida sp was the predominant species cultured from gastric and tracheal secretions of participants given pirenzepine and famotidine. In the pirenzepine and famotidine groups, an organism cultured from the stomach was grown from the tracheal secretion, 1 to 4 days later, in 6 and 5 participants, respectively |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not enough information reported on method of sequence generation |
| Allocation concealment (selection bias) | Unclear risk | Comment: not enough information reported on method of allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: unclear on blinding of personnel, although regimens and mode of administration of interventions were similar |
| Blinding (detection bias) Clinically important upper GI bleeding | Unclear risk | Comment: The physician who performed endoscopy to detect inflammation or ulceration of the upper GI tract was not blinded to the intervention. There is no clear definition of GI bleeding in the trial. Therefore, unclear on the likelihood of performance or detection bias |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: no mention of blinding technicians who cultured gastric secretions for pathogenic bacteria, radiologists who interpreted chest X‐rays, or physicians who performed the clinical examination. No clear definition of VAP provided in the trial |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Unclear risk | Comment: not enough information reported on blinding of outcome assessors for other outcomes |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All randomised participants were included in the analysis |
| Selective reporting (reporting bias) | Low risk | Comment: Trial compared participants who received prophylaxis vs participants who did not receive prophylaxis. In the intervention group, participants were randomised to receive either pirenzepine or famotidine. The outcome of interest (VAP) was reported separately for participants who were randomised to 2 different arms (pirenzepine and famotidine) |
| Other bias | Unclear risk | Comment: source of funding not clearly mentioned in the trial report. Baseline data on randomised groups unclear |
Ben‐Menachem 1994.
| Methods | Single‐blind randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 304 participants Number analysed: 300 participants Sucralfate
Cimetidine
Control
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "One hundred patients were randomly assigned to each of the treatments. The groups were similar with regard to age, gender, percentage of participants admitted from the emergency room, severity of illness, admission diagnoses, corticosteroid usage and coagulopathy. Patients often had more than one reason for ICU admission. The mean APACHE II scores for the control, sucralfate, and cimetidine groups were 16.5 ± 6.9, 16.8 ± 6.9, and 18.0 ± 8.0, respectively. Approximately one‐third of the patients in each group had APACHE II scores greater than 20.0 Comment: The 3 groups were similar with respect to demographic and other risk factors for stress haemorrhage at the beginning of the trial. The most common diagnosis on admission was pneumonia, which was reported in 89 participants. Bacterial pneumonia (control: 23; sucralfate: 26; cimetidine: 21). Non‐bacterial pneumonia was diagnosed (control: 9; sucralfate: 4; cimetidine: 6). Coagulopathy was present in 16, 14, and 21 participants in the 3 randomised groups |
|
| Interventions |
Sucralfate
Cimetidine
No prophylaxis
Adherence to regimen: Although 304 participants were randomised, only 300 (100 in each group) were part of the trial because 1 participant died 2 hours after admission, 3 participants were randomised on 2 different occasions, and second admissions were excluded. Results for only 291 participants were available when the trial was finally reviewed by the second committee. The 8 participants who did not agree to endoscopy might have been excluded from the trial denominator (unclear on the 1 remaining participant) Duration of trial: 1 February to 25 November 1992 Duration of follow up: until death or discharge from ICU |
|
| Outcomes |
Outcomes sought in review and reported in trial Primary outcomes
Note: Median time from ICU admission to onset of stress‐related haemorrhage was 5 days
Note: Noscocomial pneumonia occurred after a mean of 6.9 ± 7.2 days in the ICU (median 9 days) Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
|
|
| Notes |
Setting: Henry Ford Hospital and Health Sciences Center, Detroit, Michigan Source of funding: Quote: "Grant support: in part from the Henry Ford Hospital Research and Education Funds to Dr. Ben‐Menachem and to Dr. Fogel" Conflicts of interest: ‐ Ethics approval: Quote: "This protocol was reviewed and approved by the Henry Ford Hospital Institutional Review Board" Informed consent: Quote: "Informed consent was obtained from the patient or from legally authorized representatives when the patient could not provide consent" Clinical trials registration: ‐ Sample size calculation: Quote: "We estimated sample size to provide 80% power to detect a 75% reduction in bleeding rate, that is, a 12% bleeding rate for the control group compared with a 3% rate in either of the two treatment groups. We used an alpha value of 0.05 (two‐tailed) adjusted for the comparison of the control group with each of the prophylaxis groups. As a result of these assumptions, 160 patients were needed in each of the three groups” Additional notes: Mechanical ventilation was used in 65, 72, and 76 participants in the 3 respective groups. Respiratory failure and high dose of corticosteroid were independently associated with increased risk of stress‐related haemorrhage. According to the trial report, 43 participants satisfied the criteria for significant GI bleeding (as per the definition), and 19 of these participants did not have stress‐related bleeding (as determined by oesophagogastroduodenoscopy). Among them, 12 participants (4 controls, 3 receiving sucralfate, and 5 receiving cimetidine) had a normal result of endoscopy, suggesting that the change in haematocrit was due to fluid shifts. Seven participants bled from causes not due to stress ulceration, and 8 participants did not have endoscopy (5 did not give consent, 1 participant with lymphoma and thrombocytopaenia died of multiple‐organ system failure 10 days after the bleeding episode. Three of the 8 participants met criterion 3 of diagnosis Quote: "Two of 20 patients in the control group with coagulopathy had stress‐related haemorrhage. In the cimetidine and sucralfate groups, the incidences were 1 of 22 and 2 of 17, respectively (P > 0.05)" |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomization was by sealed envelope using the permuted block design" Comment: Method to generate a random sequence is clearly mentioned in the trial report |
| Allocation concealment (selection bias) | Low risk | Quote: "Randomization was by sealed envelope using the permuted block design" Comment: Method for allocation concealment is clearly mentioned in the trial report |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: This was a single‐blind trial, and the mode of drug administration could not have allowed blinding. Therefore, risk of performance bias is high |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Quote: "The primary study end point was substantial haemorrhage from stress gastritis. Information regarding hematocrit, haemoccult status of stool and nasogastric aspirate, and volume status [were] presented daily to two investigators who were blinded to therapy" Comment: Trial report mentions blinding of outcome assessors for the primary outcome of GI bleed, which was an objective outcome detected as per the definition used in the trial |
| Blinding (detection bias) Nosocomial pneumonia | Low risk | Comment: Outcome assessors were not blinded. However, this was an objective outcome that was diagnosed as per the definition used in the trial protocol. Therefore, the likelihood of performance or detection bias is low |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: All other outcomes of interest were objective in nature, so the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: Although 304 participants were randomised, only 300 were analysed, as 1 participant died 2 hours after admission, 3 were randomised on 2 different occasions, and second admissions were excluded. Results for only 291 participants were available when the trial was finally reviewed by the second committee. The 8 participants who did not agree to endoscopy might have been excluded from the trial denominator (unclear for the 1 remaining participant). However, intention‐to‐treat analysis was done with 100 participants in each of the 3 groups. Therefore, there was no attrition bias |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes are reported. A subgroup analysis was desirable for participants who received enteral feeds |
| Other bias | Low risk | Comment: This trial was supported in part by Henry Ford Hospital Research and Education Funds. The role of the sponsor in the conduct and reporting of this trial is unclear. No other form of bias is suspected |
Bonten 1995.
| Methods | Stratified double‐blind randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 141 participants Number analysed: 141 participants Antacids
Sucralfate
Inclusion criteria
Exclusion criteria
Baseline imbalances: Demographic characteristics of both groups, such as age, gender, and severity of illness, which was assessed by the APCHE score, were almost similar. Most participants were given a diagnosis of cardiovascular problems (30 in both groups). Neoplastic disease was more common among participants assigned to the antacid group when compared with the sucralfate group (12 and 5), whereas renal insufficiency and immunodeficiency were more common among participants in the sucralfate group (1 and 6 and 3 and 7, respectively). All other underlying conditions including presence of pneumonia (9 in antacid group and 10 in sucralfate group) on admission were similar between both groups |
|
| Interventions |
Antacids
Sucralfate
Adherence to regimen: Gastric pH was determined within 24 hours after admission via continuous intragastric monitoring. Participants were stratified into 2 groups according to gastric pH value measured on the first day of admission, which was < 3 (n = 69) or ≥ 3 (n = 72) (continuous intragastric pH in 84 and indicator papers in 57 participants; for 28 in the latter group, continuous pH monitoring was performed later). Participants were later randomised to 2 treatment arms, amongst whom 74 received antacids and 67 received sucralfate Duration of trial: August 1992 to August 1993 Duration of follow‐up: probably until discharge or death |
|
| Outcomes |
Outcomes sought in review and reported in trial
Note: The mean number of days before the first episode of VAP was 9.2 in the antacid group and 9.3 in the sucralfate group, respectively.
Outcomes sought but not reported in trial
Outcomes reported in report but not used in review
|
|
| Notes |
Setting: University Hospital Maastricht, Maastricht, The Netherlands Source of funding: Quote: "Supported by grant 28‐2125 from the Praevention Foundation" Conflicts of interest: ‐ Ethics approval: Quote: "The protocol was approved by the institutional review board of the hospital" Informed consent: Quote: "... informed consent was obtained from all participants or if this was not possible because of the clinical condition, from a representative of the family" Clinical trials registration: ‐ Sample size calculation: Quote: "The sample size of both groups of patients was calculated to detect a reduction in the incidence of VAP from an assumed 30% in the antacid group to an expected incidence of 10% in the sucralfate group (α = 0.05 and β = 0.2)" Additional notes: VAP was mainly polymicrobial in nature. Pseudomonas aeruginosa and Staphylococcus aureus were the 2 predominant pathogens cultured from participants with a diagnosis of VAP. The oropharynx was the initial site for colonisation ofEnterobacteriaceae, whereas Pseudomonas aeruginosa and Staphylococcus aureus colonised in the upper respiratory tract first |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were stratified into two groups according to, the gastric pH values measured on the first day of admission which were < 3 (n = 69) or ≥ 3 (n = 72). These patients were later randomised to two treatment arms" Comment: not enough information on method of sequence generation reported |
| Allocation concealment (selection bias) | Unclear risk | Quote: "Sucralfate and antacids were compared in a double blind fashion and analysed according to a prospectively developed plan that used definition adopted before the treatment allocation code was broken" Comment: Allocation concealment might have been in place but not enough information was provided on method of allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The study had a "double dummy model" design wherein participants receiving sucralfate received an antacid placebo at the same time when antacid was administered to the other group and vice versa" Quote: "Sucralfate and antacids were compared in a double blind fashion and analysed according to a prospectively developed plan that used definition adopted before the treatment allocation code was broken. The attending physician was unaware of the cultural results of the oropharyngeal and gastric samples, or results of pH monitoring" Comment: Personnel (and participants) were unaware of therapy and results |
| Blinding (detection bias) Clinically important upper GI bleeding | Unclear risk | Quote: "The study had a "double dummy model" design wherein participants receiving sucralfate received an antacid placebo at the same time when antacid was administered to the other group and vice versa" Comment: The trial by design inherently blinded trial personnel who were assigned to endoscopically detect upper GI bleeding; however, criteria for diagnosis of upper GI bleeding were not clearly reported |
| Blinding (detection bias) Nosocomial pneumonia | Low risk | Quote: "The study had a "double dummy model" design wherein participants receiving sucralfate received an antacid placebo at the same time when antacid was administered to the other group and vice versa" Comment: The trial by design inherently blinded trial personnel who were assigned to diagnose VAP. Criteria for diagnosis of VAP were clearly reported |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Quote: "The study had a "double dummy model" design wherein participants receiving sucralfate received an antacid placebo at the same time when antacid was administered to the other group and vice versa" Comment: The trial by design inherently blinded outcome assessors. Therefore, the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: No dropouts were reported, and all randomised participants were part of the final analysis.Therefore, there might not be an attrition bias |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were reported and were part of the analysis.. A subgroup analysis for participants who received enteral feeds was desirable |
| Other bias | Low risk | Comment: Trial was supported by grant 28‐2125 from the Praevention Foundation. The role of the sponsor in the conduct and reporting of the trial is unclear |
Borrero 1984.
| Methods | Quasi‐randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 100 participants Number analysed: 100 participants Antacids
Sucralfate
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "There was no statistically significant difference between the sucralfate treated and the antacid treated groups in number of patients, sex" Comment: Average numbers of risk factors per group were also similar; 92.5 and 83 participants had undergone an operation just before trial entry |
|
| Interventions |
Antacids (Mylanta II)
Sucralfate
Adherence to regimen: Quote: "Of the 52 patients treated with antacids, failure to achieve a pH of 3.5 or greater occurred in 8 patients initially given 30 mL of antacid. Five subsequently required 60 mL/hour and three required 120 mL/hour. All patients receiving antacids maintained a gastric pH of mote than 5" Comment: Iced saline solution lavage was given to all participants with diagnosis of upper GI bleed (by Gastroccult test) Duration of trial: August 1983 to December 1983 Duration of follow up: probably until discharge or death |
|
| Outcomes |
Outcomes sought in review and reported in trial
Note: antacids 9 and 10 hours after start of the drug and sucralfate; 8, 41, and 45 hours after initiation of prophylaxis Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
|
|
| Notes |
Setting: Queens Hospital Centre, New York, USA Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: No deaths were due to GI bleeding |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Quote: “100 patients admitted to medical and surgical intensive care units were randomised to receive either antacids and sucralfate depending on the year of birth (odd year, sucralfate; even year, antacid)” Comment: This was a quasi‐randomised trial |
| Allocation concealment (selection bias) | High risk | Comment: This was a quasi‐randomised trial, and no information on allocation concealment was reported |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: no information on blinding reported |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: Blinding was not done. However, the outcome was measured as per the definition used in the trial protocol. Moreover, owing to the objective nature of the outcome of interest, the likelihood of detection and performance bias is low |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: The trial did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Unclear risk | Comment: no information on blinding or criteria to diagnose other outcomes reported |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All randomised participants were included in the final analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: Source of funding is unclear. No other source of bias detected |
Borrero 1985.
| Methods | Quasi‐randomised trial | |
| Participants |
Baseline characteristics Number randomised: 155 participants Number analysed: 155 participants Antacids
Sucralfate
Inclusion criteria
Exclusion criteria
Baseline imbalances: no statistically significant difference between sucralfate‐treated and antacid‐treated groups in numbers, age, and gender of participants. 130 participants (61 and 69 in each group) had undergone a major operation just before trial entry |
|
| Interventions |
Antacids (Mylanta or Maalox)
Sucralfate
Adherence to regimen: not clearly mentioned in the trial report Duration of follow‐up: Quote: “Patients were continued in the trial until the onset of gastrointestinal bleeding, until they were discharged from the critical care unit, or until nasogastric suction was discontinued. The patients’ clinical course was followed until they were discharged from the hospital” |
|
| Outcomes |
Outcomes sought in review and reported in trial
Note: GI bleeding was tested for antacids 9 and 10 hours after initiating prophylaxis, and for sucralfate 8, 41, and 43 hours after initiating prophylaxis Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
|
|
| Notes |
Setting: Long Island Jewish Medical Centre and Queens Hospital Centre Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Quote: "The study protocol was approved by the institutional review boards at Long island Jewish Medical Centre and Queens Hospital Centre" Informed consent: Quote: "Informed consent was obtained from the patient or immediate relative" Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: None of the deaths were due to GI bleeding |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Quote: "Patients were randomly assigned to one of the two treatment regimens according to their date of birth"; "participants born on even days were given antacids and those born on odd days were administered sucralfate" Comment: This was a quasi‐randomised trial |
| Allocation concealment (selection bias) | High risk | Comment: This was a quasi‐randomised trial; no information on method of allocation concealment was reported |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: This was not a placebo‐controlled trial; the different interventions and their mode of administration might not have ensured blinding of personnel |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: unclear on blinding of outcome assessors. However, the outcome was measured as per the definition used in the trial protocol. Moreover, owing to the objective nature of the outcome of interest, the likelihood of detection and performance bias was judged as low |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: The trial did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: unclear on blinding of outcome assessors. However, outcomes of interest were objective in nature, so the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All randomised participants were part of the final analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: Source of funding is unclear. No other source of bias is suspected |
Borrero 1986.
| Methods | Quasi‐randomised trial | |
| Participants |
Baseline characteristics Number randomised: 50 participants Number analysed: 50 participants Antacids
Sucralfate
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "There was no statistically significant difference between the sucralfate treated and the antacid treated groups in number of patients, sex, mean age [...] or number of risk factors per patient" Comment: All participants had undergone aortobifemoral artery Dacron graft placement |
|
| Interventions |
Antacids (Mylanta II)
Sucralfate
Adherence to regimen: Comment: Iced saline solution lavage was given to all participants with diagnosed upper GI bleeding Duration of trial: August 1983 to December 1984 Duration of follow‐up: not clearly mentioned in the trial report; probably until discharge or death |
|
| Outcomes |
Outcomes sought in review and reported in trial Primary outcomes
Outcomes sought but not reported in trial report
Outcomes reported in trial but not used in review
|
|
| Notes |
Settings: Long island Jewish Medical Centre, New Hyde Park, NY 11042 Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: None of the deaths were due to GI bleeding |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Quote: "…were randomised to receive either antacid or sucralfate, depending on their year of birth (odd year sucralfate, even year antacid) Comment: quasi‐randomised trial |
| Allocation concealment (selection bias) | High risk | Comment: This was a quasi‐randomised trial; no information on method of allocation concealment was reported |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: This was not a placebo‐controlled trial; the different interventions and their mode of administration might not have ensured blinding of personnel |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: unclear on blinding of outcome assessors. However, GI bleeding was an objective outcome that was detected as per the definition used in the trial report |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: The trial did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Unclear risk | Comment: unclear on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All randomised participants were included in the final analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: Source of funding is unclear. No other source of bias is suspected |
Bresalier 1987.
| Methods | Open‐label parallel‐group trial | |
| Participants |
Baseline characteristics Number randomised: 83 participants Number analysed: 74 participants Antacids
Sucralfate
Inclusion criteria
Exclusion criteria
Baseline imbalances: The 2 groups were almost similar with respect to age, gender, and time from entry into ICU to random selection. However, the sucralfate group had more risk factors for bleeding on admission to the study when compared with the antacid group (respiratory failure was the most common ‐ 34 and 37 participants, respectively). Three participants in the antacid group had coagulopathy vs 1 participant in the antacid group. Two participants in the antacid group were given a diagnosis of pneumonia on admission |
|
| Interventions |
Antacids (Maalox Therapeutic Concentration)
Sucralfate
Adherence to regimen: Six participants from the antacid group (n = 42) and 3 from the sucralfate group (n = 41) were excluded from analysis for the following reasons: < 12 hours in the study (3 died, 2 enteral feeds, 1 severe diarrhoea, with treatment discontinued), received both interventions, had evidence of GI bleed before intervention, refused participation Duration of trial: January 1984 to September 1985 |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
|
|
| Notes |
Setting: surgical, medical, or burn intensive care units at San Francisco General Hospital, USA Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Quote: "The protocol was approved by the University of California, San Fransisco, Hum and Environmental protection committees (approval number 251701‐02)" Informed consent: "obtained from all participants. If the participant was unable to provide informed consent, then the participant's next of kin or legally authorized representative provided consent. If they could not be contacted, then the participant's attending physician was asked to provide permission" Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: One participant in each group had significant upper GI bleeding after the trial was completed |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: no information on sequence generation reported |
| Allocation concealment (selection bias) | Unclear risk | Comment: no information on allocation concealment reported |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: Participants and personnel involved in the trial were not blinded |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: GI bleeding was an objective outcome that was diagnosed as per the definition used in the trial protocol |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Trial did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Unclear risk | Comment: unclear whether outcome assessors were blinded. Moreover, criteria for diagnosis of other outcomes of interest not clearly reported |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: Not all randomised participants were included in the final analysis (83 were randomised, 74 were included in the analysis). A per‐protocol analysis was done, as 9 participants were involved in the trial for less than 12 hours. There was no imbalance between groups. Therefore, low risk of bias is due to attrition |
| Selective reporting (reporting bias) | Unclear risk | Comment: not enough information reported on outcomes of relevance in the trial |
| Other bias | High risk | Comment: Source of funding is not clearly mentioned in the trial report. No other sources of bias are suspected, but insufficient information is reported in the trial abstract |
Brophy 2010.
| Methods | Quasi‐randomised trial | |
| Participants |
Baseline characteristics Number randomised: 51 participants Number analysed: 51 participants Famotidine
Lansoprazole
Inclusion criteria
Exclusion criteria
Baseline imbalances: "There were significantly more males than females in the study. Over 75% of the patients had a Glasgow Coma Scale (GCS) < 9, and median GCS scores were similar between the two groups. All of the patients had at least two risk factors for SRMD, and each treatment group had a similar number of patients with traumatic brain injuries. The median baseline gastric pH was 3.0 for both famotidine and lansoprazole groups" Comment: There were more women in the lansoprazole group than in the famotidine group |
|
| Interventions |
Famotidine
Lansoprazole
Adherence to regimen: ‐ Duration of trial: August 1999 to April 2005 Duration of follow‐up: “Patients were followed until 24 hours after the discontinuation of SUP, the patient was discharged from the ICU, or if the patient expired, whichever came first" |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review • Duration of pH ≥ 4.0 • Percentage of time gastric residual was < 28 mL |
|
| Notes |
Setting: The Virginia Commonwealth University (VCU), 1000‐bed, academic, level 1 trauma centre Source of funding: Quote: "This study was funded by TAP Pharmaceuticals" Conflicts of interest: ‐ Ethics approval: Quote: "The Virginia Commonwealth University (VCU) Institutional Review Board approved this study prior to subject enrolment, and this study was conducted in compliance with the Declaration of Helsinki" Informed consent: Quote: "All subjects provided written informed consent prior to study commencement" Clinical trials registration: ‐ Sample size calculation: Quote: "...we assumed that on day 3 of therapy, 85% of the patients receiving lansoprazole would have pH values ≥ 4.0 for 80% of the time compared to only 40% of the patients receiving famotidine. Using these proportions, α = 0.05, β = 0.20, and a two‐way statistical test, approximately 22 patients were needed in each group to show statistical significance" Comment: This was after 30 people admitted to the neurosurgical unit were followed; it was assumed that approximately 40% of them receiving famotidine and 80% receiving lansoprazole maintained gastric pH ≥ 4 80% of the time |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Quote: "Patients admitted into the unit during odd‐numbered months received famotidine 20 mg IV every 12 hours; patients admitted during even numbered months received lansoprazole" Comment: This was a quasi‐randomised trial in which sequence generation was not done |
| Allocation concealment (selection bias) | High risk | Quote: "Patients admitted into the unit during odd‐numbered months received famotidine 20 mg IV every 12 hours; patients admitted during even numbered months received lansoprazole" Comment: This was a quasi‐randomised trial in which allocation was not concealed |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: This was not a placebo‐controlled trial; the different interventions and their mode of administration could not have made it possible to blind trial personnel |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: This was an unblinded trial in which GI bleeding was detected as per the definition used in the trial protocol |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Trial did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: This was an unblinded trial; outcomes of interest were diagnosed as described in the trial protocol |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All randomised participants were part of the final analysis. Therefore, no attrition bias is suspected |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: TAP Pharmaceuticals funded the trial. The role of the sponsor in the conduct and reporting of the trial is unclear. No other sources of bias are suspected |
Burgess 1995.
| Methods | Double‐blind parallel‐group randomised placebo‐controlled trial | |
| Participants |
Baseline characteristics Number randomised: 34 participants Number analysed: 34 participants Ranitidine
Placebo
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "All 34 patients were comatose on admission and required ventilatory support"; "No significant differences in demographic characteristics were present between the two treatment groups" Comment: The 2 groups were comparable with respect to mean Glasgow Coma Scale score (mean 8, range 4 to 10; and mean 6.7 range 3 to 10), mean Injury Severity Score (mean 32, range 25 to 41; and mean 30, range 25 to 57), and time from injury to study drug administration |
|
| Interventions |
Ranitidine
Placebo
Adherence to regimen: Quote: "All 34 patients were comatose on admission and required ventilatory support. Ten patients were withdrawn before completing 72‐hour study period. Five of these patients were in the placebo treatment group and were withdrawn from the trial because of protocol‐defined upper gastrointestinal tract bleeding. Of the remaining four patients,who were from the ranitidine group; one was withdrawn due to death secondary to severe head injury, two were withdrawn because they became combative and removed their NG tubes and pH probes, and the final patient was withdrawn from the trial when steroids were prescribed for an optic nerve injury by the attending physicians. One patient from the placebo group was removed due to withdrawal of NG tube" Comment: 24 participants completed the prescribed 72 hours of the trial, and reasons for the remaining 10 not completing the 72‐hour period are well documented Duration of trial: February 1988 to November 1988 Duration of follow‐up: 48 hours after study withdrawal |
|
| Outcomes |
Outcomes sought in review and reported in trial
Note: The 5 participants who bled belonged to the placebo group; bleeding occurred before 72 hours into the trial
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
|
|
| Notes |
Setting: Department of Surgery, University of Louisville, School of Medicine, Louisville, Kentucky, USA Source of funding: Quote: "The study was supported by a grant from Glaxo Inc. Research Institute" Conflicts of interest: ‐ Ethics approval: Quote: "The study was approved by the Human Studies Committee" Informed consent: Quote: "...informed consent was obtained from each patient's legal representative" Clinical trials registration: ‐ Sample size calculation: ‐ |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Within 24 hr of injury, each patient was randomly assigned to receive either 6.25 mg/hr continuous intravenous ranitidine infusion or a saline placebo infusion in accordance with a computer‐generated randomisation scheme" Comment: Method adopted to obtain random sequence generation is clearly mentioned in the trial report |
| Allocation concealment (selection bias) | Unclear risk | Comment: No information on allocation concealment reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "To maintain the integrity of the study blinding and to avoid potential bias, the principal investigator did not have access to the pH data" Comment: This is a double‐blind, placebo‐controlled, parallel‐group study design in which study personnel were blinded |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Quote: "To maintain the integrity of the study blinding and to avoid potential bias, the principal investigator did not have access to the pH data" Comment: The trial did investigate the relationship between intragastric pH values and the incidence of bleeding. Moreover, we are unclear whether it was the principal investigator who was also involved in outcome assessment. However, GI bleed was detected as per the study definition, and owing to the objective nature of the outcome, the likelihood of performance and detection bias is low |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Trial did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Unclear risk | Comment: Blinding for other outcome assessments is unclear. Criteria for diagnosis of other outcomes are not fully described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "All patients completed at least 8 hours of investigational therapy and were included in the analysis" Comment: All randomised participants were part of the final analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and included in the report |
| Other bias | Low risk | Comment: Trial was supported by a grant from Glaxo Inc. Research Institute; trial authors (number not sure) had affiliation with this company. No other sources of bias are suspected |
Cannon 1987.
| Methods | Open‐label randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 62 participants Number analysed: 59 participants Cimetidine
Antacids
Sucralfate
Inclusion criteria
Exclusion criteria
Baseline imbalances: The 3 groups were almost similar with respect to age, gender, number of participants, and risk factors thought to precipitate GI bleed. Most participants were medical participants with respiratory failure secondary to an intrathoracic process. There was no significant difference between groups with respect to major risk factors for GI bleeding (sepsis, peritonitis, jaundice, hypotension, and trauma). Only acute renal failure was more common with the antacid regimen than with cimetidine and sucralfate (however, the incidence of renal failure did not correlate with upper GI bleed) |
|
| Interventions |
Cimetidine
Antacids
Sucralfate
Adherence to regimen: sucralfate, H2 receptor antagonists, or antacids were used inadvertently in 3 patients who were excluded from the trial Duration of trial: October 1985 to January 1986 Duration of follow‐up: 24 hours after extubation |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial report
Outcomes reported in report but not sought in review
|
|
| Notes |
Setting: Medical‐Surgical Intensive Care Unit at Akron General Medical Centre, Ohio, USA Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Quote: "The study was approved by the institutional review board at the Akron General Medical Centre" Informed consent: Quote: "A signed consent was obtained from patients or the next of kin after the potential complications and nature of the procedure were explained" Clinical trials registration: ‐ Sample size calculation: ‐ |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Sixty‐two patients were accepted for computerised randomised study between October 1985 and January 1986" Comment: Method adopted to obtain random sequence generation is clearly mentioned in the trial report |
| Allocation concealment (selection bias) | Unclear risk | Comment: no information on allocation concealment reported |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: This was not a placebo‐controlled trial, and mode of administration of drugs could not have allowed blinding of participants and personnel involved in the trial |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: unclear if outcome assessors were blinded. However, GI bleeding was an objective outcome, which was detected as per the definition in the trial protocol |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: The trial did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Unclear risk | Comment: not enough information on criteria for diagnosis of other outcomes described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "Of the 62 patients originally entered into the study three were excluded because of the inadvertent concomitant use of sucralfate, H2 receptor antagonists or antacids in the study group" Comment: Groups to which these 3 participants belonged are unclear (ITT cannot be performed). These participants were excluded from the final analysis. Because loss to follow‐up was < 10% and appeared to be balanced across groups, this would not have introduced any attrition bias into the trial |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: Source of funding is not clearly mentioned in the trial report. No other form of bias is detected |
Chan 1995.
| Methods | Parallel‐group randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: ‐ Number analysed: 101 participants Ranitidine
Placebo
Inclusion criteria
Exclusion criteria
Baseline imbalances: The nature and location of diseases, types of operations, number of preoperative risk factors, and demographic data were comparable in the 2 groups |
|
| Interventions |
Ranitidine
Placebo
Adherence to regimen: ‐ Duration of trial: July 1988 to December 1989 Duration of follow‐up: ‐ |
|
| Outcomes |
Outcomes reported in trial and used in review
Outcomes sought but not reported in trial
Outcomes reported, but not used in review
|
|
| Notes |
Setting: Neurological Intensive Care Unit, Department of Surgery, Queen Mary Hospital, University of Hong Kong Source of funding: University of Hong Kong Research Grant and Lee Wing Tat Research Grant Conflicts of interest: ‐ Ethics approval: The protocol used in our trial was approved by the Ethics Committee of the Faculty of Medicine, at the University of Hong Kong Informed consent: Written consent was obtained from patients or their next of kin Clinical trials registration: ‐ Sample calculation: 49 patients would be required in each arm of the trial with a power of 0.8 and a 0.95 significance level by 2‐tailed test Comments: The endpoint of the trial was the development of symptomatic GD lesions defined as GD bleeding requiring blood transfusions and/or surgery for acute perforated ulcers |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomised in a standard double blind manner" Comment: not enough information on sequence generation reported |
| Allocation concealment (selection bias) | Unclear risk | Comment: no information on allocation concealment reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "All patients were randomised in a standard double‐blind manner to receive either ranitidine (50 mg) or placebo medication (normal saline) identical in appearance and volume" |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Quote: "Final outcomes were also assessed by an independent observer to ascertain whether they were a direct result of the GD lesions" "Endoscopic examination of the GD tract up to the second part of the duodenum was performed in all patients within 12 hours of surgery. Additional bolus doses of sedative and analgesic medications we re given during endoscopy. A nasogastric tube was passed into the stomach after endoscopy; its position was confirmed by radiological means, and it was connected to a bag for free drainage. Aspiration from the tube was performed at 6‐hour intervals and a pH paper was used to measure the pH of the gastric content. The total volume of daily gastric output was recorded" |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Trial did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Unclear risk | Comment: no details on criteria for diagnosis of other outcomes reported |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: no missing outcome data. All participants randomised at baseline are also included in analyses of outcomes |
| Selective reporting (reporting bias) | High risk | Comment: Outcome data for blood transfusions were not reported by treatment group, but as totals. Other outcomes were reported completely. More outcomes were reported in the Results section than in the Methods section |
| Other bias | Low risk | Comment: no other sources of bias suspected |
Cioffi 1994.
| Methods | Single‐blind randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 100 participants Number analysed: 100 participants Antacids + cimetidine
Sucralfate
Incusion criteria
Exclusion criteria
Baseline imbalance: Quote: "There were no significant differences in age, burn size, the presence of inhalation injury and requirements for intubation. The groups were compared using severity index based upon age, burn size and the presence of inhalation injury. The severity findings were not different between the groups, indicating the similarity of the participant cohorts" Comment: The 2 groups were comparable at baseline. Participants were admitted for thermal injury (burns). Inhalation injury was found in 22 and 27 participants from the cimetidine + antacid and sucralfate groups. Intubation was required in 29 and 28 participants, respectively |
|
| Interventions |
Antacids + cimetidine
Sucralfate
Adherence to regimen: 100 participants were randomised to both groups ‐ 50 each. There were 4 protocol violations (2 in each group), leaving 96 participants for evaluation. Continuous enteral feeding began from postburn day 3 onwards for participants who were not able to meet the requirements orally. The duration of intubation was longer in the sucralfate group owing to higher incidence of pneumonia. There were 9 deaths before postburn day 5 ‐ 5 in the acid neutralising group and 4 in the sucralfate group. None of these participants had pneumonia. There were 5 deaths among participants who developed pneumonia after postburn day 5 Duration of trial: March 1990 to December 1992 Duration of follow‐up: probably until death or discharge |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
|
|
| Notes |
Setting: US Army Institute of Surgical Research, Fort Sam Huston, Texas Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: Quote: "Consenting participants were randomised in a pair wise fashion" Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: There was a higher incidence of pneumonia among participants who were intubated or had inhalation injury and were administered sucralfate. Mixed pneumonia (gram‐positive and gram‐negative pathogens) was most frequent in both groups. The percentage of participants in each group who developed positive sputum cultures was 100 and 98, and for gastric cultures 96 and 83, respectively (for any bacteria) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: no information on sequence generation reported |
| Allocation concealment (selection bias) | Unclear risk | Comment: no information on allocation concealment reported |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: This was not a placebo‐controlled trial, and the mode of administration of interventions could not have permitted blinding of participants |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: Outcome assessors were not blinded. Only the presence of gross bleeding was considered in the results |
| Blinding (detection bias) Nosocomial pneumonia | Low risk | Quote: "Chest roentgenograms were reviewed by staff surgeon and radiologist who were unaware of the participants' treatment group" Comment: Outcome assessors for this particular outcome were blinded |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Unclear risk | Comment: Outcome assessors were not blinded. Criteria for diagnosis of other outcomes were not clearly reported |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: Not all participants who were randomised were part of the final analysis. Two were excluded from each arm owing to protocol violations. Because the loss to follow‐up of less than 10% is balanced between groups, this would not have introduced any attrition bias into the trial. Results were analysed in the review on the basis of ITT |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed |
| Other bias | Low risk | Comment: Source of funding is unclear. No other source of bias is suspected |
Conrad 2005.
| Methods | Double‐blind randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 359 participants Number analysed: 359 participants Omeprazole
Cimetidine
Note: 64 participants in both groups were over 65 years of age Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "The two groups were generally well matched, although the mean APACHE II score (indicating severity of critical illness) was higher in the omeprazole‐suspension group" Comment: Coagulopathy was present in 37 and 26 participants, respectively, and acute renal failure was more common in the omeprazole group (47 and 33). The 2 groups were similar to each other with respect to age, gender, baseline gastric pH, and other risk factors known to precipitate GI bleeding in the ICU |
|
| Interventions |
Omeprazole
Cimetidine
Duration of trial: June 2002 to May 2003 Duration of follow‐up: probably until death or discharge Adherence to regimen: Quote: "A total of 56 patients in the intention‐to‐treat population were excluded from the per‐protocol population (omeprazole, n = 21; cimetidine, n = 35) primarily due to failure to receive dose increases within 12 hours of observing the first of two pH values of 4 (omeprazole, n = 4; cimetidine, n = 25)" "Participation in the trial was discontinued before 14 days in the event of death, discharge from the unit, or extubation (endotracheal or gastric tube)" " ...four patients who were discontinued from the trial while actively bleeding (omeprazole, n = 1; cimetidine, n = 3)" |
|
| Outcomes |
Outcomes sought in review and reported in trial
Note: Five participants bled during the first 2 days of acid suppression therapy (omeprazole; n = 1; cimetidine; n = 4)
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
|
|
| Notes |
Setting: 47 ICUs in the United States Source of funding: supported, in part, by Santarus, San Diego, CA Conflicts of interest: ‐ Ethics approval: Quote: "This trial was approved by the institutional review board at each site" Informed consent: Quote: "Before any trial procedures were performed, each patient or his or her authorized legal representative gave written informed consent for trial participation" Clinical trials registration: ‐ Sample size calculation: Quote: "Sample size calculations were based on an assumption of a 12% rate of upper GI bleeding in cimetidine‐treated patients and a 6% rate in omeprazole suspension–treated patients. To establish the non‐inferiority of omeprazole suspension with 90% power at the one‐sided α = 0.25 level, 142 patients were required in each treatment group. Assuming that 20% of randomised patients would not satisfy per‐protocol requirements, enrolment of 178 patients per group was planned" Additional notes: An additional 75 participants had overt upper GI bleeding but did not meet the primary endpoint of clinically significant upper GI bleeding as defined by the trial protocol. Moreover all participants who met the endpoint criteria of clinically significant upper GI bleeding were in the per‐protocol population. Four of the 17 participants who had clinically significant bleeding died (omeprazole: n = 2; cimetidine: n = 2), but bleeding was not the cause of death for any of these participants |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the trial report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the trial report |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Patients, trial site personnel, and the sponsor were blinded to patient treatment assignment" "Within each site, patients were randomly assigned to receive immediate‐release omeprazole oral suspension (Zegerid, Santarus, San Diego, CA) and continuous intravenous infusion of placebo or continuous intravenous cimetidine and placebo oral suspension for up to 14 days" Comment: The presence of placebo in both arms might have ensured that participants and personnel were blinded |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Quote: "Patients, trial site personnel, and the sponsor were blinded to patient treatment assignment" Comment: The presence of placebo in both arms ensured that participants and personnel and outcome assessors were blinded. Moreover, GI bleeding was an objective outcome that was detected as per the trial protocol, so the likelihood of performance and detection bias is low |
| Blinding (detection bias) Nosocomial pneumonia | Low risk | Quote: "Patients, trial site personnel, and the sponsor were blinded to patient treatment assignment" Comment: The presence of placebo in both arms ensured that participants and personnel and outcome assessors were blinded. Moreover, nosocomial pneumonia was an objective outcome that was detected as per the trial protocol, so the likelihood of performance and detection bias is low |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Quote: "Patients, trial site personnel, and the sponsor were blinded to patient treatment assignment" Comment: The presence of placebo in both arms ensured that participants and personnel were blinded. Moreover, other outcomes of interest were objective in nature and were diagnosed according to the trial protocol, so the likelihood of performance and detection bias was low |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "The intent‐to‐treat population included all randomised patients. The per‐protocol population was used for the analysis of the primary efficacy end point; the intent‐to‐treat population was also used for the primary end point and for all other analyses" Comment: ITT analysis was performed for all outcomes of interest, so the likelihood of attrition bias was low |
| Selective reporting (reporting bias) | Low risk | Comment: All outcomes intended in the trial were analysed and reported. However, there is no mention of the presence or absence of any adverse events |
| Other bias | Unclear risk | Comment: One of the trial interventions, omeprazole oral suspension, was sponsored by Zegerid, Santarus, San Diego, CA, which is also the sponsor of the trial. Analysis of data was performed by Santarus, the sponsor of the trial. Also the analysis used was a non‐inferiority analysis, to compare rates of upper GI bleeding in the 2 treatment groups |
Cook 1998.
| Methods | Multi‐centre blinded randomised placebo‐controlled trial | |
| Participants |
Baseline characteristics Number randomised: 1200 participants Number analysed: 1200 participants Ranitidine
Sucralfate
Inclusion criteria
Exclusion criteria
Baseline imbalance: Quote: "Demographic and baseline physiologic characteristics were similar in the two groups" Comment: Mean and SD for APACHE II scores were 24.7 +/‐ 7.1 and 24.6 +/‐ 7.3 in the ranitidine and sucralfate groups. The main reasons for admission were medical: elective surgery or emergency surgery |
|
| Interventions |
Ranitidine
Sucralfate
Adherence to regimen: Quote: "No patient received active drug instead of the assigned placebo, or vice versa. Of the scheduled doses of ranitidine and sucralfate, 94.2% and 91.7%, respectively, were administered. Among patients who missed doses, the mean number of doses missed was 2.3 (median, 3; interquartile range, 2 to 3) for ranitidine and 2.9 (median, 4; interquartile range, 1 to 4) for sucralfate" Duration of trial: October 1992 to May 1996 Duration of follow‐up: probably until death or discharge |
|
| Outcomes |
Outcomes sought in review and reported in trial:
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
|
|
| Notes |
Setting: 16 ICUs: McMaster University, Hamilton, The University of Toronto, Toronto, The University of Western Ontario, London, Dalhousie University, Halifax, Memorial University, St. John’s, Newf , Queen’s University, Kingston. University of British Columbia, University of Ottawa, Ottawa, University of Alberta, Edmonton Vancouver General Hospital, Vancouver, Royal Alexandra Hospital, Edmonton, Grey Nun’s Hospital, Edmonton, Winnipeg Health Sciences Center, Winnipeg, Man., Toronto Hospital, General Division, Toronto; Wellesley Hospital, Toronto London Health Sciences Center (Victoria Campus), London, Ont., London Health Sciences Center (University Campus), London, Ont., St. Joseph’s Health Center, London, Ont., St. Joseph’s Hospital, Hamilton, Ont., Henderson Hospital, Hamilton, Ont., Kingston General Hospital, Kingston,Ont., Ottawa Civic Hospital, Ottawa, Ont., Health Sciences Center, St. John’s, Newf, Victoria General Hospital, Halifax, N.S. Source of funding: Quote: "Supported by the Medical Research Council of Canada and Hoechst Marion Roussel. Drugs were supplied by Glaxo Wellcome, Baxter, and Hoechst Marion Roussel. Dr. Cook is a Career Scientist of the Ontario Ministry of Health" Conflicts of interest: ‐ Ethics approval: Quote: "The protocol was approved by the institutional review board of all participating enters ..." Informed consent: Quote: "...and the patients or their proxies gave informed consent" Clinical trials registration: ‐ Sample size calculation: Quote: "On the basis of data published through 1991, when our study was designed, we anticipated a 25 percent incidence of pneumonia and identified a 25 percent reduction in the risk of pneumonia associated with sucralfate as being plausible and clinically important. This led to the calculation of a sample size of 1200 patients as necessary to give the study 75 percent power to detect such a difference, assuming a two‐sided significance test at the 0.05 level. We analysed all patients in the groups to which they were randomly assigned, according to the intention‐to‐treat principle" Additional notes: Gram‐negative bacilli and gram‐positive cocci were the main isolates from endotracheal aspirates from patients with ventilator‐associated pneumonia |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned to study groups in blocks of six, with stratification according to centre, by means of a computer generated random‐number table prepared at the McMaster University Methods Center and managed by the ICU study pharmacist at each site" Comment: Method adopted to obtain random sequence generation is clearly mentioned in the trial report |
| Allocation concealment (selection bias) | Low risk | Quote: "...managed by the ICU study pharmacist at each site who administered the coded drugs" Comment: Method to obtain allocation concealment is clearly mentioned in the trial report |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The patients, research nurses, and all ICU care givers were unaware of the treatment assignments for the duration of the study. Therefore, clinicians did not monitor gastric pH" "The radiologists, outcome adjudicators, all investigators, and the study statistician were also blinded until all events had been adjudicated and the analyses completed" Comment: Blinding was done, so the likelihood of performance bias and detection bias is low |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Quote: "The patients, research nurses, and all ICU care givers were unaware of the treatment assignments for the duration of the study. Therefore, clinicians did not monitor gastric pH" "The radiologists, outcome adjudicators, all investigators, and the study statistician were also blinded until all events had been adjudicated and the analyses completed" Comment: Blinding was done. Moreover the outcome of interest was objective in nature, so the likelihood of performance bias and detection bias is low |
| Blinding (detection bias) Nosocomial pneumonia | Low risk | Quote: "The patients, research nurses, and all ICU care givers were unaware of the treatment assignments for the duration of the study. Therefore, clinicians did not monitor gastric pH" "The radiologists, outcome adjudicators, all investigators, and the study statistician were also blinded until all events had been adjudicated and the analyses completed" Comment: Blinding was done. Moreover the outcome of interest was objective in nature, so the likelihood of performance bias and detection bias is low |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Quote: "The patients, research nurses, and all ICU care givers were unaware of the treatment assignments for the duration of the study..." Comment: Blinding was done. Moreover all other outcomes of interest were objective in nature |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All participants who were randomised to the 2 groups were analysed |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were reported |
| Other bias | Low risk | Comment: Hoechst Marion Roussel supported the trial and also sponsored sucralfate needed for administering to study participants. The role of the sponsor in the conduct and reporting of the trial is unclear. No other sources of bias are suspected |
Darlong 2003.
| Methods | Open‐label randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 52 participants Number analysed: 52 participants Ranitidine
Sucralfate
No prophylaxis
Inclusion criteria
Exclusion criteria
Baseline imbalances: There were no major differences in demographic profiles between study groups Comment: Participants were from medical and surgical units, in equal proportions across the 3 groups. However, the underlying reason for admission is not mentioned in the trial report |
|
| Interventions |
Ranitidine
Sucralfate
No prophylaxis
Adherence to regimen: ‐ Duration of trial: ‐ Duration of follow‐up: probably until death or discharge |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
|
|
| Notes |
Setting: All India Institute of Medical Sciences (AIIMS), New Delhi, India Source of funding: Quote: "Supported by the Medical Research Council of Canada and Hoechst Marion Roussel. Drugs were supplied by Glaxo Wellcome, Baxter, and Hoechst Marion Roussel. Dr. Cook is a Career Scientist of the Ontario Ministry of Health" Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additional Notes: Gram‐negative organisms were found in the gastric culture of 18 ranitidine participants and 6 sucralfate participants, and the control group had no pathogenic organisms. Pseudomonas was the most common organism in the gastric and BAL cultures of the ranitidine and sucralfate groups |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the trial report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the trial report |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: This was not a placebo‐controlled trial, and the mode of administration of interventions could not have permitted blinding of participants. Therefore, high risk of performance bias |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: not clearly mentioned in the trial report. However owing to the objective nature of the outcome, which was detected as per the trial protocol, the likelihood of performance and detection bias is low |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Trial did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: No other outcomes of interest were assessed in this trial |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All randomised participants were part of the analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | High risk | Comment: unclear on source of funding. Baseline imbalance on the numbers randomised to the 3 groups as the no prophylaxis arm had only 7 participants compared with 21 in the sucralfate arm and 24 in the ranitidine arm |
Davies 2012.
| Methods | Parallel‐group randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 182 participants Number analysed: 181 participants Nasojejunal nutrition
Nasogastric nutrition
Inclusion criteria
Exclusion criteria
Baseline imbalances: Baseline characteristics of the groups were similar |
|
| Interventions |
Nasojejunal nutrition
Nasogastric nutrition
Adherence to treatment: ‐ Duration of trial: ‐ Duration of follow‐up: 28 days |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
|
|
| Notes |
Setting: Medical‐Surgical ICUs, Alfred Hospital, Melbourne, Australia Sponsorship source: Cook Critical Care provided the frictional nasojejunal tubes Conflicts of interest: no conflicts of interest disclosed Comments: Other recommended treatment includes ranitidine Ethics approval: All sites received institutional human research ethics committee approval Informed consent: Prior written informed consent was obtained from the patient’s surrogate decision‐maker Clinical trials registration: ‐ Sample size calculation: 180‐patient sample size provided 80% power with 2‐sided significance of 0.05 to detect a 12% difference in mean energy delivery |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "They were randomly assigned to early nasojejunal nutrition or naso‐gastric nutrition using a computer‐generated randomisation schedule with variable block size" |
| Allocation concealment (selection bias) | Low risk | Quote: "Treatment assignments were concealed before randomisation using a centralized voice‐recognition phone system" |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "There was no blinding after randomisation" Comment: high for personnel; indifferent for patients |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Quote: "gastrointestinal haemorrhage ([was measured as] defined as major or minor based on definitions from a previous trial)" Comment: detailed description provided in supplementary material to the trial. Outcome measurement unlikely to introduce bias to the trial |
| Blinding (detection bias) Nosocomial pneumonia | Low risk | Quote: "chest radiograph images" Comment: Outcome measurement was objective and therefore was unlikely to introduce bias |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Quote: "The collected information was provided to a blinded adjudication panel of two intensivists and one respiratory physician for each patient. To maintain blinding, the radiograph images had the enteral tube shadows in the mediastinum and upper abdomen obscured. Panel members individually adjudicated the diagnosis of VAP using objective criteria (Supplemental Digital Appendix A, Supplemental Digital Content 1, http://links.lww. com/CCM/A454). Patients were formally categorized as meeting the VAP outcome if at least two of the three adjudicators determined the pres‐ ence of VAP and this occurred after the patient had been mechanically ventilated for >72 hrs. A Data Monitoring Committee regularly viewed data on safety aspects with blinding maintained and did not recommend study cessation" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "Data were analyzed according to the intention‐to‐treat principle with no imputation of missing values" Comment: No incomplete outcome data were suspected. All participants enrolled at baseline were treated and analysed |
| Selective reporting (reporting bias) | Low risk | Comment: All prespecified outcomes listed in the Methods section were reported in the Results section. No selective outcome reporting was suspected |
| Other bias | Low risk | Comment: No other sources of bias were suspected |
De Azevedo 2000.
| Methods | Randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: ‐ Number analysed: ‐ Ranitidine
Sucralfate
Omeprazole
Inclusion criteria
Exclusion criteria
Baseline imbalances: Groups were similar to each other with respect to demographic characteristics. APACHE ll scores were 55.7 ± 23.2, 54.1 ± 21.7, and 56.1 ± 23.7 in the ranitidine, sucralfate, and omeprazole groups, respectively. Pneumonia was present in 2, 3, and 3 participants in the 3 groups |
|
| Interventions |
Ranitidine
Sucralfate
Omeprazole
Adherence to regimen: ‐ Duration of trial: February 1997 to April 1998 Duration of treatment: Duration of treatment that was established was based on clinical evaluation, control of risk factors, clinical improvement, and full nutritional support, oral or enteral Duration of follow‐up: during the period that participants were in the ICU. Probably until death or discharge |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
|
|
| Notes |
Setting: Intensive Care Unit, Hospital São Domingos, Sao Luis do Maranhão, Brasil Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "The patients were randomly assigned for one of the three groups when they were admitted for the intensive unit care, they used a simple technique of randomisation, and sealed envelopes" Comment: not enough information on sequence generation reported |
| Allocation concealment (selection bias) | Unclear risk | Comment: Trial authors described that they used sealed envelopes. It was not stated whether they were numbered and opaque |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: not enough information reported |
| Blinding (detection bias) Clinically important upper GI bleeding | Unclear risk | Comment: not enough information reported on criteria for assessment of upper GI bleeding |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: not enough information reported on criteria for assessment of pneumonia |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: unclear; however all other outcomes of interest were objective outcomes. Therefore the likelihood of performance and detection bias is low |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All randomised participants were analysed |
| Selective reporting (reporting bias) | Low risk | Comment: All outcomes described in the Methods section were analysed in the Results section |
| Other bias | Low risk | Comment: no other source of bias detected |
Driks 1987.
| Methods | Parallel‐group randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 130 participants Number analysed: 130 participants Sucralfate
Antacid and/or H2 receptor antagonists
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "The two treatment groups were similar in terms of demographic characteristics and severity of illness on admission to the study" "The distribution of underlying diseases, indications for intubation and surgical procedures according to the site were similar in both the groups" "Thirty two of the 61 patients in the sucralfate group and twenty nine of the 69 participants in the antacid H2 group had infiltrates at baseline evaluation (n = 38), adult respiratory distress syndrome (n = 8), or congestive heart failure (n = 15), making it difficult to diagnose new infiltrates on a chest film" Comment: similar distribution with respect to demographic and baseline risk factors. However, a total of 35 participants (sucralfate: n = 16; antacid and/or H2: 19) had pneumonia on admission and/or had infiltrates (as mentioned above) |
|
| Interventions |
Sucralfate
Antacid and/or H2 receptor antagonists
Adherence to regimen: Quote: "Six patients who were initially assigned to sucralfate but subsequently assigned to antacids or H2 blockers by the treating physician were included in the sucralfate group (for analysis), no patients were switched from antacids or H2 blockers to sucralfate. Four of the six patients who were crossed over had evidence of upper GI tract bleeding, the other two who were switched to antacids 5 and 11 days after randomisation had no evidence of such bleeding. Two of the six patients had pneumonia after the crossover occurred but were included in the sucralfate group" "Four patients in the sucralfate group and 17 in the antacid‐H2 group underwent tracheostomy" Comment: ITT was performed for the review and not per‐protocol analysis Duration of trial: April 1986 to February 1987 Duration of follow‐up: until 48 hours after extubation |
|
| Outcomes |
Outcomes sought in review and reported in trial
Note: Pneumonia developed after 9.6 (4.6) days in sucralfate group and after 9.6 (6.3) days in antacid H2 group
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
|
|
| Notes |
Setting: surgical, medical, or coronary intensive care units, Boston City Hospital Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Quote: "The study protocol was reviewed and approved by the hospital institutional review board for human studies" Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Quantitative data were extracted for the 2 randomised groups alone (sucralfate and antacid ‐ H2 receptor groups). Data were not extracted separately for participants treated with antacid or H2 receptor alone, as this would break the randomisation. Of bacteria isolated from the tracheal aspirates of participants treated with pneumonia, gram‐negative bacilli were predominant; Pseudomonas aeruginosa in the sucralfate group; Enterobacteriaceae in antacid‐H2 group |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the trial report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the trial report |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: This was not a placebo‐controlled trial, and the mode of administration of the interventions would not have permitted blinding of participants |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: unclear whether outcome assessors were blinded, but GI bleeding was an objective outcome that was detected as per the definition in the trial protocol |
| Blinding (detection bias) Nosocomial pneumonia | Low risk | Quote: "Chest roentgenograms were interpreted by at least one of us, who had no knowledge on the patient's treatment group after randomisation" Comment: Blinding was done. Moreover, pneumonia was an objective outcome that was detected as per the definition in the trial protocol |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: unclear whether outcome assessors were blinded. Moreover the outcomes of interest were objective in nature |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: Intention‐to‐treat analysis was performed wherein all participants who were initially randomised to each of the 2 study groups were part of the final analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes have been reported |
| Other bias | Low risk | Comment: Source of funding is not clearly mentioned in the trial report. No other form of bias was detected |
Eddleston 1991.
| Methods | Parallel‐group randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 60 participants Number analysed: 60 participants Sucralfate
Ranitidine
Inclusion criteria
Exclusion criteria
Baseline imbalances: Participants in the ranitidine group were relatively older. The acute physiology and chronic health evaluation (APCHE II) score was 11.6 (1.3) for sucralfate and 12.4 (1.5) for ranitidine Admission diagnosis: More participants in the sucralfate group had both CNS injury (11/30) and trauma (8/30), and CNS injury and respiratory failure were the most common diagnoses on admission among participants in the ranitidine group (8/30) and (7/30) |
|
| Interventions |
Sucralfate
Ranitidine
Adherence to regimen: All participants were subjected to treatment within 6 hours after ICU admission.Enteral nutrition was given (exact numbers and interventional groups not specified) Duration of trial: ‐ Duration of follow‐up: probably until death or discharge |
|
| Outcomes |
Outcomes sought in review and reported in trial
Note: GI bleeding in 2 participants (day 3 in the participant from the sucralfate group and day 5 in the participant from the ranitidine group) Outcomes sought but not reported in trial
Outcomes reported in trial but not sought in review
|
|
| Notes |
Setting: ICU, Manchester Royal Infirmary, Manchester, UK Source of funding: ‐ Ethics approval: Quote: "The protocol for the study was approved by the hospital ethics committee" Informed consent: Quote: "Informed consent was obtained from the patient's next of kin" Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Retrograde colonisation from stomach to oropharynx was found in 6 participants in the sucralfate group and 14 participants in group B. From the oropharynx to the trachea, colonisation occurred in 5 of the 6 participants in group A and in 12 of the 14 participants in group B. Colonisation occurred relatively later in sucralfate‐treated participants. More pneumonia cases were reported in group B, and the pathogens were mainly similar in both groups (mainly gram‐negative bacilli) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "The patients were allocated by the use of a random sampling table into two groups" Comments: Method adopted to obtain random sequence generation is clearly mentioned in the trial report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the trial report |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering the study interventions would not have made it possible to blind the study personnel and participants. Therefore, high risk of performance bias |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Quote: "Endoscopic evolution of the gastric and the duodenal mucosa was performed within 24 hours of admission by an operator who was blinded to the randomisation and was repeated on day 4 if the participant was still receiving mechanical ventilation" Comment: Endoscopy was performed if there was fresh blood (< 50 mL) in the nasogastric aspirate, but the endoscopist was blinded. Moreover, the outcome of interest was objective in nature |
| Blinding (detection bias) Nosocomial pneumonia | Low risk | Comment: no mention of blinding of radiologists, lab technicians, or physicians. However, pneumonia was diagnosed as per the study definition and due to the objective nature of the outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: not clear on blinding assessors for other outcomes of interest. However given the objective nature of the outcomes, the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All 60 randomised participants were included in the final analysis. Therefore there is low risk of attrition bias |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were reported. No mention of any adverse events |
| Other bias | Low risk | Comment: Source of funding is not clearly mentioned in the trial report. No other sources of bias are suspected |
Eddleston 1994.
| Methods | Parallel‐group randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 26 participants Number analysed: 26 participants Sucralfate
Placebo
Inclusion criteria
Exclusion criteria
Baseline imbalances: Participants in the placebo group were relatively older. Acute Pysiology and Chronic Health Evaluation II (APCHEII) scores were 12.5 (1.7) and 14.7 (2.2), respectively. Trauma was the most common cause for admission (6 in sucralfate group and 4 in the placebo group) |
|
| Interventions |
Sucralfate
Placebo
Adherence to regimen: Treatment began within 8 hours of ICU admission. Endoscopy performed on day 3 in the placebo group revealed acute ulcerations in 5 participants, as a result of which they were withdrawn and put on sucralfate 2 g every 8 hours. On day 6, 2 more patients from placebo group were switched over to sucralfate owing to acute gastric ulceration. Mucosal deterioration and acute ulceration were significant in the placebo group. The study was halted after recruitment of 26 participants for ethical reasons Duration of trial: ‐ Duration of follow‐up: ‐ |
|
| Outcomes |
Outcomes sought in review and reported in trial
Note: GI bleeding occurred in 1 participant from placebo group on 20th day of the trial Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
|
|
| Notes |
Setting: Department of Surgery, Critical Care Unit, Department of Microbiology, Manchester Royal Infirmary, Manchester, UK Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Quote: "The protocol for the study was approved by the hospital ethics committee" Informed consent: Quote: "...informed consent was obtained from the patient's next of kin" Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Oropharyngeal retrograde colonisation occurred in 1 participant in the sucralfate group and in 2 participants in the placebo group. Tracheal colonisation occurred in 2 participants from the sucralfate group and in 1 participant from the placebo group. However only the latter developed pneumonia. Mortality in ICU was attributed to multiple organ failure |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were allocated by the use of a random sampling table into two groups..." Comment: Method adopted to obtain random sequence generation is clearly mentioned in the trial report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the trial report |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: This was a placebo‐controlled trial, and the mode of administering the interventions was similar. However, unclear whether personnel and participants were blinded |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Quote: "Endoscopic evolution of the gastric and the duodenal mucosa was performed within 24 hours of admission by an operator who was blinded to the randomisation code and was repeated on day 3, 6 and 10 if the participant was still receiving mechanical ventilation" Comment: Interim gastroscopy was performed if there was fresh blood in the nasogastric aspirate, but the endoscopist was blinded. However, the outcome of interest was objective in nature |
| Blinding (detection bias) Nosocomial pneumonia | Low risk | Comment: no mention of blinding of radiologists, lab technicians, or physicians. However, pneumonia was diagnosed as per the study definition and due to the objective nature of the outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: no clear mention of blinding of outcome assessors. However owing to the objective nature of the outcomes, the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All participants randomised were included in the analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes are reported, although unclear on adverse events |
| Other bias | Low risk | Comment: Source of funding is not clearly mentioned in the trial report. No other sources of bias are suspected |
Ephgrave 1998.
| Methods | Double‐blind randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 140 participants Number analysed: 140 participants Antacids
Sucralfate
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "The patients who were randomly assigned to receive antacid or sucralfate stress ulcer prophylaxis were similar in most risk factors for postoperative complications but they differed in the percentage of patients with a history of treatment for chronic obstructive pulmonary disease (COPD). Significantly more patients with a clinical diagnosis of COPD preoperatively were randomly assigned to receive sucralfate (COPD was found to be an independent risk factor for development of pneumonia)" Comment: Baseline age and gender distributions are not mentioned; however the distribution of all risk factors except COPD is normal across the 2 groups. Most participants from both groups were admitted for general surgery of the abdomen (23 in antacid group and 27 in sucralfate group, respectively) |
|
| Interventions |
Antacid
Sucralfate
Adherence to regimen: Quote: "One hundred twenty‐two participants received 3 or more days of stress ulcer prophylaxis with the study drugs, while only 25 received 7 or more days.Many patients were treated with intravenous H2‐receptor blockers after 3 days postoperatively, which was when the study drugs were generally discontinued" Duration of trial: June 1990 to April 1992 Duration of follow‐up: Quote: "Daily assessment for gastrointestinal tract bleeding, infections, and other postoperative complications was continued until discharge from the hospital or death for all 140 patients" |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
|
|
| Notes |
Setting: The Surgical Service, Veterans Affairs Medical Center, Iowa City, Iowa Source of funding: Quote: "This study was funded through a Veterans Affairs Merit Review grant." Conflicts of interest: ‐ Ethics approval: Quote: "...approved for human subjects by the appropriate committees of the University of Iowa College of Medicine and the Iowa City Veterans Affairs Medical Center" Informed consent: Quote: "Patients were contacted before their elective surgical procedures, and less than 10% of the patients asked to participate refused to do so. Patients who declined entry into the study were not monitored further" Clinical trials registration: ‐ Sample size calculation: Quote: "A biostatistician calculated that an 80% likelihood of detection of a 50% improvement in the expected postoperative pneumonia rate of 15% to 20% would require entry of more than 300 patients into each arm of the study. With pilot work showing that more than 50% of patients in the surgical intensive care unit harboured pathogens in their gastric contents, we determined that approximately 70 patients in each arm would give an 80% likelihood of detecting an effect by the stress ulcer prophylactic agent on the gastric microbial flora" Comment: Sample size was calculated for the primary endpoint of pneumonia Additional notes: Study endpoints of pneumonia and GI bleeding were diagnosed postoperatively. Many participants received H2 antagonists 3 days postoperatively; study drugs were discontinued by then. Pulmonary colonisation with gastric micro‐organism occurred in 16% of antacid‐treated and 15% of sucralfate‐treated participants |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomization was performed in blocks of 20 patients before the study began, to ensure that both treatment groups would be evenly represented throughout the 2 years of patient accrual" Comment: Method adopted to obtain random sequence generation is clearly mentioned in the study report |
| Allocation concealment (selection bias) | Low risk | Quote: "In keeping with the double‐blind design, the treatment group assignment of each patient was known only to the pharmacy service until all the clinical and microbial data collection was complete" Comment: Allocation concealment was done |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The sham agents were designed to be similar in colour and viscosity to the active agents, and free of acid‐neutralizing properties" Comment: This was a double‐blind study in which participants and trial personnel appear to be blinded. Therefore the likelihood of performance bias is low |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Quote: "The sham agents were designed to be similar in colour and viscosity to the active agents, and free of acid‐neutralizing properties" Comment: This was a double‐blind study in which participants and trial personnel appear to be blinded. Moreover the outcome of GI bleed was detected as per the definition in the study protocol owing to the objective nature of the outcome |
| Blinding (detection bias) Nosocomial pneumonia | Low risk | Quote: "Pneumonia was diagnosed by consensus of 2 investigators not involved with the clinical Care”; "The sham agents were designed to be similar in colour and viscosity to the active agents, and free of acid‐neutralizing properties" Comment: Blinding of outcome assessors was done. Moreover the outcome of pneumonia was detected as per the definition in the study protocol owing to the objective nature of the outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Quote: "The sham agents were designed to be similar in colour and viscosity to the active agents, and free of acid‐neutralizing properties" Comment: Blinding of outcome assessors was done. Moreover because of the objective nature of the outcomes, the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All randomised participants were analysed for the outcomes of interest |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes have been reported |
| Other bias | Low risk | Comment: Funding was through a Veterans Affairs Merit Review grant. The role of the sponsor in the conduct and reporting of the trial is unclear. No other bias was detected |
Fabian 1993.
| Methods | Open‐label randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 616 participants Number analysed: 278 participants Sucralfate
Cimetidine bolus
Cimetidine continuous
Inclusion criteria
Exclusion criteria
Baseline imbalances: After exclusion, 99 participants received sucralfate, 114 received cimetidine (bolus), and 65 received cimetidine as continuous infusion. Blunt trauma was the most common cause for admission in 72% and penetrating trauma in 28% (86% of these were gunshot wounds; 14% were stab wounds). The APACHE ll scores were 14 (5) (mean, SD) for the sucralfate group, 14 (8) (mean, SD) for the cimetidine bolus group, and 13 (6) (mean, SD) for the cimetidine infusion group |
|
| Interventions |
Sucralfate
Cimetidine bolus
Cimetidine continuous
Adherence to regimen: 616 participants were initially randomised to 3 treatment groups. Data on 338 participants were excluded from analysis for the following reasons:
The remaining 278 participants were part of the study until discharge or death Duration of trial: January 1990 to April 1991 Duration of follow‐up: Quote: "Evaluable patients were studied until discharge or death" |
|
| Outcomes |
Outcomes sought in review and reported in trial
Note: Pneumonia developed 5.6 (1.6) (mean, SD) days after injury
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
|
|
| Notes |
Setting: Department of Surgery and Clinical Pharmacy, Presley Regional Trauma Centre, University of Tennessee, Memphis Source of funding: Quote: "Supported in part by educational grant from Smith Kline Beecham Inc" Conflicts of interest: Ethics approval: Quote: "The protocol was approved by the Institutional Review Board of The University of Tennesse, Memphis" Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: Quote: "Power analysis was made using a significance level of 0.5 a power of 0.8 and a two sided alternative hypothesis" Additional notes: In the bacterial isolates of 81 participants, Staphylococcus aureus was the most predominant followed by Haemophilus influenzae. H2 receptor antagonists were combined to form a common interventional arm vs sucralfate, as the review does not aim to investigate |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned to one of the three prophylaxis groups by random number table" Comment: Method adopted to obtain random sequence generation is clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: This was not a placebo‐controlled trial. Moreover, the mode of administration of study drugs would not have permitted blinding |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: GI bleeding was an objective outcome that was diagnosed as per the definition in the study protocol |
| Blinding (detection bias) Nosocomial pneumonia | Low risk | Comment: Pneumonia was an objective outcome that was diagnosed as per the definition in the study protocol |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: All other outcomes of interest were objective in nature |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: Yes, around 55% of all participants who were randomised were excluded from the final analysis because of less time spent in the ICU (< 48 hours), adverse reactions, younger than 18 years of age (which was clearly an exclusion criterion), and other protocol violations. Although a per‐protocol analysis was done, there appears to be an imbalance between groups with respect to the number of participants available for analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes are clearly mentioned in the study report |
| Other bias | Low risk | Comment: The study was supported in part by an educational grant from Smith Kline Beecham Inc. The role of the sponsor in the conduct and reporting of the trial is unclear. No other sources of bias are suspected |
Fan 2016.
| Methods | Parallel‐group randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: participants Number analysed: participants Enteral plus parenteral nutrition
Enteral nutrition
Parenteral nutrition
Inclusion criteria
Exclusion criteria
Baseline imbalances: There was no significant difference among the 3 groups in terms of age, sex, weight, and serum‐albumin at baseline (P > 0.05) |
|
| Interventions |
Enteral plus parenteral nutrition
Enteral nutrition
Parenteral nutrition
Adherence to regimen: ‐ Duration of trial: January 2009 to May 2012 Duration of follow‐up: ‐ |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
|
|
| Notes |
Setting: Neurological Intensive Care Unit Source of funding: Natural Science Foundation of Shandong Province (Y2008C35) and Technology Supporting Program of Qingdao (12‐1‐3‐5‐(1)‐nsh) Conflicts of interest: ‐ Ethics approval: This study had been approved by the local institutional review board and the Human Ethics Committee of the Affiliated Hospital of Qingdao University Informed consent: Informed consent had been provided by patients’ guardians Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: ‐ |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Quote: "patients were assigned to EN group, PN group and EN+PN groups randomly according to the sequence of their assigned hospital record number" Comment: method of randomisation not adequate |
| Allocation concealment (selection bias) | Unclear risk | Comment: no information reported |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: no information about blinding reported |
| Blinding (detection bias) Clinically important upper GI bleeding | Unclear risk | Comment: No definition of criteria was used to diagnose upper GI bleeding as reported |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: No definition of criteria was used to diagnose nosocomial pneumonia was reported |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Unclear risk | Comment: no information reported |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: insufficient information to allow judgement; once 120 patients were enrolled, but no information on how many patients fitted eligibility criteria |
| Selective reporting (reporting bias) | Low risk | Comment: All outcomes listed in the Methods section are also reported in the Results section |
| Other bias | Low risk | Comment: no other sources of bias suspected |
Fang 2014.
| Methods | Parallel‐group randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 120 Number analysed: 120 Early onset drug treatment
Late onset drug treatment
Enteral nutrition
Inclusion criteria:
Exclusion criteria
Baseline imbalances: no baseline difference in sex, age, BMI, and causes of brain injury |
|
| Interventions |
Early‐onset drug treatment
Late‐onset drug treatment
Enteral nutrition
Adherence to regimen: ‐ Duration of follow‐up: ‐ Duration of trial: May 2011 to May 2013 |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought in review and not reported in trial
Outcomes reported in trial, but not used in review
|
|
| Notes |
Setting: Department of Nursing, First People’s Hospital of Yunnan Province, Kunming, Yunnan Province, China Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Comment: randomisation by random number tables |
| Allocation concealment (selection bias) | Unclear risk | Comment: no information reported |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: no information reported |
| Blinding (detection bias) Clinically important upper GI bleeding | Unclear risk | Comment: no definition of criteria used to diagnose upper GI bleeding as reported |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Unclear risk | Comment: no information reported |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All participants were observed and analysed after intervention |
| Selective reporting (reporting bias) | Low risk | Comment: All observed results were reported, including stress ulcer bleeding rate in group A; the correlation between gastric pH and ulcer healing time in group A; incidence of stress ulcer bleeding rate by different gastric pH and different blood sugar level in group C; clinical therapeutic effects |
| Other bias | Low risk | Comment: no other sources of bias suspected |
Fink 2003.
| Methods | Open‐label randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: not clearly mentioned in study abstract Number analysed: 189 participants Overall
Inclusion criteria Study design, inclusion and exclusion criteria similar to Martin 1993
Exclusion criteria
Baseline imbalances: not clearly mentioned in study abstract |
|
| Interventions |
Pantoprazole
Pantoprazole
Pantoprazole
Pantoprazole
Cimetidine
Adherence to regimen: ‐ Duration of trial: ‐ Duration of follow‐up: ‐ |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial report
Outcomes reported in report but not used in review
|
|
| Notes |
Setting: ‐ Adherence to regimen: ‐ Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Quote: "Overall patents with a mean APSII score of 16.8 or greater tended to die while those with 15.33 or less, tended to survive" |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in study abstract |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in study abstract |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: unblinded, open trial |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: unblinded, open trial. The definition for detecting GI bleeding is not clearly mentioned in the study abstract. However, GI bleeding was an objective outcome |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: unblinded, open trial. However owing to the objective nature of the outcomes of interest, the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "189 patients received at least 1 dose of medication and had sufficient data points to be included in the analysis" Comment: unclear on how many were randomised initially to the different arms. Therefore, unclear on attrition bias |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes appear to be reported |
| Other bias | Low risk | Comment: Source of funding remains unclear. No other sources of bias are suspected |
Fogas 2013.
| Methods | Parallel‐group randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 94 participants Number analysed: 79 participants Proton pump inhibitors
H2 receptor antagonists
Inclusion criteria
Exclusion criteria
Baseline imbalances: no significant difference (P > 0.05) between groups regarding demographics |
|
| Interventions |
Proton pump inhibitors
H2 receptor antagonists
Adherence to treatment: ‐ Duration of follow‐up: ‐ Duration of trial: ‐ |
|
| Outcomes |
Outcomes reported in trial and used in review
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
|
|
| Notes |
Setting: General ICU, University of Szeged, Hungary Sponsorship source: ‐ Conflicts of interest: ‐ Comments: conference abstract Ethics approval: Quote: "After ethics committee approval ..." Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: Quote: "However, the completion of the study on the planned 198 patients is required to come to the final conclusions" |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: no details reported |
| Allocation concealment (selection bias) | Unclear risk | Comment: no details reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: no details reported, but lack of blinding unlikely to introduce bias to the outcome and outcome measures |
| Blinding (detection bias) Clinically important upper GI bleeding | High risk | Comment: no details reported, and no diagnostic criteria described |
| Blinding (detection bias) Nosocomial pneumonia | Low risk | Comment: Pneumonia was an objective outcome that was diagnosed as per the definition in the study protocol |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Unclear risk | Comment: no details reported |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: no incomplete reporting of outcome data. Patient flow was reported transparently; attrition was unlikely to have introduced bias to the results |
| Selective reporting (reporting bias) | Low risk | Comment: All outcomes listed are reported in the Results section. Conference abstract |
| Other bias | Unclear risk | Comment: no other sources of bias suspected, but not enough details reported in conference abstract |
Friedman 1982.
| Methods | Double‐blind randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 36 participants Number analysed: 36 participants Placebo
Antacids (Mylanta)
Cimetidine
Inclusion criteria
Exclusion criteria
Baseline imbalances: The 3 groups were comparable except for a significantly higher number of participants with chronic obstructive pulmonary disease in the antacid group as compared with the control group. They were also similar with respect to the mean number of risk factors per individual in any of the groups |
|
| Interventions |
Placebo
Antacid
Cimetidine
Adherence to regimen: 6 participants were unable to complete the protocol: 5 from the antacid group (4 had severe diarrhoea, and 1 had severe hypophosphataemia and hypomagnesaemia) and 1 from the control group (severe diarrhoea). However, intention to treat was performed by study authors Duration of trial: December 1978 to May 1980 Duration of follow‐up: not clearly mentioned in the study report. Probably until death or discharge |
|
| Outcomes |
Outcomes sought in review and reported in trial
Note: Only data on overt bleeding were extracted, as they were considered to be clinically significant
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
|
|
| Notes |
Setting: Department of Medicine, University of Virginia School of Medicine, Charlottesville, VA 22908 Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Quote: "The protocol was approved by the Human Investigation Committee of the University of Virginia in February 1978" Informed consent: Quote: "Informed consent was obtained from the patients or from a member of their immediate family" Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: According to the study report, no participant died of haemorrhage and overt bleeding ceased when participants were administered cimetidine and antacids |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Quote: "If a participant developed severe diarrhoea, an independent referee broke the treatment code and substituted alternagel in those participants receiving Mylanta ll" Comment: Study must have had some sort of allocation concealment to make the above statement materialise. However, method used for allocation concealment is not clear |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "If a participant developed severe diarrhoea, an independent referee broke the treatment code and substituted Alternagel in those participants receiving Mylanta ll and the investigators were not informed of the outcomes of the referees intervention" Comment: Investigators might have been blinded in the rest of the study too because this was a placebo‐controlled trial; each treatment arm (with the actual intervention) received a placebo form of the other drug (with which it was being compared) and vice versa |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: This was a placebo‐controlled trial; each treatment arm (with the actual intervention) received a placebo form of the other drug (with which it was being compared) and vice versa. Moreover, this was an objective outcome detected as per the definition provided in the study |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: This was a placebo‐controlled trial; each treatment arm (with the actual intervention) received a placebo form of the other drug (with which it was being compared) and vice versa. Moreover, owing to the objective nature of the outcome of interest, the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All randomised participants were accounted for in the analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: unclear on source of funding. No other form of bias detected |
Groll 1986.
| Methods | Double‐blind randomised placebo‐controlled trial | |
| Participants |
Baseline characteristics Number randomised: unclear on how many were initially randomised. Study report mentions that 531 participants were eligible for the trial, of whom 221 participants completed the trial with reasons for excluding of the remaining participants Number analysed: 221 participants Cimetidine
Placebo
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "The drug and placebo groups were similar for demographic features and risk factors.The number of patients with each risk factor known to predispose to stress ulceration and the mean number of risk factors per patient were similar in both groups such as minor operative procedure, respiratory failure, renal failure, sepsis, shock, trauma, coma and liver failure" |
|
| Interventions |
Cimetidine
Placebo
Adherence to regimen: Quote: "Of the remaining 531 eligible patients 221 completed the trial (41%). The remainder were excluded because of inability to obtain early consent (n = 69), refusal to enter the study (n = 83), death within the first 24 hours (n = 48), accidental omission by house staff (n = 86), and miscellaneous reasons (n = 24)" Comment: not sure when randomisation took place and exact numbers of participants who were initially randomised Duration of trial: 21 months Duration of follow‐up: not clearly mentioned in the study report. Probably until death or discharge |
|
| Outcomes |
Outcomes sought in review and reported in trial
Note: Data were not provided separately for overt and occult bleeds. Of the 70% of participants who were in the study for 1 to 3 days, 9 of the 11 placebo and 5 of the 6 cimetidine participants bled
Outcomes sought but not reported in trial
Outcomes reported in report but not used in review
|
|
| Notes |
Setting: Kingston General Hospita, Ontario, Canada Source of funding: Quote: "This project was supported by Smith Kline and French Canada Ltd" Conflicts of interest: ‐ Ethics approval: Quote: "The study was approved by the Institutional Review Board and Grant agency at the Misnistry of Healthof Chez Republic" Informed consent: Quote: "The study was approved by the committee on human research, Department of Medicine, Queen's University" Clinical trials registration: ‐ Sample size calculation: ‐ |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Patients were randomised in a double blind method to receive injections of either cimetidine 300 mg in 20 mL normal saline or placebo prepared in an identical manner, given intravenously every 6 hours" Comment: This was a placebo‐controlled trial, and participants and personnel were blinded |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Quote: "Patients were randomised in a double blind method to receive injections of either cimetidine 300 mg in 20 mL normal saline or placebo prepared in an identical manner, given intravenously every 6 hours" Comment: This was a placebo‐controlled trial, and GI bleeding was detected as per the definition in the study owing to the objective nature of the outcome of interest |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: This was a placebo‐controlled trial, and participants and personnel were blinded. Moreover owing to the objective nature of the outcomes of interest, the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "Of the remaining 531 eligible patients 221 completed the trial (41%). The remainder were excluded because of inability to obtain early consent (n = 69), refusal to enter the study (n = 83), death within the first 24 hours (n = 48), accidental omission by house staff (n = 86), and miscellaneous reasons (n = 24)" Comment: not sure when randomisation took place and exact numbers of participants who were initially randomised. Therefore unclear on attrition bias |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: This project was supported by Smith Kline and French Canada Ltd. The role of the sponsor in the conduct and reporting of the trial is unclear. No other sources of bias suspected |
Halloran 1980.
| Methods | Double‐blind randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 50 participants Number analysed: 50 participants Cimetidine
Placebo
Inclusion criteria
Exclusion criteria
Baseline imbalances: Both groups were comparable with respect to age, gender, and underlying disorders |
|
| Interventions |
Cimetidine
Placebo
Adherence to regimen: no dropouts mentioned Duration of trial: July 1977 to March 1979 Duration of follow‐up: ‐ |
|
| Outcomes |
Outcomes sought in review and reported in trial Primary outcomes
Secondary outcomes
Outcomes sought but not reported in trial report
Outcomes reported in report but not used in review
|
|
| Notes |
Setting: Departments of Medicine and Surgery, Medical College, Virginia, USA Source of funding: Quote: "The work was supported by Smith Kline and French Laboratories.clearly mentioned in the study report" Ethics approval: Quote: "The study was approved by the committee on the conduct of clinical research.Medical College of Virginia, Virginia commonwealth university" Informed consent: Quote: "Informed consent was obtained from the patients closest relative or legal agent and later from the patient if sufficient neurologic recovery ensued" Clinical trials registration: ‐ Sample size calculation: ‐ |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Low risk | Quote: "Patients were randomly given coded medication in a standard double blind fashion" Comment: Allocation concealment might have been done |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Patients were randomly given coded medication in a standard double blind fashion" Comment: This was a placebo‐controlled double‐blind trial, so study personnel would have been blinded |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: not clearly mentioned, but this was a placebo‐controlled double‐blind trial, and GI bleeding was an objective outcome that was detected as per the study definition |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: not clearly mentioned, but this was a placebo‐controlled double‐blind trial, and outcomes of interest were objective in nature and were detected as per the study definition |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All randomised participants were part of the final analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All outcomes stated were reported in the Results |
| Other bias | Low risk | Quote: "The work was supported by Smith Kline and French Laboratories" Comment: The role of the sponsor in the conduct and reporting of the trial is unclear. No other source of bias is suspected |
Hanisch 1998.
| Methods | Double‐blind randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 827 participants Number analysed: 158 participants Ranitidine
Pirenzipine
Placebo
Notes: Age not given for all randomised participants, but only for those who were in mechanical ventilation for longer than 48 hours Inclusion criteria
Exclusion criteria
Baseline imbalances: Groups were similar with respect to age and APACHE ll score |
|
| Interventions |
Ranitidine
Pirenzipine
Placebo
Adherence to regimen: All 827 randomised patients completed the trial. However, only 158 participants who were on mechanical ventilation for ≥ 48 hours were part of the analysis, as the primary outcome was pneumonia rate Duration of trial: December 1991 to December 1992 Duration of follow‐up: not clearly mentioned in the study report. Probably until death or discharge |
|
| Outcomes |
Outcomes sought in review and reported in trial Primary outcome
Secondary outcomes
Outcomes sought but not reported in trial report
Outcomes reported in report but not used in review
|
|
| Notes |
Setting: Department of Surgery, Division of General and Trauma Surgery, Johann Wolfgang Goethe University, Frankfurt am Main, Germany Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: approved by the Ethics Committee of Johann Wolfgang Goethe University, Frankfurt/Main Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: Quote: "Assuming a pneumonia rate of 25% in ranitidine treated patients and a pneumonia rate of 15% in placebo treated patients, the complete sample size was calculated to be at least 100 patients (alpha = 0.05 and beta = 0.20)" |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "A complete and balanced randomisation schedule was generated by the institute of biomathematics of the university of Frankfurt" Comment: method adopted to obtain random sequence generation clearly mentioned in the study report |
| Allocation concealment (selection bias) | Low risk | Quote: "At the time of entering the ICU patients were assigned to a consecutive study number, the application of a blinded drug regimen was started" "...the drugs were prepared in advance by a staff who were not involved in the treatment of patients and identified by the above mentioned consecutive study number i.e. the code was located exclusively with this group during the trial'' Comment: Randomisation code was used exclusively with staff who were not involved in the treatment of patients, and these staff were responsible for preparing the 3 interventions through a similar mechanism (dissolving each in 100 mL of 5% glucose) to avoid detection during treatment |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Blinding of drugs (placebo, ranitidine, pirenzipine) was achieved by dissolving them in glucose (100 mL; 5% Braun, Melsungen, Germany) resulting in an equal infusion volume for each patient. Drugs were prepared by staff who were not involved in the treatment of patients and identified by the above mentioned consecutive study number"; "All blinded drugs (only identifiable by their consecutive study number) were given to the patients by a staff nurse in the ICU" Comment: This was a placebo‐controlled nature of the study design in which the 3 interventions were prepared in a similar manner (dissolving each in 100 mL of 5% glucose) to ensure adequate blinding because of which the details of interventions received were not known. Participants and personnel were blinded, ensuring no performance bias |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Quote: "Data documentation for each patient was performed prospectively and daily by a different staff not involved in the treatment of patients in the randomised assignment of the drugs" Comment: not clear whether they were involved in outcome assessment. Moreover, GI bleeding was detected as per the definition in the study protocol and was an objective outcome |
| Blinding (detection bias) Nosocomial pneumonia | Low risk | Quote: "Data documentation for each patient was performed prospectively and daily by a different staff not involved in the treatment of patients in the randomised assignment of the drugs" Comment: not clear whether they were involved in outcome assessment. Moreover, pneumonia was detected as per the definition in the study protocol and was an objective outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Quote: "Data documentation for each patient was performed prospectively and daily by a different staff not involved in the treatment of patients in the randomised assignment of the drugs" Comment: not clear whether they were involved in outcome assessment. However, outcomes of interest were objective in nature |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: 741 patients randomised, but only 158 completed 48 hours of mechanical ventilation and were reported in the trial |
| Selective reporting (reporting bias) | Low risk | Comment: Data on all randomised participants were not included in the results. Only outcomes for participants who were on mechanical ventilation for longer than 48 hours are reported. Of 827 randomised patients, a total of 158 were mechanically ventilated for ≥ 48 hours. Data on the remaining 669 patients were not available. However, this does not account for reporting bias, as the study intended to report its primary outcome of pneumonia only for participants who completed ≥ 48 hours on mechanical ventilation. All intended outcomes were reported only for participants who spent ≥ 48 hours in the ICU |
| Other bias | Low risk | Comment: unclear on the source of funding. No other sources of bias suspected |
Hastings 1978.
| Methods | Parallel‐group randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 100 participants Number analysed: 100 participants Antacids
No prophylaxis
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "Fifty one of the 100 participants entered into the study were randomised to receive antacid prophylaxis. Six major risk factors were analysed for each participant in each group. These risk factors were respiratory failure, extra abdominal sepsis, peritonitis, jaundice, renal failure and hypotension. There were 110 risk factors in 51 participants in the antacid group, a mean of 2.2 risk factors per participant. There were 115 risk factors in 49 participants in the group not receiving antacid prophylaxis, a mean of 2.4 risk factors per participant. All participants in respiratory failure were treated with constant‐volume ventilators. The severity of respiratory failure was judged by the alveolar‐ atrial oxygen. The mean highest value was 363+/‐ 25 mm Hg (SEM) in the participants receiving antacid and 324+/‐ 29 mm Hg in those not receiving Antacid prophylaxis (p > 0.05). The difference in mean blood urea nitrogen values (96+/‐ 10 mm/dl in the Antacid group vs. 90+/‐ 10 mm Hg in those not receiving antacid prophylaxis) was not statistically significant between the two groups. The extent of jaundice was also similar in the two groups" Comment: Both groups were similar with respect to their baseline characteristics. The major risk factor was respiratory failure |
|
| Interventions |
Antacids
No prophylaxis
Adherence to regimen: Quote: "The study was ended when oral feedings were begun and the nasogastric tube was removed, or the participant was discharged from the intensive care unit" Comment: In 2 participant who had diarrhoea, antacid was stopped, and in 2 other participants, a different antacid was administered. In 1 participant from antacid group with elevated magnesium levels, antacid was changed to a preparation that did not contain magnesium Duration of trial: March 1972 to March 1977 Notes duration of treatment: Mean duration for the entire study of 100 participants was 2.9 ± 0.29 days (SEM). Study was terminated when oral feedings were begun and the nasogastric tube was removed, or when the participant was discharged from the ICU Duration of follow‐up: ‐ |
|
| Outcomes |
Outcomes sought in review and reported in trial Primary outcome (outcomes are not categorised as primary and secondary, but from the study report we feel GI Bleeding was the primary outcome)
Outcomes sought but not reported in trial report
Outcomes reported in report but not used in review
|
|
| Notes |
Setting: Respiratory‐Surgical Intensive Care Unit of the Beth Israel Hospital, 300 Brookline Avenue, Boston, MA, 02215 Source of funding: Quote: "we are indebted to Stuart Pharmaceuticals, Division of I.C.I United States, Inc., Wilmington, DE (manufacturers of Mylanta II), whose generous contribution in part made this study possible…" Conflicts of interest: ‐ Ethics approval: Quote: "The study was approved by the Committee on Cliniccal Investigations, New procedures and new forums of therapy of the Beth Israel Hospital" Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "…the patients were randomised by a table of random numbers" Comment: Method adopted to obtain random sequence generation is clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not enough information on allocation concealment reported |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering the study interventions would not have made it possible to blind study personnel and participants |
| Blinding (detection bias) Clinically important upper GI bleeding | High risk | Comment: no clear definition for the diagnosis of upper GI bleeding or blinding of outcome assessors reported |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: All other outcomes of interest were objective in nature |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All randomised participants were part of the analysis. Therefore there is no attrition bias |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported in the study |
| Other bias | Low risk | Quote: "we are indebted to Stuart Pharmaceuticals, Division of I.C.I United States ,Inc., Wilmington, DE (manufacturers of Mylanta II), whose generous contribution in part made this study possible…" Comment: The above mentioned organisation also manufactured the antacid used as intervention in this study, which is suggestive of potential conflict of interest. The role of the sponsor in the conduct and reporting of the trial is unclear. No other sources of bias are suspected |
Hata 2005.
| Methods | Parallel‐group randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 210 participants Number analysed: 210 participants Tepterone
Ranitidine
Rabeprazole
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "Prevalence of hypertension, diabetes, renal insufficiency (serum creatinine level of more than 2.0 mg/dl), and smoking history were similar. The rates of operative characteristics such as CABG, valve replacement, and aortic surgery were also similar. There were also no differences regarding CPB duration, postoperative maximum creatine kinase (CK)‐MB levels" Comment: Participants had undergone an open heart surgery |
|
| Interventions |
Tepterone
Ranitidine
Rabenprazole
Adherence to regimen: Quote: "There was hematemesis complications in 4 patients during postoperative days 4 and 6 in each of groups I and II. Otherwise, those with active ulcers were completely healed by PPI administered for 2 weeks, and confirmed by GFS. These patients required a blood transfusion and all had emergency GFS. In contrast, in group III, no patients developed upper GI bleeding" Notes concomitant medication: Quote: "Systemic heparinization (10,000 U/day) was delayed until the second postoperative day. Patients undergoing CABG or aortic surgery had aspirin (81 mg/day) and those with valve replacement were administered Warfarin (2–3 mg/day) from the first postoperative day. Even though there was no complaints in relation to the upper GI system, gastric fibroscopy was used in all patients postoperatively during days 5 to 7" Concomitant medications: as mentioned in concurrent medication Duration of trial: January 2001 to December 2003 Duration of treatment: ‐ Duration of follow‐up: not clearly mentioned in the study report. Most probably until discharge or death (although mortality is not reported) |
|
| Outcomes |
Outcomes sought in review and reported in trial Primary outcomes
Secondary outcomes
Outcomes sought but not reported in trial report
Outcomes reported in report but not used in review
|
|
| Notes |
Setting: Department of Cardiovascular Surgery, Nihon University School of Medicine, Tokyo, Japan Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Quote: "The Ethics committee of Nihon University School of Medicine approved the current trial" Informed Consent: Quote: "All patients gave written informed consent" Clinical trials registration: ‐ Sample Size calculation: ‐ |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: This was not a placebo‐controlled trial, and the study is unclear on blinding of study personnel and participants. Therefore, it is unclear on performance bias |
| Blinding (detection bias) Clinically important upper GI bleeding | High risk | Comment: unclear on blinding of outcome assessors, and there was no clear definition for detecting clinically significant upper GI bleeding. Therefore high risk of performance and detection bias |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: unclear on blinding of outcome assessors. However owing to the objective nature of the outcome of interest, the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All randomised participants were part of the final analysis. Therefore there is no attrition bias |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported in the study |
| Other bias | Low risk | Comment: unclear on the source of funding. No other bias detected |
He 2017.
| Methods | Parallel‐group randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 120 participants Number analysed: 120 participants Naloxone
Naloxone + pantoprazole
Inclusion criteria
Exclusion criteria: ‐ Baseline imbalances: no significant difference in primary disease, sex, age, and course of disease between study group and control group |
|
| Interventions |
Naloxene
Naloxene + pantoprazole
Adherence to regimen: ‐ Duration of trial: May 2009 to May 2014 Duration of follow‐up: ‐ |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
|
|
| Notes |
Setting: ICU Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: Signed informed consent was required by all participants Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: ‐ |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: insufficient information to allow judgment |
| Allocation concealment (selection bias) | Unclear risk | Comment: insufficient information to allow judgment |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: insufficient information to allow judgment |
| Blinding (detection bias) Clinically important upper GI bleeding | High risk | Comment: no details about criteria for diagnosis of upper GI bleeding or blinding of outcome assessors reported |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Unclear risk | Comment: insufficient information to allow judgement |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: insufficient information to allow judgement |
| Selective reporting (reporting bias) | Low risk | Comment: all outcomes reported in Methods section also reported in Results |
| Other bias | Unclear risk | Comment: insufficient information to allow judgement |
Israsena 1987.
| Methods | Parallel‐group randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 86 participants Number analysed: 64 participants Sucralfate
Antacid
Inclusion criteria
Exclusion criteria
Baseline imbalances: None of the differences was statistically significant |
|
| Interventions |
Sucralfate
Antacid
Adherence to regimen: ‐ Duration of trial: April 1985 to March 1986 Duration of follow‐up: ‐ |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcome sought in review but not reported in trial
Outcomes reported in trial, but not used in review
|
|
| Notes |
Setting: Medical Intensive Care Unit, Department of Medicine, Faculty of Medicine, Chulalongkorn University, Bangkok, Thailand Sponsorship source: Siam Pharmaceutical Company, Bangkok Ethics approval: ‐ Informed consent: Failure to receive informed consent is mentioned under exclusion criteria Clinical trials registration: ‐ Sample size calculation: ‐ Comments: ‐ |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "108 subjects were divided into acute and chronic cases of respiratory failure and were allocated by means of a table of random numbers to receive either sucralfate or antacid" |
| Allocation concealment (selection bias) | Unclear risk | Comment: no details reported |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: no details about blinding reported, but lack of blinding unlikely to introduce bias to findings |
| Blinding (detection bias) Clinically important upper GI bleeding | Unclear risk | Comment: Study did not address this outcome |
| Blinding (detection bias) Nosocomial pneumonia | Low risk | Comment: GI bleeding was an objective outcome that was diagnosed as per the definition in the study protocol. Therefore the likelihood of performance or detection bias is low |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Unclear risk | Comment: no details reported |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: 16 patients were excluded from analysis in the sucralfate group and 6 from the antacid group, without a described reason |
| Selective reporting (reporting bias) | Low risk | Comment: no selective reporting of outcomes suspected; all outcomes listed in the Methods section and objectives are also reported in the Results section |
| Other bias | Low risk | Comment: funded by pharmaceutical industry. No other sources of bias suspected |
Kantorova 2004.
| Methods | Single‐center randomised placebo‐controlled trial | |
| Participants |
Baseline characteristics Number randomised: 323 participants Number analysed: 287 participants Omeprazole
Famotidine
Sucralfate
Placebo
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "At randomisation no statistically significant difference was found among the four groups in demographic and baseline physiological characters. The distribution of risk factors such as participants who had trauma, surgery or coagulopathy at ICU admission were also similar in all groups" Comment: no significant difference between the 2 groups with respect to demographic and baseline risk factors. Trauma was the main reason for admission. Coagulopaty was diagnosed in 31 participants at baseline (7, 10, 6, and 8 in each of the 4 groups, respectively) |
|
| Interventions |
Omeprazole
Famotidine
Sucralfate
Placebo
Notes for placebo intervention: A placebo group was included in the protocol design because at study initiation and during the study period, no drug was approved for prophylaxis of stress‐related upper gastrointestinal bleeding by the FDA. The European Medicines Evaluation Agency required a placebo comparison group for registration of studies in this indication Adherence to regimen: Quote: "Of the 323 randomised patients 36 were subsequently excluded from analysis (1 patient died suddenly within 2 hours after randomisation, 18 underwent mechanical ventilation < 48 hours and 16 were not assessable because of missing important data" Thus only 287 patients could be analysed. No drug‐related adverse events were seen during the whole study, possibly related adverse events were rare, and in no patient was discontinuation of therapy necessary. Duration of trial: February 2000 to June 2002 Duration of follow‐up: ‐ |
|
| Outcomes |
Outcomes sought in review and reported in trial
Secondary outcomes
Outcomes sought but not reported in trial report
Outcomes reported in trial but not used in review
|
|
| Notes |
Setting: Traumatological Hospital Brno, a Czech republic ministry of health teaching and research facility Source of funding: Quote: "The study was supported by a grant of IGA MZ CB ND 5932‐3/2000" Conflicts of interest: Ethics approval: Quote: "The study was approved by the Institutional Review Board and Grant agency at the Misnistry of Healthof Chez Republic" Informed consent: Quote: "participants not able or willing to give informed consent were eluded from the study" Clinical trials registration: not provided in the study report Sample size calculation: not mentioned in the study report Additional notes: GI bleeding occurred more commonly in participants with coagulopathies (3/31, 10% vs 4/245, 2%, P = 0.006). The most common pathogens in participants with pneumonia were Staphylococcus andStreptococcus. ICU stay and mortality were greater in participants with investigated complications of GI bleed and pneumonia but were not influenced by prophylaxis |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned to study group by means of computer generated random number table to generate a random treatment list" Comment: method adopted to obtain random sequence generation clearly mentioned in the study report |
| Allocation concealment (selection bias) | Low risk | Quote: "Treatment regimens were included in opaque sealed numbered envelopes and the envelope with the lowest number was always used for the consecutive patient" Comment: method adopted to allocation concealment clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "Blinding of the drug administered intravenously was achieved by dissolving them in 100 mL of saline solution drug were prepared by staff not involved in the study according to the randomisation. All blinded drugs were identifiable by their study number and were given to the patients by the study nurse" Comment: Blinding is mentioned only for groups that received intravenous medications (omeprazole, famotidine, and placebo), not sucralfate. Unclear whether this would have led to performance bias |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: mentioned that outcome assessment was done blinded to intervention. The presence of 1 or more macroscopic abnormalities (erythema or oedema, erosions, ulcerations, and naso‐gastric tube lesions) and GI bleeding was assessed, blinded to intervention, at endoscopy by a single experienced gastroenterologist Quote: "assessed, blinded to intervention, at endoscopy by a single experienced gastroenterologist" |
| Blinding (detection bias) Nosocomial pneumonia | Low risk | Quote: "Chest radiographs were interpreted by two independent radiologists" Comment: Blinding of outcome assessors was done. Pneumonia was an objective outcome detected as per the study protocol |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: All other outcomes of interest were objective in nature, so the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: Of the 323 randomised patients, 36 were subsequently excluded from analysis. Groups to which they were initially randomised remain unclear though. Thus only 287 participants were available for analysis. However, the number of dropouts and their characteristics were comparable in all 4 arms. Therefore there is no likelihood of risk of attrition bias |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported in the study |
| Other bias | Low risk | Comment: Study was funded by a grant of IGA MZ CB ND 5932‐3/2000. The role of the sponsor in the conduct and reporting of the trial is unclear. No other sources of bias were detected |
Kappstein 1991.
| Methods | Parallel‐group quasi‐randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 104 participants Number analysed: 104 participants Sucralfate
Cimetidine
Inclusion criteria
Exclusion criteria
Baseline imbalances: The 2 groups were largely similar according to demographic characteristics, underlying diseases, and diagnoses at admission. However, there were slight imbalances with regard to age, gender, polytrauma, and acute respiratory failure |
|
| Interventions |
Sucralfate
Cimetidine
Adherence to treatment: ‐ Duration of trial: May 1986 to January 1989 Duration of follow‐up (days): ‐ |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought in review, but not reported in trial
Outcomes reported in trial but not sought in review
|
|
| Notes |
Setting: ICU, Department of Hospital Epidemiology, Medical Biometry and Anaesthesiology, University Hospital Freiburg, Germany Sponsorship source: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Comment: ‐ |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Quote: "and tube‐fed patients were excluded. Patients were assigned to stress ulcer prophylaxis with either sucralfate or cimetidine by setting up groups of 10 patients who received one prophylactic regimen followed by the next 10 patients receiving the other regimen. Patients received either intravenous" Judgement comment: quasi‐randomisation. Patients were assigned to stress ulcer prophylaxis with either sucralfate or cimetidine by setting up groups of 10 patients who received 1 prophylactic regimen followed by the next 10 patients receiving the other regimen |
| Allocation concealment (selection bias) | Unclear risk | Judgement comment: Provided the sequence was consecutive, patients were known in each group |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Judgement comment: no blinding reported, but lack of blinding is unlikely to introduce bias. Outcome measures and outcomes are objectively measured |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Quote: "Macroscopically visible blood in gastric aspirates was taken for evidence of stress bleeding" Comment: outcome measured objectively |
| Blinding (detection bias) Nosocomial pneumonia | Low risk | Comment: criteria for the diagnosis of pneumonia clearly reported in the trial |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | High risk | Quote: "The chest x‐ray was interpreted by a radiologist who was not aware of the particular prophylactic regimen" Judgement comment: but investigators and personnel were unblinded for other criteria for pneumonia and for other outcomes |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Judgement comment: All outcomes have included all patients. No incomplete reporting of data suspected. Attrition was low and was reported transparently |
| Selective reporting (reporting bias) | Low risk | Judgement comment: no incomplete reporting of outcomes suspected. All outcomes listed in the Methods section are also reported in the Results section |
| Other bias | Low risk | Comment: no other sources of bias suspected |
Karlstadt 1990.
| Methods | Multi‐centre double‐blind randomised placebo‐controlled trial | |
| Participants |
Baseline characteristics Number randomised: 87 participants Number analysed: 87 participants Cimetidine
Placebo
Inclusion criteria Participants admitted to ICUs were eligible for entry into the trial if they had at least 1 of the following conditions generally regarded as risk factors for bleeding:
Exclusion criteria
Baseline imbalances: Quote: "Patient demographics and risk factors for bleeding on entry into the study show that the treatment groups were comparable" Comment: Participants from 14 centres were comparable with respect to demographic characteristics and baseline risk factors. Almost 40% of study participants had acute respiratory failure at baseline, of whom 14% had pneumonia |
|
| Interventions |
Cimetidine
Placebo
Adherence to regimen: Quote: "The average time in the study was longer for patients treated with cimetidine (mean, 83 hours ± 53) than for patients treated with placebo (mean, 53 hours ± 41). This difference was because more patients treated with placebo bled and left the study early" "Two patients being treated with cimetidine experienced adverse events for which they were withdrawn from the study: one developed moderate nausea and vomiting and one developed a change in mental status. In both cases, the adverse experiences were reversible. No patients treated with placebo were withdrawn from treatment, but adverse experiences attributed to placebo treatment included one case of mental confusion" Duration of trial: ‐ Duration of follow‐up: not clearly mentioned in the study report. Probably until death or discharge |
|
| Outcomes |
Outcomes sought in review and reported in trial Primary outcomes
Secondary outcomes
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
|
|
| Notes |
Setting: The study was a multi‐centric study involving 14 centres; University of Kentucky, Lexington, KY; Boston City Hospital, Boston, King Drew Medical Center, Los Angeles, CA; Winthrop University Hospital, Mineola, NY; Ellis Hospital, Schenectady, NY; Detroit Rec. Hospital, Detroit, MI; Mt. Sinai Medical Center, New York, Hartford Hospital, Hartford, CT; St. Thomas Hospital, Akron, OH; Cleveland Metro General Hospital, Cleveland, OH; University of Oklahoma Health Science Center, Oklahoma City, OK; Ravenswood Hospital, Chicago, IL; Brookdale Hospital, Brooklyn, NY; University of Texas Health Science Center, San Antonio, TX Source of funding: Study appears to have been funded by Smith Kline & French Laboratories, Philadelphia, PA, although this is not clearly mentioned in the study report Conflicts of interest: Ethics approval: Quote: "The study was approved by all institutional review boards" Comment: This study was approved by the ethics committees at all 14 centres Informed consent: Quote: " ...and all patients (or their legal guardians) provided written, informed consent" Comment: For participating in the trial, informed consent was obtained from participants or their legal guardians Clinical trials registration: not provided in the study report Sample size calculation: not clearly mentioned in the study report Additional Notes:Staphylococcus aureus was the organism found to have caused pneumonia. One participant treated with cimetidine had melena, but the upper GI source could not be detected, so this participant was not categorised as having an upper GI bleed |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not enough information about sequence generation reported |
| Allocation concealment (selection bias) | Unclear risk | Comment: not enough information about allocation concealment reported |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: This was a placebo‐controlled trial; unclear about whether participants and study personnel were blinded |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: unclear whether outcome assessors were blinded. GI bleeding was an objective outcome detected as per the definition in the study protocol |
| Blinding (detection bias) Nosocomial pneumonia | High risk | Comment: Outcome assessors were not blinded. Study had no clear mention of the definition to detect pneumonia. Moreover 14% of participants had pneumonia on entry into the study. Lack of blinding could have influenced detection of this condition, as it is mentioned that the physician did not attribute pneumonia to the study medication |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Unclear risk | Comment: Outcomes of interest were in part objective in nature; for other outcomes, the definition is not described and no blinding is in place |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "All patients who received study drugs were included in the denominator for analysis" Comment: All randomised participants were part of the final analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: Lead author is affiliated with Smith Kline & French Laboratories, Philadelphia, PA. Clear conflict of interest. No other sources of bias suspected |
Kaushal 2000.
| Methods | Open‐label randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 75 participants Number analysed: 75 participants Famotidine
Sucralfate
No prophylaxis
Inclusion criteria
Exclusion criteria
Baseline imbalances: Age, gender, and clinical features of participants in the 3 groups were comparable. Participants were people who required ICU care after head injury (mainly because if road side accident) |
|
| Interventions |
Famotidine
Sucralfate
No prophylaxis
Adherence to regimen: Quote: "Out of 56 patients who had upper GI bleeding, 39 started bleeding within 3 days of head injury" Comment: Only 1 patient in the famotidine group required escape treatment as compared with 3 in the sucralfate group and 8 in the no prophylaxis group. The exact reason for escape of treatment is not mentioned but could be attributed to untimely death or upper GI bleed that was clinically significant as per the study protocol. The 56 participants with upper GI bleed are inclusive of all grades of bleeding according to the study protocol. Interventions were discontinued if at least 3 consecutive positive occult blood readings were detected Duration of trial: ‐ Duration of follow‐up: 15 days |
|
| Outcomes |
Outcomes sought in review and reported in trial Primary outcomes
Note: Among the 56 participants who bled, 39 of them started to bleed within 3 days of head injury. Data on occult bleeding were not used in the review Secondary outcomes
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
|
|
| Notes |
Setting: Neurosurgery Unit of Medical College and Hospital, Ludhiana, India Source of funding: ‐ Ethics approval: Quote: "After approval from Hospital Ethics Committee, the study was conducted in 75 consecutive patients" Comment: The ethics committee of the concerned institution had approved the study Informed consent: Quote: "Since informed consent could not be taken directly from patients, it was taken from the next of kin" Comment: Informed consent was obtained Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: According to the study report, data on endoscopy could not be analysed because of small numbers of participants who underwent the procedure. Occult bleeding occurred in 38 participants (no prophylaxis: 11, famotidine: 12, and sucralfate: 15) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: This was not a placebo‐controlled trial, and 1 arm received no prophylaxis; this would not have made it possible to blind study personnel and participants |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: unclear whether outcome assessors were blinded. However GI bleeding was an objective outcome that was detected as per the definition in the study |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: unclear whether outcome assessors were blinded. All other outcomes of interest were objective in nature |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All randomised participants were part of the final analysis in the study report |
| Selective reporting (reporting bias) | Low risk | Comment: The intended outcome of GI bleeding was reported in the study report |
| Other bias | Low risk | Comment: unclear on the source of funding. No other form of bias detected |
Khorvash 2014.
| Methods | Parallel‐group quasi‐randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 148 participants Number analysed: 137 participants Pantoprazole
Sucralfate
Inclusion criteria
Exclusion criteria
Baseline imbalance: ‐ |
|
| Interventions |
Pantoprazole
Sucralfate
Adherence to regimen: ‐ Duration of trial: early 2010 to mid‐2011 Duration of follow‐up: ‐ |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported but not used in review
|
|
| Notes |
Setting: ICU, Infectious Diseases and Tropical Medicine Research Center, Isfahan University of Medical Sciences, Isfahan, Iran Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Study was approved by the medical universities ethics committee at Isfahan University of Medical Sciences Informed consent: Quote: "after they agreed to cooperate" Comment: included in the trial report Clinical trials registration: ‐ Sample size calculation: ‐ |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Quote: "Patients were consecutively assigned to each group. The first patient was selected randomly" Comment: no randomisation, quasi‐randomised study |
| Allocation concealment (selection bias) | High risk | Comment: consecutive pattern of allocation |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: no blinding reported, but lack of blinding is unlikely to introduce bias to outcome measures or outcomes |
| Blinding (detection bias) Clinically important upper GI bleeding | Unclear risk | Comment: Study did not address this outcome |
| Blinding (detection bias) Nosocomial pneumonia | Low risk | Quote: "Ventilator‑associated pneumonia is defined as a group of pneumonias that occur 48 hours after the patient is ventilated if the patient did not have primary signs of the infection at the time of arriving ICU. VAP is one of the most prevalent nosocomial infections and pneumonia is causes 27% of infections in ICU" Comment: Criteria for diagnosis of the outcome were reported objectively |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Unclear risk | Comment: no blinding of outcome assessors described |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: "11 patients were excluded for different reasons like not using the drug, changing the drug by the physician, and so on. Three of them were from the pantoprazole and 8 of them were from sucralfate group" |
| Selective reporting (reporting bias) | Unclear risk | Comment: no outcomes of interest described in the Methods section; assessment of selective outcome reporting difficult |
| Other bias | Low risk | Comment: no other sources of bias suspected |
Kingsley 1985.
| Methods | Randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 263 participants Number analysed: 249 participants Antacids
Cimetidine
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "The patients were randomised to the four different groups were fairly homogenous and were exposed to essentially the same risk factors" Comment: no clear mention of gender, age, and type of risk factor distribution in the study report. In all participants, a variety of illness classified II or greater by therapeutic intervention scoring system (TISS) were diagnosed |
|
| Interventions |
Antacids bolus
Antacids continuous
Cimetidine bolus
Cimetidine continuous
Adherence to regimen: Quote: "...14 patients initially admitted to the study were excluded for failure to follow protocol", "Any patient who developed severe diarrhoea was given alternagel instead of Mylanta" Comments: not sure which groups the 14 participants belonged to. Nine participants needed to be switched to alternagel Duration of trial: ‐ Duration of follow‐up: not clearly mentioned in the study report. Probably until death or discharge |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
|
|
| Notes |
Setting: Hurley Medical Centre, Michigan, USA Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Mean blood transfusion due to gastrointestinal haemorrhage was 1335 mL (range: 0 to 4500 mL). This is not mentioned separately for both groups. Irreversible haemorrhagic shock was the cause of death in 2 cimetidine participants |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: Given the mode of administration and dosing regimens, it would not have been possible to blind study personnel. Therefore unclear whether this would have caused any performance bias |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: unclear whether the outcome assessor was blinded. GI bleeding was an objective outcome that was detected as per the definition in the study protocol |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: All other outcomes of interest were objective in nature |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "...14 patients initially admitted to the study were excluded for failure to follow protocol" Comment: unclear on which groups the 14 participants were initially randomised to. But this accounts to be around 5% of total participants and is uniformly distributed across groups |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: unclear on the source of funding. No additional biases suspected |
Kitler 1990.
| Methods | Randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 401 participants Number analysed: 298 participants Overall
Inclusion criteria
Exclusion criteria
Baseline imbalances: Most participants were referred for cardiac surgical treatment. Groups were similar in demographic characteristics such as age and race. The sucralfate group had the fewest women. Ten participants assigned to receive meciadanol had a history of peptic ulcer disease (3 with a history of bleeding); 14 with sucralfate had a history of peptic ulcer disease (5 with a history of bleeding), and 13 assigned to receive antacid had a history of peptic ulcer disease (4 with bleeding) |
|
| Interventions |
Bioflavonoid (Maciadanol)
Sucralfate
Antacids (Maalox, magnesium aluminium hydroxide gel)
Adherence to regimen: Overall 401 patients had entered the study and 298 had completed the study. Those who did not complete the study could not be evaluated or were excluded because of protocol error (receiving other antacids or antiulcer drugs), by physician request, or by their own request. Participants who remained in SICU for less than 24 hours were also excluded from the study. 85 participants completed the maciadanol arm, 100 the sucralfate arm, and 113 the antacid arm Duration of trial: 16 months Duration of follow‐up: ‐ |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
|
|
| Notes |
Setting: Surgical Intensive Care Unit of Johns Hopkins Medical Institutions, Baltimore, and Francis Scott Key Medical Centre, Baltimore Source of funding: Zyma, S. A., Nyon, Switzerland Conflicts of interest: Ethics approval: Quote: "The study was approved by the institutional review board of Johns Hopkins Medical Center and the Francis Scott Key Medical Center" Informed consent: mentioned in the study report Clinical trials registration: ‐ Sample size calculation: Quote: "The power calculations resulting from study sample size were one tailed with an alpha of 0.05 and a power of 80.0 per cent..." |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomised by a table of random numbers..." Comment: Method adopted to obtain random sequence generation is clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering the study interventions would not have made it possible to blind study personnel and participants |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Quote: "All data were collected by an observer unaware of the results" Comment: not clear how this would have contributed to blinding. GI bleeding was detected as per the definition in the study. However, owing to the objective nature of the outcome of interest, the likelihood of performance and detection bias is low |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Quote: "All data were collected by an observer unaware of the results" Comment: unclear on blinding of outcome assessors. However, all other outcome data were objective in nature |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: Overall 401 patients had entered the study and 298 had completed the study. Study did a per‐protocol analysis for outcomes. Participants in the Maciadanol arm were 24% less than in the antacid arm. Unclear whether this could have influenced outcomes |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported in the study report |
| Other bias | Low risk | Comment: Source of funding is mentioned. The role of the sponsor in the conduct and reporting of the trial is unclear. No other sources of bias are suspected |
Krakamp 1989.
| Methods | Double‐blind randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 30 participants Number analysed: 30 participants Overall
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "... groups were comparable in terms of age, gender and degree of consciousness" Comment: Participants were people who were admitted to the neurosurgical ICU |
|
| Interventions |
Ranitidine + placebo
Ranitidine + pirenzepine
Adherence to regimen: no dropouts. There does not seem to be any change in dosage, nor were are any adverse events mentioned Duration of trial: ‐ Duration of follow‐up: no information given. It seems that there was no further follow‐up after the end of the study |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported but not used in review
|
|
| Notes |
Setting: Neurosurgical ICU, Cologne, Germany Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent : ‐ Clinical trials registration: ‐ Sample size calculation: ‐ |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: Study mentions that it is double‐blinded, but it does not describe clearly who was blinded or how this was done. However, because this is a placebo‐controlled trial, we assume that participants and study personnel were the ones blinded |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: Study mentions that it is double‐blinded, but it does not describe clearly who was blinded or how this was done. Moreover GI bleeding was an objective outcome that was detected as per the study definition |
| Blinding (detection bias) Nosocomial pneumonia | High risk | Comment: Study mentions that it is double‐blinded, but it does not describe clearly who was blinded or how this was done. The definition for VAP was not clearly mentioned in the study report |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: Study mentions that it is double‐blinded, but it does not describe clearly who was blinded or how this was done. Moreover outcomes were objective in nature |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All randomised participants were part of the final analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: unclear on the source of funding. No additional biases were suspected |
Kuusela 1997.
| Methods | Single‐blind randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 53 participants Number analysed: 53 participants Ranitidine
No prophylaxis
Inclusion criteria
Exclusion criteria
Baseline imbalances: The 2 groups were comparable with respect to gestational age, birth weight, Apgar score, cord blood pH, and gender. Participants were preterm and full‐term infants |
|
| Interventions |
Ranitidine
No prophylaxis
Adherence to regimen: Of 53 participants who were randomised, only 48 were analysed as there were 5 dropouts due to early death (2 participants at gestational age < 33 weeks and 1 participant at gestational age ≥ 33 weeks) and oesophageal atresia (n = 2). 3 infants belonged to the ranitidine group and 2 belonged to the control group Duration of trial: 10‐Month period Duration of treatment : 4 days; study was discontinued when significant results of the first block of 20 preterm infants randomised were available for interpretation Duration of follow‐up: Routine follow‐ups were made at 7 days, then afterwards weekly up to 4 weeks |
|
| Outcomes |
Outcomes sought in review and reported in trial
Note: The intervention was given for 4 days only, whereas endoscopic findings were done at 3 and 6 days (even after treatment), the results of which could not be extracted separately
Outcomes sought but not reported in trial
Outcomes reported but not used in review
|
|
| Notes |
Setting: Department of Neonatal Intensive Care, University Hospital, Tampere, Finland Source of funding: Quote: "Supported in part by the Finnish Foundation of Pediatric Research" Conflicts of interest: ‐ Ethics approval: Quote: "The study protocol was approved by the Ethics Committee of the Tampere University Hospital" Informed consent: Quote: "The parents received both oral and written information on the study; their informed consent was obtained" Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Randomisation of participants was performed after infants were stratified into 2 groups (infants at less than and more than 33 weeks' gestational age) because infants usually less than 33 weeks are ventilated more often. Thus balance was achieved with respect to gestational age for both groups. Study was discontinued after significant results for the first 20 preterm infants (the first block) were available.When the study was stopped, results for a second block of 10 preterm infants and a third block of 7 preterm infants were available, along with results for a block of 16 and 20 mature infants |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "The first randomisation block included 20 envelopes in random order for both groups (gestational age < 33 weeks and gestational age ≥ 33 weeks). Followed by two randomisation blocks of 10 envelops for more preterm infants. Babies closer to term were randomised in one block with 20 envelops" Comment: Method adopted to obtain random sequence generation is clearly mentioned in the study report |
| Allocation concealment (selection bias) | Low risk | Quote: "The first randomisation block included 20 envelopes in random order for both groups (gestational age < 33 weeks and gestational age ≥ 33 weeks). Followed by two randomisation blocks of 10 envelops for more preterm infants. Babies closer to term were randomised in one block with 20 envelops" Comment: Method adopted to conceal allocation is clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Only the sequence number of the patient were written in the patients records by the nurse responsible for medication; thus the attending physicians the endoscopist and the pathologist remained blinded as to the treatment group" Comment: Study personnel were blinded to treatments. Moreover outcomes of interest were objective in nature |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Quote: "Only the sequence number of the patient [was] written in the patient records by the nurse responsible for medication; thus the attending physicians, the endoscopist, and the pathologist remained blinded as to the treatment group" Comment: The endoscopist was blinded to whether or not the participant received prophylaxis. Moreover the outcome measured was objective in nature |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Quote: "Only the sequence number of the patient [was] written in the patient records by the nurse responsible for medication; thus the attending physicians, the endoscopist, and the pathologist remained blinded as to the treatment group" Comment: Outcome assessors were blinded. Moreover owing to the objective nature of the outcomes of interest, the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: 5 babies were not part of the analysis (3 in treatment and 2 in control groups). A sensitivity analysis was done in which it was assumed that the 3 dropout neonates randomised to the treatment group had mucosal abnormalities, and the 2 dropout neonates randomised to the control group had no mucosal abnormalities. Results of this assumption still indicate that ranitidine was effective in preventing gastric mucosal lesions in infants under stress |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were reported and analysed |
| Other bias | Low risk | Comment: The Finnish Foundation of Pediatric Research funded the study. The role of the sponsor in the conduct and reporting of the trial is unclear. No other sources of bias are suspected |
Lacroix 1986.
| Methods | Parallel‐group randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 40 participants Number analysed: 38 participants Cimetidine
Placebo
Inclusion criteria
Exclusion criteria
Baseline imbalances: Mechanical ventilation was used in 2 times more patients in the placebo group |
|
| Interventions |
Cimetidine
Placebo
Adherence to treatment: ‐ Duration of trial: ‐ Duration of follow‐up (days): ‐ |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcome sought in review and not reported in trial
Outcomes reported in trial and not sought in review
|
|
| Notes |
Setting: PICU, Department of Pediatrics, Pediatric intensive Care Unit, Hopital Sainte‐Justine, University of Montreal, Canada Sponsorship source: Smith Kline & French Laboratories Conflicts of interest: ‐ Ethics approval: Study was approved by the Ethics Committee of Hopital Sainte‐Justine Informed consent: Written informed consent was obtained from a parent or guardian of each patient Clinical trials registration: ‐ Sample size calculation: We calculated a necessary sample size of 37 participants |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Quote: "Allocation of treatment followed a double‐blind pattern, according to a list set up by the Smith Kline & French Laboratories from a table of random numbers" Judgement comment: list of random number set up by study sponsor. Some risk of bias suspected |
| Allocation concealment (selection bias) | Unclear risk | Quote: "Allocation of treatment followed a double‐blind pattern, according to a list set up by the Smith Kline & French Laboratories from a table of random numbers" Judgement comment: no details about allocation concealment reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: "double‐blind pattern". Procedures for blinding not explicitly reported, but lack of blinding is unlikely to introduce bias to the outcomes or outcome measures |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Quote: "UGIB noted from the nasogastric tube, which had been inserted in each patient. Outcome was assessed by the appearance of UGIB and by gastric fluid pH. Massive UGIB was defined as red or brown haemorrhage from the nasogastric tube associated with a decrease in arterial blood pressure of >20 mm Hg or with an acute decrease in haemoglobin level of >2 gm/dl; obvious UGIB was defined as red or brown haemorrhage from the nasogastric tube without a decrease in arterial blood pressure or in haemoglobin level of >2 gm/dl; slight UGIB was defined as minimal bleeding appearing only at the aspiration of gastric fluid" Comment: criteria for diagnosis of this outcome reported objectively |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Unclear risk | Comment: no details about blinding of outcome assessors reported |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: no incomplete outcome data suspected. All participants randomised at baseline were included in analyses |
| Selective reporting (reporting bias) | Low risk | Comment: All outcomes listed in the Methods section (GI bleeding) were also reported in the Results section |
| Other bias | High risk | Comment: Industry participates in providing drugs and conducting the trial. Mechanical ventilation (risk factor for GI bleeding) was used more often in placebo group |
Laggner 1988.
| Methods | Open‐label randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 84 participants Number analysed: 84 participants Ranitidine
Sucralfate
Inclusion criteria
Notes: not clear whether any at all, 1, or more of these risk factors is required to be classified as high risk Exclusion criteria
Baseline imbalance: Both groups were comparable with respect to age, underlying disorders, and factors predisposing to development of stress ulcers |
|
| Interventions |
Ranitidine
Sucralfate
Adherence to regimen: no dropouts mentioned; however, after bleeding was detected, 1 participant in the sucralfate group was switched over to ranitidine. 2 individuals in the ranitidine group who had bleeding were switched to the sucralfate group, 1 to pirenzepine; in 3, enteral feeding was started Duration of trial: 18 months Duration of treatment: until participant was discharged from ICU or when acute problem improved, so that bleeding prophylaxis was no longer deemed necessary Duration of follow‐up: until patient was discharged from ICU or prophylaxis was no longer required (unclear how this was determined) |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported but not used in review
|
|
| Notes |
Setting: ICU Medical University Hospital, Wien, Austria Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Positive bacterial cultures were obtained from bronchial secretions of (43.3% of 74 bronchial secretions in participants receiving ranitidine and 18.6% of 59 bronchial secretions in those receiving sucralfate). Only 30 participants in the ranitidine group and 25 participants in the sucralfate group needed mechanical ventilation. The duration of this was 8.6 ± 4.6 in the ranitidine group and 8.9 ± 6.1 in the sucralfate group |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants. Therefore, high risk of performance bias |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: GI bleeding was detected as per the definition in the study protocol, and the the nature of the outcome of interest is objective |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: All other outcomes of interest were objective in nature, so the likelihood of performance bias is low |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All randomised participants completed the trial and were included in the final analysis. No treatment withdrawals and no trial group changes |
| Selective reporting (reporting bias) | High risk | Quote: "There was no difference in side effects between the two medication groups" Comment: Adverse reactions due to the interventions are not clearly mentioned in the study report. This would have caused reporting bias in the study report. All other intended outcomes are reported |
| Other bias | Low risk | Comment: no mention of the source of funding. No additional biases were detected |
Laggner 1989.
| Methods | Single‐blind randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 32 participants Number analysed: 32 participants Sucralfate
Ranitidine
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "Male: female ratios and mean age of the two treatment groups appear not quite similar,but we could not detect any significant difference" Comment: All participants were long‐term ventilated persons |
|
| Interventions |
Sucralfate
Ranitidine
Adherence to regimen: ‐ Duration of trial: ‐ Note duration of treatment: Quote: "The study was terminated when the patient was dismissed from the intensive care unit or died" Duration of follow‐up: not clearly mentioned in the study report |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
|
|
| Notes |
Setting: Department of Medicine, University of Vienna, Austria Source of funding: ‐ Conflicts of interest: Ethics approval: Quote: "The study was approved by the local ethical committee" Informed consent: Quote: "...and informed consent was obtained from the next of kin after the potential complications of the procedures were explained" Comment: Consent was obtained Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Quote: "During mechanical ventilation, upper gastrointestinal bleeding developed in three sucralfate‐ and seven ranitidine‐treated patients (18.7 versus 43.7 percent, p < 0.05). Until the end of the study, bleeding developed in only three sucralfate‐ but nine ranitidine treated patients (18.7 versus 56.2 percent, p ˜ 0.05)" Comment: Data for 9 participants (in ranitidine) were taken for analysis because it was felt that this occurred when participants were still in the ICU. One death in each group was attributed to GI bleed |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: ":.. 32 mechanically ventilated patients were randomly assigned, according to a preset table, to receive either ranitidine or sucralfate" Comment: not clear on how the sequence was generated in the "preset table" |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: This was not a placebo‐controlled trial. Moreover the different modes of administering study drug would not have made it possible to blind participants. Therefore the likelihood of performance bias is high |
| Blinding (detection bias) Clinically important upper GI bleeding | High risk | Comment: unclear whether outcome assessors were blinded to this outcome. The definition of this outcome was not clearly described in the study report |
| Blinding (detection bias) Nosocomial pneumonia | Low risk | Quote: ”Analysis of chest radiographs was performed by a radiologist, who was not aware of the radiologist, who was not aware of the clinical status or the therapy of the patients” Comment: Outcome assessors for nosocomial pneumonia were blinded |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: unclear whether outcome assessors were blinded to other outcomes of interest. However, outcomes were objective in nature. Therefore the likelihood of performance and detection bias is low |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All randomised participants were part of the final analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were reported in the study |
| Other bias | Low risk | Comment: unclear on the source of funding. No other sources of bias suspected |
Lamothe 1991.
| Methods | Open‐label randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 57 participants Number analysed: 57 participants Cimetidine
Famotidine
Ranitidine
Antacids
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "There were no statistically significant difference in age distribution or sex distribution among the four treatment groups" Comment: all participants about to undergo elective coronary artery bypass graft surgery |
|
| Interventions |
Cimetidine
Famotidine
Ranitidine
Antacids (Mylanta II)
Adherence to regimen: All participants received the drugs at least 12 hours before surgery and continued to receive them postoperatively. All participants used NG tube postoperatively Duration of trial: ‐ Duration of follow‐up: until discharge from hospital |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial report
Outcomes reported in report but not used in review
|
|
| Notes |
Setting: cardiac surgery at the Medical Centre of Delware Adherence to regimen: ‐ Source of funding: ‐ Ethics approval: ‐ Informed consent: Quote: "A consent form was signed by all patient before surgery" Comment: Consent was obtained Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: H2 receptor antagonists were combined to form a common interventional arm vs antacids, as the review did not aim to investigate intraclass effects among included interventions. Blood transfusions were done but were not attributed to GI bleeding |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants. Therefore, high risk of performance bias |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: unclear on blinding of outcome assessors. GI bleeding was an objective outcome that was detected as per the definition in the study protocol |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Unclear risk | Comment: All other outcomes were objective in nature, which was detected as per the definition in the study protocol. Therefore the likelihood of performance and detection bias is low |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: All randomised participants were accounted in the final analysis |
| Selective reporting (reporting bias) | Unclear risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: unclear on source of funding. No other known form of bias detected |
Larson 1989.
| Methods | Double‐blind placebo‐controlled trial | |
| Participants |
Baseline characteristics Number randomised: 31 participants Number analysed: 31 participants Ranitidine
Placebo
Inclusion criteria
Exclusion criteria
Baseline imbalances: Comment: All participants are admitted with severe head injury |
|
| Interventions |
Ranitidine
Placebo
Adherence to regimen: Comment: 1 participant died owing to the underlying cause of severe head injury on day 2, 1 participant removed pH electrode on day 3, and 5 participants in the placebo group had upper GI bleeding Duration of trial: ‐ Duration of follow‐up: ‐ |
|
| Outcomes |
Outcomes sought in review and reported in trial Primary outcomes
Secondary outcomes
Outcomes sought but not reported in trial report
Outcomes reported in report but not used in review
|
|
| Notes |
Setting: University of Louisville, Louisville, KY Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: This was a double‐blind placebo‐controlled trial. Therefore the likelihood of performance bias is low |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: not clearly mentioned in the study report. GI bleeding was an objective outcome that was detected as per the definition in the study |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: not clearly mentioned in the study report. However, the outcome was an objective outcome. Therefore, the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All randomised participants were part of the final analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | High risk | Comment: unclear on source of funding and baseline characteristics of participants |
Lee 2014.
| Methods | Parallel‐group randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 60 participants Number analysed: 60 participants Esomeprazole
Famotidine
Inclusion criteria
Exclusion criteria
Baseline imbalances: no significant difference for sex, age, GCS, AP‐II, intracranial pressure, and operation time between these 2 groups |
|
| Interventions |
Esomeprazole
Famotidine
Adherence to regimen: ‐ Duration of trial: March 2007 to March 2010 Duration of follow‐up: 30 days |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought in review but not reported in trial
Outcome reported in trial but not used in review
|
|
| Notes |
Setting: Neurosurgical ICU, Division of Gastroenterology and Hepatology, Department of Internal Medicine, Far Eastern Memorial Hospital, New Taipei, Taiwan Source of funding: Quote: "This study was performed in the Far Eastern Memorial Hospital and was supported by the research grant FEMH‐94‐C‐016 from the Far Eastern Memorial Hospital" Conflicts of interest: Quote: "All contributing authors declare no conflicts of interest" Ethics approval: Quote: "This study was approved by the Research Ethics Review Committee of the Far Eastern Memorial Hospital" Informed consent: Quote: "After explaining the study purpose and obtaining written consent from their family members" Clinical trials registration: ‐ Sample size calculation: ‐ Sponsorship source: supported by the research grant FEMH‐94‐ C‐016 from the Far Eastern Memorial Hospital |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "The patients were randomly allocated to two groups" Comment: not enough details reported |
| Allocation concealment (selection bias) | Unclear risk | Comment: no details reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: no details on blinding reported, but lack of blinding is unlikely to introduce bias to the outcomes or outcome measures |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Quote: "[We] defined upper gastrointestinal bleeding as tarry stool, haematemesis, drainage of more than 60 mL coffee
ground substance from nasogastric tube, or decreased haemoglobin level more than 2 g/dL with proved lesions by
endoscopic examination. We also defined positive stool occult blood test as occult bleeding" Comment: Outcome was measured objectively |
| Blinding (detection bias) Nosocomial pneumonia | Low risk | Quote: "We defined ventilator‐associated pneumonia as pneumonia occurring after 48 hours of ventilator use that fulfils three or more of the following four criteria: (1) presence of persistent (> 48 hours) or new onset infiltration in CXR; (2) positive sputum smear (with < 10 epithelial cells per low‐power field, 100×, and > 25 white blood cells per low‐power field or presence of polymorphonuclear cells with phagocytosis); (3) fever with body temperature > 38.3°C; and (4) leukocytosis > 12 × 10⁹/L" Comment: Outcome was measured objectively |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Unclear risk | Comment: no details reported |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: no incomplete outcome data; all participants randomised at baseline were included in analyses |
| Selective reporting (reporting bias) | Low risk | Comment: no incomplete reporting of outcomes suspected. All outcomes listed in the Methods section were also reported in the Results section |
| Other bias | Low risk | Comment: no other sources of bias suspected |
Levy 1997.
| Methods | Open‐label randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 70 participants Number analysed: 67 participants Ranitidine
Omeprazole
Inclusion criteria
Acute Physiologic and Chronic Health Evaluation (APACHE II) scores were calculated at baseline Exclusion criteria
Baseline imbalances: Quote: "There were no statistically significant differences between the ranitidine‐treated and the omeprazole‐treated groups for age, gender, race, or APACHE II scores. Ranitidine subjects had significantly more risk factors at baseline. There were no significant differences in the number of patients who required mechanical ventilation: ranitidine, 26/35 (72%) and omeprazole 16/32 (50%)" Comment: More participants in the ranitidine group had major neurological insults or trauma. 7 and 5 participants in both groups had coagulopathy at baseline |
|
| Interventions |
Ranitidine
Omeprazole
Adherence to regimen: "Seventy patients formed the study group. Thirty five were randomised to the ranitidine treatment group, 35 received omeprazole. Three omeprazole subjects were excluded from data analysis because of errors in randomisation or enrolment criteria protocol violations, resulting in 32 omeprazole‐treated patients included in the final analysis" Duration of trial: over a 10‐month period Duration of follow up: ‐ |
|
| Outcomes |
Outcomes sought in review and reported in trial Primary outcomes
Secondary outcomes
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
|
|
| Notes |
Setting: New Hanover Regional Medical Center and the Coastal AHEC, Wilmington, North Carolina, and Department of Medicine, School of Medicine, University of North Carolina at Chapel Hill Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Quote: "This study was approved by the Institutional Review Board" Informed consent: Quote: "...informed consent was obtained from each patient or their legally authorized representative" Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Mortality was related to increased APACHE II scores. Endoscopy was performed on 27 participants (ranitidine 15 and omeprazole 15) and stress ulcers were detected in all but 2 participants (from each group). The source of bleeding in these participants could not be determined, but it was presumed to be due to stress ulcerations |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants. Therefore, high risk of performance bias |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: unclear on blinding of outcome assessors. GI bleeding was detected as per the definition in the study protocol |
| Blinding (detection bias) Nosocomial pneumonia | High risk | Comment: unclear on blinding of outcome assessors. The definition of nosocomial pneumonia is not clearly mentioned in the study report |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: unclear on blinding of outcome assessors. However, owing to the objective nature of the outcomes of interest, the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comments: Three participants (from the omeprazole group) were not part of the final analysis for legitimate reasons such as errors in randomisation or enrolment criteria protocol violations. However, we did an intention‐to‐treat analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | High risk | Comment: Source of funding was not clearly mentioned in the study report. Baseline differences between groups were detected |
Lin 2016.
| Methods | Parallel‐group randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 120 participants Number analysed: 120 participants Lansoprazole
Other medications
Inclusion criteria
Exclusion criteria
Baseline imbalances: no difference in gender, age, and mean number of comorbidities between the 2 groups. Use of ulcerogenic medications was similar between the 2 groups. There was no significant difference between the 2 groups in GCS scores, initial APACHE II score in the medical or surgical ICUs, ventilator‐dependent days before starting to be weaned, rapid shallow index (a weaning parameter), and baseline albumin and haemoglobin levels before weaning |
|
| Interventions |
Lansoprazole
Other medications
Adherence to treatment: ‐ Duration of trial: June 2009 ‐ February 2012 Duration of follow‐up: 30 days |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought in review but not reported in trial
Outcomes reported in trial but not used in review
|
|
| Notes |
Setting: Respiratory Care Center, Division of Gastroenterology and Hepatology, Far Eastern Memorial Hospital, New Taipei City, Taiwan Sponsorship source: Quote: "This study was supported by a grant from Far Eastern Memorial Hospital (FEMH) (FEMH‐2008‐C‐043)" Conflicts of interest: ‐ Ethics approval: Quote: "The protocol was approved by the Research Ethics Review Committee of the Far Eastern Memorial Hospital" Informed consent: Quote: "After obtaining written consent from their families..." Clinical trials registration: ClinicalTrials.gov ID: NCT00708149, Sample size calculation: ‐ |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Quote: "They were randomly allocated into two groups using blocked randomisation" |
| Allocation concealment (selection bias) | Unclear risk | Comment: no details reported. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "non‐double‐blind" Comment: Performance bias is a possibility, even if outcomes are objective |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: outcome measured objectively as defined in the trial report |
| Blinding (detection bias) Nosocomial pneumonia | Low risk | Comment: outcome measured objectively as defined in the trial report |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Unclear risk | Quote: "randomised, non‐double‐blind" Comment: no blinding in place, lack of blinding unlikely to influence outcome measures and outcomes |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: no incomplete reporting suspected; all participants randomised at baseline are also included in analyses |
| Selective reporting (reporting bias) | Low risk | Quote: "(ClinicalTrials.gov ID: NCT00708149). Outcome measures. The primary end point of our study was apparent UGI bleeding 2,20 within 2 weeks of enrolment, which was defined as follows: (1) a 'coffee ground' substance from the NG aspirate 60 mL; (2) fresh blood from the NG tube; or (3) passage of tarry stool. Secondary end points included clinically significant UGI bleeding 2,20 (definition: UGI bleeding with haemoglobin level decrease !2 gm/dL or in need of a blood transfusion of > 2 units), successful weaning rate, ventilator‐associated pneumonia (definition: clinical pulmonary infection score 21 > 6, a scoring system composed of body temperature, white blood cell count, purulent secretions, diffuse or localized pulmonary infiltrate in CXR, progression of infiltrate, and culture of secretions), and a 30‐day survival rate" Comment: All outcomes reported in the Methods section are also reported in the Results |
| Other bias | Low risk | Comment: no other sources of bias suspected |
Lopez‐Herce 1992.
| Methods | Open‐label randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 165 participants Number analysed: 140 participants Overall
Inclusion criteria
Exclusion criteria
Baseline imbalances: Groups were similar when compared using indexes (Theraputic Interventiobn Scoring System, Physiological Stability Index, Multi Organ System Failure Scoring System, Zinner and Tryba). Detailed baseline characteristics for each group are not provided in the study report |
|
| Interventions |
No prophylaxis
Almagate
Ranitidine
Sucralfate
Adherence to regimen: 165 randomised, 25 "excluded because of protocol reasons"; 1 patient in the Amalgamate group had watery diarrhoea and treatment was switched to ranitidine. In 6 patients from the control group (no prophylaxis) who developed GI bleed, 2 were given Amalgamate, 2 were given sucralfate, and 2 were given amalgamate Duration of trial: June 1986 to June 1988 Duration of follow‐up: ‐ |
|
| Outcomes |
Outcomes sought in review and reported in trial Primary outcome
Secondary outcomes
Outcomes sought in review but not reported in trial
Outcomes reported in trial but not used in review
|
|
| Notes |
Setting: Pediatric ICU, LaPaz Children’s Hospital, Madrid, Spain Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Quote: "The study was approved by the institutional review board" Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: One of the 7 participants in the control group with GI bleeding did not receive any treatment; the others were given the following: amalgamate to 2 participants, ranitidine to 2 participants, and sucralfate to 2 participants. The last participant needed ranitidine too to decrease the intensity of bleeding. In the antacid group, 1 child died of haemorrhage, and the other was given ranitidine and sucralfate to contain the bleeding. In the ranitidine group, 1 participant improved within the first 24 hours, without receiving any other drug. In the second participant, the intensity of bleeding decreased when amalgamate was added, and the third participant died. In the sucralfate group, the statue of the lone participant with haemorrhage improved without any addition of another drug It is mentioned that there was no incidence of nosocomial pneumonia, but under "side effects", the study reports 5 incidences of the same and goes on to say that there was a difference between groups with respect to this outcome. The incidence in each group remains unclear |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants. Therefore, high risk of performance bias |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: no blinding of outcome assessors reported, but GI bleeding was detected as per the definition in the study protocol |
| Blinding (detection bias) Nosocomial pneumonia | High risk | Comment: no blinding of outcome assessors reported, and definition for detecting pneumonia was not clearly mentioned in the study protocol. Moreover the outcome was not clearly reported in the study |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: The outcome of interest was objective in nature, so the likelihood of performance and detection bias is low |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: Although it mentions that 165 children were randomised and 25 were excluded because of various protocol violations, it is not clear to which of the 4 study groups these 25 children belonged. A per‐protocol analysis was done, and there appears to be no imbalance between groups with respect to the number of participants available for analysis. Therefore, the likelihood of bias due to attrition is low |
| Selective reporting (reporting bias) | High risk | Comment: A high mortality rate of 38.4% is found in patients with important (major) upper GI bleeding, but counts for each intervention are not given separately. Data on macroscopic upper GI bleeding, slight (microscopic) upper GI bleeding, and mortality are not reported for each intervention separately but are reported for the entire study. Data for pneumonia are not clear |
| Other bias | Low risk | Comment: Study is unclear on source of funding. No other known source of bias |
Luk 1982.
| Methods | Double‐blind randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 182 participants Number analysed: 182 participants Cimetidine
Antacids
Placebo
No prophylaxis
Inclusion criteria
Exclusion criteria: ‐ Baseline imbalance: "All 4 groups were similar in age, sex, clinical severity of illness and mortality (25%)" |
|
| Interventions |
Cimetidine
Antacids
Placebo
No prophylaxis
Adherence to regimen: ‐ Duration of trial: ‐ Duration of follow‐up: ‐ |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought in review but not reported in trial report
Outcomes reported in report but not used in review
|
|
| Notes |
Setting: Johns Hopkins University, Baltimore, Maryland, USA Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Cimetidine and placebo helped reduce the incidence of GI bleeding in participants with respiratory illness |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: This was a double‐blind placebo‐controlled trial. Study personnel were blinded.Therefore the likelihood of performance bias is low |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: This was a double‐blind placebo‐controlled trial, and GI bleed was an objective outcome that was diagnosed as per the definition in the study protocol |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: No other outcome of interest was mentioned in this study |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All randomised participants were part of the final analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: unclear on source of funding. No other sources of bias suspected |
Maasoumi 2016.
| Methods | Parallel‐group randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 90 participants Number analysed: 90 participants Ranitidine
Pantoprazole
Inclusion criteria
Exclusion criteria: ‐ Baseline imbalances: ‐ |
|
| Interventions |
Ranitidine
Pantoprazole
Adherence to regimen: ‐ Duration of trial: ‐ Duration of follow‐up: ‐ |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
|
|
| Notes |
Setting: ICU Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: ‐ |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not enough detail reported. Quote: "randomly divided" |
| Allocation concealment (selection bias) | Unclear risk | Comment: insufficient information to allow judgement |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: no information about blinding reported; no definitions for diagnosis of outcomes reported |
| Blinding (detection bias) Clinically important upper GI bleeding | Unclear risk | Comment: Study did not address this outcome |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Unclear risk | Comment: no incomplete reporting of data suspected |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: no incomplete reporting of data suspected |
| Selective reporting (reporting bias) | Unclear risk | Comment: no selective reporting of outcomes suspected. Outcome reported in the Methods section is also reported in the Results section |
| Other bias | Unclear risk | Comment: not enough detail reported in conference abstract to assess risk of other biases |
Macdougall 1977.
| Methods | Open‐label randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 75 participants Number analysed: 75 participants Antacids
No prophylaxis
Inclusion criteria
Exclusion criteria: ‐ Baseline imbalances: ‐ |
|
| Interventions |
Antacids
No prophylaxis
Adherence to regimen: The first 25 participants received either antacids or no prophylaxis. The trial was discontinued when H2 receptor antagonists became available. Of the 50 remaining participants, 10 received metiamide and 16 received cimetidine (after case reports of agranulocytosis by metiamide). The remaining 24 got no prophylaxis Duration of trial: January 1975 to July 1976 Duration of follow‐up: ‐ |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought in review but not reported in trial
Outcomes reported in trial but not used in review
|
|
| Notes |
Setting: Liver Unit, King's College Hospital and Medical School, Denmark Hill, London SES Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Only data for the comparison of antacid with no prophylaxis were extracted for the review, as it was felt that the second part of the trial, which compared H2 receptor antagonist vs no prophylaxis, was not a properly randomised trial |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: This was not a placebo‐controlled trial, and the trial was stopped and H2 receptors administered instead of antacids |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: unclear on blinding of outcome assessors, but the definition for diagnosis of GI bleeding is mentioned in the study report |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: unclear on blinding of outcome assessors, but outcomes of interest were objective in nature |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All randomised participants were part of the final analysis |
| Selective reporting (reporting bias) | High risk | Comment: Data are not reported separately for participants who received cimetidine and metiamide. Mortality data are clubbed for both groups of controls (those compared with antacids and H2 receptor antagonists) |
| Other bias | High risk | Comment: unclear on source of funding and baseline characteristics of participants |
Mahul 1992.
| Methods | Single‐blind randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 158 participants Number analysed: 145 participants Overall
Inclusion criteria
Exclusion criteria
Baseline imbalances: 48 participants had 'primitive pneumonia' on admission, 14 had tracheobronchitis, and bacterial colonisation was noted in 36 participants, respectively (not clear to which intervention these participants belonged) |
|
| Interventions |
Antacids
Sucralfate
Adherence to regimen: Quote: "A single blind randomisation was performed within 12 hours of admission according to a simultaneous and independent dual randomisation. The first randomisation involved mechanical SSD versus no‐SSD. The second randomisation involved ulcer prophylaxis with aluminium hydroxide versus sucralfate. At the end, 4 random classes were defined" "158 of them were randomly selected on the probability of intubation for more than 3 days, 13 were then excluded because of death (n = 5) or extubation (n = 8) before day 3" Duration of trial: 14 months Duration of follow‐up: not clearly mentioned in the study report |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial report
Outcomes reported in report but not used in review
|
|
| Notes |
Setting: Hospital Nord, CHRU St. Etienne, France Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "A single blind randomisation was performed within 12 hours of admission according to a simultaneous and independent dual randomisation" Comment: not clear who were blinded. Therefore, unclear on the likelihood of performance bias |
| Blinding (detection bias) Clinically important upper GI bleeding | Unclear risk | Quote: "A single blind randomisation was performed within 12 hours of admission according to a simultaneous and independent dual randomisation" Comment: unclear on blinding of outcome assessors. The definition for detecting GI bleeding is not clearly mentioned in the study report |
| Blinding (detection bias) Nosocomial pneumonia | Low risk | Comment: unclear on blinding of outcome assessors. Nosocomial pneumonia was detected as per the definition in the study protocol |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: unclear on blinding of outcome assessors. However outcomes of interest were objective in nature, so the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: Of the 158 randomised participants, only 145 were part of the final analysis. There were 13 dropouts for the reasons mentioned above. The group to which they were randomised is not clearly mentioned in the study report, and an intention‐to‐treat analysis was not done.However, the dropouts accounted for less than 10% of randomised participants and appear to be equally distributed (given the double randomisation design of the study). Therefore, the likelihood of attrition bias is low |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | High risk | Comment: source of funding and baseline imbalances of antacid and sucralfate groups unclear |
Maier 1994.
| Methods | Open‐label randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 98 participants Number analysed: 98 participants Ranitidine
Sucralfate
Inclusion criteria
Exclusion criteria
Baseline imbalances: The 2 groups were similar with respect to gender distribution, age, admission APACHE II scores, Injury Severity Score, Revised Trauma Score, and history of smoking. Overall, patterns of injury were also similar in both groups |
|
| Interventions |
Ranitidine
Sucralfate
Adherence to regimen: All 98 participants were admitted for a minimum of 72 hours in ICU Duration of trial: April 1991 to October 1993 Duration of follow‐up: up to 2 weeks |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial report
Outcomes reported in report but not used in review
|
|
| Notes |
Setting: Harborview Medical Center, 325 Ninth Avenue ZA ‐ 16, Seattle, WA 98104 Source of funding: Quote: "Supported in part by grant C#R49/CCR002570" Conflicts of interest: ‐ Ethics approval: Quote: "The study was approved by the University of Washington Human Subjects Review Board" Informed consent: Quote: "Informed consent was obtained from each patient or patient representative within 24 hours of study enrolment" Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Of the 12 participants who were classified as having gross bleeding, 7 in ranitidine and 5 in sucralfate groups had 'coffee‐ground' aspirates |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants. Therefore, high risk of performance bias |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: unclear on blinding of outcome assessors. However, GI bleeding was an objective outcome that was detected as per the definition in study protocol |
| Blinding (detection bias) Nosocomial pneumonia | Low risk | Comment: unclear on blinding of outcome assessors. However, nosocomial pneumonia was an objective outcome that was detected as per the definition in the study protocol |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: unclear on blinding of outcome assessors. However, all other outcomes of interest were objective in nature. Therefore, the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All randomised participants completed the trial and were included in the final analysis. There are no treatment withdrawals and no trial group changes. Therefore, there is no attrition bias |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: Trial is supported by a grant from the Centers for Disease and Control and Prevention CDC#R49/CCR002570. The role of the sponsor in the conduct and reporting of the trial is unclear. No other source of bias detected |
Martin 1980.
| Methods | Quasi‐randomised trial | |
| Participants |
Baseline characteristics Number randomised: 77 participants Number analysed: 77 participants Antacids
Cimetidine
Inclusion criteria
Exclusion criteria
Baseline imbalances: no difference between antacid and cimetidine groups in age and gender. The main reasons for admission were head injury and orthopaedic injury |
|
| Interventions |
Antacids
Cimetidine
Adherence to regimen: 49 participants in both groups maintained a gastric pH ≥ 4 (29 in antacid and 20 in cimetidine). Fifteen participants required increase in dosage, as they were not able to maintain a gastric pH ≥ 4 at the initial dose (6 in antacid and 7 in cimetidine). For 4 participants who were on cimetidine, an additional antacid administration was required. Nine participants failed to maintain a pH ≥ 4 despite maximum dose as per the study protocol (2 in antacid and 7 in cimetidine) Duration of trial: January 1979 to August 1979 Duration of follow‐up: Quote: "Patients were observed throughout their hospitalisation for GI bleeding and if it developed they were readmitted to the study and to intensive care unit" |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
|
|
| Notes |
Setting: Louisville General Hospital surgical intensive care unit Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: It is mentioned that 19 participants had pneumonia (7 in antacid and 12 in cimetidine), but it was not an outcome intended to be reported in the study. Not sure if this was present on admission |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Quote: "patients were assigned to either antacid or cimetidine treatment group according to odd or even date of admission" Comment: This was a quasi‐randomised trial, so sequence generation was not done |
| Allocation concealment (selection bias) | High risk | Quote: "patients were assigned to either antacid or cimetidine treatment group according to odd or even date of admission" Comment: This was a quasi‐randomised trial in which allocation was not concealed |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants. Therefore, high risk of performance bias |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: Blinding was not done ,and GI bleeding was an objective outcome that was detected as per the definition in the study objectives |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Blinding was not done, and there was no intention to report pneumonia in the study objectives. It was reported when it was diagnosed in participants and was the main cause of sepsis in participants. Unclear whether pneumonia was present on admission |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: All other outcomes of interest were objective in nature; because of this, the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All participants were included in the final analysis. There was no attrition bias |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported. Pneumonia was reported although it was not intended. However this was an outcome of interest for the review. Therefore it could have caused reporting bias |
| Other bias | Low risk | Comment: Source of funding is unclear from the study report. No other sources of bias are suspected |
Martin 1992.
| Methods | Double‐blind randomised, double‐dummy trial | |
| Participants |
Baseline characteristics Number randomised: 127 participants Number analysed: 127 participants Misoprostol
Cimetidine
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "The groups were clinically equivalent at entry with respect to age, gender race, risk factors such as hypotension, sepsis, coagulopathy, renal failure, hepatic failure, cardiac failure, adult respiratory distress syndrome (ARDS), gastric and duodenal lesions" Comment: no significant difference between the 2 groups with respect to demographic and baseline risk factors. Most participants were given a diagnosis of hypotension at baseline. Coagulopatyhy was present in 15 and 14 participants in both groups. Only 15 and 19 participants in both groups were free of any haemorrhagic gastric lesions at baseline |
|
| Interventions |
Misoprostol
Cimetidine
Adherence to regimen: Quote: "Patients meeting the above criteria for bleeding underwent an endoscopic evaluation within 12 hours and were removed from the study if a upper GI bleeding source was confirmed. All patients still in the study at 72 hours underwent a follow‐up endoscopy to determine whether the condition of the gastric or duodenal mucosa had changed. When possible, patients also underwent an endoscopy on exit from the study. If a patient underwent more than one follow‐up endoscopy, the score that represented the most severe damage was used" Duration of trial: July 1986 to January 1988. Duration of follow up: Quote: "All patients still in the study at 72 hours underwent a follow‐up endoscopy to determine whether the condition of the gastric or duodenal mucosa had changed" Notes duration of treatment: Patients were studied until 1 of 3 events occurred:
|
|
| Outcomes |
Outcomes sought in review and reported in trial
Note: GI bleeding developed 3 to 14 days after the first dose of study medication
Outcomes sought but not reported in trial
Outcomes reported in the trial but not used in review
|
|
| Notes |
Setting: 25 medical centres in United States: Medical University of South Carolina, Charleston, South Carolina; Our Lady of Mercy Center, Bronx, New York; Cook County Hospital, Chicago, Illinois; Medical Center of Central George, Macon, Georgia; VA Medical Center, Dayton, Ohio, St. Francis Medical Center, Trenton, New Jersey; Maine Medical Center, Portland, Maine; VA Medical Center, Detroit, Michigan; Buffalo General Hospital, Buffalo, New York; University of South Alabama, Mobile, Alabama; Butterworth Hospital, Grand Rapids, Michigan; Indiana University Medical Center, Indianapolis, Indiana; Brackenridge Hospital, Austin,Texas; Meharry Medical Center, NashvilleTennessee; 6196 Eagle Crest Drive, Huntington Beach, California; Humana Hospital‐University, Louisville, Kentucky; Hershey Medical Center, Hershey, Pennsylvania; University Hospital, Columbia, Missouri; Denver General Hospital, Denver, Colorado; Truman Medical Center, Kansas City, Missouri; VA Medical Center, Long Beach, California; Chicago Medical School, North Chicago, Illinois; St. Louis University Medical Center, St. Louis, Missouri; Buffalo VA Medical Center, Buffalo, New York; University of Chicago, Chicago, Illinois Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Quote: "At each of the 25 institutions, the protocol was approved by the institutional review board" Informed consent: Quote: "Written informed consent was obtained from the patient or surrogate before study entry" Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Mortality was significantly associated with adult respiratory distress syndrome (ARDS), at baseline or at subsequent development, upper GI haemorrhage and additional organ system failure. 87 participants were given diagnosis of haemorrhagic lesions, and 10 participants met the criteria of upper GI haemorrhage as per the study definition |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: This was a double‐blind 'double‐dummy study' in which placebo was administered to both groups. There personnel involved were blinded, and so the likelihood of performance bias is low |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: This was a double‐blind 'double‐dummy study' in which placebo was administered to both groups. GI bleeding was an objective outcome that was detected as per the definition in the study protocol |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: unclear whether outcome assessors were blinded. However, owing to the objective nature of the outcome of interest, the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All randomised participants completed the trial and were included in the final analysis. There are no treatment withdrawals and no trial group changes. Therefore there is no attrition bias |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: Source of funding is unclear. No other sources of bias are suspected |
Martin 1993.
| Methods | Multi‐centre double‐blind randomised placebo‐controlled trial | |
| Participants |
Baseline characteristics Number randomised: 131 participants Number analysed: 131 participants Cimetidine
Placebo
Inclusion criteria
Exclusion criteria
Baseline imbalances: The 2 treatment groups were similar in terms of demographic and clinical characteristics at baseline, including age, sex, race, type and number of risk factors for bleeding, and nasogastric pH. Most participants had major surgery (40 in cimetidine and 49 in placebo groups). Pneumonia was diagnosed at baseline in 9 and 5 participants, respectively |
|
| Interventions |
Cimetidine
Placebo
Adherence to regimen: Patients with renal failure were given 25 mg/h. Patients with pH below 4 on two different occasions (1 hour apart) 100 mg/h (if renally impaired 50 mg/h) Duration of trial: September 1988 to March 1989 Duration of follow up: In participants who developed GI bleeding, transfusion monitoring was continued for an additional 24‐hour period and a chest radiograph was taken, 48 hours after the medications were discontinued |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial report
Outcomes reported in report but not used in review
|
|
| Notes |
Setting: multi‐centre study (20 institutions) Source of funding: Smith Kline Beecham Pharmaceuticals Conflicts of interest: ‐ Ethics approval: Quote: "The study was approved by the institutional review board" Clinical trials registration: ‐ Sample size calculation: Quote: "The study was designed as a multi centred trial with 200 randomised participants. The sample size was derived from the assumption that the population failure rate for placebo infused patients would be 20% vs. 5% for cimetidine infused patients. Thus, 100 patients per group (200 patients total) would be required to provide a power of 90%, with a two sided type 1 error of 0.05" Comment: Study was designed to include 200 participants but was terminated after enrolment of 131 participants because of the statically significant reduction in bleeding among participants treated with cimetidine Additional notes: Enteral feeding was given to 5 participants (4 in cimetidine and 1 in placebo groups, and among these a participant from the cimetidine group had protocol defined GI bleeding). An intention to treat was done for the incidence of nosocomial pneumonia (although it was present at baseline in 9 and 5 participants, respectively) as the study definition required new and persistent infiltrates on the chest radiograph. Mortality was not attributed to upper GI haemorrhage |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Low risk | Quote: "All the patients received a one time, 50 mL loading dose of coded medication (cimetidine or placebo) in 5% dextrose in water that was infused over a 20 minute period. This loading dose was followed immediately by a continuous infusion of coded medication (cimetidine or placebo) in 5% dextrose in water at a rate of approximately 10.4 mL/hour, using an infusion pump" Comment: Allocation concealment might have been done since the medication is coded |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "All the patients received a one time, 50 mL loading dose of coded medication (cimetidine or placebo) in 5% dextrose in water that was infused over a 20 minute period" Comments: This was a placebo‐controlled study in which medications administered were coded and participants and personnel involved in the trial were blinded |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Quote: "To prevent bias in the interpretation of endpoints and safety data each institution designated one investigator to monitor pH and a second investigator, blinded to the pH determinations, to monitor signs and symptoms of upper GI haemorrhage, pneumonia and other safety parameters" Comment: This was a placebo‐controlled study in which medications administered were coded and the outcome assessor who monitored tsigns and symptoms of GI bleeding was unaware of the gastric pH value of respective participants. Moreover GI bleeding was an objective outcome detected as per the definition in the study protocol |
| Blinding (detection bias) Nosocomial pneumonia | Low risk | Quote: "To prevent bias in the interpretation of endpoints and safety data each institution designated one investigator to monitor pH and a second investigator, blinded to the pH determinations, to monitor signs and symptoms of upper GI haemorrhage, pneumonia and other safety parameters" Comment: This was a placebo‐controlled study in which medications administered were coded and the outcome assessor who monitored signs and symptoms for pneumonia was unaware of the gastric pH value of respective participants. Moreover, nosocomial pneumonia was an objective outcome detected as per the definition in the study protocol |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: This was a placebo‐controlled trial in which medications administered were coded. This would have ensured blinding for other outcome assessments. Moreover, outcomes of interest were objective in nature |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All randomised participants were included in the final analysis. There are no treatment withdrawals and no trial group changes |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: Smith Kline Beecham Pharmaceuticals funded part of the study. The role of the sponsor in the conduct and reporting of the trial is unclear. No other sources of bias are suspected |
Metz 1993.
| Methods | Multi‐centre double‐blind randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 167 participants Number analysed: 167 participants Ranitidine
Placebo
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "No statistically significant difference was present between treatment groups with regard to any demographic variables" Comment: The imbalance between the 2 groups was only with respect to number of people on mechanical ventilation at study entry. 65 in placebo group and 80 in the ranitidine group. The other baseline characteristics were comparable. Nosocomial pneumonia was present on entry in 2 participants in both groups, respectively. Prothrombin time > upper limit was present in 31/77 and 28/83 participants, respectively |
|
| Interventions |
Ranitidine
Placebo
Adherence to regimen: If upper GI bleeding was detected according to the definition, then participant was withdrawn from the study. All participants adhered to the prescribed study regimen Duration of trial: January 1990 to September 1991 Duration of follow‐up: probably until discharge or untimely death |
|
| Outcomes |
Outcomes sought in review and reported in trial Primary outcomes
If any of the preceding variables were present, the following 4 questions were addressed to establish the diagnosis of stress ulcer GI bleeding:
If the answer to any of the preceding 4 questions was "yes", the participant was considered to have GI bleeding Secondary outcomes
Outcomes sought but not reported in trial
Outcomes reported in report but not used in review
|
|
| Notes |
Setting: 10 ICUs from across the United States of America Source of funding: Quote: "This study was supported in part by a research grant from Glaxo Pharmaceuticals" Ethics approval: Quote: "The study was approved by the institutional review boards of all participating sites." Comment: ethics approval obtained from all 10 participating sites Informed consent: Quote: "Informed consent was obtained from patient or a legally authorized representative" Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Two participants who were diagnosed with pneumonia at baseline were excluded from the denominator of participants who subsequently developed nosocomial pneumonia during the study |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were then randomised to treatment with 6.25 mg/hour continuous ranitidine or placebo infusion according to a computer generated randomisation scheme" Comment: Method adopted to obtain random sequence generation is clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: This is a placebo‐controlled trial in which both the intervention and the control were administered at the same rate as per a computer‐generated randomisation scheme, which suggests that participants and study personnel were blinded. Therefore the likelihood of performance bias is low |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: This was a placebo‐controlled trial, and GI bleeding was an objective outcome that was detected as per the definition in the study report |
| Blinding (detection bias) Nosocomial pneumonia | Low risk | Comment: This is a placebo‐controlled trial, and nosocomial pneumonia was an objective outcome detected as per the definition in the study report |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: This is a placebo‐controlled trial, and all other outcomes of interest were objective in nature. Therefore the likelihood of performance or detection bias is low |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All randomised participants completed the trial and were included in the final analysis. There are no treatment withdrawals and no trial group changes. Therefore the likelihood of attrition bias is low |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: This study was supported in part by a research grant from Glaxo Pharmaceuticals, and some of the equipment used was provided by this organisation. The role of the sponsor in the conduct and reporting of the trial is unclear. No other form of bias was detected |
Mustafa 1994.
| Methods | Open‐label randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 31 participants Number analysed: 31 participants Ranitidine
Sucralfate
Inclusion criteria
Exclusion criteria
Baseline imbalances: no significant difference between the 2 groups with respect to demographic and baseline risk factors. Most participants were admitted post surgery |
|
| Interventions |
Ranitidine
Sucralfate
Adherence to regimen: All participants appear to have adhered to the regimen to which they were randomised Duration of trial: ‐ Duration of follow‐up: probably until discharge or untimely death of the participant |
|
| Outcomes |
Outcomes sought in review and reported in trial Primary outcomes
Secondary outcomes
Outcomes sought but not reported in trial
Outcin report but not used in review
|
|
| Notes |
Setting: Department of Intensive Care Unit, Karadeniz Teknik University, Trabzon, Turkey Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Only 3 participants treated with ranitidine and 1 with sucralfate had secondary pneumonia due to some bacterial agent isolated from the stomach. Colonistion of the oropharynx and tracheostomy were more common in participants treated with ranitidine |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: This was not a placebo‐controlled trial, and blinding of study personnel or participants was not possible owing to the different modes of administration of study drugs |
| Blinding (detection bias) Clinically important upper GI bleeding | Unclear risk | Comment: The definition for detecting GI bleeding was not clearly mentioned in the study report, and the study is unclear on blinding of outcome assessors |
| Blinding (detection bias) Nosocomial pneumonia | Low risk | Comment: The definition for detecting secondary pneumonia was clearly mentioned in the study report. Still, the study is unclear on blinding of outcome assessors |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: All other outcomes were objective in nature, so the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All randomised participants completed the trial and were included in the final analysis.There are no treatment withdrawals and no trial group changes. |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were reported |
| Other bias | Low risk | Comment: No mention of the source of funding. No additional biases were detected |
Ng 2012.
| Methods | Parallel‐group randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 313 Number analysed: 311 Esomeprazole
Famotidine
Inclusion criteria
Exclusion criteria
Baseline imbalances: The 2 treatment groups were similar with respect to baseline demographic characteristics, history of ulcers, cardiac disease, percutaneous coronary stenting, baseline haemoglobin and serum creatinine levels, and use of enoxaparin or thrombolytics |
|
| Interventions |
Esomeprazole
Famotidine
Adherence to regimen: Compliance was assessed by pill count. Good compliance with study drugs (≥ 90% ): 100% in famotidine group and 98.8% in esomeprazole group Duration of trial: July 2008 to September 2010 Duration of follow‐up: minimum of 4 weeks and maximum of 52 weeks |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought in review but not reported in trial
Outcomes reported in trial, but not used in review
|
|
| Notes |
Setting: acute medical wards, cardiac care unit, and intensive care unit, Department of Medicine and Geriatric, Ruttonjee Hospital, Hong Kong Sponsorship source: Cardiac Research Fund, Ruttonjee Hospital Conflict of interest: Quote: "Potential competing interests: None" Ethics approval: Quote: "The study protocol was approved by the Ethics Committee of the Hong Kong East Cluster" Informed consent: Quote: "All patients gave their written, informed consent" Clinical trials registration: This study was registered at http://www.clinicaltrials.gov (Identifier NCT00683111) Sample size calculation: Yes, described under statistical analysis |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Ward clerks at the four acute medical wards, the cardiac ward, and the intensive care ward generated 50 treatment codes labelled A and 50 codes labelled B. These were sealed in identical, blinded envelopes that were shuffled randomly" |
| Allocation concealment (selection bias) | Low risk | Quote: "Ward clerks at the four acute medical wards, the cardiac ward, and the intensive care ward generated 50 treatment codes labelled A and 50 codes labelled B. These were sealed in identical, blinded envelopes that were shuffled randomly. Th e investigators drew a blinded envelope randomly, and the pharmacist dispensed the repackaged medi‐ cation. The investigators and patients were blinded to the treatment‐group assignments" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The investigators drew a blinded envelope randomly, and the pharmacist dispensed the repackaged medication. The investigators and patients were blinded to the treatment‐group assignments. The treatment codes were released aft er approval of the completion of the study by the Ethics Committee" |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Quote: "overt bleeding of gastroduodenal origin (confirmed by means of upper endoscopy), overt upper GIB of unknown origin, bleeding of occult gastrointestinal origin (confirmed by means of upper gastrointestinal endoscopy), obstruction, or perforation" Comment: objective criteria for the measurement of GI bleeding reported |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Quote: "The investigators drew a blinded envelope randomly, and the pharmacist dispensed the repackaged medication. The investigators and patients were blinded to the treatment‐group assignments. The treatment codes were released after approval of the completion of the study by the Ethics Committee" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "No patients were lost to follow‐up. Premature termination occurred in 34 (20.9%) and 31 (20.9%) patients in the esomeprazole and famotidine groups, respectively. No patient in the esomeprazole group and three (2.1%) patients in the famotidine group refused to continue the study. In the famotidine group, one patient with significant dyspepsia withdrew consent, while the remaining two patients did not give specific reasons" Comment: Flow chart of participant flow is included, and no incomplete reporting of outcome data is suspected |
| Selective reporting (reporting bias) | Low risk | Comment: All outcomes that are listed in the Methods section are also reported in the Results section |
| Other bias | Low risk | Comment: no other sources of bias suspected |
Noseworthy 1987.
| Methods | Open‐label randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 86 participants Number analysed: 86 participants Ranitidine
Antacids
Inclusion criteria
Exclusion criteria
Baseline imbalances: Groups were similar with respect to age, gender, and admission diagnosis. Acute and chronic respiratory failure were the most common causes for admission |
|
| Interventions |
Ranitidine
Antacids
Adherence to regimen: Quote: "Eightysix patients were randomised, 42 received ranitidine, 44 received antacids (malox: 42 and amphojel:3), 38 receiving ranitidine completed 48 hours of study while 23 continued up to 72 hours. Of the patients receiving antacids, 39 completed 48 hours of study, while 25 continued up to 76 hours" Duration of trial: ‐ Duration of follow‐up: ‐ |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
|
|
| Notes |
Setting: Department of Adult Intensive Care, Royal Alexandria Hospitals, Edmonton, and the Division of Critical Care Medicine, University of Alberta, Edmonton, Alberta, Canada Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “Using a computer generated table of numbers, patients were assigned by restricted randomisation...” Comment: Method adopted to obtain random sequence generation is clearly mentioned in the study report |
| Allocation concealment (selection bias) | Low risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants.Therefore, high risk of performance bias |
| Blinding (detection bias) Clinically important upper GI bleeding | High risk | Comment: There was no clear definition for detecting clinically significant upper GI bleeding, and it is unclear whether the unblinded nature of the study influenced this outcome, which was otherwise objective in nature |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: All other outcomes of interest were objective in nature. Therefore the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: Although 1 participant withdrew from each group during the course of the study, all randomised participants were part of the final analysis.Therefore there was no attrition bias |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes have been reported |
| Other bias | Low risk | Comment: no mention of the source of funding. No additional biases were detected |
Ortiz 1998.
| Methods | Open‐label randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 78 participants Number analysed: 78 participants (for outcomes of interest to this review) Cimetidine bolus
Cimetidine continuous
Sucralfate
No prophylaxis
Inclusion criteria
Exclusion criteria
Baseline imbalances: Baseline characteristics for participants who met the criteria for early withdrawal from the study (for reasons mentioned below) are not mentioned. Nearly 40 participants were withdrawn from the study |
|
| Interventions |
Cimetidine bolus
Cimetidine continuous
Sucralfate
No prophylaxis
Adherence to regimen: Quote: "Patients who met criteria for early withdrawal (< 3 days) were not included in the final analysis" Comment: Withdrawal was mainly due to GI bleeding, gastric colonisation on entry, extubation, mortality, and inability to aspirate GI secretions and change to tube feedings Duration of trial: ‐ Duration of follow‐up: not clearly mentioned in the study report. Probably until death or discharge |
|
| Outcomes |
Outcomes sought in review and reported in trial (none of them were the primary outcomes in the study)
Note to 1: Only those that occurred within the first 3 days and caused withdrawal were reported Outcomes sought but not reported in trial report
Outcomes reported in report but not used in review
|
|
| Notes |
Setting: Department of Critical Care Medicine, Saint Vincent Hospital, Worcester, Massachusetts, and Department of Surgery, New England Medical Centre, Boston, Massachusetts Source of funding: Smith Kline Beecham, Inc., Philadelphia, Pennsylvania Informed consent: ‐ Ethics approval: Quote: "The study was approved by the institutional review board" Clinical trials registration: ‐ Sample size calculation: ‐ Comment: Power analysis has been done at the end of the study to detect a correlation of less than 0.4 Additional notes: Cimetidine arms were combined to form a common interventional arm as the review did not aim to investigate efficacy on the basis of dose or mode of administration in the same drug |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "All other patients were randomised (table of random numbers) into one of the three groups" Comment: Method adopted to obtain random sequence generation is clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: This is an unblinded trial, and GI bleeding was detected as per the definition in the trial protocol |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: This is an unblinded trial, and all other outcomes of interest were objective in nature |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: 40 randomised participants were withdrawn from the study (more than 50%) for protocol violations and were excluded from the final analysis. However, relevant data (for GI bleed, ICU mortality) could be obtained from the study |
| Selective reporting (reporting bias) | High risk | Comment: The duration of intubation,hospital mortality and participants requiring antibiotics are mentioned only for participants who were part of the final analysis, and this excluded earlier withdrawals. This accounts for selective reporting |
| Other bias | High risk | Comment: This study was supported by a grant from Smith Kline Beecham, and some of the equipment used was received from this organisation. However, the role of the sponsor in the conduct and reporting of the trial is unclear. Baseline characteristics for 40 participants (excluded owing to early withdrawal) are not mentioned in the study report |
Pan 2004.
| Methods | Open‐label randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 30 participants Number analysed: 30 participants Pantoprazole
Famotidine
Inclusion criteria
Exclusion criteria
Baseline imbalances: ‐ |
|
| Interventions |
Pantoprazole
Famotidine
Adherence to regimen: no loss to follow‐up; all finished treatment Duration of trial: ‐ Duration of follow‐up: not clearly mentioned in the study report. Probably until death or discharge from the hospital |
|
| Outcomes |
Outcomes sought in review and reported in trial Primary outcome
Outcomes sought but not reported in trial report
Outcomes reported in report but not used in review
|
|
| Notes |
Setting: ‐ Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: not clearly mentioned in the study report. However, this was not a placebo‐controlled trial, and modes of interventions were different. Therefore it might not have been possible to blind personnel, so the likelihood of performance bias is low |
| Blinding (detection bias) Clinically important upper GI bleeding | High risk | Comment: not clearly mentioned in the study report. However, GI bleeding was an objective outcome, but the definition was not clearly mentioned in the study report |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: No other outcomes of interest were part of this study |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All randomised participants were part of the final analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All outcomes of interest were analysed and reported |
| Other bias | Low risk | Comment: unclear on the source of funding. No other source of bias detected |
Peura 1985.
| Methods | Double‐blind randomised placebo‐controlled trial | |
| Participants |
Baseline characteristics Number randomised: 39 participants Number analysed: 39 participants Cimetidine
Placebo
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "The treatment groups were similar in the number of patients, sex and severity of illness as determined by the primary diagnosis. The cimetidine group tended to be older, but this difference was not statistically significant. Both treatment arms were also similar in the initial pretreatment endoscopic classification" Comment: The 2 groups appear to be similar in their demographic and other characteristics, suggesting no imbalance between the 2 at baseline. Mosg in both groups were diagnosed with cardiopulmonary disease |
|
| Interventions |
Cimetidine
Placebo
Adherence to regimen: ‐ Duration of trial: ‐ Duration of follow‐up: not clearly mentioned in the study report. Probably until death or discharge from the hospital |
|
| Outcomes |
Outcomes sought in review and reported in trial Primary outcome
Secondary outcomes
Outcomes sought but not reported in trial report
Outcomes reported but not used in review
|
|
| Notes |
Setting: Gastroenterology Service, Department of Medicine, Walter Reed Army Medical Center, Washington, DC Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Quote: "This protocol was approved by the Clinical Investigation and Human Use Committees of the Walter Reed Army Medical Center" Informed consent: Quote: "Each patient or guardian gave informed consent" Clinical trials registration: not provided in the study report Sample size calculation: ‐ Additional notes: On initial endoscopy, it was found that there were endoscopic signs of bleeding in 14 of 29 participants with mucosal abnormalities. 3 participants from the placebo arm developed new signs of bleeding during the study, when serial endoscopy was done. One death in the placebo group was attributed to upper GI bleed |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: “The group not receiving cimetidine received an intravenous injection of placebo every 6 hours to ensure the double blind nature of the study” Comment: This suggests that participants and personnel were blinded to the interventions |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Quote: "All endoscopic examinations were done by a single investigator and witnessed by a second investigator who observed the procedure through a lecturescope. During the endoscopy, findings were discussed and agreed on by both investigators before an entry was made on the report form. Both investigators were uninformed as to the patient’s treatment group" Comment: Outcome assessors were blinded. The definition for detecting GI bleed was not clearly mentioned. However, it was an objective outcome detected through endoscopy. Therefore the likelihood of performance or detection bias is low |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: All other outcomes of interest were objective in nature |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All randomised participants were part of the final analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were reported and analysed in the study |
| Other bias | Low risk | Comment: unclear on the source of funding. No additional biases were detected |
Phillips 1998.
| Methods | Multi‐centre randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: not clear Number analysed: 58 participants Omeprazole
Ranitidine
Inclusion criteria
Exclusion criteria: not clearly mentioned in the study report, most probably participants who did not satisfy the inclusion criteria Baseline imbalances: Quote: "APACHE II, ISS scores and other variables were comparable in both the groups" Comment: The 3 groups appeared to be comparable at baseline |
|
| Interventions |
Omeprazole
Ranitidine
Adherence to regimen: ‐ Duration of trial: ‐ Duration of follow‐up: ‐ |
|
| Outcomes |
Outcomes sought in review and reported in trial Primary outcomes
Secondary outcomes
Outcomes sought but not reported in trial
Outcomes reported but not used in review
|
|
| Notes |
Setting: University Hospitals and Clinics, Department of Surgery, 1 Hospital Drive, 65212, USA Source of funding: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: This was a poster presented at Society of Critical Care Medicine; 27th Educational and Scientific Symposium; San Antonio, Texas, USA; February 4 to 8 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants. Therefore, high risk of performance bias |
| Blinding (detection bias) Clinically important upper GI bleeding | High risk | Comment: no clear mention of blinding of outcome assessors. Moreover, there is no clear definition to detect GI bleeding |
| Blinding (detection bias) Nosocomial pneumonia | High risk | Comment: no clear mention of blinding of outcome assessors. Moreover, there is no clear definition to detect pneumonia |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: No other outcomes of interest were reported |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: Report says there were 58 evaluable participants; it does not specify the number randomised |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: unclear on source of funding. No other source of bias suspected |
Pickworth 1993.
| Methods | Open‐label quasi‐randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 92 participants Number analysed: 83 participants Sucralfate
Ranitidine
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "There were no significant differences between the two groups in demographic data, trauma score, revised trauma score, Injury severity score and APACHE II score" |
|
| Interventions |
Sucralfate
Ranitidine
Adherence to regimen: Of the 92 participants, 9 were subsequently excluded for protocol breaks: 4 patients because they did not meet age criteria, 3 patients because admitting chest radiographic films were abnormal, and 2 patients because of inadvertent extubation Duration of trial: January 1989 to August 1991 Duration of follow‐up: All patients were followed until hospital discharge or death |
|
| Outcomes |
Outcomes sought in review and reported in trial Primary outcomes
Secondary outcomes
Outcomes sought but not reported in trial report
Outcomes reported in report but not used in review
|
|
| Notes |
Setting: SICU, Grant Medical Centre, Columbus, Ohio, USA Source of funding: Quote: "Supported in part by a Roche Hospital Pharmacy Research Grant" Conflicts of interest: ‐ Ethics approval: Study was approved and monitored by the Institutional Review Board Informed consent: Consent for therapy was considered consent for study inclusion, and specific informed consent was waived by the Institutional Review Board Clinical trials registration: ‐ Sample size calculation: Quote: "The present study was designed to produce a power of 90% with a 30% difference in pneumonia rates between the groups" |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Quote: "Randomisation occurred by the use of computer generated random number table. Odd numbered patients received ranitidine 50 mg every 6 hours and even numbered patients received sucralfate 1 g dissolved in 15 mL of sterile water ..." Comment: Method adopted to obtain random sequence generation is clearly mentioned in the study report but is not adequate to generate a random sequence |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: Different modes of administering study drugs and absence of placebo would not have made it possible to blind the study |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: Study report is unclear on blinding of outcome assessors. However GI bleeding was an objective outcome detected as per the definition in the study protocol |
| Blinding (detection bias) Nosocomial pneumonia | Low risk | Comment: Study report is unclear on blinding of outcome assessors. However, nosocomial pneumonia was detected as per the definition in the study protocol |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Unclear risk | Comment: Study report is unclear on blinding of outcome assessors. However, all other outcomes were objective in nature |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "Nine patients were subsequently excluded for protocol breaks: four patients because they did not meet the age criteria, three patients because admitting chest radiographic films were abnormal and two patients had inadvertent extubation. These patients had been evenly distributed between study groups and none of the excluded patients developed pneumonia" Comment: Although 92 participants were enrolled in the study, only 83 were analysed. The interventional arms to which these 9 participants were randomised are not clearly mentioned in the study report, but it does say that participants were evenly distributed between study groups, and none of them developed pneumonia. Therefore the likelihood of attrition bias is minimal |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were part of the final analysis |
| Other bias | Low risk | Comment: Study was supported in part by a Roche Hospital Pharmacy Research Grant. However, the role of the sponsor in the conduct and reporting of the trial is unclear. No other sources of bias suspected |
Pinilla 1985.
| Methods | Parallel‐group randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 143 Number analysed: 126 Antacid
No intervention
Inclusion criteria Critically ill with 1 or more of the following risk factors for acute erosive gastritis:
Exclusion criteria
Baseline imbalances: ‐ |
|
| Interventions |
Antacid
No intervention
Adherence to regimen: ‐ Duration of follow‐up: ‐ Duration of trial: January 1981 to June 1983 |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought in review and not reported in trial
Outcomes reported in trial but not used in review
|
|
| Notes |
Setting: ICU, Department of Surgery, University Hospital and the Department of Mathematics University of Saskatchewan Sponsorship source: none Conflicts of interest: ‐ Comments: 28% of patients in the control group and 32% of patients in the treatment group were in the study for 1 day Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were assigned randomly to control or treatment group using a table of random numbers, for groups of four" |
| Allocation concealment (selection bias) | Unclear risk | Comment: no information reported |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: no details reported, but lack of blinding is unlikely to bias outcome measures and outcomes |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Quote: "The pH of the gastric aspirate was tested every 2 hours using phenaphthazine paper (Nitrazine, Squibb; or Colorphast. E. Merck), and observed and tested for blood every 4 hours using cumene hydroperoxide; 3,3', 5,5 '‐tetramethylbenzidine (Ames Lab‐stix). Upper GI bleeding was quantitated in the following manner: microscopic, defined as a small ( + ), moderate ( ++ ), or large ( +++) chemical reaction; moderate, defined as less than 200 mL of blood visible at any time; or severe, defined as more than 200 mL of blood, or as moderate bleeding recurring at least three times in 6 hours, or as moderate bleeding with worsening vital signs during 6 hours of observation" Comment: objective criteria for the diagnosis of GI bleeding reported |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Unclear risk | Comment: no details reported, but lack of blinding is unlikely to bias outcome measures and outcomes |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "Of 143 eligible patients, nine control patients and eight treatment patients were excluded because of an insufficient number of gastric pH measurements (six patients), missing protocol sheets (five patients), randomisation error (four patients), or clotting abnormalities (two patients)" Comment: Of 143 eligible patients 17 were excluded for various reasons unrelated to treatment |
| Selective reporting (reporting bias) | Unclear risk | Comment: no selective outcome reporting suspected. All outcomes listed in the Methods section were reported in the Results section |
| Other bias | Low risk | Comment: no other sources of bias suspected |
Poleski 1986.
| Methods | Single‐blind randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 44 Number analysed: 38 (for the primary outcome of GI bleed), 44 (for all other outcomes of importance to the review) Cimetidine
Antacid
Inclusion criteria
Exclusion criteria
Baseline imbalances: The 2 groups were almost similar with respect to age, gender, and number of participants. Most participants were diagnosed with respiratory failure or sepsis, and this was equally distributed in both groups |
|
| Interventions |
Cimetidine
Antacids
Adherence to regimen: Two patients in the cimetidine group were removed from the protocol (n = 23) owing to refusal of endoscopy and protocol error, respectively. From the antacid group (n = 21), 5 patients were removed; 4 could not be endoscoped (2 refused endoscopy and 2 developed cardiac complications), and 1 patient with severe ileus (developed nausea and vomiting). Amphogel was substituted for Mylanta II in patients with severe diarrhoea and renal failure Duration of trial: ‐ Duration of follow‐up: not mentioned in the study report. Probably until death or discharge |
|
| Outcomes | Outcomes were not classified as primary or secondary in the study report; however outcomes reported were: Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported but not used in review
|
|
| Notes |
Setting: McGill University, Sir Mortimer B. Davis‐Jewish Hospital, Montreal, Quebec, Canada Source of funding: Smith Kline and French Canada Ltd., and Park Davis Canada Ltd. Conflicts of interest: ‐ Ethics approval: Quote: "The study was approved by the ethics committee on clinical investigations of the Sir Mortimer B.Davis‐Jewish General Hospital" Informed consent: Quote: "A signed consent was obtained from patients or their immediate relations" Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Trial reports that NG aspirates were frequently positive for occult bleeds and were not a useful tool for determining clinically significant upper GI bleeding |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Quote: "After approximately 72 hours all patients were gastroscoped by a single endoscopist who had no knowledge about which therapy the patient was on…" Comment: GI bleeding was an objective outcome that was detected as per the definition in the study protocol; the outcome assessor was blinded |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: This was not an outcome of interest in this study |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: unclear on blinding of outcome assessors. However, all other outcomes were objective in nature, so the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: 6 participants were excluded from analysis of the primary outcome of diagnosing GI bleed endoscopically (as endoscopy could not be done on these participants) for reasons mentioned under "adherence to the regimen". An intention‐to‐treat analysis was done for other outcomes of interest |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were reported and analysed |
| Other bias | Low risk | Comment: Study was funded by Smith Kline and French Canada Ltd., and Park Davis Canada Ltd., and some of the equipment used was received from this organisation. The role of the sponsor in the conduct and reporting of the trial is unclear |
Powell 1993.
| Methods | Single‐blind randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 41 participants Number analysed: 41 participants Ranitidine
Omeprazole (bolus)
Omeprazole (infusion)
Placebo
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "There was no difference between the groups with regard to sex, age, ethnic origin, the presence of droperidol in the premedication and the drugs used after the operation" Comments: The 4 groups of participants who were scheduled for CABG appear to be similar to each other with respect to demographic characteristics and medications given |
|
| Interventions |
Ranitidine
Omeprazole (bolus)
Omeprazole (infusion)
Placebo
Adherence to regimen: ‐ Duration of trial: ‐ Duration of follow‐up: not clearly mentioned in the study record. Most probably until discharge |
|
| Outcomes |
Outcomes sought in review and reported in trial (none of these were primary outcomes for this study)
Outcomes sought but not reported in trial
Outcomes reported but not used in review
|
|
| Notes |
Setting: Department of Anaesthetics and Department of Surgery, Royal Postgraduate Medical School, Hammersmith Hospital, London W12ONN, UK Source of funding: not clearly mentioned in the study report. However, it is mentioned that Astra Clinical Research Unit, Edinburgh, supplied the drugs and supported the trial Conflicts of interest: ‐ Ethics approval: Quote: "41 patients who were scheduled for CABG were entered into the study, which was approved by the local ethics committee" Informed consent: Quote: "Informed consent was obtained from each patient on the eve of the operation" Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: The omeprazole arms were combined to form a common interventional arm as the review did not aim to investigate efficacy on the basis of dose or mode of administration of the same drug |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were assigned to one of the four treatment groups from a random list" Comment: Random sequence generation is not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Quote: "Patients were assigned to one of the four treatment groups from a random list" Comment: unclear on how allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The personnel collecting the aspirates did not know which treatment the patient received" Comment: This was a placebo‐controlled trial. Blinding of personnel was done for the primary outcomes of measuring gastric pH and volume of gastric secretion. The likelihood of performance bias is low |
| Blinding (detection bias) Clinically important upper GI bleeding | High risk | Comment: not clear whether outcome assessors were blinded. The definition for diagnosing GI bleed, which was an objective outcome, was not clearly mentioned in the study report |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: not clear whether outcome assessors were blinded. Moreover, the outcome of interest was objective in nature, so the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All randomised participants were part of the final analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: It is mentioned that Astra Clinical Research Unit, Edinburgh, supplied the drugs and supported the trial. But it is unclear whether they had any influence on the results of the trial. No other sources of bias detected |
Prakash 2008.
| Methods | Open‐label randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: Unclear Number analysed: 50 participants Ranitidine
Sucralfate
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "There were no significant differences between the groups with respect to age, sex, distribution of underlying diseases, the severity of illness, prophylactic antibiotic therapy, and gastric pH at admission" Comment: no significant difference between the 2 groups with respect to demographic and baseline risk factors. Laparotomy was the most common cause for admission in both groups. The APCHE II score was 14.21 ± 5.44 and 13.34 ± 6.03 in the ranitidine and sucralfate groups, respectively |
|
| Interventions |
Ranitidine
Sucralfate
Adherence to regimen: Quote: "Seven patients were extubated and one patient died before four days of observation and could not be analysed for the development of late onset pneumonia. 42 patients observed for more than four days" Comment: Of the initial number of participants who were randomised, only those who were eventually intubated for longer than 24 hours were part of the study analysis Duration of trial: ‐ Duration of follow‐up: Patients were followed up for a period of 7 days with daily chest radiograph, complete blood count with differential, serum electrolytes, and gastric pH measurements |
|
| Outcomes |
Outcomes sought in review and reported in trial Primary outcomes
15 early‐onset and late‐onset cases of pneumonia were diagnosed if they occurred during the first 4 days of or 4 days after initiation of mechanical ventilation, respectively. Patients observed for longer than 4 days and were evaluated for the development of late‐onset pneumonia Secondary outcomes
Outcomes sought but not reported in trial
Outcomes reported but not used in review
|
|
| Notes |
Setting: Department of Anaesthesia and Intensive Care, Vardhman Mahavir Medical College and Safdarjang Hospital, New Delhi Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Quote: "The study protocol was approved by the institutional ethics committee" Informed consent: Quote: "...informed consent was obtained from the patients or, if this was not possible because of the clinical condition, from a relative of the family" Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Of the 25 participants who developed pneumonia, 11 (44%) had the source traced to gastric colonisation (10 in ranitidine and 1 in sucralfate group). Klebsiella species was the most commonly isolated (gastric and tracheal aspirates). Late‐onset pneumonia was more common in the ranitidine group than in the sucralfate group (10 and 2; P = 0.001), and there was no significant difference in early‐onset pneumonia between the 2 groups (5 and 8; P = 0.098) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomization was done using a computer generated random number table" Comment: Method adopted to obtain random sequence generation is clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: no clear mention of blinding the outcome assessor to this outcome. But GI bleeding was an objective outcome detected as per the definition in the study report |
| Blinding (detection bias) Nosocomial pneumonia | Low risk | Quote: "Chest radiographs were interpreted by a radiologist who had no knowledge of the patients’ treatment group after randomisation" Comment: VAP was detected as per the definition in the study report, and the radiologist was blinded to the interventions |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: no clear mention of blinding outcome assessors. However, outcomes were objective in nature, so the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: Although 50 participants in each treatment arm were evaluated, we are not sure of the number of participants who were initially randomised to each of these arms |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: The source of funding is not mentioned. No additional biases were detected |
Priebe 1980.
| Methods | Open‐label randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised:75 participants Number analysed: 75 participants Cimetidine
Antacids
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "Of the 38 participants in the cimetidine group, 19 were male and 19 were female, and the mean age was 62 years. Of the 37 participants in the antacid group 22 were male and 15 were female participants. and their mean age was 63 years. The principle diagnosis of both the groups were almost similar (most of them had intra abdominal diseases ; antacid group (7) and cimetidine group (10). There was no statistically significant difference between the two groups with respect to risk factors such as major operative procedures, respiratory failure, sepsis, peritonitis, multiple trauma, renal failure, hypotension and jaundice" "Before administration of any medication, the initial gastric pH values were below 3.5 in 16 of the cimetidine treated participates and 16 of the antacid treated participants. The guaiac test was initially positive in 11 participants in cimetidine group and 9 in antacid group. None of these 20 participants had gross evidence of GI bleeding and were included in the study" Comment: Both of these groups were similar with respect to their baseline characteristics |
|
| Interventions |
Antacids
Cimetidine
Adherence to regimen: Quote: "In 38 participants treated with cimetidine, failure to achieve a pH of 3.5 or greater on one or more occasions occurred in 18 participants who were initially given 300 mg intravenously every six hours and in nine given a dose of 300 mg every four hours. Seven of eight participants who required 400 mg of cimetidine every four hours had gastric pH values below 3.5 on one or more occasions even at that dosage" "In 37 participants treated with antacids, failure to achieve a pH of 3.5 or greater occurred in nine participants initially given 30 mL of antacid, six given 60 mL, and one given 120 mL. However, during 1259 hourly administrations of 30 mL of antacids, the gastric pH remained below 3.5 only 21 times (1.7 percent)" "Patients in whom cimetidine "failed" (who had GI bleed, n = 7), were continued on cimetidine, and antacids were added at an initial dosage of 30 ml/hour and patients who bled with antacids would receive cimetidine also" "The study was ended when oral feedings were begun and the nasogastric tube was removed, or the participant was discharged from the intensive care unit" Duration of trial: January 1978 to March 1979 Duration of follow up: not mentioned in the study report. Probably until death or discharge |
|
| Outcomes | Outcomes are not categorised as primary and secondary, but from the study report, we feel GI bleeding was the primary outcome Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported but not used in review
|
|
| Notes |
Setting: Departments of Anaesthesia and Surgery, Harvard Medical School, and the Beth Israel Hospital Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Quote: "The study was approved by the Committee on Cliniccal Investigations, New Procedures and New Forums of Therapy of the Beth Israel Hospital" Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: In 1 participant from the cimetidine group, GI bleeding was the cause of death |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "…randomised by the table of random numbers to receive either cimetidine or antacids" Comment: Method adopted to obtain random sequence generation is clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: unclear whether outcome assessors were blinded. Definition of GI bleeding described in the study report |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Unclear risk | Comment: All other outcomes of interest were objective in nature |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All randomised participants were part of the final analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: Source of funding is not mentioned. No additional biases were detected |
Prod'hom 1994.
| Methods | Single‐blind randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 375 participants Number analysed: 244 participants Antacids
Ranitidine
Sucralfate
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "At randomisation, no statistically significant difference was found among the three groups in terms of age (P = 0.058), sex (P > 0.2), APACHE II scores (P > 0.2), Glasgow coma scores (P > 0.2), and other underlying characteristics such as pneumonia on admission, participants receiving antibiotic therapy or enteral nutrition" Comment: The 3 groups were similar. Among the participants from surgical ICU, 30, 28, and 33 participants in the antacid, ranitidine, and sucralfate groups were requiring emergency surgery. Most participants from surgical ICU were diagnosed with trauma requiring some form of intervention. Among participants from the medical ICU, most had some pulmonary disease; 9, 7, and 6 participants were diagnosed with pneumonia on admission to each of the respective groups |
|
| Interventions |
Antacid
Ranitidine
Sucralfate
Adherence to regimen: Quote: "375 were randomly assigned to a treatment group and 258 were eventually intubated for more than 24 hours. Fourteen were un assessable because of missing data (4, 3, and 7 patients in the antacid, ranitidine, and sucralfate groups, respectively). Thus, 244 patients could be analysed. Of these 244 patients, 81 received antacid, 80 received ranitidine, and 83 received sucralfate.The protocol had to be interrupted before extubation in 23 (28%) of the patients in the antacid group, 19 (24%) of the patients in the ranitidine group, and 17 (20%) in the sucralfate group. Renal insufficiency developed in 5, 8, and 6 patients in the antacid, ranitidine, and sucralfate groups, respectively. In the antacid group, 6 patients developed diarrhoea or ileus, which was attributed to the treatment. In the ranitidine group, one patient developed leukopaenia and another patient developed a rash. Removal of the nasogastric tube, withdrawal of supportive care, or discharge from the hospital was the other reason for premature protocol interruption. Five patients in the antacid group, 5 patients in the ranitidine group, and 8 patients in the sucralfate group had these characteristics. In addition, protocol violation prompted interruption of treatment in 7 patients in the antacid group, 4 patients in the ranitidine group, and 3 patients in the sucralfate group. For patients in whom the protocol was interrupted, the total number of assessable days before interruption was not statistically different among the three groups (P > 0.2)" Comment: It is also mentioned that physicians had to modify the anti‐stress ulcer prophylaxis regimen in 1, 2, and 3 participants in the antacid, ranitidine, and sucralfate groups, respectively, due to GI bleed Duration of trial: January 1989 to January 1991 Duration of follow up: not clearly mentioned in the study report. Probably until death or discharge |
|
| Outcomes |
Outcomes sought in review and reported in trial Primary outcomes
Note: Early‐onset pneumonia developed in 9, 8, and 7 participants in antacid, ranitidine, and sucralfate groups, respectively. Late‐onset pneumonia developed in 11, 14, and 4 participants in the antacid, ranitidine, and sucralfate groups, respectively.Three of the 4 cases of late‐onset pneumonia in the sucralfate group were observed on day 5 Note: In the antacid group, GI bleeding developed on third day for 2 and on day 18 for 1 participant. In the ranitidine group; it was diagnosed on the second day for 2 participants and on days 3, 4, and 6 for the remaining 3 participants. In the sucralfate group, it was detected on the second day for three and on days 3, 5,8,12, and 23 for the remaining participants. Outcomes sought but not reported in trial
Outcomes reported but not used in review
|
|
| Notes |
Setting: Division Autonome de Medecine Preventive Hospitaliere, Centre Hospitalier Universitaire Vaudois, CH‐1011 Lausanne, Switzerland Source of funding: by Merck and Co. Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Mortality was attributed to pneumonia in 4 participants (1 in the antacid and 3 in ranitidine groups, respectively, whereas it was attributed to GI haemorrhage in 1 participant from the sucralfate group. Staphylococcus aureus, Streptococcus pneumoniae, and Haemophilus influenzae, alone or in combination, accounted for more than half of early‐onset pneumonia cases (54%), whereas gram‐negative bacilli were most commonly isolated in late‐onset pneumonia |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomization was done using a random permutable table to generate a random treatment list" Comment: Method adopted to obtain random sequence generation is clearly mentioned in the study report |
| Allocation concealment (selection bias) | Low risk | Quote: "Treatment regimens were included in opaque, sealed envelopes" Comment: Method adopted to obtain allocation concealment is clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: There is no clear mention of blinding outcome assessors. However, GI bleeding was an objective outcome detected as per the definition in the study protocol |
| Blinding (detection bias) Nosocomial pneumonia | Low risk | Quote: "They were interpreted by a pneumologist who had knowledge of all relevant data except for the patient's stress ulcer prophylactic regimen, gastric pH, or colonization data" Comment: VAP was detected as per the definition in the study protocol by an outcome assessor who was blinded to the above mentioned participant data |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: There is no clear mention of blinding outcome assessors. However, all other outcomes of interest were objective in nature |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: Of the initial 375 randomised participants, only 258 were part of the analysis, as only these participants were in the trial for longer than 24 hours, as this was criterion was necessary to measure the outcomes of interest. Data on 14 participants were missing, and they seem to be well balanced across the 3 groups |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported in the study |
| Other bias | Low risk | Comment: The study was funded by Merck and Co. However, the role of the sponsor in the conduct and reporting of the trial is unclear. No other form of bias was detected |
Reusser 1990.
| Methods | Open label, randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 97 participants Number analysed: 40 participants Ranitidine
No prophylaxis
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "No significant differences between treatment and control groups were detected in any of the clinical and therapeutic characteristics among both the groups. However, hypotension was more frequent in the control group (12 vs. 5)" Comment: Both groups were comparable with respect to age and gender distribution, and risk factors such as disseminated intravascular coagulation in addition to severe intracranial lesion and respiratory failure. Severe head injury was the main primary diagnosis in both groups |
|
| Interventions |
Ranitidine
No prophylaxis
Adherence to regimen: For treatment of the 97 eligible participants, only 40 completed the trial and were available for analysis (19 in the treatment arm and 21 in the control arm) The remainder were excluded for the following reasons:
In the treatment group, 5 (26%) participants remained on the original ranitidine dosage of 50 mg every 8 hours throughout the study, 5 (26%) required a dosage increase to 50 mg every 6 hours, and 9 (47%) needed additional antacids Duration of trial: August 1984 to September 1986 Duration of follow up: up to 7 days after study completion |
|
| Outcomes |
Outcomes sought in review and reported in trial Primary outcome
Secondary outcomes
Outcomes sought but not reported in trial
Outcomes reported but not used in review
|
|
| Notes |
Setting: Department of Internal Medicine and Surgical Intensive Care Unit, University Hospital Basel, Switzerland Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Quote: "The study was approved by the ethical committee of the Basel University Hospital" Informed consent: Quote: "Informed consent was obtained from each patient's legal guardian" Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Data are provided on number of deaths during the study (n = 1 in the ranitidine group), which is not similar to all‐cause mortality in ICU |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "The need to monitor gastric pH to determine intensification of stress lesion prophylaxis in the treatment group prevented double‐blind study design” Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Quote: “The need to monitor gastric pH to determine intensification of stress lesion prophylaxis in the treatment group prevented double‐blind study design” Comment: Outcome assessors were not blinded. However, GI bleeding was an objective outcome that was detected as per the definition in the study protocol |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: All other outcomes of interest were objective in nature |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: It is mentioned that of the 97 eligible participants, only 40 completed the trial and were available for analysis. The remaining 43 participants were excluded for reasons mentioned under "Adherence to the regimen". Therefore, a protocol analysis was done to measure the outcomes of interest, and the number of participants appears to be balanced between groups. Therefore, the likelihood of this affecting the outcomes of interest is minimal |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: The source of funding is not clearly mentioned in the study report. No additional biases were detected |
Rohde 1980.
| Methods | Randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: ‐ Number analysed: ‐ Cimetidine
Placebo
Inclusion criteria
Exclusion criteria
Baseline imbalances: ‐ |
|
| Interventions |
Cimetidine
Placebo
Adherence to regimen
Duration of trial: March 1977 to June 1978 Duration of follow up: until death or discharge from hospital |
|
| Outcomes |
Outcome sought in review and reported in trial
Outcomes sought but not reported in trial report
Outcomes reported in report but not used in review
|
|
| Notes |
Setting: Department of Surgery, Marburg, Germany Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Quote: "The definite protocol was submitted to the local ethical committee...". Comment: Approval from the ethics committee was sought. Moreover, the trial was constantly monitored by an executive group Informed consent: Informed consent was sought from participants, as it was a criterion for inclusion/exclusion from the trial Clinical trials registration: ‐ Sample size calculation: Although the trial was planned as a double‐blind trial with a fixed sample size (100 participants) of people admitted to ICU, it was executed as a sequential single‐blind study only in 1 subgroup of participants (polytrauma) and was ended before the planned termination date for ethical reasons |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Comment: Participants were randomised in blocks of 4. The method adopted to obtain random sequence generation is clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "The study was executed as a single blind sequential trial (a deviation from the planned double blind method) due to ethical reasons" Comment: unclear on who was blinded and how it was executed |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: GI bleeding was detected as per the definition in the study protocol, not blinding the outcome assessor to this objective outcome would not have caused detection or performance bias |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: All other outcomes of interest were objective in nature, so the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: Data for only 1 strata of participants (polytrauma) are available, as the strata were terminated as mentioned above in "Adherence to regimen". This appears to be an incomplete report |
| Selective reporting (reporting bias) | High risk | Comment: Data for only 1 strata of participants (polytrauma) are available, as the strata were terminated as mentioned above in "Adherence to regimen" |
| Other bias | Low risk | Comment: The source of funding is not clearly mentioned in the study report. No additional biases were detected |
Ruiz‐Santana 1991.
| Methods | Single‐blind randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 94 participants Number analysed:73 participants Total parenteral nutrition (TNP)
TPN + sucralfate
TPN + ranitidine
Inclusion criteria
Exclusion criteria
Baseline imbalances: Groups were similar with respect to age and gender. The 2 main reasons for admission were respiratory disease (n = 26) and multiple injuries (n = 14). There is no clear mention of the distribution of clinical features across study groups |
|
| Interventions |
TPN
TPN + sucralfate
TPN + ranitidine
Adherence to regimen: Quote: "24 participant were withdrawn from the study before the sixth day on protocol with the following reasons for withdrawal: weaned from mechanical ventilation before the sixth day (n = 10), death (n = 8), acute upper GI haemorrhage (n = 5, 2 stress induced gastroduodenal ulcers, 2 chronic duodenal ulcers, 1 stomach cancer) and early tolerance to enteral feedings (n = 1)" Comment: The interventional arms to which these participants were initially randomised are not clearly mentioned in the study report Duration of trial: December 1988 to January 1990 Duration of follow up: not mentioned in the study report. Probably until death or discharge |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported but not used in review
|
|
| Notes |
Setting: ICU and gastroenterology service, Hospital del Pino, Las Palmas de Gran Canaria, Canary Islands, Spain Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Quote: "The study was approved by the institutional review board" Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Commets: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Commets: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Quote: ”All endoscopic examinations, except a few cases performed on emergency basis was done by a single investigator uninformed as to the treatment group” Comment: Outcome assessors were mostly blinded and GI bleeding was an objective outcome that was detected as per the definition in the study protocol |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: unclear on the blinding of outcome assessors. However, all other outcomes were objective in nature |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: Of the 97 participants, 24 were withdrawn from the study, as they did not complete a minimum of 6 days in the ICU as required by the protocol of this study. The interventions to which they were originally randomised were not clear from the study report. A per‐protocol analysis was done for the outcomes of interest, but there appears to be an imbalance between groups with respect to the final number of participants available for analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Unclear risk | Comment: Source of funding is not clearly stated. Baseline characteristics (clinical) are not clearly mentioned for each group. No additional biases were detected |
Ryan 1993.
| Methods | Single‐blind randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 114 participants Numner analysed:114 participants Cimetidine
Sucralfate
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "There was no significant difference in mean age, sex ratio, or number of patients admitted to medical or surgical services" Comment: The 2 groups were similar with respect to their baseline characteristics |
|
| Interventions |
Cimetidine
Sucralfate
Adherence to regimen: 114 participants who met the inclusion criteria were enrolled into the study. 25 (22%; 12 participants from cimetidine group and 13 participants from sucralfate group) participants were withdrawn from the study for the following reasons:
The remaining 89 participants constituted the study group Duration of trial: January 2009 to September 2009 Duration of follow‐up: ‐ |
|
| Outcomes |
Outcomes sought in review and reported in trial Primary outcomes
Note: Other than the CDC and prevention criteria, each participant was required to have 2 chest roentgenograms showing persistent infiltrates, with agreement by intensivist and radiologist Secondary outcomes
Outcomes sought but not reported in trial
Outcomes reported in report but not used in review
|
|
| Notes |
Setting: Medical and Surgical Intensive Care Unit, Springfield, Mass, Tufts University Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Quote: "The study was approved by the institutional review board" Informed consent: Quote: "Informed consent was obtained from each patient or guardian" Clinical trials registration: ‐ Sample size calculation: ‐ |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: ”Patients were randomised according to computer generated numbers to receive either cimetidine or sucralfate” Comment: The method adopted to obtain random sequence generation is clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: Outcome assessors were not blinded, but GI bleeding was an objective outcome detected as per the definition in the study protocol |
| Blinding (detection bias) Nosocomial pneumonia | Low risk | Quote: "The radiologist and the intensivist who assessed the chest roentgenograms for diagnosing pneumonia were both blinded to the treatment that the participant was receiving" Comment: Nosocomial pneumonia was detected as per the definition in the study protocol by an outcome assessor who was blinded to participant data |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: unclear on the blinding of outcome assessors. However, outcomes of interest were objective in nature |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "...with 10 mL of water. Twenty‐five patients (22%) were withdrawn from the study for the following reasons: two because of extubation within 48 hours of enrolment and discharge from the ICU, two because the nasogastric tube was removed (the nasogastric tube was required to instil sucralfate into the stomach and monitor bleeding), three at the request of the patient or guardian, eight because they died within 48 hours, four because of inadvertent medication change, three because of documented cases of aspiration by a witness, three because of adverse reactions from medications (two patients in the cimetidine group had confusion and neutropaenia, and one patient in the sucralfate group could not tolerate the nasogastric tube clamped). The patients who were withdrawn were equally distributed between the two treatment groups. The remaining 89 patients constituted" Comment: no incomplete reporting of outcomes suspected |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: Source of funding is not clearly mentioned in the study design. No other sources of bias suspected |
Selvanderan 2015.
| Methods | Parallel‐group randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 214 participants Number analysed: 214 participants Pantoprazole
Placebo
Inclusion criteria
Exclusion criteria: ‐ Baseline imbalances: ‐ |
|
| Interventions |
Pantoprazole
Placebo
Adherence to regimen: ‐ Duration of trial: January 2014 to January 2015 Duration of follow‐up: 90 days |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
|
|
| Notes |
Setting: ICU Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: ‐ |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not enough details reported |
| Allocation concealment (selection bias) | Unclear risk | Comment: no details reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "double blind fashion" Comment: no details on blinding reported. Lack of blinding is unlikely to introduce bias to outcome measures or outcomes |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Quote: "(haematemesis, bloody gastric aspirate, melaena or haematochezia), clinically significant bleeding (overt bleeding accompanied by a drop in mean arterial pressure > 20mmHg, or reduction in haemoglobin > 20g/L, or need for surgical intervention)" Comment: criteria for diagnosis of GI bleeding described. |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: no criteria for diagnosis of VAP described |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Unclear risk | Comment: no information about blinding of outcome assessors described |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: not enough information reported to assess incomplete outcome data. Conference abstract |
| Selective reporting (reporting bias) | Low risk | Comment: no incomplete reporting of outcomes suspected. All outcomes listed in the Methods section were also reported in the Results section briefly |
| Other bias | Unclear risk | Comment: no other sources of bias suspected, but very little information reported overall |
Selvanderan 2016.
| Methods | Parallel‐group randomised controlled trial | |
| Participants |
Baseline characteristics Overall
Inclusion criteria
Exclusion criteria: ‐ Baseline imbalances: ‐ |
|
| Interventions |
Pantoprazole
Placebo
Adherence to regimen: ‐ Duration of trial: January 2014 to January 2015 Duration of follow‐up: 90 days |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought in review but not reported in trial
Outcomes reported in trial but not used in review
|
|
| Notes |
Setting: ICU Sponsorship source: ‐ Ethics approval: ‐ Conflicts of interest: ‐ Informed consent: ‐ Study protocol: ‐ Sample size calculation: ‐ Additinal notes: conference abstract |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomly assigned" Comment: not enough details reported |
| Allocation concealment (selection bias) | Unclear risk | Comment: no details reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "double blind fashion" Comment: no details on blinding reported. Lack of blinding is unlikely to introduce bias to outcome measures or outcomes |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Quote: "(hematemesis, bloody gastric aspirate, melena or hematochezia), clinically significant bleeding (overt bleeding accompanied by a drop in mean arterial pressure > 20 mmHg, or reduction in haemoglobin > 20 g/L, or need for surgical intervention)" Comment: outcome measured objectively |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: no criteria for diagnosis of VAP described |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Unclear risk | Comment: no information about blinding of outcome assessors described |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: not enough information reported to assess incomplete outcome data. Conference abstract |
| Selective reporting (reporting bias) | Low risk | Comment: no incomplete reporting of outcomes suspected. All outcomes listed in the Methods section were also reported in the Results section briefly |
| Other bias | Unclear risk | Comment: no other sources of bias suspected, but too little information reported in this conference abstract to assess other biases |
Simms 1991.
| Methods | Open‐label randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 89 participants Number analysed: 89 participants Antacids
Cimetidine
Sucralfate
Inclusion criteria
Exclusion criteria
Baseline imbalances: ‐ |
|
| Interventions |
Antacids
Cimetidine
Sucralfate
Adherence to regimen: Quote: "Patients received cimetidine or antacids had the doses of each drug increased if the pH failed to reach 4 on any two consecutive 2‐ hours sampling patients receiving antacids alone at maximal doses whose intragastric pH < 4 received combination therapy to maintain intragastric pH > 4 and therefore were discontinued from the study" "Upper GI bleeding requiring blood transfusions were seen in two patients, one receiving cimetidine and one receiving sucralfate. These two participants were eliminated from the study and were endoscopically shown to have diffuse erosive gastritis" Duration of trial: December 1988 to January 1990 Duration of follow up: not mentioned in the study report. Probably until death or discharge |
|
| Outcomes | Outcomes were not classified as primary and secondary, but main outcomes in the study report are as follows; Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported but not used in review
|
|
| Notes |
Setting: Surgical Intensive Care Unit, Rhode Island Hospital/Brown University, Providence, USA Source of funding: Smith Kline Beecham Pharmacuticals Conflicts of interest: ‐ Ethics approval: Quote: "The study was approved by the Rhode Island Hospital Human welfare protection committee" Informed consent: Quote: "Informed consent was obtained from each participant before induction into the study" Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: The most prevalent organisms isolated from NG tube were Candida albicans, Enterococcus organisms, and β‐haemolytic streptococci; they were identical across all 3 groups |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants |
| Blinding (detection bias) Clinically important upper GI bleeding | High risk | Comment: unclear whether outcome assessors were blinded, and definition of GI bleeding not clearly mentioned in the study report |
| Blinding (detection bias) Nosocomial pneumonia | High risk | Comment: unclear whether outcome assessors were blinded, and definition of pneumonia was not clearly mentioned in the study report |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: All other outcomes of interest were objective in nature, so the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All randomised participants were part of the analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: Source of funding is not clearly mentioned in the study design. No other source of bias suspected |
Sirvent 1994.
| Methods | Randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 51 participants Number analysed: 51 participants Antacids + ranitidine
Sucralfate
Inclusion criteria
Exclusion criteria
Baseline imbalances: Groups were similar in demographic characteristics. Antacid + Ranitidine group had an APACHE II score of 14.7 ± 4.9, and sucralfate group had a score of 13.2 ± 5.1 More participants in the sucralfate group were diagnosed with polytrauma (n = 16 vs 4 in the sucralfate group) |
|
| Interventions |
Antacids + ranitidine
Sucralfate
Adherence to regimen: no change in dose/regimen mentioned. No information about dropouts Duration of trial: January 1990 to June 1991 Duration of follow‐up: ‐ |
|
| Outcomes |
Outcomes sought in review and reported in trial Primary outcomes
Secondary outcomes
Outcomes sought but not reported in trial
Outcomes reported but not used in review
|
|
| Notes |
Setting: Hospital de Bellvitge‐Princeps d’Espanya Source of funding: grant from the heath ministry Conflicts of interest: ‐ Ethics approval: Study was approved by the clinical investigations committee at the Hospital Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Two episodes of mild upper GI bleeding occurred in each group; this was not of clinical significance |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants |
| Blinding (detection bias) Clinically important upper GI bleeding | Unclear risk | Comment: Study did not address this outcome |
| Blinding (detection bias) Nosocomial pneumonia | Low risk | Comment: Study mentions that 2 outcome assessors were blinded |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: unclear on blinding of assessors for other outcomes. However owing to the objective nature of the outcomes of interest, the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All randomised participants were part of the final analysis |
| Selective reporting (reporting bias) | High risk | Comment: All‐cause mortality in the ICU was not separately mentioned for each group. However, all other intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: This study was funded by the ministry of health. The role of the sponsor in the conduct and reporting of the trial is unclear. No other sources of bias suspected |
Skillman 1984.
| Methods | Open‐label randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 60 participants Number analysed: 46 participants Antacids (Mylanta II)
Prostaglandin ((15)‐R‐15‐methyl prostaglandin E2)
Inclusion criteria
Exclusion criteria
Baseline imbalances: no significant differences between the 2 groups in baseline characteristics such as age, sex, and risk factors. Most participants were diagnosed with intra‐abdominal disease (antacids: 7 and prostaglandin: 10) |
|
| Interventions |
Antacids (Mylanta II)
Prostaglandin ((15)‐R‐15‐methyl prostaglandin E2)
Adherence to regimen: 14 patients were excluded after randomisation for reasons mentioned above under "Exclusion criteria". 11 of the 12 patients in whom prostaglandin E2 prophylaxis failed (upper GI bleeding) were switched to antacid regimen. One of the 11 patients had "double" failure because antacid also failed Duration of trial: October 1980 to August 1982 Duration of follow up: not clearly mentioned in the study report. Probably until death or discharge |
|
| Outcomes | Outcomes were not classified as primary or secondary in the study report; however outcomes reported were as follows. Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported but not used in review
|
|
| Notes |
Setting: Department of Surgery and Medicine Harvard Medical School and Beth Israel Hospital, Boston, Massachusetts Source of funding: Quote: "This study was supported in part by funds from Upjohn University, Kalamazoo, Michigan and the Charls Diana Research Institures, Beth Israel Hospital, Boston" Conflicts of interest: ‐ Ethics approval: Quote: "The study was approved by the Committee on Clinical Investigation, New Procedures and new Forms of Therapy of the Beth Israel Hospital" Comment: mentioned for only 1 centre Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “Patients were selected by a table of random numbers” Comment: Method adopted to obtain random sequence generation is clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: Outcome assessor was not blinded. GI bleeding was an objective outcome detected as per the definition in the study protocol |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: Outcomes of interest were objective in nature, so the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: 14 patients were excluded from analysis after randomisation for various reasons mentioned in "adherence to the regimen". The interventional arms to which these participants were randomised remain unclear. A per‐protocol analysis was performed, and there appears to be a balance between groups in terms of the number of participants available for final analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: This study was supported in part by a research grant from Upjohn University, Kalamazoo, Michigan, and the Charls Diana Research Institures, Beth Israel Hospital. The role of the sponsor in the conduct and reporting of the trial is unclear |
Solouki 2009.
| Methods | Double‐blind randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 129 participants Number analysed: 129 participants Ranitidine
Omeprazole
Inclusion criteria
Exclusion criteria
Baseline imbalances: Patients’ age in group A ranged from 5 to 85 years with mean age of 49.19 years. Group B patients were in the age range of 5 to 95 years with mean age of 52.41 years. In the omeprazole group. There were 32 (52.5%) males and 29 (47.5%) females. These numbers were 35 (51.5%) males and 33 (48.5%) females in the ranitidine group. The 2 groups were similar with respect to the baseline risk factors responsible for GI bleeding |
|
| Interventions |
Ranitidine
Omeprazole
Adherence to regimen: 128 participants were randomised to receive 2 interventions; Ranitidine + Placebo (68) and Omeprazole + Placebo (61). 12 participants died in the first group while 3 died in the second group Duration of trial: June 2000 to January 2001 Duration of follow up: not clearly mentioned in the study report. Probably until death or discharge |
|
| Outcomes |
Outcomes sought in review and reported in trial Primary outcome
Secondary outcomes
Outcomes sought but not reported in trial
Outcomes reported but not used in review
|
|
| Notes |
Setting: Department of Internal Medicine and Intensive Care Unit, Imam Hossein Hospital, Shahid Beheshti University, M.C., Tehran, Iran Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Quote: "The study was approved by the institutional ethics committee..." Informed consent: Quote: "Written consents signed by the patients or their family members were obtained for participation in the study" Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: In the ranitidine group, 4 in 14 participants who had overt bleeding had clinically important GI bleeding. This was 1 in 3 participants from omeprazole group |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Using the table numbers randomly, all ICU beds were divided into two groups of A and B" Comment: Method adopted to obtain random sequence generation is clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: This was a randomised double‐blind placebo‐controlled trial, and outcome assessors appeared to be blinded. GI bleeding was an objective outcome detected as per the definition in the study protocol |
| Blinding (detection bias) Nosocomial pneumonia | Low risk | Comment: This was a randomised double‐blind placebo‐controlled trial, and outcome assessors appeared to be blinded. VAP was an objective outcome detected as per the definition in the study protocol |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: This was a randomised double‐blind controlled trial, and outcome assessors seem to be blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All randomised participants were part of the final analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: Source of funding not known. No other known source of bias |
Somberg 2008.
| Methods | Multi‐centre open‐label randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 202 participants Number analysed: 202 participants Pantoprazole (40 mg per 24 hours)
Pantoprazole (40 mg per 12 hours)
Pantoprazole (80 mg per 24 hours)
Pantoprazole (80 mg per 12 hours)
Pantoprazole (80 mg per 8 hours)
Cimetidine
Inclusion criteria
Exclusion criteria
Baseline characteristics: Groups were similar with respect to age, gender, and race. Trauma was the main cause for admission, and coagulopathy was present in 2 participants from the pantoprazole group |
|
| Interventions |
Pantoprazole
Cimetidine
Adherence to regimen: Quote: "Subjects received study medication within 24 hours of the precipitating stress event and treatment was to be continued for at least 48 hours and up to 7 days. Patients were considered as completers if they received the study medication as described and participated in the study for at least 48 hours" Of 202 participants, 144 remained NPO, while 58 participants were switched over to enterally feed, mainly on day 2 (48 participants) "32 patients (16%) prematurely discontinued from the study. The most frequent reason for premature discontinuation was removal or inability to maintain tolerate NG/OG tube. Other reasons for premature discontinuation from the study included adverse events (3 in total, 2 in pantoprazole and 1 in cimetidine) or legal guardian request" Duration of trial: June 2000 to September 2001 Duration of follow up: up to 30 days |
|
| Outcomes |
Outcomes sought in review and reported in trial (none of these were primary outcomes of interest for this study)
Outcomes sought but not reported in trial
Outcomes reported but not used in review
|
|
| Notes |
Setting: 14 ICU centres across the United States Source of funding: Quote: "Supported by Wyeth Pharmaceuticals, Collegeville, PA" Ethics approval: Quote: "The study conducted according to declaration of Helsinki and its amendments. and was approved by the independent ethics committee or institutional review board at each ICU centre" Informed consent: Quote: "Written informed consent was obtained from all patients or their legal representatives before enrolment" Clinical trials registration: ‐ Sample size calculation: The sample size of 30 patients per group was chosen based on a common standard deviation of 24% for the primary end point (pH response associated with each treatment group), and there was approximately 80% power to detect a mean difference of 18% for treatment group comparisons Additonal notes:H influenzae was one of the most common bacterial isolates among participants who developed pneumonia. The pantoprazole arms were combined to form a common interventional arm vs the H2 receptor antagonist as the review did not aim to investigate efficacy on the basis of dose or mode of administration in the same drug |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "...the study centre called the central randomisation centre to obtain the treatment assignment..." Comment: Sequence generation was probably done |
| Allocation concealment (selection bias) | Low risk | Quote: "...the study centre called the central randomisation centre to obtain the treatment assignment..." Comment: Allocation was probably concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "One physician per patient remained blinded to the patients treatment assignment and pH data to assess patient’s safety” Comment: Each patient had a physician in charge, who was blinded. Unclear on how this was possible, as it is not a placebo‐controlled trial, and all other personnel were aware of the intervention |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: unclear on blinding of outcome assessors. GI bleeding was an objective outcome detected as per the definition in the study protocol |
| Blinding (detection bias) Nosocomial pneumonia | Low risk | Comment: unclear on blinding of outcome assessors. VAP was detected as per the definition in the study protocol |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: unclear on blinding of outcome assessors. However, owing to the objective nature of the outcomes of interest, the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All randomised participants completed the trial and were included in the final analysis. There were no treatment withdrawals and no trial group changes |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: Study was supported by Wyeth Pharmaceuticals, Collegeville, PA. The role of the sponsor in the conduct and reporting of the trial is unclear |
Stoehr 2006.
| Methods | Randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 30 participants Number analysed: 30 participants Sucralfate
Ranitidine
Inclusion criteria
Exclusion criteria
Baseline characteristics: Quote: "The study sample was homogeneous in terms of age and sex distribution, duration of treatment, severity of illness, inclusion criteria, and basic intensive care regimens" |
|
| Interventions |
Sucralfate
Ranitidine
Adherence to regimen: ‐ Duration of trial: ‐ Duration of follow‐up: ‐ |
|
| Outcomes |
Outcomes sought in review and reported in trial ‐ Outcomes sought but not reported in trial
Outcomes reported but not used in review
|
|
| Notes |
Setting: Surgical ICUs at a university hospital in Germany Source of funding: ‐ Ethics approval: Quote: "The study was approved by the Ethics Committee of the University of Düsseldorf" Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additonal notes: ‐ |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: no information reported |
| Allocation concealment (selection bias) | Unclear risk | Comment: no information reported |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: no information reported |
| Blinding (detection bias) Clinically important upper GI bleeding | Unclear risk | Comment: no information reported |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: This was not an outcome of the study |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Unclear risk | Comment: This was not an outcome of the study |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All participants randomised were included in the analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All outcomes described in the Methods section were included in the Results section |
| Other bias | Low risk | Comment: no other source of bias suspected. Source of funding unclear |
Stothert 1980.
| Methods | Single‐blind randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 144 participants Number analysed: 123 participants Antacids
Cimetidine
Inclusion criteria ‐ Exclusion criteria
Baseline imbalances: Quote: "No significant difference exists between the groups in relation to the risk factors such as abdominal trauma, cardiovascular disease, respiratory failure, sepsis, neurologic injury, orthopedic injury, vascular injury, renal failure, hepatic failure, alcohol or drug abuse, metastatic carcinoma, hypotension and history of peptic ulcer. Similarly, the two groups are quite comparable in age and sex ratio" Comment: There was no significant difference between the 2 groups with respect to demographic and baseline risk factors. Nine and 6 participants in both groups had a history of ulcers |
|
| Interventions |
Antacids
Cimetidine
Adherence to regimen: Quote: "One hundred forty‐four patients were included in this study. Fifty‐eight patients were randomised to the antacid treatment group and 65 patients were randomised to the cimetidine therapy group. Forty‐six patients were not included in these results because they met protocol criteria for less than 24 hours. Twenty‐one patients required no therapy because of persistent pH ≥ 4. Forty‐eight (74%) cimetidine recipients had the expected elevation of pH ≥ 4. Seventeen (26%) failed despite maximum dosage of cimetidine. All failures with cimetidine had antacids added and responded successfully. No failure of antacid therapy was seen in this study and therefore no patients crossed over from the antacid therapy group into the cimetidine therapy group.The twenty‐one patients who maintained a gastric pH ≥ 4 required no prophylaxis and were not further dealt with further in this paper. All studies continued until the patient no longer required gastric decompression or was discharged from the intensive care unit as a result of improvement or death" Comment: also stated that all patients responded to antacid therapy "Forty participants responded to 30 cc every hour to maintain gastric pH at the desired level. Only three patients required 120 cc per hour to maintain this level. In the group receiving cimetidine, 34% responded to 300 mg every six hours. An additional 40% of the participants responded to administration at a dosage level of every four or every three hours" Duration of trial: October 1978 to July 1979 Duration of follow up: not clearly mentioned in the study report. Probably until death or discharge |
|
| Outcomes |
Outcomes sought in review and reported in trial (none of these were primary outcomes of the study)
Outcomes sought but not reported in trial
Outcomes reported but not used in review
|
|
| Notes |
Setting: Harborview Medical Center Surgical Intensive Care Unit (SICU), 325 Ninth Avenue, Seattle, WA 98104 Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: It is mentioned that diarrhoea occurred in 5 of the 75 participants treated with antacids. This included 58 antacid‐treated participants and 17 participants in whom cimetidine "failed" as per the study protocol. These data are unclear on the incidence in the original denominator, so could not be analysed |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "The randomisation process was based on a random number table in a blinded fashion" Comment: Method adopted to obtain random sequence generation is clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: Not clearly mentioned in the study report. However, the baseline there is no imbalance in baseline characteristics, indicating low risk of selection bias |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Day‐to‐day clinical management was performed by an attending physician independent of the study protocol" Comment: Personnel were blinded |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: no clear mention of blinding of outcome assessors. GI bleeding was an objective outcome detected as per the definition in the study protocol |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: All other outcomes of interest were objective in nature |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: A per‐protocol analysis was performed to measure the outcomes of interest. Only 123 of 144 participants were accounted for in the analysis. Groups to which these 67 participants were randomised remains unclear. However, there is no imbalance between groups with respect to the number of participants available for final analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes stated were analysed and reported |
| Other bias | Low risk | Comment: source of funding not clear. No other known form of bias detected |
Tabeefar 2012.
| Methods | Parallel‐group randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 27 participants Number analysed: 27 participants Pantoprazole I
Pantoprazole II
Ranitidine
Inclusion criteria
Exclusion criteria
Baseline imbalances: No statistical differences in age, sex, and basal pH and SOFA were found between treatment groups |
|
| Interventions |
Pantoprazole I
Pantoprazole II
Ranitidine
Adherence to regimen: ‐ Duration of trial: April 2010 to August 2011 Duration of follow‐up: 2 days |
|
| Outcomes |
Outcomes sought in review and reported in trial ‐ Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
|
|
| Notes |
Setting: ICU, Department of Pharmacotherapy, Faculty of Pharmacy, Tehran University of Medical Sciences, Tehran Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Study protocol was approved by our institutional ethics committee Informed consent: Written consent form was obtained from each patient’s closest family member Clinical trials registration: This trial is registered in www.anzctr.org.au Sample size calculation: ‐ Additional notes: |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned to 3 study groups according to a computer‐generated table of random numbers" |
| Allocation concealment (selection bias) | Unclear risk | Comment: not enough information reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: insufficient information to allow judgement, but the outcome is unlikely to be influenced by performance bias |
| Blinding (detection bias) Clinically important upper GI bleeding | Unclear risk | Comment: Study did not address this outcome |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: insufficient information to allow judgement, but the outcome is unlikely to be influenced by detection bias, provided it was a laboratory measurement. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: no incomplete outcome data suspected. All patients randomised at baseline were also included in the analyses |
| Selective reporting (reporting bias) | Low risk | Comment: All outcomes listed in the Methods section were also reported in the Results |
| Other bias | Low risk | Comment: no other sources of bias suspected |
Terzi 2009.
| Methods | Open‐label randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 24 participants Number analysed: 20 participants Ranitidine
Pantoprazole
Inclusion criteria
Exclusion criteria
Baseline imbalances: Participants were comparable with respect to age, gender, and other clinical characteristics including the presence of H pylori, APACHE II score was 12 +/‐ 7 and 16 +/‐ 4 for ranitidine and pantoprazole groups |
|
| Interventions |
Ranitidine
Pantoprazole
Adherence to regimen: Quote: "Four patients, 3 from ranitidine group, were excluded for technical reasons” Duration of trial: ‐ Duration of treatment: ‐ Duration of follow‐up: not clearly mentioned in the trial report. Probably until discharge or an untimely event of death |
|
| Outcomes |
Outcomes sought in review and reported in trial (none of these were the primary outcomes of the trial)
Outcomes sought but not reported in trial
Outcomes reported but not used in review
|
|
| Notes |
Setting: State University of Campinas (UNICAMP), Campinas, São Paulo, Brazil Source of funding: Quote: “Support funds for research and development were provided by FAEPEX. Fundação de Apoio ao Ensino, à Pesquisa e à Extensão” Conflicts of interest: ‐ Ethics approval: Quote: "The present study was approved by the ethics committee of the State University of Campinas, Brazil (no. 035/2003, dated February 18, 2003)" Informed consent: Quote: "The informed consent form was signed by a relative of each patient included in the study, as required by Resolution no. 196/96 of the National Health Council, Brazilian Ministry of Health, concerning research involving human beings" Clinical trials registration: ‐ Sample size calculation: ‐ |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: This was not a placebo‐controlled trial, and different modes of administering the study interventions would not have made it possible to blind study personnel and participants.Therefore, high risk of performance bias |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: This was an open‐label trial, and outcome assessors were not blinded. However GI bleeding was an objective outcome that had to be detected as per the study protocol |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: This was an open‐label trial, and outcome assessors were not blinded. However the outcome of interest was an objective outcome, so the likelihood of detection bias is low. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote “Four patients, 3 from ranitidine group, were excluded for technical reasons” Comment: Nearly 16% of randomised participants were excluded from analysis. An intention to treat analysis was not performed.This would have contributed to attrition bias |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes have been reported |
| Other bias | Low risk | Comment: Funding was provided by FAEPEX. Fundação de Apoio ao Ensino, à Pesquisa e à Extensão.However, the role of the sponsor in the conduct and reporting of the trial is unclear |
Thomason 1996.
| Methods | Open‐label randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 424 participants Number analysed: 242 participants Sucralfate
Antacids
Ranitidine
Inclusion criteria
Exclusion criteria
Baseline imbalances: Participants were similar in the different groups with respect to age, gender, race, median Injury Severity Score (ISS), Trauma Score (TS), Glasgow Coma Scale Score, and APCHE II (Acute Physiology and Chronic Health Evaluation) Score and Habits (Tobacco and Caffine consumption). The only significant baseline difference observed in the 11 variables recorded for participants in the 3 drug treatment groups were median alcohol level on admission (P = 0.045) and self‐reported use of alcohol (P = 0.007) |
|
| Interventions |
Sucralfate
Antacids
Ranitidine
Adherence to regimen: Between November 1990 and May 1994, 424 patients were randomised to 1 of 3 treatment regimens. One hundred eighty‐two patients were discontinued from study participation, mainly because of the following occurring within 24 hours of study entry:
A patient was considered to have discontinued the study if gastrointestinal haemorrhage occurred before study day 7, if diet by mouth or by gastric feeding was begun before day 7, or if the study drug was discontinued or changed. A patient was considered to have completed the study if the patient developed pneumonia > 24 hours after study entry, completed study day 7, was extubated for 48 hours before study day 7, or died. Data from patients who died before study day 7 but who completed at least 24 hours on the study were included in the analysis. All patients randomised to the study were followed‐up until hospital discharge for the outcome variables of laboratory, roentgenogram, and clinical signs of pneumonia, GI bleeding, or death. Forty of the 242 patients completed all 7 study days without getting pneumonia > 24 hours after study entry or dropping out of the study Duration of trial: November 1990 to May 1994 Duration of follow up: Quote: "Mean follow up for intention to treat was 24.3 days (median, 16 days)" "The mean follow‐up time for patients in the drug treatment groups was 27.3 days, with a range of 1 to 257 days (median, 19 days)" Participants were followed up until death or discharge |
|
| Outcomes |
Outcomes sought in review and reported in trial Outcomes are not categorised as primary and secondary
Note: Greatest incidence of pneumonia developed within 7 days into the study Outcomes sought but not reported in trial
Outcomes reported but not used in review
|
|
| Notes |
Setting: Carolinas Medical Center, Charlotte, North Carolina , USA Source of funding: Quote: "This project was supported by the Health Services Foundation of Carolinas Medical Center” Conflicts of interest: ‐ Ethics approval: Quote: "The study protocol was reviewed and approved by the institutional review board at Carolinas Medical Center" Informed consent: Quote: "10 participants were excluded because consent was not signed" Clinical trials registration: ‐ Sample size calculation: Sample size analysis showed that 80 participants were needed in each treatment arm (assuming an alpha level of 0.05 and a power of 80%). This assumes a pneumonia rate of 40%, which is supported by previous studies in intensive care unit patients, and also assumes that one of the treatments would reduce the rate to 20%, enough to be meaningful clinically; therefore, our goal was to recruit 240 assessable patients Additional notes: Gram‐negative micro‐organisms were the most common tracheal isolates both in participants with and without pneumonia, and 13% of participants who developed pneumonia had retrograde colonisation from stomach or trachea |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “Patients were randomised using a computer‐generated random number table to one of the following stress ulcer prophylaxis regimens...” Comment: Method adopted to obtain random sequence generation is clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants. Therefore, high risk of performance bias |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: unclear whether outcome assessors were blinded. GI bleeding was an objective outcome that was detected as per the definition in the study protocol |
| Blinding (detection bias) Nosocomial pneumonia | Low risk | Comment: unclear whether outcome assessors were blinded. Pneumonia was an objective outcome that was detected as per the definition in the study protocol |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: All other outcomes of interest were objective in nature, so the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: 424 patients were randomised to 1 of 3 treatment regimens. One hundred eighty‐two patients were discontinued from study participation for reasons mentioned under 'adherence of regimens'. Intention to treat was done for the outcomes of pneumonia and mortality only. All participants who were randomised to the 2 groups were not analysed for all outcomes, and a per‐protocol analysis was done. The number of participants available for analysis appears to be balanced between groups. Therefore, the likelihood of bias due to attrition is low |
| Selective reporting (reporting bias) | High risk | Comment: All intended outcomes were analysed and reported, but intention to treat was done for the outcomes of pneumonia and mortality only |
| Other bias | Low risk | Comment: Source of funding was Health Services Foundation of Carolinas Medical Center. The role of the sponsor in the conduct and reporting of the trial is unclear. No other known source of bias |
Tryba 1985.
| Methods | Open‐label randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised:100 Number analysed: 100 Cimetidine
Antacids
Sucralfate
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "There were no statically significant differences among the three treatment groups on basic or laboratory parameters" Comment: The 3 groups were similar with respect to demographic data |
|
| Interventions |
Cimetidine
Antacids
Sucralfate
Adherence to regimen: Quote: "... nausea and vomiting led to the discontinuation of antacid in four patients. The interval between administration of antacid doses had to be extended to four hours in 18 patients following the removal of their stomach tubes because they refused more frequent doses. Moreover, antacids could not be administered every two hours during the night in patients without a stomach tube since it was unreasonable to awaken them repeatedly for this purpose. Nausea or vomiting occurred in nine patients receiving sucralfate following the removal of the stomach tubes. This necessitated discontinuing the drug in one patient and changing the dosage interval to eight hours in four patients" Duration of trial: September 1982 to December 1983 Duration of follow up: not clearly mentioned in the study report. Probably until death or discharge |
|
| Outcomes |
Outcomes sought in review and reported in trial Primary outcomes
Secondary outcomes
Outcomes sought but not reported in trial
Outcomes reported but not used in review
|
|
| Notes |
Setting: Department of Anaesthesiology and Biometry Hannover School of Medicine, Hannover, Federal Republic of Germany Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Quote: "The study was carried out from September 1982 to December 1983 with institutional approval and in accordance with the guidelines of the German Drug Law" Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: unclear whether outcome assessors were blinded. However, GI bleeding was an objective outcome that was detected as per the definition in the study protocol |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: All other outcomes of interest were objective in nature |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All randomised participants were part of the analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: unclear on the source of funding. No other form of bias detected |
Tryba 1987.
| Methods | Single‐blind randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 100 participants Number analysed: 100 (for all outcomes except for pneumonia, reasons mentioned below) Antacid
Sucralfate
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "Both the groups were comparable on basic clinical or laboratory parameters" Comment: Both groups were comparable with respect to the distribution of demographic characteristics and risk factors |
|
| Interventions |
Antacids
Sucralfate
Adherence to regimen: Antacid therapy was discontinued in 3 participants (vomiting after extubation (n = 2) and alkalosis on the second day (n = 1)) Duration of trial: July 1984 to November 1986 Duration of follow‐up: ‐ |
|
| Outcomes |
Outcomes sought in review and reported in trial Primary outcomes
Secondary outcomes
Outcomes sought but not reported in trial
Outcomes reported but not used in review
|
|
| Notes |
Setting: Department of Anaesthesiology, Hannover School of Medicine, Hannover, Federal Republic of Germany Source of funding: Quote: “This study was supported by a grant from E. Merck” Ethics approval: Quote: "...with the approval of the local ethical committee and in accordance with the guidelines of the German Drug Law" Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants. Therefore, high risk of performance bias |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: unclear whether outcome assessors were blinded. GI bleeding was detected as per the definition in the study protocol. Therefore, the likelihood of detection bias is low |
| Blinding (detection bias) Nosocomial pneumonia | Low risk | Quote: "The diagnosis of pneumonia was done by a physician who was unaware of the object of the study” Comment: There was clear definition for diagnosing pneumonia, and the outcome assessor was blinded to the interventions. Therefore, the likelihood of detection bias is low |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: unclear on blinding of outcome assessors. However, outcomes of interest were objective in nature, so the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: “For analysis of pulmonary infections, 39 participants were withdrawn from the study because of thoracic trauma or pneumonia at the time of admission to ICU (18 from antacid arm and 21 from sucralfate arm)” For all other outcomes, all 100 randomised participants were part of the analysis Comment: Excluding these participants is justified as it could not have led to the true estimate of the outcome (incidence of nosocomial pneumonia) due to the intervention |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: Study was supported by a grant from E. Merck. The role of the sponsor in the conduct and reporting of the trial is unclear. No other form of bias detected |
Tryba 1988.
| Methods | Randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 400 participants Number analysed: 400 participants Ranitidine
Pirenzepine
Inclusion criteria
Exclusion criteria
Baseline imbalances: no significant differences |
|
| Interventions |
Ranitidine
Pirenzepine
Adherence to regimen: ‐ Duration of trial: October 1984 to November 1986 Duration of follow‐up: unclear, but might be until discharge from ICU |
|
| Outcomes |
Outcomes sought in review and reported in trial Primary outcomes
Secondary outcomes
Outcomes sought but not reported in trial
Outcomes reported but not used in review
|
|
| Notes |
Setting: Anaesthesiology Department, Hannover and Bochum, Germany Source of funding: ‐ Conflicts of interest: ‐ Ethics Approval: Study was conducted according to the drug law. No further information Informed Consent: ‐ Clinical Trials Registration: ‐ Sample Size Calculation: ‐ |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants. Therefore, high risk of performance bias |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: unclear whether outcome assessors were blinded. GI bleeding was an objective outcome that was detected as per the definition in the study protocol |
| Blinding (detection bias) Nosocomial pneumonia | Low risk | Comment: unclear whether outcome assessors were blinded. Pneumonia was an objective outcome that was detected as per the definition in the study protocol. Outcome assessor was blinded to study aims |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Unclear risk | Comment: Outcome assessor was blinded to study aims. Therefore, low risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All randomised participants were part of the analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes have been reported |
| Other bias | Low risk | Comment: No additional biases were detected |
van den Berg 1985.
| Methods | Double‐blind randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 34 participants Number analysed: 28 participants Cimetidine
Placebo
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote "All the factors including the distribution of patients according to sex, age, nature of intensive care unit were almost same within the two groups except for the mean risk factors (it was 2.6 per person in the cimetidine group and 1.9 in the placebo group)" Comment: More people in the cimetidine group had 3 or more risk factors when compared with the placebo group |
|
| Interventions |
Cimetidine
Placebo
Adherence to regimen: 34 participants entered the study. While 28 completed the trial, 6 people dropped out for the following reasons: 1 participant died on the second day of the study from sepsis, 1 participant developed exanthema on the second day after which all medication was stopped, 1 participant was inadvertently given open cimetidine, 1 participant had bleeding duodenal ulcer proven endoscopically at the end of the study, 1 participant developed anuria, and 1 participant proved to have previous gastric surgery Duration of trial: ‐ Duration of follow‐up: not clearly mentioned in the study report. Probably until death or discharge |
|
| Outcomes |
Outcomes sought in review and reported in trial Primary outcomes
Secondary outcomes
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
|
|
| Notes |
Setting: Rotterdam University Hospital, Netherlands Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The study was performed double blind. The investigators were not involved in the treatment of the patients" Comment: This was a placebo‐controlled trial, and study personnel and investigators were blinded. Therefore, the likelihood of performance bias is low |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Quote: "The study was performed double blind. The investigators were not involved in the treatment of the patients" Comment: GI bleeding was detected as per the definition in the study protocol and the investigators were blinded. Therefore, the likelihood of detection or performance bias is low |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Quote: "The study was performed double blind. The investigators were not involved in the treatment of the patients" Comment: Study was performed as a double‐blinded randomised controlled trial, and the outcomes of interest were objective in nature. Therefore, the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: Not all randomised participants were part of the final analysed. 6 participants were excluded for reasons mentioned under "Adherence to the regimen". Moreover, it is unclear to which group these participants belonged, and an intention‐to‐treat analysis was not done. A per‐protocol analysis was performed for the outcomes of interest, and the number of participants available for analysis appears to be balanced between groups. Therefore, the likelihood of attrition bias is low |
| Selective reporting (reporting bias) | High risk | Comment: All intended outcomes were analysed and reported. All‐cause mortality in ICU was not specifically reported for each group |
| Other bias | Low risk | Comment: unclear on the source of funding. No additional biases were detected |
van Essen 1985.
| Methods | Open‐label randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 90 participants Number analysed: 58 participants Prostaglandin
Placebo
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "29 participants in both the groups were similar with regard to sex distribution, age and number and nature of risk factors" Comment: no significant difference between the 2 groups with respect to demographic and baseline risk factors |
|
| Interventions |
Prostaglandin
Placebo
Adherence to regimen: Quote: "The study lasted 3 ‐ 7 days. Treatment was discontinued if enteral feeding was given within the first three days, if gastric tube was removed or participant underwent surgery or had GI bleeding" Of 32 participants, 8 participants had gastrectomy performed within first 3 days; in 1 participant, no chromium labelling was performed, 6 participants were discharged within first 3 days from ICU 3 participants showed non‐compliance with entry criteria, 12 participants received enteral feeds or underwent gastric tube removal within first 3 days, and 9 participants died within the first 3 days Duration of trial: November 1981 to September 1983 Duration of follow‐up: ‐ |
|
| Outcomes |
Outcomes sought in review and reported in trial Primary outcomes
Secondary outcomes
Outcomes sought but not reported in trial
Outcomes reported but not used in review
|
|
| Notes |
Setting: Departments of Internal Medicine and Surgery, University Hospital, Dijkzigt, Rotterdam, The Netherlands Source of funding: Quote: "The study was supported, in part, by a grant–in‐aid from Upjohn Inc., Kalamazoo, MI" Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Patients were randomly assigned to receive placebo or prostaglandin therapy in a double blind fashion" Comment: This was a placebo‐controlled trial where most likely participants and study personnel were blinded to the interventions. Therefore, the likelihood of performance bias is low |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: unclear on blinding of outcome assessors. However, GI bleeding was an objective outcome that was detected as per the definition in the study protocol. Therefore the likelihood of detection bias is low |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: unclear on blinding of outcome assessors. However, the outcome of interest was objective in nature, so the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: Not all randomised participants were part of the final analysis. 90 were randomised, and only 58 were part of the study. However, this was a per‐protocol analysis, and the numbers were well balanced between groups. Therefore there is no serious attrition bias |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported in the study |
| Other bias | Low risk | Comment: Upjohn Inc., Kalamazoo, MI, partially funded the study. The role of the sponsor in the conduct and reporting of the trial is unclear. No other sources of bias are suspected |
Wang 2015.
| Methods | Parallel‐group randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 100 Number analysed: 100 Bowel stimulation protocol
No prophylaxis
Inclusion criteria
Exclusion criteria
Baseline imbalances: none |
|
| Interventions |
Bowel stimulation protocol
No prophylaxis
|
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought in review and not reported in trial
Outcomes reported in trial and not used in review
|
|
| Notes |
Setting: Department of ICU, the Second Hospital Affiliated to Tianjin Medical University, Tianjin, China Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Comment: mentioned in the study report that study authors sought ethics approval Informed consent: Comment: mentioned in the study report that study authors sought for informed consent Clinical trials registration: ‐ Sample size calculation: ‐ |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: Patients were randomly divided into an intervention group and a control group, with 50 patients in each group, but the detailed methods were not mentioned |
| Allocation concealment (selection bias) | Unclear risk | Comment: no information reported |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: lack of blinding of participants and healthcare providers, but outcomes were measured objectively |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: Outcome was measured objectively, and no detection bias is suspected |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Unclear risk | Comment: no information reported |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: no missing data detected |
| Selective reporting (reporting bias) | Low risk | Comment: All outcomes described in Methods were reported in Table 2 |
| Other bias | Unclear risk | Comment: Detailed diagnosis of GI dysfunction was not reported ‐ probably differential diagnostic activity between intervention group and control group |
Wee 2013.
| Methods | Parallel‐group randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: ‐ Number analysed: ‐ Famotidine
Pantoprazole
Inclusion criteria: each patient admitted to ICU/CCM Exclusion criteria: ‐ Baseline imbalances: Both groups had similar baseline characteristics including risk factors for SRMB (2.7 vs 2.7, P = 0.50); however, the pantoprazole group had higher APACHE‐II scores (23.9 vs 20.1; :P < 0.01) |
|
| Interventions |
Famotidine
Pantoprazole
Adherence to regimen: ‐ Duration of trial: December 2012 to April 2013 Duration of follow‐up: until discharge |
|
| Outcomes |
Outcomes sought in review and reported in trial
Outcomes sought in review and not reported in trial
Outcomes reported in trial, but not used in review ‐ |
|
| Notes |
Setting: ICU/CCU, Kingsbrook Jewish Medical Center, Brooklyn, NY 11203, USA, Arnold & Marie Schwartz College of Pharmacy and Health Sciences, Long Island University, Brooklyn, NY 11201, USA Sponsorship source: ‐ Conflicts of interest: ‐ Comments: 2 conference abstracts reporting on 1 study Ethical approval: Quote: "Expedited IRB approval was granted" Informed consent: Quote: "Informed consent was not required" Sample size calculation: ‐ Clinical trials registration: ‐ |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Randomized" Comment: not enough details reported |
| Allocation concealment (selection bias) | Unclear risk | Comment: no information reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: no information on blinding reported. Lack of blinding is unlikely to introduce bias to outcome measures and outcomes |
| Blinding (detection bias) Clinically important upper GI bleeding | Unclear risk | Comment: not enough details described |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Unclear risk | Comment: no information on blinding of outcome assessors reported. Lack of blinding might potentially introduce bias |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: conference abstracts. Not enough information reported to assess incomplete outcome data |
| Selective reporting (reporting bias) | Unclear risk | Comment: Secondary outcomes listed in the Methods sections of the two abstracts differ slightly. Not enough information reported to assess selective outcome reporting |
| Other bias | Unclear risk | Comment: not enough information reported to assess other sources of bias |
Weigelt 1981.
| Methods | Quasi‐randomised trial | |
| Participants |
Baseline characteristics Number randomised: 77 participants Number analysed: 77 participants Antacid
Cimetidine (300 mg every 4 hours)
Cimetidine (300 mg every 6 hours)
Cimetidine (400 mg every 4 hours)
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "A modification of Theraputic intervention scoring system (TISS) was used to grade severity of illness for patient selection and group comparison" Comment: Average TISS score was 3 for antacids and 3.16 for combined cimetidine groups |
|
| Interventions |
Antacids
Cimetidine
Adherence to regimen: Quote: "The pH control in the antacid group was achieved with an average dose of 30 mL of antacid every two hours (range 15 to 120 mL/hour). Only one patient required more than 60 mL/hour for pH control. The duration of cimetidine treatment was similar in the three groups regardless of the efficacy of pH control" Comment: According to the study report, pH was controlled in 14 cimetidine participants. The rest might have switched over to antacids according to the study protocol (but this is not clearly mentioned in the study report) Duration of trial: February 1979 to August 1979 Duration of follow up: Quote: "All patients were followed up until death, discharge from ICU or institution of enteral feedings" |
|
| Outcomes |
Outcomes sought in review and reported in trial Primary outcomes
Secondary outcomes
Outcomes sought but not reported in trial
Outcomes reported but not used in review
|
|
| Notes |
Setting: Departments of Surgery, South‐Western Medical School, University of Texas Health Science Centre, Dallas Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Cimetidine groups were combined to form a common interventional arm vs antacids, as the review did not aim to investigate efficacy among different routes of administration of the same intervention |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Quote: "Patients entering the study were randomised by hospital unit number into one of the four groups…" Comment: This was a quasi–randomised trial, and sequence was not generated |
| Allocation concealment (selection bias) | High risk | Quote: "Patients entering the study were randomised by hospital unit number into one of the four groups…" Comment: This was a quasi–randomised trial, and allocation was not concealed |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: Tthis was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants. Therefore, high risk of performance bias |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: unclear on the blinding of outcome assessors. However, GI bleeding was an objective outcome that was detected as per the study definition, so the likelihood of performance or detection bias is low |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: Outcomes of interest were objective in nature, so the likelihood of performance or detection bias is low |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All quasi‐randomised participants were part of the final analysis, so there is no attrition bias |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: unclear on source of funding. No other form of bias detected |
Yildizdas 2002.
| Methods | Open‐label randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 200 participants Number analysed: 160 participants Sucralfate
Rantidine
Omeprazole
No prophylaxis
Inclusion criteria
Exclusion criteria
Baseline imbalances: no significant difference between groups with respect to age, gender, or primary disease (sepsis and bronchitis were the most common primary diseases among participants) |
|
| Interventions |
Sucralfate
Ranitidine
Omeprazole
No prophylaxis
Adherence to regimen: Quote: "Administration of the drugs were unblinded and were begun within 6 hours of PICU admission" "40 patients were excluded from study according to the exclusion criteria" Duration of trial: August 2000 to February 2002 Duration of follow‐up: ‐ |
|
| Outcomes |
Outcomes sought in review and reported in trial Primary outcome
Secondary outcomes
Outcomes sought but not reported in trial report
Outcomes reported in report but not used in review
|
|
| Notes |
Setting: PICU of Faculty of Medicine, Cukurova University, Adana, Turkey Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: According to the study, hospital mortality was defined as deaths occurring in the PICU, and hospital stay was defined as days spent in the PICU. Pseudomonas aeruginosa and Klebsiella pneumoniae were the most common organisms cultured in participants with the diagnosis of ventilator‐associated pneumonia |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomised to one of the following stress ulcer prophylaxis regimens by using a computer generated random number table" Comment: The method adopted to generate a random sequence is clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in study report |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Administration of drugs were unblinded..." Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants. Therefore, high risk of performance bias |
| Blinding (detection bias) Clinically important upper GI bleeding | Unclear risk | Comment: not clear whether outcome assessors were blinded. Moreover, the definition for diagnosing GI bleeding, which is an objective outcome, is not clearly mentioned in the study report. Not clear whether this would have caused any detection bias |
| Blinding (detection bias) Nosocomial pneumonia | Low risk | Comment: not clear whether outcome assessors were blinded. But ventilator‐associated pneumonia was assessed as per the definition in the study report. Therefore, the likelihood of performance or detection bias is low |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: not clear whether outcome assessors were blinded. However, the other outcomes of interest were objective in nature, so the likelihood of performance or detection bias is low |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "...200 patients were randomised to one of the 4 treatment regimens. 40 patients were excluded from study according to the exclusion criteria" Comment: A per‐protocol analysis was done, and only those participants who completed ≥ 48 hours in the study were part of the final analysis. There is also a balance between groups with respect to the number of participants analysed. Therefore, the likelihood of attrition bias is low |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes have been analysed and reported |
| Other bias | Low risk | Comment: unclear on the source of funding. No additional biases were detected |
Zinner 1981.
| Methods | Open‐label randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 300 participants (the report says that 40 additional participants were entered into the randomised trial, not sure if they were randomised) Number analysed: 300 participants Cimetidine
Antacids
No prophylaxis
Inclusion criteria
Exclusion criteria
Baseline imbalances: The 3 groups were similar with respect to age, gender, and medical conditions. Most participants were admitted for cardiac or general surgery |
|
| Interventions |
Cimetidine
Antacids
No prophylaxis
Adherence to regimen: 33 participants who met the inclusion criteria were randomised (100 in each treatment arm). 40 additional participants entered into the study but were removed for the following reasons: protocol errors or request from physician (n = 31), no reason mentioned (n = 9) Duration of trial: December 1977 to December 1979 Duration of follow‐up: not clearly mentioned in the study report. Probably until death or discharge |
|
| Outcomes |
Outcomes sought in review and reported in trial Primary outcome
Secondary outcomes
Outcomes sought but not reported in trial
Outcomes reported but not used in review
|
|
| Notes |
Setting: surgical intensive care unit of the Johns Hopkins Hospital and the intensive care unit of the Baltimore City Hospitals Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: study approved by the joint committee on clinical investigation of John Hopkins Medical Institutions Informed consent: Quote: "Informed consent was obtained from all participants" clinical trials registration: ‐ Sample size calculation: ‐ |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants. Moreover, 2 arm received no prophylaxis. Therefore, high risk of performance bias |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: It is unclear whether outcome assessors were blinded. GI bleeding was an objective outcome detected as per the definition in the study protocol. Therefore, the likelihood of detection bias is low |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: It is unclear whether outcome assessors were blinded. Moreover, outcomes of interest were objective in nature. Therefore the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "Three hundred participants entered the inclusion criteria and were randomised to three treatment groups. Forty additional participants were entered into RCT but were removed from the protocol. Thirty one of these participants were excluded because of protocol errors or because of request by physician. These were evenly distributed between the treatment groups" Comment: The reason for exclusion of the remaining 9 participants is not clear. The study arms of these 40 participants are also not clearly mentioned in the study report. However, there appears to be no imbalance between study groups with respect to the number of participants available for analysis. Therefore the likelihood of bias due to attrition is low |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: Source of funding is unclear. No other known source of bias |
Zinner 1989.
| Methods | Single‐blind randomised controlled trial | |
| Participants |
Baseline characteristics Number randomised: 371 Number analysed: 281 (for primary outcome) Misoprostol
Antacid
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "There were no statistical differences between the two treatment groups with respect to age, sex, or race. They were also comparable in mean height, weight, and vital signs on admission, and these did not differ with respect to study site. Initial gastric lesion scores were 0 or 1 in 88% of the patients, and initial duodenal lesion scores were 0 or 1 in 96% of the patients, with no significant differences between the two treatment groups (p = 0.141 and 0.848, respectively)" Comment: The 2 groups were similar with respect to demographic and baseline risk factors |
|
| Interventions |
Misoprostol
Antacid
Adherence to regimen: Quote: "Comparable numbers of patients completed the 14 days of treatment or were released earlier, having met the dietary requirement. By the eighth day, only 16% of the misoprostol group and 21% of the antacid group remained in the ICU (p = not significant). Total time of nasogastric medication administration was also somewhat shorter in the misoprostol group (655 patient days vs. 731 patient days in the antacid group), but again these differences were not statistically significant.", "141 in Misoprostol and 140 in Antacid were only analysed as they met the following four criteria for other evaluations 1. completed at least three days in the study, 2. took at least 80% of the assigned medication, 3. did not withdraw from the study except for side effects of the medication, 4. had sufficient follow up endoscopy information to permit outcome evaluation" Comment: 46 participants on misoprostol and 44 participants on antacid regimens were not unavailable for analysis of the primary outcome of GI bleeding Duration of trial: ‐ Duration of follow‐up: ‐ |
|
| Outcomes |
Outcomes sought in review and reported in trial Primary outcomes
Secondary outcomes
Outcomes sought but not reported in trial report
Outcomes reported in report but not used in review
|
|
| Notes |
Setting: 16 university‐associated medical centres: King/Drew Medical Center, Los Angeles, University of Minnesota Hospitals, Minneapolis, MN, Georgetown University Hospital, Washington, DC, Denver General Hospital, Denver, Health Science Center, Brooklyn, NY: Cook County Hospital, Chicago, IL, Minneapolis VA Medical Center, Minneapolis, Hines VA Hospital, Milton S. Hershey Medical Center, VA Medical Center, Fresno, CA, W.P, Temple University Hospital, Philadelphia, PA, University of California, Irvine, CA, VA Medical Center, San Francisco, CA, University Texas Health Science Center, Dallas, TX, Johns Hopkins Medical Institutions, Baltimore Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Quote: "The Institutional Review Board at each of the study sites approved the protocol" Informed consent: Quote: "Each patient gave written informed consent" Clinical trials registration: ‐ Sample size calculation: Quote: "The protocol was designed to require a minimum of 270 fully evaluable patients to complete the study (135 in each group). This sample size is sufficient to detect differences of 20% or more between two treatment groups (p = 0.05; power = 0.90) with two‐sided tests of significance. That is, this study size should detect a clinically significant difference between misoprostol and antacid that is greater than 20%" |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: nNot clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Acceptable candidates were randomly assigned to each group and they received either tablets containing 200 mcg of misoprostol every four hours plus placebo liquid antacid every two hours, or placebo tablets every four hours plus magnesium‐aluminum hydroxide liquid antacid every two hours. Tablets were dissolved in 20 mL of water and administered six times daily through a nasogastric tube, or were given orally if no tube was in place" Comment: This was a 'double‐dummy' placebo‐controlled trial, and participants and study personnel appear to be blinded. Therefore, the likelihood of performance bias is low |
| Blinding (detection bias) Clinically important upper GI bleeding | Low risk | Comment: This was a 'double‐dummy' placebo‐controlled trial. Moreover, GI bleeding was an objective outcome that was detected as per the definition in the study protocol. Therefore, there is no likelihood of performance bias in this study |
| Blinding (detection bias) Nosocomial pneumonia | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) Adverse reactions of interventions | Low risk | Comment: This was a 'double‐dummy' placebo‐controlled trial. However, outcomes of interest were objective in nature, so the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: Not all 371 randomised participants were part of the final analysis. The study was designed in such a way that only those participants who met certain criteria for evaluation of the primary outcome (as described under 'adherence to the regimen') were included in the study report (141 participants in misoprostol and 140 participants in antacid groups). A per‐protocol analysis was done. However, the participants for whom outcomes were reported were well balanced between groups. Therefore, the likelihood of bias due to attrition is low |
| Selective reporting (reporting bias) | High risk | Comment: Study says that all randomised participants were analysed for safety of the drug, but this was true only for the adverse event of diarrhoea and not for other adverse events. The reasons for excluding some of the participants is not clearly mentioned in the study report |
| Other bias | Low risk | Comment: unclear on the source of funding. No other form of bias detected |
ADH: antidiuretic hormone.
APACHE: Acute Physiologic Assessment and Chronic Health Evaluation.
APS: acute physiology score.
BAL: bronchoalveolar lavage.
BMI: body mass index.
bw: body weight.
CABG: coronary artery bypass graft.
CNS: central nervous system.
CXR: chest X‐ray.
GCS: Glasgow Coma Scale.
GD: gastroduodenal.
GI: gastrointestinal.
ICU: intensive care unit.
IMED: infusion pump.
IQR: interquartile ratio.
ITT: intention‐to‐treat.
IV: intravenous.
IVAC: infusion pump.
NG: nasogastric.
NPO: nothing by mouth.
NSAID: non‐steroidal anti‐inflammatory drug.
OR: odds ratio.
PaO2: partial pressure of oxygen in arterial blood.
PCT: procalcitonin
PICU: paediatric intensive care unit.
PSB: protected specimen brush.
SD: standard deviation.
SE: standard error.
SICU: surgical intensive care unit.
SOFA: sepsis‐related organ failure assessment score.
SRMD: stress‐related mucosal disease.
SUP: stress‐ ulcer prophylaxis
TISS: therapeutic intervention scoring system.
VAP: ventilator‐associated pneumonia.
WBC: white blood cell.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Aanpreung 1998 | The review does not deal with comparing different drugs in the same class (H2 receptor antagonists) |
| Abe 2004 | Wrong study population |
| Al‐Quorain 1994 | The review does not deal with comparing different drugs in the same class (H2 blocker antagonists) |
| Alaniz 2014 | Wrong study design |
| Anonymous 2013 | Wrong study design |
| Anonymous 2015 | Wrong study design |
| Arora 1991 | Wrong study population |
| Baccino 1987 | Wrong study population |
| Baghaie 1995 | The review does not deal with comparing different drugs in the same class (H2 receptor antagonists) |
| Barth 1984 | The review does not deal with comparing different drugs in the same class (H2 blocker antagonists) |
| Bauer 1977 | Wrong study design |
| Bergmans 2001 | Wrong indication |
| Bhatt 2010 | Wrong setting, no ICU setting |
| Cheadle 1985 | Wrong setting |
| Chernov 1971 | Wrong study design |
| Cloud 1994 | Wrong indication/study aim (prevention of gastritis) (conference abstract) |
| Critchlow 1987 | Wrong study design |
| Dabiri 2015 | The review does not deal with comparing different drugs in the same class (proton pump inhibitors) |
| Driscoll 1993 | Wrong intervention and comparison |
| Duma 1986 | The review does not deal with comparing different drugs in the same class (H2 receptor antagonists) |
| Estruch 1991 | Wrong population |
| Fiorucci 1989 | Wrong study design |
| Forestier 2008 | Wrong intervention and study aim |
| Friedl 1985 | The review does not deal with comparing different drugs in the same class (H2 receptor antagonists) |
| Geus 1993 | The review does not deal with comparing different drugs in the same class (H2 receptor antagonists) |
| Hauer 1996 | Wrong study design |
| Heiselman 1995 | The review does not deal with comparing different drugs in the same class (H2 receptor antagonists) |
| Herrmann 1979 | Wrong study design |
| Hollander 1973 | Wrong population |
| Huang 2017 | Wrong setting |
| Kalfarentzos 1997 | The review does not deal with comparing different drugs in the same class (nutrition regimen) |
| Karlstadt 1993 | Wrong population |
| Ketterl 1984 | The review does not deal with comparing different drugs in the same class (H2 receptor antagonists) |
| Khan 1981 | Wrong study design |
| Klarin 2008 | Wrong intervention and comparison |
| Krag 2015 | Wrong study design |
| Krier 1990 | Wrong study design |
| Krueger 2002 | Wrong intervention and comparison |
| Kuusela 1998 | The review does not deal with comparing different drugs in the same class (H2 receptor antagonists) |
| Laterre 2001 | The review does not deal with comparing different drugs in the same class (proton pump inhibitors) |
| Levine 1994 | The review does not deal with comparing different drugs in the same class (H2 receptor antagonist) |
| Liu 2013 | Wrong study population |
| Madani 2014 | Wrong indication/study aim (prevention of gastritis) |
| McAlhany 1976 | Wrong study population |
| McElwee 1979 | Wrong study population |
| Metz 2010 | Wrong study design |
| Misra 2005 | Wrong study population |
| Mojtahedzadeh 2002 | The review does not deal with comparing different drugs in the same class (H2 receptor antagonist) |
| More 1985 | The review does not deal with comparing different drugs in the same class (H2 receptor antagonists) |
| Mulla 2001 | Wrong population (patients on continuous venovenous hemofiltration) |
| Olsen 1995 | The review does not deal with comparing different drugs in the same class (H2 receptor antagonists) |
| Olsen 2008 | The review does not deal with comparing different drugs in the same class (proton pump inhibitor) |
| Osteyee 1994 | The review does not deal with comparing different drugs in the same class (H2 receptor antagonist) |
| Pelfrene 1996 | The review does not deal with comparing different drugs in the same class (H2 receptor antagonist) |
| Phillips 2001 | The review does not deal with comparing different drugs in the same class (nutrition regimen) |
| Reid 1986 | The review does not deal with comparing different drugs in the same class (H2 receptor antagonist) |
| Ren 2015 | Wrong indication (management of gastrointestinal bleeding) |
| Schentag 1989 | The review does not deal with comparing different drugs in the same class (H2 receptor antagonist) |
| Simon 1984 | The review does not deal with comparing different drugs in the same class (H2 receptor antagonist) |
| Solana 2013 | The review does not deal with comparing different drugs in the same class (omeprazole) |
| Sung 2003 | Wrong study design |
| Taha 1996 | Wrong study population |
| Tofil 2008 | Wrong study design |
| Toyota 1998 | Wrong study population |
| Udd 2005 | The review does not deal with comparing different drugs in the same class (omeprazole) |
| Vaduganathan 2016 | Wrong setting |
| Vargas 1993 | The review does not deal with comparing different drugs in the same class (H2 receptor antagonist) |
| Wang 1995 | The review does not deal with comparing different drugs in the same class (H2 receptor antagonist) |
| Yao 2015 | Wrong setting, no ICU setting |
| Zhou 2002 | Wrong study population |
ICU: intensive care unit.
Characteristics of studies awaiting assessment [ordered by study ID]
Labattut 1992.
| Methods | Randomised controlled trial |
| Participants | People admitted to surgical ICU |
| Interventions | ‐ |
| Outcomes | ‐ |
| Notes | Full text could not be obtained |
Morris 2001.
| Methods | Randomised controlled trial |
| Participants | People admitted to surgical ICU |
| Interventions | ‐ |
| Outcomes | ‐ |
| Notes | Full text could not be obtained |
ICU: intensive care unit.
Characteristics of ongoing studies [ordered by study ID]
ACTRN12616000481471.
| Trial name or title | Proton pump inhibitors vs histamine‐2 receptor blockers for ulcer prophylaxis therapy in the intensive care unit |
| Methods | Multi‐centre open‐label cluster‐randomised cross‐over study |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Intervention: proton pump inhibitors Control: H2 receptor antagonists |
| Outcomes | Primary outcome
Secondary outcomes
|
| Starting date | April 2016 |
| Contact information | Dr. Paul Young, Wellington Hospital, Riddiford Street, Newtown, Wellington 6021, New Zealand; paul.young@ccdhb.org.nz |
| Notes | anzctr.org.au/Trial/Registration/TrialReview.aspx?id=370438 |
EUCTR2015‐000318‐24‐DK.
| Trial name or title | Stress ulcer prophylaxis in the intensive care unit |
| Methods | Parallel‐group double‐blind randomised controlled study |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Intervention: 4 mL pantoprazole IV Control: placebo |
| Outcomes | Primary outcomes
Secondary outcomes
|
| Starting date | August 2016 |
| Contact information | Morten Hylander; mortenhylander@gmail.com |
| Notes | clinicaltrialsregister.eu/ctr‐search/search?query=EUCTR2015‐000318‐24‐DK |
EudraCT 2007‐006102‐19.
| Trial name or title | Omeprazole treatment for prophylaxis of gastrointestinal bleeding and gastro‐oesophageal reflux in critically ill children [Tratamiento con omeprazole para la profilaxis de la hemorragia digestiva y el reflujo gastroesofágico en niños críticos] |
| Methods | ‐ |
| Participants | Infants and toddlers, children, adolescents, under 18 |
| Interventions | Intervention: 0.4 mL omeprazole IV Control: 10 mL ranitidine IV |
| Outcomes | Primary outcomes:
Secondary outcomes: ‐ |
| Starting date | February 2008 |
| Contact information | ‐ |
| Notes | clinicaltrialsregister.eu/ctr‐search/search?query=EUCTR2007‐006102‐19‐ES |
IRCT201104134578N2.
| Trial name or title | Ranitidine and pantoprazole in prevention of stress ulcer |
| Methods | Not stated |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Intervention: pantoprazole 40 mg infusion once a day until end of patient admission Control: ranitidine 50 mg infusion twice a day until end of patient admission |
| Outcomes | Primary outcome
Secondary outcome
|
| Starting date | 20 February 2011 |
| Contact information | Farshid Rahimi Bashar, Mahdiye Street, Hamedan University Of Medical Sciences, Hamedan Iran, Islamic Republic of Telephone: 00988118276295; fr_rahimibashar@yahoo.com |
| Notes | ‐ |
ISRCTN12845429.
| Trial name or title | A placebo‐controlled double‐blind randomised feasibility trial of desmopressin (DDAVP) in critical illness prior to procedures |
| Methods | Placebo‐controlled double‐blind randomised study |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Intervention: desmopressin Control: ‐ |
| Outcomes | Primary outcome
Secondary outcome ‐ |
| Starting date | November 2016 |
| Contact information | ‐ |
| Notes | clinicaltrialsregister.eu/ctr‐search/search?query=EUCTR2016‐001126‐33‐GB |
Krag 2016.
| Trial name or title | Stress ulcer prophylaxis with a proton pump inhibitor vs placebo in critically ill patients (SUP‐ICU trial): study protocol for a randomised controlled trial |
| Methods | Investigator‐initiated pragmatic international multi‐centre randomised blinded parallel‐group study |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Intervention: pantoprazole 40 mg IV (pantoprazole; Actavis, Gentofte, Denmark) Control: placebo, given once daily IV, from randomisation until ICU discharge or death for a maximum of 90 days |
| Outcomes | Primary outcomes
Secondary outcomes
|
| Starting date | January 2016 |
| Contact information | Mette Krag, Department of Intensive Care 4131, Copenhagen University Hospital, Rigshospitalet; mette.krag.01@regionh.dk |
| Notes | ‐ |
NCT00590928.
| Trial name or title | Gastric pH in critically ill patients |
| Methods | Double‐blind parallel‐group randomised controlled study |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Intervention: esomeprazole 40 mg once daily Control: ranitidine 50 mg every hour |
| Outcomes | Primary outcome
Secondary outcomes
|
| Starting date | July 2004 |
| Contact information | Christian Madl, MD, Medical University of Vienna |
| Notes | clinicaltrials.gov/ct2/show/record/NCT00590928 |
NCT00702871.
| Trial name or title | A clinico‐bacteriological study and effect of stress ulcer prophylaxis on occurrence of ventilator associated pneumonia |
| Methods | Parallel‐group open‐label randomised controlled study |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Intervention: ranitidine 50 mg IV 8‐hourly for entire duration of ICU stay Control: sucralfate 1 g via NG tube 6‐hourly for entire duration of ICU stay |
| Outcomes | Primary outcome
Secondary outcome ‐ |
| Starting date | March 2005 |
| Contact information | Rajiv Singla, MD, Maulana Azad Medical College and Lok Nayak Hospital, Delhi, India |
| Notes | clinicaltrials.gov/ct2/show/record/NCT00702871 |
NCT02157376.
| Trial name or title | Stress ulcer prophylaxis of intravenous esomeprazole in Chinese seriously ill patients (SUP) |
| Methods | Double‐blind parallel‐group randomised controlled study |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Intervention: IV esomeprazole 30 min intermittent infusions given for maximum 14 days Control: IV cimetidine 30 min bolus infusion followed by IV cimetidine continuous infusion given for maximum 14 days |
| Outcomes | Primary outcome
Secondary outcomes
|
| Starting date | July 2014 |
| Contact information | Xinyu Qin, Professor and Chairman, Department of General Surgery, Zhongshan, Hospital, Fudan University |
| Notes | clinicaltrials.gov/ct2/show/record/NCT02157376 |
NCT02290327.
| Trial name or title | Re‐evaluating the inhibition of stress erosions: gastrointestinal bleeding prophylaxis In ICU (REVISE) |
| Methods | Quadruple blind parallel‐group randomised controlled study |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Intervention: pantoprazole 40 mg in 50 mL 0.9% normal saline intravenously once daily Control: placebo 50 mL of 0.9% normal saline intravenously once daily |
| Outcomes | Primary outcomes
Secondary outcomes
|
| Starting date | May 2015 |
| Contact information | ‐ |
| Notes | clinicaltrials.gov/ct2/show/record/NCT02290327 |
NCT02718261.
| Trial name or title | Sup‐Icu RENal (SIREN) ‐ a subanalysis of the prospective SUP (stress ulcer prophylaxis)‐ICU trial on the risk of GI bleeding in ICU patients receiving renal replacement therapy |
| Methods | Parallel‐group triple‐blind randomised controlled study |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Intervention: pantoprazole IV Control: 0.9% saline IV |
| Outcomes | Primary outcome
Secondary outcomes
|
| Starting date | February 2016 |
| Contact information | Joerg Schefold; joerg.schefold@insel.ch |
| Notes | clinicaltrials.gov/ct2/show/record/NCT02718261 |
NCT03098537.
| Trial name or title | Effects of enteral nutrition on stress ulcer haemorrhage ‐ multi‐center randomised controlled trial |
| Methods | Open‐label parallel‐group randomised controlled study |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Intervention: enteral nutrition + proton pump inhibitor Control: enteral nutrition only |
| Outcomes | Primary outcome
Secondary outcome ‐ |
| Starting date | August 2016 |
| Contact information | kgundogan@erciyes.edu.tr |
| Notes | clinicaltrials.gov/ct2/show/record/NCT03098537 |
APACHE: Acute Physiologic Assessment and Chronic Health Evaluation.
aPTT: activated partial thromboplastin time.
CDI: Clostridium difficile infection.
CT: computed tomography.
DDAVP: desmopressin.
EN: enteral nutrition.
ESRD: end‐stage renal disease.
GI: gastrointestinal.
H2RA: histamine receptor‐2 antagonist.
hCG: human chorionic gonadotropin.
HIV: human immunodeficiency virus.
ICU: intensive care unit.
INR: international normalised ratio.
IV: intravenous.
NG: nasogastric.
NSAIDs: non‐steroidal anti‐inflammatory drugs.
OG: orogastric.
PLT: platelet blood test.
PPI: proton pump inhibitor.
PT: prothrombin time.
RBC: red blood cell.
RRT: rapid resolution therapy.
SAR: serious adverse reaction.
SDM: substitute decision‐maker.
TIA: transient ischaemic attack.
Differences between protocol and review
The number of participants requiring more than two units of blood was changed to not define the number of units transfused.
The outcome 'ventilator‐associated pneumonia' was changed to 'nosocomial pneumonia including ventilator‐associated pneumonia' because definitions varied across study reports.
'Potassium‐competitive acid blockers' was added to list of eligible interventions.
Contributions of authors
Ingrid Toews: searching for additional references, selecting studies, extracting data, assessing quality, locating and retrieving full‐text articles, refining and updating the review, and writing and approving the final version of the review.
Aneesh Thomas George: developing and drafting the initial version of the protocol, searching for additional references, and approving the final version of the protocol and the review.
John Victor Peter: conceptualising the review topic, developing the protocol, providing citations and full‐text articles for the background, refining the review, and approving the final version.
Richard Kirubakaran: developing the protocol and approving the final version of the protocol.
Jabez Paul Barnabas: locating and retrieving full‐text articles and approving the final version of the protocol.
Luis Eduardo S Fontes: performing study selection, data extraction, and quality assessment; locating and retrieving full‐text articles; refining and updating the review; and writing the Authors' Conclusions section.
Joerg Meerpohl: performing study selection, locating and retrieving full‐text articles, and refining and approving the final version of the review.
Sources of support
Internal sources
-
South Asian Cochrane Centre, India.
Employment and training of Dr. George, Mr. Kirubakaran and Mr. Barnabas
External sources
-
Indian Council of Medical Research, New Delhi, India.
Funding for staff and activities of the South Asian Cochrane Centre
-
Christian Medical College, Vellore, India.
Salary of Drs. Tharyan and Peter; logistic support for the South Asian Cochrane Centre
Declarations of interest
All review authors declare no conflict academic or financial interests that will influence the conduct, interpretation, or reporting of this review.
New
References
References to studies included in this review
Ali 2016 {published data only}
- Ali AY, Selvanderan S, Summers M, Finnis M, Plummer M, Anderson M, et al. Comparison of macroscopic abnormalities in patients receiving routine pantoprazole when compared to placebo. Australian Society of Anaesthetists 2016;44(2):301. [Google Scholar]
Apte 1992 {published data only}
- Apte NM, Karnad DR, Medhekar TP, Tilve GH, Morye S, Bhave GG. Gastric colonization and pneumonia in intubated critically ill patients receiving stress ulcer prophylaxis: a randomized, controlled trial. Critical Care Medicine 1992;20(5):590‐3. [PUBMED: 1572183] [DOI] [PubMed] [Google Scholar]
Barandun 1985 {published data only}
- Barandun J, Leutenegger A, Frutiger A, Ruedi T, Kobler E. Prevention of gastroduodenal stress lesions with secretion inhibitors in accident patients needing intensive care: incidence of lesions and adverse effects; consequences [Prophylaxe gastroduodenaler Stresslasionen bei intensivpflegebedürftigen Unfallpatienten mit Sekretionshemmern: Inzidenz von Lasionen und Nebenwirkungen; Konsequenzen.]. Schweizer Medizinische Wochenschrift 1985;115(29):1022‐3. [PUBMED: 3840276] [PubMed] [Google Scholar]
Bashar 2013 {published data only}
- Bashar FR, Manuchehrian N, Mahmoudabadi M, Hajiesmaeili MR, Torabian S. Effects of ranitidine and pantoprazole on ventilator‐associated pneumonia: a randomized double‐blind clinical trial. Tanaffos 2013;12(2):16‐21. [PMC free article] [PubMed] [Google Scholar]
Basso 1981 {published data only}
- Basso N, Bagarani M, Materia A, Fiorani S, Lunardi P, Speranza V. Cimetidine and antacid prophylaxis of acute upper gastrointestinal bleeding in high risk patients. Controlled, randomized trial. American Journal of Surgery 1981;141(3):339‐41. [PUBMED: 7011078] [DOI] [PubMed] [Google Scholar]
Behrens 1994 {published data only}
- Behrens R, Hofbeck M, Singer H, Scharf J, Rupprecht T. Frequency of stress lesions of the upper gastrointestinal tract in paediatric patients after cardiac surgery: effects of prophylaxis. British Heart Journal 1994;72(2):186‐9. [PUBMED: 7917695] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ben‐Menachem 1994 {published data only}
- Ben‐Menachem T, Fogel R, Patel RV, Touchette M, Zarowitz BJ, Hadzijahic N, et al. Prophylaxis for stress‐related gastric hemorrhage in the medical intensive care unit. A randomized, controlled, single‐blind study. Annals of Internal Medicine 1994;121(8):568‐75. [PUBMED: 8085688] [DOI] [PubMed] [Google Scholar]
Bonten 1995 {published data only}
- Bonten MJ, Gaillard CA, Geest S, Tiel FH, Beysens AJ, Smeets HG, et al. The role of intragastric acidity and stress ulcus prophylaxis on colonization and infection in mechanically ventilated ICU patients. A stratified, randomized, double‐blind study of sucralfate versus antacids. American Journal of Respiratory and Critical Care Medicine 1995;152(6 Pt 1):1825‐34. [DOI: 10.1164/ajrccm.152.6.8520743; PUBMED: 8520743] [DOI] [PubMed] [Google Scholar]
Borrero 1984 {published data only}
- Borrero E, Margolis IB, Bank S, Shulman N, Chardavoyne R. Antacid versus sucralfate in preventing acute gastrointestinal bleeding. A randomized trial in 100 critically ill patients. American Journal of Surgery 1984;148(6):809‐12. [PUBMED: 6391232] [DOI] [PubMed] [Google Scholar]
Borrero 1985 {published data only}
- Borrero E, Bank S, Margolis I, Schulman ND, Chardavoyne R. Comparison of antacid and sucralfate in the prevention of gastrointestinal bleeding in patients who are critically ill. American Journal of Medicine 1985;79(2C):62‐4. [PUBMED: 3898835] [DOI] [PubMed] [Google Scholar]
Borrero 1986 {published data only}
- Borrero E, Ciervo J, Chang JB. Antacid vs sucralfate in preventing acute gastrointestinal tract bleeding in abdominal aortic surgery. A randomized trial in 50 patients. Archives of Surgery 1986;121(7):810‐2. [PUBMED: 3521541] [DOI] [PubMed] [Google Scholar]
Bresalier 1987 {published data only}
- Bresalier RS, Grendell JH, Cello JP, Meyer AA. Sucralfate suspension versus titrated antacid for the prevention of acute stress‐related gastrointestinal hemorrhage in critically ill patients. American Journal of Medicine 1987;83(3B):110‐6. [PUBMED: 3499074] [DOI] [PubMed] [Google Scholar]
Brophy 2010 {published data only}
- Brophy GM, Brackbill ML, Bidwell KL, Brophy DF. Prospective, randomized comparison of lansoprazole suspension, and intermittent intravenous famotidine on gastric pH and acid production in critically ill neurosurgical patients. Neurocritical Care 2010;13(2):176‐81. [DOI: 10.1007/s12028-010-9397-3; PUBMED: 20596795] [DOI] [PubMed] [Google Scholar]
Burgess 1995 {published data only}
- Burgess P, Larson GM, Davidson P, Brown J, Metz CA. Effect of ranitidine on intragastric pH and stress‐related upper gastrointestinal bleeding in patients with severe head injury. Digestive Diseases and Sciences 1995;40(3):645‐50. [PUBMED: 7895560] [DOI] [PubMed] [Google Scholar]
Cannon 1987 {published data only}
- Cannon LA, Heiselman D, Gardner W, Jones J. Prophylaxis of upper gastrointestinal tract bleeding in mechanically ventilated patients. A randomized study comparing the efficacy of sucralfate, cimetidine, and antacids. Archives of Internal Medicine 1987;147(12):2101‐6. [PUBMED: 3500684] [PubMed] [Google Scholar]
Chan 1995 {published data only}
- Chan KH, Lai EC, Tuen H, Ngan JH, Mok F, Fan YW, et al. Prospective double‐blind placebo‐controlled randomized trial on the use of ranitidine in preventing postoperative gastroduodenal complications in high‐risk neurosurgical patients. Journal of Neurosurgery 1995;82(3):413‐7. [DOI: 10.3171/jns.1995.82.3.0413] [DOI] [PubMed] [Google Scholar]
Cioffi 1994 {published data only}
- Cioffi WG, McManus AT, Rue LW 3rd, Mason AD, McManus WF, Pruitt BA Jr. Comparison of acid neutralizing and non‐acid neutralizing stress ulcer prophylaxis in thermally injured patients. Journal of Trauma 1994;36(4):544‐7. [PUBMED: 8158717] [DOI] [PubMed] [Google Scholar]
Conrad 2005 {published data only}
- Conrad SA, Gabrielli A, Margolis B, Quartin A, Hata JS, Frank WO, et al. Randomized, double‐blind comparison of immediate‐release omeprazole oral suspension versus intravenous cimetidine for the prevention of upper gastrointestinal bleeding in critically ill patients. Critical Care Medicine 2005;33(4):760‐5. [PUBMED: 15818102] [DOI] [PubMed] [Google Scholar]
Cook 1998 {published data only}
- Cook D, Guyatt G, Marshall J, Leasa D, Fuller H, Hall R, et al. A comparison of sucralfate and ranitidine for the prevention of upper gastrointestinal bleeding in patients requiring mechanical ventilation. Canadian Critical Care Trials Group. New England Journal of Medicine 1998;338(12):791‐7. [DOI: 10.1056/NEJM199803193381203; PUBMED: 9504939] [DOI] [PubMed] [Google Scholar]
- Cook D, Heyland D, Griffith L, Cook R, Marshall J, Pagliarello J. Risk factors for clinically important upper gastrointestinal bleeding in patients requiring mechanical ventilation. Canadian Critical Care Trials Group. Critical Care Medicine 1999;27(12):2812‐7. [PUBMED: 10628631] [DOI] [PubMed] [Google Scholar]
Darlong 2003 {published data only}
- Darlong V, Jayalakhsmi TS, Kaul HL, Tandon R. Stress ulcer prophylaxis in patients on ventilator. Tropical Gastroenterology 2003;24(3):124‐8. [PUBMED: 14978984] [PubMed] [Google Scholar]
Davies 2012 {published data only}
- Davies AR, Morrison SS, Bailey MJ, Bellomo R, Cooper DJ, Doig GS, et al. A multicenter, randomized controlled trial comparing early nasojejunal with nasogastric nutrition in critical illness. Critical Care Medicine 2012;40(8):2342‐8. [DOI: 10.1097/CCM.0b013e318255d87e] [DOI] [PubMed] [Google Scholar]
De Azevedo 2000 {published data only}
- Azevedo JR, Soares MD, Silva GA, Lima PG. Prevention of stress ulcer bleeding in high risk patients. Comparison of three drugs. Gastroenterologia Endoscopia Digestiva 2000;19(6):239‐44. [Google Scholar]
Driks 1987 {published data only}
- Driks MR, Craven DE, Celli BR, Manning M, Burke RA, Garvin GM, et al. Nosocomial pneumonia in intubated patients given sucralfate as compared with antacids or histamine type 2 blockers. The role of gastric colonization. New England Journal of Medicine 1987;317(22):1376‐82. [DOI: 10.1056/Nejm198711263172204; PUBMED: 2891032] [DOI] [PubMed] [Google Scholar]
Eddleston 1991 {published data only}
- Eddleston JM, Vohra A, Scott P, Tooth JA, Pearson RC, McCloy RF, et al. A comparison of the frequency of stress ulceration and secondary pneumonia in sucralfate‐ or ranitidine‐treated intensive care unit patients. Critical Care Medicine 1991;19(12):1491‐6. [PUBMED: 1959368] [DOI] [PubMed] [Google Scholar]
Eddleston 1994 {published data only}
- Eddleston JM, Pearson RC, Holland J, Tooth JA, Vohra A, Doran BH. Prospective endoscopic study of stress erosions and ulcers in critically ill adult patients treated with either sucralfate or placebo. Critical Care Medicine 1994;22(12):1949‐54. [PUBMED: 7988131] [PubMed] [Google Scholar]
Ephgrave 1998 {published data only}
- Ephgrave KS, Kleiman‐Wexler R, Pfaller M, Booth BM, Reed D, Werkmeister L, et al. Effects of sucralfate vs antacids on gastric pathogens: results of a double‐blind clinical trial. Archives of Surgery 1998;133(3):251‐7. [PUBMED: 9517735] [DOI] [PubMed] [Google Scholar]
Fabian 1993 {published data only}
- Fabian TC, Boucher BA, Croce MA, Kuhl DA, Janning SW, Coffey BC, et al. Pneumonia and stress ulceration in severely injured patients. A prospective evaluation of the effects of stress ulcer prophylaxis. Archives of Surgery 1993;128(2):185‐92. [PUBMED: 8431119] [DOI] [PubMed] [Google Scholar]
Fan 2016 {published data only}
- Fan MC, Wang QL, Fang W, Jiang YX, Li LD, Sun P, Wang ZH. Early enteral combined with parenteral nutrition treatment for severe traumatic brain injury: effects on immune function, nutritional status and outcomes. Chinese Medical Sciences Journal 2016;31(4):213. [DOI] [PubMed] [Google Scholar]
Fang 2014 {published data only}
- Fang XW, Chang S, Zhao JH, Qian XY. Prevention and treatment of stress‐induced gastrointestinal bleeding after severe brain injury. World Chinese Journal of Digestology 2014;22(3):404‐8. [Google Scholar]
Fink 2003 {unpublished data only}
- Fink M, Karlstadt RG, Maroko RT, Field B. Intravenous pantoprazole (IVP) and continuous infusion cimetidine (C) prevent upper gastrointestinal bleeding (UGIB) regardless of APSII score (APACHE II) in high risk intensive care unit (ICU) patients. Gastroenterology 2003;124(4 Suppl 1):A625–6. [Google Scholar]
Fogas 2013 {published data only}
- Fogas JF, Kiss KK, Gyura FG, Tobias ZT, Molnar ZM. Effects of proton pump inhibitor versus H2‐receptor antagonist stress ulcer prophylaxis on ventilator‐associated pneumonia: A pilot study. Critical Care 2013;17:S150‐1. [Google Scholar]
Friedman 1982 {published data only}
- Friedman CJ, Oblinger MJ, Suratt PM, Bowers J, Goldberg SK, Sperling MH, et al. Prophylaxis of upper gastrointestinal hemorrhage in patients requiring mechanical ventilation. Critical Care Medicine 1982;10(5):316‐9. [PUBMED: 7042201] [PubMed] [Google Scholar]
Groll 1986 {published data only}
- Groll A, Simon JB, Wigle RD, Taguchi K, Todd RJ, Depew WT. Cimetidine prophylaxis for gastrointestinal bleeding in an intensive care unit. Gut 1986;27(2):135‐40. [PUBMED: 3485068] [DOI] [PMC free article] [PubMed] [Google Scholar]
Halloran 1980 {published data only}
- Halloran LG, Zfass AM, Gayle WE, Wheeler CB, Miller JD. Prevention of acute gastrointestinal complications after severe head injury: a controlled trial of cimetidine prophylaxis. American Journal of Surgery 1980;139(1):44‐8. [PUBMED: 6985776] [DOI] [PubMed] [Google Scholar]
Hanisch 1998 {published data only}
- Hanisch EW, Encke A, Naujoks F, Windolf J. A randomized, double‐blind trial for stress ulcer prophylaxis shows no evidence of increased pneumonia. American Journal of Surgery 1998;176(5):453‐7. [PUBMED: 9874432] [DOI] [PubMed] [Google Scholar]
Hastings 1978 {published data only}
- Hastings PR, Skillman JJ, Bushnell LS, Silen W. Antacid titration in the prevention of acute gastrointestinal bleeding: a controlled, randomized trial in 100 critically ill patients. New England Journal of Medicine 1978;298(19):1041‐5. [PUBMED: 25384] [DOI] [PubMed] [Google Scholar]
Hata 2005 {published data only}
- Hata M, Shiono M, Sekino H, Furukawa H, Sezai A, Iida M, et al. Prospective randomized trial for optimal prophylactic treatment of the upper gastrointestinal complications after open heart surgery. Circulation Journal 2005;69(3):331‐4. [PUBMED: 15731540] [DOI] [PubMed] [Google Scholar]
He 2017 {published data only}
- He GW, Zhao YM, Zhou J. Naloxone combined with pantoprazole for prevention of upper gastrointestinal bleeding in patients with respiratory failure. World Chinese Journal of Digestology 2017;25(1):102. [Google Scholar]
Israsena 1987 {published data only}
- Israsena S, Anantapanpong S, Kladcharoen N, Chayanand D, Limthongkul S. Sucralfate versus antacid in the prevention of stress ulcer bleeding in patients on mechanical ventilation. Journal of the Medical Association of Thailand 1987;70(12):678‐82. [PubMed] [Google Scholar]
Kantorova 2004 {published data only}
- Kantorova I, Svoboda P, Scheer P, Doubek J, Rehorkova D, Bosakova H, et al. Stress ulcer prophylaxis in critically ill patients: a randomized controlled trial. Hepato‐gastroenterology 2004;51(57):757‐61. [PUBMED: 15143910] [PubMed] [Google Scholar]
Kappstein 1991 {published data only}
- Kappstein I, Schulgen G, Friedrich T, Hellinger P, Benzing A, Geiger K, et al. Incidence of pneumonia in mechanically ventilated patients treated with sucralfate or cimetidine as prophylaxis for stress bleeding: bacterial colonization of the stomach. American Journal of Medicine 1991;91(2A):125S‐31S. [DOI: 10.1016/0002-9343(91)90464-9] [DOI] [PubMed] [Google Scholar]
Karlstadt 1990 {published data only}
- Karlstadt RG, Iberti TJ, Silverstein J, Lindenberg L, Bright‐Asare P, Rockhold F, et al. Comparison of cimetidine and placebo for the prophylaxis of upper gastrointestinal bleeding due to stress‐related gastric mucosal damage in the intensive care unit. Journal of Intensive Care Medicine 1990;5(1):26‐32. [DOI: 10.1177/088506669000500106] [DOI] [Google Scholar]
Kaushal 2000 {published data only}
- Kaushal S, Midha V, Sood A, Chopra SC, Gupta C. A comparative study of the effects of famotidine and sucralfate in prevention of upper gastro‐intestinal bleeding in patients of head injury. Indian Journal of Pharmacology 2000; Vol. 32, issue 3:246‐9. [CN‐00424209]
Khorvash 2014 {published data only}
- Khorvash F, Abbasi S, Meidani M, Dehdashti F, Ataei B. The comparison between proton pump inhibitors and sucralfate in incidence of ventilator associated pneumonia in critically ill patients. Advanced Biomedical Research 2014;3(52):1‐6. [DOI: 10.4103/2277-9175.125789] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kingsley 1985 {published data only}
- Kingsley AN. Prophylaxis for acute stress ulcers. Antacids or cimetidine. American Surgeon 1985;51(9):545‐7. [PUBMED: 3898949] [PubMed] [Google Scholar]
Kitler 1990 {published data only}
- Kitler ME, Hays A, Enterline JP, Allo M, Zuidema GD. Preventing postoperative acute bleeding of the upper part of the gastrointestinal tract. Surgery, Gynecology & Obstetrics 1990;171(5):366‐72. [PUBMED: 2237719] [PubMed] [Google Scholar]
Krakamp 1989 {published data only}
- Krakamp B, Rommel T, Edelmann M, Leidig P. Prevention of stress‐induced hemorrhage of the upper gastrointestinal tract in neurosurgical intensive care patients. A controlled, randomized double‐blind study with ranitidine alone and in combination with pirenzepine [Prophylaxe streßbedingter Blutungen aus dem oberen Gastrointestinaltrakt bei neurochirurgischen Intensivpatienten]. Medizinische Klinik 1989;84(3):133‐4. [PubMed] [Google Scholar]
Kuusela 1997 {published data only}
- Kuusela AL, Ruuska T, Karikoski R, Laippala P, Ikonen RS, Janas M, et al. A randomized, controlled study of prophylactic ranitidine in preventing stress‐induced gastric mucosal lesions in neonatal intensive care unit patients. Critical Care Medicine 1997;25(2):346‐51. [PUBMED: 9034275] [DOI] [PubMed] [Google Scholar]
Lacroix 1986 {published data only}
- Lacroix J, Infante‐Rivard C, Gauthier M, Rousseau E, Doesburg N. Upper gastrointestinal tract bleeding acquired in a pediatric intensive care unit: prophylaxis trial with cimetidine. Journal of Pediatrics 1986;108(6):1015‐8. [DOI] [PubMed] [Google Scholar]
Laggner 1988 {published data only}
- Laggner AN, Lenz K, Graninger W, Gremmel F, Grimm G, Base W, et al. Prevention of stress hemorrhage in an internal medicine intensive care station: sucralfate versus ranitidine [Stressblutungsprophylaxe auf einer internen Intensivstation: Sucralfat versus Ranitidin]. Der Anaesthesist 1988;37(11):704‐10. [PUBMED: 3063134] [PubMed] [Google Scholar]
Laggner 1989 {published data only}
- Laggner AN, Lenz K, Base W, Druml W, Schneeweiss B, Grimm G. Prevention of upper gastrointestinal bleeding in long‐term ventilated patients. Sucralfate versus ranitidine. American Journal of Medicine 1989;86(6A):81‐4. [PUBMED: 2786673] [DOI] [PubMed] [Google Scholar]
Lamothe 1991 {published data only}
- Lamothe PH, Rao E, Serra AJ, Castellano J, Woronick CL, McNicholas KW, et al. Comparative efficacy of cimetidine, famotidine, ranitidine, and mylanta in postoperative stress ulcers. Gastric pH control and ulcer prevention in patients undergoing coronary artery bypass graft surgery. Gastroenterology 1991;100(6):1515‐20. [PUBMED: 2019357] [DOI] [PubMed] [Google Scholar]
Larson 1989 {published data only}
- Larson GM, Davidson P, Brown J. Comparison of ranitidine versus placebo on 24 hour gastric pH and upper gastrointestinal (UGI) bleeding in head injuries (abstract). American Journal of Gastroenterology 1989;84:1165. [Google Scholar]
Lee 2014 {published data only}
- Lee TH, Hung FM, Yang LH. Comparison of the efficacy of esomeprazole and famotidine against stress ulcers in a neurosurgical intensive care unit. Advances in Digestive Medicine 2014;1(2):50‐3. [Google Scholar]
Levy 1997 {published data only}
- Levy MJ, Seelig CB, Robinson NJ, Ranney JE. Comparison of omeprazole and ranitidine for stress ulcer prophylaxis. Digestive Diseases and Sciences 1997;42(6):1255‐9. [PUBMED: 9201091] [DOI] [PubMed] [Google Scholar]
Lin 2016 {published data only}
- Lin CC, Hsu YL, Chung CS, Lee TH. Stress ulcer prophylaxis in patients being weaned from the ventilator in a respiratory care center: a randomized control trial. Journal of the Formosan Medical Association 2016;115(1):19‐24. [DOI] [PubMed] [Google Scholar]
Lopez‐Herce 1992 {published data only}
- Lopez‐Herce J, Dorao P, Elola P, Delgado MA, Ruza F, Madero R. Frequency and prophylaxis of upper gastrointestinal hemorrhage in critically ill children: a prospective study comparing the efficacy of almagate, ranitidine, and sucralfate. The Gastrointestinal Hemorrhage Study Group. Critical Care Medicine 1992;20(8):1082‐9. [PUBMED: 1643886] [DOI] [PubMed] [Google Scholar]
Luk 1982 {published data only}
- Luk GD, Summer WR, Messersmith JF. Cimetidine and antacid in the prophylaxis of acute gastrointestinal bleeding. A randomised,double blind, controlled study(abstract). Gastroenterology 1982;82:1121. [Google Scholar]
Maasoumi 2016 {published data only}
- Maasoumi G, Farahbakhsh F. Comparing the effects of pantoprazole and ranitidine in the prevention of post‐operative gastrointestinal complications in patients undergoing coronary artery bypass graft surgery. [Persian]. Journal of Isfahan Medical School 2016;34(376):270. [Google Scholar]
Macdougall 1977 {published data only}
- Macdougall BR, Bailey RJ, Williams R. H2‐receptor antagonists and antacids in the prevention of acute gastrointestinal haemorrhage in fulminant hepatic failure. Two controlled trials. Lancet 1977;1(8012):617‐9. [PUBMED: 66425] [DOI] [PubMed] [Google Scholar]
Mahul 1992 {published data only}
- Mahul P, Auboyer C, Jospe R, Ros A, Guerin C, Khouri Z, et al. Prevention of nosocomial pneumonia in intubated patients: respective role of mechanical subglottic secretions drainage and stress ulcer prophylaxis. Intensive Care Medicine 1992;18(1):20‐5. [PUBMED: 1578042] [DOI] [PubMed] [Google Scholar]
Maier 1994 {published data only}
- Maier RV, Mitchell D, Gentilello L. Optimal therapy for stress gastritis. Annals of Surgery 1994;220(3):353‐60; discussion 360‐3. [PUBMED: 8092901] [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Keefe GE, Gentilello LM, Maier RV. Incidence of infectious complications associated with the use of histamine2‐receptor antagonists in critically ill trauma patients. Annals of Surgery 1998;227(1):120‐5. [PUBMED: 9445119] [DOI] [PMC free article] [PubMed] [Google Scholar]
Martin 1980 {published data only}
- Martin LF, Max MH, Polk HC Jr. Failure of gastric pH control by antacids or cimetidine in the critically ill: a valid sign of sepsis. Surgery 1980;88(1):59‐68. [PUBMED: 6966835] [PubMed] [Google Scholar]
Martin 1992 {published data only}
- Martin LF, Booth FV, Reines HD, Deysach LG, Kochman RL, Erhardt LJ, et al. Stress ulcers and organ failure in intubated patients in surgical intensive care units. Annals of Surgery 1992;215(4):332‐7. [PUBMED: 1558413] [DOI] [PMC free article] [PubMed] [Google Scholar]
Martin 1993 {published data only}
- Martin LF, Booth FV, Karlstadt RG, Silverstein JH, Jacobs DM, Hampsey J, et al. Continuous intravenous cimetidine decreases stress‐related upper gastrointestinal hemorrhage without promoting pneumonia. Critical Care Medicine 1993;21(1):19‐30. [DOI: 10.1097/00003246-199301000-00009; PUBMED: 8420726] [DOI] [PubMed] [Google Scholar]
Metz 1993 {published data only}
- Metz CA, Livingston DH, Smith JS, Larson GM, Wilson TH. Impact of multiple risk factors and ranitidine prophylaxis on the development of stress‐related upper gastrointestinal bleeding: a prospective, multicenter, double‐blind, randomized trial. The Ranitidine Head Injury Study Group. Critical Care Medicine 1993;21(12):1844‐9. [PUBMED: 8252888] [DOI] [PubMed] [Google Scholar]
Mustafa 1994 {published data only}
- Mustafa NA, Akturk G, Ozen I, Koksal I, Erciyes N, Solak M. Sucralfate versus ranitidine prophylaxis in intensive care patients. Turkish Journal of Medical Sciences 1994; Vol. 22, issue 2:103‐6. [CN‐00179889]
Ng 2012 {published data only}
- Ng FH, Tunggal P, Chu WM, Lam KF, Li A, Chan K, et al. Esomeprazole compared with famotidine in the prevention of upper gastrointestinal bleeding in patients with acute coronary syndrome or myocardial infarction. American Journal of Gastroenterology 2012;107(3):389‐96. [DOI: 10.1038/ajg.2011.385] [DOI] [PubMed] [Google Scholar]
Noseworthy 1987 {published data only}
- Noseworthy TW, Shustack A, Johnston RG, Anderson BJ, Konopad E, Grace M. A randomized clinical trial comparing ranitidine and antacids in critically ill patients. Critical Care Medicine 1987;15(9):817‐9. [PUBMED: 3304837] [DOI] [PubMed] [Google Scholar]
Ortiz 1998 {published data only}
- Ortiz JE, Sottile FD, Sigel P, Nasraway SA. Gastric colonization as a consequence of stress ulcer prophylaxis: a prospective, randomized trial. Pharmacotherapy 1998;18(3):486‐91. [PUBMED: 9620099] [PubMed] [Google Scholar]
Pan 2004 {published data only}
- Pan X, Zhang W, Li Z. The preventive effects of rabeprazole on upper gastrointestinal tract haemorrhage in patients with severe acute pancreatitis. Chinese Journal of Gastroenterology 2004;1:30‐2. [Google Scholar]
Peura 1985 {published data only}
- Peura DA, Johnson LF. Cimetidine for prevention and treatment of gastroduodenal mucosal lesions in patients in an intensive care unit. Annals of Internal Medicine 1985;103(2):173‐7. [PUBMED: 3874573] [DOI] [PubMed] [Google Scholar]
Phillips 1998 {unpublished data only}
- Phillips JO, Metzler MH, Huckfeldt RE, et al. A multicenter, prospective, randomised clinical trial of continuous I.V. ranitidine vs. Omeprazole suspension in the prophylaxis of stress ulcer prophylaxis (abstract). Critical Care Medicine. 1998; Vol. 26, issue A101.
Pickworth 1993 {published data only}
- Pickworth KK, Falcone RE, Hoogeboom JE, Santanello SA. Occurrence of nosocomial pneumonia in mechanically ventilated trauma patients: a comparison of sucralfate and ranitidine. Critical Care Medicine 1993;21(12):1856‐62. [PUBMED: 8252890] [DOI] [PubMed] [Google Scholar]
Pinilla 1985 {published data only}
- Pinilla JC, Oleniuk FH, Reed D, Malik B, Laverty WH. Does antacid prophylaxis prevent upper gastrointestinal bleeding in critically ill patients?. Critical Care Medicine 1985;13(8):646‐50. [DOI] [PubMed] [Google Scholar]
Poleski 1986 {published data only}
- Poleski MH, Spanier AH. Cimetidine versus antacids in the prevention of stress erosions in critically ill patients. American Journal of Gastroenterology 1986;81(2):107‐11. [PUBMED: 3946364] [PubMed] [Google Scholar]
Powell 1993 {published data only}
- Powell H, Morgan M, Li SK, Baron JH. Inhibition of gastric acid secretion in the intensive care unit after coronary artery bypass graft. A pilot control study of intravenous omeprazole by bolus and infusion, ranitidine and placebo. Theoretical Surgery 1993; Vol. 8, issue 3:125‐30. [CN‐00182047]
Prakash 2008 {published data only}
- Prakash S, Rai A, Gogia RA. Nosocomial pneumonia in mechanically ventilated patients receiving ranitidine or sucralfate as stress ulcer prophylaxis. Indian Journal of Anaesthesia 2008;52(2):179‐84. [Google Scholar]
Priebe 1980 {published data only}
- Priebe HJ, Skillman JJ, Bushnell LS, Long PC, Silen W. Antacid versus cimetidine in preventing acute gastrointestinal bleeding. A randomized trial in 75 critically ill patients. New England Journal of Medicine 1980;302(8):426‐30. [PUBMED: 6986027] [DOI] [PubMed] [Google Scholar]
Prod'hom 1994 {published data only}
- Prod'hom G, Leuenberger P, Koerfer J, Blum A, Chiolero R, Schaller MD, et al. Nosocomial pneumonia in mechanically ventilated patients receiving antacid, ranitidine, or sucralfate as prophylaxis for stress ulcer. A randomized controlled trial. Annals of Internal Medicine 1994;120(8):653‐62. [PUBMED: 8135449] [DOI] [PubMed] [Google Scholar]
Reusser 1990 {published data only}
- Reusser P, Gyr K, Scheidegger D, Buchmann B, Buser M, Zimmerli W. Prospective endoscopic study of stress erosions and ulcers in critically ill neurosurgical patients: current incidence and effect of acid‐reducing prophylaxis. Critical Care Medicine 1990;18(3):270‐4. [PUBMED: 2302950] [DOI] [PubMed] [Google Scholar]
Rohde 1980 {published data only}
- Lorenz W, Fischer M, Rohde H, Troidl H, Reimann HJ, Ohmann C. Histamine and stress ulcer: new components in organizing a sequential trial on cimetidine prophylaxis in seriously ill patients and definition of a special group at risk (severe polytrauma). Klinische Wochenschrift 1980;58((3):653‐65. [PUBMED: 7442079] [DOI] [PubMed] [Google Scholar]
- Rohde H, Lorenz W, Fischer M. [A randomized clinical study of stress ulcer prophylaxis with cimeytidine in severe multiple injuries] [Eine randomisierte klinische Studie zur Stressulkusprophylaxe mit Cimetidin beim schweren Polytrauma.]. Zeitschrift fur Gastroenterologie 1980;18(6):328‐9. [PUBMED: 6998154] [PubMed] [Google Scholar]
Ruiz‐Santana 1991 {published data only}
- Ruiz‐Santana S, Ortiz E, Gonzalez B, Bolanos J, Ruiz‐Santana AJ, Manzano JL. Stress‐induced gastroduodenal lesions and total parenteral nutrition in critically ill patients: frequency, complications, and the value of prophylactic treatment. A prospective, randomized study. Critical Care Medicine 1991;19(7):887‐91. [PUBMED: 1905214] [DOI] [PubMed] [Google Scholar]
Ryan 1993 {published data only}
- Ryan P, Dawson J, Teres D, Celoria G, Navab F. Nosocomial pneumonia during stress ulcer prophylaxis with cimetidine and sucralfate. Archives of Surgery 1993;128(12):1353‐7. [PUBMED: 8250708] [DOI] [PubMed] [Google Scholar]
Selvanderan 2015 {published data only}
- Selvanderan SP, Summers MJ, Plummer MP, Finnis ME, Ali Abdelhamid Y, Anderson MB, et al. Withholding stress ulcer prophylaxis to mechanically ventilated enterally‐fed critically ill patients appears safe: a randomised double‐blind placebo controlled pilot study. Intensive Care Medicine Experimental 2015;3:A41. [Google Scholar]
Selvanderan 2016 {published data only}
- Deane A, Selvanderan S, Summers M, Finnis M, Plummer M, Ali AY, et al. Pantoprazole or placebo for stress ulcer prophylaxis (Popup) study. Anaesthesia and Intensive Care 2016; Vol. 44, issue 2:303‐4. [DOI] [PubMed]
- Selvanderan SP, Summers M, Finnis M, Plummer M, Abdelhamid YA, Anderson M, et al. Administration of stress ulcer prophylaxis may cause harm in critically ill patients: a randomized double blind exploratory study. Gastroenterology 2016;150(4):S882. [Google Scholar]
- Selvanderan SP, Summers MJ, Finnis ME, Plummer MP, Ali AY, Anderson MB, et al. Pantoprazole or placebo for stress ulcer prophylaxis (POP‐UP): randomized double‐blind exploratory study. Critical Care Medicine 2016;44(10):1842. [DOI] [PubMed] [Google Scholar]
Simms 1991 {published data only}
- Simms HH, DeMaria E, McDonald L, Peterson D, Robinson A, Burchard KW. Role of gastric colonization in the development of pneumonia in critically ill trauma patients: results of a prospective randomized trial. Journal of Trauma 1991;31(4):531‐6; discussion 536‐7. [PUBMED: 2020040] [PubMed] [Google Scholar]
Sirvent 1994 {published data only}
- Sirvent JM, Verdaguer R, Ferrer MJ, Avila FJ, Diaz‐Prieto A, Carratala J. Mechanical ventilation‐associated pneumonia and the prevention of stress ulcer. A randomized clinical trial of antacids and ranitidine versus sucralfate [Neumonia asociada a ventilacion mecanica y profilaxis del ulcus de estres. Ensayo clinico aleatorizado de antiacidos y ranitidina frente a sucralfato]. Medicina Clinica 1994;102(11):407‐11. [PUBMED: 8182996] [PubMed] [Google Scholar]
Skillman 1984 {published data only}
- Skillman JJ, Lisbon A, Long PC, Silen W. 15 (R)‐15‐methyl prostaglandin E2 does not prevent gastrointestinal bleeding in seriously ill patients. American Journal of Surgery 1984;147(4):451‐5. [PUBMED: 6370007] [DOI] [PubMed] [Google Scholar]
Solouki 2009 {published data only}
- Solouki M, Marashian SM, Kouchak M, Mokhtari M, Nasiri E. Comparison between the preventive effects of ranitidine and omeprazole on upper gastrointestinal bleeding among ICU patients. Tanaffos 2009;8(4):37‐42. [Google Scholar]
- Solouki M, Marashian SM, Kouchak M, Mokhtari M, Nasiri E. Ventilator‐associated pneumonia among ICU patients receiving mechanical ventilation and prophylaxis of gastrointestinal bleeding. Iranian Journal of Clinical Infectious Diseases 2009;4(3):177‐80. [Google Scholar]
Somberg 2008 {published data only}
- Somberg L, Morris J Jr, Fantus R, Graepel J, Field BG, Lynn R, et al. Intermittent intravenous pantoprazole and continuous cimetidine infusion: effect on gastric pH control in critically ill patients at risk of developing stress‐related mucosal disease. Journal of Trauma 2008;64(5):1202‐10. [PUBMED: 18469642] [DOI] [PubMed] [Google Scholar]
Stoehr 2006 {published data only}
- Stoehr G, Luebbers K, Wilhelm M, Hoelzer J, Ohmann C. Aluminum load in ICU patients during stress ulcer prophylaxis. European Journal of Internal Medicine 2006;17(8):561‐6. [PUBMED: 17142175] [DOI] [PubMed] [Google Scholar]
Stothert 1980 {published data only}
- Stothert JC Jr, Simonowitz DA, Dellinger EP, Farley M, Edwards WA, Blair AD, et al. Randomized prospective evaluation of cimetidine and antacid control of gastric pH in the critically ill. Annals of Surgery 1980;192(2):169‐74. [PUBMED: 7406571] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tabeefar 2012 {published data only}
- Tabeefar H, Beigmohammadi MT, Javadi MR, Abdollahi M, Mahmoodpoor A, Ahmadi A, et al. Effects of pantoprazole on systemic and gastric pro‐and anti‐inflammatory cytokines in critically ill patients. Iranian Journal of Pharmaceutical Research 2012;11(4):1051‐8. [PMC free article] [PubMed] [Google Scholar]
Terzi 2009 {published data only}
Thomason 1996 {published data only}
- Thomason MH, Payseur ES, Hakenewerth AM, Norton HJ, Mehta B, Reeves TR, et al. Nosocomial pneumonia in ventilated trauma patients during stress ulcer prophylaxis with sucralfate, antacid, and ranitidine. Journal of Trauma 1996;41(3):503‐8. [PUBMED: 8810971] [DOI] [PubMed] [Google Scholar]
Tryba 1985 {published data only}
- Tryba M, Zevounou F, Grabhoefer P, Seifert V, Torok M. Prevention of acute stress hemorrhages of the upper gastrointestinal tract with pirenzepine and antacids. A controlled comparison between pirenzepine and cimetidine [Prophylaxe akuter Stressblutungen des oberen Gastrointestinaltraktes mit Pirenzepin und Antazida. Ein kontrollierter Vergleich zwischen Pirenzepin und Cimetidin]. Anasthesie, Intensivtherapie, Notfallmedizin 1984;19(5):240‐4. [PubMed] [Google Scholar]
- Tryba M, Zevounou F, Torok M, Zenz M. Prevention of acute stress bleeding with sucralfate, antacids, or cimetidine. A controlled study with pirenzepine as a basic medication. American Journal of Medicine 1985;79(2C):55‐61. [DOI: 10.1016/0002-9343(85)90574-1; PUBMED: 3876031] [DOI] [PubMed] [Google Scholar]
Tryba 1987 {published data only}
- Tryba M. Risk of acute stress bleeding and nosocomial pneumonia in ventilated intensive care unit patients: sucralfate versus antacids. American Journal of Medicine 1987;83(3B):117‐24. [PUBMED: 3310626] [DOI] [PubMed] [Google Scholar]
Tryba 1988 {published data only}
- Tryba M, Zevounou F, Wruck G. Stress bleeding and postoperative pneumonia in intensive care patients with ranitidine or pirenzepine [Streßblutungen und postoperative Pneumonien bei Intensivpatienten unter Ranitidin oder PirenzepinM.]. Deutsche Medizinische Wochenschrift 1988;113(23):930‐6. [DOI: 10.1055/s-2008-1067744] [DOI] [PubMed] [Google Scholar]
van den Berg 1985 {published data only}
- Berg B, Blankenstein M. Prevention of stress‐induced upper gastrointestinal bleeding by cimetidine in patients on assisted ventilation. Digestion 1985;31(1):1‐8. [PUBMED: 3979676] [DOI] [PubMed] [Google Scholar]
van Essen 1985 {published data only}
- Essen HA, Blankenstein M, Wilson JH, Berg B, Bruining HA. Intragastric prostaglandin E2 and the prevention of gastrointestinal hemorrhage in ICU patients. Critical Care Medicine 1985;13(11):957‐60. [PUBMED: 3902362] [DOI] [PubMed] [Google Scholar]
Wang 2015 {published data only}
- Wang YQ. Impact of comprehensive intestinal irritation on digestive function in mechanically ventilated critically ill patients. World Chinese Journal of Digestology 2015;23(34):5544‐8. [Google Scholar]
Wee 2013 {published data only}
- Wee B, Liu CH, Cohen H, Kravchuk S, Reddy K, Mukherji R. IV famotidine vs. IV pantoprazole for stress ulcer prevention in the ICU: a prospective study. Critical Care Medicine 2013;41(12):A181. [DOI: 10.1097/01.ccm.0000439969.36301.c9] [DOI] [Google Scholar]
- Wee BC, Liu M, Cohen H, Kravchuk S, Reddy K, Mukherji R. Efficacy and safety of intravenous famotidine versus intavenous pantoprazole for stress ulcer prophylaxis in the critically ill: a prospective randomized study. Journal of Pharmacy Practice 2013;26(3):299‐300. [Google Scholar]
Weigelt 1981 {published data only}
- Weigelt JA, Aurbakken CM, Gewertz BL, Snyder WH 3rd. Cimetidine vs antacid in prophylaxis for stress ulceration. Archives of Surgery 1981;116(5):597‐601. [PUBMED: 7016067] [DOI] [PubMed] [Google Scholar]
Yildizdas 2002 {published data only}
- Yildizdas D, Yapicioglu H, Yilmaz HL. Occurrence of ventilator‐associated pneumonia in mechanically ventilated pediatric intensive care patients during stress ulcer prophylaxis with sucralfate, ranitidine, and omeprazole. Journal of Critical Care 2002;17(4):240‐5. [DOI: 10.1053/jcrc.2002.36761; PUBMED: 12501151] [DOI] [PubMed] [Google Scholar]
Zinner 1981 {published data only}
- Zinner MJ, Zuidema GD, Smith P[L, Mignosa M. The prevention of upper gastrointestinal tract bleeding in patients in an intensive care unit. Surgery, Gynecology & Obstetrics 1981;153(2):214‐20. [PUBMED: 7017982] [PubMed] [Google Scholar]
Zinner 1989 {published data only}
- Zinner MJ, Rypins EB, Martin LR, Jonasson O, Hoover EL, Swab EA, et al. Misoprostol versus antacid titration for preventing stress ulcers in postoperative surgical ICU patients. Annals of Surgery 1989;210(5):590‐5. [PUBMED: 2510618] [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies excluded from this review
Aanpreung 1998 {published data only}
- Aanpreung P, Vanprapar N, Susiva C, Parkpreaw C, Boonyachart C. A randomized clinical trial comparing the efficacy of ranitidine and famotidine on intragastric acidity in critically ill pediatric patients. Journal of the Medical Association of Thailand 1998;81(3):185‐9. [PUBMED: 9623009] [PubMed] [Google Scholar]
Abe 2004 {published data only}
- Abe Y, Inamori M, Togawa J, Kikuchi T, Muramatsu K, Chiguchi G, et al. The comparative effects of single intravenous doses of omeprazole and famotidine on intragastric pH. Journal of Gastroenterology 2004;39(1):21‐5. [PUBMED: 14767730] [DOI] [PubMed] [Google Scholar]
Alaniz 2014 {published data only}
- Alaniz C, Hyzy RC. Time to declare a moratorium on stress ulcer prophylaxis in critically Ill. Critical Care Medicine 2014;42(9):e636‐7. [DOI] [PubMed] [Google Scholar]
Al‐Quorain 1994 {published data only}
- Al‐Quorain A, Ammar A, Al‐Awami M, Hegazi M, El‐Munshid KA, Ibrahim EM, et al. Comparison of intravenous famotidine and ranitidine in suppressing gastric acid secretion in critically ill patients. Current Therapeutic Research 1994;55(10):1263‐70. [Google Scholar]
Anonymous 2013 {published data only}
- Anonymous. 32nd Congress of the Scandinavian Society of Anaesthesiology and Intensive Care Medicine, Focusing on the Brain. Acta Anaesthesiologica Scandinavica, Supplement 2013;57:no pagination. [DOI] [PubMed] [Google Scholar]
Anonymous 2015 {published data only}
- Anonymous. 33rd Congress of the Scandinavian Society of Anaesthesiology and Intensive Care Medicine. Acta Anaesthesiologica Scandinavica 2015;59:no pagination. [DOI] [PubMed] [Google Scholar]
Arora 1991 {published data only}
- Arora A, Tandon RK, Acharya SK, Tandon BN. The role of sustained achlorhydria in bleeding peptic ulcer. Journal of Clinical Gastroenterology 1991;13(2):147‐53. [PUBMED: 2033221] [DOI] [PubMed] [Google Scholar]
Baccino 1987 {published data only}
- Baccino E, Boles JM, Guillou M, Geier B, Garo B, Robaszkiewicz M, et al. Attempt at preventive treatment of esophagitis caused by intubation during intensive care [Tentative de traitement preventif des oesophagites sur sonde en reanimation.]. Gastroenterologie Clinique et Biologique 1987;11(1):24‐8. [PUBMED: 3556957] [PubMed] [Google Scholar]
Baghaie 1995 {published data only}
- Baghaie AA, Mojtahedzadeh M, Levine RL, Fromm RE Jr, Guntupalli KK, Opekun AR Jr. Comparison of the effect of intermittent administration and continuous infusion of famotidine on gastric pH in critically ill patients: results of a prospective, randomized, crossover study. Critical Care Medicine 1995;23(4):687‐91. [PUBMED: 7712759] [DOI] [PubMed] [Google Scholar]
Barth 1984 {published data only}
- Barth HO, Brunner G, Berg F, Dammann HG, Friedl W, Franken FH. Ranitidine versus cimetidine in preventing acute gastroduodenal bleeding: a randomized trial in 193 critically ill patients ‐ a multicentre study in Germany. Intensivmedizin 1984;21:15‐8. [Google Scholar]
Bauer 1977 {published data only}
- Bauer H, Doenicke A, Holle F. Prevention and treatment of upper gastrointestinal haemorrhage with cimetidine and somatostatin in intensive care patients (author's transl) [Kasuistische Mitteilung uber Moglichkeiten der Prophylaxe und Therapie gastrointestinaler Blutungen mit Cimetidin oder Somatostatin bei Schwerstkranken.]. Der Anaesthesist 1977;26(12):662‐4. [PUBMED: 304692] [PubMed] [Google Scholar]
Bergmans 2001 {published data only}
- Bergmans DC, Bonten MJ, Gaillard CA, Paling JC, Geest S, Tiel FH, et al. Prevention of ventilator‐associated pneumonia by oral decontamination: a prospective, randomized, double‐blind, placebo‐controlled study. American Journal of Respiratory and Critical Care Medicine 2001;164(3):382‐8. [PUBMED: 11500337] [DOI] [PubMed] [Google Scholar]
Bhatt 2010 {published data only}
- Bhatt DL, Cryer BL, Contant CF, Cohen M, Lanas A, Schnitzer TJ, et al. Clopidogrel with or without omeprazole in coronary artery disease. New England Journal of Medicine 2010;363(20):1909‐17. [DOI: 10.1056/NEJMoa1007964] [DOI] [PubMed] [Google Scholar]
Cheadle 1985 {published data only}
- Cheadle WG, Vitale GC, Mackie CR, Cuschieri A. Prophylactic postoperative nasogastric decompression. A prospective study of its requirement and the influence of cimetidine in 200 patients. Annals of Surgery 1985;202(3):361‐6. [PUBMED: 4037908] [DOI] [PMC free article] [PubMed] [Google Scholar]
Chernov 1971 {published data only}
- Chernov MS, Hale HW Jr, Wood M. Prevention of stress ulcers. American Journal of Surgery 1971;122(5):674‐7. [PUBMED: 5112074] [DOI] [PubMed] [Google Scholar]
Cloud 1994 {published data only}
- Cloud ML, Offen W. Continuous infusions of nizatidine are safe and effective in the treatment of intensive care unit patients at risk for stress gastritis. The Nizatidine Intensive Care Unit Study Group. Scandinavian Journal of Gastroenterology. Supplement 1994;206:29‐34. [PUBMED: 7863249] [DOI] [PubMed] [Google Scholar]
Critchlow 1987 {published data only}
- Critchlow JF. Comparative efficacy of parenteral histamine (H2)‐antagonists in acid suppression for the prevention of stress ulceration. American Journal of Medicine 1987;83(6a):23‐8. [DOI] [PubMed] [Google Scholar]
Dabiri 2015 {published data only}
- Dabiri Y, Fahimi F, Jamaati H, Hashemian SMR. The comparison of extemporaneous preparations of omeprazole, pantoprazole oral suspension and intravenous pantoprazole on the gastric pH of critically ill‐patients. Indian Journal of Critical Care Medicine 2015;19(1):21‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Driscoll 1993 {published data only}
- Driscoll DM, Cioffi WG Jr, Molter NC, McManus WF, Mason AD Jr, Pruitt BA Jr. Intragastric pH monitoring. Journal of Burn Care & Rehabilitation 1993;14(5):517‐24. [PUBMED: 8245105] [DOI] [PubMed] [Google Scholar]
Duma 1986 {published data only}
- Duma S. Prevention of stress ulcers with cimetidine and ranitidine. Comparative studies within the scope of cardiosurgical interventions [Prophylaxe von Stressulzera mit Cimetidin und Ranitidin. Vergleichende Untersuchungen im Rahmen von kardiochirurgischen Eingriffen]. Wiener Medizinische Wochenschrift (1946) 1986;136(18):467‐72. [PUBMED: 3798932] [PubMed] [Google Scholar]
Estruch 1991 {published data only}
- Estruch R, Pedrol E, Castells A, Masanes F, Marrades RM, Urbano‐Marquez A. Prophylaxis of gastrointestinal tract bleeding with magaldrate in patients admitted to a general hospital ward. Scandinavian Journal of Gastroenterology 1991;26(8):819‐26. [PUBMED: 1771386] [DOI] [PubMed] [Google Scholar]
Fiorucci 1989 {published data only}
- Fiorucci S, Clausi GC, Farinelli M, Santucci L, Pelli MA, Morelli A. Intragastric pH monitoring during antisecretory therapy in patient with gastrointestinal bleeding. American Journal of Gastroenterology 1989;84(11):1416‐20. [PUBMED: 2816875] [PubMed] [Google Scholar]
Forestier 2008 {published data only}
- Forestier C, Guelon D, Cluytens V, Gillart T, Sirot J, Champs C. Oral probiotic and prevention of Pseudomonas aeruginosa infections: a randomized, double‐blind, placebo‐controlled pilot study in intensive care unit patients. Critical Care (London, England) 2008;12(3):R69. [PUBMED: 18489775] [DOI] [PMC free article] [PubMed] [Google Scholar]
Friedl 1985 {published data only}
- Friedl W, Krier C, Dammann HG, Muller P, Simon B. I.V. famotidine versus I.V. ranitidine: intragastric pH behavior in surgical intensive care patients [I.V. Famotidin versus I.V. ranitidine: intragastrales pH‐Verhalten bei chirurgischen Intensivpatienten]. Zeitschrift fur Gastroenterologie 1985;23(11):603‐7. [PUBMED: 2868579] [PubMed] [Google Scholar]
Geus 1993 {published data only}
- Geus WP, Vinks AA, Smith SJ, Westra P, Lamers CB. Comparison of two intravenous ranitidine regimens in a homogeneous population of intensive care unit patients. Alimentary Pharmacology & Therapeutics 1993;7(4):451‐7. [PUBMED: 8218759] [DOI] [PubMed] [Google Scholar]
Hauer 1996 {published data only}
- Hauer T, Lacour M, Gastmeier P, Schulgen G, Schumacher M, et al. A prevalence survey of nosocomial infections in intensive care units [Nosokomiale Infektionen auf Intensivstationen]. Anaesthesist 1996;45:1184‐91. [DOI] [PubMed] [Google Scholar]
Heiselman 1995 {published data only}
- Heiselman DE, Hulisz DT, Fricker R, Bredle DL, Black LD. Randomized comparison of gastric pH control with intermittent and continuous intravenous infusion of famotidine in ICU patients. American Journal of Gastroenterology 1995;90(2):277‐9. [PUBMED: 7847300] [PubMed] [Google Scholar]
Herrmann 1979 {published data only}
- Herrmann V, Kaminski DL. Evaluation of intragastric pH in acutely ill patients. Archives of Surgery 1979;114(4):511‐4. [PUBMED: 35135] [DOI] [PubMed] [Google Scholar]
Hollander 1973 {published data only}
- Hollander D, Harlan J. Antacids vs placebos in peptic ulcer therapy. A controlled double‐blind investigation. Journal of the American Medical Association 1973;226(10):1181‐5. [PUBMED: 4584411] [PubMed] [Google Scholar]
Huang 2017 {published data only}
- Huang JX, Liu XK. Proton pump inhibitor therapy for prevention of gastrointestinal bleeding after percutaneous coronary intervention [PCI治疗术后应用质子泵抑制剂预防消化道出血效果]. World Chinese Journal of Digestology 2017;25(11):1012‐5. [DOI: 10.11569/wcjd.v25.i11.1012] [DOI] [Google Scholar]
Kalfarentzos 1997 {published data only}
- Kalfarentzos F, Kehagias J, Mead N, Kokkinis K, Gogos CA. Enteral nutrition is superior to parenteral nutrition in severe acute pancreatitis: results of a randomized prospective trial. British Journal of Surgery 1997;84(12):1665‐9. [PUBMED: 9448611] [PubMed] [Google Scholar]
Karlstadt 1993 {published data only}
- Karlstadt RG, Hedrich DA, Asbel‐Sethi NR, Palmer RH. Acid‐suppression profile of two continuously infused intravenous doses of cimetidine. Clinical Therapeutics 1993;15(1):97‐106. [PUBMED: 8458059] [PubMed] [Google Scholar]
Ketterl 1984 {published data only}
- Ketterl R, Holscher AH, Weiser HF, Siewert JR. Control of the intragastric pH value in infection and peritonitis by ranitidine versus cimetidine. A double‐blind study [Kontrolle des intragastralen pH‐Wertes bei Sepsis bzw. Peritonitis durch Ranitidin versus Cimetidin‐‐Eine Doppelblindstudie]. Zeitschrift fur Gastroenterologie 1984;22(10):602‐8. [PUBMED: 6095549] [PubMed] [Google Scholar]
Khan 1981 {published data only}
- Khan F, Parekh A, Patel S, Chitkara R, Rehman M, Goyal R. Results of gastric neutralization with hourly antacids and cimetidine in 320 intubated patients with respiratory failure. Chest 1981;79(4):409‐12. [DOI] [PubMed] [Google Scholar]
Klarin 2008 {published data only}
- Klarin B, Molin G, Jeppsson B, Larsson A. Use of the probiotic Lactobacillus plantarum 299 to reduce pathogenic bacteria in the oropharynx of intubated patients: a randomised controlled open pilot study. Critical Care (London, England) 2008;12(6):R136. [PUBMED: 18990201] [DOI] [PMC free article] [PubMed] [Google Scholar]
Krag 2015 {published data only}
- Krag M, Perner A, Wetterslev J, Wise MP, Borthwick M, Bendel S, et al. Prevalence and outcome of gastrointestinal bleeding and use of acid suppressants in acutely ill adult intensive care patients. Intensive Care Medicine 2015;41(5):833‐45. [DOI] [PubMed] [Google Scholar]
Krier 1990 {published data only}
- Krier C, Bohrer H, Jurs G, Warth S, Just OH. Continuous intragastric pH measurement in intensive care patients treated with ranitidine and tube feeding [Kontinuierliche intragastrale pH‐Wert‐Messung bei Intensivepatienten unter Ranitidin‐ und Sondenkostgabe]. Anasthesie, Intensivtherapie, Notfallmedizin 1990;25 Suppl 1:10‐3. [PUBMED: 2106804] [PubMed] [Google Scholar]
Krueger 2002 {published data only}
- Krueger WA, Lenhart FP, Neeser G, Ruckdeschel G, Schreckhase H, Eissner HJ, et al. Influence of combined intravenous and topical antibiotic prophylaxis on the incidence of infections, organ dysfunctions, and mortality in critically ill surgical patients: a prospective, stratified, randomized, double‐blind, placebo‐controlled clinical trial. American Journal of Respiratory and Critical Care Medicine 2002;166(8):1029‐37. [PUBMED: 12379544] [DOI] [PubMed] [Google Scholar]
Kuusela 1998 {published data only}
- Kuusela AL. Long‐term gastric pH monitoring for determining optimal dose of ranitidine for critically ill preterm and term neonates. Archives of Disease in Childhood. Fetal and Neonatal Edition 1998;78(2):F151‐3. [PUBMED: 9577289] [DOI] [PMC free article] [PubMed] [Google Scholar]
Laterre 2001 {published data only}
- Laterre PF, Horsmans Y. Intravenous omeprazole in critically ill patients: a randomized, crossover study comparing 40 with 80 mg plus 8 mg/hour on intragastric pH. Critical Care Medicine 2001;29(10):1931‐5. [PUBMED: 11588454] [DOI] [PubMed] [Google Scholar]
Levine 1994 {published data only}
- Levine RL, Fromm RE Jr, Mojtahedzadeh M, Baghaie AA, Opekun AR Jr. Equivalence of litmus paper and intragastric pH probes for intragastric pH monitoring in the intensive care unit. Critical Care Medicine 1994;22(6):945‐8. [PUBMED: 7911416] [DOI] [PubMed] [Google Scholar]
Liu 2013 {published data only}
- Liu BL, Li B, Zhang X, Fei Z, Hu SJ, Lin W, et al. A randomized controlled study comparing omeprazole and cimetidine for the prophylaxis of stress‐related upper gastrointestinal bleeding in patients with intracerebral hemorrhage. Journal of Neurosurgery 2013;118(1):115‐20. [PUBMED: 23061387] [DOI] [PubMed] [Google Scholar]
Madani 2014 {published data only}
- Madani S, Kauffman R, Simpson P, Lehr VT, Lai ML, Sarniak A, et al. Pharmacokinetics and pharmacodynamics of famotidine and ranitidine in critically ill children. Journal of Clinical Pharmacology 2014;54(2):201‐5. [DOI] [PubMed] [Google Scholar]
McAlhany 1976 {published data only}
- McAlhany JC Jr, Colmic L, Czaja AJ, Pruitt BA Jr. Antacid control of complications from acute gastroduodenal disease after burns. Journal of Trauma 1976;16(08):645‐8. [PUBMED: 785019] [DOI] [PubMed] [Google Scholar]
McElwee 1979 {published data only}
- McElwee HP, Sirinek KR, Levine BA. Cimetidine affords protection equal to antacids in prevention of stress ulceration following thermal injury. Surgery 1979;86(4):620‐6. [PUBMED: 483170] [PubMed] [Google Scholar]
Metz 2010 {published data only}
- Metz DC, Fulda GJ, Olsen KM, Monyak JT, Simonson SG, Sostek MB. Intravenous esomeprazole pharmacodynamics in critically ill patients. Current Medical Research Opinion 2010;26(5):1141‐8. [DOI: 10.1185/03007991003694308] [DOI] [PubMed] [Google Scholar]
Misra 2005 {published data only}
- Misra UK, Kalita J, Pandey S, Mandal SK, Srivastava M. A randomized placebo controlled trial of ranitidine versus sucralfate in patients with spontaneous intracerebral hemorrhage for prevention of gastric hemorrhage. Journal of the Neurological Sciences 2005;239(1):5‐10. [PUBMED: 16182311] [DOI] [PubMed] [Google Scholar]
Mojtahedzadeh 2002 {published data only}
- Mojtahedzadeh M, Rastegarpanah M, Rouini MR, Malekzadeh R, Khalili H, Ganji MR, et al. A comparative study of bolus administration and continuous infusion of ranitidine on gastric pH with intragastric pH‐probe. Daru 2002;10(4):153‐7. [Google Scholar]
More 1985 {published data only}
- More DG, Raper RF, Munro IA, Watson CJ, Boutagy JS, Shenfield GM. Randomized, prospective trial of cimetidine and ranitidine for control of intragastric pH in the critically ill. Surgery 1985;97(2):215‐24. [PUBMED: 3881838] [PubMed] [Google Scholar]
Mulla 2001 {published data only}
- Mulla H, Peek G, Upton D, Lin E, Loubani M. Plasma aluminium levels during sucralfate prophylaxis for stress ulceration in critically ill patients on continuous venovenous hemofiltration: a randomized, controlled trial. Critical Care Medicine 2001;29(2):267‐71. [PUBMED: 11246304] [DOI] [PubMed] [Google Scholar]
Olsen 1995 {published data only}
- Olsen KM, Hiller FC, Ackerman BH, Crisp‐Landwehr K, San Pedro GS. Effect of single intravenous doses of histamine2 receptor antagonists on volume and pH of gastric acid secretions in critically ill patients. Health Advance 1995;56(8):756‐68. [Google Scholar]
Olsen 2008 {published data only}
- Olsen KM, Devlin JW. Comparison of the enteral and intravenous lansoprazole pharmacodynamic responses in critically ill patients. Alimentary Pharmacology & Therapeutics 2008;28(3):326‐33. [PUBMED: 19086331] [DOI] [PubMed] [Google Scholar]
Osteyee 1994 {published data only}
- Osteyee JL, Banner W Jr. Effects of two dosing regimens of intravenous ranitidine on gastric pH in critically ill children. American Journal of Critical Care 1994;3(4):267‐72. [PUBMED: 7920954] [PubMed] [Google Scholar]
Pelfrene 1996 {published data only}
- Pelfrene E, Vandewoude K, Vogelaers D, Elewaut A, Colardyn F. The effect of cimetidine versus ranitidine on the gastric emptying rate of intensive care unit patients sustained on artificial respiration. Acta Gastro‐enterologica Belgica 1996;59(4):229‐33. [PUBMED: 9085622] [PubMed] [Google Scholar]
Phillips 2001 {published data only}
- Phillips JO, Olsen KM, Rebuck JA, Rangnekar NJ, Miedema BW, Metzler MH. A randomized, pharmacokinetic and pharmacodynamic, cross‐over study of duodenal or jejunal administration compared to nasogastric administration of omeprazole suspension in patients at risk for stress ulcers. American Journal of Gastroenterology 2001;96(2):367‐72. [PUBMED: 11232677] [DOI] [PubMed] [Google Scholar]
Reid 1986 {published data only}
- Reid SR, Bayliff CD. The comparative efficacy of cimetidine and ranitidine in controlling gastric pH in critically ill patients. Canadian Anaesthetists' Society Journal 1986;33(3 Pt 1):287‐93. [DOI] [PubMed] [Google Scholar]
Ren 2015 {published data only}
- Ren XS, Chen YF. Pantoprazole for prevention of post‐traumatic stress ulcer [泮托拉唑应用于预防创伤后应激性溃疡的临床价值]. World Chinese Journal of Digestology 2015;23(7):1145‐8. [DOI: 10.11569/wcjd.v23.i7.1145] [DOI] [Google Scholar]
Schentag 1989 {published data only}
- Schentag JJ, Carter CA, Welage LS. Safety and acid‐suppressant properties of histamine2‐receptor antagonists for the prevention of stress‐related mucosal damage in critical care patients. DICP: Annals of Pharmacotherapy 1989;23(10 Suppl):S36‐9. [PUBMED: 2573208] [DOI] [PubMed] [Google Scholar]
Simon 1984 {published data only}
- Simon B. Ranitidine versus cimetidine in preventing acute gastroduodenal bleeding: a randomised trial in 193 critically ill patients ‐ a multi centre study in Germany. Intensivmedizin 1984;21(1):15‐8. [Google Scholar]
Solana 2013 {published data only}
- Solana MJ, Lopez‐Herce J, Botran M, Urbano J, Castillo J, Garrido B. Hemodynamic effects of intravenous omeprazole in critically ill children. Anales de Pediatria 2013;78(3):167‐72. [DOI] [PubMed] [Google Scholar]
Sung 2003 {published data only}
- Sung JJ. The role of acid suppression in the management and prevention of gastrointestinal hemorrhage associated with gastroduodenal ulcers. Gastroenterology Clinics of North America 2003;32(3 Suppl):S11‐23. [PUBMED: 14556432] [DOI] [PubMed] [Google Scholar]
Taha 1996 {published data only}
- Taha AS, Hudson N, Hawkey CJ, Swannell AJ, Trye PN, Cottrell J, et al. Famotidine for the prevention of gastric and duodenal ulcers caused by nonsteroidal antiinflammatory drugs. New England Journal of Medicine 1996;334(22):1435‐9. [PUBMED: 8618582] [DOI] [PubMed] [Google Scholar]
Tofil 2008 {published data only}
- Tofil NM, Benner KW, Fuller MP, Winkler MK. Histamine 2 receptor antagonists vs intravenous proton pump inhibitors in a pediatric intensive care unit: a comparison of gastric pH. Journal of Critical Care 2008;23(3):416‐21. [PUBMED: 18725049] [DOI] [PubMed] [Google Scholar]
Toyota 1998 {published data only}
- Toyota N, Takada T, Yasuda H, Amano H, Yoshida M, Isaka T, et al. The effects of omeprazole, a proton pump inhibitor, on early gastric stagnation after a pylorus‐preserving pancreaticoduodenectomy: results of a randomized study. Hepato‐gastroenterology 1998;45(22):1005‐10. [PUBMED: 9755997] [PubMed] [Google Scholar]
Udd 2005 {published data only}
- Udd M, Toyry J, Miettinen P, Vanninen E, Mustonen H, Julkunen R. The effect of regular and high doses of omeprazole on the intragastric acidity in patients with bleeding peptic ulcer treated endoscopically: a clinical trial with continuous intragastric pH monitoring. European Journal of Gastroenterology & Hepatology 2005;17(12):1351‐6. [PUBMED: 16292089] [DOI] [PubMed] [Google Scholar]
Vaduganathan 2016 {published data only}
- Vaduganathan M, Bhatt DL, Cryer BL, Liu, Y, Hsieh, WH, Doros G, et al. Proton‐pump inhibitors reduce gastrointestinal events regardless of aspirin dose in patients requiring dual antiplatelet therapy. Journal of the American College of Cardiology 2016;67(14):1661‐71. [DOI] [PubMed] [Google Scholar]
Vargas 1993 {published data only}
- Vargas V, Castro J, Solar F, Sanhueza H, Aguila R, Luppi M, et al. Comparative study of famotidine vs ranitidine in critically ill patients in mechanical ventilation [Estudio comparativo de famotidina vs ranitidina en pacientes criticos en ventilacion mecanica]. Revista Medica de Chile 1993;121(7):746‐51. [PUBMED: 8296077] [PubMed] [Google Scholar]
Wang 1995 {published and unpublished data}
- Wang SC, Chen MF, Bullard MJ, Liao CC. The effectiveness of gastric pH control with bolus famotidine versus cimetidine for stress ulcer prophylaxis in a surgical intensive care unit. Current Theraputic Research 1995;56(5):530‐41. [Google Scholar]
Yao 2015 {published data only}
- Yao D‐K, Chen H, Wang L, Li H‐W, Wang L‐X. Comparison of intravenous plus oral pantoprazole therapy and oral pantoprazole alone for preventing gastrointestinal bleeding in acute coronary syndrome patients with high bleeding risk. Heart, Lung & Circulation 2015;24(9):885‐90. [DOI] [PubMed] [Google Scholar]
Zhou 2002 {published data only}
- Zhou Y, Qiao L, Wu J, Hu H, Xu C. Comparison of the efficacy of octreotide, vasopressin, and omeprazole in the control of acute bleeding in patients with portal hypertensive gastropathy: a controlled study. Journal of Gastroenterology and Hepatology 2002;17(9):973‐9. [PUBMED: 12167118] [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
Labattut 1992 {published data only}
- Labattut AG, Santolalla PM, Andres AP, Ortigosa AM, Del MM, Serrano G, et al. Efficacy of sucralfate in the prevention of upper gastrointestinal stress bleeding in intensive care patients: comparison vs a control group. Clinical Intensive Care 1992;3(5 Suppl):19‐25. [Google Scholar]
Morris 2001 {published data only}
- Morris J, Karlstadt R, Blatcher D, et al. Intermittent intravenous pantoprazole rapidly achieves and maintains gastric pH ≥ 4.0 compared with continuous infusion H2‐receptor antagonist in intensive care unit patients. Critical Care Medicine 2001;29:A147. [Google Scholar]
References to ongoing studies
ACTRN12616000481471 {published data only}
- ACTRN12616000481471. Proton pump inhibitors vs. histamine‐2 receptor blockers for ulcer prophylaxis therapy in the intensive care unit [A multi‐centre, cluster randomised, crossover, registry trial comparing the safety and efficacy of proton pump inhibitors with histamine‐2 receptor blockers for ulcer prophylaxis in intensive care patients requiring invasive mechanical intervention]. anzctr.org.au/ACTRN12616000481471.aspx (first received 7 April 2016).
EUCTR2015‐000318‐24‐DK {published data only}
- EUCTR2015‐000318‐24‐DK. Stress ulcer prophylaxis in the intensive care unit. clinicaltrialsregister.eu/ctr‐search/search?query=EUCTR2015‐000318‐24‐DK (first received 11 March 2015).
EudraCT 2007‐006102‐19 {published data only}
- EudraCT 2007‐006102‐19. Omeprazole treatment for prophylaxis of gastrointestinal bleeding and gastroesophagic reflux in critically ill children [Tratamiento con omeprazol para la profilaxis de la hemorragia digestiva y el reflujo gastroesofágico en niños críticos]. clinicaltrialsregister.eu/ctr‐search/trial/2007‐006102‐19/ES (first received 23 January 2008).
IRCT201104134578N2 {published data only}
- IRCT201104134578N2. Ranitidine and pantoprazole in prevention of stress ulcer. http://apps.who.int/trialsearch/Trial.aspx?TrialID=IRCT201104134578N2 (accessed 24 Feburary 2012).
ISRCTN12845429 {published data only}
- ISRCTN12845429. DRIVE ‐ desmopressin for procedures or radiological interventions [A placebo‐controlled double blind, randomised feasibility trial of Desmopressin (DDAVP) in critical illness prior to procedures]. isrctn.com/ISRCTN12845429 (first received 30 January 2017).
Krag 2016 {published data only}
- Krag M, Perner A, Wetterslev J, Lange T, Wise MP, Borthwick M, et al. Stress ulcer prophylaxis in the intensive care unit trial: detailed statistical analysis plan. Acta Anaesthesiologica Scandinavica 2017;61(7):859‐68. [DOI] [PubMed] [Google Scholar]
- Krag M, Perner A, Wetterslev J, Wise MP, Borthwick M, Bendel S, et al. Stress ulcer prophylaxis with a proton pump inhibitor versus placebo in critically ill patients (SUP‐ICU trial): study protocol for a randomised controlled trial. Trials 2016;17(1):205. [DOI] [PMC free article] [PubMed] [Google Scholar]
NCT00590928 {published data only}
- NCT00590928. Gastric pH in critically ill patients [Effect of intravenous esomeprazole versus ranitidine on gastric pH in critically ill patients ‐ a prospective, randomized, double‐blind study]. clinicaltrials.gov/show/NCT00590928 (first received 11 January 2008).
NCT00702871 {published data only}
- NCT00702871. A clinico‐bacteriological study and effect of stress ulcer prophylaxis on occurrence of ventilator associated pneumonia [A clinico‐bacteriological study and effect of stress ulcer prophylaxis on occurrence of ventilator associated pneumonia: a randomized prospective study]. clinicaltrials.gov/show/NCT00702871 (first received 20 June 2008).
NCT02157376 {published data only}
- NCT02157376. Stress ulcer prophylaxis of intravenous esomeprazole in Chinese seriously ill patients (SUP) [Effect of intravenous esomeprazole versus cimetidine in prevention of stress ulcer prophylaxis in Chinese seriously ill patients ‐ a randomized, double‐blind, parallel‐group study]. clinicaltrials.gov/show/NCT02157376 (first received 6 June 2014).
NCT02290327 {published data only}
- NCT02290327. Re‐evaluating the inhibition of stress erosions: gastrointestinal bleeding prophylaxis In ICU (REVISE). clinicaltrials.gov/show/NCT02290327 (first received 14 November 2014).
NCT02718261 {published data only}
- NCT02718261. Sup‐Icu RENal (SIREN) [Sup‐Icu RENal (SIREN) ‐ a sub‐analysis of the prospective SUP (Stress Ulcer Prophylaxis)‐ICU trial on the risk of GI‐bleeding in ICU patients receiving renal replacement therapy]. clinicaltrials.gov/show/NCT02718261 (first received 24 March 2016).
NCT03098537 {published data only}
- NCT03098537. Effects of enteral nutrition on stress ulcer hemorrage. Multicenter randomized controlled trial. clinicaltrials.gov/show/NCT03098537 (first received 31 March 2017).
Additional references
AHSP 1999
- ASHP Commission on Therapeutics. ASHP therapeutic guidelines on stress ulcer prophylaxis. American Journal of Health‐System Pharmacy 1999;56(4):347‐79. [DOI] [PubMed] [Google Scholar]
Alhazzani 2013
- Alhazzani W, Alenezi F, Jaeschke RZ, Moayyedi P, Cook DJ. Proton pump inhibitors versus histamine 2 receptor antagonists for stress ulcer prophylaxis in critically ill patients: a systematic review and meta‐analysis.. Critical Care Medicine 2013;41:693‐705. [DOI: 10.1097/CCM.0b013e3182758734] [DOI] [PubMed] [Google Scholar]
Alhazzani 2017
- Alhazzani W, Alshamsi F, Belley‑Cote E, Heels‑Ansdell D, Brignardello‑Petersen R, Alquraini M, et al. Efficacy and safety of stress ulcer prophylaxis in critically ill patients: a network meta‑analysis of randomized trials. Intensive Care Medicine 2017 Dec 4 [Epub ahead of print]. [DOI: 10.1007/s00134-017-5005-8] [DOI] [PMC free article] [PubMed]
Alquraini 2017
- Alquraini M, Alshamsi F, Hylander Moller M, Belley‐Cote E, Almenawer S, Jaeschke R, et al. Sucralfate versus histamine 2 receptor antagonists for stress ulcer prophylaxis in adult critically ill patients: a meta‐analysis and trial sequential analysis of randomized trials. Journal of Critical Care 2017;40:21‐30. [DOI] [PubMed] [Google Scholar]
Alshamsi 2016
- Alshamsi F, Belley‐Cote E, Cook D, Almenawer S, Alqahtani Z, Perri D, et al. Efficacy and safety of proton pump inhibitors for stress ulcer prophylaxis in critically ill patients: a systematic review and meta‐analysis of randomized trials. Critical Care 2016;20(1):120. [DOI: 10.1186/s13054-016-1305-6; PUBMED: 27142116] [DOI] [PMC free article] [PubMed] [Google Scholar]
Armstrong 1999
- Armstrong TA, Coursin DB, Devlin J, Duke JS, Fish D, Gonzalez ER, et al. ASHP therapeutic guidelines on stress ulcer prophylaxis. American Journal of Health‐System Pharmacy 1999;56(4):347‐79. [DOI] [PubMed] [Google Scholar]
Arnold 2007
- Arnold DM, Donahoe L, Clarke FJ, Tkaczyk AJ, Heels‐Ansdell D, Zytaruk N, et al. Bleeding during critical illness: a prospective cohort study using a new measurement tool. Clinical & Investigative Medicine 2007;30(2):93‐102. [DOI] [PubMed] [Google Scholar]
Arriola 2016
- Arriola V, Tischendorf J, Musuuza J, Barker A, Rozelle JW, Safdar N. Assessing the risk of hospital‐acquired Clostridium difficile infection with proton pump inhibitor use: a meta‐analysis. Infection Control and Hospital Epidemiology 2016;37(2):1408‐17. [DOI: 10.1017/ice.2016.194] [DOI] [PMC free article] [PubMed] [Google Scholar]
Atherton 1978
- Atherton ST, White DJ. Stomach as source of bacteria colonising respiratory tract during artificial ventilation. Lancet 1978;2:968‐9. [DOI] [PubMed] [Google Scholar]
Augustyn 2007
- Augustyn B. Ventilator‐associated pneumonia risk factors and prevention. Critical Care Nurse 2007;27(4):32‐40. [PubMed] [Google Scholar]
Balshem 2011
- Balshem H, Helfand M, Schunemann H, Oxman AD, Kunz R, Brozek J, et al. GRADE guidelines: 3. Rating the quality of evidence. Journal of Clinical Epidemiology 2011;64(4):401‐6. [DOI: 10.1016/j.clinepi.2010.07.015] [DOI] [PubMed] [Google Scholar]
Bardou 2015
- Bardou M, Quenot JP, Barkun A. Stress‐related mucosal disease in the critically ill patient. Nature Reviews Gastroenterology and Hepatology 2015;12(2):98‐107. [DOI] [PubMed] [Google Scholar]
Barkun 2012
- Barkun AN. Bardou M, Pham CQD, Martel M. Proton pump inhibitors vs. histamine 2 receptor antagonists for stress‐related mucosal bleeding prophylaxis in critically ill patients: a meta‐analysis. American Journal of Gastroenterology 2012;107(4):507‐20. [DOI] [PubMed] [Google Scholar]
CDC 2003
- Centres for Disease Control and Prevention. Guidelines for preventing health‐care associated pneumonia, 2003: recommendations of CDC and the Heathcare Infection Control Practices Advisory Committee. http://www.cdc.gov/ncidod/dhqp/pdf/guidelines/CDCpneumo_guidelines.pdf (accessed 1 May 2009). [PubMed]
Chastre 2002
- Chastre J, Fagon JY. Ventilator‐associated pneumonia. American Journal of Respiratory and Critical Care Medicine 2002;165(7):867‐903. [DOI] [PubMed] [Google Scholar]
Collard 2003
- Collard HR, Saint S, Matthay MA. Prevention of ventilator associated pneumonia: an evidence‐based systematic review. Annals of Internal Medicine 2003;138:494‐501. [DOI] [PubMed] [Google Scholar]
Cook 1991
- Cook DJ, Laine LA, Guyatt GH, Raffin TA. Nosocomial pneumonia and the role of gastric pH. A meta‐analysis. Chest 1991;100:7‐13. [DOI] [PubMed] [Google Scholar]
Cook 1994b
- Cook DJ, Reeve BK, Scholes LC. Histamine‐2‐receptor antagonists and antacids in the critically ill population: stress ulceration versus nosocomial pneumonia. Infection Control and Hospital Epidemiology 1994;15(7):437‐42. [DOI] [PubMed] [Google Scholar]
Cook 1995
- Cook DJ. Stress ulcer prophylaxis: gastrointestinal bleeding and nosocomial pneumonia. Scandinavian Journal of Gastroenterology 1995;30(1):48‐52. [DOI] [PubMed] [Google Scholar]
Cook 1995b
- Cook DJ. Stress ulcer prophylaxis: gastrointestinal bleeding and nosocomial pneumonia. Best evidence synthesis. Scandinavian Journal of Gastroenterology 1995;210(Suppl):48‐52. [DOI] [PubMed] [Google Scholar]
Cook 1996
- Cook DJ, Reeve BK, Guyatt GH, Heyland Dk, Griffith LE, Buckingham L, et al. Stress ulcer prophylaxis in critically ill patients. Resolving discordant meta‐analyses. Journal of the American Medical Association 1996;275(4):308‐14. [PubMed] [Google Scholar]
Cook 1998a
- Cook D, Guyatt G, Marshall J, Leasa D, Fuller H, Hall R, et al. A comparison of sucralfate and ranitidine for the prevention of upper gastrointestinal bleeding in patients requiring mechanical ventilation. New England Journal of Medicine 1998;338(12):791‐7. [DOI] [PubMed] [Google Scholar]
Cook 1998b
- Cook DJ, Walter SD, Cook RJ, Griffith LE, Guyatt GH, Leasa D, et al. Incidence of and risk factors for ventilator‐associated pneumonia in critically ill patients. Annals of Internal Medicine 1998;129(6):433‐40. [DOI] [PubMed] [Google Scholar]
Craven 1986
- Craven DE, Kunches LM, Kilinsky V, Lichtenberg DA, Make BJ, McCabe WR. Risk factors for pneumonia and fatality in patients receiving continuous mechanical ventilation. American Review of Respiratory Disease 1986;133:792‐6. [PubMed] [Google Scholar]
Cunningham 2003
- Cunningham R, Dale B, Undy B, Gaunt N. Proton pump inhibitors as a risk factor for Clostridium difficile diarrhoea. Journal of Hospital Infection 2003;54(3):243‐5. [DOI] [PubMed] [Google Scholar]
Deeks 2011
- Deeks JJ, Higgins JPT, Altman DG (editors). Chapter 9: Analysing data and undertaking meta‐analyses. Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org. [Google Scholar]
Dorland 1995
- Dorland WA. Dorland's Pocket Medical Dictionary. 25. New Delhi: Oxford and IBH Publishing, 1995. [Google Scholar]
Goldhil 2002
- Goldhil. Levels of critical care for adult patients. Standards and guidelines. Intensive Care Society Standards. London: Intensive Care Society, 2002:1‐7. [Google Scholar]
GRADEpro 2015 [Computer program]
- McMaster University (developed by Evidence Prime). GRADEpro GDT. Version accessed 31 May 2018. Hamilton (ON): McMaster University (developed by Evidence Prime), 2015.
Guillamondegui 2008
- Guillamondegui OD, Gunter OL, Bonadies JA, Coates JE, Kurek SJ, Moya MA, et al. Practice Management Guidelines for Stress Ulcer Prophylaxis. EAST Practice Management Guidelines Committee. Chicago, IL: Eastern Association for the Surgery of Trauma (EAST), 2008:24. [Google Scholar]
Hammond 2017
- Hammond DA, Kathe N, Shah A, Martin BC. Cost‐effectiveness of histamine2 receptor antagonists versus proton pump inhibitors for stress ulcer prophylaxis in critically ill patients. Pharmacotherapy 2017;31(1):43‐53. [DOI: 10.1002/phar.1859] [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Altman DG, Sterne JAC (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Higgins 2011a
- Higgins JPT, Deeks JJ, Altman DG. Chapter 16: Special topics in statistics. In: Higgins JPT, Green S editor(s). Cochrane Handbook for Systematic Reviews of Interventions. The Cochrane Collaboration, 2011. [Google Scholar]
Hinds 1999
- Hinds CJ, Fletcher SN. Ranitidine reduced clinically important gastrointestinal bleeding in patients who required mechanical ventilation. Gut 1999;44:10‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Krag 2014
- Krag M, Perner A, Wetterslev J, Wise M, Hylander Moller M. Stress ulcer prophylaxis versus placebo or no prophylaxis in critically ill patients. A systematic review of randomised clinical trials with meta‐analysis and trial sequential analysis. Intensive Care Medicine 2014;40:11‐22. [DOI] [PubMed] [Google Scholar]
Kwok 2012
- Kwok CS, Arthur AK, Anibueze CI, Singh S, Cavallazzi R, Loke YK. Risk of Clostridium difficile infection with acid suppressing drugs and antibiotics: meta‐analysis. American Journal of Gastroenterology 2012;107(7):1011‐9. [DOI: 10.1038/ajg.2012.108] [DOI] [PubMed] [Google Scholar]
Lin 2010
- Lin PC, Chang CH, Hsu PI, Pi‐Lai Tseng MS, Yaw‐Bin H. The efficacy and safety of proton pump inhibitors vs histamine‐2 receptor antagonists for stress ulcer bleeding prophylaxis among critical care patients: a meta‐analysis. Critical Care Medicine 2010;30(4):1197‐206. [DOI: 10.1097/CCM.0b013e3181d69ccf] [DOI] [PubMed] [Google Scholar]
MacLaren 2014
- MacLaren R, Campbell J. Cost‐effectiveness of histamine receptor‐2 antagonist versus proton pump inhibitor for stress ulcer prophylaxis in critically ill patients. Critical Care Medicine 2014;42(4):809‐15. [DOI: 10.1097/CCM.0000000000000032] [DOI] [PubMed] [Google Scholar]
Masterton 2008
- Masterton RG, Galloway A, French G, Street M, Armstrong J, et al. Guidelines for the management of hospital‐acquired pneumonia in the UK: report of the Working Party on Hospital‐Acquired Pneumonia of the British Society for Antimicrobial Chemotherapy. Journal of Antimicrobial Chemotherapy 2008;62:5‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mayhall 2001
- Mayhall CG. Ventilator‐associated pneumonia or not? Contemporary diagnosis. Emerging Infectious Diseases 2001;7(2):200‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
McClave 2009
- McClave SA, Martindale RG, Vanek VW, McCarthy M, Roberts P, Taylor B, et al. Guidelines for the provision and assessment of nutrition support therapy in the adult critically ill patient: Society of Critical Care Medicine (SCCM) and American Society for Parenteral and Enteral Nutrition (A.S.P.E.N.). JPEN. Journal of Parenteral and Enteral Nutrition 2009;33:277‐316. [DOI] [PubMed] [Google Scholar]
Messori 2000
- Messori A, Trippoli S, Vaiani M, Gorini M, Corrado A. Bleeding and pneumonia in intensive care patients given ranitidine and sucralfate for prevention of stress ulcer: metaanalysis of randomised controlled trials. British Medical Journal 2000;321:1103‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mutlu 2001
- Mutlu GM, Mutlu EA, Factor P. GI complications in patients receiving mechanical ventilation. Chest 2001;119:1222‐41. [DOI] [PubMed] [Google Scholar]
Neligan 2006
- Neligan P. Critical care medicine tutorials. Stress ulcer prophylaxis in critical care. http://www.ccmtutorials.com/support/ulcers/index.htm (accessed 1 September 2008).
Penner 2005
- Penner RM, Brindely PG, Jacka MJ. Best evidence in critical care medicine: stress ulcer prophylaxis in the intensive care unit: damned if you do, damned if you don't. Canadian Journal of Anesthesia 2005;52:650‐1. [DOI] [PubMed] [Google Scholar]
Pfeffer 2007
- Pfeffer MA, Galindo MS. Prevention of complications in hospitalized patients part III: upper gastrointestinal stress ulcers. Proceedings of UCLA Healthcare 2007;2:1‐5. [Google Scholar]
Pilkington 2012
- Pilkington KB, Wagstaff MJD, Greenwood JE. Prevention of gastrointestinal bleeding due to stress ulceration: a review of current literature. Anaesthesia and Intensive Care 2012;40:253‐9. [DOI] [PubMed] [Google Scholar]
Rahbar 2006
- Rahbar M. Detection and quantitation of the etiologic agents of ventilator‐associated pneumonia in endotracheal tube aspirates from patients in Iran. Infection Control and Hospital Epidemiology 2006;27(8):884‐5. [DOI] [PubMed] [Google Scholar]
Review Manager (RevMan) 5.2 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.2. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2011.
Rorbaek Madsen 2014
- Rorbaek Madsen K, Lorentzen K, Clausen N, Oberg E, Kierkegaard PRC, Maymann‐Holler N, et al. Guideline for stress ulcer prophylaxis in the intensive care unit. Danish Medical Journal 2014;61(3):C4811. [PUBMED: 24814922] [PubMed] [Google Scholar]
Safdar 2005
- Safdar N, Dezfulian C, Collard HR, Saint S. Clinical and economic consequences of ventilator‐associated pneumonia: a systematic review. Critical Care Medicine 2005;33(10):2184‐93. [DOI] [PubMed] [Google Scholar]
Schunemann 2008
- Schünemann HJ, Oxman AD, Vist GE, Higgins JPT, Deeks JJ, Glasziou P, et al. Interpreting results drawing conclusions. In: Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.0.1 (updated September 2008). 5th Edition. London: Wiley, 2008. Available from www.cochrane‐handbook.org. [Google Scholar]
Spirt 2006
- Spirt MJ, Stanley S. Update on stress ulcer prophylaxis in critically ill patients. Critical Care Nurse 2006;26(1):18‐28. [PubMed] [Google Scholar]
Tryba 1991
- Tryba M. Sucralfate versus antacids or H2‐antagonists for stress ulcer prophylaxis: a meta‐analysis on efficacy and pneumonia rate. Critical Care Medicine 1991;19:942‐9. [DOI] [PubMed] [Google Scholar]
Tryba 1991b
- Tryba M. Prophylaxis of stress‐ulcer bleeding. A meta‐analysis. Journal of Clinical Gastroenterology 1991;13(Suppl 2):S44‐55. [PubMed] [Google Scholar]
Tryba 1995
- Tryba M, Cook DJ. Gastric alkalinization, pneumonia, and systemic infections: the controversy. Scandinavian Journal of Gastroenterology 1995;210:53‐9. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
George 2010
- George AT, Tharyan P, Peter JV, Kirubakaran R, Barnabas JP. Interventions for preventing upper gastrointestinal bleeding in people admitted to intensive care units. Cochrane Database of Systematic Reviews 2010, Issue 9. [DOI: 10.1002/14651858.CD008687] [DOI] [PMC free article] [PubMed] [Google Scholar]