

Abstract
The mechanisms by which breast cancers progress from relatively indolent ductal carcinoma in situ (DCIS) to invasive ductal carcinoma (IDC) are not well understood. However, this process is critical to the acquisition of metastatic potential. MAPK-interacting serine/threonine-protein kinase 1 (MNK1) signaling can promote cell invasion. NODAL, a morphogen essential for embryogenic patterning, is often reexpressed in breast cancer. Here we describe a MNK1/NODAL signaling axis that promotes DCIS progression to IDC. We generated MNK1 knockout (KO) or constitutively active MNK1 (caMNK1)-expressing human MCF-10A-derived DCIS cell lines, which were orthotopically injected into the mammary glands of mice. Loss of MNK1 repressed NODAL expression, inhibited DCIS to IDC conversion, and decreased tumor relapse and metastasis. Conversely, caMNK1 induced NODAL expression and promoted IDC. The MNK1/NODAL axis promoted cancer stem cell properties and invasion in vitro. The MNK½ inhibitor SEL201 blocked DCIS progression to invasive disease in vivo. In clinical samples, IDC and DCIS with microinvasion expressed higher levels of phospho-MNK1 and NODAL versus low-grade (invasion-free) DCIS. Cumulatively, our data support further development of MNK1 inhibitors as therapeutics for preventing invasive disease.
Significance:
These findings provide new mechanistic insight into progression of ductal carcinoma and support clinical application of MNK1 inhibitors to delay progression of indolent ductal carcinoma in situ to invasive ductal carcinoma.
Introduction
Over the last 40 years, there has been a significant increase in the diagnosis of breast ductal carcinoma in situ (DCIS) due to the implementation of mammography screening (1). DCIS has the versely, caMNK1 induced NODAL expression and promoted IDC. The MNK1/NODAL axis promoted cancer stem cell properties and invasion in vitro. The MNK1/2 inhibitor SEL201 blocked DCIS progression to invasive disease in vivo. In clinical samples, IDC and DCIS with microinvasion expressed higher levels of phospho-MNK1 and NODAL versus low-grade (invasion-free) DCIS. Cumulatively, our data support further development of MNK1 inhibitors as therapeutics for preventing invasive disease. potential to, but does not always, progress into invasive ductal carcinoma (IDC). The signaling pathways that mediate the transition from noninvasive to invasive disease remain largely uncharacterized.
MAPK-interacting serine/threonine-protein kinases 1 and 2 (MNK1/2) are ubiquitously expressed serine/threonine kinases downstream of the ERK1/2 and p38 (2) pathways. The best characterized function of MNK1/2 is to phosphorylate eukaryotic initiation factor 4E (eIF4E) at Ser209 (3). For a variety of reasons, there is clinical interest in characterizing the role of MNK1/2 signaling in tumor initiation and progression. First, MNKs are often linked to prosurvival signals in response to a wide variety of stimuli (4, 5). Second, loss of MNK function impairs both cell migration and VIMENTIN expression (6). Third, previous work has described the significance of the MNK/eIF4E pathway in regulating oncogenic mRNA translation in various hematologic and solid malignancies (7–10). Finally, lack of MNK1 decreases the oncogenic potential of leukemia (11), gliomas (12), melanoma (13), ovarian cancer (14), and malignant peripheral nerve sheath tumors (15).
In this study, we take advantage of CRISPR-Cas9 technology and a new pharmacologic tool to provide insight into the role of MNK1 signaling as a regulator of the progression of DCIS to invasive disease. Furthermore, we identify NODAL as an important downstream effector of MNK1 signaling. NODAL is a TGFβ family morphogen, and promotes invasiveness during primitive streak formation and mammary gland development (16). Abnormal reexpression of NODAL has been observed in various malignancies (17), and increased NODAL expression has been positively correlated with the transition from local (stage I) to invasive (stage II and above) disease (18). NODAL maintains the self-renewal capacity of cancer stem cells (CSC), and promotes the invasiveness of several solid tumors, including breast cancer (19). Importantly, while MNK1 has been shown to lie downstream of TGFβ1 signaling (9), interactions between MNK1 and NODAL have not been considered.
MNKs are potential therapeutic targets in various malignancies, and there are now efforts to identify selective small-molecule inhibitors that can be used in the clinic (13). We were therefore prompted to interrogate whether MNK1 could serve as a target for therapeutic intervention in preinvasive disease.
Materials and Methods
Cells and reagents
The MCF10DCIS.com (DCIS) cell line was purchased from Wake Forest University (authenticated by short tandem repeat analysis, Genetica DNA Laboratories, 2018). DCIS cells were first tagged with luciferase (DCIS-Luc). DCIS-Luc CTL and MNK1 knock-out (KO) cell lines were generated using the CRISPR/Cas9 system (20). DCIS-Luc cells were transfected with Cas9/sgRNA- GFP plasmids. GFP+ single cells were sorted 48 hours after transfection. Single-cell clones were expanded and Western blotting was performed to verify whether MNK1 expression was successfully ablated in each clone. DCIS-Luc cells were also genetically modified to express either MNK1T332D (constitutively active MNK1, caMNK1) or the corresponding empty vector, pBABE (21). The pBABE and caMNK1 plasmids were kind gifts from Dr. J.A. Cooper (Fred Hutchinson Cancer Research Center, Seattle, WA). All modified DCIS cell lines were cultured in DMEM/ F12 supplemented with 5% horse serum, 10 mmol/L HEPES, 1.05 mmol/L CaCl2, and antibiotics. All experiments with DCIS- Luc CTL/MNK1-KO and DCIS-Luc pBABE/caMNK1 cells were done with passages 3–6. SUM225 cells (provided by Dr. Fariba Behbod, University of Kansas Medical Center, Kansas City, KS), were cultured in DMEM/F12 supplemented with 5% FBS, 10 mmol/L HEPES, 5 mg/mL insulin, and 1 mg/mL hydrocortisone and antibiotics. MDA-MB-468 and 66cl4 cells were kindly provided by Dr. Josie Ursini-Siegel (McGill University, Montreal, Quebec, Canada). MDA-MB-468 cells were cultured in DMEM supplemented with 10% FBS and antibiotics. 66cl4 cells were cultured in RPMI supplemented with 10% FBS and antibiotics. Cell lines were generally maintained at low passages before use. The DCIS and 66cl4 cell lines were tested for Mycoplasma (Venor GeM Mycoplasma Detection Kite, Sigma-Aldrich) and were negative. No other cell lines were tested.
Growth curves
Growth and viability of cells was determined via Trypan blue exclusion using a hemocytometer.
Mammosphere formation assay
Mammosphere formation assay was performed as described previously (22). Briefly, 60,000 cells were seeded per well in 6-well low-adherent plates. Bright-field images were collected at day 7 and colonies with a diameter greater than 50 μm were recorded. Colonies were then sorted based on size as described previously (23).
Western blotting
Cells were lysed with RIPAbuffer (150 mmol/LTris-HCl, pH 7, 150 mmol/L NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS) supplemented with protease and phosphatase inhibitors (Roche) as described previously (24). Equal amount of protein samples were loaded and separated on 10% SDS-PAGEs. p-eIF4E, p-MNK1, eIF4E, MNK1, NODAL, and VIMENTIN were probed with corresponding antibodies. GAPDH was probed to confirm equal protein loading. Antibody information is listed in Supplementary Table S1.
Quantitative PCR
RNA was prepared using TRIzol (Invitrogen). cDNA was prepared from 1 mg of total RNA, using iScript cDNA Synthesis Kit (Bio-Rad). NODAL expression (forward primer: 5’-AGG-GCGAGTGTCCTAATCCT-3’; reverse primer: 5’-CAAAGCTA-GAGCCCTGTCCC-3’) was quantified using the Applied Biosystems 7500 Fast Real-Time PCR System with SYBR Green. Ribosomal protein lateral stalk subunit P0 [RPLP0 (gene encoding 36B4); forward primer: 5’-GGCACCGAGGCAACAGTT-3’; reverse primer: 5’-TCATCCAGCAGGTGTTTGACA-3’] was used as the internal control.
Clonogenic assay
All DCIS cells were seeded at 300 cells per well in 6-well plates and treated as indicated. After 14 days, cells were fixed and stained with 0.5% crystal violet in 70% ethanol. SUM225 cells were seeded at 2,000 cells per well in 6-well plates and treated as indicated. After 21 days, cells were fixed and stained with 0.5% crystal violet in 70% ethanol. Visible colonies were counted using a Gel Count colony counter (Oxford Optronix).
Aldefluor assay
The enzymatic activity of ALDH was detected using the ALDEFLUOR Staining Kit (StemCell Technologies). A total of 1 × 106 trypsinized single cells were suspended in the ALDEFLUOR assay buffer and incubated with 1.5 μmol/L BODIPY-aminoacetaldehyde (BAAA) for 30 minutes at 37°C. As a negative control to establish background fluorescence level, a separate sample was treated with 15 μmol/L diethyla-minobenzaldehyde, a selective ALDH inhibitor. Necrotic cells were excluded by incubating each sample with 2.5 μL of 7-aminoactinomycin D for 15 minutes prior to FACS analysis. Cell fluorescence was measured using BD Biosciences FACScalibur flow cytometer, and data were analyzed using Flowjo Software Version.10 from LCC.
Migration and invasion assay
Cells were seeded at 1 × 106 cells per 10-cm dish on day 1 in full media, then switched to serum-free media on day 2 and starved overnight. On day 3, the transwells were coated with Collagen I (20 μg/mL) as reported previously (24). A total of 2 × 105 cells were seeded into the transwells (Corning) and were allowed to migrate and invade for 16 hours. Migrated cells were fixed with 5% glutaradehyde (Sigma) and stained with 0.5% crystal violet (Sigma) as reported previously (24).
IHC
Human breast cancer tissue was obtained in collaboration with M. Brackstone and the project was approved by the Research Ethics Board at the University of Western Ontario (REB 102254). Written informed consent from all patients was obtained in accordance with the Declaration of Helsinki.
IHC and hematoxylin and eosin staining was performed as described previously (25). Briefly, human and mouse tumor sections were stained for p63, MNK1, p-MNK1, NODAL, VIMEN-TIN, p-histone H3, Ki67, and cleaved caspase-3 and counter-stained with 20% Harris-modified hematoxylin (Thermo Fisher Scientific). Antibody information is listed in SupplementaryTable S1. Slides were scanned and assessed using Spectrum (Aperio Technologies). All the animal and patient IHC samples were scored for intensity by two pathologists (J. Torres and N.V. Giannakopoulos), as described previously (26). Three sections were analyzed for each case and the average score was taken for both invasive and DCIS lesions. Staining intensity was scored as 0, 1, 2, 3, 4, and 5 (0 as negative staining, 5 as the strongest staining intensity). All pathologists were blinded to all clinical data and antibodies used for IHC. The specificity of phospho-MNK1 and MNK1 antibody staining was validated on samples from DCIS xenografts and breast cancer tissue microarrays with adjacent breast tissue.
Orthotopic mouse model
Athymic nude mice, severe combined immunodeficiency (SCID) mice and NOD/SCID mice were purchased from Charles River Laboratory. A total of 1 × 105 modified DCIS cells were resuspended in 50% Matrigel and were injected into the mammary fat pad of athymic nude mice. DCIS-Luc CTL and MNK1-KO xenografts were allowed to grow for 5 weeks. DCIS-Luc pBABE and caMNK1 tumors were allowed to grow for 1.5 weeks. The xenograft, mammary fat pad, and adjacent lymph nodes were then removed surgically. The mice were monitored for potential tumor relapse and metastasis. In parallel, 1 × 105 modified DCIS-Luc pBABE and caMNK1 cells were resuspended in 1× PBS and were injected into the mammary glands of SCID mice. Tumors were allowed to grow for 8 weeks.
For the limiting dilution experiment, 10,000, 1,000, or 100 DCIS-Luc pBABE and caMNK1 MCFDCIS.com cells were resuspended in 1× PBS, and injected into the mammary glands of NOD/SCID mice. Tumors were monitored using IVIS imaging and allowed to grow for 60 days.
A total of 50,000 SUM225 cells were resuspended in PBS and injected through the nipples into the mammary ducts of NOD/SCID mice (kindly provided by M. Alaoui-Jamali) with a 33-gauge Hamilton syringe. Tumors were allowed to grow for 9 weeks. Animal experiments were conducted following protocols approved by the McGill University Animal Care and Use Committee.
Statistical analysis
Prism software (GraphPad) was used to perform statistical analysis. Three independent experiments were performed for all in vitro work. The numbers of samples for all in vitro and in vivo work are listed in Supplementary Table S2. The significance of differences between groups by applying either unpaired Student t test, Wilcoxon-Mann-Whitney, one-way or two-way ANOVA, Kruskal-Wallis test, or χ2test of independence as appropriate. The specific statistical analysis for each figure is listed in Supplementary Table S2. P values < 0.05 were considered significant.
Results
MNK1 activity is elevated in high grade and IDC compared with low-grade DCIS clinical samples
To determine whether the activity of MNK1 is tumor grade-dependent in patients with breast cancer, we assessed the expression of phospho-MNK1 by IHC in a series of human breast samples, comprising low-grade DCIS (13 cases), high-grade DCIS (12 cases), and IDC (15 cases). High-grade DCIS usually presents with significant variation in the size and shape of nuclei, as well as comedo necrosis. Phospho-MNK1 levels were significantly elevated in high-grade DCIS/IDC lesions, compared with low-grade DCIS (Fig. 1A; Supplementary Fig. S1A). Phospho-MNK1 levels were scored from 0 to 5, with 0–2 corresponding to low levels, and 3–5 corresponding to high levels (Fig. 1B). The percentage of samples with high phospho-MNK1 levels increases from low- grade DCIS to high-grade DCIS/IDC. Specifically, there are only 8.3% samples with high phospho-MNK1 levels in low-grade DCIS, but it is elevated to 55% in high-grade DCIS/IDC samples (Fig. 1B). Moreover, our analysis of high-grade DCIS samples revealed a higher expression of MNK1 in 30.4% of analyzed samples, compared with 0% of low-grade DCIS samples analyzed (Supplementary Fig. S1B and S1C). Thus, our results show that increased expression of phospho-MNK1 and MNK1 occurs in a larger percentage of high-grade DCIS/IDC samples, compared with low-grade DCIS specimens.
Figure 1.
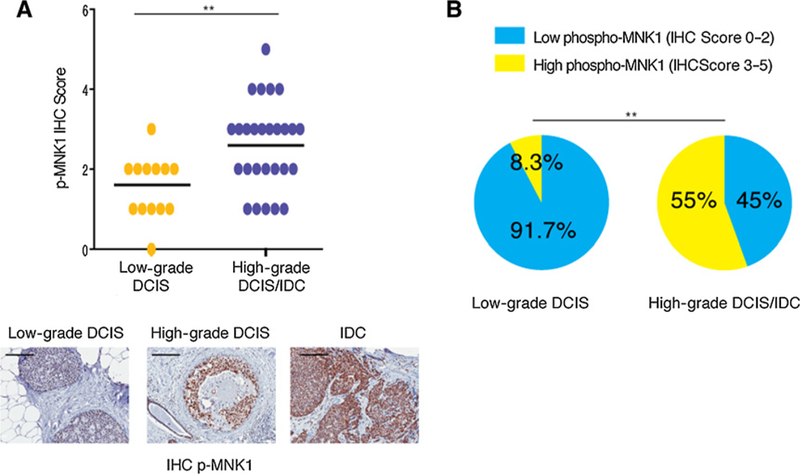
Increased MNK1 activity in IDC and high-grade DCIS. A, Phospho-MNK1 levels are higher in high-grade DCIS/IDC samples than low-grade DCIS samples. Scale bar,200 μm. B, Increased percentage of samples with high phospho-MNK1 staining in high-grade DCIS/IDC compared with low-grade DCIS. _**, P < 0.01.
MNK1 knockout impairs DCIS proliferation and DCIS to IDC conversion
MCF10DCIS.com (DCIS) cells are a basal-like breast cancer cell line, commonly employed to model human DCIS (27–29). We used this model to examine whether MNK1 deficiency would impair DCIS tumorigenesis, invasion, and metastasis. We therefore generated luciferase-tagged DCIS-Luc Cas9 control (CTL) and MNK1 knockout (MNK1-KO) cells, using CRISPR-Cas9 technology. MNK1-KO in two independent clones was confirmed by Western blotting (Supplementary Fig. S2A). The proliferation rate and colony formation capacities were measured in the MNK1-KO clones compared with CTL cells. Both MNK1-KO clones have decreased proliferation, and an impaired ability to form colonies compared with CTL cells (Supplementary Fig. S2B and S2C).
When orthotopically injected into the mammary glands of mice, DCIS cells form lesions that resemble human DCIS and that can progress to IDC (27, 30). To assess the impact of loss of MNK1 activity on the progression from DCIS to IDC in vivo, CTLs or MNK1-KO cells were mixed with 50% Matrigel and injected into the mammary fat pad of athymic nude mice. A previous study using this methodology reported that DCIS xenografts should remain DCIS after 4 weeks of injection, but progress to IDC after 5 weeks (27). Hypothesizing that MNK1 activity would accelerate the conversion of DCIS to IDC, we resected the xenografts, including surrounding mammary gland tissue, 5 weeks after DCIS tumor cell injection (Supplementary Fig. S2D). Tumor outgrowth was monitored by luciferin injection and IVIS imaging. MNK1-KO tumor initiation and proliferation was significantly slower than observed with CTL-derived tumors (Fig. 2A). MNK1 deficiency in tumor xenografts was verified by IHC (Fig. 2B). DCIS is characterized by tubular-like structures with an intact myoepithelial layer of cells that stain positive for p63, while IDC is characterized by structures that lack intact myoepithelial layers (27). A histologic analysis demonstrated that 80% of MNK1-KO xenograftsretained a DCIS morphology, while all CTL tumors progressed into IDC (Fig. 2C). In addition, we observed tissue necrosis in 25% of the CTL tumors, while none of the MNK1-KO xenografts presented with necrosis (Fig. 2D). This could be important clinically, as the presence of necrosis in breast cancer is related to increased invasiveness and poor prognosis (31). Animals were kept alive after primary tumor resection, to monitor for potential tumor recurrence, metastasis, and survival rates. Although not statistically significant, mice that had received MNK1-KO cells showed a trend toward better overall survival, than those animals that were implanted with CTL cells (Fig. 2E). Furthermore, 80% of mice that received CTL cells had relapsed metastatic disease, while no mice that received MNK1- KO cells had metastasis (Fig. 2F). The metastatic CTL tumors were found in the chest area and abdominal cavity, next to the pancreas, small bowel, kidneys, and rib bones (Fig. 2F and G; Supplementary Table S3). Full necropsy and histologic analysis also revealed metastatic cancer in the lungs (Fig. 2G). The percentage of mice with metastasis to the lungs, chest cavity, or abdomen is graphed in Fig. 2G.
Figure 2.
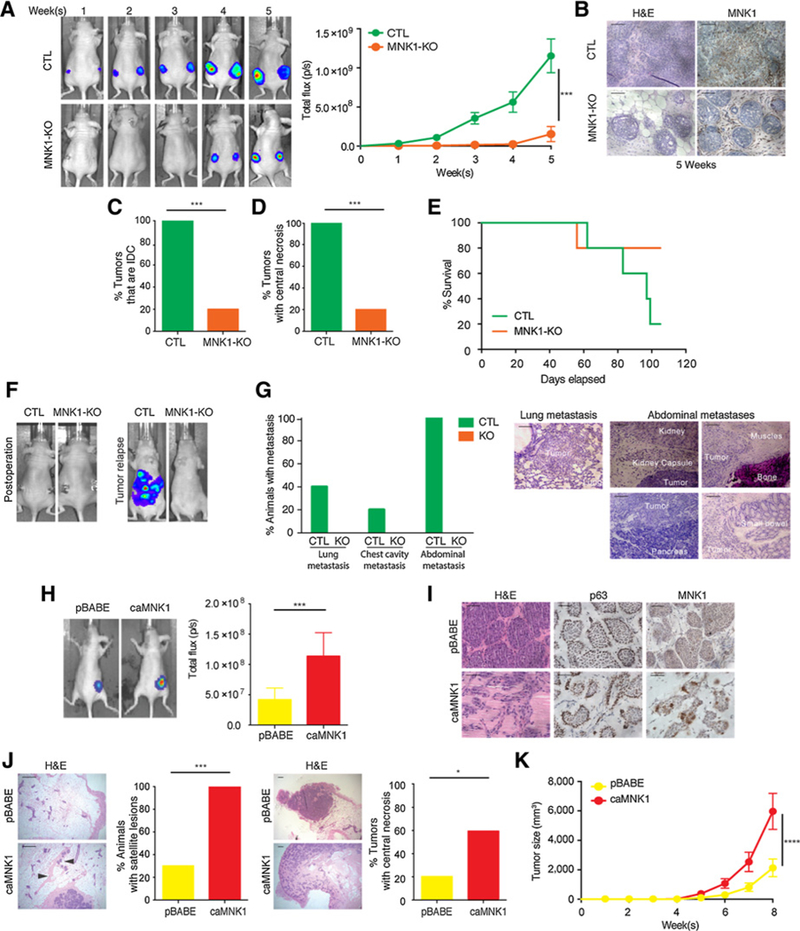
MNK1 regulates the DCIS-IDC transition in vivo. A, DCIS-Luc CTL and MNK1-KO tumor outgrowth is measured by IVIS imaging. B, MNK1 knockout is retained in the DCIS-Luc MNK1-KO xenografts as confirmed by IHC. Scale bar, 50 μm. C, All DCIS-Luc CTL xenografts have progressed into IDC, while only 20% DCIS-Luc MNK1-KO tumors have progressed to an IDC-like morphology. D, All DCIS-Luc CTL xenografts have central necrosis, while only 20% DCIS-Luc MNK1-KO tumors have central necrosis. E, Survival curve of mice receiving DCIS-Luc CTL/MNK1-KO cells. F, Representative IVIS imaging showing complete tumor removal postoperation and tumor recurrence in animals receiving DCIS-Luc CTL cells. G, Percentage of animals presented with metastasis at different sites and representative images of metastasis in various tissues of mice receiving DCIS-Luc CTL/MNK1-KO cells. Scale bar, 200 μm. H, Tumor outgrowth is measured by IVIS imaging. I, DCIS-Luc pBABE xenografts maintain DCIS morphology, while DCIS-Luc caMNK1 tumors have progressed into a mixed morphology of DCIS/IDC. caMNKI overexpression is maintained in the xenografts as confirmed by IHC. Scale bar, 50 μm. J, 100% of mice with DCIS-Luc caMNK1 tumors have micrometastasis in the mammary gland, while 30% of DCIS-Luc pBABE have micrometastasis. Arrows, micrometastases. Sixty percent DCIS-Luc caMNK1 and 20% DCIS-Luc pBABE tumors have central necrosis. Scale bar, 200 μm. K, DCIS-Luc caMNK1 xenografts present with growth advantage over DCIS-Luc pBABE controls. H&E, hematoxylin and eosin. ***, P < 0.001; ****, P< 0.0001.
Next, we hypothesized that constitutive activation of MNK1 would promote the DCIS to IDC transition. Thus, we generated DCIS-luciferase-tagged cells that stably express empty vector pBABE (DCIS-luc pBABE) or MNK1T332D (constitutively active MNK1, DCIS-Luc caMNK1; ref. 21). caMNK1 expression in DCIS was validated to be functional by detecting increased phosphorylation of one of its best studied substrates, eIF4E (Supplementary Fig. S2E). pBABE- and caMNK1-derived cell lines showed similar proliferation rates and clonogenic capacities (Supplementary Fig. S2F and S2G).
We characterized the in vivo tumorigenic effects of increased MNK1 activation in DCIS cells. DCIS-luc pBABE or caMNK1 cells were mixed with 50% Matrigel and injected into the mammary fat pad of athymic nude mice (Supplementary Fig. S2H). We resected the xenografts at 1.5 weeks after DCIS tumor cell injection, expecting that caMNK1 xenografts might transition faster to IDC, compared with vector control-derived tumors. Tumors are not palpable after 1.5 weeks of implantation, thus tumor outgrowth was estimated by IVIS imaging. Unlike the lack of proliferative advantage observed in 2D culture, caMNK1-expressing tumors were larger compared with pBABE control expressing tumors (Fig. 2H). caMNK1 expression in the tumor xenografts was confirmed by IHC for MNK1 (Fig. 2I). As expected, DCIS-luc pBABE cells formed lesions that were characteristic ofDCIS, with positive staining for p63 (Fig. 2I). DCIS-luc caMNK1 cells on the other hand, formed lesions that consisted of a mixture of tubular-like and irregular structures, resembling IDC (Fig. 2I). Consistent with the previously reported role of MNK1 in promoting tumor invasion, we also found that all mice that had been injected with caMNK1-expressing DCIS cells had satellite lesions throughout the mammary fat pad, compared with only 3 of 10 mice receiving pBABE cells (Fig. 2J). In addition, 6 of the 10 mice injected with caMNK1 cells have tumors with central necrosis, while only 2 of 10 mice injected with pBABE cells present with tumors with central necrosis (Fig. 2J). Finally, we sought to confirm whether the injection of caMNK1-expressing DCIS cells resulted in enhanced tumor proliferation. To this end, we performed a second animal experiment, wherein DCIS-luc pBABE and caMNK1 cells were injected into the mammary fat pads of SCID mice, and tumor outgrowth was monitored, via IVIS imaging, over the course of 8 weeks. Tumor formation in the orthotopic mammary fat pad was significantly increased in mice injected with caMNK1 cells compared with those injected with the control pBABE cells (Fig. 2K; Supplementary Fig. S2I). We further interrogated whether the increase in tumor formation associated with caMNK1 expression was due to an increase in proliferation or a decrease in apoptosis. Using IHC, we stained pBABE and caMNK1-expressing tumor xenografts for phospho-histone H3, a marker of proliferation (32), and showed no difference between the pBABE and caMNK1 tumor groups (Supplementary Fig. S2J). We also included in our analysis, IHC staining for the proliferative marker Ki67, and this too showed no difference in pBABE versus caMNK1-expressing tumor xenografts (Supplementary Fig. S2J). However, we did find that our IHC staining for the levels of cleaved caspase-3, a marker of cell death, trended toward decreased expression in caMNK1-derived tumors (Supplementary Fig. S2J). These data suggest that caMNK1-derived tumors are larger than their pBABE counterparts, due to an evasion of apoptotic cell death. Cumulatively, the data presented here demonstrate that modulation of MNK1 influences the DCIS to IDC transition in vivo.
MNK1 regulates NODAL morphogen to control DCIS progression
We next investigated the molecular mechanism underlying the regulation of the DCIS to IDC transition by activated MNK1. Strizzi and colleagues previously showed that NODAL protein levels are increased in IDC relative to in DCIS (18). As NODAL has been shown to be downstream of p38 (33), a major upstream activator of MNK1, we hypothesized that NODAL may be regulated in our model system. Consistent with the previous report from Strizzi and colleagues (18), we observed that NODAL levels are higher in the DCIS patient samples with microinvasion, as compared with pure DCIS lesions (Fig. 3A). MNK1-KO DCIS xenografts express reduced NODAL, as detected by IHC staining (Fig. 3B; Supplementary Fig. S3A). Conversely, the expression of constitutively activated MNK1 in DCIS cells results in an increased expression in NODAL, as determined by IHC for NODAL in pBABE- and caMNK1-derived xenografts (Fig. 3C; Supplementary Fig. S3B).
Figure 3.
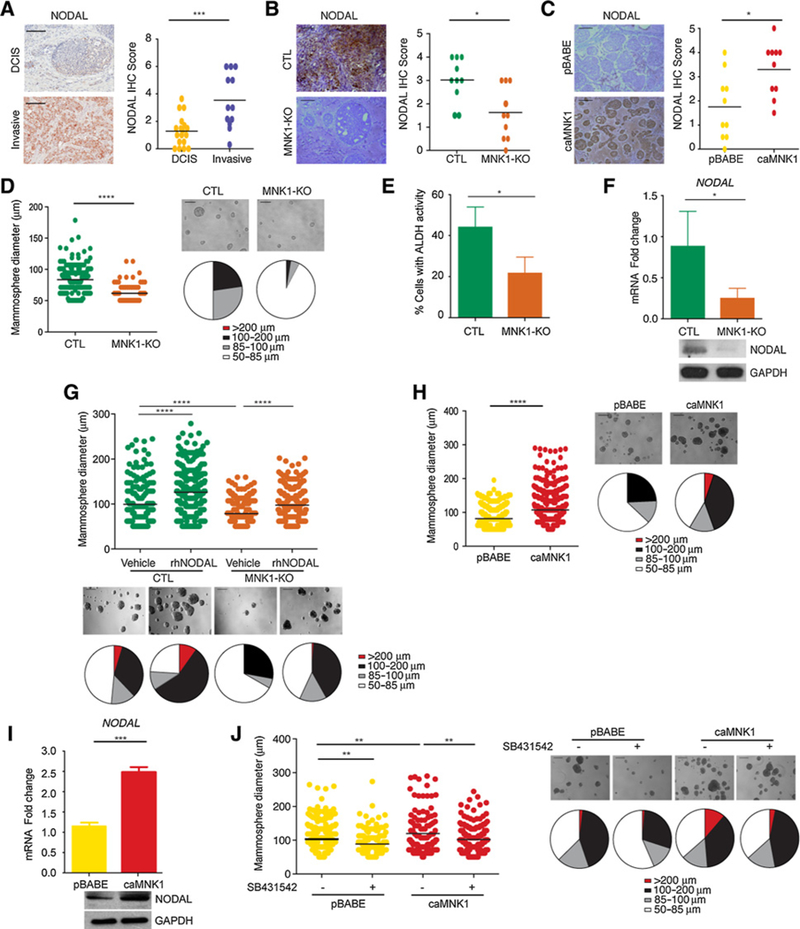
MNK1 expression regulates NODAL morphogen expression. A, NODAL expression in DCIS versus invasive human breast samples. Scale bar, 200 μm. B, DCIS-Luc MNK1-KO xenografts have decreased NODAL levels compared with DCIS-Luc CTL tumors. Representative images are shown. Scale bar, 200 μm. C, DCIS-Luc caMNK1 xenografts have increased NODAL levels compared with DCIS-Luc pBABE tumors. Representative images are shown. Scale bar, 200 μm. D, DCIS-Luc MNK1-KO cells have mammosphere size in low adherent culture. Scale bar, 200 μm E, DCIS-Luc MNK1-KO cells have reduced ALDH+ populations. F, DCIS-Luc MNK1-KO mammospheres express lower NODAL mRNA levels. G, rhNODAL treatment increases mammosphere size in DCIS-Luc CTL/MNK1-KO cells. Scale bar, 200 μm. H, DCIS-Luc caMNK1 overexpression increases mammosphere size in low adherent culture. Scale bar, 200 μm. I, DCIS-Luc caMNK1 mammospheres express higher NODAL mRNA levels. J, DCIS-Luc pBABE/caMNK1 mammosphere sizes can both be reduced by 10 μmol/LSB431542, a NODAL pathway inhibitor. Scale bar, 200 μm. *, P < 0.05;**, P< 0.01; ***, P< 0.001; ****, P < 0.0001.
Examination of DCIS clinical specimens and cell lines has revealed the existence of stem cell-like subpopulations (22, 34). When grown on low adherent plates, DCIS cells can form mammospheres, which are formed by cells with the ability to survive anoikis and to self-renew (35); two functional properties of cancer stem cells (CSC; ref. 22). As NODAL signaling can promote self-renewal and tumorigenicity of cancer stem cells (36), we next examined whether MNK1 regulates DCIS mammosphere formation. MNK1-KO-derived mammospheres were smaller than those formed by their CTL counterparts (Fig. 3D). Another hallmark of CSCs is enhanced activity of aldehyde dehydrogenase (ALDH; ref. 22). MNK1-KO mammospheres showed decreased ALDH activity (Fig. 3E), as assessed by flow cytometry, compared with their Cas9 CTL counterparts. Moreover, we determined whether depletion of MNK1 could reduce the tumor-initiating cell subpopulation by performing FACS analysis for CD44hi/ CD24lo populations in the MCFDCIS.com cells knocked out for MNK1. Our data showed that loss of MNK1 levels causes the percentage of CD44hi/CD24lo cells in the DCIS population to decrease (Supplementary Fig. S3C).
In agreement with our in vivo data, the MNK1-KO-derived mammospheres also express less NODAL protein and mRNA (Fig. 3F). We next determined the interplay between MNK1 and NODAL in the formation of mammospheres. When we added recombinant human NODAL (rhNODAL) to CTL DCIS cells, the mammosphere size increased (Fig. 3G). Moreover, rhNODAL treatment of MNK1-KO cells partially rescued the decrease in mammosphere size observed when MNK1 expression is ablated (Fig. 3G). Together, these results suggest that MNK1-dependent regulation of NODAL is needed to regulate mammosphere size. To confirm and extend our results of MNK1-dependent regulation of NODAL expression, we also determined the impact of MNK1 deficiency on NODAL expression in two breast cancer cell lines, 66cl4 and MB-MDA-468. Similar to our results in DCIS cells, depleting MNK1 in 66cl4 cells using CRISPR-Cas9 technology, and by siRNA in MB-MDA-468 cells, caused these cells to express less NODAL (Supplementary Fig. S3D and S3E). Moreover, biological data in MNK1-null 66cl4 cells shows a decrease in mammosphere growth, colony formation, and invasion (Supplementary Fig. S3F). Similarly, concomitant with the reduced NODAL levels observed when we silence MNK1 in MDA-MB-468 cells, their invasion is also impaired (Supplementary Fig. S3G).
caMNK1 expression increased both the size of DCIS mammospheres (Fig. 3H), and the expression of NODAL mRNA and protein in these spheres (Fig. 3I). Consistent with our observations in the caMNK1 tumor xenografts (Supplementary Fig. S2J), we see no change in Ki67 levels, but a decrease in the expression of cleaved caspase-3 in the caMNK1-derived mammospheres (Supplementary Fig. S3H). We also assessed tumor formation from limiting dilutions of inoculated pBABE orcaMNK1 cells, wherein we injected 10,000, 1,000, or 100 cells. As shown on the graph in Supplementary Fig. S3I, the caMNK1-expressing cells have an increased tumor-initiating capability, compared with their pBABE control counterparts. Sixty days postinjection, the 100 cell-injected cohort of mice, did not show any signal by IVIS imaging. Finally, we determined whether the induction of NODAL expression by activated MNK1 was responsible for increased DCIS mammosphere formation. We utilized SB431542, an inhibitor routinely used to block NODAL signaling (37). Mammosphere size was significantly reduced in pBABE and caMNK1 cells, indicating again the importance of NODAL signaling to the increased and mammosphere size when MNK1 is constitutively activated (Fig. 3J).
MNK1 and NODAL regulate DCIS tumor invasion
MNK1 and NODAL can facilitate tumor invasion and metastasis, with the regulation of known mediators of invasion such as VIMENTIN (3, 8, 9). Here we observed that MNK1-KO tumor xenografts express less VIMENTIN, compared with the CTL- derived tumors (Fig. 4A; Supplementary Fig. S4A). In contrast, caMNK1 tumors show stronger staining for VIMENTIN, compared with the pBABE-derived tumors (Fig. 4B; Supplementary Fig. S4B). The changes in VIMENTIN expression that we observed by modulating MNK1 levels, led us to investigate the invasive characteristics of our cell models. MNK1-KO cells express less VIMENTIN (Fig. 4C), and MNK1 deficiency restrained the invasion of DCIS cells (Fig. 4C). In comparison, caMNK1 overexpression promoted the invasion of DCIS cells, and increased VIMEN-TIN expression (Fig. 4D). Next, we tested the role that VIMENTIN might play in the increased invasion seen in cells that express a constitutively activated MNK1 (caMNK1). When VIMENTIN expression is repressed using siRNA, the caMNK1 cells lose their ability to invade (Fig. 4E), suggesting a role for VIMENTIN in the enhanced invasion phenotype related to constitutive MNK1 activity.
Figure 4.
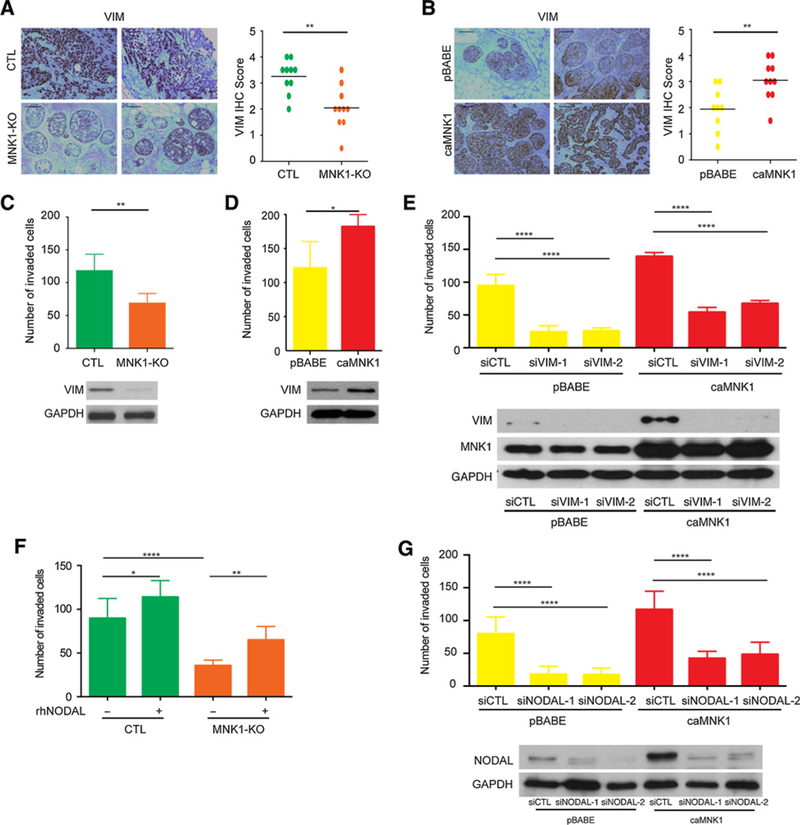
The MNK1/NODAL axis regulates migration and invasion. A, DCIS-Luc MNK-KO xenografts have decreased VIMENTIN (VIM) levels compared with DCIS-Luc CTL tumors. Representative images from two tumors are shown. Scale bar, 200 μm. B, DCIS-Luc caMNK1 xenografts have increased VIMENTIN (VIM) levels compared with DCIS-Luc pBABE control tumors. Representative images from two tumors are shown. Scale bar, 200 μm. C, DCIS-Luc MNK1-KO cells have impaired capacity to migrate and to invade through Collagen I in transwells. D, DCIS-LuccaMNK1 cells showed increased capacity to migrate and invade through Collagen I in transwells. E, VIMENTIN knockdown by siRNA decreases the invasive capacity of DCIS-Luc pBABE/caMNK1 cells. F, rhNODAL treatment increases the migration and invasion of DCIS-Luc MNK1-KO cells. G, Transient knockdown of NODAL decreases the invasive capacity of DCIS-Luc pBABE/caMNK1 cells. *, P< 0.05;**, P < 0.01; ****, P < 0.0001.
Finally, to assess whether MNK1-dependent regulation of NODAL contributes to cell invasion, CTL and MNK1-KO cells were treated with rhNODAL. We observed that rhNODAL treatment can enhance the invasion of DCIS CTL cells, and partially overcome the decrease in cell invasion observed in MNK1-depleted cells (Fig. 4F). In addition, we next looked at cell invasion using pBABE and caMNK1 cells that had been transiently transfected with NODAL siRNA. As expected, caMNK1 cells transfected with control siRNA (siCTL) are more invasive than pBABE siCTL cells. NODAL knockdown reduced the invasion of both pBABE and caMNK1 cells, but caMNK1 siNODAL cells are still more invasive than their pBABE siNODAL counterparts (Fig. 4G).
MNK1 can be pharmacologically targeted to inhibit the DCIS to IDC transition
We next determined whether the effects we observed with ablation of MNK1 in DCIS cells would be phenocopied using SEL201, an orally bioavailable small-molecule inhibitor of MNK1/2 activity (13). SEL201 induces a G2-M cell cycle arrest and inhibits proliferation of both pBABE and caMNK cells in a dose-dependent manner (Supplementary Fig. S5A and S5B). Consistent with genetic ablation of MNK1 expression, SEL201 treatment efficiently reduced the number of colonies (Fig. 5A), ALDH activity (Fig. 5B), and the size of mammospheres (Fig. 5C), derived from pBABE and caMNK1 cells. SEL201 treatment also suppressed invasion of both pBABE and caMNK1-expressing DCIS cells, concomitant with reduced levels of NODAL (Fig. 5D).
Figure 5.
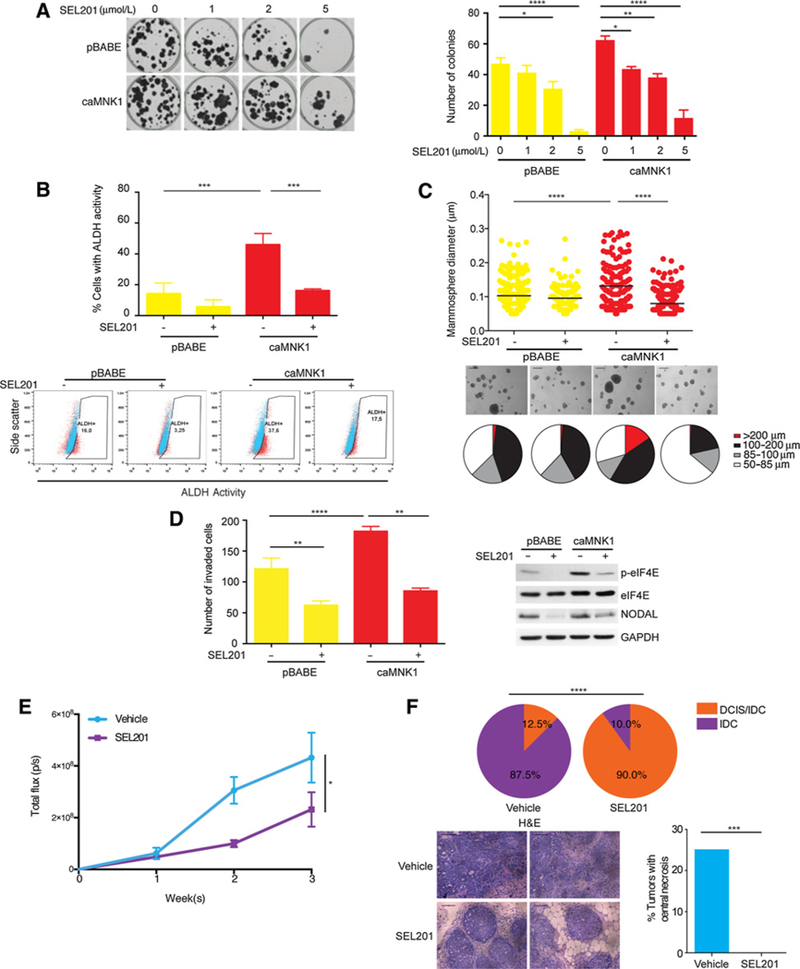
Pharmacologically targeting MNK1 inhibits the DCIS/IDC transition A. SEL201 inhibits DCIS-Luc pBABE/caMNK1 colony formation in a dose-dependent manner. B, Two μmol/L SEL201 decreases the ALDH+ population in DCIS-Luc caMNK1 cells. C, Five μmol/L SEL201 decreases mammosphere size formed by DCIS-Luc caMNK1 cells. Scale bar, 200 μm D, Two μmol/L SEL201 decreases the invasive capacity of DCIS-Luc caMNK1 cells. E, Primary tumor outgrowth over 3 weeks in vehicle versus SEL201-treated animals injected with DCIS-Luc CTL cells. F, MNK inhibitor SEL201 treatment slows down DCIS to IDC progression in nude mice injected with DCIS-Luc CTL cells and decreases the percentage of tumors with detectable central necrosis. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Having shown that MNK1 regulates the DCIS to IDC conversion in animal models, we next investigated whether pharmacologically blocking MNK1 would inhibit the progression of DCIS to invasive disease. DCIS-Luc CTL cells were orthotopically injected into the mammary glands of nude mice, and then the mice were randomized to either vehicle control or SEL201 treatment groups (Supplementary Fig. S5C). SEL201 was delivered by gavage at a dose of 75 mg/kg per day, which we have previously shown concomitantly suppresses MNK1 activity and lung metastasis (13). SEL201 significantly decreased the primary tumor size after a three-week treatment (Fig. 5E). Furthermore, xenografts from the SEL201 group showed a reduced percentage of IDC (10% of tumors) compared with the vehicle group (87.5% of tumors; Fig. 5F). Consistent with our previous work (13), SEL201 showed no overt systemic toxicity, as evidenced by body weight (Supplementary Fig. S5D) and tests for liver function (ALT and AST; Supplementary Fig. S5D). SUM225 cells are also commonly used to model DCIS, and consistent with the data shown thus far, reducing MNK1 levels using siRNA in SUM225 induces a loss of NODAL expression, while SUM225-expressing caMNK1 express increased levels ofNODAL (Supplementary Fig. S5E). Compared with MCFDCIS.com cells, SUM225 cells slowly transition from DCIS to IDC following intraductal injections, a technique that minimizes disturbance to the mammary gland microenvironment (38). SEL201 also inhibits colony formation of SUM225 cells in vitro, and slows down the transition from DCIS to IDC in the SUM225 intraductal model of DCIS (Supplementary Fig. S5F and S5G). In summary, our results provide evidence to show the feasibility of inhibiting MNK1 to slow the conversion of DCIS to IDC.
Discussion
The mechanism underlying the transition of DCIS to IDC remains poorly understood. Previous studies have identified several potential factors that facilitate the conversion to inva- sivebreast cancer, such as p63/MT1-MMP (39), SMARCE1 (40), and Singleminded-2s (SIM2s; ref. 29). Unfortunately, many of these proteins have proven difficult to therapeutically target as a means to block disease progression in pre-clinical models. Herein, we identify the MNK1/NODAL signaling axis as a key molecular pathway regulating the progression of DCIS to IDC and breast cancer recurrence as metastatic disease (Fig. 6). Moreover, MNK1 inhibitors such as SEL201 could therapeutically block NODAL signaling in patients diagnosed with NODAL-positive DCIS as a viable means to suppress invasive disease.
Figure 6.
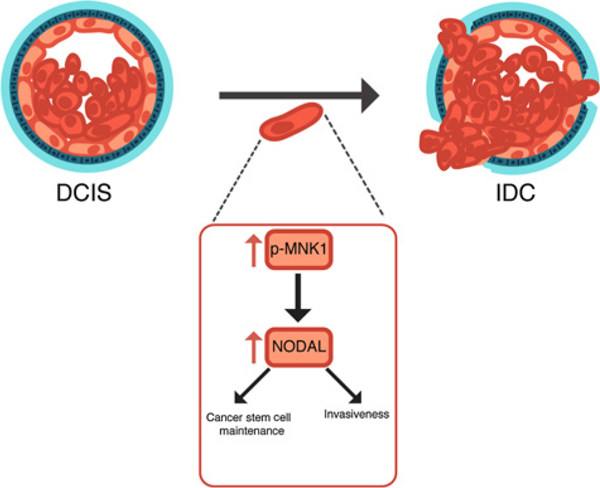
A model depicting the MNK1/NODAL axis during the DCIS to IDC transition. Our data suggest that high MNK1 activity promotes the expression of NODAL. NODAL, in turn, promotes cell invasion and cancer stem cell maintenance.
Our data show a positive correlation between the activity of MNK1 and the expression of NODAL and VIMENTIN, two critical regulators of invasion and metastasis. The results presented here suggest that the activation of the MNK/NODAL axis leads to a partial, but not full, induction of an EMT. Similar results were previously shown in response to Twist1 expression, which induces a partial EMT and dedifferentiation toward “sternness” (41). Thus, while we cannot conclude that a full EMT is driving the invasive phenotype seen in our caMNK1-expressing cells, it is likely that a partial EMT is sufficient. This would be consistent with the hypothesis that tumor cells exhibiting a partial EMT pose a higher metastatic risk (42).
Although the significance of MNK/eIF4E in breast cancer tumorigenesis, metastasis, and therapeutic resistance is recognized (43), MNK1 has other downstream substrates whose role in breast cancer is not well characterized. MNK1 also regulates the expression of proinflammatory and protumorigenic cytokines, including TNFα, IL6, TGFB (44). Our results have not only added NODAL as a novel downstream cytokine controlled by MNK1, but also defines a role for MNK1/NODAL signaling in controlling CSC-like phenotypes. Although previous research has reported on the potential role of MNK1 in maintaining CSC properties in acute myeloid leukemia (AML), chronic myeloid leukemia (11), and glioblastoma (45), very little is known about the function of MNK1 in breast CSCs. Using CRISPR/Cas9 technology to selectively knockout MNK1 expression in human DCIS cells, we show that MNK1 deficiency is sufficient to impair high ALDH enzymatic activity, and to reduce the percentage of CD44hi/CD24lo cells, both known characteristics of cancer stem cells in numerous malignancies (46). This could be clinically important as DCIS CSC subpopulations are thought to be responsible for resistance to radiotherapy and potential disease recurrence (47,48). We thus speculate that pharmacologically inhibiting MNK1 will limit the plasticity of DCIS cells via downregulation of NODAL and forestall resistance to radiotherapy.
Our study has shown that NODAL mRNA levels are controlled by MNK1, and MNK1/2 have a traditional role as regulators of oncogenic mRNA translation. Therefore, it is possible that MNK1 is regulating NODAL on the level of mRNA translation. In fact, our work and that of other laboratories, has shown that NODAL works with a positive feedback loop to activate its own transcription (49, 50). We posit that MNK1 may inhibit NODAL protein synthesis, and this in turn would lead to a corresponding suppression in NODALmRNA. It is also possible that MNK1 regulates NODAL by a yet to be ascribed role in the nucleus. MNK1/2 are also found in the nucleus, suggesting activities beyond their role in protein synthesis (44,51). Interestingly, we have observed nuclear localization of MNK1 in our murine xenografts and clinical breast samples, in this study (Fig. 1A), and in our melanoma work (13). This observation is line with MNK1 containing both nuclear localization and export signals, and prior work showing that MNK1 can shuttle between the nucleus and cytoplasm (52, 53). Access to CRISPR/Cas9 technology for selectively deleting MNK1 and MNK2 will no doubt lead to defining novel biological functions of these kinases.
A question raised by our work is identifying the molecular mechanism by which MNK1 signaling is activated in invasive disease. Our data show that NODAL lies downstream of MNK1; however, it is entirely possible that NODAL can itself feedback to activate MNK1. TGFβ super-family cytokines, such as NODAL and Activin, utilize noncanonical MAPK cascades to regulate breast tumor progression (15, 24, 51); however, it remains unclear whether MNK1 cooperates with NODAL to drive DCIS toward invasive disease. Our results also demonstrated that exogenous NODAL treatment can increase DCIS mammosphere size and it is tempting to speculate that this cytokine can also induce MNK1 phosphorylation. Indeed, NODAL has been shown to activate ERK1/2, immediately upstream of MNK1, in breast cancer cells (37).
We have demonstrated the feasibility of pharmacologically blocking MNK1/2 activity in vivo, resulting in blocking of the progression of DCIS to invasive disease. Together, our data contribute to the increasing discussion about the clinical applications of MNK1/2 inhibitors in cancer.
Supplementary Material
Acknowledgments
This research is funded by grants from the Cancer Research Society (PIN 20239 to W. Miller), the Canadian Institutes for Health Research (MOP-142281 to W. Miller), the Canadian Institutes for Health Research (PJT- 156269 to W. Miller and S.V. del Rincón), the Canadian Cancer Society (703811 to W. Miller and S.V. del Rincón), the Rossy Cancer Network (to W. Miller), and the Israel Cancer Research Fund (to W. Miller and S.V. del Rincón). Q. Guo was supported by a Cole Foundation Ph.D fellowship, McGill Integrated Cancer Research Training Program (MICRTP) graduate studentship, and a McGill Faculty of Medicine graduate studentship. V.Z. Li was supported by a MICRTP undergraduate studentship. J. Nichol was supported by a fellowship from the Rossy Cancer Network. W. Yang and F. Huang were supported by MICRTP graduate studentships. Y. Zhan was supported by a Toronto-Dominion Bank/Lady Davis Institute (LDI) Ph.D fellowship and the Israel Cancer Research Fund. We thank Selvita S.A. for supplying SEL201 and the Flow Cytometry and Animal Core Facilities of the Lady Davis Institute for their support.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
References
- 1.Martinez-Perez C, Turnbull AK, Ekatah GE, Arthur LM, Sims AH, Thomas JS, et al. Current treatment trends and the need for better predictive tools in the management of ductal carcinoma in situ of the breast. Cancer Treat Rev 2017;55:163–72. [DOI] [PubMed] [Google Scholar]
- 2.Wang X, Flynn A, Waskiewicz AJ, Webb BL, Vries RG, Baines IA, et al. The phosphorylation of eukaryotic initiation factor eIF4E in response to phorbol esters, cell stresses, and cytokines is mediated by distinct MAP kinase pathways. J Biol Chem 1998; 273:9373–7. [DOI] [PubMed] [Google Scholar]
- 3.Bhat M, Robichaud N, Hulea L, Sonenberg N, Pelletier J, Topisirovic I. Targeting the translation machinery in cancer. Nat Rev Drug Discov 2015; 14:261–78. [DOI] [PubMed] [Google Scholar]
- 4.Adesso L, Calabretta S, Barbagallo F, Capurso G, Pilozzi E, Geremia R, et al. Gemcitabine triggers a pro-survival response in pancreatic cancer cells through activation of the MNK2/eIF4E pathway. Oncogene 2013;32:2848–57. [DOI] [PubMed] [Google Scholar]
- 5.Chrestensen CA, Shuman JK, Eschenroeder A, Worthington M, Gram H, Sturgill TW. MNK1 and MNK2 regulation in HER2-overexpressing breast cancer lines. J Biol Chem 2007;282:4243–52. [DOI] [PubMed] [Google Scholar]
- 6.Beggs JE, Tian S, Jones GG, Xie J, Iadevaia V, Jenei V, et al. The MAP kinase- interacting kinases regulate cell migration, vimentin expression and eIF4E/ CYFIP1 binding. Biochem J 2015;467:63–76. [DOI] [PubMed] [Google Scholar]
- 7.Furic L, Rong L, Larsson O, Koumakpayi IH, Yoshida K, Brueschke A, et al. eIF4E phosphorylation promotes tumorigenesis and is associated with prostate cancer progression. Proc Natl Acad Sci U S A 2010;107:14134–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Konicek BW, Stephens JR, McNulty AM, Robichaud N, Peery RB, Dumstorf CA, et al. Therapeutic inhibition of MAP kinase interacting kinase blocks eukaryotic initiation factor 4E phosphorylation and suppresses outgrowth of experimental lung metastases. Cancer Res 2011;71:1849–57. [DOI] [PubMed] [Google Scholar]
- 9.Robichaud N, Del Rincon SV, Huor B, Alain T, Petruccelli LA, Hearnden J, et al. Phosphorylation of eIF4E promotes EMT and metastasis via translational control of SNAIL and MMP-3. Oncogene 2015;34:2032–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wendel HG, Silva RL, Malina A, Mills JR, Zhu H, Ueda T, et al. Dissecting eIF4E action in tumorigenesis. Genes Dev 2007;21:3232–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lim S, Saw TY, Zhang M, Janes MR, Nacro K, Hill J, et al. Targeting of the MNK-eIF4E axis in blast crisis chronic myeloid leukemia inhibits leukemia stem cell function. Proc Natl Acad Sci U S A 2013;110: E2298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ueda T, Sasaki M, Elia AJ, Chio II, Hamada K, Fukunaga R, et al. Combined deficiency for MAP kinase-interacting kinase 1 and 2 (Mnkl and Mnk2) delays tumor development. Proc Natl Acad SciU SA2010;107:13984–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhan Y, Guo J, Yang W, Goncalves C, Rzymski T, Dreas A, et al. MNK1/2 inhibition limits oncogenicity and metastasis of KIT-mutant melanoma. J Clin Invest 2017;127:4179–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu S, Zha J, Lei M. Inhibiting ERK/Mnk/eIF4E broadly sensitizes ovarian cancer response to chemotherapy. Clin Transl Oncol 2017;20:374–81. [DOI] [PubMed] [Google Scholar]
- 15.Lock R, Ingraham R, Maertens O, Miller AL, Weledji N, Legius E, et al. Cotargeting MNK and MEK kinases induces the regression of NF1-mutant cancers. J Clin Invest 2016;126:2181–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hendrix MJ, Seftor EA, Seftor RE, Kasemeier-Kulesa J, Kulesa PM, Postovit LM. Reprogramming metastatic tumour cells with embryonic microenvironments. Nat Rev Cancer 2007;7:246–55. [DOI] [PubMed] [Google Scholar]
- 17.Kirsammer G, Strizzi L, Margaryan NV, Gilgur A, Hyser M, Atkinson J, et al. Nodal signaling promotes a tumorigenic phenotype in human breast cancer. Semin Cancer Biol 2014;29:40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Strizzi L, Hardy KM, Margaryan NV, Hillman DW, Seftor EA, Chen B, et al. Potential for the embryonic morphogen Nodal as a prognostic and predictive biomarker in breast cancer. Breast Cancer Res 2012;14: R75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bodenstine TM, Chandler GS, Seftor RE, Seftor EA, Hendrix MJ. Plasticity underlies tumor progression: role of Nodal signaling. Cancer Metastasis Rev 2016;35:21–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013;339: 819–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Waskiewicz AJ, Johnson JC, Penn B, Mahalingam M, Kimball SR, Cooper JA. Phosphorylation of the cap-binding protein eukaryotic translation initiation factor 4E by protein kinase Mnk1 in vivo. Mol Cell Biol 1999; 19:1871–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Q, Eades G, Yao Y, Zhang Y, Zhou Q. Characterization of a stem-like subpopulation in basal-like ductal carcinoma in situ (DCIS) lesions. J Biol Chem 2014;289:1303–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.West NR, Murray JI, Watson PH. Oncostatin-M promotes phenotypic changes associated with mesenchymal and stem cell-like differentiation in breast cancer. Oncogene 2014;33:1485–94. [DOI] [PubMed] [Google Scholar]
- 24.Pettersson F, Del Rincon SV, Emond A, Huor B, Ngan E, Ng J, et al. Genetic and pharmacologic inhibition of eIF4E reduces breast cancer cell migration, invasion, and metastasis. Cancer Res 2015;75:1102–12. [DOI] [PubMed] [Google Scholar]
- 25.Quail DF, Walsh LA, Zhang G, Findlay SD, Moreno J, Fung L, et al. Embryonic protein nodal promotes breast cancer vascularization. Cancer Res 2012;72:3851–63. [DOI] [PubMed] [Google Scholar]
- 26.Yu L, Harms PW, Pouryazdanparast P, Kim DS, Ma L, Fullen DR. Expression of the embryonic morphogen Nodal in cutaneous melanocytic lesions. Mod Pathol 2010;23:1209–14. [DOI] [PubMed] [Google Scholar]
- 27.Hu M, Yao J, Carroll DK, Weremowicz S, Chen H, Carrasco D, et al. Regulation of in situ to invasive breast carcinoma transition. Cancer Cell 2008;13:394–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rakha EA, Reis-Filho JS, Ellis IO. Basal-like breast cancer: a critical review. J Clin Oncol 2008;26:2568–81. [DOI] [PubMed] [Google Scholar]
- 29.Scribner KC, Behbod F, Porter WW. Regulation of DCIS to invasive breast cancer progression by Singleminded-2s (SIM2s). Oncogene 2013;32: 2631–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miller FR, Santner SJ,Tait L, Dawson PJ. MCF10DCIS.comxenograftmodel of human comedo ductal carcinoma in situ. J Natl Cancer Inst 2000;92: 1185–6. [DOI] [PubMed] [Google Scholar]
- 31.Gilchrist KW, Gray R, Fowble B, Tormey DC, Taylor SGt. Tumor necrosis is a prognostic predictor for early recurrence and death in lymph node-positive breast cancer: a 10-year follow-up study of 728 Eastern Cooperative Oncology Group patients. J Clin Oncol 1993;11:1929–35. [DOI] [PubMed] [Google Scholar]
- 32.Gerring Z, Pearson JF, Morrin HR, Robinson BA, Harris GC, Walker LC. Phosphohistone H3 outperforms Ki67 as a marker of outcome for breast cancer patients. Histopathology 2015;67:538–47. [DOI] [PubMed] [Google Scholar]
- 33.Bradham CA, McClay DR. p38 MAPK is essential for secondary axis specification and patterning in sea urchin embryos. Development 2006; 133:21–32. [DOI] [PubMed] [Google Scholar]
- 34.Currie MJ, Beardsley BE, Harris GC, Gunningham SP, Dachs GU, Dijkstra B, et al. Immunohistochemical analysis of cancer stem cell markers in invasive breast carcinoma and associated ductal carcinoma in situ: relationships with markers of tumor hypoxia and microvascularity. Hum Pathol 2013; 44:402–11. [DOI] [PubMed] [Google Scholar]
- 35.Farnie G, Clarke RB, Spence K, Pinnock N, Brennan K, Anderson NG, et al. Novel cell culture technique for primary ductal carcinoma in situ: role of Notch and epidermal growth factor receptor signaling pathways. J Natl Cancer Inst 2007;99:616–27. [DOI] [PubMed] [Google Scholar]
- 36.Quail DF, Taylor MJ, Postovit LM. Microenvironmental regulation of cancer stem cell phenotypes. Curr Stem Cell Res Ther 2012;7:197–216. [DOI] [PubMed] [Google Scholar]
- 37.Quail DF, Zhang G, Findlay SD, Hess DA, Postovit LM. Nodal promotes invasive phenotypes via a mitogen-activated protein kinase-dependent pathway. Oncogene 2014;33:461–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Behbod F, Kittrell FS, LaMarca H, Edwards D, Kerbawy S, Heestand JC, et al. An intraductal human-in-mouse transplantation model mimics the sub-types of ductal carcinoma in situ. Breast Cancer Res 2009;11:R66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lodillinsky C, Infante E, Guichard A, Chaligne R, Fuhrmann L, Cyrta J, et al. p63/MT1-MMP axis is required for in situ to invasive transition in basal- like breast cancer. Oncogene 2015;35:344–57. [DOI] [PubMed] [Google Scholar]
- 40.Sokol ES, Feng YX, Jin DX, Tizabi MD, Miller DH, Cohen MA, et al. SMARCE1 is required for the invasive progression of in situ cancers. Proc Natl Acad Sci U S A2017;114:4153–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu Y, Lee DK, Feng Z, Xu Y, Bu W, Li Y, et al. Breast tumor cell-specific knockout ofTwist1 inhibits cancer cell plasticity, dissemination, and lung metastasis in mice. Proc Natl Acad Sci U S A 2017;114:11494–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu M, Bardia A, Wittner BS, Stott SL, Smas ME, Ting DT, et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013;339:580–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wheater MJ, Johnson PW, Blaydes JP. The role of MNK proteins and eIF4E phosphorylation in breast cancer cell proliferation and survival. Cancer Biol Ther 2010;10:728–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Joshi S, Platanias LC. Mnk kinases in cytokine signaling and regulation of cytokine responses. Biomol Concepts 2015;6:85. [DOI] [PubMed] [Google Scholar]
- 45.Bell JB, Eckerdt FD, Alley K, Magnusson LP, Hussain H, Bi Y, et al. MNK Inhibition Disrupts Mesenchymal Glioma Stem Cells and Prolongs Survival inaMouse Model of Glioblastoma. Mol CancerRes 2016;14:984–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, et al. ALDH1isa marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007;1: 555–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Williams KE, Bundred NJ, Landberg G, Clarke RB, Farnie G. Focal adhesion kinase andWnt signaling regulate human ductal carcinoma in situ stem cell activity and response to radiotherapy. Stem Cells 2015;33:327–41. [DOI] [PubMed] [Google Scholar]
- 48.Farnie G, Johnson RL, Williams KE, Clarke RB, Bundred NJ. Lapatinib inhibits stem/progenitor proliferation in preclinical in vitro models of ductal carcinoma in situ (DCIS). Cell Cycle 2014;13:418–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saijoh Y, Oki S, Tanaka C, Nakamura T, Adachi H, Yan YT, et al. Two nodal- responsive enhancers control left-right asymmetric expression of Nodal. Dev Dyn 2005;232:1031–6. [DOI] [PubMed] [Google Scholar]
- 50.Topczewska JM, Postovit LM, Margaryan NV, Sam A, Hess AR, Wheaton WW, et al. Embryonic and tumorigenic pathways converge via Nodal signaling: role in melanoma aggressiveness. Nat Med 2006;12:925–32. [DOI] [PubMed] [Google Scholar]
- 51.Rannou Y, Salaun P, Benaud C, Khan J, Dutertre S, Giet R, et al. MNK1 kinase activity is required for abscission. J Cell Sci 2012;125:2844–52. [DOI] [PubMed] [Google Scholar]
- 52.Parra-Palau JL, Scheper GC, Wilson ML, Proud CG. Features in the N and C termini ofthe MAPK-interacting kinase Mnk1 mediate its nucleocytoplas- mic shuttling. J Biol Chem 2003;278:44197–204. [DOI] [PubMed] [Google Scholar]
- 53.Scheper GC, Parra JL, Wilson M, Van Kollenburg B, Vertegaal AC, Han ZG, et al. The N and C termini of the splice variants of the human mitogen- activated protein kinase-interacting kinase Mnk2 determine activity and localization. Mol Cell Biol 2003;23:5692–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.