

Abstract
Background
Impaired mucociliary clearance characterises lung disease in cystic fibrosis (CF). Hypertonic saline enhances mucociliary clearance and may lessen the destructive inflammatory process in the airways. This is an update of a previously published review.
Objectives
To investigate efficacy and tolerability of treatment with nebulised hypertonic saline on people with CF compared to placebo and or other treatments that enhance mucociliary clearance.
Search methods
We searched the Cochrane Cystic Fibrosis and Genetic Disorders Group's Cystic Fibrosis Trials Register, comprising references identified from comprehensive electronic database searches, handsearches of relevant journals and abstract books of conference proceedings. We also searched ongoing trials databases.
Date of most recent searches: 08 August 2018.
Selection criteria
Randomised and quasi‐randomised controlled trials assessing hypertonic saline compared to placebo or other mucolytic therapy, for any duration or dose regimen in people with CF (any age or disease severity).
Data collection and analysis
Two authors independently reviewed all identified trials and data, and assessed trial quality. The quality of the evidence was assessed using GRADE.
Main results
A total of 17 trials (966 participants, aged 4 months to 63 years) were included; 19 trials were excluded, three trials are ongoing and 16 are awaiting classification. We judged 14 of the 17 included trials to have a high risk of bias due to participants ability to discern the taste of the solutions.
Hypertonic saline 3% to 7% versus placebo
At four weeks, we found very low‐quality evidence from three placebo‐controlled trials (n = 225) that hypertonic saline (3% to 7%, 10 mL twice‐daily) increased the mean change from baseline of the forced expiratory volume at one second (FEV1) (% predicted) by 3.44% (95% confidence interval (CI) 0.67 to 6.21), but there was no difference between groups in lung clearance index in one small trial (n = 10). By 48 weeks the effect was slightly smaller in one trial (n = 134), 2.31% (95% CI ‐2.72 to 7.34) (low‐quality evidence). No deaths occurred in the trials. Two trials reporting data on exacerbations were not combined as the age difference between the participants in the trials was too great. One trial (162 adults) found 0.5 fewer exacerbations requiring antibiotics per person in the hypertonic saline group; the second trial (243 children, average age of two years) found no difference between groups (low‐quality evidence). There was insufficient evidence reported across the trials to determine the rate of different adverse events such as cough, chest tightness, tonsillitis and vomiting (very low‐quality evidence). Four trials (n = 80) found very low‐quality evidence that sputum clearance was better with hypertonic saline.
A further trial was performed in adults with an acute exacerbation of lung disease (n = 132). The effects of hypertonic saline on short‐term lung function, 5.10% higher (14.67% lower to 24.87% higher) and the time to the subsequent exacerbation post‐discharge, hazard ratio 0.86 (95% CI 0.57 to 1.30) are uncertain (low‐quality evidence). No deaths were reported. Cough and wheeze were reported but no serious adverse events (very low‐quality evidence).
Hypertonic saline versus mucus mobilising treatments
Three trials compared a similar dose of hypertonic saline to recombinant deoxyribonuclease (rhDNase); two (61 participants) provided data for inclusion in the review. There was insufficient evidence from one three‐week trial (14 participants) to determine the effects of hypertonic saline on FEV1 % predicted, mean difference (MD) 1.60% (95% CI ‐7.96 to 11.16) (very low‐quality evidence). In the second trial, rhDNase led to a greater increase in FEV1 % predicted than hypertonic saline (5 mL twice daily) at 12 weeks in participants with moderate to severe lung disease, MD 8.00% (95% CI 2.00 to 14.00) (low‐quality evidence). One cross‐over trial (47 participants) reported 15 exacerbations during treatment with hypertonic saline and 18 exacerbations in the rhDNase group (low‐quality evidence). Increased cough was reported in 13 participants using hypertonic saline and 17 on daily rhDNase in one cross‐over trial of 47 people (low‐quality evidence). There was insufficient evidence to assess rates of other adverse events reported. No deaths were reported.
One trial (12 participants) compared hypertonic saline to amiloride and one (29 participants) to sodium‐2‐mercaptoethane sulphonate. Neither trial found a difference between treatments in any measures of sputum clearance; additionally the comparison of hypertonic saline and sodium‐2‐mercaptoethane sulphonate reported no differences in courses of antibiotics or adverse events (very low‐quality evidence).
One trial (12 participants) compared hypertonic saline to mannitol but did not report lung function at relevant time points for this review; there were no differences in sputum clearance, but mannitol was reported to be more 'irritating' (very low‐quality evidence).
Authors' conclusions
Regular use of nebulised hypertonic saline by adults and children over the age of 12 years with CF results in an improvement in lung function after four weeks (very low‐quality evidence from three trials), but this was not sustained at 48 weeks (low‐quality evidence from one trial). The review did show that nebulised hypertonic saline reduced the frequency of pulmonary exacerbations (although we found insufficient evidence for this outcome in children under six years of age) and may have a small effect on improvement in quality of life in adults.
Evidence from one small cross‐over trial in children indicates that rhDNase may lead to better lung function at three months; qualifying this we highlight that while the study did demonstrate that the improvement in FEV1 was greater with daily rHDNase, there were no differences seen in any of the secondary outcomes.
Hypertonic saline does appear to be an effective adjunct to physiotherapy during acute exacerbations of lung disease in adults. However, for the outcomes assessed, the quality of the evidence ranged from very low to at best moderate, according to the GRADE criteria.
Plain language summary
Hypertonic saline (salt water with at least 3% salt) nebulised as a fine mist through a mask or mouthpiece for cystic fibrosis
Review question
We reviewed the evidence for treatment with nebulised hypertonic saline compared to placebo or other agents for improving mucus clearance in the lungs of people with cystic fibrosis (CF).
Background
People with CF produce large amounts of thick mucus which is difficult to clear and blocks up their airways. Chest physiotherapy or medication e.g. hypertonic saline, or both combined, are used to try and clear this mucus from the airways. Hypertonic saline is water with a concentration of 3% to 7% salt and is inhaled as a fine mist. This is an update of an earlier review.
Search date
The evidence is current to: 08 August 2018.
Trial characteristics
We included 17 trials with 966 participants with CF aged between 4 months and 63 years. Eleven trials compared hypertonic saline to isotonic saline (water with 0.12 to 0.9% salt (described as placebo (a dummy treatment)); one trial compared isotonic saline and voluntary cough to hypertonic saline or mannitol 300 mg; three trials compared hypertonic saline to rhDNase (Pulmozyme®); one trial compared hypertonic saline to amiloride; and one trial compared hypertonic saline to Mistabron®. Trials assessed different concentrations of hypertonic saline with different nebulisers and different treatment schedules; the most common treatment was twice‐daily 7% hypertonic saline and the most common nebuliser was ultrasonic. Most trials treated people with a bronchodilator to widen the airways before giving the hypertonic saline.
Key results
Hypertonic saline 3% to 7% versus placebo
In three trials (225 people) lung function improved after four weeks, but only one trial (164 people) reported results after 48 weeks, and showed no difference in lung function. No deaths were reported. One adult trial reported fewer exacerbations needing antibiotics with hypertonic saline than with placebo, but a trial in children found no difference in this outcome. There was not enough information to properly assess adverse events such as cough, chest tightness, tonsillitis and vomiting. In four trials (80 participants) sputum clearance was better with hypertonic saline.
One trial in 132 adults with an exacerbation reported uncertain effects of hypertonic saline on short‐term lung function and the time to the next exacerbation after discharge from hospital. No deaths were reported. Side effects such as cough and wheeze were reported, but there were no serious side effects.
Hypertonic saline versus mucus mobilising treatments
We could analyse data from two of the three trials comparing hypertonic saline to rhDNase (61 participants). In one trial there was no difference in lung function at three weeks, but the second reported rhDNase led to a greater increase in lung function at 12 weeks in people with moderate to severe disease. One trial (47 participants) reported no difference in the number of exacerbations, but there was increased cough with hypertonic saline compared to rhDNase. There was not enough information to assess other side effects. No deaths were reported.
One trial (12 participants) compared hypertonic saline to amiloride and one (n = 29) to Mistabron®. Neither trial found a difference between treatments in any measures of sputum clearance. The trial comparing hypertonic saline and Mistabron® also reported no differences in how many antibiotic courses were prescribed or in side effects.
The trial comparing hypertonic saline to mannitol (12 participants) did not report lung function at relevant time points for this review; there were no differences in sputum clearance, but mannitol was reported to be more 'irritating' .
Quality of the evidence
The risks of bias due to people not being randomly chosen to receive different treatments or due to not all results being reported range from low to unclear, but there is a high risk that people knew which treatment they were receiving in half the trials as they could taste the difference between the solutions.
The quality of the evidence was low or very low. Besides the risks of bias, the main problems were the small numbers of participants in trials combined with a wide variation in results; also, some trials limited participants to those who could tolerate hypertonic saline or to certain age groups.
Summary of findings
Background
Description of the condition
Cystic fibrosis (CF) is the most common life‐limiting autosomal recessive genetic disorder in populations of Northern European descent. In 1989 the gene responsible was identified on the long arm of chromosome 7 (Kerem 1989). This gene encodes for a protein named the cystic fibrosis transmembrane conductance regulator (CFTR) which functions as a chloride channel on the surface of epithelial cells. The altered CFTR is thought to result in defects of electrolyte transport which then cause increased water reabsorption across respiratory epithelia. This may lead to dehydration of the airway surface liquid, which in turn may prevent normal clearance of mucus (Davis 1996), although the precise mechanism by which CFTR causes abnormal mucus is still unknown.
Description of the intervention
Improvement of sputum clearance is a major therapeutic aim in CF. Treatments to improve mucus clearance in CF include chest physiotherapy, with and without the addition of agents that enhance mucus clearance. Treatment with nebulised recombinant deoxyribonuclease (rhDNase) has been widely accepted to be of benefit in CF (Yang 2018) and is thought to exert its major effect by enhancing sputum clearance. However, treatment with rhDNase is relatively expensive and its use in most countries is restricted as a consequence. Hypertonic saline may represent a potential alternative or supplementary therapy to improve mucociliary clearance in the context of long‐term maintenance therapy or during times of acute worsening of lung disease in CF.
How the intervention might work
In vitro deposition of hypertonic saline onto the airway surface improves mucus clearance. Dasgupta demonstrated that the addition of 3% hypertonic saline improved measures of sputum clearance and that hypertonic saline had a greater effect on mucus clearance in vitro than rhDNase (Dasgupta 1995). The postulated molecular mechanism of this effect is as follows:
hypertonic saline breaks the ionic bonds within the mucus gel, which could reduce the degree of cross linking and entanglements and lower viscosity and elasticity (Ziment 1978);
with chronic infection the mucin macromolecules develop fixed negative charges, causing increased repulsion; the addition of hypertonic saline increases the ionic concentration of the mucus and causes a conformational change by shielding the negative charges and thereby reducing repulsion ‐ this would result in a more compact mucus macromolecule that would allow more effective clearance (Robinson 1997);
in addition hypertonic saline induces an osmotic flow of water into the mucus layer, rehydrating secretions and thereby improving mucus rheology (Robinson 1997).
Why it is important to do this review
In the long term, improvement in mucociliary function may reduce bacterial load and chronic inflammation within the airways and therefore reduce the decline in lung function that is consequent to this. Hypertonic saline is easy and inexpensive to produce. Therefore, it is important to determine if nebulised hypertonic saline improves outcomes in CF, and to determine the frequency of adverse effects. This is an update of a previously published review (Wark 1999; Wark 2000; Wark 2003; Wark 2009).
Objectives
To investigate efficacy and tolerability of nebulised hypertonic saline treatment in people with CF compared to placebo and or other treatments that enhance mucociliary clearance.
Methods
Criteria for considering studies for this review
Types of studies
Controlled clinical trials. Both random allocation and quasi‐random allocation (e.g. where there is alternate allocation to treatment and control groups) were included.
Types of participants
People of all ages and of both sexes with CF diagnosed clinically or by sweat and genetic testing, including all degrees of disease severity.
Types of interventions
Nebulised hypertonic saline (defined as any concentration of saline greater than or equal to 3% delivered via a mask or mouthpiece with a nebuliser pump) compared to either placebo or usual treatment or any other mucus‐mobilising treatments (including, but not limited to, physical airway clearance techniques and medications which demonstrate improved mucus clearance e.g. rhDNase). Miniumum treatment duration considered in this review is a single dose. Trials comparing hypertonic saline used in conjunction with another intervention would be considered if the comparator group also received the second intervention.
Types of outcome measures
Primary outcomes
-
Lung function (absolute change and change in per cent (%) predicted)
forced expiratory volume at one second (FEV1)
forced vital capacity (FVC)
lung volume (residual volume (RV) and total lung capacity (TLC))
FEV0.5
lung clearance index (LCI)
Mortality
Secondary outcomes
Measures of sputum clearance (including measures of mucociliary clearance)
Measures of exercise capacity
Measures of quality of life (QoL) and symptoms
-
Pulmonary exacerbations (where a clear definition is described demonstrating an increase in symptoms or a decline in pulmonary function)
frequency
admission to hospital
duration of hospital stay (post hoc change)
outpatient treatments (hospital in the home, unscheduled visits to the doctor)
use of antibiotics, either intravenous, oral or inhalational
Medication delivery time (minutes)
Cost of treatment
Adherence to treatment with hypertonic saline along with other treatments after hypertonic saline is added
Bacteriology in pulmonary secretions, including sputum culture, culture from cough swab or bronchial lavage (post hoc change)
Adverse effects such as bronchospasm, cough and acute decline in pulmonary function (acute decline will be limited to the immediate phase of receiving treatment with hypertonic saline to within the first three hours and described separately to longer‐term lung function data as it represents acute bronchospasm provoked by hypertonic saline)
Search methods for identification of studies
We searched for all relevant published and unpublished trials without restrictions on language, year or publication status.
Electronic searches
Relevant trials were identified from the Cochrane Cystic Fibrosis and Genetic Disorders Group's Cystic Fibrosis Trials Register using the term: hypertonic saline.
The Cystic Fibrosis Trials Register is compiled from electronic searches of the Cochrane Central Register of Controlled Trials (CENTRAL) (updated each new issue of theCochrane Library), weekly searches of MEDLINE, a search of Embase to 1995 and the prospective handsearching of two journals ‐ Pediatric Pulmonology and the Journal of Cystic Fibrosis. Unpublished work is identified by searching the abstract books of three major cystic fibrosis conferences: the International Cystic Fibrosis Conference; the European Cystic Fibrosis Conference and the North American Cystic Fibrosis Conference. For full details of all searching activities for the register, please see the relevant sections of the Group's website.
Date of the most recent search of the Group's Cystic Fibrosis Trials Register: 08 August 2018.
We also searched the following trials registries:
US National Institutes of Health Ongoing Trials Register Clinicaltrials.gov (www.clinicaltrials.gov; searched 08 August 2018);
World Health Organization International Clinical Trials Registry Platform (WHO ICTRP) (apps.who.int/trialsearch; searched 08 August 2018).
For details of our search strategies, please see Appendix 1.
Searching other resources
We checked the bibliographies of included studies and any relevant systematic reviews identified for further references to relevant trials.
Data collection and analysis
Selection of studies
The authors (PW and VMM) independently selected the abstracts found during the searches. They then discussed potential and excluded abstracts to reach consensus. If trials were only in abstract form, the review authors contacted the trial authors for additional information. Both review authors then independently reviewed the full trials and, by consensus, included them if they were suitable or excluded them, documenting reasons for exclusion.
Data extraction and management
Two authors (PW and VMM) independently extracted data on trial characteristics and results using standard data acquisition forms. The authors entered the data into the Review Manager software (RevMan 2014).
The authors considered data reported up to and including three months to be short term and data reported at over three months to be long term.
The authors obtained additional data for one trial from the original investigators (Dentice 2016). Where an author of this Cochrane Review was a co‐author on an included trial, a third party performed the data extraction and assessment of quality (both risk of bias and GRADE) for that trial. This occurred when both the current review authors were co‐investigators in the National Hypertonic Saline in Cystic Fibrosis Study trial and Ashley Jones and a second person from the editorial base extracted the data and assessed the risk of bias (Elkins 2006a). This was also the case when one author (PW) was an author on one further trial, when VP and a second person from the editorial base extracted the data and assessed the risk of bias (Dentice 2016).
Assessment of risk of bias in included studies
Two authors assessed the risk of bias of each trial using the Cochrane Handbook for Systeamtic Reviews of Interventions (Higgins 2011). In particular, they examined details of the generation of allocation sequence, the concealment of treatment allocation schedule, whether the trial was blinded, whether intention‐to‐treat (ITT) analyses were possible from available data and if the number of participants lost to follow‐up or subsequently excluded from the trial was recorded.
Measures of treatment effect
If the review authors find that trials do not use a ITT analysis, then they will seek data on the number of participants with each outcome event, by allocated treated group, irrespective of adherence and whether or not the participant was later thought to be ineligible or otherwise excluded from treatment or follow‐up. With regards to dichotomous outcome measures, currently none of the trials report on mortality. For adverse event data, the authors have calculated a pooled estimate of the treatment effect for each outcome across the studies and determined the risk ratio. For the outcome of an improvement of over 10% in FEV1 reported by two cross‐over trials, the authors used the generic inverse variance to analyse the data and present the odds ratio (OR).
For continuous outcomes, the authors recorded either a mean change from baseline for each group or mean post‐treatment or post‐intervention values and standard deviation (SD) for each group. They calculated a pooled estimate of treatment effect for each of these individually by calculating the mean difference (MD) and 95% confidence intervals (CIs) where appropriate. Where the SD was not reported or available to use, we used the mean difference and 95% CI for each group to calculate the SDs.
We report costs of treatment narratively.
Unit of analysis issues
Where trials measured data longitudinally, the authors based the analysis on the final time point results. Methods do exist to carry out a meta‐analysis of aggregate longitudinal data, where individual patient data (IPD) are not available but these are not available at the moment in RevMan.
For trials with a cross‐over design, at least one week was required to allow sufficient washout of effect, at least for the measures of short‐term outcomes. For these trials of cross‐over design, the authors planned to carry out the analysis using results from a paired analyses, as recommended by Elbourne (Elbourne 2002). This was only possible in one trial (Suri 2001). For the remaining cross‐over trials, the data that were provided in the trial report were not sufficient to carry out this type of analysis. For these trials, we chose to ignore the cross‐over design and treat the results from the two periods as if they were independent (Adde 2004; Amin 2010; Ballmann 1998; Chadwick 1997; Laube 2009; Riedler 1996; Robinson 1996; Robinson 1997; Robinson 1999; Weller 1980). Elbourne reports that using this approach is conservative, due to the fact that it ignores the within‐patient correlation (Elbourne 2002).
Dealing with missing data
The authors originally planned to include missing participants due to dropouts in an ITT analysis The authors attempted to obtain any missing statistics (such as standard deviations (SDs) or correlation coefficients) from the trial authors, or they obtained the original data and determined the statistics. The authors were only able to obtain additional data from two trials (Adde 2004; Dentice 2016). The authors made two attempts to contact authors for missing data before accepting that the additional data would not be made available. However, if the trial authors contact us with data in the future we will add the information to the review at the following update.
Assessment of heterogeneity
The authors tested for heterogeneity between studies using a standard Chi² test and I² statistic (Higgins 2003). The Chi² test is a statistical test for heterogeneity, whereas I² assesses the quantity of inconsistency across studies in the meta‐analysis. The authors accepted a P value of below 0.1. They used the following I² ranges to interpret heterogeneity:
0% to 40%: might not be important;
30% to 60%: may represent moderate heterogeneity;
50% to 90%: may represent substantial heterogeneity;
75% to 100%: considerable heterogeneity.
Assessment of reporting biases
Due to the chronic nature of the disease, in many CF trials investigators collect data longitudinally at different time points throughout the course of the trial. In all the trials the authors examined when data were collected during the trial and also which data were reported in the trial publication. If it appeared that time points were missing, that the review authors would expect to have been reported (based on clinical and biologic plausibility) the review authors would have reported this. The authors also planned to assess publication bias by constructing funnel plots if they had been able to include a sufficient number of trials .
Data synthesis
The authors have used fixed‐effect analyses in this review. For future updates, when appropriate, where between‐trial variability is statistically significant, the authors plan to carry out random‐effects analyses.
Subgroup analysis and investigation of heterogeneity
For future updates, where possible, the authors plan to investigate heterogeneity using subgroup analysis if the I² statistic is over 40%; they will consider the following subgroups:
strength of hypertonic saline (comparing a concentration of 3% to 7% versus a concentration greater than 7%);
volume of hypertonic saline (less than 5 mL versus 5 mL to 10 mL versus more than 10 mL).
Sensitivity analysis
For a future update, when possible, the authors plan to perform a sensitivity analysis based on risk of bias of the trials, excluding those with a high risk of performance bias and including and excluding quasi‐randomised trials.
Summary of findings and quality of the evidence
In a post hoc change in line with current Cochrane guidance, at the 2018 update we added a summary of findings table for each comparison presented in the review (Table 1; Table 2; Table 3; Table 4; Table 5; Table 6). We selected the following seven outcomes to report (chosen based on relevance to clinicians and consumers).
Summary of findings for the main comparison. Hypertonic saline 3% to 7% versus isotonic saline for cystic fibrosis (stable lung disease).
| Hypertonic saline 3% to 7% versus isotonic saline for cystic fibrosis (stable lung disease) | ||||||
|
Patient or population: adults and children with cystic fibrosis (stable lung disease) Settings: outpatients Intervention: hypertonic saline 3% to 7% Comparison: isotonic saline | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (trials) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Isotonic saline | Hypertonic saline 3% to 7% | |||||
|
FEV1 (% predicted) change from baseline, short term Follow‐up: 4 weeks |
The mean change in FEV1 (% predicted) ranged from ‐1.42 to 2.8 in the isotonic saline groups. | The mean change in FEV1 (% predicted) was 3.44 higher (0.67 higher to 6.21 higher) in the hypertonic saline group. | NA | 225 (3 trials)1 |
⊕⊝⊝⊝ very low2, 4, 5, 6 | |
|
FEV1 (% predicted) change from baseline, long term Follow‐up: 48 weeks |
The mean change in FEV1 (% predicted) was 2.44 in the isotonic saline group. | The mean change in FEV1 (% predicted) was 2.31 higher (2.72 lower to 7.34 higher) in the hypertonic saline group. | NA | 134 (1 trial) |
⊕⊕⊝⊝ low2, 3 | The included trial also measured change in FEV1 (% predicted) at: 12 weeks, MD 4.10 (95% CI ‐0.08 to 8.28); 24 weeks, MD 5.37 (95% CI 1.03 to 9.71); and 36 weeks, MD 3.63 (95% CI ‐1.56 to 8.82). |
|
LCI Follow‐up: 4 weeks |
The mean LCI was 8.89 in the isotonic saline group. | The mean LCI was 1.03 lower (2.76 lower to 0.70 higher) in the hypertonic saline group. | NA | 10 (1 trial) |
⊕⊝⊝⊝ very low5, 7 | Trial had a cross‐over design. |
| Mortality | Outcome not reported. | NA | NA | NA | ||
|
Measures of sputum clearance Follow‐up: up to 24 hours |
The trials used radio‐labelled aerosol clearance and an 'area under the curve' measure to assess mucociliary clearance. Both measures significantly favoured treatment with hypertonic saline. |
NA | 80 (4 trials) | ⊕⊝⊝⊝ very low2, 4, 5 | All trials had cross‐over design. | |
|
Pulmonary exacerbations Follow‐up: up to 48 weeks |
One trial showed that there were fewer exacerbations per year requiring intravenous antibiotic therapy in the hypertonic saline group than in the isotonic saline group and that the interval during which participants remained free of exacerbations was also significantly longer in the hypertonic saline group. The second trial found no significant differences in the mean number of exacerbations per year. There was no difference reported in hospitalisation rates between the hypertonic saline group and the controls. |
NA | 415 (2 trials) |
⊕⊕⊝⊝ low2, 8 | ||
|
Adverse events Follow up: up to 48 weeks |
There were no significant difference between treatment groups in adverse events including cough, chest tightness, pharyngitis, haemoptysis, sinusitis, sneezing, tonsillitis and vomiting | NA | 589 (6 trials)9 |
⊕⊝⊝⊝ very low2, 4, 5 | ||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; FEV1: forced expiratory volume in 1 second;LCI: lung clearance index; MD: mean difference; NA: not applicable. | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
1. 1 trial (n = 19) was of a cross‐over design. 2. Downgraded once due to applicability: results apply only to those who can tolerate hypertonic saline. 3. Downgraded once due to imprecision; small sample size which did not achieve the targeted sample size generated by the power calculation. 4. Downgraded once due to risk of bias: high risk of detection bias as participants could discern the taste of the intervention and also limited information about trial methods. 5. Downgraded once due to imprecision: cross‐over trials analysed as a parallel trials (due to available data) which is likely to over‐estimate the within study variability and increase imprecision. 6. Downgraded once due to inconsistency: substantial heterogeneity (I² = 67%) which may have originated from different age groups recruited in the trials or different baseline levels of lung function. 7. Downgraded once due to applicability: results apply only to those who can tolerate hypertonic saline and the trial only included children aged 6 to 18 years, so results may not apply to adults. 8. Downgraded once due to risk of bias: one trial was at high risk of detection bias as participants could discern the taste of the intervention. 9. 4 trials (n = 104) were of a cross‐over design.
Summary of findings 2. Hypertonic saline 3% to 7% versus isotonic saline for cystic fibrosis (during acute exacerbations of lung disease).
| Hypertonic saline 3% to 7% versus isotonic saline for cystic fibrosis (during acute exacerbations of lung disease) | ||||||
|
Patient or population: adults and children with cystic fibrosis (during acute exacerbations of lung disease) Settings: hospitalised patients and outpatients Intervention: hypertonic saline 3% to 7% Comparison: isotonic saline | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (trials) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Isotonic saline | Hypertonic saline 3% to 7% | |||||
|
FEV1 (% predicted) change from baseline, short term Follow‐up: approximately 14 days (at time of hospital discharge) |
The mean % change in FEV1 (% predicted) was 32.3% in the isotonic saline group. | The mean % change in FEV1 (% predicted) was 5.10% higher (14.67% lower to 24.87% higher) in the hypertonic saline 3% to 7% group. | NA | 132 (1 trial) |
⊕⊕⊝⊝ low1,2 | |
|
FEV1 (% predicted) change from baseline, long term Follow‐up: NA |
Outcome not reported. | NA | NA | NA | ||
|
LCI Follow‐up: NA |
Outcome not reported. | NA | NA | NA | ||
|
Mortality Follow‐up: NA |
No deaths were reported in either trial. | NA | 142 (2 trials) |
⊕⊕⊝⊝ low2,3 | 1 trial had a cross‐over design. | |
|
Measures of sputum clearance Follow‐up: NA |
Outcome not reported. | NA | NA | NA | ||
|
Pulmonary exacerbations Follow‐up: up to 1 year |
There was no significant difference between the groups in time until the next pulmonary exacerbation requiring hospitalisation. | HR 0.86 (95% CI 0.57 to 1.30) | 132 (1 trial) |
⊕⊕⊝⊝ low1,2 | ||
|
Adverse events Follow up: up to 1 year |
Adverse events reported were cough and wheeze. No serious adverse events were reported. |
NA | 142 (2 trials) |
⊕⊝⊝⊝ very low2,3,4, | 1 trial had a cross‐over design. | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; FEV1: forced expiratory volume in 1 second; HR: hazard ratio;LCI: lung clearance index; MD: mean difference; NA: not applicable. | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
1. Downgraded once due to risk of bias: high risk of selection bias due to sequential allocation. 2. Downgraded once due to applicability: results apply only to those who can tolerate hypertonic saline and the trial included only adults so results may not apply to children. 3. Downgraded once due to risk of bias: first trial was at high risk of detection bias as participants could discern the taste of the intervention, second trial was at high risk of selection bias due to sequential allocation. 4. Downgraded once due to imprecision: no numerical data provided and small sample size.
Summary of findings 3. Hypertonic saline compared with rhDNase with for cystic fibrosis.
| Hypertonic saline compared with rhDNase with for cystic fibrosis | ||||||
|
Patient or population: adults and children with cystic fibrosis Settings: outpatients Intervention: hypertonic saline (daily) Comparison: rhDNase (daily)1 | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (trials) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| rhDNase | Hypertonic saline | |||||
|
FEV1 (% predicted) change from baseline, short term Follow‐up: 3 weeks |
The mean change from baseline in FEV1 (% predicted) was 1.6% higher (7.96% lower to 11.16% higher) in the hypertonic saline group compared to the daily rhDNase group.2 | NA | 14 (1 trial) | ⊕⊝⊝⊝ very low3,4,5 | Trial had a cross‐over design. No significant difference in the primary outcome (lung function) at this time‐point, with improvements only in secondary outcomes. |
|
|
FEV1 (% predicted) change from baseline, long term Follow‐up: 3 months |
The mean change from baseline in FEV1 (% predicted) was 8% higher (2% higher to 14% higher) in the hypertonic saline group compared to the daily rhDNase group.2 | NA | 47 (1 trial) | ⊕⊝⊝⊝ very low2,6,7 | Trial had a cross‐over design. An additional cross‐over trial of 18 participants found no difference between treatments in FEV1 after 10 weeks (no data presented). |
|
| LCI | Outcome not reported. | NA | NA | NA | ||
| Mortality | Outcome not reported. | NA | NA | NA | ||
| Measures of sputum clearance | Outcome not reported. | NA | NA | NA | ||
|
Pulmonary exacerbations Follow‐up: NA |
15 episodes occurring during treatment with hypertonic saline and 18 with daily rhDNase, there was no statistical difference between treatments (see comment). | NA | 47 (1 trial) | ⊕⊝⊝⊝ very low2,6,7 | Trial had a cross‐over design. Number of episodes reported rather than the number of participants with exacerbations (leading to a unit of analysis issue) so data not entered into the analysis. |
|
|
Adverse events Follow up: 3 months |
Increased cough was reported in 13 participants using hypertonic saline and 17 on daily rhDNase. There were similar rates of other adverse events between treatment arms (see comment). | NA | 47 (1 trial) | ⊕⊝⊝⊝ very low2,6,7 | Trial had a cross‐over design, so data not entered into analysis. | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; FEV1: forced expiratory volume in 1 second; LCI: lung clearance index; MD: mean difference; NA: not applicable. | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
1. An alternate day rhDNase group was also included in one of the trials (Suri 2001), but to allow a comparison across the trials, only results from the rhDNase daily group are presented in the tables. 2. Data analysed as MD between treatment groups via generic inverse variance due to cross‐over design of the trial, therefore an estimate of the assumed risk is not available. 3. Downgraded once due to risk of bias: high risk of detection bias as participants could discern the taste of the intervention and limited information was provided about the methodological design of the trial. 4. Downgraded once due to applicability: results apply only to those who can tolerate hypertonic saline. 5. Downgraded once due to imprecision: cross‐over trial analysed as a parallel trial due to available data, this approach is likely to over‐estimate the within study variability and increase imprecision, also small sample size. 6. Downgraded once due to applicability: results apply only to those who can tolerate hypertonic saline and the trial included only participants under the age of 18 so results may not apply to adults. 7. Downgraded once due to imprecision: small sample size.
Summary of findings 4. Hypertonic saline compared with amiloride for cystic fibrosis.
| Hypertonic saline compared with amiloride for cystic fibrosis | ||||||
|
Patient or population: adults and children with cystic fibrosis Settings: outpatients Intervention: hypertonic saline Comparison: amiloride | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Amiloride | Hypertonic saline | |||||
| FEV1: change from baseline, short term | Outcome not reported. | NA | NA | NA | ||
| FEV1: change from baseline, long term | Outcome not reported. | NA | NA | NA | ||
| LCI | Outcome not reported. | NA | NA | NA | ||
| Mortality | Outcome not reported. | NA | NA | NA | ||
|
Measures of sputum clearance Follow‐up: 60 minutes |
There was no significant difference between treatment groups. | NA | 12 (1 trial) | ⊕⊝⊝⊝ very low1,2,3 | Trial had cross‐over design. | |
| Pulmonary exacerbations | Outcome not reported. | NA | NA | NA | ||
| Adverse events | Outcome not reported. | NA | NA | NA | ||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; FEV1: forced expiratory volume in 1 second; LCI: lung clearance index; NA: not applicable. | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
1 Downgraded once due to risk of bias: high risk of detection bias as participants could discern the taste of the intervention and and limited information was provided about the trial methods (including whether a washout period was used). 2 Downgraded once due to applicability: results apply only to those who can tolerate hypertonic saline and the trial included only adults so results may not apply to children. 3 Downgraded once due to imprecision: no numerical data provided and small sample size.
Summary of findings 5. Hypertonic saline compared with sodium‐2‐mercaptoethane sulphonate (Mistabron®) for cystic fibrosis.
| Hypertonic saline compared with sodium‐2‐mercaptoethane sulphonate (Mistabron®) for cystic fibrosis | ||||||
|
Patient or population: adults and children with cystic fibrosis Settings: outpatients Intervention: hypertonic saline Comparison: sodium‐2‐mercaptoethane sulphonate | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (trials) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Sodium‐2‐mercaptoethane sulphonate | Hypertonic saline | |||||
| FEV1: short term | Outcome not reported. | NA | NA | NA | ||
| FEV1: long term | Outcome not reported. | NA | NA | NA | ||
| LCI | Outcome not reported. | NA | NA | NA | ||
| Mortality | Outcome not reported. | NA | NA | NA | ||
|
Measures of sputum clearance Follow‐up: 2 months |
No significant difference in sputum volume, colour or cough frequency between the groups. | NA | 29 (1 trial) | ⊕⊝⊝⊝ very low1,2,3 | Trial had cross‐over design. | |
|
Pulmonary exacerbations Follow‐up: 2 months |
See comment. | NA | 29 (1 trial) | ⊕⊝⊝⊝ very low1,2,3 | Trial had cross‐over design. The only information provided relevant to this outcome was that there was no change in the number of courses of antibiotics prescribed. |
|
|
Adverse events Follow‐up: 2 months |
See comment. | NA | 29 (1 trial) | ⊕⊝⊝⊝ very low1,2,3 | Trial had cross‐over design. Participants in both treatment groups described coughing at the beginning of their inhalations. No serious adverse events occurred during the trial. |
|
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; FEV1: forced expiratory volume in 1 second; LCI: lung clearance index; NA: not applicable. | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
1 Downgraded once due to risk of bias: high risk of detection bias as participants could discern the taste of the intervention and limited information was provided about the trial design. 2 Downgraded once due to applicability: results apply only to those who can tolerate hypertonic saline and the trial included only children aged 6 to 15 years so results may not apply to other age groups. 3 Downgraded once due to imprecision: no numerical data provided and small sample size.
Summary of findings 6. Hypertonic saline compared with mannitol for cystic fibrosis.
| Hypertonic saline compared with mannitol for cystic fibrosis | ||||||
|
Patient or population: adults and children with cystic fibrosis Settings: outpatients Intervention: hypertonic saline Comparison: mannitol | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (trials) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Mannitol | Hypertonic saline | |||||
|
FEV1: short term Follow‐up: up to 95 minutes |
See comment. | NA | 12 (1 trial) | ⊕⊝⊝⊝ very low1,2,3 | Trial had cross‐over design. FEV1 was assessed in the included trial at 5 minutes and 95 minutes post‐intervention. These very short‐term time‐points are not of clinical relevance to this review. Change from baseline within‐groups was reported but no between‐group data. |
|
| FEV1: long term | Outcome not reported. | NA | NA | NA | ||
| LCI | Outcome not reported. | NA | NA | NA | ||
| Mortality | Outcome not reported. | NA | NA | NA | ||
|
Measures of sputum clearance Follow‐up: up to 95 minutes |
There was no significant difference between treatment groups for matched voluntary cough. | NA | 12 (1 trial) | ⊕⊝⊝⊝ very low1,2,4 | Trial had cross‐over design. | |
| Pulmonary exacerbations | Outcome not reported. | NA | NA | NA | ||
|
Adverse events Follow up: up to 95 minutes |
See comment. | NA | 12 (1 trial) | ⊕⊝⊝⊝ very low1,2,4 | Trial had cross‐over design. Mannitol was considered to be a more 'irritating' treatment than other treatments (4‐armed trial); no specific data given. |
|
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; FEV1: forced expiratory volume in 1 second; LCI: lung clearance index; NA: not applicable. | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
1. Downgraded once due to risk of bias: high risk of detection bias as participants could discern the taste of the intervention and no washout period was used. 2. Downgraded once due to applicability: results apply only to those who can tolerate hypertonic saline and the trial included only participants over the age of 16 so results may not apply to younger children. 3. Downgraded once due to applicability: the outcome measured only at very short‐term time‐points (minutes after intervention), which are not of clinical relevance to this review. 4. Downgraded once due to imprecision: no numerical data provided and small sample size.
FEV1 (short‐term change (up to and including three months))
FEV1 (long‐term change (longer than three months))
LCI
Mortality
Measures of sputum clearance
Pulmonary exacerbations
Adverse events
We determined the quality of the evidence using the GRADE approach; and downgraded evidence in the presence of a high risk of bias in at least one trial, indirectness of the evidence, unexplained heterogeneity or inconsistency, imprecision of results, high probability of publication bias. We downgraded evidence by one level if they considered the limitation to be serious and by two levels if very serious.
Results
Description of studies
The trials included in this review were heterogenous in terms of age, severity of underlying lung disease, colonisation of microorganisms, other interventions, as well as the dose, timing and delivery of hypertonic saline.
Results of the search
The searches identified 55 potentially eligible trials. A total of 17 trials were included (Adde 2004; Amin 2010; Ballmann 1998; Cardinale 2003; Chadwick 1997; Dentice 2016: Elkins 2006a; Eng 1996; Laube 2009; Mainz 2015; Riedler 1996; Robinson 1996; Robinson 1997; Robinson 1999; Rosenfeld 2012; Suri 2001; Weller 1980) and 19 trials were excluded (Brivio 2016; Buonpensiero 2010; DeCono 2008; Dentice 2012; Donaldson 2006; Elkins 2006b; EUCTR2007‐002707‐40‐BE; Genkova 1998; King 1997; Kobylyansky 2000; NCT01094704; O'Neill 2017; Ros 2012; San Miguel 2016; Van Ginderdeuren 2008; Van Ginderdeuren 2011; Vanlaethem 2008). There are 16 trials currently listed as 'Awaiting classification' until more information is available to allow a judgement regarding eligibility (Amin 2016; Balinotti 2015; Brown 2010; Donaldson 2013; Dwyer 2013; Hofmann 1997; NCT00928135; NCT01355796; NCT01377792; NCT01619657; NCT02378467; NCT03391414; Nenna 2017; Palacio 2014; PRESIS 2018) and three trials have been identified which are ongoing (NCT02276898; NCT02343445; NCT02950883).
Included studies
There were 17 trials which met the inclusion criteria with a total of 966 participants (Adde 2004; Amin 2010; Ballmann 1998; Cardinale 2003; Chadwick 1997; Dentice 2016: Elkins 2006a; Eng 1996; Laube 2009; Mainz 2015; Riedler 1996; Robinson 1996; Robinson 1997; Robinson 1999; Rosenfeld 2012; Suri 2001; Weller 1980). Of these, 12 were published as full papers and four were reported in abstract form only (Adde 2004; Cardinale 2003; Chadwick 1997; Laube 2009), but additional data were provided by one of these investigators (Adde 2004).
Trial design
Five trials were of parallel design (Cardinale 2003; Dentice 2016, Elkins 2006a; Eng 1996; Rosenfeld 2012). There were 12 trials that were of cross‐over design (Adde 2004; Amin 2010; Ballmann 1998; Chadwick 1997; Laube 2009; Mainz 2015, Riedler 1996; Robinson 1996; Robinson 1997; Robinson 1999; Suri 2001; Weller 1980) and two of these had a four‐arm cross‐over design (Robinson 1997; Robinson 1999). A washout period was not stated in four cross‐over trials (Chadwick 1997; Laube 2009; Robinson 1996; Robinson 1997); there was no washout (interventions given on single days consecutively) in one trial (Mainz 2015). Where there was a washout period described, this ranged from two weeks (Adde 2004; Suri 2001) up to eight weeks (Weller 1980).
The number of participants varied between trials from 10 (Riedler 1996; Robinson 1997) to 321 (Rosenfeld 2012).
Seven trials were multicentre (Amin 2010; Dentice 2016; Elkins 2006a; Eng 1996; Mainz 2015; Rosenfeld 2012; Suri 2001) and three trials were single centre (Adde 2004; Laube 2009; Riedler 1996); it was unclear whether the remaining seven trials were multicentre or single centre. Seven trials were run in Australia (Dentice 2016; Elkins 2006a; Eng 1996; Riedler 1996; Robinson 1996; Robinson 1997; Robinson 1999). Five trials were run in Europe ‐ two trials were run in Germany (Ballmann 1998; Mainz 2015), two in the UK (Suri 2001; Weller 1980) and one in Italy (Cardinale 2003). One trial was run in Canada (Amin 2010) and one trial in the USA (Laube 2009); a further trial was run in centres across both Canada and the USA (Rosenfeld 2012). One trial was run in Brazil (Adde 2004). One trial did not clearly state where it was run (Chadwick 1997).
Participants
The age of participants ranged from four months (Rosenfeld 2012) to 63 years (Robinson 1999), but details of age were not given in three studies (Ballmann 1998; Cardinale 2003; Chadwick 1997). Most studies only recruited participants over the age of five or six years, but Rosenfeld recruited only children aged from 4 to 60 months (Rosenfeld 2012). All trials recruited both males and females equally.
The diagnostic criteria for CF in the participants was stated in eight trials which confirmed CF on the basis of a positive sweat chloride test or the presence of two common genetic mutations (Dentice 2016; Elkins 2006a; Eng 1996; Laube 2009; Mainz 2015Rosenfeld 2012; Suri 2001; Weller 1980). In the remaining nine trials it was only stated that the participants had CF.
Selection by tolerance for hypertonic saline
Three trials stated they tested for tolerance to hypertonic saline (Dentice 2016; Elkins 2006a; Rosenfeld 2012). The Rosenfeld trial excluded those who were intolerant to their test dose of hypertonic saline (Rosenfeld 2012) and Elkins excluded participants who demonstrated bronchial reactivity following hypertonic saline defined by a fall in FEV1 of 15% following tolerability testing (Elkins 2006a). Three trials stated that prior use of hypertonic saline was an exclusion criteria (Ballmann 1998; Elkins 2006a; Rosenfeld 2012). Additionally two trials excluded participants who had previously used rhDNase (Ballmann 1998; Suri 2001).
Baseline microbiology
Baseline sputum microbiology was stated in 11 trials (Amin 2010; Ballmann 1998; Elkins 2006a; Laube 2009; Mainz 2015; Riedler 1996; Robinson 1997; Robinson 1999; Rosenfeld 2012; Suri 2001; Weller 1980). Weller mentioned bacterial growth, but no details were given (Weller 1980). The Dentice trial measured change in bacterial density for Pseudomonas aeruginosa and Staphlycoccus aureus, but did not state baseline microbiology (Dentice 2016).
P aeruginosa
The presence of P aeruginosa was described in 11 trials. In one trial, seven (37%) participants were described as colonised with P aeruginosa (Amin 2010) and in the later Robinson trial 10 out of 12 participants were colonised with P aeruginosa (Robinson 1999). In the Laube trial, P aeruginosa was cultured in 17% of participants (Laube 2009). In the Rosenfeld trial 60 participants (38%) and 69 participants (42.3) were colonised with P aeruginosa in the hypertonic saline group and isotonic saline groups respectively (Rosenfeld 2012). Ballmann reported that three out of the 14 participants were chronically colonised with P aeruginosa (Ballmann 1998). Elkins reported the presence of P aeruginosa in 79 of the 83 participants in the hypertonic saline group and 78 of the 81 control participants (Elkins 2006a). In each of two further trials, all of the 10 participants had P aeruginosa in their sputum (Riedler 1996; Robinson 1997). In the Suri trial, 48% of participants had P aeruginosa (Suri 2001). Finally, Mainz reported the presence of P aeruginosa in 23 (33%) participants (Mainz 2015).
S aureus
The presence of S aureus was described in five trials. Laube reported that S aureus was cultured in 42% of participants (Laube 2009). Elkins reported that S aureus was present in 44 of the 83 participants in the hypertonic saline group and 47of the 81 control participants (Elkins 2006a). Robinson reported that 5 out of 10 participants in the 1997 trial and 7 out of 12 in the 1999 trial had S aureus (including two who also had P aeruginosa) (Robinson 1997; Robinson 1999). Suri reported that 39% of participants were colonised with S aureus (Suri 2001).
Other pathogens
Three studies excluded participants if they were colonised with Burkholderia cepacia complex (Amin 2010; Elkins 2006a; Suri 2001). Amin further excluded any participant who had positive sputum cultures for non‐tuberculosis mycobacteria in the past year (Amin 2010). Three trials reported that no participants in either group had B cepacia (Riedler 1996; Robinson 1997; Rosenfeld 2012).
Robinson also reported in the 1999 trial that 4 out of 12 participants had Aspergillus fumigatus (Robinson 1999), and Suri reported that 2% of participants were infected with Stenotrophomonas maltophilia (Suri 2001). Mainz reported a wide range of pathogens (Mainz 2015).
Baseline clinical severity
Most trials recruited participants with stable disease; in one of the Robinson trials, it was clearly stated that people with CF who were clinically unstable (defined as an exacerbation in the previous four weeks) were excluded (Robinson 1999). Two further trials excluded participants who were experiencing or had recently experienced an acute respiratory exacerbation (Ballmann 1998; Rosenfeld 2012). Rosenfeld also excluded any participant with a secondary chronic lung condition not related to their CF, or other major organ dysfunction (Rosenfeld 2012). The Elkins trial required participants to be clinically stable (Elkins 2006a). Amin included only participants with a baseline FEV1 of greater than 80% predicted and a room air oxyhaemoglobin saturation of greater than 90% (Amin 2010). In the Eng trial, participants were required to have an FEV1 greater than 20% predicted at baseline and to be on stable medications for the previous 14 days (Eng 1996). In the trial by Laube, the children had FEV1 and FVC greater than 90% predicted (Laube 2009). In two trials by Robinson, participants needed to be in a stable clinical condition without any change to their medications (Robinson 1996; Robinson 1997); only the 1997 Robinson trial included a participant with an FEV1 % predicted of less than 30% (Robinson 1997). Suri required participants to have an FEV1 less than 70% predicted (people with CF with at least moderate lung disease) and be clinically stable with no exacerbations or change in medications in the last 14 days (consequently, these participants have more severe lung disease at baseline, mean FEV1 % predicted 48% (range 14 to 77%)) (Suri 2001). Weller stated that all participants received routine treatment for five years (Weller 1980). Others used mean FEV1 as a % predicted value or FVC as a % predicted value to assess disease severity (Adde 2004; Cardinale 2003; Chadwick 1997). Mainz recruited participants with clinical symptoms of rhinosinusitis, but did not state the clinical severity of lung disease or whether they were stable clinically at the recruitment (Mainz 2015).
Two trials recruited participants who were experiencing an acute exacerbation (Dentice 2016; Riedler 1996). Dentice enrolled participants a confirmed diagnosis of CF within 24 hours of a hospital admission for management of a pulmonary exacerbation (defined as at least 4 out of 12 criteria described by Fuchs for a minimum of seven days (Fuchs 1994)) (Dentice 2016). The Riedler trial selected 10 consecutive adolescents admitted with an exacerbation of their lung disease who all had productive coughs (Riedler 1996).
Interventions
An ultrasonic nebuliser was used to deliver hypertonic saline in seven trials (Amin 2010; Cardinale 2003; Eng 1996; Robinson 1996; Robinson 1997; Robinson 1999; Suri 2001); while six trials used a high‐output jet nebuliser (Adde 2004; Ballmann 1998; Dentice 2016; Elkins 2006a; Riedler 1996). One trial used a Pari LC Sprint Sinus nebuliser (Mainz 2015).
Different concentrations of hypertonic saline were used in the trials ranging from 3.5% to 7% and this is outlined in detail in the tables (Characteristics of included studies).
In 13 trials isotonic (0.9%) saline was used as a control (Amin 2010; Cardinale 2003; Chadwick 1997; Dentice 2016; Elkins 2006a; Eng 1996; Laube 2009; Mainz 2015; Riedler 1996; Robinson 1996; Robinson 1997; Robinson 1999; Rosenfeld 2012). Three of these trials compared hypertonic saline 7% with isotonic saline 0.9% twice daily (Amin 2010; Elkins 2006a; Rosenfeld 2012) and one trial compared hypertonic saline 6% to isotonic saline twice daily (Eng 1996). Two trials added quinine sulphate (0.25 mg per mL) to both solutions to mask the taste (Dentice 2016; Elkins 2006a). One trial administered 6% hypertonic saline or isotonic saline once per day, approximately 1 mL to each nostril (Mainz 2015). Five trials used a single administration of nebulised hypertonic saline compared to isotonic saline (Laube 2009; Riedler 1996; Robinson 1996; Robinson 1997; Robinson 1999;). In the 1996 trial, Robinson compared a single administration of nebulised hypertonic saline (7%), amiloride (0.3% in 0.12% NaCl) and a combination of amiloride and hypertonic saline to isotonic saline (0.9%) (Robinson 1996), while in the 1997 trial Robinson compared differing concentrations of nebulised hypertonic saline (3%, 7%, and 12%) with isotonic saline and voluntary cough (Robinson 1997). In the 1999 trial, Robinson compared hypertonic saline 6% to 0.9% isotonic saline with matched voluntary cough, mannitol 300 mg, and placebo capsules with matched voluntary cough (Robinson 1999). In Laube participants attended for two visits at least one week apart. They received either 5 mL 0.12% isotonic saline or 5 mL 7% hypertonic saline, with the order of treatment randomised (Laube 2009). Two trials did not state the frequency of nebulisation (Cardinale 2003; Chadwick 1997).
Three trials compared hypertonic saline to rhDNase (Adde 2004; Ballmann 1998; Suri 2001). Adde used a regimen of hypertonic saline 6% (10 mL) compared to 2.5 mg rhDNase twice daily (Adde 2004); Ballmann compared nebulised 5.75% saline (10 mL) to 2.5 mg rhDNase twice daily (Ballmann 1998); and Suri compared hypertonic saline 7% (5 mL) twice daily to rhDNase 2.5 mg daily or to rhDNase 2.5 mg alternate daily (Suri 2001).
Weller compared hypertonic saline 7% (3 mL) to Mistabron® 20% (a mucolytic agent) (Weller 1980).
Additional treatments were also used in association with the hypertonic saline. All the trials with the exception of Chadwick, Mainz and Weller pre‐treated participants with short‐acting beta‐agonists (Chadwick 1997; Mainz 2015; Weller 1980). In the Suri trial, the only pre‐treated participants were those who were already using bronchodilators or whose FEV1 fell by more than 15% after the test dose of hypertonic saline (Suri 2001). Pre‐treatment was not stated by Laube (Laube 2009).
In the Eng trial participants performed physiotherapy at home and received hypertonic saline or isotonic saline prior to their regular physiotherapy session (Eng 1996). The place of chest physiotherapy is likely to be an important contributor to mucolytic therapy, but its role as a confounder was not addressed.
Two trials used hypertonic saline or isotonic saline as an adjunct to physiotherapy and an exercise programme while hospitalised for a pulmonary exacerbation when all participants also received intravenous antibiotics (Dentice 2016; Riedler 1996). In one trial participants received hypertonic saline 7%, three times a day (Dentice 2016) and in the second trial they received hypertonic saline 6% as a single treatment (Riedler 1996).
Outcomes
Lung function was the most common outcome measured, but this was reported in a number of ways. Nine trials reported on FEV1 (Adde 2004; Amin 2010; Ballmann 1998; Dentice 2016; Elkins 2006a; Eng 1996; Riedler 1996; Robinson 1996; Suri 2001); seven trials reported on FVC (Amin 2010; Dentice 2016; Elkins 2006a; Eng 1996; Riedler 1996; Suri 2001; Weller 1980); and three reported on FEF25‐75 (Amin 2010; Riedler 1996; Rosenfeld 2012). A number of trials reported on less common measures of lung function (see the tables for further details (Characteristics of included studies) and one trial simply reported on general lung function (Cardinale 2003). Mucociliary clearance was reported in five trials (Laube 2009; Riedler 1996; Robinson 1996; Robinson 1997; Robinson 1999) and two reported on sputum production (Riedler 1996; Weller 1980). Pulmonary exacerbations were reported in four trials (Dentice 2016; Elkins 2006a; Rosenfeld 2012; Suri 2001) and Rosenfeld additionally reported additional antibiotics for all causes (Rosenfeld 2012). Sputum cultures were reported in two trials (Adde 2004; Rosenfeld 2012) and one trial reported changes in quantitative microbiology (Dentice 2016). A further four trials reported symptom scores (Adde 2004; Dentice 2016; Eng 1996; Weller 1980) and two reported on satisfaction or preference (Adde 2004; Ballmann 1998). One trial reported on sinus and nasal symptoms (Mainz 2015). Tolerability was only reported on by one trial (Rosenfeld 2012) and one trial reported on nebulization time (Ballmann 1998). Linked to this two trials reported adherence to treatment (Rosenfeld 2012; Suri 2001). Five trials reported on QoL (Amin 2010; Dentice 2016; Elkins 2006a; Rosenfeld 2012; Suri 2001) and five trials on adverse events (Cardinale 2003; Dentice 2016; Eng 1996; Mainz 2015; Rosenfeld 2012). Two trials reported data relating to cost in comparison to rhDNase (Ballmann 1998; Suri 2001).
Excluded studies
There were 19 trials excluded from the review (Brivio 2016; Buonpensiero 2010; DeCono 2008; Dentice 2012; Donaldson 2006; Elkins 2006b; EUCTR2007‐002707‐40‐BE; Genkova 1998; Grasemann 2013; IRCT20180307038994N1; King 1997; Kobylyansky 2000; NCT01094704; O'Neill 2017; Ros 2012; San Miguel 2016; Van Ginderdeuren 2008; Van Ginderdeuren 2011; Vanlaethem 2008).
Four trials were not randomised in design and were therefore excluded (DeCono 2008; EUCTR2007‐002707‐40‐BE; IRCT20180307038994N1; NCT01094704) and one was excluded as there was no comparison group (Genkova 1998). One trial was excluded as it was performed in a non‐CF population (Kobylyansky 2000) and a further trial was performed in vitro (King 1997). Eight trials were excluded as they studied hypertonic saline in conjunction with other therapies, but did not include a comparator group without hypertonic saline (Dentice 2012; Donaldson 2006; Elkins 2006b; O'Neill 2017; San Miguel 2016; Van Ginderdeuren 2011; Van Ginderdeuren 2008; Vanlaethem 2008). One trial compared the timing of the delivery of hypertonic saline (Dentice 2012) and another the frequency of delivery (Elkins 2006b). One trial compared hypertonic saline with or without pre‐treatment with amiloride (Donaldson 2006). Two trials did not compare hypertonic saline to control, instead they compared different sequences of autogenic drainage (Van Ginderdeuren 2008; Van Ginderdeuren 2011) and two used other airway clearance techniques in participants using hypertonic saline (O'Neill 2017; Vanlaethem 2008) and one compared the use of hypertonic saline with or without physiotherapy (San Miguel 2016). One trial compared isotonic saline to L‐arginine and did not use hypertonic saline (Grasemann 2013). The three remaining trials compared two different formulations of 7% hypertonic saline (with and without hyaluronic acid) (Brivio 2016; Buonpensiero 2010; Ros 2012).
Studies awaiting classification
There are 16 trials are currently listed as 'Awaiting classification' until more information is available to allow a judgement regarding eligibility (Amin 2016; Balinotti 2015; Brown 2010; Corcoran 2017; Donaldson 2013; Dwyer 2013; Hofmann 1997; NCT00928135; NCT01355796; NCT01377792; NCT01619657; NCT02378467; NCT03391414; Nenna 2017; Palacio 2014; PRESIS 2018).
Twelve trials are described as randomised controlled trials, but with no or only few details of the methodology (Balinotti 2015; Donaldson 2013; Dwyer 2013; Hofmann 1997; NCT00928135; NCT01355796; NCT01377792; NCT01619657; NCT02378467; Nenna 2017; Palacio 2014; PRESIS 2018); one trial is a controlled clinical trial but it is not clear if there was any form of randomisation employed in the trial (Brown 2010). Three trials employed a randomised cross‐over design in 21 participants (Amin 2016; Corcoran 2017; NCT03391414).
The duration of trials ranged from single exposure (Corcoran 2017; Hofmann 1997) to 52 weeks (NCT01619657; PRESIS 2018); one trial gave no information on the duration (Brown 2010).
Fifteen trials compared hypertonic saline at a concentration ranging from 3% to 7% to isotonic saline (Amin 2016; Balinotti 2015; Brown 2010; Corcoran 2017; Donaldson 2013; Dwyer 2013; NCT00928135; NCT01355796; NCT01377792; NCT01619657; NCT02378467; NCT03391414; Nenna 2017; Palacio 2014; PRESIS 2018) and one trial did not state the concentration (NCT01377792). Most trials (n = 9) utilised hypertonic saline at a concentration of 7% (Amin 2016; Brown 2010; Corcoran 2017; NCT00928135; NCT01355796; NCT02378467; NCT03391414; Nenna 2017; Palacio 2014); five trials used hypertonic saline 6% (Balinotti 2015; Donaldson 2013; Dwyer 2013; NCT01619657; PRESIS 2018); one trial randomised participants to three concentrations of saline, 0.9%, 3% and 6% and assessed tolerability; this trial also added quinine sulphate to all intervention arms and the control arm (Dwyer 2013). One trial compares amiloride in hypertonic saline (5.85%) to amiloride in isotonic saline (Hofmann 1997).
Nine trials were conducted in children (Amin 2016; Balinotti 2015; Brown 2010; Donaldson 2013; NCT01619657; NCT02378467; Nenna 2017; Palacio 2014; PRESIS 2018) and three in adults (Corcoran 2017; Hofmann 1997; NCT01355796) and three in mixed age groups (Dwyer 2013; NCT00928135; NCT01377792; NCT03391414).
Ongoing studies
There are three ongoing trials identified from trials registries (NCT02276898; NCT02343445; NCT02950883). All three were described as randomised controlled trials, two of parallel design (NCT02343445; NCT02950883) and one of cross‐over design (NCT02276898). Duration of the three trials was a single administration (NCT02276898), 15 days (NCT02343445) and 48 weeks (NCT02950883). Two trials used hypertonic saline 7% (NCT02276898; NCT02950883) and one trial compared P‐1037 solution for inhalation in hypertonic saline (4.2%) to P‐1037 solution for inhalation in 0.17% isotonic saline (NCT02343445).
The long‐term trial was in children only (NCT02950883), but the remaining two trials recruited both children and adults (NCT02276898; NCT02343445).
Risk of bias in included studies
Please refer to the risk of bias figure (Figure 1).
1.
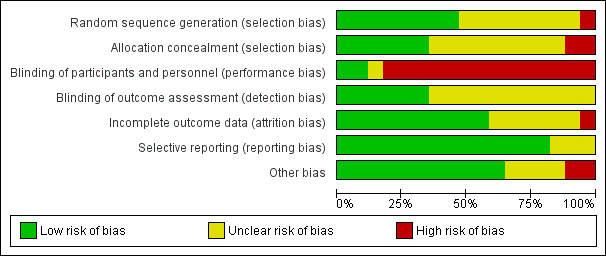
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Allocation
Generation of the randomisation sequence
No details of the randomisation process were provided by 10 trials (Adde 2004; Cardinale 2003; Chadwick 1997; Laube 2009; Mainz 2015; Robinson 1996; Robinson 1997; Robinson 1999; Suri 2001; Weller 1980). For nine of these we judged the risk of bias to be unclear; however, additional data received from Dr Adde confirmed that a random numbers table was used to generate the randomisation sequence (Adde 2004), as such we judged this trial to have a low risk of bias.
We also judged a further six trials to have a low risk of bias (Amin 2010; Ballmann 1998; Elkins 2006a; Eng 1996; Riedler 1996, Rosenfeld 2012). Computer‐generated randomisation lists were used in two trials (Amin 2010; Elkins 2006a), one trial used random permuted block allocation (Rosenfeld 2012), one reported that participants drew lots to decide treatment (Ballmann 1998). Eng stated the use of random number tables (Eng 1996) and Riedler used a coin toss to randomise participants (Riedler 1996).
We judged there to be a high risk of bias in the Dentice trial as participants were enrolled sequentially upon admission to hospital (Dentice 2016).
Allocation concealment
No details were published regarding methods of allocation concealment for 10 trials (Adde 2004; Ballmann 1998; Cardinale 2003; Chadwick 1997; Eng 1996; Laube 2009; Mainz 2015; Robinson 1996; Robinson 1997; Weller 1980). We judged nine of these to have an unclear risk of bias. Additional data received from Dr Adde confirmed that the sequence of treatment was put into numbered envelopes which were kept in the hospital pharmacy and not opened until after participants were recruited (Adde 2004); thus we judged this trial to have a low risk of bias.
A further six trials were also judged to have a low risk of bias (Amin 2010; Elkins 2006a; Riedler 1996; Robinson 1999; Rosenfeld 2012; Suri 2001). Four trials concealed the allocation sequence either by using investigators off‐site, investigators not otherwise involved in the trial or a secure website (Amin 2010; Elkins 2006a; Rosenfeld 2012; Suri 2001). Randomisation was coded such that investigators were blinded to the identity of the intervention at the time of analysis in one trial (Robinson 1999) and in another each participant was assigned to order of treatment by a coin toss (Riedler 1996).
One trial used sequential alternate allocation and thus we did not judge the allocation to be adequately concealed (high risk of bias) (Dentice 2016).
Blinding
With the exception of three trials where the interventions were described as "blinded" (Adde 2004; Dentice 2016; Elkins 2006a), most trials further stated that it was not possible to blind participants due to the discernible taste of hypertonic saline. These three trials, however, reported adding quinine sulphate to the solutions to mask the taste and were judged to have a low risk of bias with regards to blinding of participants (Adde 2004; Dentice 2016; Elkins 2006a). We judged three trials to have an unclear risk of bias (Cardinale 2003; Chadwick 1997; Robinson 1996). Chadwick described the trial as single‐blind and did not address the issue of the taste of hypertonic saline (Chadwick 1997). Robinson described the participants in the 1996 trial as being blinded, but admitted they may have been able to discern which group they were in due to the taste and duration of nebulization for the different interventions (Robinson 1996). The remaining trials were judged to have a high risk of bias due to the discernible taste of hypertonic saline (Amin 2010; Ballmann 1998; Eng 1996; Laube 2009; Riedler 1996; Robinson 1997; Robinson 1999; Rosenfeld 2012; Suri 2001; Weller 1980).
With regards to the blinding of the investigators, seven trials reported that researchers were blinded and so had a low risk of bias (Amin 2010; Dentice 2016; Elkins 2006a; Eng 1996; Robinson 1996; Robinson 1999; Rosenfeld 2012). A lack of information led nine trials to be judged as having an unclear risk of bias (Adde 2004; Cardinale 2003; Chadwick 1997; Laube 2009; Mainz 2015; Riedler 1996; Robinson 1997; Suri 2001; Weller 1980). Riedler and Chadwick were described as single‐blind and stated that participants could discern the taste of hypertonic saline, thus implying that the researchers were blinded (Riedler 1996) and Weller was described as double‐blind although the participants could discern the taste (Weller 1980), but there were no definite statements on the blinding of trial investigators. The Ballman trial was judged to have a high risk of bias with regards to the blinding of investigators (Ballmann 1998). This trial also reported the use of different volumes of liquid in the two groups (Ballmann 1998).
Incomplete outcome data
Seven trials were judged to have an unclear risk of bias due to incomplete outcome data (Adde 2004; Ballmann 1998; Cardinale 2003; Chadwick 1997; Riedler 1996; Robinson 1997; Robinson 1999). Adde did not report withdrawals; additional data provided by the trial investigators stated that one participant (not included in the analysis) had to stop treatment with hypertonic saline due to severe dyspnoea during its nebulization (Adde 2004). Ballmann provided no information about whether an ITT was used (Ballmann 1998); and three trials did not state as whether an ITT approach had been used and did not describe any withdrawals (Riedler 1996; Robinson 1997; Robinson 1999). The Chadwick trial had no description of dropouts (Chadwick 1997). Cardinale stated that no adverse events were reported with hypertonic saline, but did not give any information for the placebo group (Cardinale 2003).
The remaining 10 trials were judged to be at low risk (Amin 2010; Dentice 2016; Elkins 2006a; Eng 1996; Laube 2009; Robinson 1996; Rosenfeld 2012; Mainz 2015; Suri 2001; Weller 1980). Two trials stated there were no withdrawals (Dentice 2016; Robinson 1996). The remaining trials gave details of withdrawals and the reasons for these. Amin described details of missing data for three participants; two due to uninterpretable LCI results and one due to an inability to adhere to the trial protocol (Amin 2010). In the Elkins trial, two participants (one from each group) withdrew voluntarily after randomisation and before the first dose; a clear description of withdrawals after randomisation was given by group and with reasons, with a total of 82 participants in the hypertonic saline group and 80 in control group included in an ITT analysis (Elkins 2006a). Laube described two dropouts following randomisation and accounted for this by replacing them (Laube 2009). Mainz detailed six dropouts due to non‐adherence (Mainz 2015). Rosenfeld gave details (with reasons) of withdrawals which were in roughly equal numbers across groups and reported an ITT analysis of 158 participants in the hypertonic saline group and 163 in the control group (Rosenfeld 2012). Suri provided details of withdrawals with reasons and used an ITT analysis; an additional report of airway inflammatory changes following treatment stated that only 28 of the 48 participants were able to perform induced sputum and be included (Suri 2001). Two trials gave details of withdrawals, but did not state whether an ITT analysis had been performed (Eng 1996; Weller 1980). In the Eng trial, six participants withdrew in total (three from each group) with reasons given (Eng 1996). In the Weller trial there was a clear description of dropouts and withdrawals (Weller 1980).
Selective reporting
Three trials were reported only in abstract form, hence the availability of data concerning the outcomes which were planned to have been reported was limited and we judged these to have an unclear risk of bias (Adde 2004; Cardinale 2003; Chadwick 1997). In the remaining 14 trials, the outcomes stated in the 'Methods' section were reported in the 'Results' section (Amin 2010; Ballmann 1998; Dentice 2016; Elkins 2006a; Eng 1996; Laube 2009; Mainz 2015; Riedler 1996; Robinson 1996; Robinson 1997; Robinson 1999; Rosenfeld 2012; Suri 2001; Weller 1980).
Other potential sources of bias
Sample size calculations were described in seven trials and were judged to be at low risk of bias (Amin 2010; Dentice 2016; Elkins 2006a; Eng 1996; Laube 2009; Rosenfeld 2012; Suri 2001); however, the remaining 10 trials did not undertake such calculations and were judged to have a high risk (at increased risk of a type 1 error).
A cross‐over design was used in 12 trials, but six of these either did not use or did not state a washout period between treatment arms (Chadwick 1997; Mainz 2015; Riedler 1996; Robinson 1996; Robinson 1997; Robinson 1999). Trials that reported a washout period of less than one week would be judged at high risk of bias; the stated wash‐out periods were one week (Laube 2009), two weeks (Adde 2004; Suri 2001), three weeks (Ballmann 1998), four weeks (Amin 2010) and eight weeks (Weller 1980). These six trials were judged at low risk of bias.
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5; Table 6
The quality of the evidence has been graded for those outcomes included in the summary of findings tables. For the definitions of these gradings, please refer to the summary of findings tables (Table 1; Table 2; Table 3; Table 4; Table 5; Table 6).
Hypertonic saline 3% to 7% versus isotonic saline in stable lung disease
This comparison included 11 trials (n = 795) (Amin 2010; Cardinale 2003; Chadwick 1997; Elkins 2006a; Eng 1996; Laube 2009; Mainz 2015; Robinson 1996; Robinson 1997; Robinson 1999; Rosenfeld 2012). A summary of the results and our judgements with regards to the quality of the evidence can be found in the tables (Table 1).
In the analysis, due to data limitations, data from cross‐over trials have been entered and analysed as if they were from parallel trials. One trial assessed children with stable lung disease and chronic sinuitis and looked at the effect of the intervention on sinus symptoms (Mainz 2015).
Primary outcomes
1. Lung function
a. FEV1
Three trials examined the effect of hypertonic saline 3% to 7% compared to isotonic saline on the change in FEV1 % predicted after four weeks treatment (Amin 2010; Elkins 2006a; Eng 1996). The combined analysis demonstrated a significant increase in FEV1 % predicted, MD 3.44% (95% CI 0.67 to 6.21) (Analysis 1.1) (very low‐quality evidence).
1.1. Analysis.
Comparison 1 Hypertonic saline 3% to 7% versus isotonic saline, Outcome 1 Change in FEV1 (% predicted).
Amin selected children aged 6 to 18 years old with essentially normal lung function as measured by spirometry with an FEV1 greater than 80% predicted and found no difference compared to isotonic saline, MD ‐0.42% (95% CI ‐7.45 to 6.61) (Amin 2010) (Analysis 1.1).
The two earlier trials recruited participants with greater impairment in lung function (Elkins 2006a; Eng 1996). When the data from these two trials were pooled and analysed, there was a significant improvement in FEV1 % predicted at four weeks, MD 4.15% (95% CI 1.14 to 7.16) (Analysis 1.1). The I² value at this time point for all participants is 67% (according to our definition this may represent substantial heterogeneity); however, if the Eng trial is removed from the meta‐analysis I² reverts to zero. The participants in the Eng trial had a lower FEV1 % predicted at baseline than in the other trials, which may account for this difference (Eng 1996). Similarly, for the analysis of data for participants aged over 14 years, which combines data for just two trials (Elkins 2006a; Eng 1996), the lower baseline lung function values of the participants in the Eng trial could account for this heterogeneity.
Only Elkins examined the mean % change in FEV1 % predicted at time points after four weeks (4, 12, 24, 36 and 48 weeks). Trial results failed to demonstrate a significant benefit over isotonic saline at any time‐point, other than at 24 weeks, MD 5.37% (95% CI 1.03 to 9.71) (Elkins 2006a) (low‐quality evidence).
Cardinale stated that the trial measured lung function, but did not report any results other than to state there was no difference in lung function results (Cardinale 2003).
Mainz also measured FEV1% predicted at day 1 and day 29, with no differences seen between the groups. However as hypertonic saline was not delivered to the airways, these outcomes are not included in our analysis (Analysis 1.1).
b. FVC
We analysed data from three trials for the mean change in FVC % predicted at four weeks (Amin 2010; Elkins 2006a; Eng 1996). When the data were pooled they did not demonstrate a significant improvement in FVC, MD 1.07% (95% CI ‐1.63 to 3.78) (Analysis 1.2).
1.2. Analysis.
Comparison 1 Hypertonic saline 3% to 7% versus isotonic saline, Outcome 2 Change in FVC (% predicted).
Again, only Elkins measured the change in FVC % predicted at 48 weeks (Elkins 2006a). Results failed to reach significance for either group, although the control group showed no improvement over this period of time (Analysis 1.2).
c. Lung volumes
These were not reported as outcomes in any of the included trials.
d. Change in FEV0.5
Rosenfeld assessed infant lung function in a subset of participants (73 out of 158), which included FEV0.5, and found a small difference (but with wide CIs) following treatment with hypertonic saline compared to isotonic saline at 48 weeks, MD 41.00 mL (95% CI 0.96 to 81.04) (Rosenfeld 2012) (Analysis 1.3).
1.3. Analysis.
Comparison 1 Hypertonic saline 3% to 7% versus isotonic saline, Outcome 3 Mean change in FEV0.5 (mL).
e. LCI
The Amin trial used LCI as its primary outcome (Amin 2010). In the original trial analysis investigators found hypertonic saline improved LCI compared to isotonic saline at four weeks, although in our analysis of the mean difference this just failed to reach significance, MD ‐1.03 (95% CI ‐2.76 to 0.70) (Analysis 1.4) (very low‐quality evidence).
1.4. Analysis.
Comparison 1 Hypertonic saline 3% to 7% versus isotonic saline, Outcome 4 Lung clearance index (LCI).
2. Mortality
No trials reported on this as an outcome.
Secondary outcomes
1. Measures of sputum clearance
Four trials used radio‐labelled aerosol clearance to assess mucociliary clearance (Laube 2009; Robinson 1996; Robinson 1997; Robinson 1999) and we judged the quality of the evidence to be very low. In this method the participant was given the radio‐labelled aerosol from an ultrasonic nebuliser and serial lung scans were performed. Two of the Robinson trials showed that hypertonic saline increased radioisotope clearance compared to isotonic saline controls, P < 0.05 (Robinson 1996) and P < 0.01 (Robinson 1997). The 1997 Robinson trial showed that increasing concentrations of hypertonic saline also had an effect, with a significant difference between hypertonic saline 3% and hypertonic saline 12% favouring the higher concentration; but no significant difference between hypertonic saline 7% and hypertonic saline 12% was reported (Robinson 1997). A comparison was made between three trials for isotope clearance at 60 to 120 minutes (Laube 2009; Robinson 1997; Robinson 1999). This favoured treatment with hypertonic saline, MD 6.14 (95% CI 2.56 to 9.72) (Analysis 1.5).
1.5. Analysis.
Comparison 1 Hypertonic saline 3% to 7% versus isotonic saline, Outcome 5 Radiolabelled isotope clearance.
Two of the Robinson trials reported measuring mucociliary clearance as 'area under the curve' (AUC), where the lower the value of AUC is the faster the clearance (Robinson 1996; Robinson 1997). Robinson showed that hypertonic saline 7% and hypertonic saline 12% were significantly different from isotonic saline (Robinson 1997). In the 1996 Robinson trial, the results for AUC showed hypertonic saline and hypertonic saline with amiloride were significantly different from cough, isotonic saline and amiloride alone (Robinson 1996). Combined analysis of the two trials favoured treatment with an MD of ‐212.06 (95% CI ‐271.64 to ‐152.48) (Robinson 1996; Robinson 1997) (Analysis 1.6).
1.6. Analysis.
Comparison 1 Hypertonic saline 3% to 7% versus isotonic saline, Outcome 6 Mucociliary clearance measured as area under the curve.
The participants in the Robinson trials had moderate to severe airflow obstruction; those in the 1996 trial had a mean FEV1 % predicted of 60.8% (range 27% to 112%) (Robinson 1996); those in the 1997 trial had a mean FEV1 % predicted of 52% (range 31% to 84%) (Robinson 1997); and in the 1999 trial, the mean FEV1 was 60% (range 27% to 112%) (Robinson 1999). This differed from the Laube trial, who included only participants with normal lung function (greater than 90% predicted FEV1 and FVC). In this trial there was no difference in mucociliary clearance measured at 20, 60, 90 minutes and 24 hours (Laube 2009).
2. Measures of exercise capacity
Eng demonstrated a significant improvement in exercise tolerance, the paper used a visual analogue scale (VAS) and during week 1 reported a mean (SD) improvement with hypertonic saline of 2.05 (1.3) and with isotonic saline of 1.7 (1.25) (P = 0.015); during week 2 the mean (SD) rise with hypertonic saline was 2.76 (1.45) and with isotonic saline 1.75 (1.6) (P = 0.02) (Eng 1996). When analysed, these data significantly favour hypertonic saline; week 1, MD 0.88 (95% CI 0.19 to 1.57) and week 2, MD 1.01 (95% CI 0.18 to 1.84) (Analysis 1.7).
1.7. Analysis.
Comparison 1 Hypertonic saline 3% to 7% versus isotonic saline, Outcome 7 Exercise capacity (using a subjective visual analogue score).
3. Measures of QoL and symptom scores
Three trials assessed health‐related QoL (Amin 2010; Elkins 2006a; Rosenfeld 2012). Amin assessed the CFQ‐R for both parent and participant (Amin 2010) and Rosenfeld used the the CFQ‐R for parents only (Rosenfeld 2012). The Elkins trial measured QoL using the SF‐36 questionnaire and the Cystic Fibrosis questionnaire (CFQ) for adults and for parents (Elkins 2006a).
The CFQ‐R domain for parents or participants was assessed in three trials and this demonstrated no statistically significant improvement in the hypertonic saline group, MD 1.62 (95% CI ‐1.69 to 4.92) (Amin 2010; Rosenfeld 2012; Elkins 2006a) (Analysis 1.9). There is moderate heterogeneity for CFQ Parent data (three trials) according to our definition above; the I² value is 47%. This value reverts to zero if the data from the Rosenfeld trial are removed. This trial recruited young children under the age of five years in contrast to the other two trials, which may account for the heterogeneity (Rosenfeld 2012).
1.9. Analysis.
Comparison 1 Hypertonic saline 3% to 7% versus isotonic saline, Outcome 9 Quality of life (change from baseline).
Elkins reported a significantly higher (better) score in the mental health domain of the SF‐36 in participants over 14 years, MD 7.77 (95% CI 1.86 to 13.68); however, the overall score for the CFQ and SF‐36 was not significantly different in those under 14 years of age, MD 2.84 (95% CI ‐7.90 to 13.58) (Elkins 2006a) (Analysis 1.9). There were significantly better results in the domains of role (P = 0.04), emotion (P = 0.03) and health (P = 0.01) in the CFQ Adult compared to the control group. In participants under the age of 14, the digestion domain was significantly better (P = 0.02) in the control group compared to the hypertonic saline group using the CFQ for parents. This trial also measured absenteeism from work and school; and participants in the hypertonic saline group experienced fewer days off work, school or days they were unable to participate in usual activity; seven days in the hypertonic saline group as compared to 24 days in the control group (P < 0.001) (Elkins 2006a).
Two trials assessed symptoms using a VAS with 10 cm scales ranging from ‐5 cm to +5 cm (Eng 1996; Riedler 1996). One trial found significant improvements in symptoms for quality of sleep and feeling of cleared chest measured after one and two weeks of treatment with hypertonic saline 6% (Eng 1996). The Riedler trial looked at a similar VAS for feeling of cleared chest alone which was measured four days after treatment and demonstrated a significant difference in their first block of 10 participants between hypertonic saline 6% and isotonic saline (Riedler 1996). The results of the two trials were pooled and demonstrated a result favouring treatment, MD of 0.97 (95% CI 0.35 to 1.60) (Analysis 1.8).
1.8. Analysis.
Comparison 1 Hypertonic saline 3% to 7% versus isotonic saline, Outcome 8 Feeling of cleared chest (using a subjective visual analogue scale).
One trial compared hypertonic saline versus isotonic saline delivered via a pulsating nebuliser (designed to enhance aerosol deposition into the nose and sinus cavities) for improving nasal and sinus symptoms assessed using the validated SNOT‐20 symptom score (Mainz 2015). This tool is a disease‐specific, health‐related, 20‐item QoL measure for people with rhinosinusitis focusing on rhinogenous as well as on general discomforts. Scores range between 0 and 5 for each item, with higher scores indicating a greater health burden. On the first day of treatment the hypertonic saline group described significantly different worsened symptoms scores, mean (SD) 23.0 (10.4) compared to 24.8 (11.0) in the isotonic saline group (P < 0.005). By day 29 however, there was no difference between the groups; hypertonic saline 20.7 (10.1) and isotonic saline 19.4 (9.6).
4. Pulmonary exacerbations
a. frequency
Two trials reported on this outcome, but although the definition of exacerbations in both were comparable we elected not to pool the data, as the age difference between the participants in the trials was so great (Elkins 2006a; Rosenfeld 2012). We judged the quality of the evidence to be low.
Elkins reported pulmonary exacerbations as a secondary outcome (Elkins 2006a). The trial found there were fewer exacerbations per year requiring intravenous antibiotic therapy in the hypertonic saline group than in the control group, MD ‐0.46 (95% CI ‐0.82 to ‐0.10) (Analysis 1.10) (Figure 2). It was also reported that the mean number of days on which participants met this definition of an exacerbation was 17 days in the control group and six days in the hypertonic saline group (difference, 11 days (95% CI 3 to 19) (P = 0.02)). The interval during which participants remained free of exacerbations was also significantly longer in the hypertonic saline group than in the control group (P = 0.03) (Elkins 2006a). The Elkins trial also found that there was no difference in the mean number of visits not requiring antibiotics, MD ‐0.25 (95% ‐0.84 to 0.34) (Analysis 1.10) (Figure 2).
1.10. Analysis.
Comparison 1 Hypertonic saline 3% to 7% versus isotonic saline, Outcome 10 Average number of exacerbations.
2.
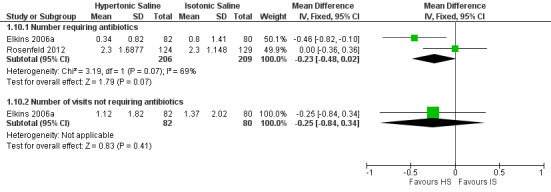
Forest plot of comparison: 1 Hypertonic saline 3% to 7% versus isotonic saline, outcome: 1.10 Average number of exacerbations.
Elkins stated in the original trial report that exacerbations, defined according to signs and symptoms alone, regardless of treatment, were also less frequent in the hypertonic saline group compared to control: 1.32 per participant and 2.74 per participant respectively (difference, 1.42 (95% CI 0.86 to 1.99); P < 0.001. The mean number of days during which participants met criteria for a symptom‐defined exacerbation was 69 days in the control group and 22 days in the hypertonic saline group (difference, 47 days (95% CI 30 to 63); P < 0.001). The time participants remained free of exacerbations was significantly longer in the hypertonic saline group (P < 0.001), with a 48‐week exacerbation‐free survival rate of 41% in the hypertonic saline group and 16% in the control group (Elkins 2006a).
Rosenfeld used protocol‐defined exacerbations as their primary outcome (Rosenfeld 2012). They found no difference in the mean (SD) number of exacerbations; those with hypertonic saline experienced 2.3 (1.69) events per year, compared to isotonic saline 2.3 (1.15) events per year, MD 0.00 (95% CI ‐0.36 to 0.36) (Analysis 1.10).
b. admission to hospital
Elkins reported that there was no difference reported in hospitalisation rates between the hypertonic saline group and the controls (Analysis 1.11).
1.11. Analysis.
Comparison 1 Hypertonic saline 3% to 7% versus isotonic saline, Outcome 11 Average number of hospital admissions per participant.
c. duration of hospital stay (post hoc change)
Neither trial reported the duration of hospitalisation due to pulmonary exacerbations (Elkins 2006a; Rosenfeld 2012).
d. outpatient treatments (hospital in the home, unscheduled visits to the doctor)
Neither trial reported details of outpatient treatments due to pulmonary exacerbations (Elkins 2006a; Rosenfeld 2012).
5. Medication delivery time
No trials reported on this as an outcome.
6. Cost
No trials reported on this as an outcome.
7. Adherence
Three trials judged treatment adherence by the number of returned ampoules or vials (Amin 2010; Elkins 2006a; Rosenfeld 2012). In the cross‐over trial by Amin, the adherence was 95.3% for the hypertonic saline period and 84.5% for the isotonic saline period, this was not statistically significant (P = 0.26) (Amin 2010). Elkins reported adherence as 63% in the control group compared to 64% in the hypertonic saline group (Elkins 2006a). Rosenfeld reported no difference in adherence between those receiving hypertonic saline and isotonic saline, with an overall mean adherence of 75.2% (95% CI 72.2% to 78.2%) (Rosenfeld 2012).
8. Bacteriolgy
Elkins also measured the bacterial load of sputum (Elkins 2006a); but when analysed, there was no significant difference between groups in the concentration of P aeruginosa or S aureus from baseline to 48 weeks (Analysis 1.13).
1.13. Analysis.
Comparison 1 Hypertonic saline 3% to 7% versus isotonic saline, Outcome 13 Change in log10 colony forming units (GFU)/g from baseline at final visit.
Rosenfeld found no new bacterial pathogens and the pathogens that were identified did not differ between the groups (Rosenfeld 2012).
9. Adverse events
Adverse events were reported in six trials (Amin 2010; Chadwick 1997; Elkins 2006a; Eng 1996; Robinson 1999; Rosenfeld 2012). Where appropriate data are available, these are presented in the analyses (Analysis 1.12; Analysis 1.13; Analysis 1.14). None of the results were statistically significant and the quality of the evidence was very low.
1.12. Analysis.
Comparison 1 Hypertonic saline 3% to 7% versus isotonic saline, Outcome 12 Adverse events: acute fall in lung function.
1.14. Analysis.
Comparison 1 Hypertonic saline 3% to 7% versus isotonic saline, Outcome 14 Adverse events.
The trial by Amin reported the overall number of adverse events and adverse events related to the trial drugs, rather than the number of participants experiencing an adverse event. There were no significant differences in cough, hoarseness or chest pain (P = 0.17); however, there were significantly more overall adverse events in the hypertonic saline group for the symptoms of increased sputum production, fever, rhinorrhoea, malaise and ear infections (P = 0.0035) (Amin 2010). Amin further reported a mean (SD) drop in FEV1 % predicted after the first inhalation of hypertonic saline 116 (140) mL and after isotonic saline 41 (88) mL, MD 75.00 (95% CI ‐31.49 to 181.49) (Analysis 1.12).
The Chadwick trial demonstrated that participants with an FEV1 % predicted of 40% to 70% at baseline experienced a significant fall in FEV1 following isotonic saline, while none of the participants fell significantly with hypertonic saline (Chadwick 1997).
The Elkins trial reported the number of adverse events (not participants experiencing adverse events) as pulmonary exacerbations (see above), chest pain, gastro‐intestinal symptoms, headache, joint pain, pharyngitis and tonsillitis; these were significantly fewer in the hypertonic saline group (Elkins 2006a). Adverse drug reactions were also reported, events that in the opinion of the investigator were related directly to the trial medication. These were significantly higher in the hypertonic saline group (n = 14) than the control group (n = 1), P = 0.01, and included cough, chest tightness, pharyngitis, haemoptysis, sinusitis, sneezing, tonsillitis and vomiting (Elkins 2006a).
In the Eng trial there were similar reports of increased cough and haemoptysis; one participant in the hypertonic saline group had to withdraw because of haemoptysis, although it was not clear if this was directly related to treatment (Eng 1996). In the hypertonic saline group, one participant complained of chest tightness and one of throat irritation, with none in the isotonic saline group complaining of these symptoms (Analysis 1.14).
In the 1999 Robinson trial, despite participants being pre‐treated with terbutaline, there was a tendency for those who received hypertonic saline to have a larger fall in FEV1 % predicted within five minutes of receiving hypertonic saline, although the difference was not significant, MD 5.20% (95% CI ‐0.59 to 10.99) (Analysis 1.12). Participants who received hypertonic saline described higher scores for throat irritation on a VAS compared to isotonic saline control, though the number describing throat irritation was not stated. Frequency of cough in those treated with hypertonic saline could not be directly compared to the days when they received the isotonic saline control as to they were encouraged to cough on the control days to match the active day's cough so as not to confound the results of the mucociliary clearance data (Robinson 1999).
Rosenfeld also reported adverse events related to the trial interventions and found that symptoms of cough and found there was no difference in serious adverse events (Rosenfeld 2012).
Hypertonic saline 3% to 7% versus isotonic saline during acute exacerbations of lung disease
Two trials (n = 142) assessed the effect of hypertonic saline during an acute exacerbation of lung disease (Dentice 2016; Riedler 1996). One was of parallel design and we were able to enter data into the analysis (Dentice 2016). The second trial used a cross‐over design and due to data limitations we have reported the results narratively (Riedler 1996). A summary of the results and our judgements with regards to the quality of the evidence can be found in the tables (Table 2).
Primary outcomes
1. Lung function
a. FEV1
Only the Dentice trial assessed the change in lung function, and this was as a secondary outcome (Dentice 2016). Spirometry was performed daily and the investigators reported that FEV1 was higher in the hypertonic saline arm using a mixed‐effects model in the first 10 days of treatment. We were able to assess the change from baseline in FEV1 % predicted after the authors supplied the raw data. There were no differences between those treated with hypertonic saline versus isotonic saline either at treatment day 7, MD 3.95% (95% CI ‐16.69 to 24.59); at treatment day 10, MD ‐2.70% (95% CI ‐24.69 to 19.29); or at time of discharge, MD 5.10% (95% CI ‐14.67 to 24.87) (Analysis 2.1) (low‐quality evidence). Dentice also showed that participants treated with hypertonic saline were more likely to return to their pre‐exacerbation FEV1 than those treated with isotonic saline, 75% versus 57% (number needed to treat was 6 (95% CI 3 to 65)) (Dentice 2016).
2.1. Analysis.
Comparison 2 Hypertonic saline 3% to 7% versus isotonic saline in acute lung disease, Outcome 1 Change in FEV1 from baseline (% predicted).
b. FVC
Only Dentice measured the change in FVC as a secondary outcome in a similar manner to FEV1 (Dentice 2016). We were able to assess the change from baseline in FVC % predicted after the authors supplied the raw data. Again, there were no differences between groups when assessed at treatment day 7, MD 1.10% (95% CI ‐5.74 to 7.94); at day 10, MD 7.90% (95% CI ‐1.86 to 17.66); or at discharge, although significance was nearly reached at this time point, MD 6.1% (95% CI ‐0.68 to 12.88) (Analysis 2.2).
2.2. Analysis.
Comparison 2 Hypertonic saline 3% to 7% versus isotonic saline in acute lung disease, Outcome 2 Change in FVC from baseline (% predicted).
2. Mortality
There were no deaths reported in either short‐term trial (Dentice 2016, Riedler 1996) (low‐quality evidence).
Secondary outcomes
1. Measures of sputum clearance
This outcome was not reported in either trial (Dentice 2016, Riedler 1996).
2. Exercise tolerance
Dentice assessed exercise tolerance using the shuttle walk test comparing the groups at day 7; the results just failed to demonstrate a difference between groups, MD 46.00 m (95% CI ‐14.81 to 106.81) (Analysis 2.3).
2.3. Analysis.
Comparison 2 Hypertonic saline 3% to 7% versus isotonic saline in acute lung disease, Outcome 3 Shuttle walk test.
3.. Measures of QoL and symptom score
Riedler used a VAS to report a feeling of cleared chest in a short‐term cross‐over trial (Riedler 1996). The paper reports median (interquartile range) scores, which we are unable to analyse and so report narratively. The score was significantly higher after hypertonic saline, median (range) 2.0 (0.0 to 3.0) compared to after isotonic saline 0.5 (‐2.0 to 2.3) (P = 0.04), but this was not the case after participants had crossed over to the alternate arm when the median (range) score for hypertonic saline was 2.0 (1.0 to 3.0) and for isotonic saline 1.0 (0.0 to 3.0) (P = 0.463) (Riedler 1996). Dentice also used a VAS (100 mm scale) to measure changes in chest congestion, sleep disturbance and dyspnoea (both daily and at the time of discharge) and analysed the data using a mixed effect model (Dentice 2016). At discharge, the hypertonic saline group had significantly less severe sleep disturbance by 15 mm (95% CI 6 to 23), chest congestion by 9 mm (95% CI 4 to 14) and dyspnoea by 6 mm (95% CI 1 to 12). Dentice also measured QoL using the SF36 and the CFQ at day 7 and at discharge, but no differences were seen between the groups (Dentice 2016).
4. Pulmonary exacerbations
Dentice assessed the length of time until the next pulmonary exacerbation requiring hospitalisation and duration of hospitalisation.
b. admission to hospital
There was no significant difference between treatment groups in the length of time until the next pulmonary exacerbation requiring hospitalisation, hazard ratio 0.86 (95% CI 0.57 to 1.30) (Dentice 2016) (low‐quality evidence).
c. duration of hospital stay
This was the primary outcome in the Dentice trial (Dentice 2016). The length of stay was 12 days in the hypertonic saline group and 13 days in the isotonic saline group, the paper reported this with a MD of 1 day (95% CI 0 to 2), P = 0.07. The mean estimate of one day was below the two‐day difference nominated in the sample size calculation.
5. Medication delivery time
This outcome was not assessed by either trial (Dentice 2016; Riedler 1996).
6. Cost of treatment
This outcome was not assessed by either trial (Dentice 2016; Riedler 1996).
7. Adherence
In Dentice 2016, both treatment arms demonstrated very good adherence; only 6% of the hypertonic saline group and 14% of the control group were less than 75% adherent to the allocated intervention (Dentice 2016). Adherence was not reported by Riedler (Riedler 1996).
8. Bacteriology
Dentice found no difference between the groups in terms of bacterial density when comparing bacterial cultures taken on admission with cultures taken at day 7 (Dentice 2016). There was no difference between the groups for participants who were positive for P aeruginosa on admission and who were negative on day 7 (10% in the hypertonic saline group and 6% in the isotonic saline group). Clearance of S aureus was higher in both groups but still did not show a difference between groups, 25% in the hypertonic group and 24% in the control group. This outcome was not reported by Riedler (Riedler 1996).
9. Adverse events
Both trials reported on this outcome, but the quality of the evidence was judged to be very low. Dentice reported that no participants had an acute fall greater than 15% in FEV1 or oxygen desaturation after their first dose of hypertonic saline (with salbutamol before treatment) (Dentice 2012). There were reports of mild cough and wheeze that resolved in 15 minutes; but there were no serious adverse events reported in either group (Dentice 2012).
Riedler reported that most participants coughed "substantially more" when inhaling hypertonic saline compared to isotonic saline, especially during the first few minutes of inhalation, but this usually resolved before the end of the inhalation period (Riedler 1996).
Hypertonic saline versus mucus mobilising treatments
Hypertonic saline versus rhDNase
Three trials were eligible for inclusion in this comparison (n = 80) (Adde 2004; Ballmann 1998; Suri 2001). All three were cross‐over trials; due to data limitations we analysed two of these as if they were parallel trials (Adde 2004; Ballmann 1998), but were able to analyse the Suri data using the generic inverse variance (Suri 2001). A summary of the main results and judgements on the quality of the evidence are presented in the tables (Table 3).
Primary outcomes
1. Lung function
a. FEV1
All three trials reported on this outcome (Adde 2004; Ballmann 1998; Suri 2001). Suri measured the mean increase in FEV1 % predicted from baseline comparing hypertonic saline 3% 5 mL (3% increase) to daily rhDNase (16% increase) and alternate‐daily rhDNase (14% increase) (Suri 2001). Comparisons between daily hypertonic saline and daily rhDNase have been presented in this review; these were reported by Ballmann and Suri. The results have not been pooled because the duration of the interventions in the two trials was very different; three weeks for the Ballmann trial and three months for the Suri trial (Ballmann 1998; Suri 2001).
After three weeks Ballmann did not demonstrate a significant difference between hypertonic saline and rhDNase, MD 1.60% (95% CI ‐7.96 to 11.16) (Analysis 3.1) (Ballmann 1998) (very low‐quality evidence). However, after three months, Suri found that hypertonic saline showed a lower change in FEV1 % predicted compared to rhDNase, MD 8.00% (95% CI 2.00 to 14.00) (Analysis 3.1) (low‐quality evidence). Both Ballmann and Suri compared the number of participants who improved their FEV1 by 10% or more from baseline after treatment (Ballmann 1998; Suri 2001). At three weeks Ballman reported in the paper that those treated with hypertonic saline were less likely to increase their FEV1 by 10% but our analysis does not show a statistical difference between treatments, OR 1.00 (95% CI 0.25 to 4.00); this was also true for the Suri trial at three months, OR 0.38 (95% CI 0.14 to 1.08) (Analysis 3.2).
3.1. Analysis.
Comparison 3 Hypertonic saline versus rhDNase, Outcome 1 Change in FEV1 (% predicted).
3.2. Analysis.
Comparison 3 Hypertonic saline versus rhDNase, Outcome 2 Improvement in FEV1 >10%.
Adde reported the change in FEV1 % predicted and found no difference between treatments. The results have not been presented in a meta‐analysis as they did not report a MD and standard error (Adde 2004).
b. FVC
Suri reported that there was no statistical difference between daily rhDNase and hypertonic saline, MD 0.03 (95% CI ‐0.06) (Analysis 3.3) (Suri 2001).
3.3. Analysis.
Comparison 3 Hypertonic saline versus rhDNase, Outcome 3 Change in FVC (% predicted).
2. Mortality
None of the trials reported on this outcome.
Secondary outcomes
1. Measures of sputum clearance
None of the trials reported on this outcome.
2. Measures of exercise capacity
Suri measured exercise tolerance using a three‐minute step test at the end of each treatment period. As part of the step test, the changes in the saturation of haemoglobin with oxygen in arterial blood (SaO2), the VAS score and the 'fifteen count breathlessness score' (FCS) were recorded (Suri 2001). They reported no significant differences between the groups for either oxygen saturation, MD ‐0.06 (95% CI ‐0.95 to 0.83), VAS for breathlessness, MD 0.38 (95% CI ‐0.16 to 0.92), or FCS, MD ‐0.05 (95% CI ‐0.44 to 0.34) (Analysis 3.4; Analysis 3.5; Analysis 3.6).
3.4. Analysis.
Comparison 3 Hypertonic saline versus rhDNase, Outcome 4 Exercise tolerance ‐ oxygen saturation.
3.5. Analysis.
Comparison 3 Hypertonic saline versus rhDNase, Outcome 5 Exercise tolerance ‐ VAS for breathlessness.
3.6. Analysis.
Comparison 3 Hypertonic saline versus rhDNase, Outcome 6 Exercise tolerance ‐ FCS.
3. Measures of QoL and symptom scores
Two trials reported on this outcome; however, the results have not been pooled as the outcome measures were not standardised (Adde 2004; Suri 2001). Suri assessed symptoms using the quality of well being self‐administered form 1.04 (Suri 2001). They reported there was no significant difference in scores between the groups, MD 0.03 (95% CI ‐0.01 to 0.07) (Analysis 3.7). Adde assessed symptom scores using a five‐point Likert scale and also reported no significant difference between groups (Adde 2004).
3.7. Analysis.
Comparison 3 Hypertonic saline versus rhDNase, Outcome 7 Mean percentage change in quality of life score.
4. Pulmonary exacerbations
Suri described pulmonary exacerbations during the trial, with 15 episodes occurring during treatment with hypertonic saline and 18 with daily rhDNase (Suri 2001). The authors of the paper report that there was no statistical difference between treatments (low‐quality evidence).
5. Medication delivery time
Ballmann compared delivery time in minutes between hypertonic saline 5.85% 10 mL twice‐daily and rhDNase 2.5 mg twice‐daily and found that hypertonic saline took significantly longer to nebulise, MD ‐31.00 minutes (95% CI ‐37.56 to ‐24.44) (Analysis 3.8). The large difference in nebulisation time relates to the difference in volumes nebulised using the same Pari master or Pari LL nebuliser (Ballmann 1998).
3.8. Analysis.
Comparison 3 Hypertonic saline versus rhDNase, Outcome 8 Delivery time (minutes).
6. Cost
Two trials compared the cost of treatment between rhDNase and hypertonic saline (Ballmann 1998; Suri 2001). Ballmann compared one month of hypertonic saline treatment with rhDNase (Deutschmark (DM) 2427) to hypertonic saline (DM 86) (Ballmann 1998). Suri compared total healthcare cost for the treatments incorporating not just drug cost but also admission, outpatient review, cost of investigations and the cost of utilising community resources (Suri 2001).
Suri investigated the mean cost difference between daily rhDNase and hypertonic saline and alternate‐day rhDNase at 12 weeks. As reported in the original paper, the drug cost per day was reported to be GBP 0.38 for hypertonic saline, GBP 20.39 for once‐daily rhDNase and GBP 10.20 for alternate‐day rhDNase. The average total cost of an occupied bed per day ranged from GBP 280 to GBP 397 (Suri 2001). The mean annual drug cost of daily rhDNase was GBP 1755 compared with GBP 37 for hypertonic saline and the MD in the total health service cost between daily rhDNase and hypertonic saline was GBP 1409.00 (95% CI GBP 440.00 to GBP 2318.00). The MD in total cost between daily rhDNase and alternate‐day rhDNase was GBP 513.00 (95% CI GBP ‐546.00 to GBP 1510.00) (Suri 2001).
7. Adherence
Only one trial assessed adherence; Suri reported the number of returned treatment packs (Suri 2001). Those on rhDNase had compliance rates of 84%, with those on hypertonic saline having 93% compliance.
8. Bacteriology
Suri assessed sputum microbiology throughout the trial and did not identify any new pathogens acquired during the course of the trial amongst individuals (Suri 2001).
Adde compared P aeruginosa growth and found no difference in bacterial load using hypertonic saline compared to rhDNase (Adde 2004).
9. Adverse events
Suri found similar rates of adverse events between all treatment arms, but the quality of the evidence was judged to be low (Suri 2001). Three participants had to withdraw because of a fall of 15% or greater in FEV1 after receiving hypertonic saline despite pre‐treatment with bronchodilators. Increased cough was very common with all treatments and reported in 13 participants using hypertonic saline, 17 on daily rhDNase and 23 on alternate day rhDNase.
Hyperontic saline versus amiloride
One trial was eligible for inclusion in this comparison (n = 12) (Robinson 1996). The main results and the quality of the evidence are presented in the tables (Table 4).
Primary outcomes
1. Lung function
The included trial did not report on this as an outcome.
2. Mortality
The included trial did not report on this as an outcome.
Secondary outcomes
1. Measures of sputum clearance
The included trial looked at the effect of amiloride with hypertonic saline and amiloride alone compared to isotonic saline (Robinson 1996). There was no additional difference with amiloride and amiloride alone was not significantly different from isotonic saline (very low‐quality evidence).
2. Measures of exercise capacity
The included trial did not report on this as an outcome.
3. Measures of QoL and symptom scores
The included trial did not report on this as an outcome.
4. Pulmonary exacerbations
The included trial did not report on this as an outcome.
5. Medication delivery time
The included trial did not report on this as an outcome.
6. Cost
The included trial did not report on this as an outcome.
7. Adherence
The included trial did not report on this as an outcome.
8. Bacteriology
The included trial did not report on this as an outcome.
9. Adverse events
The included trial did not report on this as an outcome.
Hypertonic saline versus sodium‐2‐mercaptoethane sulphonate (Mistabron®)
One trial was eligible for inclusion in this comparison (n = 29) (Weller 1980). Only 3 mL of 7% hypertonic saline was used in the Weller trial (Weller 1980). We were unable to enter data into the graphs, because SDs were not reported in the paper. The main results and the judgements on the quality of the evidence are presented in the tables (Table 5).
Primary outcomes
1. Lung function
Weller compared sodium‐2‐mercaptoethane sulphonate 20% 3 mL twice‐daily to hypertonic saline 7% 3 mL twice‐daily (Weller 1980). Participants were divided into sputum producers and non‐sputum producers. The sputum producers who were given sodium‐2‐mercaptoethane sulphonate increased peak expiratory flow (PEF) (change from baseline of 7 L/min) compared to hypertonic saline (change from baseline of ‐2 L/min, P < 0.02). There was no significant difference in PEF in the non‐sputum producers. Furthermore, FVC was not significantly different in either group. The Vmax 50% vital capacity (VC) increased in the sputum producers with sodium‐2‐mercaptoethane sulphonate (+10) compared to hypertonic saline (0, P < 0.005). In the non‐sputum producers, hypertonic saline improved Vmax 50% VC (+14) compared to sodium‐2‐mercaptoethane sulphonate (‐5), but this was not significant. In the sputum producers group residual volume (RV) and total lung capacity (TLC) improved with hypertonic saline (+1) compared to sodium‐2‐mercaptoethane sulphonate (‐5, P < 0.05). In the non‐sputum producers group, sodium‐2‐mercaptoethane sulphonate had no effect (0) on RV or TLC, whilst hypertonic saline had some effect (‐6), again this did not reach significance.
2. Mortality
The included trial did not report on this as an outcome.
Secondary outcomes
1. Measures of sputum clearance
Weller described no significant difference in sputum volume, colour or cough frequency between the groups (Weller 1980) (very low‐quality evidence).
2. Measures of exercise capacity
The included trial did not report on this as an outcome.
3. Measures of QoL and symptom scores
The included trial did not report on this as an outcome.
4. Pulmonary exacerbations
There was no change in sputum bacteriology or the number of courses of antibiotics prescribed (Weller 1980) (very low‐quality evidence).
5. Delivery time
The included trial did not report on this as an outcome.
6. Cost
The included trial did not report on this as an outcome.
7. Adherence
The included trial did not report on this as an outcome.
8. Bacteriology
The included trial did not report on this as an outcome.
9. Adverse events
In the Weller trial, the group given sodium‐2‐ mercaptoethane sulphonate and hypertonic saline described coughing at the beginning of their inhalations, no other serious adverse events occurred (Weller 1980) (very low‐quality evidence).
Hypertonic saline versus mannitol
One trial was eligible for inclusion in this comparison (n = 12) (Robinson 1999). In the analysis, due to data limitations, data from this cross‐over trial have been entered and analysed as if they were from a parallel trial. The main results and the quality judgements on the evidence are presented in the tables (Table 6).
Primary outcomes
1. Lung function
The change from the pre‐intervention FEV1 was reported at five minutes after inhalation and again 95 minutes later (Robinson 1999). The paper reported the mean (standard error) value for each intervention and their respective controls at each time‐point, but only reported the level of significance compared to control. At five minutes post inhalation the mean (standard error) change in FEV1 for hypertonic saline was ‐5.8 (1.2) and for mannitol ‐7.3 (2.5); at the later time‐point the mean (standard error) difference was ‐2.0 (0.7) for hypertonic saline and ‐1.8 (2.7) for mannitol (Robinson 1999) (very low‐quality evidence).
2. Mortality
The included trial did not report on this as an outcome.
Secondary outcomes
1. Measures of sputum clearance
In the included trial, there was no significant difference between mannitol at 300 mg and hypertonic saline for matched voluntary cough (Robinson 1999) (very low‐quality evidence).
2. Measures of exercise capacity
The included trial did not report on this as an outcome.
3. Measures of QoL and symptom scores
In the 1999 trial, Robinson used a VAS to assess the need to cough and reported that there was no difference between hypertonic saline 6% and mannitol 300 mg (Robinson 1999).
4. Pulmonary exacerbations
The included trial did not report on this as an outcome.
5. Medication delivery time
Robinson compared the time taken to nebulise hypertonic saline 6% (4.4 mL) to mannitol 300 mg and found hypertonic saline took less time, MD ‐6.10 min (95% CI ‐7.32 to ‐4.88) (Analysis 4.1) (Robinson 1999).
4.1. Analysis.
Comparison 4 Hypertonic saline versus mannitol, Outcome 1 Delivery time (mins).
6. Cost
The included trial did not report on this as an outcome.
7. Adherence
The included trial did not report on this as an outcome.
8. Bacteriology
The included trial did not report on this as an outcome.
9. Adverse events
In the 1999 Robinson trial, mannitol was regarded as more irritating than the control on VAS. While FEV1 fell significantly five minutes after treatment with both mannitol and hypertonic saline 6% compared to control (P = 0.004), by 95 minutes there was no significant difference between the groups (Robinson 1999) (very low‐quality evidence).
Discussion
Summary of main results
Hypertonic saline versus control (stable disease)
Eleven trials evaluated hypertonic saline compared to control in participants with stable lung disease (Amin 2010; Cardinale 2003; Chadwick 1997; Elkins 2006a; Eng 1996; Laube 2009; Mainz 2015; Robinson 1996; Robinson 1997; Robinson 1999; Rosenfeld 2012). This review has shown that, compared to placebo, the regular use of nebulised hypertonic saline by adults and children over the age of 12 years with CF and stable lung disease appears to lead to a modest improvement in lung function (FEV1 % predicted) after four weeks of treatment (very low‐quality evidence) (Amin 2010; Elkins 2006a; Eng 1996), but this effect does not result in a sustained improvement in lung function after 48 weeks of treatment (low‐quality evidence) (Elkins 2006a). However, this improvement at four weeks may not be seen in those with normal lung function or mild obstruction as measured by spirometry. There were mixed results for other measures of lung function. Our analysis of one paediatric trial showed no difference in LCI at four weeks, although the original investigators reported a difference in favour of hypertonic saline (very low‐quality evidence) (Amin 2010). No trial reported on mortality.
Hypertonic saline improved mucociliary clearance compared to control (very low‐quality evidence) (Laube 2009; Robinson 1996; Robinson 1997; Robinson 1999). Two trials assessed symptom improvement after short‐term treatment using simple VAS and found an improvement in feelings of better chest clearance, exercise tolerance and quality of sleep (Eng 1996; Riedler 1996). In the long‐term trials (48 weeks), Elkins showed treatment may improve some aspects of QoL in adults but not in children, while Rosenfeld showed no improvement in parent‐reported QoL scores (Elkins 2006a; Rosenfeld 2012). Elkins also reported decreased absenteeism from work or school (Elkins 2006a). When delivered by a Pari Sinus nebuliser, hypertonic saline did not improve nasal and sinus symptoms in one trial (Amin 2010).
We found low‐quality evidence from two trials for the outcome of pulmonary exacerbations (Elkins 2006a; Rosenfeld 2012). One multicentre trial (164 adults) showed that nebulised hypertonic saline reduced the frequency of pulmonary exacerbations (Elkins 2006a). However, in a paediatric multicentre trial (321 children under six years of age with stable lung disease) hypertonic saline did not reduce the frequency of pulmonary exacerbations when compared to placebo (Rosenfeld 2012). Our analysis of the adult trial demonstrated a reduction in exacerbations requiring antibiotics, though no difference was seen in hospitalisations (Elkins 2006a). This finding has been felt to be important enough to result in an increasing uptake in treatment with hypertonic saline and led Rosenfeld to determine if early intervention in young children (under six years of age) would be beneficial. The Rosenfeld trial was powered to assess an impact on well‐defined pulmonary exacerbations, but no benefit was seen after 48 weeks of treatment (Rosenfeld 2012).
No trial assessed medication delivery time or the cost of treatment. Limited reporting from three trials indicated no difference between hypertonic saline or control for adherence (Amin 2010; Elkins 2006a; Rosenfeld 2012).
Six trials reported details of adverse events (Amin 2010; Chadwick 1997; Elkins 2006a; Eng 1996; Robinson 1999; Rosenfeld 2012; ). In the long‐term trials there was no increase in serious adverse events, though cough and throat irritation do appear to be more frequent in hypertonic saline compared to control, but this does not appear to have been a serious enough side effect to have led to participant withdrawals (Elkins 2006a; Rosenfeld 2012). Hypertonic saline does not increase the bacterial load of P aeruginosa or S aureus (Elkins 2006a). In the Eng trial, one participant did withdraw from the hypertonic saline group because of haemoptysis; although it is not proven that this was a consequence of the treatment (Eng 1996).
Hypertonic saline versus control (acute exacerbation)
Two trials assessed hypertonic saline in participants with an acute exacerbation (Dentice 2016; Riedler 1996). Hypertonic saline remains the only reported mucus clearing therapy that has been used in the context of acute exacerbations (Dentice 2016; Riedler 1996). In adults admitted to hospital with an acute exacerbation of CF lung disease, hypertonic saline did not lead to a greater improvement in lung function as measured by FEV1 (low‐quality evidence) and FVC, although a greater proportion of those treated with hypertonic saline regained their pre‐exacerbation FEV1 (Dentice 2016). No deaths were reported in either trial (low‐quality evidence). Neither trial assessed measures of sputum clearance. Hypertonic saline did lead to improvements in symptoms and QoL as well as a modest reduction in length of hospital stay, although it did not lengthen the time until the next exacerbation (low‐quality evidence). No trial assessed medication delivery time or the cost of treatment. Only one trial reported on adherence and assessed this as very good in both treatment arms (Dentice 2016), The same trial reported no difference in any measures of bacteriology. Even though these trials treated participants with acute exacerbations of lung disease, Dentice reported there were no serious adverse events and the intervention was well‐tolerated (Dentice 2016); Riedler reported that initially most participants coughed "substantially more" when inhaling hypertonic saline compared to isotonic saline, but this usually resolved before the end of the inhalation period (Riedler 1996).
Hypertonic saline versus rhDNase
Three trials compared hypertonic saline to rhDNase (Adde 2004; Ballmann 1998; Suri 2001). In comparison with rhDNase, there was no difference between treatments at three weeks (very low‐quality evidence) (Ballmann 1998), but at three months hypertonic saline is less likely to result in an improvement in lung function in those with stable lung disease (very low‐quality evidence) (Suri 2001). Neither trial assessed LCI, mortality or measures of sputum clearance. One trial reported at 12 weeks on the change in exercise tolerance, dyspnoea, oxygen saturation during exercise and symptom score and found no differences between those treated with rhDNase and hypertonic saline (Suri 2001). In the same trial, all treatment arms experienced a high frequency of pulmonary exacerbations, but this may be a reflection of the severity of the groups underlying lung disease (very low‐quality evidence). In terms of cost both Ballmann and Suri found hypertonic saline to be less expensive compared to rhDNase (Ballmann 1998; Suri 2001). One trial found that hypertonic saline took significantly longer than rhDNase to administer (Ballmann 1998) and this may have implications for adherence to treatment. Suri specifically examined adherence, which appeared high and comparable to rhDNase in the three‐month Suri trial (Suri 2001). Only one trial assessed adverse events and found similar rates of events between groups (Suri 2001) (very low‐quality evidence). Acute bronchospasm remains a concern with hypertonic saline. Despite pre‐treatment of participants with bronchodilators, three were excluded from the Suri trial due to a fall in FEV1 greater than 15% (Suri 2001).
Hypertonic saline versus amiloride
One small trial evaluated this comparison, but did not report on most of our outcomes (Robinson 1996). The trial did report that there was no difference between hypertonic saline and amiloride in terms of sputum clearance (very low‐quality evidence).
Hypertonic saline compared with sodium‐2‐mercaptoethane sulphonate (Mistabron®)
One small trial evaluated this comparison, but did not provide data we were able to analyse (Weller 1980). The trial did not report FEV1, but reported mixed results for other lung function measurements. There was no difference between groups in measures of sputum clearance, sputum bacteriology or the number of courses of antibiotics prescribed (very low‐quality evidence). All participants described coughing at the beginning of their inhalations, no other serious adverse events occurred (Weller 1980) (very low‐quality evidence).
Hypertonic saline versus mannitol
One cross‐over trial compared hypertonic saline to mannitol (Robinson 1999). Investigators assessed FEV1 at up to 95 minutes, which is not of clinical relevance to this review (very low‐quality evidence), and reported within‐group changes from baseline but no data for between‐group comparisons. There was no difference in sputum clearance between groups (very low‐quality evidence). The trial also reported no difference between groups in symptoms (the need to cough) and that hypertonic saline took less time to nebulise than mannitol. However, mannitol was considered to be more irritating than hypertonic saline (very low‐quality evidence). No other outcomes from this review were reported.
Overall completeness and applicability of evidence
The rationale behind the use of hypertonic saline is the inherent defect in the CFTR that results in abnormal airway mucus and reduced mucus clearance. This in turn is likely to predispose to recurring infection which continues to have the greatest impact on mortality and morbidity in CF. Treatment to improve mucociliary clearance has been proposed to at least retard this progressive destructive process, to provide an adjunct to physical therapies and to reduce the reliance on antimicrobial use.
A series of small proof‐of‐concept trials included in this review initially showed that hypertonic saline resulted in an improvement in measures of mucociliary clearance over isotonic saline and cough alone (Riedler 1996; Robinson 1996; Robinson 1997; Robinson 1999). They showed that a dose of 7% hypertonic saline was more effective than 3%, but there was no significant advantage gained by increasing the dose to 12% or by adding amiloride. While they investigated the dose‐response effect of varying concentrations of saline, they did not look at the impact of the volume nebulised.
The evidence available for hypertonic saline in stable CF lung disease has increased since this review was commenced. However, the conclusions remain limited to a small number of relatively large multicentre trials, that have examined the issue in heterogenous patient populations and unfortunately there are no data on the effects of hypertonic saline in combination with rhDNase. The Elkins trial, while not demonstrating a sustained improvement in lung function, remains promising by demonstrating a reduction in exacerbation frequency and a small improvement in QoL (Elkins 2006a). This effect was not seen in in children without airflow limitation as measured by spirometry (FEV1 over 80% predicted) (Amin 2010). It did improve mucociliary clearance and ventilation inhomogeneity as measured by the LCI in those with mild lung disease, though the long‐term implications of this finding are unclear. When delivered following a bronchodilator it is an inexpensive additional therapy for people with CF, which does not appear to be associated significant adverse effects (Amin 2010). In younger children hypertonic saline did not improve measures of infant lung function or reduce pulmonary exacerbations (Rosenfeld 2012).
We chose lung function as our primary outcome because of its relationship in the long term with mortality in CF and by inference to see if hypertonic saline would alter this decline (Courtney 2007). Elkins performed the largest trial in adults and children over six years of age, to address whether hypertonic saline (7%) compared to isotonic saline (0.9%) would improve FEV1 after 48 weeks of treatment (Elkins 2006a). They failed to demonstrate a significant improvement in lung function over 48 weeks, although our pooled analysis of both the smaller trial by Eng together with the Elkins trial did demonstrate a small improvement in FEV1 after four weeks of treatment (Elkins 2006a; Eng 1996).
One limitation of using lung function markers such FEV1 is that, despite their association with long‐term outcomes, they may be relatively insensitive to small changes in CF, especially over time given the progressive nature of the disease. Also, those with intact lung function who may benefit most in the long term from hypertonic saline, would be less likely to demonstrate an improvement. With this in mind, Amin used the LCI, which is a novel measure of ventilation inhomogeneity using an inert gas multiple‐breath washout technique and which has been validated in CF and shown to be more sensitive in detecting early airways disease (Gustafsson 2008). Amin proposed that in those with only mild airflow limitation this would be more sensitive to change with hypertonic saline, and demonstrated this after four weeks of treatment, although there was no improvement seen with FEV1 or even improvement in symptom scores (Amin 2010). It remains to be determined if this change will result in significant long‐term improvement in health for individuals with CF or how this may relate to the progression of CF lung disease.
It is important to realise that before participants started the trials, they were pre‐treated with salbutamol and lung function measured to determine whether they could tolerate hypertonic saline and only those who did tolerate it were allowed to carry on. This means that the results of the trials only apply to those people with CF who can tolerate hypertonic saline.
Ballmann compared hypertonic saline 5.75% 10 mL twice‐daily to nebulised rhDNase 2.5 mg and found that in both groups FEV1 improved to a similar degree in three weeks (Ballmann 1998). In the Suri trial, twice‐daily 5 mL hypertonic saline 7% was compared to daily and alternate‐day rhDNase 2.5 mg (Suri 2001). When treated with either rhDNase regimen, participants had a significant improvement in lung function from baseline, but when treated with hypertonic saline there was an increase of only 3% from baseline, less than seen in the Ballmann and Eng trials (Ballmann 1998; Eng 1996). In both of these trials 10 mL of hypertonic saline was used compared to 5 mL as used by Suri and Weller (Suri 2001; Weller 1980). This raises the possibility that the effectiveness of treatment may also depend on the total volume of saline nebulised and this may account for the lower effect size seen by Suri (Suri 2001). In addition, Suri found a wide variation in response to treatment with both rhDNase and hypertonic saline, with over 50% of participants demonstrating a more than 10% increase in FEV1 with rhDNase and 35% of participants with hypertonic saline. Thus, despite the overall reduced effect seen with hypertonic saline on lung function in individuals, both rhDNase and hypertonic saline have the potential to substantially improve lung function in the medium term. The wide variation seen in response to both hypertonic saline and rhDNase raises the possibility that there may be subgroups of people with CF who are more likely to respond to efforts to improve mucociliary clearance. It also suggests that some individuals may respond better to one treatment compared to the other and physicians may wish to consider this particularly in individuals who fail to respond to rhDNase.
Quality of the evidence
Using GRADE, we judged the quality of the evidence from this review to be of very low to low quality, depending on the outcome measured (Table 1; Table 2; Table 3; Table 4; Table 5; Table 6).
We do not think that the way the trials were designed affected the results, with even the cross‐over trials allowing for adequate washout at least in terms of lung function change. We judged that all participants had equal chances of being in either of the treatment groups. However, in all but two trials (Dentice 2016; Elkins 2006a), the taste was not masked and participants would be able to identify the hypertonic saline. The participants who dropped out of the trials appear to have been accounted for and are unlikely to influence the results.
Potential biases in the review process
Data were extracted, using predefined data extract sheets, independently by each of the authors and then a consensus was reached in regard to inclusion or exclusion. In addition, both the authors were involved in one trial (Elkins 2006a) and Wark was involved in a further trial (Dentice 2016). Where one or more author(s) of this Cochrane Review was a co‐author on an included trial, a third party performed the data extraction and assessment of quality for that trial.
Agreements and disagreements with other studies or reviews
The first therapy used to enhance mucus clearance was rhDNase; it is effective in improving lung function in CF and has been adopted in most countries (Ramsey 1994). A separate systematic review found that when compared with placebo rhDNase improves lung function in people with CF in trials lasting from one month to two years (Yang 2018). There was a decrease in pulmonary exacerbations in trials of six months or longer. Voice alteration and rash were the only adverse events reported with increased frequency in randomised controlled trials. Although investigators also concluded that there that there was not enough evidence to firmly conclude if rhDNase is superior to hyperosmolar agents such as hypertonic saline in improving lung function. Nebulised rhDNase is a relatively expensive treatment and in many countries its use is restricted to those who have degrees of impairment of lung function and who demonstrate an improvement in pulmonary function tests during a trial period (Ramsey 1994).
Similar to the authors of the Cochrane Review of rhDNase (Yang 2018), we do not believe there is sufficient evidence to conclusively show superiority of rhDNase over hypertonic saline; however, the effect size does appear to be greater for the former.
The other hyperosmolar agent that has been adapted for use as a treatment in CF has been inhaled mannitol. This treatment is the subject of a Cochrane Review which found that at least for up to six months, mannitol led to an improvement in lung function (Nevitt 2018). The review did not find that it improved QoL, but it did reduce exacerbation frequency. While mannitol was assessed against control, against rhDNase and in combination with rhDNase, it was not assessed against hypertonic saline.
Authors' conclusions
Implications for practice.
There is low to very low‐quality evidence showing the benefits of hypertonic saline compared to placebo in those over the age of 12 years with cystic fibrosis (CF) in terms of improvement in lung function at least in the short term, but it should not be used in preference to dornase alfa (rhDNase). At this stage the benefit appears to be a modest reduction in frequency of pulmonary exacerbations, and an improvement in chest symptoms, though evidence does not exist to say in whom it works best. In children under the age of 12 years we have not found sufficient evidence to justify its routine use. In those over the age of six years, an improvement in exacerbation frequency was not seen, and in this cohort of children with milder lung disease, the modest benefits seen with hypertonic saline in earlier trials could not be detected. Improvement in one small cross over trial in 10 children (very low‐quality evidence) showed a small improvement this was not seen in our analysis . A larger trial would be needed to determine if this improvement can be related to clinical outcomes in children under 12 years of age.
The variation in response seen in individuals to both rhDNase and hypertonic saline raises the possibility that certain individuals will respond better to one agent compared to the other. In order to assess the individual response to therapy it would therefore be reasonable to conduct a modified N of 1 trial in people with CF where the person acts as their own control, on and off treatment. Suitable individuals to consider therapy are those in the clinic who fail to respond, are ineligible or are unable to tolerate rhDNase; with hypertonic saline used as an alternative agent to increase mucociliary clearance. Unfortunately there are no data on the effects of hypertonic saline in combination with rhDNase.
In the majority of trials hypertonic saline was used after pre‐treatment with bronchodilators and as an adjunct to chest physiotherapy; in both cases this may be important to ensure its efficacy. When delivered following a bronchodilator, hypertonic saline is an inexpensive and safe therapy for people with CF.
Implications for research.
The effect hypertonic saline has on mucus clearance and consequently short‐term improvements in lung function is relatively small. Future assessments of efficacy for agents that improve mucociliary clearance should be assessed in robust longer‐term randomised controlled trial designs. These should consider alternative primary outcomes, such as pulmonary exacerbations or validated quality of life (QoL) measures, as these appear to be more responsive to change for this intervention than conventional lung function tests. Measurement of pulmonary exacerbation rates are difficult and would benefit from tighter clinical definitions, especially around milder exacerbations. The emergence of more sensitive measures of early disease such as lung clearance index (LCI) or chest computer tomography (CT), may provide novel outcomes that are a more sensitive index to change. It may be possible, using these outcomes, to determine in longitudinal trials if hypertonic saline increases the time to next exacerbation and if there is a sustained reduction in exacerbation frequency. It may then be possible to see if this translates to a more compelling improvement in QoL. Such long‐term trials are difficult to undertake in CF, but data could be obtained in retrospect from national data registries.
There is also a good case for undertaking equivalence trials comparing hypertonic saline to other interventions, as hypertonic saline is likely to remain less costly, especially where new treatments tend to be associated with increasing costs.
Consideration should be given to defining if there are groups of individuals who will respond better to hypertonic saline or other mucociliary clearance agents, especially rhDNase so as to better tailor treatment. Any future trials of hypertonic saline should determine who will benefit most from this intervention. Trials that combine mucociliary agents and or physical therapy also need to be considered to assess efficacy.
What's new
| Date | Event | Description |
|---|---|---|
| 11 September 2018 | New search has been performed | A major update of the review was conducted with 90 new references identified from searches of the Cochrane Cystic Fibrosis and Genetic Disorders Trials Register, clinicaltrials.gov and the WHO ICTRP. New trials Five important new trials (24 references) have been included (Amin 2010; Dentice 2016; Laube 2009; Mainz 2015; Rosenfeld 2012). This increased the number of participants in the trial from 442 to 966. Nine new trials (27 references) have also been excluded (Brivio 2016; Buonpensiero 2010; DeCono 2008; Dentice 2012; EUCTR2007‐002707‐40‐BE; Grasemann 2013; IRCT20180307038994N1; NCT01094704; O'Neill 2017; Ros 2012; San Miguel 2016; Van Ginderdeuren 2008; Van Ginderdeuren 2011). 16 newly identified trials (21 references) are currently awaiting assessment until further information is available to allow inclusion or exclusion (Amin 2016; Balinotti 2015; Brown 2010; Corcoran 2017; Donaldson 2013; Dwyer 2013; Hofmann 1997; NCT00928135; NCT01355796; NCT01377792; NCT01619657; NCT02378467; NCT03391414; Nenna 2017; Palacio 2014; PRESIS 2018). Three new trials (four references) are listed as ongoing (NCT02276898; NCT02343445; NCT02950883). Previously identified trials 11 additional references have been added to five already included trials (Elkins 2006a; Eng 1996; Robinson 1997; Robinson 1999; Weller 1980). One additional reference has been added to an already excluded trial (Donaldson 2006). Two trials (two references) which were previously listed as 'Awaiting classification' have been excluded (Elkins 2006b; Vanlaethem 2008). An additional reference to the Elkins trial was identified in the latest searches. One trial (one reference) previously excluded has been moved to 'Awaiting classification' pending further information (Hofmann 1997). |
| 11 September 2018 | New citation required and conclusions have changed | The conclusions have been amended in light of the new evidence. The addition of new data has allowed us to conclude that hypertonic saline appears to be an effective adjunct to physiotherapy during acute exacerbations of lung disease in adults. |
History
Protocol first published: Issue 3, 1998 Review first published: Issue 2, 1999
| Date | Event | Description |
|---|---|---|
| 26 April 2010 | Amended | Contact details updated. |
| 31 December 2008 | New citation required and conclusions have changed | A large multi‐centre trial has been included in the review, resulting in the conclusions being updated (Elkins 2006a). |
| 31 December 2008 | New search has been performed | A search of the Group's trials register identified two new eligible trials (Cardinale 2003; Elkins 2006a). These trials have now been included in the review. A trial previously listed in Studies awaiting classification has now been included in the review (Adde 2004). Two studies identified by the searches have now been excluded from the review (Donaldson 2006; Kobylyansky 2000). A further reference to the already included Suri trial has been added (Suri 2001). Two trials have been added to Studies awaiting classification (Elkins 2006b; Vanlaethem 2008). |
| 12 November 2008 | Amended | Converted to new review format. |
| 2 May 2005 | New citation required but conclusions have not changed | No new trials have been included. One trial is currently awaiting assessment (Adde 2004). Individual patient data have kindly been provided by Dr Adde and will be included in a future update of the review. Further data have been included for the Suri trial (Suri 2001). The layout of the analysis has been changed from the previous updates to ensure clarity for the reader. |
| 2 October 2003 | New search has been performed | Six additional references to the already included Suri trial have been added (Suri 2001). This did not add greatly to the data already present. Two of the references looked at the cost of treatment with HS and rhDNase and additional data has been added. One other reference looked at the effect of rhDNase and HS on airway inflammation and the data has now been included.
The other references did not add to the data already presented. One additional reference to the already included Ballmann trial has been added (Ballmann 1998). No additional data has been included in the review. One additional reference to the already included Riedler trial has been added (Riedler 1996). No additional data has been included in the review. |
| 3 November 2002 | New search has been performed | An additional trial was found and incorporated in the review (Suri 2001). This was a relatively large clinical trial comparing hypertonic saline and rhDNase. Significant changes have been made to the review. |
| 3 November 2001 | New search has been performed | With this update significant changes to style were made particularly in the order of the outcomes and the presentation of the results. The Suri trial was added and this contained a large amount of additional information particularly concerning the effect of hypertonic saline versus DNase on lung function (Suri 2001). |
Acknowledgements
The authors would like to thank Dr G Ryan, Sir Charles Gardiner Hospital Perth Western Australia for editorial advice on the previous versions of the review.
We would also like to thank the authors of seven of our included trials for supplying additional data and clarifying points of fact (Adde 2004; Amin 2010; Dentice 2016; Elkins 2006a; Donaldson 2006; Robinson 1996; Rosenfeld 2012; Suri 2001). We further thank Dr Adde and Ruth Dentice for providing individual patient data from their trials.
We would also like to thank Mr Ashley Jones for his contribution to the review (as author), from Issue 3, 2005 to Issue 1, 2009, inclusive, Professor P Gibson of the Priority Research Centre for Asthma and Respiratory Disease at The University of Newcastle and Ms J Roberts previously of the Airways research Centre John Hunter Hospital Australia for technical assistance with the first analysis.
We would like to thank Mrs Nikki Jahnke and Ms Tracey Remington for their considerable assistance with preparation of this review.
This project was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Cochrane Cystic Fibrosis and Genetic Disorders Group. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS or the Department of Health.
Appendices
Appendix 1. Electronic Searches
| Database or Resource | Strategy |
| Clinicaltrials.gov | [Advanced Search Form] Other Terms: hypertonic Study Type: Interventional Studies Condition/ Disease: cystic fibrosis |
| WHO ICTRP | cystic fibrosis AND hypertonic |
Data and analyses
Comparison 1. Hypertonic saline 3% to 7% versus isotonic saline.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Change in FEV1 (% predicted) | 3 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.1 At 2 ‐ 4 weeks (all participants) | 3 | 225 | Mean Difference (IV, Fixed, 95% CI) | 3.44 [0.67, 6.21] |
| 1.2 At 2 ‐ 4 weeks (participants aged 14 years and over) | 2 | 205 | Mean Difference (IV, Fixed, 95% CI) | 4.15 [1.14, 7.16] |
| 1.3 At 2 ‐ 4 weeks (children FEV1 > 80% predicted) | 1 | 20 | Mean Difference (IV, Fixed, 95% CI) | ‐0.42 [‐7.45, 6.61] |
| 1.4 At 12 weeks | 1 | 149 | Mean Difference (IV, Fixed, 95% CI) | 4.1 [‐0.08, 8.28] |
| 1.5 At 24 weeks | 1 | 140 | Mean Difference (IV, Fixed, 95% CI) | 5.37 [1.03, 9.71] |
| 1.6 At 36 weeks | 1 | 134 | Mean Difference (IV, Fixed, 95% CI) | 3.63 [‐1.56, 8.82] |
| 1.7 At 48 weeks | 1 | 134 | Mean Difference (IV, Fixed, 95% CI) | 2.31 [‐2.72, 7.34] |
| 2 Change in FVC (% predicted) | 3 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2.1 At 2 ‐ 4 weeks | 3 | 225 | Mean Difference (IV, Fixed, 95% CI) | 1.07 [‐1.63, 3.78] |
| 2.2 At 12 weeks | 1 | 141 | Mean Difference (IV, Fixed, 95% CI) | 4.56 [0.79, 8.33] |
| 2.3 At 24 weeks | 1 | 140 | Mean Difference (IV, Fixed, 95% CI) | 3.64 [0.17, 7.11] |
| 2.4 At 36 weeks | 1 | 134 | Mean Difference (IV, Fixed, 95% CI) | 3.40 [‐0.82, 7.62] |
| 2.5 At 48 weeks | 1 | 134 | Mean Difference (IV, Fixed, 95% CI) | 2.76 [‐1.09, 6.61] |
| 3 Mean change in FEV0.5 (mL) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 3.1 At 48 weeks | 1 | 45 | Mean Difference (IV, Fixed, 95% CI) | 41.0 [0.96, 81.04] |
| 4 Lung clearance index (LCI) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 4.1 At 4 weeks | 1 | 19 | Mean Difference (IV, Fixed, 95% CI) | ‐1.03 [‐2.76, 0.70] |
| 5 Radiolabelled isotope clearance | 3 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5.1 At 60 mins | 3 | 68 | Mean Difference (IV, Fixed, 95% CI) | 6.14 [2.56, 9.72] |
| 6 Mucociliary clearance measured as area under the curve | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 6.1 Single dose | 2 | 44 | Mean Difference (IV, Fixed, 95% CI) | ‐212.06 [‐271.64, ‐152.48] |
| 7 Exercise capacity (using a subjective visual analogue score) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 7.1 At Week 1 | 1 | 52 | Mean Difference (IV, Fixed, 95% CI) | 0.88 [0.19, 1.57] |
| 7.2 At Week 2 | 1 | 52 | Mean Difference (IV, Fixed, 95% CI) | 1.01 [0.18, 1.84] |
| 8 Feeling of cleared chest (using a subjective visual analogue scale) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 8.1 At up to Week 1 | 2 | 72 | Mean Difference (IV, Fixed, 95% CI) | 0.97 [0.35, 1.60] |
| 9 Quality of life (change from baseline) | 3 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 9.1 CFQ parent | 3 | 365 | Mean Difference (IV, Fixed, 95% CI) | 1.62 [‐1.69, 4.92] |
| 9.2 CFQ 14+ | 1 | 91 | Mean Difference (IV, Fixed, 95% CI) | 7.77 [1.86, 13.68] |
| 9.3 SF36 | 1 | 91 | Mean Difference (IV, Fixed, 95% CI) | 2.84 [‐7.90, 13.58] |
| 10 Average number of exacerbations | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 10.1 Number requiring antibiotics | 2 | 415 | Mean Difference (IV, Fixed, 95% CI) | ‐0.23 [‐0.48, 0.02] |
| 10.2 Number of visits not requiring antibiotics | 1 | 162 | Mean Difference (IV, Fixed, 95% CI) | ‐0.25 [‐0.84, 0.34] |
| 11 Average number of hospital admissions per participant | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 11.1 At 48 weeks | 1 | 162 | Mean Difference (IV, Fixed, 95% CI) | ‐0.13 [‐0.48, 0.22] |
| 12 Adverse events: acute fall in lung function | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 12.1 Acute fall in FEV₁ % predicted | 1 | 24 | Mean Difference (IV, Fixed, 95% CI) | 5.2 [‐0.59, 10.99] |
| 12.2 Acute fall in FEV₁ mL | 1 | 19 | Mean Difference (IV, Fixed, 95% CI) | 75.0 [‐31.49, 181.49] |
| 13 Change in log10 colony forming units (GFU)/g from baseline at final visit | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 13.1 P. aeruginosa | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 13.2 S. aureus | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 14 Adverse events | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 14.1 Cough | 2 | 373 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.03 [0.78, 1.35] |
| 14.2 Pharyngitis | 1 | 52 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.79 [0.12, 65.38] |
| 14.3 Chest tightness | 2 | 373 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.66 [0.11, 3.98] |
| 14.4 Haemoptysis | 1 | 52 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.21, 4.17] |
| 14.5 Vomiting | 1 | 321 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.52 [0.13, 2.03] |
| 14.6 Wheezing | 1 | 321 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.06 [0.38, 11.11] |
| 14.7 Fever | 1 | 321 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.99 [0.58, 1.68] |
| 14.8 Nasal congestion | 1 | 321 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.76 [0.42, 1.37] |
| 14.9 Ear infection | 1 | 321 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.40 [0.78, 2.51] |
| 14.10 Acquisition of Burkholderia cepacia | 1 | 321 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.03 [0.15, 7.23] |
Comparison 2. Hypertonic saline 3% to 7% versus isotonic saline in acute lung disease.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Change in FEV1 from baseline (% predicted) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.1 At day 7 | 1 | 130 | Mean Difference (IV, Fixed, 95% CI) | 3.95 [‐16.69, 24.59] |
| 1.2 At day 10 | 1 | 99 | Mean Difference (IV, Fixed, 95% CI) | ‐2.70 [‐24.69, 19.29] |
| 1.3 At discharge | 1 | 130 | Mean Difference (IV, Fixed, 95% CI) | 5.10 [‐14.67, 24.87] |
| 2 Change in FVC from baseline (% predicted) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2.1 At day 7 | 1 | 130 | Mean Difference (IV, Fixed, 95% CI) | 1.10 [‐5.74, 7.94] |
| 2.2 At day 10 | 1 | 99 | Mean Difference (IV, Fixed, 95% CI) | 7.90 [‐1.86, 17.66] |
| 2.3 At discharge | 1 | 130 | Mean Difference (IV, Fixed, 95% CI) | 6.10 [‐0.68, 12.88] |
| 3 Shuttle walk test | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 3.1 At day 7 | 1 | 132 | Mean Difference (IV, Fixed, 95% CI) | 46.0 [‐14.81, 106.81] |
Comparison 3. Hypertonic saline versus rhDNase.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Change in FEV1 (% predicted) | 2 | Treatment difference (Fixed, 95% CI) | Totals not selected | |
| 1.1 At 3 weeks | 1 | Treatment difference (Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 At 3 months | 1 | Treatment difference (Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Improvement in FEV1 >10% | 2 | Odds Ratio (Fixed, 95% CI) | Totals not selected | |
| 2.1 At 3 weeks | 1 | Odds Ratio (Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 At 3 months | 1 | Odds Ratio (Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Change in FVC (% predicted) | 1 | Treatment difference (Fixed, 95% CI) | Totals not selected | |
| 3.1 At 3 months | 1 | Treatment difference (Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Exercise tolerance ‐ oxygen saturation | 1 | Treatment difference (Fixed, 95% CI) | Totals not selected | |
| 4.1 At 3 months | 1 | Treatment difference (Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Exercise tolerance ‐ VAS for breathlessness | 1 | Treatment difference (Fixed, 95% CI) | Totals not selected | |
| 5.1 At 3 months | 1 | Treatment difference (Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Exercise tolerance ‐ FCS | 1 | Treatment difference (Fixed, 95% CI) | Totals not selected | |
| 6.1 At 3 months | 1 | Treatment difference (Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7 Mean percentage change in quality of life score | 1 | Treatment difference (Fixed, 95% CI) | Totals not selected | |
| 7.1 At 3 months | 1 | Treatment difference (Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Delivery time (minutes) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected |
Comparison 4. Hypertonic saline versus mannitol.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Delivery time (mins) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Adde 2004.
| Methods | Open‐label, randomised trial. Design: cross‐over with 4 weeks in each treatment arm and a 2‐week washout period in between. Location: Brazil (single centre). |
|
| Participants | Total participants: n = 18, 5 male and 13 female. CF diagnosis: not stated. Age: mean (SD) 14.8 (4.8) years, range 8.7 to 25.8 years. Baseline characteristics Airway colonization:
Shwachman score: median (range) 65 (55 – 90). Pancreatic insufficiency: n = 17. Previous use of DNase: n = 15. Continuous inhaled gentamicin therapy: n = 11. |
|
| Interventions |
Group 1: HS 6% 10 mL. Group 2: rhDnase 2.5 mg 2x daily. Both trial drugs were delivered by a Pari LC Plus nebulizer with a Proneb® compressor, by a mouthpiece. The first inhalation was done in the hospital, for technique supervision, checking of immediate side effects, post‐medication PFT and measurement of nebulization time. Salbutamol (400 mcg) was given prior to inhalation, twice daily, in both arms of the trial. All the other treatment for CF was unchanged. While in the rhDnase arm of the trial the participant was asked to do a normal saline nebulization (5 mL) at another time during the day. |
|
| Outcomes | Change in FEV1, sputum culture bacterial growth, invitro studies of mucus, symptom score, satisfaction with treatment. | |
| Notes | Abstract only, but some additional information from authors. No sample size calculation stated. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | No detail provided in abstract, but additional information from authors confirmed use of a random numbers table. |
| Allocation concealment (selection bias) | Low risk | No detail provided in abstract, but additional information from authors confirmed that for each potential participant the selected sequence of treatment was defined and written in a piece of paper which was then put into numbered envelopes that were kept in the hospital pharmacy. After participants were recruited and informed consent obtained the first envelope was opened and read to see the allocated treatment. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Double blind, participant blinding was attempted by masking the taste of the solutions with quinine sulphate but additional info states "as participants were well aware of the medication flavour of HS this could not be masked". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not discussed. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Did not report withdrawals. Additional data provided stated that 1 participant (not included in the analysis) had to stop treatment with HS due to severe dyspnoea during its nebulization. |
| Selective reporting (reporting bias) | Unclear risk | Abstract only. |
| Other bias | Low risk | 2‐week washout period in between treatment arms. No sample size calculation stated. |
Amin 2010.
| Methods | Randomised trial. Design: cross‐over trial with 4 weeks treatment in each arm and 4‐week washout period. Location: Canada (multicentre). |
|
| Participants | Total participants: n = 20 randomised, 1 excluded from analysis. 7 males and 12 females. CF diagnosis ? Mean (SD) age at baseline = 10.6 (3.1) years. Baseline characteristics Pseudomonas aeruginosa +ve: n = 7 BMI: mean (SD) 17.0 (3.0) Pancreatic insufficient: 84% DF508/DF508: 42% DF508 compound heterozygous: 21% Lung function:
|
|
| Interventions |
Group 1: 4 mL HS 7% 2x daily. Group 2: 4 mL IS 0.9% 2x daily. The solutions were administered using the PARI LC Star nebulizer. 2x 100 mg puffs of salbutamol (Ventolin) were administered before each inhalation of study solution using a holding chamber. |
|
| Outcomes | LCI, CFQ‐R. FEV1, FVC, FEF 25‐75. | |
| Notes | Investigators calculated the sample size required for testing using HS as the main exposure variable and the LCI as the primary outcome variable. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Concealed computer‐generated randomisation. |
| Allocation concealment (selection bias) | Low risk | Randomisation concealed as performed by a research pharmacist not otherwise involved in the trial. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | The solutions were indistinguishable from each other in appearance but participants could discern a difference in taste between solutions. High risk with this cross‐over design. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Blinding of researchers. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 20 recruited, details of missing data for 3 participants described (as follows), complete cross‐over data were therefore available for 17 participants.
|
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the 'Methods' section reported in the 'Results' section. |
| Other bias | Low risk | 4‐week washout period. Investigators calculated the sample size. |
Ballmann 1998.
| Methods | Randomised controlled trial. Design: cross‐over with 2 treatment periods of 3 weeks each and a washout period of 3 weeks in between. Location: Germany. |
|
| Participants | Total participants: n = 14, 8 males and 6 females.
FEV1 % predicted had to be greater than 40%. Baseline characteristics: FEV1 % predicted: mean (SD) 75.6% (14%). Pseudomonas aeruginosa:
|
|
| Interventions | Pre‐treated salbutamol 200 mcg MDI inhaled (2 puffs).
Group 1: 10 mL HS 5.85% 2x daily. Group 2: 2 mL Pulmozyme 2.5 mg 2x daily. Routine medication not altered during trial. |
|
| Outcomes | Change in FEV1 as a % of predicted, nebulisation time, comparison of cost (in Deutschmarks), preference. | |
| Notes | There was a 3‐week washout between interventions. No sample size calculation stated. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants randomised into groups of 4 and drew lots to start with HS or rhDNase. |
| Allocation concealment (selection bias) | Unclear risk | No details provided as to how lots concealed allocation. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Taste of HS and difference in volume made blinding not possible. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not discussed. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No information provided whether an ITT was used. |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the 'Methods' section reported in the 'Results' section. |
| Other bias | Low risk | Washout period of 3 weeks. No sample size calculation stated. |
Cardinale 2003.
| Methods | Randomised placebo‐controlled trial. Design: parallel. Location: Italy. |
|
| Participants | Total participants: n = 25. Treatment group: n = 12. Placebo group: n = 13. Baseline characteristics: stable disease. |
|
| Interventions |
Group 1: HS 7%. Group 2: IS 0.09%. Frequency of nebulization not stated. |
|
| Outcomes | Change in lung function and exhaled nitric oxide measures, cough, sputum production. Measurements at baseline and 2 weeks. |
|
| Notes | Abstract only. No sample size calculation stated. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No detail provided. |
| Allocation concealment (selection bias) | Unclear risk | No detail provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No detail provided. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No detail provided. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Cardinale stated that no adverse events were reported with HS, but no information given for IS group. |
| Selective reporting (reporting bias) | Unclear risk | Abstract only, no detail provided. |
| Other bias | Unclear risk | No sample size calculation stated. |
Chadwick 1997.
| Methods | Randomised trial. Design: cross‐over with 3 arms. |
|
| Participants | Total participants: n = 15. Groups stratified according to FEV1 (over 70% and 40% ‐ 70%). |
|
| Interventions |
Group 1: IS. Group 2: HS 3.5%. Group 3: hypotonic saline. |
|
| Outcomes | Change in FEV1 % predicted, nebulisation. | |
| Notes | Abstract only. No sample size calculation stated. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No detail provided. |
| Allocation concealment (selection bias) | Unclear risk | No detail provided. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Stated as single blind, possibly assessors who were blinded due to difficulties in masking taste of intervention, but not clear. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Stated as single blind, possibly assessors who were blinded due to difficulties in masking taste of intervention, but not clear. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No description of any dropouts given. |
| Selective reporting (reporting bias) | Unclear risk | Abstract only, no detail provided. |
| Other bias | Unclear risk | No sample size calculation stated. No washout period stated. |
Dentice 2016.
| Methods | Randomised controlled trial. Design: parallel. Duration: 3‐day study which began towards the end of hospitalisation for an exacerbation (approximately 14 days) with follow up for 1 year. If readmission within that year, participant invited to repeat 3‐day study. Location: Australian multicentre trial. |
|
| Participants | 132 adults with CF admitted to hospital with a respiratory exacerbation. Age: mean (SD) 28 (9) years. 49% were female. FEV1 % predicted: mean (SD) 48 (20)%. |
|
| Interventions |
Group 1: 3x daily nebulisation of 4 mL HS 7%. Group 2: IS 0.12% (taste‐masked). Interventions given immediately before or during physiotherapy. |
|
| Outcomes | Length of hospital stay, lung function, oxygenation, bacterial load, symptom scores, QoL, exercise tolerance, time to relapse. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Done sequentially upon admission. |
| Allocation concealment (selection bias) | High risk | Allocation was sequential. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Taste of intervention and control masked by quinine. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Investigators blinded to intervention. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No drop outs. |
| Selective reporting (reporting bias) | Low risk | All outcomes reported. |
| Other bias | Low risk | None apparent. |
Elkins 2006a.
| Methods | Randomised, double‐blind trial. Design: parallel. Location: 16 adult or paediatric hospitals in Australia. Duration: 48‐weeks. The trial was conducted between September 2000 and November 2003. |
|
| Participants | Total participants: n = 164, 93 males and 71 females. Age: over 6 years. HS Group (n = 83) Mean (SD) age: 18.4 (9.3) years. Gender split: females 46%. BMI mean (SD): 19.9 (3.9). Lung function (mean (SD))
Sputum: Pseudomonas aeruginosa: 79%; Staphylococcus aureus: 44%. Control Group (n = 81) Mean (SD) age: 18.7 (9.2) years Gender split: females 42% BMI mean (SD): 20.1 (3.6) Lung function (mean (SD))
Sputum: Pseudomonas aeruginosa: 78%; Staphylococcus aureus: 47%. |
|
| Interventions |
Group 1: 4 mL HS 7% 2x daily. Group 2: 4 mL IS 0.09% 2x daily. Solutions were prepared by Pfizer, quinine sulphate (0.25 mg per mL) was added as a taste‐masking agent. Solutions were nebulized with a Pari LC PLUS jet nebulizer and a Pari Proneb Turbo compressor. A bronchodilator was administered before each inhalation of the trial solution. All other standard care was maintained throughout the trial. |
|
| Outcomes | Mean change in FEV1 and FVC at 4, 12, 36 and 48 weeks. QOL and pulmonary exacerbations. | |
| Notes | Sample size calculation undertaken. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Concealed computer randomisation with minimisation algorithm to balance for age, FEV1 and long‐term treatment with rhDNase, use of physiotherapy and trial centre. |
| Allocation concealment (selection bias) | Low risk | Randomisation performed by a person not otherwise involved in the trial. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double blind, participant blinding was achieved by masking the taste of the solutions with quinine sulphate. Participants and their clinicians, remained unaware of the treatment assignments throughout the trial. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The research assistants and the trial coordinator remained unaware of the treatment assignments throughout the trial. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 2 participants (1 from each group) withdrew voluntarily after randomisation and before first dose. 82 in HS group and 80 in control group included in ITT analysis. Withdrawals described as follows: HS Group: 15 in total withdrawn: 7 lost to follow‐up (2 owing to time constraints; 2 owing to insufficient perceived benefit from trial solution; 2 owing to adverse reaction to trial solution (cough); 1 provided no reason) and 8 stopped inhalations but continued visits (4 had adverse reaction to trial solution; 1 had cough and vomiting; 1 had pharyngitis and wheezing; 1 had voice changes; 1 had chest tightness; 2 could not tolerate taste of trial solution; 1 had insufficient benefit from trial solution; 1 lost interest). Control Group: 17 in total withdrawn: 10 Lost to follow‐up (5 owing to time constraints; 3 owing to insufficient perceived benefit from trial solution; 1 failed to attend; 1 provided no reason) and 7 stopped inhalations but continued visits (3 owing to time constraints; 2 had adverse reaction to trial solution (tonsillitis in 1 and lethargy in 1); 1 had insufficient benefit from trial solution; 1 provided no reason. |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the methods were described in the results. |
| Other bias | Low risk | Sample size calculation undertaken, no other potential bias identified. |
Eng 1996.
| Methods | Open‐label, randomised trial. Design: parallel. Duration: 2 weeks. Location: 2 centres in Australia (one children's hospital and one adult unit). |
|
| Participants | Total participants: n = 58 randomised, 6 withdrew during trial. Inclusion criteria Diagnosis of CF with positive sweat chloride test. Able to do pulmonary function tests. Cough and daily sputum production. Regular chest physiotherapy at home. Reasonable distance from clinic. On stable medications regime for last 14 days. Exclusion criteria > 20% fall in FEV1 at baseline assessment. Exacerbation of CF in last 4 weeks requiring admission to hospital. Exacerbation requiring admission to hospital during trial period. HS Group (n = 27). Gender split: 18 males, 9 females. Mean (range) age: 16.1 (7 to 25) years. Height (mean (SD)): 155 (17.7) cm. Weight (mean (SD)): 47 (14.5) kg. FEV1 % predicted (mean (SD)): 50.0 (9.7). FVC % predicted (mean (SD)): 73.5 (15.9). IS Group (n = 25). Gender split: 13 males, 12 females. Mean (range) age: 16.7 (8 to 36) years. Height (mean (SD)): 150 (19.6) cm. Weight (mean (SD)): 43 (16.2) kg. FEV1 % predicted (mean (SD)): 53.7 (7.8). FVC % predicted (mean (SD)):77.2 (9.8). |
|
| Interventions | Pre‐treated salbutamol 600 mcg MDI and volumatic spacer device.
Group 1: 10 mL HS 6% 2x daily. Group 2: 10 mL IS 2x daily. Inhaled using ultrasonic nebuliser (Omron NE‐U 07). First and last inhalations administered in clinic under a doctor's supervision. |
|
| Outcomes | Mean change in FEV1 at 2 weeks; mean change FVC at 2 weeks; VAS for cleared chest at 1 and 2 weeks; VAS for dyspnoea, fatigue, appetite, exercise tolerance, quality of sleep, general well‐being; adverse effects (increased cough, haemoptysis, chest tightness and pharyngitis). | |
| Notes | Sample size calculation undertaken. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random number tables used. |
| Allocation concealment (selection bias) | Unclear risk | No detail provided. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open label, participants not told which group they were in, but not able to disguise trial drug due to salty taste. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Technician measuring lung function was blinded to treatment assignment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 6 participants withdrew in total (3 from each group) due to: non‐compliance with clinic visit (1 in IS group); exacerbation of respiratory infection requiring hospital admission (2 IS group, 1 HS group); irritating cough during inhalation (1 HS group); increased haemoptysis which occurred 3 hours after first treatment (1 HS group). No specific mention of whether an ITT analysis had been performed. |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the methods were described in the results. |
| Other bias | Low risk | Sample size calculation undertaken, no other bias identified. |
Laube 2009.
| Methods | Randomised double‐blind trial. Design: cross‐over. Duration: single dose of each treatment, washout period not stated. Location: USA (single centre). |
|
| Participants | Total participants: n = 12. Age: median (range) 10.5 (8.9 ‐ 12.4) years, 5 males. Normal pulmonary function (FEV1 and FVC > 90% of predicted values). |
|
| Interventions |
Group 1: HS 7%. Group 2: 0.12% saline. Treatment followed by radio‐labelled isotopes. |
|
| Outcomes | Mucociliary clearance at 20, 60 and 90 minutes and 24 hours. | |
| Notes | Mearsurements of MMC following inhalation of 0.12% saline were compared to 9 healthy adult controls. Abstract only. No sample size calculation stated. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The method of randomisation is not stated. |
| Allocation concealment (selection bias) | Unclear risk | Not stated in methods. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No attempt was made to blind to taste of the solution. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated in methods. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Low risk as dropouts were replaced. |
| Selective reporting (reporting bias) | Low risk | All reported outcomes were given. |
| Other bias | Low risk | Sample size was estimated from data based on adolescents |
Mainz 2015.
| Methods | Double‐blind randomised controlled trial. Design: cross‐over design. Duration: 1st arm of 28 days followed by a 28‐day washout period and then alternative treatment for 28 days. Location: multicentre in Germany. |
|
| Participants | 69 people with CF. | |
| Interventions | Group 1: HS 6% via PariSinusTM. Group 2: IS via PariSinusTM. |
|
| Outcomes | Primary outcome: Sinonasal outcome test (SNOT‐20) upper airway symptoms/disease‐specific QoL. Secondary outcomes: rhinoscopy, rhinomanometry, cytokines in nasal lavage. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Stated but not defined. |
| Allocation concealment (selection bias) | Unclear risk | This was stated but not described. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No attempt was made to blind the hypertonic saline solution taste. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Stated but not described in detail. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All drop outs clearly defined. |
| Selective reporting (reporting bias) | Low risk | All outcomes that were stated were described. |
| Other bias | Low risk | No other major source of bias detected. |
Riedler 1996.
| Methods | Randomised controlled trial. Design: cross‐over. Duration: first treatment on day 1, alternative treatment on second day. Location: single centre in Australia. |
|
| Participants | Total participants: n = 10, 3 males and 7 females.
Age: mean (range) 16.5 (13 ‐ 20) years. Severity of lung disease:
Participants were recruited as they were admitted with exacerbations of their lung disease with cough productive of tenacious sputum. |
|
| Interventions | Pre‐treated with 4 mL nebulised salbutamol 5 mg via jet nebuliser.
Group 1: HS 6%. Group 2: IS. Single treatment via ultrasonic nebuliser (Timeter Compuneb MP500) for 10 min. |
|
| Outcomes | Sputum weight, VAS to assess feeling of cleared chest, spirometry (FEV1, FVC, FEF25‐75, PEF). | |
| Notes | 7 participants were treated for a second block of treatment, but it was not defined who these were. Sputum expectoration and score changes not distributed normally. No sample size calculation stated. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Assigned by coin toss. |
| Allocation concealment (selection bias) | Low risk | Each participant assigned by coin toss to order of treatment ‐ no one could foresee allocation. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Single blinded, taste could be discerned by participants. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Single blinded, possibly assessors who were blinded due to difficulties in masking taste of intervention, but not clear. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Did not discuss whether an intention‐to‐treat approach had been used and there was no description of withdrawals. |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the methods were described in the results. |
| Other bias | Unclear risk | No washout period. No sample size calculation stated. |
Robinson 1996.
| Methods | Randomised controlled trial. Design: cross‐over trial with the order of the 4 interventions being randomised. The last 9 participants underwent a cough study day, this was not included on the randomisation order and was always performed on the last day. Duration: single dose of each intervention, study days generally a week apart. Location: Australia. |
|
| Participants | Total participants: n = 12, 9 males and 3 females. Age mean (SEM) (range): 21.9 (3.0) (18 ‐ 28) years. Height mean (SEM): 173.4 (11.6) cm. Weight mean (SEM): 64.7 (11.3) kg. Stable disease. |
|
| Interventions | Pre‐treated with nebulised salbutamol 5 mg in 2.5 mL saline via ultrasonic nebuliser (Omron NE‐U06).
Single inhalations of: Group 1: 7 mL HS 7%. Group 2: 7 mL amiloride 3 mg/mL. Group 3: 7 mL HS plus 7 mL amiloride 3 mg/mL. Group 4: 7 mL IS 0.9%. Voluntary cough single episode. All done 1 week apart (control group 2). |
|
| Outcomes | Sputum isotope clearance 60 minutes, mucociliary clearance rate, change in FEV1. | |
| Notes | Participants acted as own controls.
Spirometry measures were taken immediately after inhalation and are not a long‐term outcome measure. Paper reported lung function (mean values for all trial days):
No sample size calculation stated. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No details provided. |
| Allocation concealment (selection bias) | Unclear risk | No details provided. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Participants and all but 1 of investigators blinded to solutions, but participants may have been able to discern taste of intervention and work out the longer nebulization times for the combination of amiloride and HS (although none made reference to this). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | All but 1 of investigators blinded to solutions, but not clear which of investigators knew and whether they were assessing outcomes. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No withdrawals or dropouts. |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the methods were described in the results. |
| Other bias | High risk | Washout period not clear. No sample size calculation stated. The time course of the effect of the interventions on the outcomes is not clear. |
Robinson 1997.
| Methods | Randomised controlled trial. Design: cross‐over with 4 arms (no information on any washout period). Duration: each intervention given on 1 study day. Location: Australia. |
|
| Participants | Total participants: n = 10, 7 males and 3 females. Age mean (SD) (range): 22.1 (3.8) (19 ‐ 28) years. FEV1 % predicted mean (SD) (range): 52.0% (6.7) (31 ‐ 84%). All participants were chronically colonisedPseudomonas aeruginosa. In addition, 5 of the participants had Staphylococcus aureus. Participants in a stable clinical condition and baseline medications were not altered throughout the trial period. |
|
| Interventions | Pre‐treated with nebulised salbutamol 5 mg in 2.5 mL saline via an ultrasonic nebuliser. Group 1: HS 3% single dose. Group 2: HS 7% single dose. Group 3: HS 12% single dose. Group 4: Voluntary cough and IS combined as the control. Each participant took part in each arm. |
|
| Outcomes | Sputum isotope % clearance at 30 minutes, sputum isotope clearance at 90 minutes, mucociliary clearance. | |
| Notes | No sample size calculation stated. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No detail provided. |
| Allocation concealment (selection bias) | High risk | There was no random allocation to treatment. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Participants could discern taste for intervention. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not discussed. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No discussion of whether an intention‐to‐treat analysis had been used or of any withdrawals. |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the methods were described in the results. |
| Other bias | High risk | Washout period not clear. No sample size calculation stated. The time course of the effect of the interventions on the outcomes is not clear. |
Robinson 1999.
| Methods | Randomised controlled trial. Design: 4‐way cross‐over trial (no information on any washout period). Duration: single day for each intervention. Participants were initially randomised to receive either the mannitol or HS. On the second day they were randomised to either the remaining active or the control for the first day. On the third day they were randomised to receive either the remaining active or either of the controls. The final day was the remaining control. Location: Australia. |
|
| Participants | Total participants: n = 12, 5 males and 7 females.
Age mean (SD) (range): 29.9 (9.4) (16 ‐ 46) years. BMI mean (SD) (range): 21.0.(1.8) (18 ‐ 24). 10/12 participants were colonised with Pseudomonas aeruginosa, 7/12 had Stapylocococcus aureus (including the 2 participants without Pseudomonas aeruginosa) and 4/12 had Aspergillus fumigatus. |
|
| Interventions | Pre‐treated with terbutaline 1000 mcg (turbulhaler). Group 1: single dose 7 mL HS 6%. Group 2: IS (0.9%) plus matched voluntary cough. Group 3: mannitol 300 mg (encapsulated dry powder). Group 4: empty capsules with matched voluntary coughs. | |
| Outcomes | Sputum isotope % clearance at 30 minutes, sputum isotope clearance at 90 minutes, mucociliary clearance. | |
| Notes | Isotope clearance was reported in this paper as occurring at 60 minutes. This is actually the same time period as the 90‐minute clearance reported in 1997 paper. The terminology had been changed. Lung function data are the mean values for all trial days: FEV1 % predicted mean (SD) (range): 60.2.(16.5) (42 ‐ 87). FVC % predicted mean (SD) (range): 78.8.(16.5) (47 ‐ 102). FEF25‐75 % predicted mean (SD) (range): 32.5.(21.1) (11 ‐ 77). No sample size calculation stated. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No detail provided. |
| Allocation concealment (selection bias) | Unclear risk | The allocation concealment process was not described. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Participants could discern taste. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The trials were coded such that the investigators were blinded to the identity of the intervention at the time of data analysis. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No discussion of whether an ITT analysis had been used or of any withdrawals. |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the methods were described in the results. |
| Other bias | Unclear risk | Washout period not clear. No sample size calculation stated. The time course of the effect of the interventions on the outcomes is not clear. |
Rosenfeld 2012.
| Methods | Double‐blind randomised controlled trial. Design: parallel. Duration: 48 weeks. Location: 30 centres in USA and Canada. |
|
| Participants | Total participants: n = 321 (176 males, 145 females) aged 4 ‐ 60 months with an established diagnosis of CF (details of diagnosis given in supplementary paper). Age (mean (SD)): HS group 2.2 (1.4) years; control group 2.3 (1.5) years. Gender split: HS group 84 males (53%); control group 92 males (56%). Weight, mean (SD) kg; HS group 12.2 (4.1) kg; control group 12.5 (4.1) kg. Weight percentile, mean (SD): HS group 39.7 (28.1); control group 43.0 (29.1). Height, mean (SD) cm: HS group 84.8 (14.8) cm; control group 85.7 (15.0) cm. Height percentile, mean (SD): HS group 36.9 (27.0); control group 39.9 (28.1). Positive respiratory culture (Pseudomonas aeruginosa isolated from respiratory culture at or at any time prior to randomisation. For other organisms, positive culture at or within 24 months prior to randomisation):
|
|
| Interventions | Pre‐treatment: all participants received albuterol or levalbuterol prior to each trial drug dose ‐ 2 puffs via metered dose inhaler via a valved holding chamber with face mask or by nebulizer (distinct from the nebulizer used to administer the trial drug) and PARI Proneb® Ultra compressor. Group 1 (n = 158): HS 7% 2x daily. Group 2 (n = 163): IS 0.9% 2x daily. Both treatments administered via Proneb Ultra compressor with a Sprint Jr nebulizer equipped with a Baby face mask or mouthpiece ‐ participants under 36 months used a facemask and those over 36 months used a mouthpiece, but this was individualized as developmentally appropriate. |
|
| Outcomes | Pulmonary exacerbation rate (events per person‐year; defined as treatment with oral, inhaled, or intravenous antibiotics for 1 or more prespecified signs and symptoms). Number of treatment days and number of courses of antibiotics. Time to first exacerbation. Lung function (FEV 0.5, FEF 75, FEF25‐75, FRC, RV/TLC). CFQ‐R ‐ Parent and reported. Change in height and weight. Change in resting respiratory rate, pulse oximetry and parent‐reported cough. Rate of intolerance to the test dose of hypertonic saline at enrolment. Adverse events and withdrawal rates. Treatment‐emergent respiratory cultures positive for CF pathogens detected through clinical cultures performed at each site’s microbiology laboratory. Adherence to treatment was assessed by (1) the number of used drug vials returned, (2) the Treatment Adherence Questionnaire completed quarterly, and (3) the weekly parent questionnaire. |
|
| Notes | Clinicaltrials.gov Identifier: NCT00709280. Trial visits occurred at enrolment/randomization and 4, 12, 24, 36, and 48 weeks after randomization. At the enrolment visit, after pre‐treatment with albuterol or levalbuterol, all participants were evaluated for intolerance to a test dose of 7% hypertonic saline according to predefined criteria. Participants who tolerated the test dose were randomized. Sample size calculation was undertaken. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomised 1:1 based on random permuted blocks, stratified by age and site (4 to 29 months, 30 to 60 months) via a secure website. |
| Allocation concealment (selection bias) | Low risk | Randomisation was done via a secure website. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | HS (Hyper‐Sal; PARI Respiratory Equipment) and 0.9% IS supplied by Catalent Pharma Solutions as identically packaged 4‐mL blow‐fill‐seal plastic ampoules, but taste could be discerned as different. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Researchers blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ITT analysis of 158 in HS group and 163 in control group. Details of withdrawals given as follows: HS group: 15 withdrew in total (follow‐up range, 2 ‐ 44 weeks): 5 lost to follow‐up; 4 treatment burden; 2 intolerant to trial drug; 1 time constraints; 3 other. Control group: 14 withdrew in total (follow‐up range, 0 ‐ 42 weeks): 2 lost to follow‐up; 3 treatment burden; 2 insufficient perceived benefit from trial drug; 1 intolerant to trial drug; 1 time constraints; 1 other adverse event; 4 other. |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the methods were described in the results. |
| Other bias | Low risk | Sample size calculation was done. No other bias identified |
Suri 2001.
| Methods | Prospective open‐label randomised controlled trial. Design: cross‐over with 3 treatment arms. 48 children were randomised, 8 to each of the 6 possible treatment orders. Duration: each arm lasted 12 weeks with a 2‐week washout period between treatments. Location: multicentre in UK (2 institutions). |
|
| Participants | Total participants recruited: n = 48, but 1 withdrew without commencing treatment, so trial population is 47.
Age (mean (SD)): 12.6 (2.8) years, range (7.3 ‐ 17 years). Gender split: 19 (40%) males; 28 (60%) females. Weight (mean (SD)): 40·0 (12·6) kg, range (18·8 ‐ 77). Spirometry:
Lung microbiology (number of children with 3 positive cultures of the organism in the previous year):
|
|
| Interventions | Pre‐treated with bronchodilators. All treatments were administered with a Sidestream nebuliser and Porta‐Neb compressor (Medic‐Aid, Bognor Regis, UK). Group 1: 5 mL HS 7% 2x daily immediately before the participant's regular physiotherapy session. Group 2: rhDNase 2.5 mg 1x daily at least 1 hour before physiotherapy. Group 3: 2.5 mg alternate daily at least 1 hour before physiotherapy. Routine medication and physiotherapy were continued throughout the trial. |
|
| Outcomes | Primary outcome: % change in FEV1 from baseline. Secondary outcomes: FVC; number of pulmonary exacerbations (defined as a previously outlined protocol for respiratory‐tract infections); weight gain; exercise tolerance (3‐min step test and oxygen saturation monitored); QoL (quality of well‐being scale self‐administered form 1·04, filled out by the participant and guardian together); total health‐care cost (hospital and community‐health‐service perspective, so participants' costs excluded from analysis; resources included covered hospital admissions (inpatient, outpatient, and day case), radiological investigations, blood tests, drug use, and the use of community services (visits to general practitioners, district nurses, and physiotherapists)); adherence (count of unused bottles of HS and empty vials of rhDNase; each participant was also given a diary to record the treatment doses taken). |
|
| Notes | Sample size calculation undertaken. Before starting the hypertonic‐saline treatment period, each participant received a test dose of HS in hospital so that he or she could be monitored for bronchoconstriction. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Telephone randomisation to an independent trials coordinating unit, stratified by hospital and balanced after each group of 12 children. |
| Allocation concealment (selection bias) | Low risk | Telephone randomisation to an independent trials coordinating unit, |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No attempt to conceal taste, paper states masking impossible because HS can easily be distinguished from rhDNase by its salty taste and timing in relation to physiotherapy. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No statement on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | An ITT approach was used within this trial, 43 children are included in the comparison of daily and alternate‐day rhDNase, and 40 in the comparison of daily rhDNase and hypertonic saline. Details of withdrawals as follows:
In the additional report of airway inflammatory changes following treatment, only 28 of the 48 participants were able to perform induced sputum and be included. It is not evident that any attempts were made to adjust for missing data from those participants unable to do induced sputum. |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the methods were described in the results. |
| Other bias | Low risk | We judged a 2‐week washout period to be low risk. Sample size calculation undertaken. |
Weller 1980.
| Methods | Double‐blind randomised controlled trial. Design: cross‐over with 2 arms. Duration: 2‐month baseline periods preceding and following 2x 8‐week treatment periods. Location: UK. |
|
| Participants | Total participants recruited: n = 29. Diagnosis was confirmed in all by history, examination, and a sweat test. 27 (13 males, 14 females) completed the trial aged 6 to 15 years (mean 10.7 years); of these 27 participants, 22 were chronic sputum producers. Baseline characteristics Age (mean (SD)); sputum producers 10.9 (2.1) years, range (6.5 ‐ 15); sputum non‐producers 9.8 (2.8) years range (6.1 ‐ 12.25). FVC (% predicted) (mean (SD)): sputum producers 76% (15.9), range (38 ‐ 101); sputum non‐producers 88% (10.1), range (76 ‐ 104). PEFR (% predicted) (mean (SD)): sputum producers 80% (21.5), range (27 ‐ 113); sputum non‐producers 96% (11.6), range (78 ‐ 107). |
|
| Interventions | No reported pre‐treatment. Group 1: 3 mL sodium‐2‐mercaptoethane sulphonate (Mistabron) 20% 2x daily after physiotherapy. Group 2: 3 mL HS 7% 2x daily after physiotherapy. Inhalations via a Wright nebuliser operated by an air compressor (Aerolyser Electric Inhaler, Aerosol Products (Colchester) Ltd) producing a flow of 8 L/min. |
|
| Outcomes | PEFR, FVC, V max 50% VC, RV/TLC. Diary card to record sputum volume, sputum colour, and cough frequency. At monthly clinic visits: sputum cultures, pulmonary function tests (PEFR, FVC, Vmax50%VC, RV/TLC). At beginning and end of trial: chest radiographs (Chrispin Norman score), full blood count, liver function tests and plasma electrolytes including creatinine. |
|
| Notes | Participants were divided into sputum producers and non‐sputum producers. Sample size calculation not undertaken. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Stated that order of treatment was randomised, but no details given as to randomisation process. |
| Allocation concealment (selection bias) | Unclear risk | No detail provided. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Described as double‐blind, but participants could discern difference in taste of interventions. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Described as double‐blind, but not clear whether outcome assessors were one of the parties blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Clear description of dropouts and withdrawals (see below) but it was not stated if an ITT analysis had been performed. 2 participants withdrawn from trial:
|
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the methods were described in the results. |
| Other bias | Low risk | Washout period: 2‐month baseline periods preceding and following 2x 8‐week treatment periods. Sample size calculation not undertaken. |
CF: cystic fibrosis FEV1: forced expiratory volume at one second FVC: forced vital capacity HA: hyaluronic acid HS: hypertonic saline IS: isotonic saline ITT: intention‐to‐treat LCI: lung clearance index MDI: metered dose inhaler PEFR: peak expiratory flow rate QOL: quality of life rhDNase: deoxyribonuclease RV: residual volume SEM: standard error of the mean TLC: total lung capacity VAS: visual analogue scale VC: vital capacity
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Brivio 2016 | Comparison of HS to HS plus HA (no control without HS). |
| Buonpensiero 2010 | Comparison of HS to HS plus HA (no control without HS) |
| DeCono 2008 | Not randomised. |
| Dentice 2012 | Did not compare HS to a control group. Instead the trial sought out the optimum timing of physiotherapy to HS use, before, during or after. |
| Donaldson 2006 | Did not compare to a control group that did not include HS, comparison of HS with or without pre‐treatment with amiloride. |
| Elkins 2006b | Comparison of dose frequency of HS; all participants received HS, but randomised to 2 or 4 times daily. |
| EUCTR2007‐002707‐40‐BE | Not randomised. |
| Genkova 1998 | Did not compare to a control treatment; did not report any results. |
| Grasemann 2013 | The trial did not include an arm with HS. It compared inhaled L‐arginine with isotonic saline as a control. |
| IRCT20180307038994N1 | Not randomised. |
| King 1997 | In vitro trial only. |
| Kobylyansky 2000 | Trial was not performed in a CF population. |
| NCT01094704 | Not randomised; reporting results at different time points after a single dose of HS. |
| O'Neill 2017 | Comparison of timing of HS in reference to airway clearance. No comparator group that did not include HS. |
| Ros 2012 | Comparison of 2 formulations of 7% HS, with or without HA. |
| San Miguel 2016 | Comparison of 2 arms both using HS with and without physiotherapy interventions. |
| Van Ginderdeuren 2008 | Did not compare HS to a control group. Instead the trial sought to evaluate the efficacy of an autogenic drainage treatment combined with HS. |
| Van Ginderdeuren 2011 | Did not compare HS to a control group. Instead the trial sought to evaluate the efficacy of an autogenic drainage treatment combined with HS. |
| Vanlaethem 2008 | Comparison of HS in conjunction with airway clearance techniques. |
CF: cystic fibrosis HA: hyaluronic acid HS: hypertonic saline
Characteristics of studies awaiting assessment [ordered by study ID]
Amin 2016.
| Methods | RCT. Cross‐over design (1‐week washout between treatments). Duration: single inhalation of each treatment. Multicentre (2 centres) in Canada. Concealed, computer‐generated randomisation performed by a research pharmacist not otherwise involved in the study; clinicians and research personnel remained unaware of the treatment assignments throughout the study, including the primary efficacy analysis. The solutions were indistinguishable from each other in appearance, but not in taste. Sample size calculation was undertaken. |
| Participants |
Inclusion criteria: confirmed diagnosis of CF; informed consent and verbal assent (as appropriate); at least 6 years of age at enrolment; able to perform reproducible spirometry meeting American Thoracic Society standards; pre‐bronchodilator FEV1% predicted ≥ 40% predicted; able to perform reproducible LCI maneuvers at screening. Exclusion criteria: airway cultures yielding Burkholderia cepacia complex in the previous 2 years or non‐tuberculous mycobacteria in the past year; oral corticosteroid use; oxygen supplementation; lung transplantation; intravenous antibiotics or oral quinolones within 14 days of enrolment; or investigational drugs within 30 days of enrolment. Participants who experienced a drop in FEV1 of 20% or more after study drug administration were also excluded from further study participation. 242 people with CF identified for the study (134 from St Michael's Hospital and 108 from the Hospital for Sick Children). 113 (47%) were excluded because they were currently using HS. 129 were approached of which 21 (16%) people consented and were enrolled in the trial. 21 participants randomised; 16 completed all of the time point measurements; 18 participants contributed to the intention‐to‐treat analysis and 3 participants were excluded. Age median (range): 140 (7 ‐ 56) months. Gender n (%): 11 (61.1%) females. LCI median (range): 122 (6.5 ‐ 21.1). FEV1% predicted median (range): 86.9 (47.4 ‐ 102.8). FVC % predicted median (range): 97.9 (61.9 ‐ 121.8). FEF25‐75% predicted median (range): 67.1 (19.5 ‐ 99.2). Delta F508 homozygous n (%): 16 (88.9%). Pseudomonas aeruginosa positive sputum culture n (%): 17 (94.4%). |
| Interventions |
Intervention: single inhalation of 4 mL HS (7%). Control: single inhalation of 4 mL IS (0.9%). The solutions were administered using the PARI LC Star nebuliser (Pari, Midlothian, Virginia, USA). 2x 100 ug puffs of salbutamol were administered before each inhalation of study solution using a holding chamber (Aerochamber Max, Trudell, London, Canada). |
| Outcomes | MBW and spirometry were performed at each visit ‐ at baseline, 1, 2, 4 and 24 h post inhalation. Primary outcome LCI change from baseline to 24 h post inhalation. Secondary outcomes LCI SF6 change from baseline to 24 h post inhalation. Spirometry (FEV1, FEF25‐75) change from baseline to 24 h post inhalation. Changes in LCIN2, LCISF6 and spirometric outcomes over 24 h (i.e. slope analysis of all time points). Adverse events. |
| Notes | All tests were performed in the research pulmonary function laboratory at the Hospital for Sick Children, Toronto Canada between April 2012 and September 2014. The trial was stopped because of difficulty with recruitment. Dr. Felix Ratjen has acted as a consultant for Novartis Pharmaceuticals. Funding support as well as the hypertonic saline used in the study was provided by Novartis Pharmaceuticals. |
Balinotti 2015.
| Methods | Prospective, randomised, double‐blind controlled trial. Duration: 1 year. |
| Participants | Children over 6 years of age with a diagnosis of CF made by 6 months of age. 19 participants randomised: 3 excluded, 9 participants in HS group, 7 participants in IS group. |
| Interventions |
Intervention: HS 3% plus 0.25 mg/kg salbutamol 2x daily. Control: IS 0.9% plus 0.25 mg/kg salbutamol 2x daily. |
| Outcomes | Primary outcome: maximal flow at functional residual capacity (change from baseline). Secondary outcomes: pulmonary exacerbations, respiratory rate, nutritional status, adverse events. |
| Notes |
Brown 2010.
| Methods | Controlled trial ‐ randomisation not clear. |
| Participants | Children with CF under 6 years. |
| Interventions |
Intervention: single inhalation of HS 7%. Control: single inhalation of IS 0.9%. |
| Outcomes | Change in LCI. |
| Notes | To study the feasibility of using lung clearance index to assess treatment effect outcomes in CF trials, especially in children under 6 years. We are waiting for further data to judge whether this should be included. |
Corcoran 2017.
| Methods | RCT. Cross‐over design with washout period (participants "discontinued use of HS for 72 h prior to each study visit"). Duration: 2 overnight visits, 1 for each treatment arm Location: USA. |
| Participants |
Inclusion criteria: adults with CF with FEV1 40% ‐110% of predicted and a BMI < 30kg/m². Exclusion criteria: currently experiencing an acute upper or lower respiratory tract infection or requiring treatment with antibiotics or systemic corticosteroids within 28 days of enrolment (not including normal maintenance antibiotics), diagnosis of sleep apnoea, symptoms of allergic rhinitis, previous diagnosis of Burkholderia cepacia, smokers, or pregnant or nursing. All participants performed a 30 min tolerance test of pulmosal inhalation from the tPAD device prior to the overnight visits and were excluded if they demonstrated a > 10% reduction from pre‐dose value in FEV1, measured 30 min after completion. 12 participants included. Age, mean (SD): 31.8 (9.7) years. Gender: 8 males, 4 females. |
| Interventions | Participants discontinued use of HS for 72 h prior to each visit. Long‐acting beta agonists were halted for 12 h and short‐acting beta agonists for 6 h prior. Inhaled antibiotics were allowed prior to, or after tPAD use. The tPAD device includes a 2 LPM air pump, an Aeroneb ProNebulizer (Aerogen,Galway, Ireland), a 250 mL liquid reservoir, a liquid collection chamber, and an integrated nebulization chamber where the aerosol is combined with the flow from the air pump. It also includes electronics allowing for timed device operation for up to 8 h. A system‐specific nasal cannula is attached to the integrated nebulization chamber and used to deliver the aerosol. For this trial the cannula was secured to the participant using a Wisp nasal mask frame and headgear (Philips Respironics, Murraysville, PA). Overnight HS visit: inhaled HS* from the tPAD device from 11:30 pm to 7:30 am in the clinical research unit; the tPAD was loaded with an initial HS dose of 240 mL and reloaded 5.5 h later with 60 ‐ 80mL of HS. Overnight sham visit: participants wore the cannula as they did during the HS visit and the tPAD was powered on, but the nebuliser and the air pump were non‐operational and no air flowed through the cannula. * Pulmosal 7% (pH+) sodium chloride inhalation solution was used for all trials except 1 where it was not available. Pulmosal is sterile, non‐pyrogenic, preservative‐free, and balanced to a pH of 7.4. In participant 9 another 7% HS product for inhalation was used. |
| Outcomes |
Primary outcome: MCC. Secondary outcomes: safety assessments, absorptive clearance, and change from baseline in Sino‐Nasal Symptoms (SNOT‐14) and modified Leeds Sleep Evaluation Questionnaires (mLSEQ). Pulse oximetry was monitored throughout the night while the participants slept and the tPAD operated continuously. |
| Notes | Participants who did not complete both nights were not included in the analysis and were replaced until a total of 12 participants had completed both visits. |
Donaldson 2013.
| Methods | RCT. Location: USA. Duration: 4 weeks. |
| Participants | Children with CF and stable lung disease (FEV1 > 60%) aged between 5 and 18 years. 24 screened, 23 randomised, 20 completed treatment and follow‐up. |
| Interventions |
Intervention: HS 6% 3x daily via eFlow nebuliser. Control: IS 0.12% 3x daily via eFlow nebuliser. |
| Outcomes |
Primary outcome Mucociliary clearance. Secondary outcomes Spirometry. QoL (CFQ‐R). |
| Notes | We are waiting for further data to judge whether this trial should be included. |
Dwyer 2013.
| Methods | RCT. Parallel design. Duration: 16 weeks. Location: Australia. Sequence generation: random number generation with block allocation. Treatment allocation is stratified for: age, gender, FEV1. Allocation concealment: random allocation to 1 of 3 groups by the Trial Pharmacist at the trial co‐ordinating centre. Treatment allocation recorded at the Trial Pharmacy and the random allocation lists, randomisation procedure and the unblinded treatment allocation is to be concealed from all other trial staff and the participant. Blinded. |
| Participants |
Inclusion criteria: informed consent, diagnosis of CF (positive sweat test or genotyping), best FEV1 in the previous six months > 20% of predicted normal value FEV1 > 85% of best in the previous 6 months, no non‐routine antibiotics in the last 14 days, minimum age 6 years, both genders eligible. Exclusion criteria: colonisation with Burkholderia cepacia, major haemoptysis within the last 12 months, pregnant or lactating females, investigational drugs within the last 30 days, previous lung transplant, hypertonic saline within the last 14 days, inhaled mannitol within the last 14 days. |
| Interventions |
Intervention 1: 2x daily inhalation of 4 mL of nebulised 6% HS + 0.25 mg/mL quinine sulphate. Intervention 2: 2x daily inhalation of 4 mL of nebulised 3% HS + 0.25 mg/mL quinine sulphate. Control: 2x daily inhalation of 4 mL of nebulised 0.9% IS + 0.25mg/mL quinine sulphate. |
| Outcomes |
Primary outcome Lung function as measured by the change in FEV1 % predicted (measured at baseline, Week 1, Week 4, Week 8 and Week 16 (end of trial)). Secondary outcomes Lung function as measured by the change in FVC % predicted (measured at baseline, Week 1, Week 4, Week 8 and Week 16 (end of trial)). Lung function as measured by the change in FEF25‐75 % predicted (measured at baseline, Week 1, Week 4, Week 8 and Week 16 (end of trial)). QoL as measured by the Cystic Fibrosis Questionnaire‐Revised (CFQ‐R) (measured at baseline, Week 1, Week 4, Week 8 and Week 16 (end of trial)). QoL as measured by the Medical Outcomes Survey Short Form‐36 (SF‐36) (measured at baseline, Week 1, Week 4, Week 8 and Week 16 (end of trial)). Exercise capacity as measured by the total distance covered in the Modified Shuttle Test‐25 (MST‐25) (measured at baseline, Week 4 and Week 16 (end of trial)) Exercise capacity as measured by the total exercise time in the Endurance Shuttle Test‐25 (EST‐25) (measured at baseline, Week 4 and Week 16 (end of trial)). Sputum bacterial diversity as measured by the acquisition or loss of bacterial organisms in expectorated sputum as measured by routine microscopy culture and sensitivity (M/C/S) (measured at baseline and Week 16 (end of trial)). Tolerability of nebulised trial solution as measured by participant on a 10‐point visual analogue scale (measured at baseline, Week 1, Week 4, Week 8 and Week 16 (end of trial)). Medication use as measured by number of doses of each prescribed medication (measured at baseline and weekly during trial (Week 1 to Week 16)). Pulmonary exacerbations as measured by the Fuchs exacerbation criteria (measured weekly during trial (Week 1 to Week 16)). Adverse events (such as intolerable cough, sore throat, bronchospasm, haemoptysis, nausea, pulmonary exacerbation) as measured by presence of new symptoms, likelihood of being related to trial solution, severity of symptoms and time to resolution of symptoms (measured weekly during trial (Week 1 to Week 16)). Adherence to nebulisation of trial solution as measured weekly by self‐report in patient diary and count of returned unused ampoules of trial solution at the end of the trial (week 16) Administration time of nebulised trial solution as measured by stop watch from start to completion of 1 dose of trial inhalation solution (measured at baseline, Week 1, Week 4, Week 8 and Week 16 (end of trial)). |
| Notes | Saline at lower tonicity in cystic fibrosis (SALTI‐CF) trial. Funding source: Australian Cystic Fibrosis Research Trust. |
Hofmann 1997.
| Methods | Open label RCT. Single dose. |
| Participants | 20 adults with CF. |
| Interventions |
Intervention: amiloride in HS 5.58%. Control: amiloride in IS. |
| Outcomes | Change in nasal potential difference. |
| Notes | Trial to determine the effects of HS and amiloride on change in ion flow by nasal potential difference. Abstract only. |
NCT00928135.
| Methods | RCT. Parallel design. Duration; 14 days. Triple‐blind (participant, care provider, investigator). |
| Participants | Estimated enrolment: 60 participants. Both genders eligible. Inclusion criteria: diagnosed with CF (medical record evidence of CFTR mutation or sweat chloride test or nasal voltage difference, and 1 or more clinical findings of CF); 12 years or older; FEV1 > 30% predicted (within the last 14 days and oxygen saturation > 90% on FiO2 ≤ 50%); admitted for an exacerbation; use of effective contraception in women; written informed consent. Exclusion criteria: pregnancy; history of asthma based on methacholine challenge or bronchial hyper‐responsiveness on PFT; haemoptysis more than 60 mL within the last 30 days; use of any investigational study drug within the last 30 days; initiation of hypertonic saline within the last 30 days; a serum creatinine 2 mg/dL or more; active malignancy in the last year; antibiotics for CF exacerbation as an outpatient in the last 2 weeks; Burkholderia cepacia colonisation; waiting list for lung transplant; lack of FEV1 data from the last 14 days; previous participation in this study. |
| Interventions |
Intervention: aerosolized 7% HS (5 mL) 2x daily. Control: 15% xylitol (5 mL) 2x daily. |
| Outcomes |
Primary outcomes FEV1 (change from baseline). Adverse events. Respiratory symptom score. Secondary outcomes Density of colonisation per g of sputum. Time to next exacerbation. Sputum cytokines. CFQ‐R. |
| Notes | Principal Investigator: Joseph Zabner, M.D. Study Director: Lakshmi Durairaj, M.D. Study Chair: Jan L Launspach, R.N., CCRC. Study Start Date : June 2009. Estimated Primary Completion Date : January 2018. Estimated Study Completion Date : January 2018. |
NCT01355796.
| Methods | Randomised, cross‐over trial. Open label. Duration: 2 weeks. |
| Participants | Estimated enrolment: n = 30 (stated that recruitment complete). Inclusion criteria: documented diagnosis of CF (medical record evidence of 2 identified CFTR mutations or a positive sweat chloride test or nasal voltage difference, and 1 or more clinical findings of CF); 16 years or older; FEV1 over 30% predicted; oxygen saturation at least 90% on room air; clinically stable, without evidence of pulmonary exacerbation for at least 2 weeks prior to screening (defined as use of oral or intravenous antibiotics for CF exacerbation); use of effective contraception in women; ability to provide written informed consent and assent; successful completion of the trial doses of study drugs. Exclusion criteria: pregnancy; haemoptysis more than 100 mL within the last 30 days; change in chronic medication within the last 30 days; history of elevated serum creatinine (at least 2 mg/dL) within 30 days or at screening; history of lung and other solid organ transplantation; wait‐listed for lung or other solid organ transplant; known intolerance to inhaled HS. |
| Interventions |
Intervention 1: aerosolised xylitol (5 mL) 2x daily for 14 days. Intervention 2: aerosolised 7% HS (4 mL) 2x daily for 14 days. |
| Outcomes |
Primary outcomes: FEV1 change from baseline; adverse events; and respiratory symptom score. Secondary outcomes: density of colonisation per gram of sputum; time to next exacerbation; sputum cytokines; and CFQ‐R. All outcomes measured at 98 days. |
| Notes | Sponsored by University of Iowa. Collaborators: Ann & Robert H Lurie Children's Hospital of Chicago Northwestern University. U01HL102288 (US NIH Grant/Contract). |
NCT01377792.
| Methods | Open‐label RCT. Parallel design. Duration: 48 weeks. Multicentre (3 centres) in Madrid, Spain. |
| Participants | Actual enrolment : 71 participants. Inclusion criteria: clinical diagnosis of CF; over 6 years old; FEV1 over 30% predicted; able to perform spirometry; able to perform induced sputum; able to tolerate the maximum dose of 10 mL HS; no oral or intravenous treatment within the previous 2 weeks before the beginning of the study; no treatment with HS in the 2 weeks before study entry. Exclusion criteria: no clinical diagnosis of CF; not able to tolerate 10 mL HS; positive pregnancy test; not able to tolerate beta 2‐agonist; treatment with corticosteroids; FEV1 < 30% predicted; liver or lung transplantation, or both; oxygen treatment; hospital admission within the 4 previous weeks; oral or intravenous antibiotic treatment within the 2 previous weeks; smokers; pulmonary colonisation with Burkholderia cepacia complex. |
| Interventions |
Intervention: 10 mL HS. Control: 5 mL HS. |
| Outcomes |
Primary outcomes Time free from pulmonary exacerbation (days) at 12 months. Secondary outcomes Changes in lung function at 12 months. Changes in inflammatory markers in induced sputum at 12 months. QoL at 12 months (CFQ‐R). |
| Notes | Principal Investigator: Dr Adelaida Lamas, Cystic Fibrosis Unit. Ramón y Cajal University Hospital, Madrid, Spain. Study Start Date : March 2009. Primary Completion Date : May 2011. Study Completion Date : September 2012. |
NCT01619657.
| Methods | Blinded (participant, care provider, investigator, outcomes assessor) RCT. Parallel design. Duration: 1 year. Multicentre in Germany. |
| Participants | Actual enrolment : 42 participants. Inclusion criteria: confirmed diagnosis of CF; age at enrolment is 0 ‐ 4 months; ability of child and parents to comply with medication use, trial visits, and trial procedures as judged by the investigator; written consent from participants' parents or legal guardians. Exclusion criteria: born < 30 weeks gestation; prolonged mechanical ventilation in the first 3 months of life; significant medical disease or condition other than CF likely to interfere with the child's ability to complete the entire protocol; previous major surgery except for meconium ileus; other major organ dysfunction, excluding pancreatic or hepatic dysfunction or another condition due to CF; physical findings that would compromise the safety of the participant or the quality of the study data as determined by investigator; history of adverse reaction to sedation; known hypersensitivity to study treatment; participation in other interventional studies at the same time. |
| Interventions |
Intervention: 6% HS 4 mL inhaled 2x daily. Control: 0.9% IS 4 mL inhaled 2x daily. Both treatments inhaled via PARI LC SPRINT® Junior nebulizer with a baby bend, size‐adapted PARI® Baby face mask size 0 ‐ 3, connection tubing (2.2m) and a PARI JuniorBOY® SX compressor. |
| Outcomes |
Primary outcomes Number of participants with AEs and serious AEs. Safety of inhalation with HS and IS in newborns and infants with CF assessed by proportion of AEs and serious AEs. Secondary outcomes Protocol‐defined pulmonary exacerbations requiring treatment with oral, inhaled or intravenous antibiotics. Morphological or functional changes (or both) due to CF lung disease according to MRI chest score and chest x‐ray. Chrispin‐Norman score. Extent and severity of bronchial dilatation after MRI and chest x‐ray scores. Impairments in lung function determined via multiple breath washout. Severity of impairment in lung function test. Health‐related QoL (as assessed by CFQ‐R, German version). Change in anthropometric and basic respiratory parameters (weight, height, BMI, weight‐for‐height, resting respiratory rate, and room air oxygen saturation). New isolates of CF pathogens from clinically collected respiratory cultures. Time to first isolation of a CF pathogen. |
| Notes | Sponsors and Collaborators: Heidelberg University, German Center for Lung Research. Principal Investigator: Marcus A Mall, MD University Hospital Heidelberg. Study Start Date : June 2012. Primary Completion Date : November 2016. Study Completion Date : October 2017. |
NCT02378467.
| Methods | Blinded (participant, care provider, investigator, outcomes assessor) RCT. Parallel design. Duration: 48 weeks. Multicentre in USA and Canada. |
| Participants | Actual enrolment : 150 participants (both genders). Inclusion criteria: confirmed diagnosis of CF; written informed consent by parent or legal guardian; age ≥ 36 months and ≤72 months at screening visit; able to comply with medication use, trial visits and trial procedures as judged by the site investigator; able to perform technically acceptable multiple breath washout measurements at the screening and enrolment visits. Exclusion criteria: acute intercurrent respiratory infection, defined as an increase in cough, wheezing, or respiratory rate with onset within 3 weeks preceding screening or enrolment visit; acute wheezing at screening or enrolment visit; oxygen saturation < 95% (<90% in centres located above 4000 feet elevation) at screening or enrolment visit; physical findings that would compromise the safety of the participant or the quality of the trial data as determined by site investigator; investigational drug use within 30 days prior to at screening or enrolment visit; treatment with inhaled HS at any concentration within 30 days prior to screening or enrolment visit; chronic lung disease not related to CF; inability to tolerate first treatment dose at the enrolment visit. |
| Interventions |
Intervention: 7% HS inhaled 2x daily. Control: 0.9% IS inhaled 2x daily. The delivery system for both groups is a PARI Sprint Junior nebulizer with a PARI Baby face mask or mouthpiece driven by a PARI Vios® compressor. |
| Outcomes |
Primary outcomes Change from baseline in LCI measured by N2 multiple breath washout. Secondary outcomes Change from baseline in FEV 0.75 measured by preschool spirometry. Protocol‐defined pulmonary exacerbation rate. Health‐related QoL measured by the modified parent‐reported CFQ‐R for preschoolers. Respiratory signs as measured by the Cystic Fibrosis Respiratory Sign Diary for ages 0 ‐ 6 (CFRSD0‐6). Treatment‐emergent CF respiratory pathogens from clinical respiratory cultures. |
| Notes | Principal Investigators: Stephanie Davis, MD Indiana University; Richard A Kronmal, PhD University of Washington; Felix Ratjen, MD, PhD, FRCPC Hospital for Sick Kids, Toronto; Margaret Rosenfeld, MD, MPH Seattle Children's Hospital. Study Start Date : March 2015. Estimated Primary Completion Date : August 2018. Estimated Study Completion Date : February 2019. |
NCT03391414.
| Methods | RCT (phase I). Cross‐over design. Location: USA. |
| Participants |
Inclusion criteria: aged 12 years and over; FEV1 greater than 50% predicted; able to spontaneously expectorate sputum (with or without chest physiotherapy); stable disease as defined by clinician assessment and no use of IV antibiotics in the past 4 weeks, no changes in CF‐related medications in the 4 weeks prior to study screening and SpO2 < 94% on room air or use of supplemental oxygen. Exclusion criteria: reactive airway disease; use of inhaled hypertonic saline in the past 28 days; use of IV antibiotics in the past 4 weeks; changes in CF‐related medications in the 4 weeks prior to screening; SpO2 < 94% on room air or use of supplemental oxygen; presence of untreated gastroesophageal reflux disease or residual acid reflux symptoms more than 3 times per week; pregnant or nursing females. 12 participants enrolled. |
| Interventions |
Hypertonic bicarbonate group: inhaled solution of 8.4% hypertonic bicarbonate by nebuliser. HS group: inhaled solution of 7% HS by nebuliser. |
| Outcomes |
Primary outcomes: change in exhaled breath condensate pH at 4 hours; change in pH after inhalation of 2 doses on 1 day. Secondary outcomes: change in expectorated sputum at 4 hours; change in sputum wet‐to‐dry ratio after inhalation of 2 doses on 1 day; change in spirometry at 4 hours; FEV1 before and after inhalation of 2 doses on 1 day. |
| Notes | Principal Investigator: Joseph M PIlewski, MD, University of Pittsburgh. Collaborators: Cystic Fibrosis Foundation Therapeutics. Start date: August 2014. Completion date: July 2016. Listed retrospectively on clinicaltrials.gov in 2018. No publication to date. |
Nenna 2017.
| Methods | Double‐blind RCT. Cross‐over design. Duration: 16 weeks in each treatment arm; total duration 32 weeks. Single centre in Italy. |
| Participants |
Inclusion criteria: aged 4 ‐ 6 years; diagnosed with CF; clinically stable; undergoing a simple therapy based on bronchodilators and physiotherapy; no respiratory infections during the treatment or 2 weeks before. Exclusion criteria: children with instable medical conditions 12 participants randomised. Age, mean (SD): 5.7 (0.8) years. Gender split: 6 males, 6 females. |
| Interventions |
Intervention: 4 mL HS (7%) inhaled 2x daily. Control: 4 mL IS (0.9%) inhaled 2x daily. There is no washout period between the 2 treatments, and children are followed up after 4, 16, 20 and 32 weeks. |
| Outcomes |
Primary outcomes Airways resistance measured using interrupter resistance technique at baseline, 4, 16, 20, 32 weeks Lung function (FVC, FEV1 and FEF25‐75) measured using spirometry at baseline, 4, 16, 20, 32 weeks Secondary outcomes Side effects measured using a standardized questionnaire created for the purpose of this study throughout the 31 week study period by healthcare providers. |
| Notes | Results published in 2017 (www.ncbi.nlm.nih.gov/pubmed/28709466). |
Palacio 2014.
| Methods | Open‐label RCT. Design: parallel group. Location: 2 centres in Argentina. Duration: 24 weeks. |
| Participants | 27 children aged 3 to 6 years with CF. 21 completed trial. |
| Interventions |
Intervention 1: HS 7% nebulised 2x daily. Control: IS 0.9%, nebulised 2x daily. Salbutamol given prior to each dose. |
| Outcomes |
Primary outcome Lung function. Secondary outcomes Respiratory symptoms. Anthropometric measures. New isolation of Pseudomonas aeruginosa. Rate of exacerbations. AEs. Adherence. |
| Notes | We are waiting for further data to judge whether this should be included. |
PRESIS 2018.
| Methods | RCT. Parallel assignment. Blinding of participants, care providers, investigators and outcome assessors. |
| Participants |
Inclusion criteria: confirmed diagnosis of CF established in neonatal period either via CF newborn screening or because of symptoms typical for CF (e.g. meconium ileus), positive family history or positive prenatal screening and fulfilling at least 1 of 3 criteria (sweat chloride ≥ 60mEq/L, 2 CF causing mutations of CFTR gene alterations of transepithelial potential difference of nasal or rectal epithelia typical for CF); age at enrolment is 0 ‐ 4 months; participant's and parent's ability to comply with medication use, trial visits, and trial procedures as judged by the investigator (therefore parents have to understand the character of the study and individual consequences); participation is voluntary so only participants, whose parents or legal guardians gave written consent, are included. Exclusion criteria: born < 30 weeks gestation; prolonged mechanical ventilation in the first 3 months of life; a significant medical disease or condition other than CF likely to interfere with the child's ability to complete the entire protocol; previous major surgery except for meconium ileus; other major organ dysfunction, excluding pancreatic or hepatic dysfunction or another condition due to CF; physical findings that would compromise the safety of the participant or the quality of the trial data as determined by investigator; history of adverse reaction to sedation; known hypersensitivity to treatment; participation in other interventional trials at the same time. Criteria, which lead to a displacement of the procedures in sedation until the child has recovered: clinically significant upper airway obstruction as determined by investigator (e.g. severe laryngomalacia, markedly enlarged tonsils, significant snoring, diagnosed obstructive sleep apnoea); acute intercurrent respiratory infection, defined as an increase in cough, wheezing, or respiratory rate with onset in 2 weeks preceding visit; oxygen saturation < 95% before initial pulmonary function test or initial MRI; severe gastroesophageal reflux, defined as persistent frequent emesis despite anti‐reflux therapy. 42 participants enrolled (originally aimed for 40) aged up to 4 months. |
| Interventions |
Intervention group: 4 mL HS 6% (MucoClear® 6%) administered via inhalation 2x daily for 52 weeks. Control group: 4 mL IS 0.9% administered via inhalation 2x daily for 52 weeks. Both interventions delivered using the PARI LC SPRINT® Junior nebulizer with a baby bend, size‐adapted PARI® Baby face mask size 0‐3, connection tubing (2.2m) and a PARI JuniorBOY® SX compressor. |
| Outcomes |
Primary outcome Number of participants with AEs and serious AEs at end of trial. Secondary outcomes Rate of protocol‐defined pulmonary exacerbations requiring treatment with oral, inhaled or intravenous antibiotics. Time to first pulmonary exacerbation. Change from baseline in proportion of children with morphological or functional changes, or both, due to CF lung disease according to magnetic resonance imaging (MRI) chest score and chest x‐ray (CXR) Chrispin‐Norman score (at end of trial). Change in extent and severity of bronchial dilatation after MRI and CXR scores at end of trial. Proportion of children with impairments in lung function determined via multiple breath washout at baseline, after 3, 6, 9, and 12 months of inhalation. Severity of impairment in lung function test at baseline, after 3, 6, 9, and 12 months. Health‐related QoL as assessed by scores from the CFQ‐R (German version), administered quarterly. Change in anthropometric and basic respiratory parameters (weight, height, BMI, weight‐for‐height, resting respiratory rate, and room air oxygen saturation) at end of trial. Proportion of participants with new isolation of CF pathogen from clinically collected respiratory cultures among participants from whom Pseudomonas aeruginosa or other CF pathogens were not isolated from respiratory cultures prior to enrolment. Time to first isolation of a CF pathogen. |
| Notes | Principal investigator: Marcus A. Mall, MD, Heidelberg University, Germany. Collaborator: German Center for Lung Research. Start date: June 2012. Final data collection: November 2016. |
AE: adverse events BMI: body mass index CF: cystic fibrosis CFQ‐R: cystic fibrosis questionnaire ‐ revised CFTR: cystic fibrosis transmembrane conductance regulator FEV1: forced expiratory volume at one second FEF25‐75: mid peak expiratory flow FiO2: fraction of inspired oxygen HS: hypertonic saline IS: isotonic saline IV: intravenous. LCI: lung clearance index MBW: multiple breath washout MCC: mucociliary clearance MRI: magnetic resonance imaging PFT: pulmonary function test QoL: quality of life RCT: randomised controlled trial SpO2: peripheral capillary oxygen saturation
Characteristics of ongoing studies [ordered by study ID]
NCT02276898.
| Trial name or title | A randomized‐controlled trial of inhaled hypertonic saline (7%) to evaluate the lung clearance index as a short‐term pharmacodynamic biomarker in patients with cystic fibrosis. |
| Methods | Double‐blind (participant, investigator) RCT. Cross‐over design. Duration: single administration. 2 centres in Canada. |
| Participants | Target enrolment: 24 participants. Inclusion criteria: confirmed diagnosis of CF; informed consent and verbal assent (as appropriate) provided by the participant's parent or legal guardian and the participant; at least 6 years of age at enrolment; able to perform reproducible spirometry meeting American Thoracic Society standards; pre‐bronchodilator FEV1 % predicted > or equal to 40 % predicted; ability to perform a reproducible LCI maneuver at screening. Exclusion criteria: known respiratory culture positive for Burkholderia cepacia; previous lung transplantation; use of intravenous antibiotics within 14 days of screening; use of oral antibiotics including prophylactic antibiotics (e.g., augmentin, tetracycline, cloxacillin, cephalosporins, septra, bactrim) within 14 days of screening; initiation of a new maintenance (e.g. high‐dose ibuprofen, Pulmozyme®, aerosolized antibiotics) within 14 days of screening; use of systemic corticosteroids within 14 days of screening; investigational drug use within 30 days of screening; use of hypertonic saline (7%) < 4 weeks before screening or outside of the study protocol; participation in any therapeutic clinical study < 4 weeks or, 5 half‐lives, whichever is longer, before screening; smoking < 3 months before screening; presence of a condition or abnormality that in the opinion of the site investigator would compromise the safety of the participant or the quality of the data. |
| Interventions |
Intervention: single inhalation of 4 mL 7% HS. Control: single inhalation of 4 mL 0.9% IS. |
| Outcomes |
Primary outcomes LCI (change from baseline to 24 hours post inhalation). Secondary outcomes PFTs (FEV1 % predicted, FVC % predicted and FEF 25‐75 % predicted). LCI (measured using mass spectroscopy: MBW). LCI (measured using nitrogen washout). |
| Starting date | Trial Start Date : November 2011. Primary Completion Date : September 2014. Study Completion Date : September 2014. |
| Contact information | Principal Investigator: Reshma Amin, MD The Hospital for Sick Children, Toronto, Canada. |
| Notes |
NCT02343445.
| Trial name or title | Clearing lungs with ENAC inhibition in cystic fibrosis (CLEAN‐CF) |
| Methods | Blinded (participant, care provider, investigator, outcomes assessor) RCT. Parallel design. 3 arms. Duration: 15 days. Multicentre (33 locations) in the USA. |
| Participants | Actual enrollment : 142 participants, both genders. Inclusion criteria: aged 12 years or older; diagnosis of CF as determined by the 1997 CF consensus criteria; non‐smoker; FEV1 at Screening Visit 1 between 40% and 90%; stable regimen of CF medications and chest physiotherapy for the 28 days prior to screening; willing to discontinue use of HS for the duration of the trial; clinically stable for at least 2 weeks; all females of child‐bearing potential must have a negative serum pregnancy test and if sexually active must agree to practice a highly effective form of contraception throughout the trial and for 28 days after the last dose of trial medication. Exclusion criteria: history of any organ transplantation or any significant disease or disorder; use of diuretics (including amiloride) or renin‐angiotensin antihypertensive drugs or trimethoprim in the 28 days prior to Screening; history of significant intolerance to inhaled HS, as determined by the investigator; known hypersensitivity to the trial drug or amiloride; any clinically significant laboratory abnormalities at Screening Visit 1 as judged by the investigator (or any of the following: potassium ≥ 5 mEq/L; abnormal renal function; abnormal liver function, defined as ≥ 3 x upper limit of normal; haemoglobin level < 10.0 g/dL); female who is pregnant or lactating; history of sputum or throat swab culture yielding Burkholderia species or Mycobacterium abscessus within 2 years of screening; previous participation in an investigational trial involving administration of any investigational compound or use of an investigational device with 28 days prior to Screening; currently being treated with any ivacaftor containing regimen. |
| Interventions |
Intervention 1: P‐1037 solution for inhalation, 85 μg (28.3 μg/mL) in HS (4.2%). Intervention 2: P‐1037 Solution for Inhalation, 85 μg BID (28.3 μg/mL) in 0.17% saline. Control: placebo (0.17% saline). All treatments were inhaled 2x daily. |
| Outcomes |
Primary outcomes Adverse events related to P‐1037 in treatment groups. FEV1 (change from from pre‐dosing to 1 hour post dosing). Secondary outcomes FEV1 (absolute change from baseline to Day 15). FVC (absolute change from baseline to Day 15). CFQ‐R. FEF25‐75% (absolute change from baseline to Day 15). |
| Starting date | Trial Start Date : April 2015. Primary Completion Date : February 2016. Study Completion Date : February 2016. |
| Contact information | Vertex Pharmaceuticals Incorporated. |
| Notes | Sponsors and Collaborators: Vertex Pharmaceuticals Incorporated, Parion Sciences. |
NCT02950883.
| Trial name or title | Saline hypertonic in preschoolers + CT (SHIP‐CT). |
| Methods | Blinded (participant, care provider, investigator, outcomes assessor) RCT. Parallel design. Duration: 48 weeks. |
| Participants | Estimated enrolment: 120 participants. Inclusion criteria: confirmed diagnosis of CF; informed consent by parent or legal guardian; age ≥ 36 months and ≤ 72 months at screening visit; able to comply with medication use, trial visits and trial procedures as judged by the site investigator; able to cooperate with chest CT at the enrolment visit as determined by the lung function technician. Exclusion criteria: chest CT within 8 months prior to the Screening visit; acute intercurrent respiratory infection, defined as an increase in cough, wheezing, or respiratory rate with onset within 3 weeks preceding screening or enrolment visit; acute wheezing at screening or enrolment visit; oxygen saturation < 95% (< 90% in centres located above 4000 feet elevation) at screening or enrolment visit; other major organ dysfunction, excluding pancreatic dysfunction; physical findings that would compromise the safety of the participant or the quality of the trial data as determined by site investigator; investigational drug use within 30 days prior to screening or enrolment visit; treatment with inhaled HS at any concentration within 30 days prior to screening or enrolment visit; initiation (i.e. new prescription) of any inhaled hydrating agent such as mannitol or mucolytic agents such as dornase alfa within 30 days prior to the screening or enrolment visit; chronic lung disease not related to CF; inability to tolerate first treatment dose at the enrolment visit. |
| Interventions |
Intervention: 7% HS 4 mL inhaled 2x daily. Control: 0.9% IS 4 mL inhaled 2x daily. The delivery system is a PARI Sprint Junior nebulizer with a PARI Baby face mask or mouthpiece driven by a PARI compressor (PARI Vios® Pro in USA, PARI BOY SX in Australia and Europe). |
| Outcomes |
Primary outcomes Chest CT (PRAGMA‐CF %Dis between groups at 48 weeks, adjusted for baseline, measured from standardised chest CT). Secondary outcomes PRAGMA‐CF sub‐scores: change in %Bx (the volume proportion of the lung with bronchiectasis) and %TA (the volume proportion of the lung with trapped air); absolute number of airways, airway dimensions and AA ratios from TLC CTs, acquired at the 48‐week visit. LCI (change from baseline to 48 weeks measured by N2 MBWs). Cross‐sectional and longitudinal relationships between primary and secondary PRAGMA‐CF outcomes (%Dis, %Bx and %TA) and MBWs (LCI), airway dimensions and PRAGMA‐CF and MBW outcomes, as well as CFQ‐R scores and PRAGMA‐CF and MBWs. |
| Starting date | Trial Start Date : August 2016. Estimated Primary Completion Date : February 2019. Estimated Study Completion Date : April 2019. |
| Contact information | |
| Notes | SHIP‐CT is a parallel study to SHIP001 (ClinicalTrials.gov Identifier NCT02378467). |
CF: cystic fibrosis CFQ‐R: cystic fibrosis questionnaire ‐ revised CT: computed tomography FEF: forced expiratory flow rate FEV1: forced expiratory volume in one second FVC: forced vital capacity HS: hypertonic saline IS: isotonic saline LCI: lung clearance index MBW: multiple breath washout PFT: pulmonary function test PRAGMA‐CF: Perth‐Rotterdam annotated grid morphometric analysis for cystic fibrosis RCT: randomised controlled trial
Differences between protocol and review
Update 2017 The updated review now includes FEV0.5 and the lung clearance index (LCI) which were both added as additional outcomes. We also added the outcome of duration of hospital stay due to pulmonary exacerbations and bacteriology. We feel these new outcomes are useful and of interest to clinicians and patients alike.
We have removed the outcome FEF25‐75 from the review, since this outcome is not deemed clinically important in this population and is not a reliable measure of lung function. We have also removed the outcome of exhaled nitric oxide as we do not think this is an important outcome for people with CF.
Summary of findings tables have been added to the review.
Contributions of authors
Two authors (PW and VMM) selected the trials that were included in this review and each author independently assessed the methodological quality (risk of bias) for each trial. PW and VMM wrote the review
Peter Wark acts as guarantor of the review.
Sources of support
Internal sources
No sources of support supplied
External sources
-
National Institute for Health Research, UK.
This systematic review was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Cochrane Cystic Fibrosis and Genetic Disorders Group.
Declarations of interest
Peter Wark: I or my institution have received money in support of investigator initiated research, participation in advisory boards or fees for speaking from GSK, Astra Zeneca, Bohringer Ingelheim, Vertex and Menarini; none of which are relevant to this review. I was an investigator and an author on one trial included in the review (Dentice 2016) and a co‐investigator on a further trial, the National Hypertonic Saline in Cystic Fibrosis Study trial (Elkins 2006a).
Vanessa McDonald: I have received honorariums for participation in educational meetings from GSK, AstraZeneca and Menarini and was a co‐investigator in the National Hypertonic Saline in Cystic Fibrosis Study trial (Elkins 2006a).
New search for studies and content updated (conclusions changed)
References
References to studies included in this review
Adde 2004 {published data only}
- Adde FV, Borges KTL, Hatanaka ACF, Nakaie CMA, Cardieri JMA, Oliveira RC, et al. Hypertonic saline X recombinant human DNase: a randomised crossover study in 18 cystic fibrosis patients [abstract]. Journal of Cystic Fibrosis 2004;3(Suppl 1):S66. [CFGD Register: BD113] [Google Scholar]
Amin 2010 {published data only}
- Amin. The Effect of Hypertonic Saline on the Lung Clearance Index in Patients With Cystic Fibrosis. NCT00635141 August 2013.
- Amin R, Subbarao P, Jabar A, Balkovec S, Jensen R, Kerrigan S, et al. Hypertonic saline improves the LCI in paediatric patients with CF with normal lung function. Thorax 2010;65(5):379‐83. [CFGD Register: BD158b] [DOI] [PubMed] [Google Scholar]
- Amin R, Subbarao P, Jabar A, Balkovec S, Jensen R, Kerrigan S, et al. Inhaled hypertonic saline (7%) improves LCI in paediatric CF patients with normal lung function [abstract]. Pediatric Pulmonology 2009;44 Suppl 32:289, Abstract no: 223. [CFGD Register: BD158a] [Google Scholar]
Ballmann 1998 {published data only}
- Ballmann M, Hardt H. Hypertonic saline and recombinant human DNase: a randomised cross‐over pilot study in patients with cystic fibrosis. Journal of Cystic Fibrosis 2002;1(1):35‐7. [CFGD Register: BD70b] [DOI] [PubMed] [Google Scholar]
- Ballmann M, Hardt H. Hypertonic saline and recombinant human DNase: a randomised cross‐over pilot study in patients with cystic fibrosis [abstract]. Proceedings of the 22nd European Cystic Fibrosis Conference; 1998 June 13‐19; Berlin. 1998:80. [CFGD Register: BD70a] [DOI] [PubMed]
Cardinale 2003 {published data only}
- Cardinale F, Manca A, Mappa L, Tesse R, Cavallone R, Loffredo MS, et al. Effects of exhaled nitric oxide levels and lung function of ultrasonically nebulized hypertonic saline solution in cystic fibrosis [abstract]. European Respiratory Journal 2003;22(Suppl 45):231s. [CFGD Register: BD115] [Google Scholar]
Chadwick 1997 {published data only}
- Chadwick SL, Moss SJ, Bott J, Geddes DM, Alton EWFW. Effect of hypertonic saline, isotonic saline and water challenges on the airways of cystic fibrosis patients. Thorax 1997;52(Suppl 6):A43. [CFGD Register: BD94; ] [Google Scholar]
Dentice 2016 {published data only}
- Dentice R, Elkins M, Bye P. A randomised controlled trial of hypertonic saline inhalation to enhance airway clearance physiotherapy in adults hospitalised with Cystic Fibrosis (CF) [abstract]. Physiotherapy (United Kingdom) 2015;101:eS356. [CENTRAL: 1126510; CRS: 5500050000000302; EMBASE: 72114064; BD185c] [Google Scholar]
- Dentice R, Elkins M, Bye P. A randomised trial of hypertonic saline nebulisation during hospitalisation for pulmonary exacerbation in adults with cystic fibrosis [abstract]. Pediatric Pulmonology 2012;47 Suppl:257‐8, Abstract no: 102. [CENTRAL: 1030604; CFGD Register: BD185b; CRS: 5500050000000301; EMBASE: 70891850] [Google Scholar]
- Dentice R, Elkins M, Middleton P, Bishop J, Wark P, Dorahy D, et al. A randomised controlled trial of the effect of hypertonic saline (HS) inhalation on exacerbation resolution, hospital length of stay and time to relapse in adults with cystic fibrosis [abstract]. Journal of Cystic Fibrosis 2013;12 Suppl 1:S38, Abstract no: WS19.4. [CFGD Register: BD185a] [Google Scholar]
- Dentice RL, Elkins MR, Middleton PG, Bishop JR, Wark PAB, Dorahy DJ, et al. A randomised trial of hypertonic saline during hospitalisation for exacerbation of cystic fibrosis. Thorax 2016;71(2):141‐7. [CENTRAL: 1138352; CFGD Register: BD185d; CRS: 5500050000000401; EMBASE: 20160158105] [DOI] [PubMed] [Google Scholar]
Elkins 2006a {published data only}
- Bye P, Elkins M, Robinson M, Moriarty C, Rose B, Harbour C, et al. Long‐term inhalation of hypertonic saline in patients with cystic fibrosis ‐ a randomised controlled trial [abstract]. Pediatric Pulmonology 2004;38(Suppl 27):329. [CFGD Register: BD114a] [Google Scholar]
- Elkins M, Robinson M, Moriarty C, Sercombe J, Rose B, Harbour C, et al. Three methods of monitoring adherence in a long‐term trial in cystic fibrosis [abstract]. Journal of Cystic Fibrosis 2006;5 Suppl:S111. [CFGD Register: BD114d] [Google Scholar]
- Elkins MR, Robinson M, Moriarty C, Sercombe J, Rose B, Harbour C. Three methods of monitoring adherence in the national hypertonic saline trial in cystic fibrosis [abstract]. Pediatric Pulmonology 2005;40(Suppl 28):268. [CFGD Register: BD114c] [Google Scholar]
- Elkins MR, Robinson M, Rose BR, Harbour C, Moriarty C, Marks GB, et al. A controlled trial of long‐term hypertonic saline in patients with cystic fibrosis. New England Journal of Medicine 2006;354(3):229‐40. [CFGD Register: BD114b] [DOI] [PubMed] [Google Scholar]
- NCT00271310. The effects of long term inhalation of hypertonic saline in subjects with cystic fibrosis [The effects of long term inhalation of hypertonic saline in subjects with cystic fibrosis]. clinicaltrials.gov/ct2/show/NCT00271310 (first received 30 December 2005).
- Ratjen F. Inhaled hypertonic saline produces small increases in lung function in patients with cystic fibrosis. Journal of Pediatrics 2006;149(1):142. [CENTRAL: 1253122; CFGD Register: BD114e; CRS: 5500135000001675; PUBMED: 17243308] [DOI] [PubMed] [Google Scholar]
Eng 1996 {published data only}
- Button BM, Riedler J, Eng P, Robertson CF. Inhaled hypertonic saline as an adjunct to chest physiotherapy in cystic fibrosis; the three year clinical experience [abstract]. Pediatric Pulmonology 1996;Suppl 13:306. [CFGD Register: BD71d] [Google Scholar]
- Eng P, Morton J, Douglas J, Riedler J, Wilson J, Robertson CF. Efficacy of short‐term ultrasonically nebulised hypertonic saline in patients with cystic fibrosis [abstract]. European Respiratory Journal 1995;8(Suppl 19):510s. [CFGD Register: BD71c] [Google Scholar]
- Eng PA, Morton J, Douglass J, Riedler J, WIlson J, Robertson C. Short‐term administration of ultrasonically nebulised hypertonic saline improves pulmonary function in patients with cystic fibrosis. Australian & New Zealand Journal of Medicine 1995;25:440. [CFGD Register: BD71b] [Google Scholar]
- Eng PA, Morton J, Douglass JA, Riedler J, Wilson J, Robertson CF. Short‐term efficacy of ultrasonically nebulized hypertonic saline in cystic fibrosis. Pediatric Pulmonology 1996;21(2):77‐83. [CFGD Register: BD71a] [DOI] [PubMed] [Google Scholar]
Laube 2009 {published data only}
- Laube BL, Sharpless G, Carson KA, Kelly A, Mogayzel PJ. Acute inhalation of hypertonic saline does not improve mucociliary clearance in all children with cystic fibrosis. BMC Pulmonary Medicine 2011;11:45. [CENTRAL: 806034; CFGD Register: BD157b; CRS: 5500050000000304; PUBMED: 21896198] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laube BL, Sharpless GJ, Kelly AE, Mogayel PJ. Effects of hypertonic saline on mucocilliary clearance in children with cystic fibrosis [abstract]. Pediatric Pulmonology 2009;44(Suppl 32):Abstract 77. [CFGD Register: BD157a] [Google Scholar]
- NCT01293084. Hypertonic saline and mucociliary clearance in children [Acute inhalation of hypertonic saline does not improve mucociliary clearance in all children with cystic fibrosis]. clinicaltrials.gov/ct2/show/NCT01293084 (first received 10 February 2011).
Mainz 2015 {published data only}
- Mainz JG, Schaedlich K, Hentschel J, Schumacher U, Koitschev C, Koitschev A, et al. Sinonasal inhalation of isotonic vs. hypertonic saline (6.0%) in CF patients with chronic rhinosinusitis ‐ results of a multicentre, double‐blind, controlled prospective trial [abstract]. Journal of Cystic Fibrosis : Official Journal of the European Cystic Fibrosis Society 2015;14 Suppl 1:S95, Abstract no: 146. [CENTRAL: 1081480; CFGD Register: CO59; CRS: 5500135000000026] [DOI] [PubMed] [Google Scholar]
- Mainz JG, Schumacher U, Schadlich K, Hentschel J, Koitschev C, Koitschev A, et al. Sino nasal inhalation of isotonic versus hypertonic saline (6.0%) in CF patients with chronic rhinosinusitis ‐ Results of a multicenter, prospective, randomized, double‐blind, controlled trial. Journal of Cystic Fibrosis : Official Journal of the European Cystic Fibrosis Society 2016 June 04 [Epub ahead of print]. [CENTRAL: 1155399; CFGD Register: CO59b; CRS: 5500135000001522; DOI: 10.1016/j.jcf.2016.05.003; PUBMED: 27267518] [DOI] [PubMed]
Riedler 1996 {published data only}
- Button BM, Riedler J, Eng P, Roberston CF. Inhaled hypertonic saline as an adjunct to chest physiotherapy in cystic fibrosis; the three year clinical experience [abstract]. Pediatric Pulmonology 1996;Suppl 13:306. [CFGD Register: BD69a] [Google Scholar]
- Riedler J, Reade T, Button B, Robertson CF. A pilot study of inhaled hypertonic saline to increase sputum production in adolescents with cystic fibrosis [abstract]. European Respiratory Journal 1994;7(Suppl 18):430s. [CFGD Register: BD69c] [Google Scholar]
- Riedler J, Reade T, Button B, Robertson CF. Inhaled hypertonic saline increases sputum expectoration in cystic fibrosis. Journal of Paediatric Child Health 1996;32(1):48‐50. [CFGD Register: BD69b] [DOI] [PubMed] [Google Scholar]
Robinson 1996 {published data only}
- Robinson M, Regnis JA, Bailey DL, King M, Bautovich GJ, Bye PT. Effect of hypertonic saline, amiloride, and cough on mucociliary clearance in patients with cystic fibrosis. American Journal of Respiratory and Critical Care Medicine 1996;153(5):1503‐9. [CFGD Register: BD83; ] [DOI] [PubMed] [Google Scholar]
Robinson 1997 {published data only}
- Robinson M, Hemming A, Regnis J, Wong A, Bailey D, Bautotvich G, et al. Effect of increasing doses of hypertonic saline on mucociliary clearance inpatients with cystic fibrosis. Thorax 1997;52(10):900‐3. [CFGD Register: BD45b] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M, Hemming AL, Bailey DL, Mellis CM, Bautovich GJ, Bye PTP. Effect of increasing doses of hypertonic saline on mucociliary clearance in patients with cystic fibrosis [abstract]. American Journal of Respiratory and Critical Care Medicine 1996;153(Suppl):A70. [CFGD Register: BD45a; ] [DOI] [PubMed] [Google Scholar]
- Robinson M, Parsons S, Hemming A, Bautovich G, Mellis CM, Bye PTP. Effect of increasing doses of hypertonic saline on mucociliary clearance in patients with cystic fibrosis. [abstract]. Australian and New Zealand Journal of Medicine 1997;27:258. [CFGD Register: BD45c; ] [Google Scholar]
Robinson 1999 {published data only}
- Robinson M, Daviskas E, Eberl S, Baker J, Anderson SD, Bye PT. Effect of inhaling a dry powder of mannitol on mucociliary clearance in adults with cystic fibrosis [abstract]. Pediatric Pulmonolgy 1998;Suppl 17:280. [CFGD Register: BD84a] [Google Scholar]
- Robinson M, Daviskas E, Eberl S, Baker J, Chan H, Anderson, S, et al. The effect of inhaled mannitol on bronchial mucus clearance in cystic fibrosis patients: a pilot study. European Respiratory Journal 1999;14(3):678‐85. [CFGD Register: BD84b] [DOI] [PubMed] [Google Scholar]
- Robinson M, Daviskas E, Eberl S, Baker J, Chan HK, Anderson SD, Bye PT. The effect of inhaled mannitol on bronchial mucus clearance in cystic fibrosis patients: a pilot study [abstract]. Pediatric Pulmonology 2000;29(6):484. [CFGD Register: BD84c] [DOI] [PubMed] [Google Scholar]
Rosenfeld 2012 {published data only}
- Alpern AN, Brumback LC, Ratjen F, Rosenfeld M, Davis SD, Quittner AL. Initial evaluation of the Parent Cystic Fibrosis Questionnaire‐‐Revised (CFQ‐R) in infants and young children. Journal of Cystic Fibrosis : Official Journal of the European Cystic Fibrosis Society 2015;14(3):403‐11. [CENTRAL: 1130846; CFGD Register: BD175i ; CRS: 5500135000001461; PUBMED: 25443473] [DOI] [PubMed] [Google Scholar]
- Brumback LC, Baines A, Ratjen F, Davis SD, Daniel SL, Quittner AL, et al. Pulmonary exacerbations and parent‐reported outcomes in children <6 years with cystic fibrosis. Pediatric Pulmonology 2015;50:236‐43. [CENTRAL: 1046755; CRS: 5500131000000349; GR:: U01 HL092931/HL/NHLBI NIH HHS/United States; JID:: 8510590; PMCID:: PMC4213320; PUBMED: 24777957; U01: HL092932/HL/NHLBI NIH HHS/United States] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis SD, Ratjen F, Brumback LC, Johnson RC, Filbrun AG, Kerby GS, et al. Infant lung function tests as endpoints in the ISIS multicenter clinical trial in cystic fibrosis. Journal of Cystic Fibrosis : Official Journal of the European Cystic Fibrosis Society 2016;15(3):386‐91. [CFGD Register: BD175l; CRS: 5500135000001676; PUBMED: 26547590] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis SD, Rosenfeld M, Brumback L, Donaldson S, Johnson R, Rowbotham R, et al. Infant PFTs as an endpoint in the infant study of inhaled saline randomized controlled trial [abstract]. Pediatric Pulmonology 2012;47(S35):302, Abstract no: 225. [CFGD Register: BD175d; ] [Google Scholar]
- NCT01293084. Hypertonic saline and mucociliary clearance in children [Acute inhalation of hypertonic saline does not improve mucociliary clearance in all children with cystic fibrosis]. clinicaltrials.gov/ct2/show/NCT01293084 (first received 10 February 2011).
- Ratjen F, Rosenfeld M, Brumback L, Daniel S, Rowbotham R, Kronmal R, et al. Efficacy of hypertonic saline in infants and young children: the ISIS study [abstract]. Journal of Cystic Fibrosis 2012;11 (Suppl 1):S50. [CFGD Register: BD175a] [Google Scholar]
- Ratjen F, Salazar J, Jensen R, Amin R, Gustafsson P, Stanojevic S, et al. Alternative multiple breath washout outcomes for clinical trials in cystic fibrosis [abstract]. European Respiratory Journal 2012;40 Suppl:Abstract no: 1261. [CENTRAL: 1100495; CFGD Register: BD175j ; CRS: 5500050000000311; EMBASE: 71925430] [Google Scholar]
- Rosenfeld M. Hypertonic saline should be the first drug used in CF [summary]. Pediatric Pulmonology 2013;48 Suppl 36:157, Summary: S12.3. [CENTRAL: 921667; CRS: 5500125000000393] [Google Scholar]
- Rosenfeld M, Ratjen F, Brumback L, Daniel S, Rowbotham R, McNamara S, et al. Inhaled hypertonic saline in infants and children younger than 6 years with cystic fibrosis: the ISIS randomized controlled trial. JAMA 2012;307(21):2269‐77. [CFGD Register: BD175b] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld M, Ratjen F, Brumback L, Daniel S, Rowbotham R, McNamara S, et al. Online supplement to "Inhaled hypertonic saline in infants and children younger than 6 years with cystic fibrosis: the ISIS randomized controlled trial" [online]. JAMA 2012;307(21):2269‐77. [CENTRAL: 1155401; CFGD Register: BD175k; CRS: 5500135000001526] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld M, Ratjen F, Brumback L, Kronmal R, Davis SD. Hypertonic saline in children with CF <6 years of age: Results of the ISIS trial [abstract]. Pediatric Pulmonology 2012;47:172. [CENTRAL: 1030608; CFGD Register: BD175h ; CRS: 5500050000000298; EMBASE: 70891711] [Google Scholar]
- Subbarao P, Stanojevic S, Brown M, Jensen R, Rosenfeld M, Davis S, et al. Lung clearance index as an outcome measure for clinical trials in young children with cystic fibrosis. A pilot study using inhaled hypertonic saline. American Journal of Respiratory and Critical Care Medicine 2013;188(4):456‐60. [CRS: 5500127000000014; GR:: Canadian Institutes of Health Research/Canada; JID:: 9421642; OID: [Other ID]: NLM: PMC3778738; PMCID:: PMC3778738; PUBMED: 23742699; SI:: ClinicalTrials.gov/NCT00709280] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbararo P, Stanojevic S, Brown M, Jensen R, McDonald N, Gent K, et al. Effect of hypertonic saline on lung clearance index in infants and preschool children with CF: a pilot study [abstract]. Pediatric Pulmonology 2012;47(S35):301. [CFGD Register: BD175c] [Google Scholar]
Suri 2001 {published data only}
- Grieve R, Thompson S, Normand C, Suri R, Bush A, Wallis C. A cost‐effectiveness analysis of rhDNase in children with cystic fibrosis. International Journal of Technology Assessment in Health Care 2003;19(1):71‐9. [CFGD Register: BD96h] [DOI] [PubMed] [Google Scholar]
- Suri R, Grieve R, Normand C, Metcalfe C, Thompson S, Wallis C, et al. Effects of hypertonic saline, alternate day and daily rhDNase on healthcare use, costs and outcomes in children with cystic fibrosis. Thorax 2002;57(10):841‐6. [CFGD Register: BD96e] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suri R, Grieve R, Normand C, Metcalfe C, Thompson S, Wallis C, et al. Effects of hypertonic saline, alternate day and daily rhDNase on healthcare use, costs and outcomes in children with cystic fibrosis [abstract]. Thorax 2001;56(Suppl 3):iii84. [CFGD Register: BD96f] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suri R, Marshall LJ, Wallis C, Metcalfe C, Bush A, Shute JK. Effects of recombinant human DNase and hypertonic saline on airway inflammation in children with cystic fibrosis. American Journal of Respiratory and Critical Care Medicine 2002;166(3):352‐5. [CFGD Register: BD96d] [DOI] [PubMed] [Google Scholar]
- Suri R, Metcalfe C, Lees B, Flather M, Normand C, Thompson S, et al. A cross‐over comparative study of hypertonic saline alternate day and daily rhDNase in children with Cystic Fibrosis [abstract]. Thorax 2000;55:A75. [CFGD Register: BD96g] [Google Scholar]
- Suri R, Metcalfe C, Lees B, Grieve R, Flather M, Normand C, et al. Comparison of hypertonic saline and alternate day or daily recombinant deoxyribonuclease in children with cystic fibrosis: a randomised trial. Lancet 2001;358(9290):1316‐21. [CFGD Register: BD96b] [DOI] [PubMed] [Google Scholar]
- Suri R, Metcalfe C, Wallis C, Bush A. Assessing the usefulness of outcomes measured in a cystic fibrosis treatment trial. Respiratory Medicine 2007;101(2):254‐60. [CFGD Register: BD96k] [DOI] [PubMed] [Google Scholar]
- Suri R, Metcalfe C, Wallis C, Bush A. Predicting response to rhDNase and hypertonic saline in children with cystic fibrosis children with cystic fibrosis. Pediatric Pulmonology 2004;37(4):305‐10. [CFGD Register: BD96i] [DOI] [PubMed] [Google Scholar]
- Suri R, Wallis C, Bush A. In vivo use of hypertonic saline in cystic fibrosis [abstract]. Pediatric Pulmonology 2000;Suppl 20:125‐6. [CFGD Register: BD96a] [Google Scholar]
- Suri R, Wallis C, Bush A, Thompson S, Normand C, Flather M, et al. A comparative study of hypertonic saline, daily and alternate‐day rhDNase in children with cystic fibrosis. Health Technology Assessment 2002; Vol. 6, issue 34:iii, 1‐60. [CFGD Register: BD96j] [PubMed]
- Suri R, Wallis C, Metcalfe C, Thompson S, Bush A, Shute J. Effects of rhDNase and hypertonic saline on airway inflammation in children with cystic fibrosis [abstract]. Pediatric Pulmonology 2001;Suppl 22:281. [CFGD Register: BD96c] [Google Scholar]
Weller 1980 {published data only}
- Weller P, Ingram D, Preece M, Matthew D. A controlled trial of intermittent aerosol therapy in patients with cystic fibrosis [abstract]. Proceedings of the 9th Meeting European Working Group for Cystic Fibrosis; 1979 June 12‐13; Noordwijkerhout, The Netherlands. 1979:58. [CFGD Register: BD26b]
- Weller P, Ingram D, Preece M, Matthew D. Controlled trial of intermittent aerosol therapy with sodium 2‐mercaptoethane sulphonate in cystic fibrosis. Thorax 1980;35(1):42‐6. [CFGD Register: BD26a] [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies excluded from this review
Brivio 2016 {published data only}
- Brivio A, Ceruti C, Gambazza S, Colombo C. Randomized double‐blind monocentric trial on tolerability, acceptability and efficacy of two formulations of inhaled 7% hypertonic saline with and without hyaluronic acid in reducing airways inflammation in patients with cystic fibrosis ‐ preliminary results [abstract]. Journal of Cystic Fibrosis : Official Journal of the European Cystic Fibrosis Society 2013;12 Suppl 1:S104, Abstract no: 217. [CENTRAL: 999881; CFGD Register: BD208a; CRS: 5500127000000003] [Google Scholar]
- Brivio A, Conese M, Gambazza S, Biffi A, Tirelli AS, Russo M, et al. Pilot Randomized Controlled Trial Evaluating the Effect of Hypertonic Saline With and Without Hyaluronic Acid in Reducing Inflammation in Cystic Fibrosis. Journal of Aerosol Medicine and Pulmonary Drug Delivery 2016 May 15 [Epub ahead of print]. [CENTRAL: 1155400; CFGD Register: BD208b; CRS: 5500135000001523; PUBMED: 27149365] [DOI] [PubMed]
Buonpensiero 2010 {published data only (unpublished sought but not used)}
- Buonpensiero P, Gregorio F, Sepe A, Pasqua A, Ferri P, Siano M, et al. Hyaluronic acid improves "pleasantness" and tolerability of nebulized hypertonic saline in a cohort of patients with cystic fibrosis. Advances in Therapy 2010;27(11):870‐8. [CENTRAL: 781252; CFGD Register: BD156c; CRS: 5500100000003514; EMBASE: 2010642492; PUBMED: 20953746] [DOI] [PubMed] [Google Scholar]
- Buonpensiero P, Gregorio F, Sepe A, Pasqua A, Ferri P, Siano M, et al. Hyaluronic acid improves tolerability of hypertonic saline in CF patients [abstract]. Journal of Cystic Fibrosis 2010;9 Suppl 1:S63, Abstract no: 245. [CENTRAL: 775087; CFGD Register: BD156b; CRS: 5500100000003507] [Google Scholar]
- Buonpensiero P, Gregorio F, Sepe A, Pasqua A, Ferri P, Siano M, et al. Inhaled hyaluronic acid improves pleasantness and tolerability of nebulised hypertonic saline in patients with cystic fibrosis [abstract]. Pediatric Pulmonology 2009;44 Suppl 32:243, Abstract no: 93. [CFGD Register: BD156a] [Google Scholar]
- NCT01658449. Comparison of the tolerability of two formulations of hypertonic saline in cystic fibrosis patients. clinicaltrials.gov/ct2/show/NCT01658449 (first posted 07 August 7 2012).
DeCono 2008 {published data only}
- Cono N, Schelstraete P, Haerynck F, daele S, Sanders M, Baets F. Hypertonic saline: effect on mucus rheology and spirometry. Journal of Cystic Fibrosis 2008;7(Suppl 2):S24, Abstract 93. [CFGD Register: BD130] [Google Scholar]
Dentice 2012 {published data only}
- ACTRN12611000673943. A randomised trial of the effect of timing of nebulised hypertonic saline in relation to airway clearance physiotherapy, in people with cystic fibrosis lung disease [Randomised trial of the effect of timing of nebulised hypertonic saline in relation to airway clearance physiotherapy, on subjective efficacy, tolerability and overall satisfaction, and lung function parameters in adults with cystic fibrosis lung disease]. apps.who.int/trialsearch/Trial3.aspx?trialid=ACTRN12611000673943 (first received 04 July 2011).
- Dentice R, Elkins M, Bye P. Hypertonic saline before vs during vs after physiotherapy techniques for airway clearance in people with cystic fibrosis: A randomised trial [abstract]. Physiotherapy (United Kingdom) 2011;97:eS308‐eS309, Abstract no: RR‐PL‐3136. [CENTRAL: 1089301; CFGD Register: BD160d; CRS: 5500050000000300; EMBASE: 71882596] [Google Scholar]
- Dentice R, Elkins MR, Bye PT. A randomised trial of the effect of timing of hypertonic saline inhalation in relation to airway clearance physiotherapy in adults with cystic fibrosis [abstract]. Pediatric Pulmonology 2010;45 Suppl 33:384, Abstract no: 459. [CFGD Register: BD160a] [Google Scholar]
- Dentice RL, Elkins MR, Bye P. Adults with cystic fibrosis prefer hypertonic saline before or during airway clearance techniques: a randomised crossover trial. Journal of Physiotherapy 2012;58(1):33‐40. [CFGD Register: BD160b] [DOI] [PubMed] [Google Scholar]
- Dentice RL, Elkins MR, Bye PT. Online Supplementary Material ‐ Table 3 : Individual outcome data to "Adults with cystic fibrosis prefer hypertonic saline before or during airway clearance techniques: a randomised crossover trial" [online]. Journal of Physiotherapy 2012;58(1):33‐40 Online. [CFGD Register: BD160c] [DOI] [PubMed] [Google Scholar]
Donaldson 2006 {published data only}
- Donaldson SH, Bennett W, Zeman K, Knowles MR, Boucher RC. Efficacy of amiloride and hypertonic saline in cystic fibrosis [abstract]. Pediatric Pulmonology 2003;Suppl 25:251. [CFGD Register: BD110a] [Google Scholar]
- Donaldson SH, Bennett WD, Zeman KL, Knowles MR, Tarran R, Boucher RC. Mucus clearance and lung function in cystic fibrosis with hypertonic saline. New England Journal of Medicine 2006;354(3):241‐50. [CFGD Register: BD110b] [DOI] [PubMed] [Google Scholar]
- Donaldson SH, Bennett WD, Zeman KL, Knowles MR, Tarran R, Boucher RC. Online supplement to 'Mucus clearance and lung function in cystic fibrosis with hypertonic saline'. New England Journal of Medicine 2006;354(3):241‐50 Online. [CFGD Register: BD110c] [DOI] [PubMed] [Google Scholar]
- NCT00274391. Efficacy of amiloride and hypertonic saline in cystic fibrosis [Efficacy of amiloride and hypertonic saline in cystic fibrosis]. clinicaltrials.gov/ct2/show/NCT00274391 (first posted 10 January 2006).
Elkins 2006b {published data only}
- ACTRN12606000053527. Pilot trial of the tolerability of hypertonic saline when delivered by a high‐output nebuliser twice or four times daily to people with cystic fibrosis [Pilot trial of the tolerability of hypertonic saline when delivered by a high‐output nebuliser twice or four times daily to people with cystic fibrosis]. apps.who.int/trialsearch/Trial3.aspx?trialid=ACTRN12606000053527 (first received 07 February 2006).
- Elkins MR, Tingpej P, Moriarty CP, Yozghatlian V, Rose BR, Harbour C, et al. Tolerability of hypertonic saline when delivered rapidly via the Eflow® rapid nebulizer in subjects with cystic fibrosis [abstract]. Pediatric Pulmonlogy 2006;41 (Suppl 29):292. [CFGD Register: BD124] [Google Scholar]
EUCTR2007‐002707‐40‐BE {unpublished data only}
- EUCTR2007‐002707‐40‐BE. The effect of inhalation with hypertonic saline (7%) on lung function and sputum rheology in cystic fibrosis patients. www.clinicaltrialsregister.eu/ctr‐search/search?query=2007‐002707‐40 (first received 28 June 2007).
Genkova 1998 {published data only}
- Genkova N, Bosheva M, Ivancheva D. Inhaled hypertonic saline solution in cystic fibrosis [abstract]. Proceedings of the 22nd European Cystic Fibrosis Conference; 1998 June 13‐19; Berlin. 1998.
Grasemann 2013 {published and unpublished data}
- Grasemann H, Tullis E, Ratjen F. A randomized controlled trial of inhaled L‐arginine in patients with cystic fibrosis. Journal of Cystic Fibrosis 2013;12(5):468‐74. [DOI] [PubMed] [Google Scholar]
- NCT00405665. The short term safety and efficacy of inhaled L‐arginine in patients with cystic fibrosis [Pilot Study of the Short Term Safety and Efficacy of Inhaled L‐arginine in Patients With Cystic Fibrosis]. clinicaltrials.gov/ct2/show/NCT00405665 (first posted 30 November 2006).
IRCT20180307038994N1 {published data only}
- IRCT20180307038994N1. Comparison of the nebulized mannitol with nebulized hypertonic saline on pulmonary function in patients with cystic fibrosis. apps.who.int/trialsearch/Trial2.aspx?TrialID=IRCT20180307038994N1 (first received 15 April 2018).
King 1997 {published data only}
- King M, Dasgupta B, Tomkiewicz RP, Brown NE. Rheology of cystic fibrosis sputum after in vitro treatment with hypertonic saline alone and in combination with recombinant human deoxyribonuclease. American Journal of Respiratory and Critical Care Medicine 1997;156(1):173‐7. [DOI] [PubMed] [Google Scholar]
Kobylyansky 2000 {published data only}
- Kobylyansky VI, Gembitskaya TE. Study of the mucociliary and cough clearance in patients with mucoviscidosis and evaluation of hypertonic sodium chorlide solution influence on them [abstract]. European Respiratory Journal 2000;16(Suppl 31):121S. [CFGD Register: BD111; ] [Google Scholar]
NCT01094704 {published data only}
- NCT01094704. Durability of hypertonic saline for enhancing mucociliary clearance in cystic fibrosis. clinicaltrials.gov/show/NCT01094704 (first posted 29 March 2010).
O'Neill 2017 {published data only}
- NCT01753869. Timing of hypertonic saline inhalation relative to airways clearance in cystic fibrosis [Timing of hypertonic saline inhalation relative to airways clearance in cystic fibrosis]. clinicaltrials.gov/ct2/show/NCT01753869 (first posted 20 December 2012).
- O'Neill K, Moran F, Tunney MM, Elborn JS, Bradbury I, Downey DG, et al. Timing of hypertonic saline and airway clearance techniques in adults with cystic fibrosis during pulmonary exacerbation: pilot data from a randomised crossover study. BMJ Open Respiratory Research 2017;4(1):e000168. [DOI: 10.1136/bmjresp-2016-000168] [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill K, Moran F, Bradbury I, Downey DG, Rendall J, Tunney MM, et al. Exploring the timing of hypertonic saline (HTS) and airways clearance techniques (ACT) in cystic fibrosis (CF): a cross over study. Thorax 2016;71(Suppl 3):A134‐5. [Google Scholar]
Ros 2012 {published data only}
- Ros M, Casciaro R, Lucca F, Alatri F, Salonini E, Favilli F, et al. Tolerability and acceptability in patients with cystic fibrosis (CF) of two formulations of 7% hypertonic saline: a prospective multicenter clinical study [abstract]. Pediatric Pulmonology 2012;47(S35):364, Abstract no: 390. [CFGD Register: BD181a; ] [Google Scholar]
- Ros M, Casciaro R, Lucca F, Troiani P, Salonini E, Favilli F, et al. Hyaluronic acid improves the tolerability of hypertonic saline in the chronic treatment of cystic fibrosis patients: a multicenter, randomized, controlled clinical trial. Journal of Aerosol Medicine and Pulmonary Drug Delivery 2014;27(2):133‐7. [CFGD Register: BD181b] [DOI] [PubMed] [Google Scholar]
San Miguel 2016 {published data only}
- Herrero Cortina B, San Miguel Pagola M, Cebria i Ranzo MA, Gomez Romero M, Diaz Gutierrez F, Reychler G. Short‐term effects of hypertonic saline nebulization combined with oscillatory positive expiratory pressure in cystic fibrosis:randomised crossover trial. Journal of Cystic fibrosis 2016;15 Suppl 1:S33. [Abstract no.: WS21.3; CFGD Register: BD229a] [DOI] [PubMed] [Google Scholar]
- NCT02303808. Positive expiratory pressure during inhalation of hypertonic saline in patients with cystic fibrosis [Effect of introducing a positive expiratory pressure device during inhalation of hypertonic saline in patients with cystic fibrosis: a randomized crossover trial]. clinicaltrials.gov/ct2/show/NCT02303808 (first received 01 December 2014).
- San Miguel Pagola M, Herrero Cortina B, Cebria i Iranzo MA, Gomez Romero M, Diaz Gutierrez F, Reychler G. Hypertonic saline nebulization combined with oscillatory positive expiratory pressure accelerate sputum clearance in cystic fibrosis: A randomised crossover trial. European Respiratory Journal 2016;48(Suppl 60):PA1369. [CFGD Register: BD229b] [DOI] [PubMed] [Google Scholar]
Van Ginderdeuren 2008 {published and unpublished data}
- Ginderdeuren F, Verbanck S, Cauwelaert K, Vanlaethem S, Schuermans D, Vincken W, et al. Chest physiotherapy in cystic fibrosis: short‐term effects of autogenic drainage preceded by wet inhalation of saline versus autogenic drainage preceded by intrapulmonary percussive ventilation with saline. Respiration 2008;76(2):175‐80. [CFGD Register: BD178] [DOI] [PubMed] [Google Scholar]
Van Ginderdeuren 2011 {published data only}
- Ginderdeuren F, Vanlaethem S, Eyns H, Schutter I, Dewachter E, Malfroot A. Influence of inhaled hypertonic saline (NaCl 6%) before or during autogenic drainage on sputum weight, oxygen saturation, heart frequency and dyspnea in cystic fibrosis patients [abstract]. Journal of Cystic Fibrosis 2011;10(Suppl 1):S62. [CFGD Register: BD164] [Google Scholar]
Vanlaethem 2008 {published data only}
- Vanlaethem S, Ginderdeuren F, Eyns H, Malfroot A. Influence of inhaled hypertonic saline combined with airway clearance on SpO2, heart rate, dyspnoea and wet sputum weight in hospitalised CF patients [abstract]. Journal of Cystic Fibrosis 2008;7(Suppl 2):S71. [CENTRAL: 643104; CFGD Register: BD129; CRS: 5500100000003217] [Google Scholar]
References to studies awaiting assessment
Amin 2016 {published data only}
- Amin R, Stanojevic S, Kane M, Webster H, Ratjen F. A randomized controlled trial to evaluate the lung clearance index as an outcome measure for early phase studies in patients with cystic fibrosis. Respiratory Medicine 2016;112:59‐64. [CFGD Register: BD228; CRS: 5500135000001524; PUBMED: 26856191] [DOI] [PubMed] [Google Scholar]
Balinotti 2015 {published data only}
- Balinotti J, Rodriguez V, Zaragoza S, Lubovich S, Kofman C, Garcia Bournissen F. Effect of early intervention with inhaled hypertonic saline on lung function in infants and toddlers with cystic fibrosis diagnosed by neonatal screening [abstract]. American Journal of Respiratory and Critical Care Medicine 2015;191(Meeting Abstracts):A3342. [CENTRAL: 1127230; CFGD Register: BD227b; CRS: 5500050000000303] [Google Scholar]
- Teper A, Balinotti J, Rodriguez V, Zaragoza S Lubovich S, Kofman C, Garcia Bournissen F. Effect of early intervention with inhaled hypertonic saline on lung function in infants and toddlers with cystic fibrosis (CF) diagnosed by neonatal screening [abstract]. Journal of Cystic Fibrosis : Official Journal of the European Cystic Fibrosis Society 2015;14 Suppl 1:S49, Abstract no: ePS04.8. [CFGD Register: BD227a; CRS: 5500135000001305] [Google Scholar]
Brown 2010 {published data only}
- Brown AW, Laube BL, Zeman K, Lechtzin N, Sharpless G, Wu J, et al. Durability of hypertonic saline for enhancing mucociliary clearance in cystic fibrosis [abstract]. Pediatric Pulmonology 2010;45 Suppl 33:303, Abstract no: 237. [CFGD Register: BD173; ] [Google Scholar]
Corcoran 2017 {published data only}
- Corcoran TE, Godovchik JE, Donn KH, Busick DR, Goralski J, Locke LW, et al. Overnight delivery of hypertonic saline by nasal cannula aerosol for cystic fibrosis. Pediatric Pulmonology 2017;52(9):1142‐9. [CFGD Register: BD207; CTG: NCT02141191] [DOI] [PMC free article] [PubMed] [Google Scholar]
Donaldson 2013 {published data only}
- Donaldson SH, Samulski D, LaFave C, Wu J, Zeman K, Salazar C, et al. Sustained effect of hypertonic saline on mucociliary clearance in CF children with mild lung disease. Pediatric Pulmonology 2013;48 Suppl 36:280. [Abstract no.: 210; CENTRAL: 999885; CFGD Register: BD206; CRS: 5500127000000007] [Google Scholar]
- NCT01031706. Effect of hypertonic saline on mucus clearance in children ages 5‐12 with cystic fibrosis [Sustained impact of hypertonic saline on mucociliary clearance in young children with cystic fibrosis]. clinicaltrials.gov/ct2/show/study/NCT01031706 (first received 15 December 2009).
Dwyer 2013 {unpublished data only}
- ACTRN12610000754044. Trial of different concentrations of nebulised saline for cystic fibrosis saline at lower tonicity in cystic fibrosis (SALTI‐CF) trial [The effect of nebulised 0.9% vs 3% vs 6% saline on lung function and quality of life in people with cystic fibrosis]. apps.who.int/trialsearch/Trial3.aspx?trialid=ACTRN12610000754044 (first received 13 September 2010).
- Dwyer T, Elkins M, Dentice R, Forbes S, MacArthur M, Cooper P, et al. Saline at a lower tonicity in cystic fibrosis (SALTI‐CF) trial ‐ a randomised controlled trial comparing 0.9% v 3% v 6% nebulised saline. Journal of Cystic Fibrosis 2013;12 Suppl 1:S19. [Abstract no.: WS9.5; CFGD Register: BD186] [Google Scholar]
Hofmann 1997 {published data only}
- Hofmann T, Senier I, Ziersch A, Geidel C, Regnis J, Lindemann H. Additive effects of amiloride and hypertonic saline on respiratory ion transport in cystic fibrosis [abstract]. Proceedings of the 21st European Cystic Fibrosis Conference; 1997; Davos. 1997:119. [CFGD Register: BD75]
NCT00928135 {unpublished data only}
- NCT00928135. Aerosolized hypertonic xylitol versus hypertonic saline in cystic fibrosis (CF) subjects [Randomized controlled study of aerosolized hypertonic xylitol versus hypertonic saline in hospitalized patients with exacerbation of cystic fibrosis]. clinicaltrials.gov/ct2/show/NCT00928135 (first received 25 June 2009).
NCT01355796 {unpublished data only}
- NCT01355796. Inhaled xylitol versus saline in stable subjects with cystic fibrosis [Randomized cross over study of inhaled hypertonic xylitol versus hypertonic saline in stable subjects with cystic fibrosis]. clinicaltrials.gov/ct2/show/NCT01355796 (first posted 18 May 2011).
NCT01377792 {published data only}
- NCT01377792. Study of long‐term treatment with hypertonic saline in patients with cystic fibrosis [Phase 4 study of the efficacy of long‐term treatment with hypertonic saline on pulmonary exacerbations in patients with cystic fibrosis]. clinicaltrials.gov/ct2/show/NCT01377792 (first received 21 June 2011).
NCT01619657 {unpublished data only}
- NCT01619657. Preventive inhalation of hypertonic saline in infants with cystic fibrosis (PRESIS). clinicaltrials.gov/ct2/show/NCT01619657 (first received 14 June 2012).
NCT02378467 {published data only}
- NCT02378467. Saline hypertonic in preschoolers (SHIP) [Saline hypertonic in preschoolers]. clinicaltrials.gov/ct2/show/NCT02378467 (first received 04 March 2015).
NCT03391414 {published data only}
- NCT03391414. Effects of inhaled bicarbonate on airway pH in cystic fibrosis. clinicaltrials.gov/show/NCT03391414 (first received 05 January 2018).
Nenna 2017 {published data only}
- ISRCTN13412080. Effects of inhaled hypertonic saline in children with cystic fibrosis [Effects of inhaled hypertonic (7%) saline on lung function test in preschool children with cystic fibrosis: a crossover, randomized clinical trial]. apps.who.int/trialsearch/Trial3.aspx?trialid=ISRCTN13412080 (first received 06 July 2016).
- Nenna R, Midulla F, Lambiase C, Castro G, Zicari AM, Indinnimeo L, et al. Effects of inhaled hypertonic (7%) saline on lung function test in preschool children with cystic fibrosis: results of a crossover, randomized clinical trial. Italian Journal of Pediatrics 2017;43(1):60. [CFGD Register: BD238; DOI: 10.1186/s13052-017-0376-6] [DOI] [PMC free article] [PubMed] [Google Scholar]
Palacio 2014 {published data only}
- Palacio S, Giugno H, Diaz Casaux A, Lucero B, Smith S, Giorgetti M, et al. Inhaled 7% hypertonic saline treatment in preschool children with cystic fibrosis [abstract]. Journal of Cystic Fibrosis 2014;13 Suppl 2:S7, Abstract no: WS3.5. [CENTRAL: 1000053; CFGD Register: BD205; CRS: 5500131000000003] [Google Scholar]
PRESIS 2018 {published data only}
- Stahl M, Graeber SY, Sommerburg O, Ricklefs I, Diekmann G, Dopfer C, et al. Randomized double blind controlled pilot study on safety and efficacy of hypertonic saline as preventive inhalation therapy in infants with CF (PRESIS). Atemwegs‐ und Lungenkrankheiten. Conference: 40th annual joint meeting of the society for pediatric pneumology. Austria 2018;44(2):60‐1. [CFGD Register: BD249b] [Google Scholar]
- Stahl M, Ricklefs I, Dopfer C, Barth S, Schlegtendal A, Graeber SY, et al. Randomised, double‐blind, controlled pilot study on safety and efficacy of hypertonic saline as preventive inhalation therapy in infants with CF (PRESIS). Journal of Cystic Fibrosis 2018;17(Suppl 3):S25‐6. [CFGD Register: BD249a] [Google Scholar]
References to ongoing studies
NCT02276898 {unpublished data only}
- NCT02276898. A randomized‐controlled trial of inhaled hypertonic saline (7%) to evaluate the lung clearance index [A randomized‐controlled trial of inhaled hypertonic saline (7%) to evaluate the lung clearance index as a short‐term pharmacodynamic biomarker in patients with cystic fibrosis]. clinicaltrials.gov/ct2/show/NCT02276898 (first received 28 October 2014).
NCT02343445 {unpublished data only}
- NCT02343445. Clearing lungs with ENAC inhibition in cystic fibrosis (CLEAN‐CF) [A randomized, double‐blind, placebo‐controlled, parallel‐group study to evaluate the safety and efficacy of P‐1037 solution for inhalation in patients with cystic fibrosis (CF)]. clinicaltrials.gov/ct2/show/NCT02343445 (first received 22 January 2015).
NCT02950883 {unpublished data only}
- ACTRN12615001067561. Saline hypertonic in preschoolers with cystic fibrosis and lung structure as measured by computed tomography (CT) [A Phase 3 randomised, double‐blind, controlled trial of inhaled 7% hypertonic saline versus 0.9% isotonic saline for 48 weeks on lung structure in patients with cystic fibrosis at 3‐6 years of age, in parallel with the North American SHIP clinical trial, as measured by computed tomography (CT)]. apps.who.int/trialsearch/Trial3.aspx?trialid=ACTRN12615001067561 (first received 13 October 2015).
- NCT02950883. Saline hypertonic in preschoolers + CT (SHIP‐CT) [Saline hypertonic in preschoolers with cystic fibrosis and lung structure as measured by computed tomography (CT)]. clinicaltrials.gov/ct2/show/NCT02950883 (first received 01 November 2016).
Additional references
Courtney 2007
- Courtney JM, Bradley J, McCaughan J, O'Connor TM, Shortt C, Bredin CP, et al. Predictors of mortality in adults with cystic fibrosis. Pediatric Pulmonolgy 2007;42(6):525‐32. [DOI] [PubMed] [Google Scholar]
Dasgupta 1995
- Dasgupta B, Tomkiewicz RP, Brown NE, King M. Combined effects of hypertonic saline and rhDNase on cystic fibrosis sputum in‐vitro. Pediatric Pulmonology 1995;20(Suppl 12):A201‐A236. [DOI] [PubMed] [Google Scholar]
Davis 1996
- Davis P, Drumm M, Konstan W. Cystic fibrosis. American Journal of Respiratory and Critical Care Medicine 1996;154(5):1229‐56. [DOI] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne DR, Altman DG, Higgins JPT, Curtin F, Worthington HV, Vail A. Meta‐analyses involving cross‐over trials: methodological issues . International Journal of Epidemiology 2002;31(1):140‐9. [DOI] [PubMed] [Google Scholar]
Fuchs 1994
- Fuchs HJ, Borowitz DS, Christiansen DH, Morris EM, Nash ML, Ramsey BW, et al. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. New England Journal of Medicine 1994;331(10):637‐42. [DOI] [PubMed] [Google Scholar]
Gustafsson 2008
- Gustafsson PM, DeJong PA, Tiddens HA. Multiple‐breath inert gas washout and spirometry versus structural structural lung disease in cystic fibrosis. Thorax 2008;63(2):129‐34. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327(7414):557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Altman DG (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Kerem 1989
- Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, Buchwald M, Tsui LC. Identification of the cystic fibrosis gene: Genetic analysis. Science 1989;245:1073‐1080. [DOI] [PubMed] [Google Scholar]
Nevitt 2018
- Nevitt SJ, Thornton J, Murray CS, Dwyer T. Inhaled mannitol for cystic fibrosis. Cochrane Database of Systematic Reviews 2018, Issue 2. [DOI: 10.1002/14651858.CD008649.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ramsey 1994
- Ramsey BW, Dorkin HL, for the Consensus Committee. Consensus conference: practical applications of Pulmozyme. September 22, 1993. Pediatric Pulmonology 1993;17(6):404‐8. [DOI] [PubMed] [Google Scholar]
RevMan 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Yang 2018
- Yang C, Montgomery M. Dornase alfa for cystic fibrosis. Cochrane Database of Systematic Reviews 2018, Issue 9. [DOI: 10.1002/14651858.CD001127.pub4] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ziment 1978
- Ziment I. Respiratory pharmacology and therapeutics. Philadelphia: WB Saunders, 1978:60‐104. [Google Scholar]
References to other published versions of this review
Wark 1999
- Wark P, McDonald V. Nebulised hypertonic saline for cystic fibrosis. Cochrane Database of Systematic Reviews 1999, Issue 2. [DOI: 10.1002/14651858.CD001506] [DOI] [PubMed] [Google Scholar]
Wark 2000
- Wark P, McDonald V. Nebulised hypertonic saline for cystic fibrosis. Cochrane Database of Systematic Reviews 2000, Issue 2. [DOI: 10.1002/14651858.CD001506] [DOI] [PubMed] [Google Scholar]
Wark 2003
- Wark P, McDonald V. Nebulised hypertonic saline for cystic fibrosis. Cochrane Database of Systematic Reviews 2003, Issue 1. [DOI: 10.1002/14651858.CD001506.pub2] [DOI] [PubMed] [Google Scholar]
Wark 2009
- Wark, P, McDonald VM. Nebulised hypertonic saline for cystic fibrosis. Cochrane Database of Systematic Reviews 2009, Issue 2. [DOI: 10.1002/14651858.CD001506.pub3] [DOI] [PubMed] [Google Scholar]