

Abstract
Background
Early diagnosis and treatment of lower respiratory tract infections are the mainstay of management of lung disease in cystic fibrosis. When sputum samples are unavailable, treatment relies mainly on cultures from oropharyngeal specimens; however, there are concerns regarding the sensitivity of these to identify lower respiratory organisms.
Bronchoscopy and related procedures (including bronchoalveolar lavage) though invasive, allow the collection of lower respiratory specimens from non‐sputum producers. Cultures of bronchoscopic specimens provide a higher yield of organisms compared to those from oropharyngeal specimens. Regular use of bronchoscopy and related procedures may help in a more accurate diagnosis of lower respiratory tract infections and guide the selection of antimicrobials, which may lead to clinical benefits.
This is an update of a previous review.
Objectives
To evaluate the use of bronchoscopy‐guided antimicrobial therapy in the management of lung infection in adults and children with cystic fibrosis.
Search methods
We searched the Cochrane Cystic Fibrosis Trials Register, compiled from electronic database searches and handsearching of journals and conference abstract books. Date of latest search: 30 August 2018.
We also searched three registries of ongoing studies and the reference lists of relevant articles and reviews. Date of latest search: 10 April 2018.
Selection criteria
We included randomized controlled studies including people of any age with cystic fibrosis, comparing outcomes following therapies guided by the results of bronchoscopy (and related procedures) with outcomes following therapies guided by the results of any other type of sampling (including cultures from sputum, throat swab and cough swab).
Data collection and analysis
Two review authors independently selected studies, assessed their risk of bias and extracted data. We contacted study investigators for further information. The quality of the evidence was assessed using the GRADE criteria.
Main results
The search identified 11 studies, but we only included one study enrolling infants with cystic fibrosis under six months of age and diagnosed through newborn screening (170 enrolled); participants were followed until they were five years old (data from 157 children). The study compared outcomes following therapy directed by bronchoalveolar lavage for pulmonary exacerbations with standard treatment based on clinical features and oropharyngeal cultures.
We considered this study to have a low risk of bias; however, the statistical power to detect a significant difference in the prevalence of Pseudomonas aeruginosa was limited due to the prevalence (of Pseudomonas aeruginosa isolation in bronchoalveolar lavage samples at five years age) being much lower in both the groups compared to that which was expected and which was used for the power calculation. The sample size was adequate to detect a difference in high‐resolution computed tomography scoring. The quality of evidence for the key parameters was graded as low except high‐resolution computed tomography scoring and cost of care analysis, which were graded as moderate quality.
At five years of age, there was no clear benefit of bronchoalveolar lavage‐directed therapy on lung function z scores or nutritional parameters. Evaluation of total and component high‐resolution computed tomography scores showed no significant difference in evidence of structural lung disease in the two groups.
In addition, this study did not show any difference between the number of isolates of Pseudomonas aeruginosa per child per year diagnosed in the bronchoalveolar lavage‐directed therapy group compared to the standard therapy group. The eradication rate following one or two courses of eradication treatment was comparable in the two groups, as were the number of pulmonary exacerbations. However, the number of hospitalizations was significantly higher in the bronchoalveolar lavage‐directed therapy group, but the mean duration of hospitalizations was significantly less compared to the standard therapy group.
Mild adverse events were reported in a proportion of participants, but these were generally well‐tolerated. The most common adverse event reported was transient worsening of cough after 29% of procedures. Significant clinical deterioration was documented during or within 24 hours of bronchoalveolar lavage in 4.8% of procedures.
Authors' conclusions
This review, limited to a single, well‐designed randomized controlled study, shows no clear evidence to support the routine use of bronchoalveolar lavage for the diagnosis and management of pulmonary infection in pre‐school children with cystic fibrosis compared to the standard practice of providing treatment based on results of oropharyngeal culture and clinical symptoms. No evidence was available for adult and adolescent populations.
Plain language summary
Using samples obtained by bronchoscopy to decide how to treat lung infections in people with cystic fibrosis
Review question
We reviewed the evidence about whether to use samples obtained by bronchoscopy when deciding how to treat lung infections in people with cystic fibrosis.
Background
Breathing problems in people with cystic fibrosis are mainly due to repeated lung infections. Growing bugs from samples of mucus coughed up from the lower airways often allows doctors to quickly identify the bug causing the infection and start treatment early. If people can not cough up mucus, swabs are taken from the upper throat to identify the bug causing infection in the lower airways‐but this may not be the most reliable method.
During a bronchoscopy, clinicians examine the lower airways using a long, thin flexible tube with a light and camera at one end; they may also collect mucus. The person needs to be sedated or have a general anaesthetic. We do not know if treatment based on samples taken during a bronchoscopy is better than treatment based on throat swabs. This is an update of an already published review.
Search date
The evidence is current to: 10 April 2018.
Study characteristics
We searched for studies of people of any age, but this review only includes one study in which 170 babies with cystic fibrosis, aged less than six months, were divided into two groups completely at random. One group was given antibiotics based on samples taken by bronchoscopy and the other group based on samples taken from throat. The investigators measured outcomes at five years of age. A total of 157 children completed the study.
Key results
This study did not show any difference between the groups in terms of lung function, weight, body mass index or in the score calculated by a CT scan of the lungs at five years of age. There were no differences in how many children in each group had an infection with Pseudomonas aeruginosa at five years of age, or per year of follow up, or how often a child was unwell with respiratory symptoms. Children in the bronchoscopy group were admitted to hospital more often although admissions were generally shorter than the other group. There was no difference in the overall cost of care between the two groups.
Side effects reported during, and after bronchoscopy, were not serious; the most common side effect was increased coughing (in one third of children).
There is currently not enough evidence to support the regular use of bronchoscopy to diagnose and treat lung infections in children with cystic fibrosis.
Quality of the evidence
Evidence was limited to only one well‐designed study. Overall quality of evidence was of low (for most outcomes) to moderate quality (for high‐resolution computed tomography scoring and cost of care analysis). Quality limitations were due to fewer children taking part in the study than the statisticians thought were needed to show true results for some outcomes. Since the treatment of a first infection with Pseudomonas aeruginosa is highly successful, larger and longer studies are needed to detect small differences between the groups. Conducting such large studies is extremely difficult. Also, the study only included young children and we do not know if the results would be the same in other age groups.
Summary of findings
Summary of findings for the main comparison. BAL‐directed therapy versus standard therapy for cystic fibrosis.
| BAL‐directed therapy versus standard therapy for cystic fibrosis | ||||||
| Patient or population: people with pulmonary exacerbations in cystic fibrosis Settings: hospital Intervention: BAL‐directed therapy Comparison: standard therapy | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Standard therapy | BAL‐directed therapy | |||||
| Z score FEV1 Follow up: 5 years | The mean (SD) z score for FEV1 in the standard therapy group was ‐0.41 (1.23). | The mean (SD) z score FEV1 in the intervention group ‐0.56 (1.25) that was 0.15 lower (0.54 lower to 0.24 higher) than the standard therapy group. | 157 (1 study) | ⊕⊕⊝⊝ low1, 2 | FEV1 and FVC were measured using standard spirometer after bronchodilatation. The mean difference between the two groups represents the difference in the mean z scores for each parameter. Z scores for FEV1, FVC were calculated from British reference values (www.lungfunction.org/growinglungs). |
|
| Z score FVC Follow up: 5 years | The mean (SD) z score for FVC in the standard therapy group was 0.01 (1.2). | The mean (SD) z score FVC in the intervention groups was ‐0.04 (1.31) that was 0.05 higher (0.44 lower to 0.34 higher) than the standard therapy group. | 157 (1 study) | ⊕⊕⊝⊝ low1, 2 | FEV1 and FVC were measured using standard spirometer after bronchodilatation. The mean difference between the two groups represents the difference in the mean z scores for each parameter. Z scores for FEV1, FVC were calculated from British reference values (www.lungfunction.org/growinglungs). |
|
| HRCT score (Brody‐II) Follow up: 5 years | The mean (SD) HRCT score in the standard therapy group was 2.83 (3.5). | The mean (SD) HRCT score (Brody‐II) in the intervention group was 3.02 (3.48) 0.19 higher (0.93 lower to 1.31 higher) than the standard therapy group. | 152 (1 study) | ⊕⊕⊕⊝ moderate2 | The study had adequate power to detect a difference in HRCT score. HRCT scans were assesses by an independent assessor who was blinded to subject allocation using an updated version of Brody‐II score. | |
| Z score for weight Follow up: 5 years | The mean (SD) z score for weight was ‐0.21 (0.82) in the standard therapy group. | The mean z score for weight in the intervention group was ‐0.15 that was 0.06 higher (0.21 lower to 0.33 higher) than the standard therapy group. | 157 (1 study) | ⊕⊕⊝⊝ low1, 2 | The z scores for weight and BMI were calculated from the 2000 CDC Growth Reference Charts (http://cdc.gov/growthcharts). The mean difference between the two groups represents the difference in their z scores for each parameter. | |
| Z score BMI Follow up: 5 years | The mean (SD) z score for BMI was 0.01 (0.83) in the standard therapy group. | The mean (SD) z score BMI in the intervention group was 0.03 (0.93) that was 0.02 higher (0.26 lower to 0.30 higher) than the standard therapy group. | 157 (1 study) | ⊕⊕⊝⊝ low1, 2 | The z scores for weight and BMI were calculated from the 2000 CDC Growth Reference Charts (http://cdc.gov/growthcharts). The mean difference between the two groups represents the difference in their z scores for each parameter. | |
| Number of hospitalizations per participant per year Follow‐up: 5 years | The number of hospitalizations per participant per year was 1.08 in the standard therapy group. | The number of hospitalizations per participant per year in the intervention group was 1.52 that was 1.4 times higher (1.08 lower to 1.82 higher) than the standard therapy group. | 157 (1 study) | ⊕⊕⊝⊝ low1, 2 | ||
|
Overall cost of care per participant (AUD) Follow up: 5 years |
The total cost of care for each participant was 90,958 AUD (SD 110,255) in the standard therapy group. | The total cost of care in the intervention group was 92,860 AUD (SD 73,378) that was 1902 AUD higher (27,508.98 lower to 31,312.98 higher) than the standard therapy group. | 157 (1 study) | ⊕⊕⊕⊝ moderate2 | The cost of hospital admissions per participant over the total study duration in the intervention group was 9288 AUD lower in the intervention group (34,996.37 lower to 16,420.37 higher). | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). AUD: Australian dollars; BAL: bronchoalveolar lavage; BMI: body mass index (weight in kg/height in metres squared); CI: Confidence interval; FEV1: forced expiratory volume at one second; FVC: forced vital capacity; HRCT: high‐resolution computed tomography | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1. Downgraded once because the study had low statistical power and research is needed to provide definitive answers.
2. Downgraded once due to indirectness (only age group studied was under five years of age).
Background
Please note: there is a glossary of terms provided in the appendices (Appendix 1)
Description of the condition
Cystic fibrosis (CF) is one of the most common life‐shortening autosomal recessive disorders. The prevalence is highest in populations of northern European descent, with around 1 in 2500 in the UK (Dodge 2007) and the condition is increasingly being recognised as being important in other populations (Bobadilla 2002; Heim 2001). It affects multiple systems including the lungs, intestines and the digestive system. Respiratory disease is the cause of most morbidity and is a major contributor towards mortality related to CF. Respiratory disease is characterised by repeated episodes of pulmonary infections, termed as exacerbations, which manifest as acute deterioration of respiratory symptoms or signs. These exacerbations are usually related to infection and associated with worsening of lung function or loss of weight (Rabin 2004). Over a period of time, these exacerbations lead to progressive structural lung damage and poor lung function.
Respiratory infections in CF are caused by a limited range of organisms. At an early age, the commonest organisms are Staplylococcus aureus and Haemophilus influenzae which are followed very soon after by Pseudomonas aeruginosa. Over a period of time, the initial intermittent infections by P aeruginosa establish into chronic infection (Li 2005), which is the case with more than 60% of the adult CF population (UK CF Trust 2004). Chronic P aeruginosa infection is associated with increased morbidity and mortality (Frederiksen 1997; Gibson 2003; Li 2005; Nixon 2001; UK CF Trust 2004).
The management of CF‐lung disease incorporates different aspects such as the prevention and treatment of infections, chest physiotherapy and mucus clearance techniques. The isolation of infecting organisms is important as these are more readily eradicated at an early stage in the infectious process. For example, eradication of an initial infection with P aeruginosa can be achieved (Langton Hewer 2017; Stuart 2010) and is recommended in guidelines (UK CF Trust 2004). However, chronic pulmonary infection with P aeruginosa cannot be eradicated. Eradication has become a major treatment goal in people who are P aeruginosa‐naïve as it delays the onset of chronic infection (Hansen 2008). To achieve early identification of organisms, routine surveillance is conducted which includes regular two‐ to three‐monthly microbiological cultures with isolation of organisms mainly from the cultures of respiratory specimens; these include either spontaneously produced or induced sputum. Very young children, many older children and even some adults are unable to expectorate sputum. In such cases, the standard practice has been to use upper respiratory swabs including throat or oropharyngeal swabs (a cotton‐tipped swab is rotated in the back of the throat or tonsils) or 'cough swabs' (a cough swab is taken by holding a sterile swab at the back of the throat and asking the individual, who is often a child, to cough). The same methods are used to guide the treatment of infective exacerbations.
Description of the intervention
Bronchoscopy is an endoscopic procedure which helps visualize the upper and lower airways using a bronchoscope. A bronchoscope is a slender tubular instrument with a light at the end; it can be either flexible or rigid. Flexible bronchoscopy was first introduced by Ikeda in 1968 (Ikeda 1968) and was first reported to be used in children in 1978 (Wood 1978). Since then the technique has been significantly improved and safety of the procedure has been described (de Blic 2002; Nussbaum 2002; Picard 2000).
Several special bronchoscopic procedures are used to aid diagnosis. Bronchoalveolar lavage (BAL) and protected bronchial brush (PBB) sampling have been used to obtain specimens from the lower respiratory tract. The use of BAL involves the instillation of small aliquots of saline into bronchi followed by its recovery by suction; whereas PBB sampling allows direct sampling from the airway mucosal surface.
How the intervention might work
The newborn screening programme was started for CF with one of its aims being the improvement of clinical outcome for people with CF by diagnosing them soon after birth and by providing early interventions. It has been shown to have an effect on nutritional status (Farrell 1997a), but no significant difference has been documented in the status of the lung disease (Dankert‐Roelse 2009; Farrell 1997b; Southern 2009).
There is an increasing recognition that the onset of lung disease happens very early in the course of CF and infection and inflammation have been documented in BAL in the first few months of life by many groups, including asymptomatic infection with P aeruginosa (Armstrong 1995; Khan 1995; Sly 2009). Early identification of the correct organism should allow appropriate treatment and the use of eradication regimens (where appropriate e.g. for P aeruginosa), with the aim of avoiding chronic infection and related morbidity. Improving the accuracy of microbiological testing is therefore a logical goal.
Traditional surveillance methods to guide the treatment of acute exacerbations and to prevent the development of chronic infection include the identification of pathological organisms from sputum and, in non‐expectorating patients, of organisms from the oropharynx, e.g. from a throat swab or a 'cough swab'. However, the upper respiratory tract is inhabited by a large number of bacteria as normal commensals (passive inhabitants which do not cause active disease); this leads to contamination of the upper respiratory specimens and can affect the culture results. It has been shown that oropharyngeal cultures, particularly in very young children, have poor sensitivity for lower respiratory tract infection (Rosenfeld 1999). The most reliable method for obtaining lower respiratory secretions is BAL; and some studies have shown cultures based on these samples have a higher yield of organisms as compared to standard methods (Baughman 1997; Hilliard 2007; Rosenfeld 1999; Stafler 2011). This could potentially be useful in routine surveillance for people with CF, but it comes with the drawback of being an invasive procedure with the need for anaesthesia and a hospital visit.
Why it is important to do this review
There has been a change in practice in many CF centres, based on the need for early identification of infections; and bronchoscopy has been used to collect lower respiratory specimens routinely from young children and from adults in some specific situations. Studies have suggested that this practice has led to the identification of new organisms and use of different antibiotics in situations where bronchoscopy has been performed both in symptomatic individuals (Baughman 1997) and in asymptomatic individuals (Sly 2009). No systematic review has been undertaken to assess the effect of routine bronchoscopy‐guided antimicrobial management of acute and chronic respiratory disease in children and adults with CF compared to standard management (which is directed by culture of oropharyngeal swab or sputum, as described above).
This is an update of a previously published review (Jain 2013; Jain 2016).
Objectives
To evaluate the use of bronchoscopy‐guided antimicrobial therapy in the management of lung infection in people (both adults and children) with CF.
Methods
Criteria for considering studies for this review
Types of studies
Randomized controlled studies.
Types of participants
Children and adults with CF (according to standard definitions, clinical features of CF plus a positive sweat test or the presence of two genes known to be associated with CF (Rosenstein 1998)).
Types of interventions
We included studies comparing outcomes following therapies guided by the results of bronchoscopy (with BAL or PBB sampling) with outcomes following therapies guided by the results of any other type of sampling (including but not limited to cultures from sputum, throat swab and cough swab).
Types of outcome measures
Primary outcomes
-
Lung function
-
conventional spirometry
per cent (%) predicted forced expiratory volume at one second (FEV1) change and absolute values in litres (L)
% predicted forced vital capacity (FVC) change and absolute values in L
-
infant lung function‐squeeze
% predicted FEV0.5, forced expiratory flow from 25% to 75% of vital capacity (FEF25‐75) and FEF75 changes and absolute values in litres
% predicted FVC change and absolute values in L
lung clearance index (LCI)
-
High‐resolution computed tomography (HRCT) appearances using a recognised scoring system (e.g. Brody 2004; Loeve 2009)
-
Nutritional parameters
weight (in kg or percentile)
body mass index (BMI) percentile
Secondary outcomes
-
Number of positive isolates per child per year of follow‐up (for BAL defined as a positive culture of the growths from 1000 CFU/ml)
P aeruginosa
S aureus
H influenza
Burkholderia cepacia complex
Stenotrophomonous maltophilia
Achromobacter xylosoxidans
non‐tuberculous mycobacteria
Aspergillus species
any other organism
Clearance of the organism from the cultures
Time to first infection with P aeruginosa*
-
Time to chronic infection using any recognized definition e.g. (Lee 2003)*
with P aeruginosa
withS aureus
Complications and adverse effects related to bronchoscopy (e.g. fever, hypoxaemia and increased cough, unplanned admissions and other serious adverse events etc.)
Quality of life (QoL) measured using a validated tool such as Cystic Fibrosis Questionnaire‐Revised version (CFQ‐R) (Quittner 2009) and Cystic Fibrosis Quality of Life Questionnaire (CFQoL) (Gee 2000)
-
Hospitalisations
number of hospitalisations per participant per year
days as inpatient per participant per year
cost of care
-
Number of courses of antibiotics prescribed per participant per year (not including prophylactic antibiotics)
intravenous
oral
Number of pulmonary exacerbations (requiring oral or intravenous antibiotics) per participant per year (diagnosis based on clinical judgement or any approved or published definition e.g. Rabin 2004, Rosenfeld 2001)
* In the studies where the follow up was started just after the birth, we considered the age of first acquisition of infection and age of establishing chronic infection. In studies in older people where follow up is started later in life, we considered time to acquire the first infection and time to acquire chronic infection.
Search methods for identification of studies
We did not apply any language or date restrictions.
Electronic searches
We identified relevant studies from the Cochrane Cystic Fibrosis and Genetic Disorders Group's Cystic Fibrosis Trials Register using the term: sampling techniques. The Cystic Fibrosis Trials Register is compiled from electronic searches of the Cochrane Central Register of Controlled Trials (CENTRAL) (updated each new issue of theCochrane Library), weekly searches of MEDLINE, a search of Embase to 1995 and the prospective handsearching of two journals ‐ Pediatric Pulmonology and the Journal of Cystic Fibrosis. Unpublished work is identified by searching the abstract books of three major cystic fibrosis conferences: the International Cystic Fibrosis Conference; the European Cystic Fibrosis Conference and the North American Cystic Fibrosis Conference. For full details of all searching activities for the register, please see the Cochrane Cystic Fibrosis and Genetic Disorders Group website.
Date of the latest search of the Group's Cystic Fibrosis Trials Register: 30 August 2018.
We also searched the ongoing study registers on 10 April 2018 (Appendix 2).
Searching other resources
We checked the reference lists of primary studies and review articles for additional references.
Data collection and analysis
We conducted the review according to the recommendations from theCochrane Handbook of Systematic Reviews of Interventions (Higgins 2011a).
Selection of studies
Two review authors (KJ and CW) independently screened the titles and abstracts of all the studies identified by the primary search and then obtained the full text of relevant studies. Both authors independently went through the studies while considering inclusion criteria to decide whether to include or exclude the studies. The authors planned to contact the investigators for more information for any study where the criteria for inclusion were unclear. They planned to resolve any disagreement by discussion, referral to the third review author and consensus. They have described the reasons for exclusion of the studies, initially considered eligible for inclusion according to title and abstract.
Data extraction and management
One review author (KJ ) extracted the data from the only included study; the second review author (CW) was familiar with the data (being the author of the study). They completed a pre‐decided data extraction form. They resolved any disagreements by discussion, referral to the third review author and consensus. Extracted information included:
administrative details including authors, year of publication, country of study;
participant characteristics including number of participants in each group, age, gender, weight, BMI, concomitant morbidities and other baseline characteristics mentioned in the studies;
study characteristics including design of study, inclusion and exclusion criteria, duration of follow up, co‐morbidities, primary and secondary outcome measures;
details of intervention techniques used for the collection of specimens and the description of any adverse effects;
data for primary and secondary outcome measures.
The review authors created a study flow diagram following the template described in the PRISMA statement (Moher 2009). They also completed a table of 'Characteristics of included studies' including information about study design, relevant information on the demographics and health of participants and a list of interventions and outcome measures.
If suitable data were available, they would have undertaken a meta‐analysis for each outcome. Where possible, they have presented the results in a graph, or otherwise in a narrative way.
The authors planned to report data at two weeks (after finishing the antibiotic course), three months, six months, one year and annually thereafter. If there were studies reporting data at other time points, they would have considered reporting those as well. In the current version of the review, the authors have presented data at the five‐year time‐point.
Assessment of risk of bias in included studies
Two authors independently assessed the studies fulfilling the inclusion criteria for risk of bias as per guidelines from theCochrane Handbook of Systematic Reviews of Interventions (Higgins 2011b). They assessed studies for risk of bias according to a standardised set of questions covering the following domains:
random sequence generation;
allocation concealment;
blinding of outcome assessment for the primary outcomes for HRCT assessment and lung function parameters;
incomplete data outcome;
selective reporting bias;
other sources of bias.
They classified the responses from each category into one of three grades (low risk of bias, high risk of bias and unclear risk of bias) and generated a risk of bias graph.
Measures of treatment effect
The authors planned to analyse binary data using risk ratio (RR) and 95% confidence intervals (CIs). They used the mean difference (MD) with 95% CIs to analyse continuous data. The MD measures the absolute difference between the mean value in two groups when outcome measurements in all studies use the same scale; they would have used the standardized MD when the studies were assessing the same outcome but measuring it in a variety of ways. We used time‐to‐event analysis using hazard ratios (HRs) with 95% CIs, e.g. for time to acquisition of chronic infection with P aeruginosa. With reference to count data, e.g. for the number of isolates of organisms, we treated the data as continuous data and measured the intervention effect using the MD and 95% CIs between the groups. By convention, the changes during the study were reported as the effect measured post‐intervention minus that measured pre‐intervention; and differences between the study arms were reported as treatment arm effects minus control arm effects.
Unit of analysis issues
If the review authors identify further studies in the future, they plan to include cluster‐randomised studies if the clustering has been taken into account and the intra‐cluster coefficient is included. For the analysis process they will calculate the design effect to allow calculation of an effective sample size or an inflated standard error. They will consider estimates from an appropriately analysed cluster‐randomised controlled study, e.g. effects from a mixed model. They plan to use generic inverse variance output method in these contexts.
The review authors planned not to include cross‐over studies in the review, since CF is a chronic disorder with progressive worsening of lung disease following repeated infections. Some of the infections like P aeruginosa, once established, cannot be eradicated and are associated with worse outcome. In addition, for outcome measures such as change in lung function and structural lung damage, it may not be possible to revert completely to the same level, even with treatment. In view of this, the authors felt it was justified not to include cross‐over studies.
For events happening multiple times, such as pulmonary infections, the unit of analysis will be individual participants (except in some cases e.g. the number of isolates of organisms per year, where we will analyse the number of events).
Dealing with missing data
In the included study, there were no outcome data available for fewer than 10% of the recruited participants. In future, when the authors include more studies, in case of missing data, they will attempt to contact the primary author of any such studies and if they do not receive any responses, they will perform an intention‐to‐treat analysis where possible.
Assessment of heterogeneity
If the authors identify more studies in future, they will use the Chi² test in the forest plot and also the I² test for assessing the heterogeneity of results (Higgins 2003).
According to chapter 9 of theCochrane Handbook of Systematic Reviews of Interventions (Deeks 2011), a rough guide to interpretation of thresholds for the I² statistic is as follows:
0% to 40%: might not be important;
30% to 60%: may represent moderate heterogeneity*;
50% to 90%: may represent substantial heterogeneity*:
75% to 100%: considerable heterogeneity*.
* The importance of the observed value of I² depends on (i) magnitude and direction of effects and (ii) strength of evidence for heterogeneity (e.g. P value of Chi² test or a CI for I²).
Assessment of reporting biases
Two review authors (KJ and AS) examined the included study for any evidence of selective outcome reporting bias by comparing the outcomes described in the protocol with the results published. In future, when they include more studies, they will construct a matrix indicating which outcomes were recorded in each study to establish if any studies omitted any key outcomes. They will compare the protocols (if available) or the 'Methods' section of published articles fulfilling the inclusion criteria with their 'Results' sections to establish selective reporting of outcomes that were pre‐specified. When there is suspicion of, or direct evidence of, selective outcome reporting, they will contact the study authors to provide the study protocol and full information for the outcomes reported inadequately.
If there are a sufficient number of studies included and combined (at least 10), then the review authors will construct a funnel plot and assess this to help identify evidence of publication bias or any other type of bias.
Data synthesis
The authors followed the guidelines from theCochrane Handbook of Systematic Reviews of Interventions to conduct the comparison between the bronchoscopy‐guided and the standard treatment groups covering all the primary and secondary outcome measures (Higgins 2011a).
If they had combined more than a single data set and had identified no heterogeneity or only mild to moderate heterogeneity based on I² test results (Higgins 2003), they would have used a fixed‐effect analysis model; if the value of I² had been between 50% and 75% they would have performed a random‐effects analysis. If the I² values had been greater than 75%, they would not have considered it suitable to pool the studies together.
For each outcome for which the authors were able to extract suitable data, they constructed a forest plot to display effect estimates and 95% CIs using a fixed‐effect model of analysis.
Summary of findings and quality of the evidence
In a post hoc change, and in line with current Cochrane guidance, the authors have summarized the evidence in a summary of findings table (Table 1). They have presented the primary outcomes lung function, HRCT and nutritional parameters along with the secondary outcomes of hospitalisations and cost of care after five years of follow‐up. The authors determined the quality of the evidence using the GRADE approach; and downgraded evidence for most outcomes as for those the study had low statistical power and more research is needed to provide definitive answers. High quality evidence was available for effect of BAL‐directed therapy on radiological features (HRCT) and cost of care.
Subgroup analysis and investigation of heterogeneity
In future, if the authors include and combine a sufficient number of studies, they plan to undertake the following subgroup analyses:
age groups: 0 to 5 years, 6 to 16 years and 17 years and above;
chronic or intermittent isolation of P aeruginosa versus no P aeruginosa;
different methods of sampling, e.g. BAL versus PBB or sputum culture versus throat swab.
Sensitivity analysis
In future, if the authors include and combine a sufficient number of studies, they plan to undertake the following sensitivity analyses:
to analyse the effect of bias including effects of sequence generation, allocation concealment, intention‐to‐treat analysis and reporting bias which are not resolved after contacting the authors;
to assess the differences between fixed‐effect and random‐effects analysis on the results.
Results
Description of studies
Results of the search
The searches identified a total of 34 references to 11 separate studies. Of these, only one study (11 references) met our inclusion criteria and we excluded 10 studies (23 references). The process of assessing the search results is shown in the PRISMA diagram (Figure 1).
1.

Study flow diagram.
Included studies
Methods
The only included study was a multicentre, randomised controlled study of parallel design (Wainwright 2011). This study was conducted across eight CF centres in Australia and New Zealand and recruited infants between June 1999 to April 2005. After consent, the participants were randomly assigned in a 1:1 ratio to two groups. The study was completed in December 2009.
Participants
Infants younger than six months, diagnosed with CF through the newborn screening program with a confirmed diagnosis of classic CF (two of the following: two CF mutations; sweat chloride level over 60 mEq/L; pancreatic insufficiency; or meconium ileus) were eligible for inclusion.
Of the total 267 infants eligible for study, 170 infants were recruited. Eighty‐four infants were randomized to receive BAL‐directed therapy and 86 randomised to receive standard therapy. All the participants randomised to the BAL group received the intended therapy, however, 4 out of 84 participants did not complete the study and were excluded from the primary analysis. Of the 86 participants randomised to standard therapy group, 84 participants received the intended therapy and 77 of these (92%) completed the study to be included in the primary analysis.
The mean (SD) age of the participants was 3.8 (1.6) months in the BAL‐directed therapy group and 3.7 (1.7) months in the standard therapy group. In both the groups, 44 participants were male. Mean (SD) weight at enrolment was 5.7 (1.40) kg in BAL‐directed therapy group and 5.6 (1.5) kg in the standard therapy group. The number of participants with homozygous ΔF508 mutation was 57 (68%) in the BAL‐directed therapy group and 54 (64%) in the standard therapy group. The number of participants with pancreatic insufficiency was 73 (87%) in the BAL‐directed therapy group and 71 (85%) in the standard therapy group. The number of participants with meconium ileus was 17 (20%) in the BAL‐directed therapy group and 16 (19%) in the standard therapy group. The number of participants born pre‐term (under 37 week gestation) was eight (10%) in the BAL‐directed therapy group and nine (11%) in the standard therapy group. History of exposure to tobacco smoke during pregnancy was present in 22 (26%) and 13 (15%) participants from the BAL‐directed therapy group and standard therapy group respectively. History of concurrent smoking in the household was present in 30 (36%) participants in the BAL‐directed therapy and 23 (28%) of participants from standard therapy group.
Interventions
Participants received treatment of pulmonary exacerbations directed either by results of BAL or according to standard policy of treatment (based on clinical features and oropharyngeal cultures).
The standard therapy included oropharyngeal swabs at following time points:
when a child was unwell with a change in respiratory symptoms from baseline (pulmonary exacerbation);
at the end of the antibiotic eradication treatment for P aeruginosa.
In addition, the participants in the BAL‐directed therapy groups also underwent BAL at following times:
before six months of age when well;
when hospitalised for a pulmonary exacerbation (unwell with change in respiratory symptoms from baseline);
if P aeruginosa was cultured from oropharyngeal specimens;
following P aeruginosa eradication therapy.
At five years of age, all the participants underwent BAL, HRCT scan (to compute CF‐CT scores), pulmonary function testing and anthropometric assessments.
Outcomes
The primary outcome measures were prevalence of P aeruginosa infection (defined as greater than or equal to 10³ colony forming units (CFU)/ml) in the BAL cultures) and evidence of structural lung disease assessed by total CF‐CT score (as percentage of a maximum score) on HRCT at age five years. Secondary outcome measures, also assessed at five years age, included z scores for weight, height and BMI; lung function parameters; CF‐CT components; respiratory exacerbation rate; number and duration of hospitalizations for respiratory exacerbations not associated with P aeruginosa infection; number of episodes of P aeruginosa infection per child per year; and final BAL microbiology and inflammatory indices.
Excluded studies
We excluded 10 studies (23 references) after screening. Four studies were excluded as they were investigating a different intervention which was not relevant to this review: cough plates (Jyothish 2005; Maiya 2004); induced sputum (Chmiel 2015); and throat swabs and nasopharyngeal suction (Taylor 2006). One study looked at the effect of dornase alfa on the surface of the lungs using BAL (Paul 2004) and one study was to establish levels of tobramycin, not a comparison of therapy depending on sampling technique (Rosenfeld 2006). One study compared sputum induction, BAL and expectorated sputum to identify pathogens, but did not lead to a comparison of treatment (Henig 2001). Two studies compared the pathogen yield of various sampling methods, but were not randomised (ISRCTN 12473810; NCT02363764). The remaining study was excluded as it had a cross‐over design to compare inflammatory marker in the samples obtained by sputum induction and bronchoscopy but did not follow any comparison of treatment (McGarvey 2002).
Risk of bias in included studies
A summary of the risk of bias for the included study is shown in the figures (Figure 2).
2.
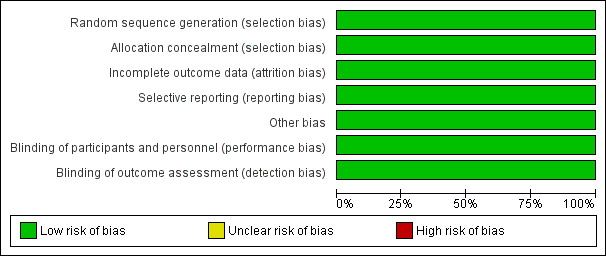
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Allocation
Sequence generation
A central computer‐generated sequence was used to randomise the participants to either group in blocks that were stratified by CF centre and gender. A randomisation number was generated centrally, away from the local investigators. This domain was judged to have a low risk of bias.
Concealment of allocation
The randomisation key was concealed and held remotely; and allocation was disclosed on the telephone after confirmation of recruitment to the study. The procedure was judged to have low risk of bias.
Blinding
The participants and the personnel were not blinded to the randomisation; however, the risk of bias was judged to be low for this item because the primary outcome measures were unlikely to be influenced by the lack of blinding. These were evaluated by the assessors who were blinded to treatment allocation and were not directly involved in participant care (a microbiologist assessed BAL and an expert scientist scored the HRCT).
Incomplete outcome data
Although, the study was set up to be analysed on an intention‐to‐treat basis, the primary analysis was based only on the participants who provided final outcome data, analysed according to randomised groups. The risk of bias was considered moderate to low as less than 10% of the data were missing, and the number of participants with missing outcome data and the reasons of their exclusion were balanced across both treatment arms. In the BAL‐directed therapy group, 4 out of 84 participants did not complete the study (three protocol violations and one withdrawal); and of the 86 participants randomised to standard therapy group, 77 participants (92%) completed the study (four protocol violations and three withdrawals).
Selective reporting
The outcomes reported in the study protocol were compared with the reported results. Only the difference in cost between treatment arms was not reported; however, study investigators informed us that economic analysis would be reported in a separate paper in future. The review will monitor published literature for this promised publication.
Other potential sources of bias
No other potential sources of bias could be identified for the study.
Effects of interventions
See: Table 1
In the summary of findings tables, the quality of the evidence has been graded for pre‐defined outcomes (see above) and definitions of these gradings provided. The quality of evidence for the key parameters was graded as low except HRCT scoring and cost of care analysis, which were graded as moderate quality (Table 1).
Primary outcomes
1. Lung function
a. conventional spirometry
i. % predicted FEV1 (change and absolute values in L)
Wainwright reported z score for FEV1 and no significant difference was found between the groups for this outcome at five years (n = 157), MD ‐0.15 (95% CI ‐0.54 to 0.24) (low‐quality evidence) (Analysis 1.1).
1.1. Analysis.

Comparison 1 BAL‐directed therapy versus standard therapy, Outcome 1 Z score FEV1.
ii. % predicted FVC (change and absolute values in L)
The study reported z score for FVC at five years. There was no significant difference between the groups (n = 157), MD ‐0.05 (95% CI ‐0.44 to 0.34) (Analysis 1.2) (low‐quality evidence)
1.2. Analysis.
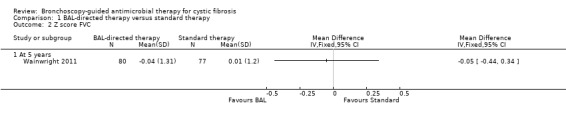
Comparison 1 BAL‐directed therapy versus standard therapy, Outcome 2 Z score FVC.
b. infant lung function‐squeeze
Infant lung function was not assessed in this study.
c. LCI
This outcome was not measured in this study.
2. HRCT appearances
Wainwright scored HRCT appearances according to an upgraded version of Brody‐II score (referred to as CF‐CT score) (Wainwright 2011). Five different components were used to calculate the total CF‐CT score; four were calculated on inspiratory images (bronchiectasis score, parenchymal disease score, mucous plugging score, airway wall thickening score) and air trapping score was assessed on expiratory images. The results were reported as percentages of the total possible score for each component and for the maximum total score. The components were scored according to the distribution of changes into the following groups: 0%, 0% to 5% and more than 5% for inspiratory images; 0%, 0% to 20% and more than 20% for expiratory images.
There was no significant difference between the groups for total CF‐CT score at five years (n = 152), MD 0.19 (95% CI ‐0.93 to 1.31) (Analysis 1.3) (moderate‐quality evidence). The scores across different components did not show any significant difference between the two groups with the risk of having a score of more than 5% for inspiratory images and more than 20% for expiratory images being the following: bronchiectasis score, MD ‐0.10 (95% CI ‐1.53 to 1.33); parenchymal disease score, MD ‐0.49 (95% CI ‐1.36 to 0.38); mucous plugging score, MD 1.38 (95% CI ‐0.36 to 3.12); airway wall thickening score, MD 0.71 (95% CI ‐0.16 to 1.58); air trapping score, MD 0.43 (95% CI ‐4.82 to 5.68) (Analysis 1.4).
1.3. Analysis.

Comparison 1 BAL‐directed therapy versus standard therapy, Outcome 3 Total CF‐CT score (Brody‐II).
1.4. Analysis.
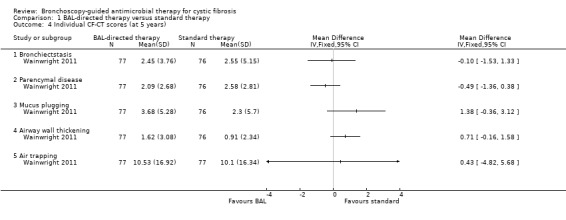
Comparison 1 BAL‐directed therapy versus standard therapy, Outcome 4 Individual CF‐CT scores (at 5 years).
3. Nutritional parameters
a. weight (in kg or percentile)
Wainwright reported z score for weight at five years (n = 157) (Wainwright 2011). There was no significant difference between the groups, MD 0.06 (95% CI ‐0.21 to 0.33) (Analysis 1.5) (low‐quality evidence).
1.5. Analysis.
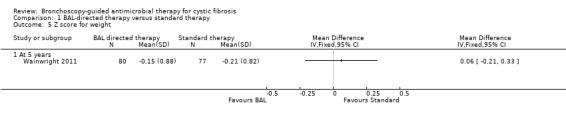
Comparison 1 BAL‐directed therapy versus standard therapy, Outcome 5 Z score for weight.
b. BMI percentile
The study found no significant difference in the z score for BMI between the two groups at five years (n = 157), MD 0.02 ( 95% CI ‐0.26 to 0.30) (Analysis 1.6) (low‐quality evidence).
1.6. Analysis.
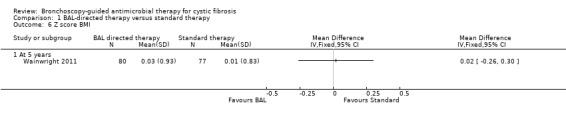
Comparison 1 BAL‐directed therapy versus standard therapy, Outcome 6 Z score BMI.
Secondary outcomes
1. Number of positive isolates per child per year of follow up
a. P aeruginosa
The number of positive isolates of P aeruginosa per participant per year of follow up was reported using the intervention specific to the randomizations, i.e. using BAL cultures for the BAL‐directed group and oropharyngeal cultures for the standard therapy group. There was no significant difference between the groups, RR 0.77 (95% CI 0.52 to 1.16) (Analysis 1.7).
1.7. Analysis.

Comparison 1 BAL‐directed therapy versus standard therapy, Outcome 7 Positive P aeruginosa isolates per patient per year.
At age five years, P aeruginosa infection was diagnosed in BAL of 8 out of 79 (10%) participants and 9 out of 76 (12%) participants from BAL directed therapy group and standard therapy groups respectively, RR 0.86 (95% CI 0.35 to 2.10) (Analysis 1.8). We conducted a sensitivity analysis after including the missing data in the respective groups and assessed the influence on P aeruginosa prevalence considering hypothetical situations where the risk of excluded participants being positive at five years was 40% for the BAL‐directed therapy group and and 5% for the standard therapy group (Analysis 1.9) and vice versa (Analysis 1.10). In both cases the result was not significant, RR 1.11 (95% CI 0.48 to 2.60) and RR 0.67 (95% CI 0.29 to 1.55) respectively.
1.8. Analysis.

Comparison 1 BAL‐directed therapy versus standard therapy, Outcome 8 Prevalence of P aeruginosa in BAL at 5 years age.
1.9. Analysis.

Comparison 1 BAL‐directed therapy versus standard therapy, Outcome 9 Sensitivity analysis ‐ Prevalence of P aeruginosa in BAL at 5 years age (40% vs 5%).
1.10. Analysis.

Comparison 1 BAL‐directed therapy versus standard therapy, Outcome 10 Sensitivity analysis ‐ Prevalence of P aeruginosa in BAL at 5 years age (5% vs 40%).
b. Other organisms
Data specific to the number of positive isolates per participant per year of follow up were not available for S aureus, H influenza, B cepacia, S maltophilia, A xylosidans, non‐tuberculous mycobacteria, aspergillus species or any other organisms.
2. Clearance of the organism from the cultures
There was no significant difference between the two groups in the proportion of children who were cleared of P aeruginosa infection following one or two courses of eradication treatment, RR 1.08 (95% CI 0.96 to 1.21) (Wainwright 2011) (Analysis 1.11).
1.11. Analysis.

Comparison 1 BAL‐directed therapy versus standard therapy, Outcome 11 Clearance of P aeruginosa after 1 or 2 eradication treatments.
3. Time to first infection with P aeruginosa
There was no statistically significant difference in the age of first acquisition of P aeruginosa infection between the two groups, RR 0.81 (95% CI 0.53 to 1.23) (Analysis 1.12).
1.12. Analysis.

Comparison 1 BAL‐directed therapy versus standard therapy, Outcome 12 Age at first acquisition of P aeruginosa infection.
4. Time to chronic infection
a. with P aeruginosa
There was no statistical analysis of the number of children who developed chronic infection in either group. Only one child in the BAL‐directed therapy group developed chronic infection (confirmation by BAL) at 56 months of age. In the standard therapy group, four children developed chronic infection (confirmed by cough swab) between 33 months and 61 months of age (however, the BAL at five years age was negative for P aeruginosa in all four participants) (Wainwright 2011).
b. with S aureus
Data specific to the time to chronic infection with S aureus were not available in this study.
5. Complications and adverse effects
At present data are not available for adverse events except those relating to BAL or use of medications. These data were collected and have been recorded, but not yet published. The review authors have requested the relevant information from the statisticians working on the included study and will include these in a future update of the review.
A number of adverse events could be attributed to both bronchoscopy and general anaesthesia. Significant clinical deterioration was noted during or following the BAL in 25 of 524 procedures (4.8%). During the procedure, seven children developed haemoglobin desaturation of less than 90% which lasted for more than 60 seconds and needed intervention. Post‐operatively, six children required supplemental oxygen, one required noninvasive ventilation and three developed stridor. The most common adverse event was transient worsening of cough noted in 96 children after 151 of 524 procedures (29%); however, this was a temporary effect and did not lead to hospitalizations. Fever with a temperature higher than 38.5°C which occurred within 24 hours of BAL was reported following 40 (7.6%) procedures and fever with a temperature below 38.5°C following 52 (9.9%) procedures. In the post‐BAL period, 12 (2.3%) participants required unplanned hospital admissions. Contamination of bronchoscope was reported on two incidences. Overall, the procedure was well‐tolerated with mild adverse effects to the participants; however, these were in addition to the inconvenience associated with hospitalizations and fasting pre‐ and post‐procedure.
6. QoL
Data specific to QoL were not available for this study.
7. Hospitalisations
a. number of hospitalisations per participant per year
The total number of hospital admissions per participant per year were higher in the BAL‐directed therapy group compared to the standard therapy group (n = 157), RR 1.40 (95% CI 1.08 to 1.82) (Analysis 1.13) (data not published but provided by the authors) (low‐quality evidence). For non‐P aeruginosa exacerbations, the children in BAL‐directed therapy group had 218 hospitalisations (0.57 per person‐year), whereas the children in the standard therapy group had fewer admissions reported as 140 admissions (0.37 per person‐year), RR 1.52 (95% CI 0.98 to 2.35) (Analysis 1.14).
1.13. Analysis.

Comparison 1 BAL‐directed therapy versus standard therapy, Outcome 13 Number of hospital admissions per patient per year.
1.14. Analysis.

Comparison 1 BAL‐directed therapy versus standard therapy, Outcome 14 Number of hospitalizations per person per year due to non‐P aeruginosa exacerbations.
b. days as inpatient per participant per year
Mean duration of hospital admissions for non‐P.aeruginosa respiratory exacerbations was reported to be significantly shorter in the BAL‐directed therapy group compared to standard therapy, MD ‐3.50 days (95% CI ‐5.67 to ‐1.33) (Analysis 1.15). However, there was no significant difference between the hospital admission days per participant per year for each group, risk difference 0.08 (95% CI ‐3.31 to 3.47) (Analysis 1.16) (data not published, provided by the authors).
1.15. Analysis.

Comparison 1 BAL‐directed therapy versus standard therapy, Outcome 15 Duration of hospital admissions due to non‐P aeruginosa exacerbations.
1.16. Analysis.
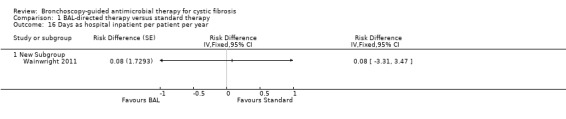
Comparison 1 BAL‐directed therapy versus standard therapy, Outcome 16 Days as hospital inpatient per patient per year.
c. cost of care
There was no difference in the total cost of care for the children in each group over the study duration (five years), MD 1902.00 Australian dollars (AUD) (95% CI 27508.98 to 31312.98) (Analysis 1.17). There was no difference in the total cost of hospital admissions per child over the five‐year study duration (n = 157) (Wainwright 2011), MD ‐9288.00 AUD (95% CI 34996.37 to 16420.37) (Analysis 1.18) (moderate‐quality evidence).
1.17. Analysis.
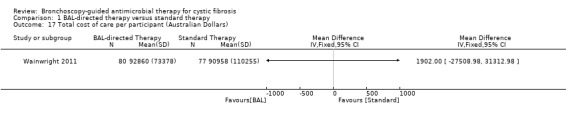
Comparison 1 BAL‐directed therapy versus standard therapy, Outcome 17 Total cost of care per participant (Australian Dollars).
1.18. Analysis.

Comparison 1 BAL‐directed therapy versus standard therapy, Outcome 18 Mean hospital admissions cost per patient.
8. Number of courses of antibiotics prescribed per participant per year (not including prophylactic antibiotics)
a. intravenous
Data for the number of courses of intravenous antibiotics prescribed per participant per year were not available.
b. oral
Data for the number of courses of oral antibiotics prescribed per participant per year were not available.
9. Number of pulmonary exacerbations (requiring oral or intravenous antibiotics) per participant per year
There was no significant difference in the number of pulmonary exacerbations diagnosed in either group, RR 1.01 (95% CI 0.85 to 1.19) (Analysis 1.19).
1.19. Analysis.
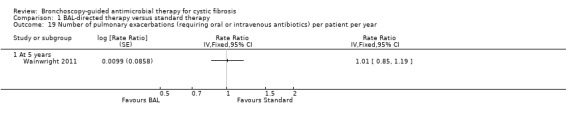
Comparison 1 BAL‐directed therapy versus standard therapy, Outcome 19 Number of pulmonary exacerbations (requiring oral or intravenous antibiotics) per patient per year.
Discussion
Summary of main results
Our review included one randomised controlled study comparing outcomes following BAL‐directed therapy with standard therapy. At age five years, there was no clear benefit of BAL‐directed therapy on lung function or nutritional parameters; and there was no significant difference between the two groups regarding evidence of structural lung disease diagnosed by HRCT scan. The number of positive isolates for P aeruginosa per child per year as well as the age at development of first infection were similar in the two groups. In addition, the overall morbidity concerning the number of pulmonary exacerbations was similar and there was no difference in the proportion of participants who cleared the infection after one or two courses of eradication therapy. A higher number of hospital admissions in the BAL‐directed therapy group was balanced by the shorter duration of hospitalisations in this group. There was no difference in the overall cost of care and cost of hospital admissions.
Overall completeness and applicability of evidence
As only one study could be included in the review, a meta‐analysis could not be conducted but this study provided highly relevant evidence to answer the review question. Although, no evidence was available for adults with CF, this study did include infants and young children, which is the group mostly unable to provide sputum samples. Low to moderate quality evidence was available for most outcomes; however, regarding the microbiological outcomes, we should be aware that two different treatment goals were compared (e.g. clearance of P aeruginosa from throat swab versus clearance from BAL). None of the four children who were defined to have developed chronic P aeruginosa infection by sampling via throat swabs, had the organism isolated from the BAL at the five‐year time‐point. While this highlights the difference in sensitivities of the methods involved, it also indicates that we should be relying more on highly sensitive clinical parameters rather than microbiological outcomes for such studies. Most adults who do not expectorate spontaneously and many, quite young, children respond to hypertonic saline to provide satisfactory sputum samples. This process is less invasive as well as more acceptable than repeated BAL. No randomised controlled studies were available which compared treatment outcomes following induced sputum with bronchoscopy and BAL from single or multiple lobes. The available evidence was mainly relevant to P aeruginosa infection. This is the most significant infection associated with CF lung disease, however, many other organisms are also increasingly being implicated in the disease progression. No study is available regarding the effect of the intervention on other organisms.
As there is no difference in the number of pulmonary exacerbations and the number of P aeruginosa isolates per participant per year between the two treatment arms, the significant difference observed with regards to the number and duration of hospital admissions appears to be mainly protocol‐driven and related to short admissions for BALs at different stages of the study. Data on the economic impact of conducting repeated bronchoscopies and frequent hospital admissions, however, did not show any difference between the two groups.
Quality of the evidence
The main strengths of the study were the quality of methods and also the low attrition rate (despite a prolonged follow‐up period and invasive intervention procedure). Though the study was set up for analysis on an intention‐to‐treat basis, it was analysed on an available‐case basis as the outcome data were available for more than 90% of participants. A sensitivity analysis for prevalence of P aeruginosa at five years of age, based on best and worst‐case scenarios, led to similar results. The prevalence of P aeruginosa and the establishment of chronic infection are clinically relevant outcomes considering the influence that chronic infection with this organism can have on progressive CF lung disease. However, this study highlights the difficulties with prolonged longitudinal studies, particularly when the outlook is changing consistently as with CF.
The main limitation of the study was its reduced statistical power for its primary microbiological outcome. The study was designed with an anticipated prevalence of P aeruginosa infection at five years of age to be 30% in the control group. The sample size of the study was revised during the study; the current sample size was predicted to provide adequate statistical power for HRCT scoring and prevalence of P aeruginosa observed during the middle of the study. However, the latter was found to be only 12% at five years age. Many other centres have also reported reduced prevalence of P aeruginosa infection and a delay in the onset of chronic infection following improved management practices and the institution of early eradication therapy against P aeruginosa. Defining time to onset of chronic infection was difficult in this study as the comparison was between different methods of diagnosis, i.e. BAL‐based cultures versus oropharyngeal cultures. The participants diagnosed with chronic infection (by using only oropharyngeal cultures to define it) were very low in number, too low to allow any meaningful comparison; only one participant in the BAL‐directed therapy group developed chronic infection compared to four in the standard therapy group. The overall quality of evidence for the key parameters was graded as low, except HRCT scoring and cost of care analysis, which were graded as moderate quality (Table 1).
Only 64% of the eligible participants could be recruited to the study; however, the age and sex distribution of the eligible participants who could not be enrolled in the study was similar to those who participated. In addition, the prevalence of bronchiectasis in the study participants was similar to that reported in other parts of Australia. These points suggest generalisability of the results to the Australian CF population.
A crucial point that can act as a confounding factor while carrying out multicentre studies is the practice of different treatment protocols at different centres. As the issue of an effect of anti‐staphylococcal prophylaxis on acquisition of P aeruginosa infection is not yet resolved, it is important to mention that participants from three of the eight centres were using oral flucloxacillin prophylactic therapy until their first birthday. However, this study randomised and stratified the participants by site to equalize the bias for both the groups, hence the aforementioned difference is unlikely in practice to have affected the validity of the comparison.
A very detailed economic analysis of the cost of care for participants in the two groups was presented which took into account various aspects such as pharmaceutical cost (both hospital and home treatment), cost of procedures and investigations, cost of hospital admission and professional attendances. Acknowledging the difficulties of obtaining such large amount of data from various centres for a period spanning half a decade, a potential confounding factor in the economic analysis was the use of assumption and estimates for costs of some aspects of care (such as cost of professional attendances or consultations from some centres). However, this was a small part of the overall picture and is likely to have been balanced over the two arms to some extent.
Potential biases in the review process
One of the review authors was also the lead author of the only included study. To avoid any influence of this fact on the interpretation of bias in the study, the remaining two review authors (KJ and AS) assessed the study for all potential sources of biases and agreed on the interpretations used to generate risk of bias graph and any related comments.
Agreements and disagreements with other studies or reviews
In many non‐randomised studies, bronchoscopy‐based cultures have been found to give a higher yield of microorganisms. However, this review, which is limited to the evidence from a single study raises several questions.
On the one hand, oropharyngeal cultures have been shown to have a low positive predictive value but a high negative predictive value for a lower airway P aeruginosa infection. Does that imply that by taking oropharyngeal swabs more frequently, we are successful at ruling out positive cases while treating (or may be over‐treating) some people who are negative?
On the other hand, BAL has been described as a 'gold standard' method to diagnose lower airway infections. However, there is still no evidence‐based answer to the question of what constitutes an infection for a BAL culture and whether the presence of any number of organisms in BAL specimens from young people who are P aeruginosa‐naïve should be considered as evidence of infection. This study used a cut‐off point of greater than 103organisms which was a balanced approach between the two extremes of detection of any organisms and greater than 105organisms. In children younger than five years age, the sensitivity of oropharyngeal cultures to detect lower respiratory infections improves as the lower limit of bacterial density to define a positive culture is increased (Rosenfeld 1999).
Could the lack of effect be related to the sensitivity of BAL for diagnosing lower airway infections? There is evidence to suggest that sampling two lobes for BAL is better than sampling only one; however, the microbiological yield is shown to improve further if samples are taken from all the lobes (Gilchrist 2011). It is uncertain whether this will make a real difference to the overall outcome for all people with CF. Analysis of the microbiology of airway cultures during pulmonary exacerbations suggests negative CF‐related bacterial cultures in a significant proportion of exacerbations. Does a negative BAL culture indicate no infection or signify the lack of ability to detect the complex range of organisms comprising the CF airway microbial community?
Eradication therapy for P aeruginosa in CF is highly effective. To achieve still greater and more prolonged efficacy in eradication, more sensitive diagnostic tests for P aeruginosa will be needed, particularly for children and individuals who do not produce sputum. The Wainwright study used HRCT score as one outcome measure, which is shown to be a highly sensitive parameter for detecting structural lung disease at the cost of increased radiation exposure. Though pre‐school children are able to provide reliable lung function results, the LCI detects abnormal lung function more readily compared to spirometry (Aurora 2005). It is shown to act in a complementary fashion to HRCT and can be a better alternative to spirometry to investigate early differences in lung function. However, even if a minor improvement in outcome was to be disclosed with these measures, it has to be enough to justify subjecting all patients to an invasive procedure with the potential risk of mild adverse effects.
Authors' conclusions
Implications for practice.
This review, limited to only one included study, shows that there is insufficient evidence to support use of BAL routinely for the diagnosis and management of pulmonary infections in pre‐school children with CF compared to the standard practice of providing treatment based on results of oropharyngeal cultures and clinical symptoms. There are no data available with regard to the adolescents and adult populations.
Implications for research.
More research needs to be oriented towards the effect of incorporating BAL at certain stages in the management of CF, e.g. in asymptomatic patients at diagnosis of CF. A comparison of outcomes following the use of other less invasive alternatives, such as induced sputum, needs to be made. Highly sensitive outcome measures like HRCT and LCI individually or a composite score of these need to be explored further. Considering the improved health of young people with CF, in order to identify any difference in outcomes, larger studies, including other age groups are required to be conducted for a longer duration of follow‐up.
What's new
| Date | Event | Description |
|---|---|---|
| 30 August 2018 | New citation required but conclusions have not changed | No new data have been added to the review so our conclusions remain the same. |
| 30 August 2018 | New search has been performed | A search of the Cochrane Cystic Fibrosis and Genetic Disorders Reveiw Group's Cystic Fibrosis Trials Register identified three references potentially eligible for inclusion in this review. Two references were additional references to the included study (Wainwright 2011) and one reference was an additional reference to an already excluded study (Chmiel 2015). The search of three registries of ongoing studies identified two references potentially eligible for inclusion in this review. Both the references were excluded from the analysis after screening (ISRCTN 12473810; NCT02363764). |
History
Protocol first published: Issue 12, 2011 Review first published: Issue 12, 2013
| Date | Event | Description |
|---|---|---|
| 14 January 2016 | New search has been performed | A search of the Cochrane Cystic Fibrosis and Genetic Disorders Group's Cystic Fibrosis Register identified an single additional reference to the already included study and added some additional economic data (Wainwright 2011). We have also added a summary of findings table. |
| 14 January 2016 | New citation required but conclusions have not changed | Since no new studies were added to the review, our conclusions remain the same. |
Acknowledgements
We would like to thank Mrs Nikki Jahnke, the Managing Editor of Cochrane Cystic Fibrosis and Genetic Disorders Group for her assistance in the development of this review.
This project was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Cochrane Cystic Fibrosis and Genetic Disorders Group. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS or the Department of Health.
Appendices
Appendix 1. Glossary of terms
| Term | Explanation |
| autosomal recessive | a type of genetic condition that requires the presence of two copies of a gene mutation (one from each parent) in order to express its features; this specifically refers to the genes on one of the 22 pairs of autosomes (non‐sex chromosomes) |
| morbidity | a disease state or poor health due to any cause |
| respiratory | refers to breathing system; includes nose, breathing passages and lungs |
| pulmonary | relating to the lungs |
| microbiology | study of microorganisms (microscopic organisms) and their effect on humans |
| oropharynx | the first part of the throat just behind the mouth which connects the mouth to the throat; includes the back of the tongue, the back of the roof of the mouth, tonsils and the back wall of the throat |
| endoscopy |
a medical procedure to see inside the body using a long tubular instrument with a camera at the end (called endoscope); depending on which part of the body is to be seen, there are different names for it such as laparoscopy, bronchoscopy, colonoscopy etc. ‐ bronchoscopy is the procedure to look inside the airways (breathing tract) |
| pathological | pertaining to any disease or deviation from health |
Appendix 2. Additional searches
| Database/Resource | Search terms | Latest date searched |
| clinicaltrials.gov (https://clinicaltrials.gov/) |
cystic fibrosis AND bronchoscopy, cystic fibrosis AND bronchoalveolar lavage, cystic fibrosis AND sampling technique | 10/04/2018 |
| International Standard Randomised Controlled Trial Number (ISRCTN) Register (www.isrctn.org) |
cystic fibrosis AND bronchoscopy, cystic fibrosis AND bronchoalveolar lavage, cystic fibrosis AND sampling technique | 10/04/2018 |
| WHO ICTRP (http://apps.who.int/trialsearch/) |
cystic fibrosis AND bronchoscopy, cystic fibrosis AND bronchoalveolar lavage, cystic fibrosis AND sampling technique | 10/04/2018 |
Data and analyses
Comparison 1. BAL‐directed therapy versus standard therapy.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Z score FEV1 | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 1.1 At 5 years | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Z score FVC | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 2.1 At 5 years | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Total CF‐CT score (Brody‐II) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 3.1 At 5 years | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Individual CF‐CT scores (at 5 years) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 4.1 Bronchiectstasis | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 Parencymal disease | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 Mucus plugging | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.4 Airway wall thickening | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.5 Air trapping | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Z score for weight | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 5.1 At 5 years | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Z score BMI | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 6.1 At 5 years | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7 Positive P aeruginosa isolates per patient per year | 1 | Rate Ratio (Fixed, 95% CI) | Totals not selected | |
| 7.1 At 5 years | 1 | Rate Ratio (Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Prevalence of P aeruginosa in BAL at 5 years age | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 9 Sensitivity analysis ‐ Prevalence of P aeruginosa in BAL at 5 years age (40% vs 5%) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 10 Sensitivity analysis ‐ Prevalence of P aeruginosa in BAL at 5 years age (5% vs 40%) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 11 Clearance of P aeruginosa after 1 or 2 eradication treatments | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 11.1 At 5 years | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 12 Age at first acquisition of P aeruginosa infection | 1 | Rate Ratio (Fixed, 95% CI) | Totals not selected | |
| 12.1 At 5 years | 1 | Rate Ratio (Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 13 Number of hospital admissions per patient per year | 1 | Rate Ratio (Fixed, 95% CI) | Totals not selected | |
| 13.1 At 5 years | 1 | Rate Ratio (Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 14 Number of hospitalizations per person per year due to non‐P aeruginosa exacerbations | 1 | Rate Ratio (Fixed, 95% CI) | Totals not selected | |
| 14.1 At 5 years | 1 | Rate Ratio (Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 15 Duration of hospital admissions due to non‐P aeruginosa exacerbations | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 15.1 New Subgroup | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 16 Days as hospital inpatient per patient per year | 1 | Risk Difference (Fixed, 95% CI) | Totals not selected | |
| 16.1 New Subgroup | 1 | Risk Difference (Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 17 Total cost of care per participant (Australian Dollars) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 18 Mean hospital admissions cost per patient | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 19 Number of pulmonary exacerbations (requiring oral or intravenous antibiotics) per patient per year | 1 | Rate Ratio (Fixed, 95% CI) | Totals not selected | |
| 19.1 At 5 years | 1 | Rate Ratio (Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Wainwright 2011.
| Methods | Multicentre (8 CF centres in Australia and New Zealand), randomised controlled study. | |
| Participants | 170 Infants younger than 6 months age, with confirmed diagnosis of CF, diagnosed through newborn screening programs. 84 infants were randomised to receive BAL‐directed therapy (80 completed study) and 86 randomised to receive standard therapy (77 completed study). BAL‐directed group Mean age (SD) 3.8 (1.6) years. Gender split: 44 male/40 female. Mean (SD) weight at enrolment: 5.7 kg (1.40). Number of participants with homozygous ΔF508 mutation: 57 (68%). Number of participants with pancreatic insufficiency: 73 (87%). Number of participants with meconium ileus: 17 (20%). Number of participants born pre‐term (under 37 week gestation): 8 (10%). History of exposure to tobacco smoke during pregnancy present in: 22 (26%). History of concurrent smoking in the household present in: 30 (36%). Standard therapy group Mean age (SD) 3.7 (1.7) years. Gender split: 44 male/42 female. Mean weight (SD) at enrolment: 5.6 kg (1.5). Number of participants with homozygous ΔF508 mutation: 54 (64%). Number of participants with pancreatic insufficiency: 71 (85%). Number of participants with meconium ileus: 16 (19%). Number of participants born pre‐term (under 37‐week gestation): 9 (11%). History of exposure to tobacco smoke during pregnancy present in: 13 (15%). History of concurrent smoking in the household present in: 23 (28%). |
|
| Interventions |
Intervention: BAL‐directed therapy for pulmonary exacerbations until age 5 years. The participants in the BAL‐directed therapy groups underwent BAL at following times:
Control: Standard therapy (directed by clinical features and oropharyngeal swab cultures) for pulmonary exacerbations until age 5 years. The standard therapy included oropharyngeal swab at following time points:
|
|
| Outcomes | Reported at at 5 years age. Primary outcome measures
Secondary outcome measures
|
|
| Notes | The BAL‐directed therapy group had BAL before age 6 months when well, when hospitalised for pulmonary exacerbations, when P aeruginosa was cultured from their oropharyngeal specimens and following P aeruginosa therapy. The standard therapy included taking oropharyngeal swabs when having pulmonary exacerbation and at the end of antibiotic therapy. Children in both groups had BAL and HRCT scan of chest at 5 years age. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | After consent, the participants were randomly assigned in 1:1 ratio to 2 groups by a centralized computer‐generated schedule with stratification by site and sex. |
| Allocation concealment (selection bias) | Low risk | The allocation was done by a centralized computer‐generated schedule; the randomisation key was concealed and held remotely. Allocation was revealed by telephone after confirmed recruitment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Although the study was set up to be analysed on intention‐to‐treat basis, the participants with missing outcomes were not included in the primary analysis. The risk of bias is considered moderate to low as less than 10% of the data were missing and the reasons of exclusions were balanced across both groups. |
| Selective reporting (reporting bias) | Low risk | The economic analysis was planned but was not reported initially but now has been published. It has been included in the updated version of the review. All other outcomes planned to be assessed in the protocol were reported. |
| Other bias | Low risk | No other potential source of bias was identified. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | The participants and the personnel were not blinded to the randomisation (which might not have been possible in this study setting). However, the risk of bias is low as the primary outcome measures were unlikely to be influenced by the lack of blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Risk of bias is low as the outcome assessors were blinded for both the primary outcome measures. |
BAL: bronchoalveolar lavage BMI: body mass index CF: cystic fibrosis CFU: colony forming units CT: computer tomography P aeruginosa: Pseudomonas aeruginosa SD: standard deviation
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Chmiel 2015 | Study of induced sputum and not bronchoscopy. |
| Henig 2001 | 3‐way cross‐over study of single sample from sputum induction, bronchoalveolar lavage and expectorated sputum to identify pathogens; did not lead to comparison of treatment. |
| ISRCTN 12473810 | Non‐randomized comparison of pathogen yield by different sampling methods; no comparison of treatments or clinical outcomes |
| Jyothish 2005 | A different intervention (cough plates) was studied. |
| Maiya 2004 | A different intervention (cough plates) was studied. |
| McGarvey 2002 | 2‐way cross‐over study of induced sputum and BAL to compare inflammatory markers; no comparison of treatment. |
| NCT02363764 | Non‐randomized comparison of microbiological yield of different sampling methods; did not lead to comparison of treatments |
| Paul 2004 | Study of the effect of dornase alfa on lungs using bronchoalveolar lavage; all participants underwent bronchoalveolar lavage. |
| Rosenfeld 2006 | Study to establish levels of tobramycin, not comparison of therapy depending on sampling technique. |
| Taylor 2006 | Comparison of throat swabs and nasopharyngeal suction specimens not bronchoscopy. |
Differences between protocol and review
2016 update
In line with Cochrane guidance, we added a summary of findings table to the 2016 update.
Contributions of authors
| Roles and responsibilities | |
| TASK | WHO WILL UNDERTAKE THE TASK? |
| Protocol stage: draft the protocol | KJ, CW, AS |
| Review stage: select which studies to include (2 + 1 arbiter) | KJ, CW, AS |
| Review stage: extract data from studies (2 people) | KJ, CW |
| Review stage: enter data into RevMan | KJ |
| Review stage: carry out the analysis | KJ |
| Review stage: interpret the analysis | KJ, CW, AS |
| Review stage: draft the final review | KJ, CW, AS |
| Update stage: update the review | KJ, CW, AS |
Sources of support
Internal sources
No sources of support supplied
External sources
-
National Institute for Health Research, UK.
This systematic review was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Cochrane Cystic Fibrosis and Genetic Disorders Group.
Declarations of interest
Kamini Jain declares no known potential conflict of interest.
Claire Wainwright has received research grants from the NHMRC, GlaxoSmithKline and Novo Nordisk Pharmaceuticals P/L. Her institution has received income on a per patient basis from pharmaceutical studies (Vertex Pharmaceuticals Inc., Boehringer‐Ingelheim & Ablynx NV). Dr Wainwright has been a consultant on the Vertex Physician Pediatric CF Advisory Board and the Vertex Innovation Awards (VIA) Grants Committee and is currently on the board of the International Advisory Board Vertex Pharmaceuticals P/L. She has received travel and accommodation expenses from Vertex Pharmaceuticals Inc and Novartis Pharmaceuticals. Payments for all items listed above have been made into an institutional consultancy fund account.
Alan R Smyth declares relevant activities of membership of consultancies for Vertex and PTC, and lectures paid for by PTC, Vertex, Teva, Novartis.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Wainwright 2011 {published data only}
- Byrnes CA, Vidmar S, Cheney JL, Carlin JB, Armstrong DS, Cooper PJ, et al. Prospective evaluation of respiratory exacerbations in children with cystic fibrosis from newborn screening to 5 years of age. Thorax 2013;68(7):643‐51. [CFGD Register: PE167i] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheney J, Vidmar S, Grimwood K, Carlin JB, Wainwright C, on behalf of ACFBAL Study Group. Interim outcomes of a Pseudomonas aeruginosa (Pa) eradication protocol in young children in the Australian Cystic Fibrosis Bronchoalveolar Lavage (ACFBAL) Study. Journal of Cystic Fibrosis 2009;8(Suppl 2):S39. [Abstract no.: 157; CFGD Register: PE167c] [Google Scholar]
- Cheney J, Wainwright C, ACFBAL SG. Trials, tribulations and triumphs of a cystic fibrosis study ‐ a behind the scenes look at the workings of an international multi‐centre study. Journal of Cystic Fibrosis 2010;9 Suppl 1:S118. [Abstract no.: 452; CFGD Register: PE167g; ] [Google Scholar]
- Kidd TJ, Ramsay KA, Vidmar S, Carlin JB, Bell SC, Wainwright CE, et al. Pseudomonas aeruginosa genotypes acquired by children with cystic fibrosis by age 5 years. Journal of Cystic Fibrosis 2015;14(3):361‐9. [CFGD Register: PE167k] [DOI] [PubMed] [Google Scholar]
- Moodie M, Lal A, Vidmar S, Armstrong DS, Byrnes CA, Carlin JB, et al. Costs of bronchoalveolar lavage‐directed therapy in the first 5 years of life for children with cystic fibrosis. Journal of Pediatrics 2014;165(3):564‐9. [CFGD Register: PE167j; CRS: 5500131000000365; JID:: 0375410; PUBMED: 24996984; SI:: ANZCTR/ACTRN12605000665639] [DOI] [PubMed] [Google Scholar]
- Wainwright C, Carlin J, Cooper P, Byrnes C, Martin J, Grimwood K, et al. Australasian cystic fibrosis BAL study interim analysis. Pediatric Pulmonology 2006;41 Suppl 29:317. [CFGD Register: PE167a] [Google Scholar]
- Wainwright CE, Carlin J, Cooper P, Byrnes C, Whitehead B, Martin J, et al. Early infection with Pseudomonas aeruginosa can be cleared in young children with cystic fibrosis. Pediatric Pulmonology 2002;34 Suppl 24:300‐1. [CFGD Register: PE167d] [Google Scholar]
- Wainwright CE, Kidd TJ, Ramsey KA, Bell SC, Grimwood K. Australasian CF bronchoalveolar lavage (ACFBAL) study: P.Aeruginosa (PA) genotypes in pre‐school CF children. Pediatric Pulmonology 2011;46 Suppl 34:320. [Abstract no.: 299; CFGD Register: PE167h; ] [Google Scholar]
- Wainwright CE, Vidmar S, Armstrong DS, Byrnes CA, Carlin JB, Cheney J, et al. Effect of bronchoalveolar lavage‐directed therapy on Pseudomonas aeruginosa infection and structural lung injury in children with cystic fibrosis: a randomized trial. JAMA 2011;306(2):163‐71. [CFGD Register: PE167e] [DOI] [PubMed] [Google Scholar]
- Wainwright CE, Vidmar S, Armstrong DS, Byrnes CA, Carlin JB, Cheney J, et al. Online Supplement to 'Effect of bronchoalveolar lavage‐directed therapy on Pseudomonas aeruginosa infection and structural lung injury in children with cystic fibrosis: a randomized trial' [online]. JAMA 2011;306(2):163‐171 Online. [CFGD Register: PE167f; ] [DOI] [PubMed] [Google Scholar]
- Wainwright CE, Grimwood K, Carlin JB, Vidmar S, Cooper PJ, Francis PW, et al. Safety of bronchoalveolar lavage in young children with cystic fibrosis. Pediatric Pulmonology 2008;43(10):965‐72. [CFGD Register: PE167b] [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Chmiel 2015 {published data only}
- Chmiel JF, Konstan MW, Accurso FJ, Lymp J, Mayer‐Hamblett N, VanDevanter DR, et al. Use of ibuprofen to assess inflammatory biomarkers in induced sputum: implications for clinical trials in cystic fibrosis. Journal of Cystic Fibrosis 2015;14(6):720‐6. [CFGD Register: PI218b] [DOI] [PubMed] [Google Scholar]
- Chmiel JF, Konstan MW, Lymp J, Mayer‐Hamblett N, Hilliard KA, Accurso FJ, et al. Assessment of induced sputum as a tool to evaluate anti‐inflammatory agents in CF. Pediatric Pulmonology 2007;42 Suppl 30:228. [CFGD Register: PI218a] [Google Scholar]
Henig 2001 {published data only}
- Henig NR, Tonelli MR, Pier MV, Burns JL, Aitken ML. Sputum induction as a research tool for sampling the airways of subjects with cystic fibrosis. Pediatric Pulmonology 1999;28 Suppl 19:312‐3. [CFGD Register: PI225] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henig NR, Tonelli MR, Pier MV, Burns JL, Aitken ML. Sputum induction as a research tool for sampling the airways of subjects with cystic fibrosis. Thorax 2001;56(4):306‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
ISRCTN 12473810 {unpublished data only}
- ISRCTN 12473810. Induced sputum in children with cystic fibrosis. www.isrctn.com/ISRCTN12473810 (first received 03 January 2018).
Jyothish 2005 {published data only}
- Jyothish D, Desai M, Clarke JR, Weller PH, Gray J. Comparison of Cough Plates with Cough Swabs for identifying lower respiratory tract pathogens in children with Cystic Fibrosis. American Thoracic Society International Conference; 2005 May 20‐25; San Diego, USA. 2005:C16. [CFGD Register: PI192a]
- Jyothish D, Desai M, Clarke JR, Weller PH, Gray J. Use of cough plates for identifying lower respiratory tract pathogens in children with cystic fibrosis. Journal of Cystic Fibrosis 2005;4 Suppl:S40. [CFGD Register: PI192b] [Google Scholar]
Maiya 2004 {published data only}
- Maiya S, Desai M, Baruah A, Clarke J, Weller P, Gray J. Cough plate versus cough swab. Journal of Cystic Fibrosis 2002;1 Suppl 1:S122. [CFGD Register: PI168a] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiya S, Desai M, Baruah A, Weller P, Clarke JR, Gray J. Cough plate versus cough swab in patients with cystic fibrosis; a pilot study. Archives of Disease in Childhood 2004;89(6):577‐9. [CFGD Register: PI168b] [DOI] [PMC free article] [PubMed] [Google Scholar]
McGarvey 2002 {published data only}
- McGarvey LPA, Dunbar K, Martin SL, Brown V, MacMahon J, Ennis M, et al. Cytokine concentrations and neutrophil elastase activity in bronchoalveolar lavage and induced sputum from patients with cystic fibrosis, mild asthma and healthy volunteers. Journal of Cystic Fibrosis 2002; Vol. 1, issue 4:269‐75. [CFGD Register: PI217] [DOI] [PubMed]
NCT02363764 {unpublished data only}
- NCT02363764. The challenge of obtaining qualitative bacterial cultures in non‐expectorating cystic fibrosis patients. clinicaltrials.gov/ct2/show/NCT02363764 (first received 16 February 2016).
Paul 2004 {published data only}
- Griese M, Essl R, Schmidt R, Ballmann M, Paul K, Rietschel E, et al. Sequential analysis of surfactant, lung function and inflammation in cystic fibrosis patients. Respiratory Research 2005;6:133. [CFGD Register: IB21i] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung A, Shute JK, Chen CIU, Ballmann M, Griese M, Ratjen F, et al. Influence of long‐term inhaled rhDNase on free and total IL‐8 concentration in BAL fluid in cystic fibrosis. 12th European Respiratory Society Annual Congress; 2002 Sept 14‐18; Stockholm. 2002:P3285. [CFGD Register: IB21d]
- Paul K, Ballmann M, Griese M, Rietschel E, Chen C, Schink T, et al. Effect of rhDNase on endobronchial inflammation in CF patients with mild lung disease: Results of the multi‐center BEAT study. Pediatric Pulmonology 2002;34 Suppl 24:310‐1. [CFGD Register: IB21c] [Google Scholar]
- Paul K, Rietschel E, Ballmann M, Griese M, Worlitzsch D, Shute J, et al. Effect of treatment with dornase alpha on airway inflammation in patients with cystic fibrosis. American Journal of Respiratory and Critical Care Medicine 2004;169(6):719‐25. [CFGD Register: IB21f] [DOI] [PubMed] [Google Scholar]
- Paul KP, Ballmann M, Döring G, Griese M, Kleinau I, Ratjen F, et al. Airway inflammation in CF patients with good pulmonary function ‐ baseline data from the multicenter "BEAT" study. Pediatric Pulmonology 1998;26 Suppl 17:383‐4. [CFGD Register: IB21a] [Google Scholar]
- Paul KP, Rietschel E, Ballmann M, Griese M, Chen CI, Schink T, et al. Longitudinal evaluation of airway inflammation in stable cystic fibrosis (CF) patients by bronchoalveolar lavage (BAL) and influence of rhDNase. Proceedings of ATS 99th International Conference, May 16‐21, Seattle, USA. 2003:D041 [Poster: E42]. [CFGD Register: IB21e]
- Ratjen F, Griese M, Ballmann M, Rietschel E, Paul K, Beat Study Group. Dornase alpha in pediatric CF patients with normal lung function ‐ effects on airway inflammation. Pediatric Pulmonology 2005;40 Suppl 28:143. [CFGD Register: IB21h] [Google Scholar]
- Ratjen F, Paul K, Rietschel E, Nikolaizik W. Treatment with rhDNase reduces DNA levels in BAL fluid of CF patients with mild lung disease [abstract]. Pediatric Pulmonology 2002;34 Suppl 24:310. [CFGD Register: IB21b] [Google Scholar]
- Ratjen F, Paul K, Koningsbruggen S, Breitenstein S, Rietschel E, Nikolaizik W. DNA concentrations in BAL fluid of cystic fibrosis patients with early lung disease: Influence of treatment with dornase alpha. Pediatric Pulmonology 2005;39(1):1‐4. [CFGD Register: IB21g] [DOI] [PubMed] [Google Scholar]
Rosenfeld 2006 {published data only}
- Rosenfeld M, Emerson J, Uh D, Anderson G, Genatossio A, McNamara S, et al. Does tobramycin accumulate in respiratory secretions with repeated aerosol administration: a pilot study. Pediatric Pulmonology 2006;41 Suppl 29:327. [CFGD Register: PI203] [Google Scholar]
Taylor 2006 {published data only}
- Taylor L, Corey M, Matlow A, Geremia J, Sweezey N. Nasopharyngeal auger suction vs throat swab cultures in CF patients [abstract]. Pediatric Pulmonology 1998;26 Suppl 17:399. [CFGD Register: PI147a] [Google Scholar]
- Taylor L, Corey M, Matlow A, Sweezey NB, Ratjen F. Comparison of throat swabs and nasopharyngeal suction specimens in non‐sputum‐producing patients with cystic fibrosis. Pediatric Pulmonology 2006;41(9):839‐43. [CFGD Register: PI147b] [DOI] [PubMed] [Google Scholar]
Additional references
Armstrong 1995
- Armstrong DS, Grimwood K, Carzino R, Carlin JB, Olinsky A, Phelan PD. Lower respiratory infection and inflammation in infants with newly diagnosed cystic fibrosis. BMJ 1995;310(6994):1571‐2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Aurora 2005
- Aurora P, Bush A, Gustafsson P, Oliver C, Wallis C, Price J, et al. Multiple‐breath washout as a marker of lung disease in preschool children with cystic fibrosis. American Journal of Respiratory and Critical Care Medicine 2005;171(3):249‐56. [DOI] [PubMed] [Google Scholar]
Baughman 1997
- Baughman RP, Keeton DA, Perez C, Wilmott RW. Use of bronchoalveolar lavage semi‐quantitative cultures in cystic fibrosis. American Journal of Respiratory and Critical Care Medicine 1997;156(1):286‐91. [DOI] [PubMed] [Google Scholar]
Bobadilla 2002
- Bobadilla JL, Macek M Jr, Fine JP, Farrell PM. Cystic fibrosis: a worldwide analysis of CFTR mutations‐‐correlation with incidence data and application to screening. Human Mutation 2002;19(6):575‐606. [DOI] [PubMed] [Google Scholar]
Brody 2004
- Brody AS, Klein JS, Molina PL, Quan J, Bean JA, Wilmott RW. High‐resolution computed tomography in young patients with cystic fibrosis: distribution of abnormalities and correlation with pulmonary function tests. Journal of Pediatrics 2004;145(1):32‐8. [DOI] [PubMed] [Google Scholar]
Dankert‐Roelse 2009
- Dankert‐Roelse JE, Merelle ME. Review of outcomes of neonatal screening for cystic fibrosis versus non‐screening in Europe. Journal of Pediatrics 2009;147(3 Suppl):S15‐20. [DOI] [PubMed] [Google Scholar]
de Blic 2002
- Blic J, Marchac V, Scheinmann P. Complications of flexible bronchoscopy in children: prospective study of 1,328 procedures. European Respiratory Journal 2002;20(5):1271‐6. [DOI] [PubMed] [Google Scholar]
Deeks 2011
- Deeks J, Higgins J, Altman D. Chapter 9 Analysing data and undertaking meta‐analysis. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Dodge 2007
- Dodge JA, Lewis PA, Stanton M, Wilsher J. Cystic fibrosis mortality and survival in the UK: 1947‐2003. European Respiratory Journal 2007;29(3):522‐6. [DOI] [PubMed] [Google Scholar]
Farrell 1997a
- Farrell PM, Kosorok MR, Laxova A, Shen G, Koscik RE, Bruns WT, et al. Nutritional benefits of neonatal screening for cystic fibrosis. Wisconsin Cystic Fibrosis Neonatal Screening Study Group. New England Journal of Medicine 1997;337(14):963‐9. [DOI] [PubMed] [Google Scholar]
Farrell 1997b
- Farrell PM, Shen G, Splaingard M, Colby CE, Laxova A, Kosorok MR, et al. Acquisition of Pseudomonas aeruginosa in children with cystic fibrosis. Pediatrics 1997;100(5):E2. [DOI: 10.1542/peds.100.5.e2] [DOI] [PubMed] [Google Scholar]
Frederiksen 1997
- Frederiksen B, Koch C, Hoiby N. Antibiotic treatment of initial colonization with Pseudomonas aeruginosa postpones chronic infection and prevents deterioration of pulmonary function in cystic fibrosis. Pediatric Pulmonology 1997;23(5):330‐5. [DOI] [PubMed] [Google Scholar]
Gee 2000
- Gee L, Abbott J, Conway S, Etherington C, Webb A. Development of a disease specific health related quality of life measure for adults and adolescents with cystic fibrosis. Thorax 2000;55(11):946‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
Gibson 2003
- Gibson RL, Burns JL, Ramsey BW. Pathophysiology and management of pulmonary infections in cystic fibrosis. American Journal of Respiratory and Criticial Care Medicine 2003;168(8):918‐51. [DOI] [PubMed] [Google Scholar]
Gilchrist 2011
- Gilchrist FJ, Salamat S, Clayton S, Peach J, Alexander J, Lenney W. Bronchoalveolar lavage in children with cystic fibrosis: how many lobes should be sampled?. Archives of Disease in Childhood 2011;96(3):215‐7. [DOI] [PubMed] [Google Scholar]
Hansen 2008
- Hansen CR, Pressler T, Hoiby N. Early aggressive eradication therapy for intermittent Pseudomonas aeruginosa airway colonization in cystic fibrosis patients: 15 years experience. Journal of Cystic Fibrosis 2008;7(6):523‐30. [DOI] [PubMed] [Google Scholar]
Heim 2001
- Heim RA, Sugarman EA, Allitto BA. Improved detection of cystic fibrosis mutations in the heterogeneous U.S. population using an expanded, pan‐ethnic mutation panel. Genetics in Medicine: Official Journal of the American College of Medical Genetics 2001;3(3):168‐76. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327(7414):557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011a
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Higgins 2011b
- Higgins JPT, Altman DG, Sterne JAC on behalf of the Cochrane Statistical Methods Group and the Cochrane Bias Methods Group (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Hilliard 2007
- Hilliard TN, Sukhani S, Francis J, Madden N, Rosenthal M, Balfour‐Lynn I, et al. Bronchoscopy following diagnosis with cystic fibrosis. Archives of Disease in Childhood 2007;92(10):898‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ikeda 1968
- Ikeda S, Yani N, Ishikawa S. Flexible bronchofiberscope. Keio Journal of Medicine 1968;17(1):1‐16. [DOI] [PubMed] [Google Scholar]
Khan 1995
- Khan TZ, Wagener JS, Bost T, Martinez J, Accurso FJ, Riches DW. Early pulmonary inflammation in infants with cystic fibrosis. American Journal of Respiratory and Critical Care Medicine 1995;151(4):1075‐82. [DOI] [PubMed] [Google Scholar]
Langton Hewer 2017
- Langton Hewer SC, Smyth AR. Antibiotic strategies for eradicating Pseudomonas aeruginosa in people with cystic fibrosis. Cochrane Database of Systematic Reviews 2017, Issue 4. [DOI: 10.1002/14651858.CD004197.pub5] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lee 2003
- Lee TW, Brownlee KG, Conway SP, Denton M, Littlewood JM. Evaluation of a new definition for chronic Pseudomonas aeruginosa infection in cystic fibrosis patients. Journal of Cystic Fibrosis 2003;2(1):29‐34. [DOI] [PubMed] [Google Scholar]
Li 2005
- Li Z, Kosorok MR, Farrell PM, Laxova A, West SE, Green CG, et al. Longitudinal development of mucoid Pseudomonas aeruginosa infection and lung disease progression in children with cystic fibrosis. JAMA 2005;293(5):581‐8. [DOI] [PubMed] [Google Scholar]
Loeve 2009
- Loeve M, Hal PT, Robinson P, Jong PA, Lequin MH, Hop WC, et al. The spectrum of structural abnormalities on CT scans from patients with CF with severe advanced lung disease. Thorax 2009;64(10):876‐82. [DOI] [PubMed] [Google Scholar]
Moher 2009
- Moher D, Liberati A, Tetzlaff J, Altman DG, The PRISMA Group. Preferred Reporting Items for Systematic Reviews and Meta‐Analyses: The PRISMA Statement. Journal of Clinical Epidemiology 2009;62(10):1006‐12. [DOI] [PubMed] [Google Scholar]
Nixon 2001
- Nixon GM, Armstrong DS, Carzino R, Carlin JB, Olinsky A, Robertson CF, et al. Clinical outcome after early Pseudomonas aeruginosa infection in cystic fibrosis. Journal of Pediatrics 2001;138(5):699‐704. [DOI] [PubMed] [Google Scholar]
Nussbaum 2002
- Nussbaum E. Pediatric fiberoptic bronchoscopy: Clinical experience with 2,836 bronchoscopies. Pediatric Critical Care Medicine 2002;3(2):171‐6. [DOI] [PubMed] [Google Scholar]
Picard 2000
- Picard E, Schwartz S, Goldberg S, Glick T, Villa Y, Kerem E. A prospective study of fever and bacteremia after flexible fiberoptic bronchoscopy in children. Chest 2000;117(2):573‐7. [DOI] [PubMed] [Google Scholar]
Quittner 2009
- Quittner AL, Modi AC, Wainwright C, Otto K, Kirihara J, Montgomery AB. Determination of the minimal clinically important difference scores for the Cystic Fibrosis Questionnaire‐Revised respiratory symptom scale in two populations of patients with cystic fibrosis and chronic Pseudomonas aeruginosa airway infection. Chest 2009;135(6):1610‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Rabin 2004
- Rabin HR, Butler SM, Wohl ME, Geller DE, Colin AA, Schidlow DV, et al. Pulmonary exacerbations in cystic fibrosis. Pediatric Pulmonology 2004;37(5):400‐6. [DOI] [PubMed] [Google Scholar]
Rosenfeld 1999
- Rosenfeld M, Emerson J, Accurso F, Armstrong D, Castile R, Grimwood K, et al. Diagnostic accuracy of oropharyngeal cultures in infants and young children with cystic fibrosis. Pediatric Pulmonology 1999;28(5):321‐8. [DOI] [PubMed] [Google Scholar]
Rosenfeld 2001
- Rosenfeld M, Emerson J, Williams‐Warren J, Pepe M, Smith A, Montgomery AB, et al. Defining a pulmonary exacerbation in cystic fibrosis. Journal of Pediatrics 2001;139(3):359‐65. [DOI] [PubMed] [Google Scholar]
Rosenstein 1998
- Rosenstein BJ, Cutting GR. The diagnosis of cystic fibrosis: a consensus statement. Cystic Fibrosis Foundation Consensus Panel. Journal of Pediatrics 1998;132(4):589‐95. [DOI] [PubMed] [Google Scholar]
Sly 2009
- Sly PD, Brennan S, Gangell C, Klerk N, Murray C, Mott L, et al. Lung disease at diagnosis in infants with cystic fibrosis detected by newborn screening. American Journal of Respiratory and Critical Care Medicine 2009;180(2):146‐52. [DOI] [PubMed] [Google Scholar]
Southern 2009
- Southern KW, Merelle Marieke ME, Dankert‐Roelse JE, Nagelkerke AD. Newborn screening for cystic fibrosis. Cochrane Database of Systematic Reviews 2009, Issue 1. [DOI: 10.1002/14651858.CD001402.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Stafler 2011
- Stafler P, Davies J C, Balfour‐Lynn I M, Rosenthal M, Bush A. Bronchoscopy in cystic fibrosis infants diagnosed by newborn screening. Pediatric Pulmonology 2011;46(7):696‐700. [DOI] [PubMed] [Google Scholar]
Stuart 2010
- Stuart B, Lin J H, Mogayzel P J Jr. Early eradication of Pseudomonas aeruginosa in patients with cystic fibrosis. Paediatric Respiratory Reviews 2010;11(3):177‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
UK CF Trust 2004
- UK CF Trust. Pseudomonas aeruginosa infection in people with cystic fibrosis. Report of UK CF Trust Infection Control group 2004.
Wood 1978
- Wood RE, Fink RJ. Applications of flexible fiberoptic bronchoscopes in infants and children. Chest 1978;73(5 Suppl):737‐40. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Jain 2013
- Jain K, Wainwright C, Smyth AR. Bronchoscopy‐guided antimicrobial therapy for cystic fibrosis. Cochrane Database of Systematic Reviews 2013, Issue 12. [DOI: 10.1002/14651858.CD009530.pub2] [DOI] [PubMed] [Google Scholar]
Jain 2016
- Jain K, Wainwright C, Smyth AR. Bronchoscopy‐guided antimicrobial therapy for cystic fibrosis. Cochrane Database of Systematic Reviews 2016, Issue 1. [DOI: 10.1002/14651858.CD009530.pub3] [DOI] [PubMed] [Google Scholar]