

Abstract
Constraining σ3-P compounds in nontrigonal, entatic geometries has proven to be an effective strategy for promoting biphilic oxidative addition reactions more typical of transition metals. Although qualitative descriptions of the impact of structure and symmetry on σ3-P complexes have been proposed, electronic structure variations responsible for biphilic reactivity have yet to be elucidated experimentally. Here we report P K-edge XANES data and complementary TDDFT calculations for a series of structurally-modified P(N)3 complexes that both validate and quantify electronic structure variations proposed to give rise to biphilic reactions at phosphorus. These data are presented alongside experimentally-referenced electronic structure calculations that reveal nontrigonal structures predicted to further enhance biphilic reactivity in σ3-P ligands and catalysts.
Keywords: biphilic, phosphorus, X-ray absorption spectroscopy, main group, TDDFT
Tricoordinate phosphorus (σ3-P) compounds are indispensable tools in many fields of synthetic chemistry.1,2,3,4,5.Despite a diversity of applications, the reactivity of most typical trigonal pyramidal σ3-P compounds falls within a narrowly prescribed range, governed overwhelmingly by the nucleophilic reactivity of the stereochemically active lone pair (Figure 1).6,7. Complementary electrophilic reactivity of σ3-P compounds is comparatively rare;8 within trigonal symmetry the phosphorus-based unfilled acceptor orbitals for compounds R3P are σ*P–R in character and therefore rather high in energy.9–12
Figure 1.
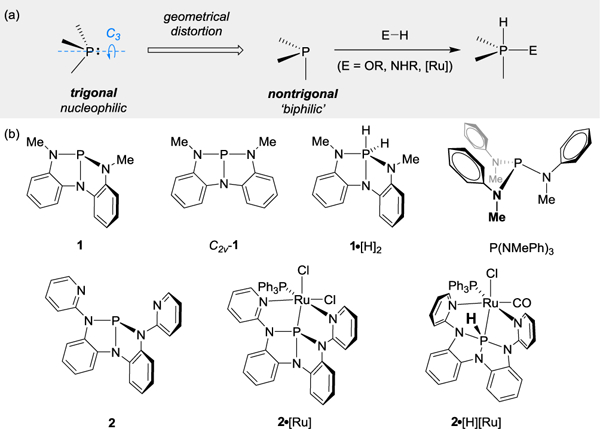
(a) An illustration of the biphilic hypothesis of nontrigonal σ3-P. (b) P(N)3 compounds investigated in this study.
Motivated by the perception that the restriction of phosphorus to local (pseudo)trigonal symmetry places an unnecessary check on the synthetic performance of σ3-P compounds,13,14,15,16,17,18,19,20,21,22,23 we have advanced the hypothesis that descent from trigonal symmetry should colocalize both donor and acceptor reactivity at a single σ3-P site (i.e. biphilic24 reactivity). Indeed, we have shown that nontrigonal σ3-P compounds (viz. 1 and 2, Figure 1a) evolve to pentacoordinate phosphorus (σ5-P) products through oxidative addition of E–H bonds (E=OR, NHR, Ru),25–26,27,28 in some cases reversibly.29–35
Although applications of the biphilic character of nontrigonal σ3-P compounds such as 1 and 2 have emerged in reactivity, a direct experimental demonstration of how the C3v→Cs structural deformation controls σ3-P frontier orbital energies has not been previously presented. With a view toward establishing a rational schema for biphilic σ3-P design, we provide here the first explicit spectroscopic evidence validating the descent-in-symmetry hypothesis of biphilicity at σ3-P. Through phosphorus K-edge X-ray absorption near-edge structure (XANES) spectroscopy and supporting (TD)DFT demonstrations, we quantify the impact of nontrigonal distortion (C3v→Cs→C2v) on the electronic structure of a series of compositionally related compounds and show that nontrigonal σ3-P compounds present inherently contracted frontier orbital energy gaps that colocalize electron–donor and –acceptor behavior at a single σ3-P site, engendering biphilic character. Overall, these results codify how structural modifications control σ3-P electronic structure and provide a forward-looking blueprint for the development of next generation nontrigonal P(III) catalysts and ligands with exceptional biphilic reactivity.
Phosphorus K-edge XANES36 is an ideal element-specific technique for extracting information concerning electronic structure and chemical bonding in diverse phosphorus compounds.37,38,39,40,41–42,43,44 Unoccupied frontier orbital energies are measured by excitation of core P 1s electrons, and transitions are governed by the dipole selection rule so that peak intensity correlates to P 3p character in the associated virtual orbitals.45-46 For tris-methylanilide P(NMePh)347, a benchmark crystalline C3-symmetric phosphorous triamide, the P K-edge XANES spectrum exhibits an intense pre-edge feature at 2147.0 eV (Figure 2a, blue dashes). TDDFT simulation (B3LYP-D3/6–31G(d,p)) and subsequent natural transition orbital analysis48 (NTOs; Figure S12) assigned this transition as an excitation of a core 1sP electron into excited states with mixing of MOs 94/95 and 100/101, the latter of which correspond to the degenerate e-symmetry σ*P–N orbitals assuming rigorous C3 point group symmetry. The magnitude of the calculated oscillator strength for each transition stems primarily from the large calculated 3pP character of MOs 100 and 101 (47.4% and 53.2%, respectively). This is consistent with the analysis of the NTO for the first transition in which the particle state is 53.6% 3pP. In contrast, MOs 93 (HOMO), 94 (LUMO), and 95 (LUMO+1) have smaller calculated 3pP character (4.6% - 7.7%) and are best described as aryl π* (Figure S6). Correspondingly, the P lone pair is better matched to MO 87 (Figure 2d), as reflected by its increased P 3sP (10.7%) and 3pP (18.4%) character compared to the HOMO (4.4% and 4.6% for 3sP and 3pP, respectively).
Figure 2.
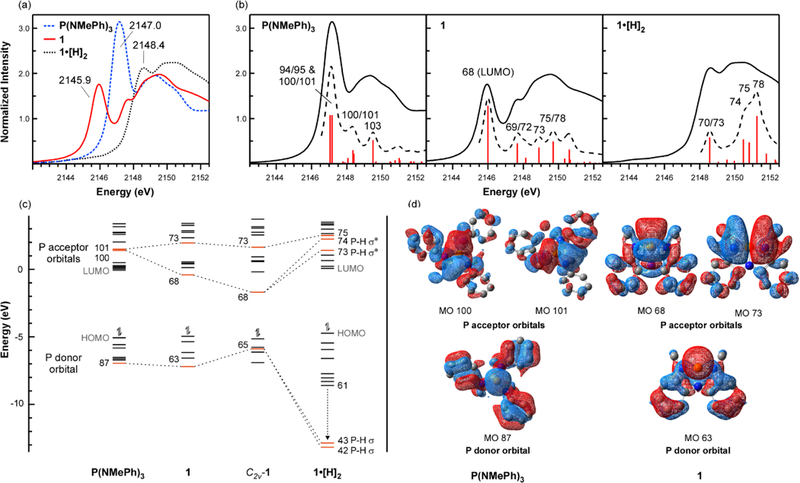
(a) P K-edge XANES spectra for P(NMePh)3, 1, and 1•[H]2. (b) Experimental (solid line) and simulated (dashed line) P K-edge XANES spectra for P(NMePh)3, 1, and 1•[H]2. Individual transitions are represented by red bars and indicate relative differences in calculated oscillator strength. The transitions are labeled according to the predominant calculated MOs mixing in the excited states. The labels correspond to those provided in Figure 2c and Table S15. (c) Truncated MO correlation diagram of molecular P(N)3 complexes. Frontier orbitals relevant to the XAS results and reactivity at phosphorus are highlighted in red and connected by dashed lines to show how they transform across the series. (d) Selected donor-acceptor Kohn-Sham orbitals from DFT calculations of P(NMePh)3 and 1 (isovalue = 0.0200; hydrogen atoms not shown). Kohn Sham orbitals for the remaining MOs labelled in the MO diagram are included in the SI.
Despite the apparent compositional similarity in their local σ3-P environments, the P K-edge XANES spectrum of nontrigonal σ3-P 1 (Figure 2a, red) revealed a dramatic bathochromic shift of about 1.1 eV for the first pre-edge peak (2145.9 eV) compared to P(NMePh)3. This difference in XANES pre-edge energies cannot be attributed to variance in P oxidation state; as expected for P compounds with identical valency and local bonding environment, there is only a small (0.1 eV) difference in the calculated 1sP orbital energies. Rather, the 1.1 eV difference between the first peak energies for 1 and P(NMePh)3 stems primarily from lowering of frontier phosphorus acceptor orbital energy by the nontrigonal distortion of σ3-P compound 1. Upon descent in symmetry, the nearly degenerate P acceptor MOs in P(NMePh)3 splits in 1 to yield a new unoccupied orbital at lower energy (MO 68; Figure 2d); the energy of the remaining P acceptor orbital (MO 73) is relatively unaffected by comparison. TDDFT simulation of the K-edge absorption confirms excitation to MO 68, a transition best described as 1sP → σ*P–N (Figure 2b). In effect, the C3→Cs structure change yields a pronounced decrease in σ*P–N energy that is consistent with increased biphilicity of 1 relative to P(NMePh)3.
Notably, further deformation of Cs-symmetric 1 to a C2v-symmetry structure (C2v-1) is predicted by DFT to cause the LUMO energy to decrease by an additional 1.3 eV (Figure 2c). This decrease in LUMO energy is significant because it suggests that the P-acceptor orbital becomes more susceptible to substrate binding upon distortion from nontrigonal Cs to C2v symmetry. To further analyze how these MOs are transformed upon biphilic oxidative addition reactions, a previously unreported σ5-P compound (1•[H]2, Figure 1b) with a nearly planar PN3 framework was synthesized.47
By comparison to both 1 and P(NMePh)3, the P K-edge XANES absorption edge for σ5-P compound 1•[H]2 appeared at much higher energy (2148.4 eV; Figure 2b, right). The large energy increase (2.5 eV) for the absorption edge in 1•[H]2 relative to 1 is consistent with oxidation of P(III) to P(V), which lowers the energy of the 1sP orbital due to the increased effective nuclear charge (Zeff) at P. Indeed, the calculated 1sP orbital for 1•[H]2 is stabilized by 1.2 eV relative to 1. Equally important, TDDFT spectral simulations and NTO analysis for 1•[H]2 revealed that the resulting excited state contains mixtures of orbitals that have appreciable σ*P–H character (MOs 70 and 73; Figure S9), so the transition is assigned as 1sP → σ*P–H. These data indicate that the P-based frontier donor/acceptor electronic structure of C2v-1 is transformed upon formation of the σ5-P P–H bonds, evolving into the σP–H and σ*P–H MOs of divergent energy (Figure 2c).
The same qualitative electronic features observed for 1 and 1•[H]2 are also evident when nontrigonal σ3-P complexes are bound to transition metals. For instance, the P K-edge XANES spectrum of 2•[Ru], in which the nontrigonal σ3-P behaves as an L-type donor ligand with respect to the metal, shows three features: a shoulder at 2145.9 eV and two prominent peaks at 2146.8 and 2148.1 eV (Figure 3a). DFT calculations revealed that the phosphorus acceptor orbital on the free ligand 2 mixes with the 4dz2 on Ru to form a new MO that has some σ*Ru–P character but is best defined as the P acceptor orbital (MO 210; Figures 3c and 3d), as reflected in the calculated MO composition (43.3% P 3p and 11.2% Ru 4d; Table S16). For comparison, a complementary σRu–P bonding MO was identified at lower energy (MO 151; Figure 3c) with lower 3pP mixing due to the absence of P acceptor character (9.7% P 3p and 5.4% Ru 4d). The remaining frontier orbitals calculated for 2•[Ru] have < 5% 3pP character stemming from 2, and the HOMO and LUMO are best described as Ru-Cl and Ru-N π* (Figure S10). TDDFT calculations revealed that the intense transition at 2146.8 eV in the P K-edge XANES spectrum of 2•[Ru] shown in Figure 3b is assigned to an excited state containing mixtures of MOs 194 – 199, 201, and 210 due to the low molecular symmetry. However, NTO calculations (Figure S13) reveal that the particle state for this transition is 32.8% 3pP consistent with a large contribution from MO 210 due to its significantly higher 3pP character (43.3%; Table S16) compared to the other MOs (<4.4%). Weaker transitions centered around 2146 eV are associated with unoccupied frontier orbitals containing smaller amounts of 3pP character from PPh3, whereas transitions at higher energy (ca. 2148 eV) arise from higher energy MOs that have small amounts of Ru-P π* consistent with Ru→P backbonding (MOs 212, 215, and 216; Figure S10). However, these MOs also contain significant amounts of diffuse 5pRu character (Table S16) suggestive of valence and Rydberg orbitals.49,50
Figure 3.
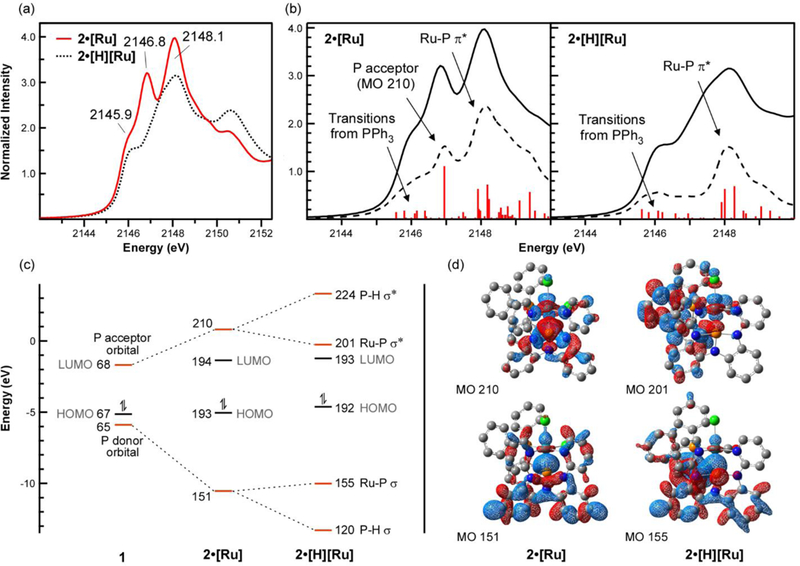
(a) Comparison of P K-edge XANES spectra for 2•[Ru] and 2•[H][Ru]. (b) Comparison of experimental (solid line) and simulated (dashed line) P K-edge XANES spectra for 2•[Ru] and 2•[H][Ru]. Individual transitions are represented by red bars and indicate relative differences in calculated oscillator strength. The transitions are labeled with calculated MOs mixing in the excited states.(c) Truncated MO correlation diagram of 1, 2•[Ru], and 2•[H][Ru]. Frontier orbitals relevant to the XAS results and reactivity at phosphorus are highlighted in red in Figure 3c and connected by dashed lines to show how they transform across the series. (d) Selected donor-acceptor Kohn-Sham orbitals from DFT calculations of 2•[Ru], and 2•[H][Ru] (isovalue = 0.0270; hydrogen atoms not shown). Kohn Sham orbitals for the remaining MOs labelled in the MO diagram are included in the SI.
The P K-edge XANES spectrum of 2•[H][Ru] exhibits similar features at 2145.9 and 2148.2 eV, as confirmed by TDDFT calculations, but the intense peak at 2146.8 eV in 2•[Ru] is no longer present (Figure 3b). Inserting P into the Ru–H bond in 2•[H][Ru] yields σRu–P and σ*Ru–P orbitals at similar energies to those calculated for 2•[Ru] (MOs 155 and 201; Figure 3c), but the P acceptor character in the unoccupied orbital is extinguished (MO 201 has only 2.5% P 3p compared to 43.3% in 2•[Ru]). The complementary σRu–P orbital in 2•[H][Ru] (MO 155) has 8.3% 3pP and 3.4% 4dRu character – slightly less than that observed in 2•[Ru]. Two new MOs assigned to σP–H and σ*P–H (MOs 120 and 224, respectively) were also identified for 2•[H][Ru], as expected (Figure S11). Overall, the XANES and TDDFT data for 2•[Ru] and 2•[H][Ru] mirror observations made for 1 and 1•[H]2. The low-lying P acceptor orbital in nontrigonal σ3-P compounds is clearly transformed upon biphilic chemical reaction, whether in the absence of presence of a transition metal host.
The results of the forgoing spectroscopic investigations are usefully contextualized by a qualitative molecular orbital diagram tracing the nontrigonal distortion coordinate. In threefold symmetry (Figure 4, left), orbital 2a1 defines the lone pair and orbitals 2e are the doubly degenerate set of phosphorus-substituent antibonding orbitals. Descent to Cs symmetry does not dramatically alter the lone pair energy but lifts the degeneracy of the unfilled orbitals and lowers the frontier energy gap by decreasing destructive overlap of the substituents with respect to px. Further transit along this distortion coordinate culminates at a C2v-symmetric T-shaped structure possessing orthogonal frontier orbitals, the precise energetic ordering of which depends on the nature of the substituent and its electronegativity.
Figure 4.
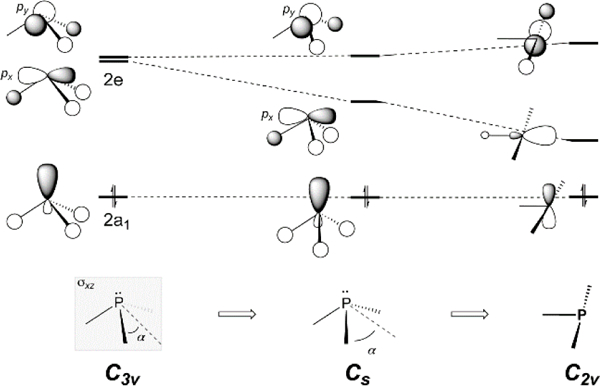
Qualitative frontier molecular orbital diagrams depicting the electronic structure arising from nontrigonal perturbation of a model σ3–P compound.
Moving beyond discrete structures such as 1 and C2v-1, a more general evaluation of the effect of nontrigonal distortion on the frontier orbital energies of σ3-P was undertaken. The PN3 framework of model tricoordinate phosphorous triamide P(NH2)3 was parameterized within Cs symmetry (Figure 5a) and a relaxed potential energy surface scan was conducted at the B3LYP/6–31G* level for two independent driving coordinates (bond angle θ and dihedral angle φ). At this level of theory, the overall electronic energy plot (Figure 5b) describes a minimum for P(NH2)3 that is nearly trigonal (θ=93°, φ=113°),51 with electronic energy increasing upon planarization (φ→180°) but varying only modestly for excursions of θ from equilibrium. Whereas HOMO (i.e. nP) energies largely correlate with the total electronic energy (Figure S14) in accord with predictions from Walsh’s rules, the plot of ELUMO (Figure 5c) shows a qualitative inversion in energetic ordering with respect to Etot. Specifically, structures close to the trigonal minimum for P(NH2)3 exhibit the energetically highest-lying LUMOs while the lowest-lying LUMO energies are found for planarized structures (φ→180°).
Figure 5.
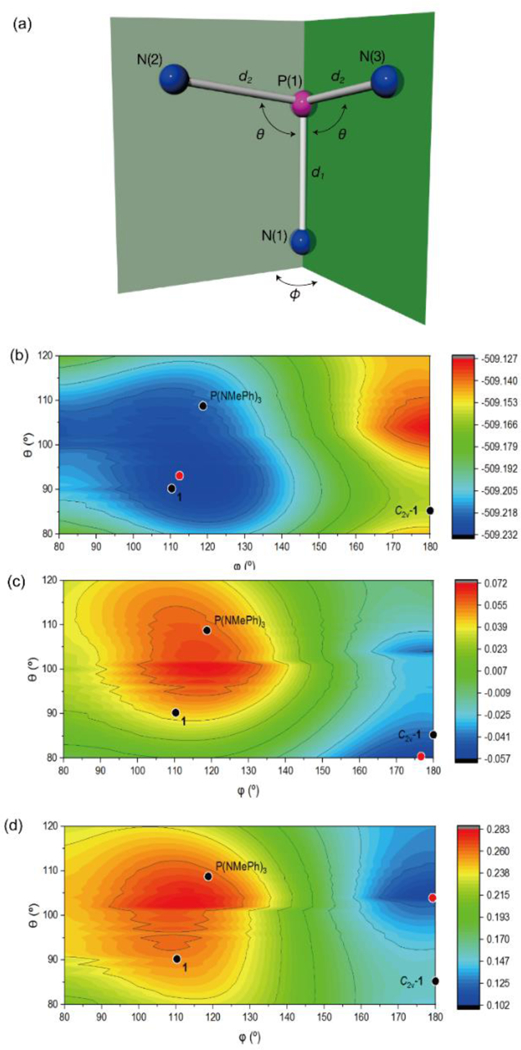
(a) Definition of geometrical parameters used to index the computational structures of P(NH2)3 within local Cs symmetry. Hydrogens are omitted for clarity. (b), (c), (d) Contour maps depicting the (b) total electronic energy (Etot), (c) LUMO energies (ELUMO), (d) ΔEHOMO/LUMO for P(NH2)3 structures with Cs-symmetry (N=4141 discrete input structures in the range 80° ≤ θ ≤ 120° and 80° ≤ φ ≤ 180° at 1° increments). Energies are shown in units of Hartrees. Points corresponding to the structures of P(NMePh)3, 1 and C2v-1 are superimposed as black points. The red points correspond to the absolute minimum.
Most decisively for the biphilic hypothesis of nontrigonal σ3-P, a plot of the frontier orbital energy gap ΔEHOMO/LUMO is given in Figure 5d. Here, trigonal structures maximize ΔEHOMO/LUMO. In terms of reactivity, this large gap in frontier orbital energies diminishes the propensity for biphilic reactivity, instead accentuating a dominant nucleophilicity from the HOMO lone pair. By complement, nontrigonal structures such as 1 and C2v–1 decrease energy gaps ΔEHOMO/LUMO consistent with an increased biphilicity for these species relative to compositionally related trigonal phosphorous triamides. The difference in ΔEHOMO/LUMO between 1 and C2v–1 conforms with the orbital analysis in Figure 2c and suggests an increase in biphilic reactivity of nontrigonal σ3–P through planarization. Indeed, given that the barrier to interconversion of 1 and C2v-1 is known to be low,32 it is plausible the reported biphilic reactivity of 1 is expressed through a preequilibrium deformation to C2v-1.
Finally, further inspection of Figure 5d shows that C2v-1 does not represent the minimum ΔEHOMO/LUMO within the surveyed range. Rather, a planar structure with slightly obtuse bond angles (i.e. θ=104°, φ=180°) defines the absolute minimum for ΔEHOMO/LUMO. Consequently, if one were able to access such a structure (presumably through appropriate molecular constraint), then the biphilicity of the phosphorus center would be predicted to be maximized within this local PN3 substitution pattern.
We conclude that contracted frontier orbital energy gaps and dense spatial orbital array are an inherent consequence of nontrigonal deformation for σ3-P compounds. This electronic feature allows for colocalized electron–donor and –acceptor behavior at a single phosphorus site that manifests the biphilic reactivity underpinning the insertion of σ3–P to E–H and M–H bonds. Our spectroscopic and theoretical results represent a rare example where a nontrigonal σ3-P compound is evaluated side-by-side with a compositionally-related trigonal analogue, allowing a direct quantitative comparison of their electronic structures in a way that illuminates aspects of their differential reactivity. These insights are now guiding our design of next-generation nontrigonal σ3-P ligands and biphilic catalysts.
Supplementary Material
Acknowledgements
ATR acknowledges support from NIH NIGMS (GM114547) for the synthesis of 2, NSF (CHE-1724505) for synthesis and characterization of 1•[H]2, and MIT. SRD acknowledges support from ACS PRF (55989-DNI3) for collection of the XANES data. JMK thanks Colgate University for computational resources. CMD would like to thank the U.S. Department of Education for a Graduate Assistance in Areas of National Need (GAANN) fellowship. Portions of this research were carried out at the Stanford Synchrotron Radiation Laboratory (SSRL), a national user facility supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under contract number DE-AC02–76SF00515. AT recognizes fellowship support from the Funai Foundation for Information Technology. We thank Mr. J. Connor Gilhula (MIT) for support with Python scripting, Mr. Nozomu Tanushi (Univ. of Tokyo) for figure editing, and Dr. Charlene Tsai (MIT) for assistance with crystallographic data collection.
Footnotes
Supporting information for this article is given via a link at the end of the document
References
- 1.Rowley AG, Organophosphorus Reagents in Organic Synthesis. Cadogan, J. I. G., Ed. Academic Press: London, 1979. [Google Scholar]
- 2.(a) Methot JL, Roush WR, Adv. Synth. Catal. 2004, 346, 1035. [Google Scholar]; (b) Ye L-W, Zhou J, Tang Y, Chem. Soc. Rev. 2008, 37, 1140. [DOI] [PubMed] [Google Scholar]; (c) Lu X, Zhang C, Xu Z, Acc. Chem. Res. 2001, 34, 535. [DOI] [PubMed] [Google Scholar]; (d) Fan YC, Kwon O, Chem. Commun. 2013, 49, 11588–11619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stephan DW, J. Am. Chem. Soc. 2015, 137, 10018–10032. [DOI] [PubMed] [Google Scholar]
- 4.(a) Kamer PCJ, van Leeuwen PWNM, Phosphorus(III) Ligands in Homogeneous Catalysis: Design and Synthesis; Wiley: Hoboken, N.J, 2012. [Google Scholar]; (b) Grabulosa A, P-Stereogenic Ligands in Enantioselective Synthesis; RSC Publishing: Cambridge, 2011. [Google Scholar]
- 5.van Berkel SS, van Eldijk MB, van Hest JCM, Angew. Chem. Int. Ed. 2011, 50, 8806–8827. [DOI] [PubMed] [Google Scholar]
- 6.Denmark SE, Beutner GL, Angew. Chem., Int. Ed. 2008, 47, 1560–1638. [DOI] [PubMed] [Google Scholar]
- 7.Gilheany DG, Chem. Rev. 1994, 94 (5), 1339–1374. [DOI] [PubMed] [Google Scholar]
- 8.Dillon KB, Chem. Rev. 1994, 94 (5), 1441–1456. [Google Scholar]
- 9.Dillon KB, Platt AWG, Schmidpeter A, Zwaschka F, Sheldrick WSZ, Anorg. Allg. Chem. 1982, 488, 7–26. [Google Scholar]
- 10.Burford N, Ragogna PJ, Coordination Chemistry of Phosphorus(III) as a Lewis Acceptor In Modern Aspects in Main Group Chemistry. Lattman M, Kemp RA, Eds. American Chemical Society Press: Washington D.C., 2006. [Google Scholar]
- 11.Holmes RR, J. Phys. Chem. 1960, 64, 1295. [Google Scholar]
- 12.(a) Wang Y, Xie Y, Wei P, King RB, Schaefer I, Schleyer P. v. R., Robinson GH, J. Am. Chem. Soc. 2008, 130, 14970. [DOI] [PubMed] [Google Scholar]; (b) Wang Y, Robinson GH, Inorg. Chem. 2011, 50, 12326. [DOI] [PubMed] [Google Scholar]
- 13.Schmiedekamp A, Skaarup S, Pulay P, Boggs JE, J. Chem. Phys. 1977, 66, 5769–5776. [Google Scholar]
- 14.Burdett JK, Molecular Shapes: Theoretical Models of Inorganic Stereochemistry; Wiley: New York, 1980. [Google Scholar]
- 15.Marynick DS, J. Chem. Phys. 1980, 73, 3939–3943. [Google Scholar]
- 16.Arduengo AJ, Dixon DA, Roe DC, J. Am. Chem. Soc. 1986, 108, 6821–6823. [DOI] [PubMed] [Google Scholar]
- 17.Dixon DA, Arduengo AJ, Fukunaga T, J. Am. Chem. Soc. 1986, 108, 2461–2462. [DOI] [PubMed] [Google Scholar]
- 18.Dixon DA, Arduengo AJ, J. Am. Chem. Soc. 1987, 109, 338–341. [Google Scholar]
- 19.Dixon DA, Arduengo AJ, J. Chem. Soc. Chem. Commun. 1987, 498–500.Dixon, [Google Scholar]
- 20.Dixon DA, Arduengo AJ, Lappert MF, Heteroatom Chem. 1991, 2, 541–544. [Google Scholar]
- 21.Schwerdtfeger P, Laakkonen LJ, Pyykkö P, J. Chem. Phys. 1992, 96, 6807–6819. [Google Scholar]
- 22.Pelzer S, Wichmann K, Wesendrup R, Schwerdtfeger P, J. Phys. Chem. A. 2002, 106, 6387–6394. [Google Scholar]
- 23.Varga Z, Verma P, Truhlar DG, J. Phys. Chem. A. 2019, 123, 301–312. [DOI] [PubMed] [Google Scholar]
- 24.Kirby AJ; Warren SG The Organic Chemistry of Phosphorus; Elsevier: Amsterdam, 1967, p 20. [Google Scholar]
- 25.Arduengo AJ, Stewart CA, Chem. Rev. 1994, 94, 1215–1237. [Google Scholar]
- 26.Robinson TP, De Rosa DM, Aldridge S, Goicoechea Angew JM. Chem., Int. Ed. 2015, 54, 13758–13763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Robinson TP, Lo S-K, De Rosa DM, Aldridge S, Goicoechea JM, Chem. Eur. J. 2016, 22, 15712–15724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tanushi A, Radosevich AT, J. Am. Chem. Soc. 2018, 140, 8114–8118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wolf R, Pure App. Chem. 1980, 52, 1141–1150. [Google Scholar]
- 30.Dunn NL, Ha M, Radosevich AT, J. Am. Chem. Soc. 2012, 134, 11330–11333. [DOI] [PubMed] [Google Scholar]
- 31.McCarthy SM, Lin YC, Devarajan D, Chang JW, Yennawar HP, Rioux RM, Ess DH, Radosevich AT, J. Am. Chem. Soc. 2014, 136, 4640–4650. [DOI] [PubMed] [Google Scholar]
- 32.Zhao W, McCarthy SM, Lai TY, Yennawar HP, Radosevich AT, J. Am. Chem. Soc. 2014, 136, 17634–17644. [DOI] [PubMed] [Google Scholar]
- 33.Lin YC, Hatzakis E, McCarthy SM, Reichl KD, Lai TY, Yennawar HP, Radosevich AT, J. Am. Chem. Soc. 2017, 139, 6008–6016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pistner AJ, Moon HW, Silakov A, Yennawar HP, Radosevich AT, Inorg. Chem. 2017, 56, 8661–8668. [DOI] [PubMed] [Google Scholar]
- 35.Lin Y-C, Gilhula JC, Radosevich AT, Chem. Sci. 2018, 9, 4338–4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Donahue CM, Daly SR, Comments Inorg. Chem. 2018, 38, 54–78. [Google Scholar]
- 37.Adhikari D, Mossin S, Basuli F, Huffman JC, Szilagyi RK, Meyer K, Mindiola DJ, J. Am. Chem. Soc. 2008, 130, 3676–3682. [DOI] [PubMed] [Google Scholar]
- 38.Harkins SB, Mankad NP, Miller AJM, Szilagyi RK, Peters JC, J. Am. Chem. Soc. 2008, 130, 3478–3485. [DOI] [PubMed] [Google Scholar]
- 39.Mankad NP, Antholine WE, Szilagyi RK, Peters JC, J. Am. Chem. Soc. 2009, 131, 3878–3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mossin S, Tran BL, Adhikari D, Pink M, Heinemann FW, Sutter J, Szilagyi RK, Meyer K, Mindiola DJ, J. Am. Chem. Soc. 2012, 134, 13651–13661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Donahue CM, McCollom SP, Forrest CM, Blake AV, Bellott BJ, Keith JM, Daly SR, Inorg. Chem. 2015, 54, 5646–5659. [DOI] [PubMed] [Google Scholar]
- 42.Lee K, Wei H, Blake AV, Donahue CM, Keith JM, Daly SR, Dalton Trans. 2016, 45, 9774–9785. [DOI] [PubMed] [Google Scholar]
- 43.Blake AV, Wei H, Lee K, Donahue CM, Keith JM, Daly SR, Eur. J. Inorg. Chem. 2018,, 2267–2276. [DOI] [PubMed] [Google Scholar]
- 44.Lee K, Wei H, Blake AV, Donahue CM, Keith JM, Daly SR, Inorg. Chem. 2018, 57, 10277–10286. [DOI] [PubMed] [Google Scholar]
- 45.Solomon EI, Hedman B, Hodgson KO, Dey A, Szilagyi RK, Coord. Chem. Rev. 2005, 249, 97–129. [Google Scholar]
- 46.MacMillan SN, Lancaster KM, ACS Catal. 2017, 7, 1776–1791. [Google Scholar]
- 47.See Supporting Information for experimental details, including synthesis, spectroscopic characterization, and X-ray crystallographic structure determination.
- 48.Martin RL, J. Chem. Phys. 2003, 118, 4775–4777. [Google Scholar]
- 49.Van Meer R, Gritsenko OV, Baerends EJ, Chem. Theory Comput. 2014, 10, 4432–4441 [DOI] [PubMed] [Google Scholar]
- 50.Stohr J, “NEXAFS Spectroscopy” Springer Series in Surface Science 25, (1992). [Google Scholar]
- 51.For a discussion of P(NR2)3 geometry, see: Mitzel NW, Smart BA, Dreihäupl K-H, Rankin DWH, Schmidbaur H, J. Am. Chem. Soc. 1996, 118, 12673–12682. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.