

Abstract
Liver cirrhosis is associated with increased morbidity and mortality with important health and social consequences; however, an effective treatment has not been found yet. Previous reports have shown some beneficial effects of stevioside (SVT) in different diseases, but the ability of SVT to inhibit liver cirrhosis has not been reported. Therefore, we studied the potential of this diterpenoid to inhibit liver cirrhosis induced by thioacetamide, a model that shares many similarities with the human disease, and investigated the possible underlying molecular mechanism using in vivo and in vitro approaches. Cirrhosis was induced in male Wistar rats by chronic thioacetamide administration (200 mg/kg) intraperitoneally three times per week. Rats received saline or SVT (20 mg/kg) two times daily intraperitoneally. In addition, co-cultures were incubated with either lipopolysaccharide or ethanol. Liver fibrosis, hepatic stellate cells activation, metalloproteinases activity, canonical and non-canonical Smads pathway and expression of several profibrogenic genes were evaluated. Thioacetamide activated hepatic stellate cells and distorted the liver parenchyma with the presence of abundant thick bands of collagen. In addition, thioacetamide upregulated the protein expression of α-smooth muscle actin, transforming growth factor-β1, metaloproteinases-9,−2 and −13 and overstimulate the canonical and non-canonical Smad pathways. SVT administration inhibited all of these changes. In vitro, SVT inhibited the upregulation of several genes implicated in cirrhosis when cells were exposed to lipopolysaccharides or ethanol. We conclude that SVT inhibited liver damage by blocking hepatic stellate cells activation, downregulating canonical and non-canonical profibrotic Smad pathways.
Keywords: Hepatic stellate cell, fibrosis, stevioside, Smad, co-culture
INTRODUCTION
Chronic liver disease, causing progressive loss of liver function, represents an enormous challenge for clinicians. In particular, liver cirrhosis is associated with increased morbidity and mortality, with important health and social consequences (1). Hepatic fibrosis, which eventually leads to cirrhosis, is a dynamic process characterized by the net accumulation of extracellular matrix (ECM) proteins as a result of chronic liver injury of any aetiology (2). Hepatic stellate cells (HSCs) are the main producer of ECM proteins in the liver, following their direct activation by profibrogenic factors, such as transforming growth factor beta 1 (TGF-β1) as well as by independent activation (2,3).
Because there is not yet an effective treatment for cirrhosis, research on compounds with potential beneficial effects on the liver needs to be encouraged. In this regard, the Stevia rebaudiana (Bertoni) plant seems to offer several pharmacological properties suitable for fighting chronic liver disease (4). Stevioside (SVT) is a natural non-calorie sweetener present in the stevia plant, which is 200–300 times sweeter than sugar. Several therapeutic benefits have been attributed to SVT, including lowering blood pressure and the glucose level, potentiating insulin secretion and improving insulin sensitivity and anti-inflammatory, anti-tumour, anti-diarrhoeal, diuretic, antioxidant, vasodilator and bradycardic effects. The safety of SVT as a sweetener has been evaluated by the Joint Food and Agricultural Organization and the World Organization Expert Committee on Food Additives. SVT has been used in the food industry since 2011 (5–8).
Alcohol is one of the main causes of liver cirrhosis in developed countries; unfortunately, alcohol administration does not produce cirrhosis in rodents. Several factors impair the establishment of alcohol-induced liver cirrhosis in rats, such as natural aversion to ethanol (EtOH), a higher rate of EtOH metabolism, and a greater tolerance to EtOH. To overcome this problem, we produced cirrhosis by chronic administration of thioacetamide (TAA) in rats, which is one of the most useful experimental models to mimic human cirrhosis in rodents because it induces the formation of portal-portal or portal-central fibrosis and, in the long-term, cirrhosis (9). On the other hand, recombinant cell lines, such as VL-17A, which are Hep G2 cells that constitutively express CYP2E1 (from human origin) and alcohol dehydrogenase (ADH, from murine origin) genes (10) offer a good tool for studying the mechanism involved in EtOH injury. Moreover, lipopolysaccharide (LPS), a molecule involved in human immunological response after bacterial infection, was used as inductor of profibrogenic molecules in co-cultured cells (9,11). These models are powerful tools to investigate the potential hepatoprotective effects of compounds with putative beneficial properties in the liver. Therefore, the objective of this study was to investigate whether SVT was able to inhibit TAA-induced cirrhosis in rats and if so, to search for the mechanisms involved in in vivo and in vitro.
MATERIALS AND METHODS
The study was conducted in accordance with Basic & Clinical Pharmacology & Toxicology policy for experimental and clinical studies (12).
Animal study design
All animal protocols were reviewed and approved by the Unit of Production and Experimentation of Laboratory Animals (UPEAL) at the Center for Research and Advanced Studies of the National Polytechnic Institute (Cinvestav-IPN) and comply with the Mexican official regulation (NOM-062-ZOO-1999) and the National Institute of Health guide for the care and use of laboratory animals (NIH Publications No. 8023, revised 1978). Wistar male rats weighing 100–120 g and approximately two weeks of age upon arrival were obtained from the UPEAL and were allowed to acclimatize for one week before manipulation. Rats were housed in polycarbonate cages under controlled conditions (21±1 °C, 50–60% relative humidity, and 12-hr dark-light cycles), were fed with Purina® rat chow and drank water ad libitum. Male Wistar rats (100–120 g initial weight) were randomly divided into four groups of 8 rats each. Cirrhosis was induced by intraperitoneal administration of TAA (200 mg/kg of body weight) three times per week for 8 weeks. Previously, we reported that 100 mg/kg of stevia leaves power significantly inhibited inflammation, oxidative stress and serum markers of liver damage in acute and chronic CCl4-intoxicated rats (13). Approximately, the content of SVT in stevia leaves is 20% (14), thus it seemed reasonable to administer 20 mg/kg of SVT to TAA-treated rats to evaluate its antifibrotic effect. Therefore, to determine the ability of SVT to attenuate liver cirrhosis, the diterpenoid was administered intraperitoneally at a dose of 20 mg/kg of body weight, two times daily, during TAA treatment. Appropriate vehicle, TAA and SVT groups were formed. Animals were euthanized under light ketamine/xylazine anaesthesia. Blood was collected by cardiac puncture, and the liver was rapidly removed. All samples were kept at −70°C until analysis.
Cell lines and culture conditions
Human HSC (hHSC) were isolated, as described elsewhere, using collagenase and pronase digestion and fractionation on an OptiPrep (Sigma, St Louis, MO, USA) gradient (15,16), from human liver biopsies from patients with morbid obesity who were subjected to bypass surgery by an approved protocol (IRB 070701). Written informed consent was obtained from the study participants, and the study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki, as was reflected in prior approval by the Institutional Review Committee.
VL-17A cells were kindly provided by Dr. Dahn L. Clemens. Briefly, Hep G2 cells were obtained from the American Type Culture Collection, and VL-17A cells were developed by transfecting VA-13 cells (ADH+; expressing the murine class I ADH) with the expression vector pIV-G2. This vector contains the coding region of the human cytochrome CYP2E1 that was kindly provided by Dr. Frank Gonzalez (National Cancer Institute, Bethesda, MD, USA). VA-13 cells were transfected using Lipo TAXI (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions. Recombinant cells were selected in a culture medium containing G418 and zeocin, both at 400 µL/mL.
Co-cultures of hHSC/VL-17A cells were created with a ratio of 1:10, closely mimicking the conditions of the liver in vivo (17). Cells were treated with SVT at 1, 5, 10, 20 and 100 mM and with 200 µL/20 mL (1 mg/1 mL) LPS or 100 mM EtOH, along with SVT or alone and were maintained in an incubator at 37°C and 5% CO2.
LPS and Et-OH concentrations were chosen from a report by Arellanes-Robledo et al., 2018 that demonstrated that LPS at 1 1µg/mL and EtOH at 100 mM in these cells produced a suitable response to investigate compounds with putative immunomodulatory activity such as SVT (18).
Chemicals used
SVT, TAA, ethanol and lipopolysaccharide, were purchased from Sigma-Aldrich (St Louis, MO, USA). Methanol, toluene, eosin, and formaldehyde were obtained from J. T. Baker (Mexico City, Mexico). All reagents were of analytical or biological grade, when appropriate.
Histology
Liver slices were taken from all the animals and fixed with 10% formaldehyde in PBS for 24 hr. Tissue samples were washed with tap water, dehydrated in alcohol, and embedded in paraffin. Five-micrometer-thick sections were mounted on glass slides covered with silane. Staining was performed with Masson’s trichrome stain.
Western blot assays
Western blot assays were performed, as previously described (19). Volumes equivalent to 50 and 120 µg of protein were transferred onto 15, 12 and 10% polyacrylamide gels. Separated proteins were transferred onto an Immuno-Blot™ PVDF membrane (BIO-RAD, Hercules, CA, USA). Then, blots were blocked for 1 or 2 hr at room temperature and incubated independently overnight at 4°C with the follow primary antibodies: metalloproteinase (MMP)-13 and TGF-β1 (EMD Millipore, Billerica, MA, USA), collagen (Col)-1α and α-smooth muscle actin (SMA) (Sigma-Aldrich, St Louis, MO, USA), Smad7, p-Smad3L, p-JNK, p-p38 (Abcam, Cambridge, MA, USA), then exposed to a secondary peroxidase-labelled antibody in the blocking solution at room temperature for 2 hr. Blots were stripped and incubated with a monoclonal antibody against β-actin (Ambion, Austin, TX, USA) and were used as a control to normalize cytokine protein expression levels. Images were digitalised and then analysed densitometrically using the ImageJ® software (National Institutes of Health, Bethesda, MD, USA).
Immunohistochemistry assays
Immunohistochemical (IHC) staining was performed, as previously described (16). Briefly, endogenous peroxidase was blocked with methanol-peroxidase for 1 hr (46 mL of MeOH + 4 mL of H2O2). Subsequently, the liver sections were incubated with primary antibodies mentioned above and diluted in 3% FBS overnight, and then incubated with the appropriate secondary antibody diluted in 3% foetal bovine serum for 2 hr at room temperature. Once washed, a DAB-H2O2 1:10 solution was added and incubated for 30 min. Hematoxylin was added as a counterstain for the samples and mounted with resin.
Quantification of α-SMA, TGF-β1 and MMP-13 was performed as follows: liver samples were taken from the right lobe of each treatment group and fixed with 10% formaldehyde. Then, tissues were processed for paraffin embedding and 5-μm thick sections were obtained and placed on glass slides covered with 3% silane. Five blank (unstained) slides were reacted with each of the respective antibodies and observed with a light microscope. Micrographs of representative areas were taken at a magnification of 10× for α-SMA, 40× for TGF-β1 and 20× for MMP-13. For each slide, at least 5 photos were obtained, and morphometric analysis was performed with an ImageJ software (NIH, Bethesda, MD, USA).
RNA isolation and quantitative RT-qPCR assays
Total RNA isolation was performed, as previously described (20). qPCR was performed using the Applied Biosystems™ StepOnePlus Real-Time PCR system. Taq polymerase (TaqMan, Universal Master Mix Prod # 4304437, Applied Biosystems, CA, USA) was used for the analysis of relative expression for α-SMA (gene ID: 81633) (Rn01759928_g1) and TGF-β1 (gene ID: 116487) (Rn01442102_m1). Probes were purchased from Thermo Fisher Scientific. A no template control, a target reagent sample, and a negative sample were included on the plate. GAPDH (gene ID: 24383) (Rn01775763_g1) was used as the internal control, and the relative gene expression was calculated using the Livak method (18), as follows:
For co-cultures, RNA isolation and determination of integrity and concentration was performed, as mentioned above. All reagents were purchased from Invitrogen (Grand Island, NY, USA). qPCR was performed using the Applied Biosystems™ 7900HT fast real-time system. SYBR Green (Power SYBR™ Green PCR master mix, Prod # 4368702, Applied Biosystems, CA, USA) was used for the analysis of relative expression for the genes tested. The primers used for qPCR amplification are the following: α-SMA (gene ID: 59) (F:5’-CTG AGC GTG GCT ATT CCT TC-3’, R: 5’-GCA GTG GCC ATC TCA TTT TC-3’), PDGF-B receptor (gene ID: 5159) (F: 5’-GGC TAC ATG GAC ATG AGC AA-3’, R: 5’-TCG GCA GGT CCT CTC AG-3’), TGF-β1 (gene ID: 7040) (F: 5’-TGA ACC GGC CTT TCC TGC TTC TCA TG-3’, R: 5’-GCG GAA GTC AAT GTA CAG CTG CCG C-3’), Smad3 (gene ID: 4088) (F: 5’-GGA GAA ATG GTG CGA GAA GG-3’, R: 5’-GAA GGC GAA CTC ACA CAG C-3’), and c-Myc (gene ID: 4609) (F: 5’-TCA AGA GGC GAA CAC ACA AC-3’, R: 5’-GGC CTT TTC ATT GTT TTC CA-3’). A no template control, a target reagent sample, and a negative sample were included on the plate. β-actin (gene ID: 60) (F: 5’-GGA GAA TGG CCC AGT CCT C-3’, R: 5’-GGG CAC GAA GGC TCA TCA T-3’) was used as the internal control, and the results were expressed as a ratio relative to the control, as mentioned above.
Collagen determination
Total collagen amount determination was performed as previously described (19), using the method by Prockop and Udenfriend (21).
Zymography assay
Proteolytic activity was measured as previously described (19) using gelatin-substrate gels. Images were digitalised and then analysed densitometrically using the ImageJ® software.
DNA synthesis assay
A colourimetric immunoassay based on incorporation of bromodeoxyuridine (BrdU) into the cellular DNA was performed following the manufacturer’s protocol (Abcam Prod # ab126556; Cambridge, MA, USA).
Statistical analysis
All data are expressed as the mean values ±SEM. Comparisons were carried out by 1-way analyses of variance, followed by a post-hoc Tukey Honest Significant Difference test, as appropriate, using GraphPad Prism version 6.0 for Windows (La Jolla, CA, USA; www.graphpad.com). Differences were considered statistically significant when P was ≤ 0.05.
RESULTS
SVT administration inhibits liver fibrogenesis through multitarget mechanisms
We assessed the ability of SVT to interfere with the fibrogenesis process when this compound was administered along with TAA. As shown in Fig. 1, chronic TAA treatment significantly elevated the amount of collagen in the liver parenchyma (Fig. 1B and E) compared with the control group (Fig. 1 A and E); importantly, SVT co-administration (Fig. 1 C and E) partially inhibited the accumulation of collagen fibres in TAA-treated rats. To confirm the histopathological analysis, total collagen in the liver was measured as the tissue hydroxyproline content (Fig. 1F), as well as by western blotting of Col-1α (Fig. 1G). In all cases, TAA significantly elevated the total content of collagen compared with the control rats; notably, administration of SVT inhibited the fibrotic process. SVT treatment of control rats did not alter normal collagen content.
Figure 1. STV inhibits collagen deposition in liver parenchyma.

Liver section of control rat (A); TAA-treated rat (B); STV+TAA-treated rat (C); rat administered with STV alone (D). Masson’s trichrome stain. The histogram depicts the percentage of positively stained collagen in liver slices stained with Masson’s trichrome (E) (n = 3). Hydroxyproline content obtained from liver sections (F) (n = 8). Protein expression levels of Col-1α were measured by western blot from liver slices of control rats (G) (n=3). β-actin was used as a control. The values are expressed as a fold increase of optical densitometry normalized to the control group values (control = 1). Each bar represents the mean value of experiments conducted in duplicate assays ± SEM. ap < 0.05 vs control. bp < 0.05 vs TAA.
SVT blocks HSC activation
Activated HSCs are the major cell type mediating ECM protein deposition and are, ultimately, the main responsible cells for the establishment of fibrosis and cirrhosis; in this scenario, HSCs appear to be the most attractive pharmacological target to block fibrogenesis. Therefore, to determine if the antifibrotic effect of SVT (Fig. 1) involved the inhibition of HSC activation, α-SMA, which is expressed as a transdifferentiation marker and is closely related to HSC activation, contraction and migration, was measured (16). Fig. 2A-D shows liver IHC slices with α-SMA antibody. The expression of α-SMA was significantly higher in the TAA group (Fig. 2B) compared to the control group (Fig. 2A). However, treatment with SVT significantly ameliorated the induced up-regulation of α-SMA (Fig. 2C), compared to the cirrhotic animals. Fig. 2E shows the percentage of positivity for α-SMA obtained from 3 liver slices. qRT-PCR (Fig. 2F) and western blotting (Fig. 2G) of α-SMA confirmed that TAA induced significant increases in α-SMA mRNA and protein, while SVT treatment completely inhibited this effect. SVT treatment of the control rats produced no effect on α-SMA expression.
Figure 2. Participation of STV in the downregulation of α-SMA.

Representative immunohistochemistry for α-SMA obtained from livers of control rats (A); TAA-treated rats (B); STV+TAA-treated rats (C) and rats administered with STV alone (D). Histogram depicts the percentage of positivity of α-SMA obtained from immunohistochemistry slices (E). Increase in relative mRNA expression of α-SMA (F) is shown. Protein expression level of α-SMA was measured by western blot from liver slices (G). β-actin was used as a control. The values are expressed as a fold increase of optical densitometry normalized to the control group values (control = 1). Each bar represents the mean value of experiments conducted in duplicate assays ± SEM (n = 3). ap < 0.05 vs control. bp < 0.05 vs TAA.
SVT downregulates TGF-β1
We sought to investigate if the inhibition of HSC activation by SVT was due to the downregulation of the TGF-β1 pathway. As shown in Fig. 3B and E, TAA treatment significantly increased the TGF-β1 positive area compared with the control group (Fig. 3A and E). It is worth noting that SVT treatment (Fig. 3C and E) completely inhibited the overexpression of TGF-β1. Moreover, as shown in Fig. 3F and 3G, the administration of the hepatotoxin increased the mRNA and protein levels compared to the control, whereas SVT significantly inhibited this increase. In all cases, SVT administration in control rats produced no effect.
Figure 3. STV downregulates TGF-β1 in fibrotic livers.

Immunohistochemistry TGF-β1 obtained from livers of control rats (A); TAA-treated rats (B); STV+TAA-treated rats (C) and rats administered with STV alone (D). Histogram depicts the percentage of positivity TGF-β1 obtained from immunohistochemistry slices (E). Increase in relative mRNA expression of TGF-β1 (F) is shown. Protein expression level of TGF-β1 was measured by western blot from liver slices (G). β-actin was used as a control. The values are expressed as a fold increase of optical densitometry normalized to the control group values (control = 1). Each bar represents the mean value of experiments conducted in duplicate assays ± SEM (n = 3). ap < 0.05 vs control. bp < 0.05 vs TAA.
SVT inhibits MMPs
The degradation of ECM components is normal and an important feature of tissue repair and remodelling and is thus a dynamic process. However, in liver cirrhosis, the hepatic architecture is lost by the generation of dense fibrous septa that delineate nodules of damaged liver parenchyma. As shown in Fig. 4A, TAA administration increased the activities of metalloproteinase (MMP)-9 and MMP-2, compared with control group, whereas the administration of SVT totally inhibited these increments. Similar results were found when we measured MMP-13 protein levels, where the hepatotoxicity increased its levels and SVT completely inhibited this effect (Fig. 4B-F). SVT treatment in control rats did not exhibit any effect.
Figure 4. STV preserves normal metalloproteinase (MMP) levels and activity.

MMP-9 and MMP-2 activities were analysed by zymography using gelatin-substrate gels in liver sections of control, TAA, STV+TAA, and STV only rats. Histograms depict signal intensities that were determined by densitometric analysis for MMP-9 and MMP-2 (A). Representative immunohistochemistry for MMP-13 obtained from control rats (B); TAA-treated rats (C); STV+TAA-treated rats (D) and rats administered with STV alone (E). Histograms depict the percentage of positivity of MMP-13 obtained from immunohistochemistry slices and protein expression levels of pro-MMP-13 and active MMP-13 by western blot from liver slices (F). β-actin was used as a control. The values are expressed as a fold increase of optical densitometry normalized to the control group values (control = 1). Each bar represents the mean value of experiments conducted in duplicate assays ± SEM (n = 3). ap < 0.05 vs control. bp < 0.05 vs TAA.
SVT downregulates canonical and non-canonical Smad pathways and increases Smad7
The canonical and non-canonical Smad pathways are critical in inducing ECM deposition and the development of fibrosis and cirrhosis. Therefore, these pathways constitute valuable targets for developing new and effective drugs to prevent liver fibrosis. In this regard, we investigated the ability of SVT to interfere with canonical and non-canonical Smad profibrogenic pathways to search for other mechanisms to explain the potent antifibrotic effect of SVT. As shown in Fig. 5A, chronic TAA intoxication significantly increased the levels of p-JNK, the active form of the kinase, and SVT completely inhibited this increase. Moreover, p-p38, which also phosphorylates Smad3 in the linker region, was increased by TAA treatment and, again, SVT completely inhibited this effect (Fig. 5B). As a result, the highly profibrogenic p-Smad3L (Fig. 5C) was increased by TAA and SVT inhibited this increase. In addition, we determined the role of the inhibitory protein of the canonical pathway, Smad7, in the antifibrogenic response elicited by SVT. As shown in Fig. 5D, TAA treatment decreased the levels of the Smad7 protein, compared with the control group, whereas SVT administration significantly inhibited this diminution. In all cases, SVT alone exhibited no effect.
Figure 5. SVT inhibits the phosphorylation of Smad3 in the linker region by inhibiting the activation of JNK and p38 and preserves Smad7.

Western blot analysis of pJNK (A), pp38 (B), pSmad3L (C), and Smad7 (D) in samples of liver tissue from control, thioacetamide (TAA)-treated, STV + TAA-treated, and STV-only treated rats. β-actin was used as a control. Values are expressed as the fold increase of OD normalized to the control group values (control = 1). Each bar represents the mean value of three rats ± SE (n = 3 ap < 0.05 vs control. bp < 0.05 vs TAA.
SVT inhibits the upregulation of profibrogenic genes induced by LPS or EtOH in co-cultures
To confirm the in vivo results, the antifibrotic properties were also characterized in co-cultures of hHSC/VL-17A cells. As observed in Fig. 6, α-SMA, TGF-β1, Smad3, PDGFR and c-Myc mRNAs were significantly elevated, either by LPS or EtOH. It is worth noting that incubation with SVT effectively inhibited increased mRNA expression of these highly profibrogenic genes. The incubation with SVT in the control group did not produce any effect.
Figure 6. SVT antifibrogenic mechanism assessed in cocultured cells.

Increase in relative mRNA expression of α-SMA (A, B); TGF-β1 (C, D); Smad3 (E, F); PDGFR (G, H); and c-Myc (I, J) in samples of cocultured cell lines of hHSC and VL-17A from control, LPS and EtOH treated, STV + LPS or EtOH treated, and STV-only treated cocultured cells are shown. β-actin was used as a control. STV concentration was 40 mM. Each bar represents the mean value of experiments conducted in duplicate assays ± SEM (n = 3). ap < 0.05 vs control. bp < 0.05 vs TAA.
SVT exhibits a dose-dependent anti-proliferative effect
In order to explore a possible anti-proliferative effect of SVT, a BrdU assay was performed. Fig. 7 shows a concentration-dependent anti-proliferative effect of SVT on co-cultured cells. It was found that 100 mM of SVT produced the highest anti-proliferative effect, which is consistent with previous results inhibiting c-Myc oncogene overexpression (Fig. 6).
Figure 7. Effect of STV on proliferative assay.
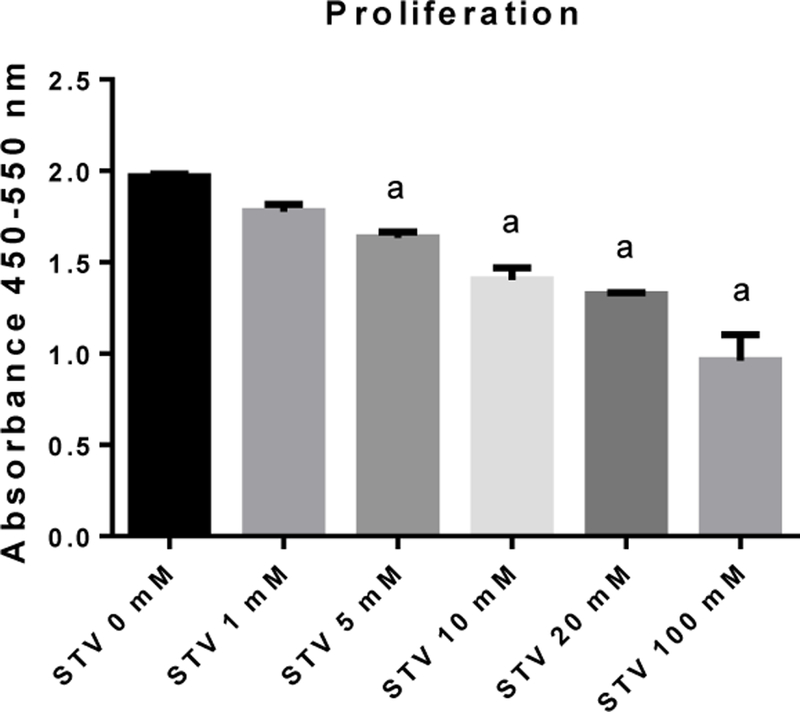
Histogram depicts a dose-dependent response in co-culture cell lines of hHSC and VL-17A. Each bar represents the mean value of experiments conducted in duplicate assays ± SEM (n = 3). ap < 0.05.
DISCUSSION
Previously, we found that stevia leaves powder inhibited chronic liver damage induced by CCl4 administration by reducing hepatic myofibroblasts and modulating some molecular profibrotic pathways (22). Herein, we found that SVT, a diterpenoid present in stevia leaves, inhibited liver cirrhosis induced by TAA in rats by modulating MMPs, inhibiting the activation of HSCs, downregulating TGF-β1 and the canonical and non-canonical Smad pathways. Moreover, in this study, we found that, in vitro, SVT inhibited the upregulation of several genes implicated in cirrhosis when cells were exposed to LPS or Et-OH. Interestingly, the previous and present results provide evidence of the hepatoprotective actions of stevia and SVT in CCl4, TAA, Et-OH and LPS models of liver damage and, notably, illuminate the involved action mechanisms. Therefore, altogether, our results suggest that stevia/SVT may be useful to treat human liver diseases and that research effort may be focused on toxicological and controlled clinical trials before proposing SVT as a treatment for fibrosis and cirrhosis in humans.
A preliminary dose-response study on the effect of SVT on TAA-induced acute liver damage was performed, and it was found that the dose of 20 mg/kg afforded the best hepatoprotective effects without evident toxicity (data not shown); therefore, in this work, we decided to study the SVT effects on chronic TAA toxicity utilizing this dose.
Hereby, we investigated the benefits of SVT in a model of cirrhosis that shares some characteristic with the human condition. The profibrogenic actions of TAA have been well-established and are mainly caused by the metabolite of TAA, thioacetamide-S-oxide, a free radical that promotes necrosis of hepatocytes and, in the long term, leads to prominent liver damage and persistent fibrosis, similar to liver damage and fibrosis in humans (9).
The susceptibility of liver to TAA-chronic intoxication was evaluated by the changes in liver parenchyma leading to the formation of regenerative nodules and thick bands of collagen fibres. Notably SVT was able to inhibit these alterations.
During chronic injury, TGF-β1 mediates the proliferation and transdifferentiation of HSCs into activated myofibroblasts that express α-SMA and display contractile pro-inflammatory and profibrotic features (20,21). In addition, it is well known that TGF-β1 is a strong inducer of the transdifferentiation of HSCs to produce an excessive accumulation of ECM proteins (23,24). Interestingly, our results suggest that the antifibrotic effects of SVT are due, at least in part, by downregulating TGF-β1, leading to the consequent inactivation of HSCs, the main collagen-producing cells.
ECM is an important component of the liver environment, providing a surface for cell adhesion, a reservoir for signalling molecules and space for cell growth and migration. It is important to mention that remodelling by MMPs is necessary for the degradation of ECM components by liberating active profibrogenic and mitogenic cytokines and other factors (25,26). Activated HSCs are sources of MMP-9 and MMP-2, where MMP-9 has been found in scar areas of active fibrogenesis, both cleaving TGF-β1 from the ECM and promoting HSC activation, migration and proliferation (17,26,27). MMP upregulation has been associated with the change from normal to abnormal matrix turnover in a CCl4 model; in addition, MMP-13 gene deletion leads to retarded resolution in CCl4-induced fibrosis (16,27–29). Therefore, it seems likely that the antifibrotic effect of SVT may be due, partially, to downregulation of MMPs, leading to the inhibition of HSC activation, mobilization, migration, proliferation and production of fibrous tissue.
The canonical Smad pathway starts with the binding of TGF-β1 to the serine/threonine kinase receptor known as TβRII, which is a homodimer that provides a stable structural interface for binding with another serine/threonine receptor homodimer named TβRI, thus forming a heterodimeric receptor-ligand complex. Once the complex is formed, the constitutively active TβRII undergoes an autophosphorylation and promotes the transphosphorylation of TβRI, activating its kinase activity (30,31). In this pathway, Smads 2 and 3 are important mediators, serving as transcriptional factors that constantly shuttle between the cytoplasm and nucleus, once they are complexed to Smad4 and are phosphorylated at the C-terminal by TβRI. Nevertheless, once this pathway is over-stimulated, a negative feedback response is released, mediated by Smad7, antagonizing the signalling cascade of TGF-β1 (19,32). There is also a non-canonical pathway in which PDGF activates JNK, which, in turn, phosphorylates Smad3 in the linker domain to generate p-Smad3L, which rapidly translocates to the nucleus where it stimulates HSC proliferation (33,34). Our results suggest that SVT counterattacks the progression of liver fibrosis by downregulating p-JNK and p-p38, which, in turn, cannot phosphorylate Smad3 in the linker domain, as well as by preserving a negative feedback pathway through Smad7. In this study, the results obtained in cirrhotic rats provide, for the first time, strong evidence on the antifibrotic effects of SVT. As previously mentioned, α-SMA is an indicator of transdifferentiation or activation of HSCs, and TGF-β1 is a key factor to induce HSC activation and ECM production (30). In addition, Smad3 plays a major role in fibrogenesis by inducing the production of collagen (31); thus, these proteins are essential in the fibrogenic process that ultimately leads to cirrhosis. Moreover, proteins involved in HSC proliferation, such as PDGFR, that enhance inflammatory and fibrogenic responses via MAPK, as well as NF-κB and c-Myc, a potent mitogen that can be promoted by PDGF and pro-inflammatory cytokines via pSmad3L, contribute to the exacerbation of the fibrogenic response (32–34).
In contrast to primary hepatocyte cultures that lose their ability to metabolize ethanol with time, VL17A cells express active CYP2E1 and ADH1 and thereby are similar to hepatocytes in alcoholics (35); in addition, HSC do not express CYP2E1 (36); as a result, co-cultures of VL17A cells and HSC constitute a suitable model to study alcohol-induced liver damage, which is a common disease that eventually leads to cirrhosis because metabolism of EtOH produces acetaldehyde and ROS that in turn activate HSC. Thus, this co-culture represents a closer link with in the vivo model of TAA-induced cirrhosis that shares some similarities with human cirrhosis originated by excessive EtOH consumption. In the present report, we utilized a human co-culture system consisting of primary HSC and VL17A cells to further investigate the action mechanism of SVT to block inflammation and fibrosis. In fact, by utilizing this in vitro model, we found that SVT inhibited the upregulation of several genes implicated in cirrhosis when cells were exposed to LPS or EtOH. Therefore, the in vitro results are in agreement with the observations obtained from TAA-induced cirrhosis, further reinforcing the important anti-profibrogenic effect of SVT in chronic liver injury and providing multitarget mechanisms of action.
Several reports have established that Myc genes are transcribed in similar rates as cell cycle when cells differentiate from quiescent to a proliferative state (37–40). In agreement, the results from c-Myc correlated with co-cultures proliferation. Therefore, our results with BrdU suggest that another antifibrotic effect of SVT may be mediated by inhibiting HSC proliferation.
In general, most parameters, such as collagen content and activation of HSCs in vivo were inhibited partially but significantly, however, others, such as MMPs and Smad exhibited a total protection from TAA when SVT was administered; some profibrotic pathways were completely blocked and others only partially and the final result was that fibrosis, which is the most important feature after chronic liver damage, was partially inhibited by SVT. Therefore, we conclude that the hepatoprotective effect of STV was partial.
CONCLUSION
Our current results present robust evidence of the antifibrotic effects of SVT and provide important information about the multitarget mechanism of the anti-cirrhotic actions of this diterpenoid. Notably, SVT exhibits a reasonable safety profile and may, therefore, be utilized in the clinical setting. However, further studies are required in animal models to provide a rationale for subsequently performing safety and short- and long-term studies in cirrhotic patients.
ACKNOWLEDGEMENTS
The authors thank Silvia Galindo Gómez, Karla M. Gil Becerril, Veronica Reyes Olivares, Rafael Leyva, Benjamín E. Chavez, and Ricardo Gaxiola for their excellent technical assistance. The authors also acknowledge the Animal Lab Facility, UPEAL-Cinvestav and Dr. Jorge Fernández-Hérnandez.
Statement of financial support:
This research was supported by the National Council of Science and Technology (Conacyt) of Mexico, grant No. 253037 to Muriel P, by fellowship No. 355795 to Sael Casas-Grajales from Conacyt, and by the NIAAA/NIH grant No. K01AA025140-02.
Footnotes
Conflict of interest:
The authors declare that they have no conflicts of interest.
REFERENCES
- 1.Muriel P The Liver: General aspects and epidemiology. In: Muriel P, (ed). Liver Pathophysiology: Therapies and Antioxidants Waltham, MA, USA: Elsevier Inc; 2017; 3–22. [Google Scholar]
- 2.Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol 2017;14:397–411. [DOI] [PubMed] [Google Scholar]
- 3.Kershenobich D, Gutiérrez-Reyes DG. Is human cirrhosis a reversible disease? In: Muriel P, (ed). Liver Pathophysiology: Therapies and Antioxidants Waltham, MA, USA: Elsevier Inc; 2017; 259–65. [Google Scholar]
- 4.Ramos-Tovar E, Muriel P. Stevia as a putative hepatoprotector. In: Muriel P, (ed). Liver Pathophysiology: Therapies and Antioxidants Waltham, MA, USA: Elsevier Inc; 2017; 715–27. [Google Scholar]
- 5.Ragone MI, Bonazzola P, Colareda GA, Lazarte ML, Bruno F, Consolini AE. Cardioprotection of stevioside on stunned rat hearts: A mechano-energetical study. Phytomedicine 2017;35:18–26. Doi: 10.1016/j.phymed.2017.08.022 [DOI] [PubMed] [Google Scholar]
- 6.Ilić V, Vukmirović S, Stilinović N, Čapo I, Arsenović M, Milijašević B. Insight into anti-diabetic effect of low dose of stevioside. Biomed Pharmacother 2017;90:216–21. [DOI] [PubMed] [Google Scholar]
- 7.Panagiotou C, Mihailidou C, Brauhli G, Katsarou O, Moutsatsou P. Effect of steviol, steviol glycosides and stevia extract on glucocorticoid receptor signaling in normal and cancer blood cells. Mol Cell Endocrinol 2018;460:189–99. Doi: 10.1016/j.mce.2017.07.023 [DOI] [PubMed] [Google Scholar]
- 8.Wang Z, Xue L, Guo C, Han B, Pan C, Zhao S, et al. Stevioside ameliorates high-fat diet-induced insulin resistance and adipose tissue inflammation by downregulating the NF-κB pathway. Biochem Biophys Res Commun 2012;417:1280–5. Doi: 10.1016/j.bbrc.2011.12.130 [DOI] [PubMed] [Google Scholar]
- 9.Muriel P, Ramos-Tovar E, Montes-Páez G, Buendía-Montaño LD. Experimental models of liver damage mediated by oxidative stress. In: Muriel P, (ed). Liver Pathophysiology: Therapies and Antioxidants Waltham, MA, USA: Elsevier Inc; 2017; 529–46. [Google Scholar]
- 10.Donohue TM, Osna NA, Clemens DL. Recombinant Hep G2 cells that express alcohol dehydrogenase and cytochrome P450 2E1 as a model of ethanol-elicited cytotoxicity. Int J Biochem Cell Biol 2006;38:92–101. [DOI] [PubMed] [Google Scholar]
- 11.S L, Chaudhary S, R.S R. Hydroalcoholic extract of Stevia rebaudiana bert. leaves and stevioside ameliorates lipopolysaccharide induced acute liver injury in rats. Biomed Pharmacother 2017;95:1040–50. Doi: 10.1016/j.biopha.2017.08.082 [DOI] [PubMed] [Google Scholar]
- 12.Tveden-Nyborg P, Bergmann TK, Lykkesfeldt J. Basic & Clinical Pharmacology & Toxicology Policy for Experimental and Clinical studies. Basic Clin Pharmacol Toxicol 2018;123:233–5. [DOI] [PubMed] [Google Scholar]
- 13.Ramos-Tovar E, Hernández-Aquino E, Casas-Grajales S, Buendia-Montaño LD, Galindo-Gómez S, Camacho J, et al. Stevia prevents acute and chronic liver injury induced by carbon tetrachloride by blocking oxidative stress through Nrf2 upregulation. Oxid Med Cell Longev 2018;2018:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abou-Arab A, Abou-Arab E, Abou-Arab A, Abu-Salem MF. Physico-chemical assessment of natural sweeteners steviosides produced from Stevia rebaudiana Bertoni plant. Afr J Food Sci 2010;4:269–281. [Google Scholar]
- 15.Nakamura A, Ueno T, Yagi Y, Okuda K, Ogata T, Nakamura T, et al. Human primary cultured hepatic stellate cells can be cryopreserved. Med Mol Morphol 2010;43:107–15. [DOI] [PubMed] [Google Scholar]
- 16.Reyes-Gordillo K, Shah R, Popratiloff A, Fu S, Hindle A, Brody F, et al. Thymosin-β4 (Tβ4) blunts PDGF-dependent phosphorylation and binding of AKT to actin in hepatic stellate cells. Am J Pathol 2011;178:2100–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.History Geerts A., heterogeneity, developmental biology, and functions of quiescent hepatic stellate cells. Semin Liver Dis 2001;21:311–35. [DOI] [PubMed] [Google Scholar]
- 18.Arellanes-Robledo J, Reyes-Gordillo K, Ibrahim J, Leckey L, Shah R, Lakshman MR. Ethanol targets nucleoredoxin/dishevelled interactions and stimulates phosphatidylinositol 4-phosphate production in vivo and in vitro. Biochem Pharmacol 2018;156:135–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hernández-Aquino E, Zarco N, Casas-Grajales S, Ramos-Tovar E, Flores-Beltrán RE, Arauz J, et al. Naringenin prevents experimental liver fibrosis by blocking TGFβ-Smad3 and JNK-Smad3 pathways. World J Gastroenterol 2017;23:4354–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 2001;25:402–8. [DOI] [PubMed] [Google Scholar]
- 21.Prockop DJ, Udenfriend S. A specific method for the analysis of hydroxyproline in tissues and urine. Anal Biochem 1960;1:228–39. [DOI] [PubMed] [Google Scholar]
- 22.Ramos-Tovar E, Buendia-Montaño LD, Galindo-Gómez S, Hernández-Aquino E, Tsutsumi V, Muriel P. Stevia prevents experimental cirrhosis by reducing hepatic myofibroblasts and modulating molecular profibrotic pathways. Hepatol Res 2018. In press. Available from: http://doi.wiley.com/10.1111/hepr.132754 [DOI] [PubMed] [Google Scholar]
- 23.Wallace MC, Friedman SL, Mann DA. Emerging and disease-specific mechanisms of hepatic stellate cell activation. Semin Liver Dis 2015;35:107–18. [DOI] [PubMed] [Google Scholar]
- 24.Koyama Y, Brenner DA. Liver inflammation and fibrosis. J Cinical Investig 2017;127:55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bataller R, Brenner DA. Liver fibrosis. J Cinical Investig 2005;115:209–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schuppan D, Afdhal NH. Liver cirrhosis. Lancet 2008;371:838–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baiocchini A, Montaldo C, Conigliaro A, Grimaldi A, Correani V, Mura F, et al. Extracellular matrix molecular remodeling in human liver fibrosis evolution. PLoS One 2016;11:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duarte S, Baber J, Fujii T, Coito AJ. Matrix metalloproteinases in liver injury, repair and fibrosis. Matrix Biol 2015;44–46:147–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Han Y-P. Matrix metalloproteinases, the pros and cons, in liver fibrosis. J Gastroenterol Hepatol 2006;21:88–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee YA, Wallace MC, Friedman SL. Pathobiology of liver fibrosis: A translational success story. Gut 2015;64:830–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zi Z, Chapnick DA, Liu X. Dynamics of TGF-β/Smad signaling. FEBS Lett 2012;586:1921–8. Doi: 10.1016/j.febslet.2012.03.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matsuzaki K, Seki T, Okazaki K. TGF-β signal shifting between tumor suppression and fibro-carcinogenesis in human chronic liver diseases. J Gastroenterol 2014;49:971–81. [DOI] [PubMed] [Google Scholar]
- 33.Higashi T, Friedman SL, Hoshida Y. Hepatic stellate cells as key target in liver fibrosis. Adv Drug Deliv Rev 2017;121:27–42. Doi: 10.1016/j.addr.2017.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matsuzaki K Smad phosphoisoform signals in acute and chronic liver injury: similarities and differences between epithelial and mesenchymal cells. Cell Tissue Res 2012;347:225–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Donohue TM, Osna NA, D.L. Clemens DL. Recombinant Hep G2 cells that express alcohol dehydrogenase and cytochrome P450 2E1 as a model of ethanol-elicited cytotoxicity. Int J Biochem Cell Biol 2006;38:92–101. [DOI] [PubMed] [Google Scholar]
- 36.Casini A, Pellegrini G, Ceni E, Salzano R, Parola M, Robino G, et al. Human hepatic stellate cells express class I alcohol. J Hepatol 1998;28:40–45. [DOI] [PubMed] [Google Scholar]
- 37.Bouchard C, Staller P, Eilers M. Control of cell proliferation and growth by Myc proteins. Trends Cell Biol 1998;8:202–6. [DOI] [PubMed] [Google Scholar]
- 38.Niu Z, Liu H, Zhou M, Wang H, Liu Y, Li X, et al. Knockdown of c-Myc inhibits cell proliferation by negatively regulating the Cdk/Rb/E2F pathway in nasopharyngeal carcinoma cells. Acta Biochim Biophys Sin 2015;47:183–91. [DOI] [PubMed] [Google Scholar]
- 39.Bretones G, Delgado MD, León J. Myc and cell cycle control. Biochim Biophys Acta-Gene Regul Mech 2015;1849:506–16. [DOI] [PubMed] [Google Scholar]
- 40.Dang CV c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell 1999;19:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]