

Abstract
Background and Purpose
The δ‐opioid receptor is an emerging target for the management of chronic pain and depression. Biased signalling, the preferential activation of one signalling pathway over another downstream of δ‐receptors, may generate better therapeutic profiles. BMS 986187 is a positive allosteric modulator of δ‐receptors. Here, we ask if BMS 986187 can directly activate the receptor from an allosteric site, without an orthosteric ligand, and if a signalling bias is generated.
Experimental Approach
We used several clonal cell lines expressing δ‐receptors, to assess effects of BMS 986187 on events downstream of δ‐receptors by measuring G‐protein activation, β‐arrestin 2 recruitment, receptor phosphorylation, loss of surface receptor expression, ERK1/ERK2 phosphorylation, and receptor desensitization.
Key Results
BMS 986187 is a G protein biased allosteric agonist, relative to β‐arrestin 2 recruitment. Despite showing direct and potent G protein activation, BMS 986187 has a low potency to recruit β‐arrestin 2. This appears to reflect the inability of BMS 986187 to elicit any significant receptor phosphorylation, consistent with low receptor internalization and a slower onset of desensitization, compared with the full agonist SNC80.
Conclusions and Implications
This is the first evidence of biased agonism mediated through direct binding to an allosteric site on an opioid receptor, without a ligand at the orthosteric site. Our data suggest that agonists targeting δ‐receptors, or indeed any GPCR, through allosteric sites may be a novel way to promote signalling bias and thereby potentially produce a more specific pharmacology than can be observed by activation via the orthosteric site.
What is already known
The δ‐receptor is a potential drug target for the management of pain and depression.
BMS 986197 is a positive allosteric modulator of the δ‐receptor.
What this study adds
BMS 986187 directly activates δ‐receptors via an allosteric site.
The direct δ‐receptor agonism of BMS 986187 is biased for G‐protein activation over β‐arrestin recruitment.
What is the clinical significance
δ‐receptor orthosteric agonists can be proconvulsant.
Targeting δ‐receptors with a G‐protein‐biased allosteric agonist may be a potentially safer therapeutic strategy.
Abbreviations
- NTI
naltrindole
- PAM
positive allosteric modulator
1. INTRODUCTION
Chronic pain and depression are two of the most common medical ailments experienced worldwide and are often co‐morbid. For example, an estimated 25% of the United States population (75 million people) experience moderate‐to‐severe chronic pain (Reinke, 2014), whereas an estimated 15–20% experience depression (Kessler & Bromet, 2013). Opioid analgesics that target the μ ‐opioid receptor are the most widely prescribed drugs for both chronic and acute pain but suffer from serious side effects including respiratory depression and abuse liability (McNicol et al., 2017; Przewłocki & Przewłocka, 2001). Treatments for depression are varied, but under the best circumstances, only an estimated 50% of patients show full remission (Rush et al., 2006). Mounting evidence suggests that agonists targeting the δ‐opioid receptor, a GPCR, are effective in preclinical models of chronic pain and depression and could provide for new therapies (Bie & Pan, 2007; Cahill, Holdridge, & Morinville, 2007; Jutkiewicz, Kaminsky, Rice, Traynor, & Woods, 2005; Kabli & Cahill, 2007; Saitoh & Yamada, 2012).
The development of δ‐receptor agonists, such as BW373U86, SNC80, and related compounds, as medications has been limited due to on‐target side effects, namely, a propensity to cause convulsions and the rapid development of tolerance in both rodent and non‐human primate models (Danielsson et al., 2006; Jutkiewicz et al., 2005; Lutz & Brigitte, 2014; Pradhan, Smith, Kieffer, & Evans, 2012). Until recently, all compounds developed as δ‐receptor agonists targeted the orthosteric site on the receptor. However, the discovery of allosteric modulators that act at δ‐receptors, in particular BMS 986187 (Burford, Livingston, et al., 2015), presents an opportunity to interrogate this receptor in a novel way. An allosteric modulator is a compound that binds to a site on a GPCR other than the orthosteric site, where the endogenous ligand binds, and by doing so modulates the affinity and/or efficacy of an orthosteric ligand. Allosteric modulators can be positive (PAM), negative (NAM), or silent with regard to their effect on orthosteric ligands (Conn, Christopoulos, & Lindsley, 2010; Keov, Sexton, & Christopoulos, 2011). Modulators may also possess direct intrinsic pharmacological activity themselves. Such compounds are commonly referred to as “ago‐PAMs” or “ago‐NAMs” depending on the nature of this activity (Kenakin, 2007; Langmead & Christopoulos, 2006).
One potential benefit of allosteric modulators is to engender biased agonism or functional selectivity (Kenakin & Christopoulos, 2013). Biased agonism is the preferential activation, or inhibition, of certain downstream signalling cascades over others, classically G‐protein activation over β‐arrestin recruitment (Kenakin & Christopoulos, 2013; Kenakin, Watson, Muniz‐Medina, Christopoulos, & Novick, 2012; Schmid et al., 2017; Whalen, Rajagopal, & Lefkowitz, 2011). Thus, in theory, a drug could promote downstream effectors associated with beneficial actions while bypassing the effectors associated with the unwanted effects. Multiple studies have suggested biased agonism stemming from orthosteric activation of the δ‐receptors, although pertinent rigorous bias calculations are rarely performed (Audet et al., 2008; see Pradhan et al., 2012). Evidence suggests that β‐arrestin 2‐mediated internalization of the δ‐receptors might be associated with some of the negative effects of δ‐receptor agonists. Indeed, a number of ligands that fail to internalize the δ‐receptors, such as ARM390, despite potent G‐protein activation, have shown reduced tolerance (Pradhan et al., 2009, 2010, 2012) and reduced propensity to cause convulsions (Pradhan, Befort, Nozaki, Gavériaux‐Ruff, & Kieffer, 2011) in animal models. This suggests an agonist that preferentially activates G‐protein over β‐arrestin 2 recruitment may have reduced on‐target side effects that have limited the utility of other δ‐receptor ligands, such as SNC80. However, this does conflict with recent experiments in β‐arrestin knockout mice, suggesting that β‐arrestin 2 recruitment does not contribute significantly to the onset of convulsions (Dripps et al., 2017).
To date, no studies have examined the role played by allosteric modulation in functional selectivity at the δ‐receptors. BMS 986187 shows probe dependence at the δ‐receptors, and our prior work suggests it may be a directly acting allosteric agonist, as shown by its ability to inhibit AC in the absence of an orthosteric agonist (Burford, Livingston, et al., 2015; Burford, Traynor, & Alt, 2015). To this end, we set out to elucidate the nature of this ago‐PAM activity. We found that BMS 986197 was an allosteric agonist with biased signalling towards G‐protein pathways over the recruitment of β‐arrestin 2.
2. METHODS
2.1. Animals
All animal care and experimental procedures complied with the US National Research Council's Guide for the Care and Use of Laboratory Animals (National Research Council, 1996). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010) and with the recommendations made by the British Journal of Pharmacology. Male mice were used for all experiments. C57BL/6N (RRID:MGI:5659255) mice were obtained from Envigo (formerly Harlan, Indianapolis, IN, USA). The Oprd1 tm1Kff/J mouse strain (Oprd1tm1Kff/J, RRID:IMSR_JAX:007557) was obtained from The Jackson Laboratory (Bar Harbor, ME, USA; https://www.jax.org/strain/007557; Filliol et al., 2000). Mice were group housed with a maximum of five animals per cage in clear polypropylene cages with corn cob bedding and nestlets as enrichment. For breeding of the Oprd1 tm1Kff/J mice, heterozygote pairs were employed. Mice had free access to food and water at all times. Animals were housed in pathogen‐free rooms maintained between 68°F and 79°F and humidity between 30% and 70% humidity with a 12‐hr light/dark cycle with lights on at 07:00 a.m.
2.2. Cell lines
HEK (HEK293, RRID:CVCL_0045) cells stably expressing a tTA‐dependent luciferase reporter and a β‐arrestin 2‐TEV fusion gene (HTLA cells; Thermo Fisher Scientific) were maintained in DMEM supplemented with 10% FBS, 1% penicillin, and 100 μg·ml−1 streptomycin, 2 μg·ml−1 puromycin, and 100 μg·ml−1 hygromycin B at 37°C and 5% CO2. HEK293 (ATCC Cat# CRL‐1573, RRID:CVCL_0045) cells expressing N‐terminally FLAG‐tagged human δ‐receptors (HEK‐hDOPr cells) were cultured in DMEM containing 10% FBS and 1% penicillin and streptomycin and maintained in 0.8 mg·ml−1 G418. The HEK‐hDOPr cells used for ERK1/ERK2 imaging studies were generated as previously described and maintained in DMEM supplemented with 10% FBS (Shiwarski, Darr, Telmer, Bruchez, & Puthenveedu, 2017). CHO (ATCC Cat# CCL‐61, RRID:CVCL_0214) cells stably expressing wild‐type human δ‐receptors (CHO‐hDOPr) were grown in DMEM containing 10% FBS and 1% penicillin and streptomycin and maintained in 0.4 mg·ml−1 G418 as previously described (Burford, Livingston, et al., 2015; Burford, Traynor, & Alt, 2015).
2.3. Preparation of membrane homogenates
Cells were harvested, and membrane homogenates were prepared as previously described (Clark, Harrison, Zhong, Neubig, & Traynor, 2003). Briefly, cells were washed with ice‐cold PBS, pH 7.4, and detached from plates by incubation in harvesting buffer (0.68 mM EDTA, 150 mM NaCl, and 20 mM HEPES at pH 7.4) and pelleted by centrifugation at 200x g for 3 min. Cells were resuspended in ice‐cold 50 mM Tris (pH 7.4), homogenized using a Tissue Tearor (Dremel; Mount Prospect, IL, USA), and centrifuged at 20,000x g at 4°C for 20 min. The pellet was then resuspended, homogenized, and centrifuged a second time. This final pellet was resuspended in ice‐cold 50 mM Tris (pH 7.4) and homogenized using a glass Dounce homogeniser to give a protein concentration of 0.5–1.5 mg·ml−1 and stored at −80°C. Protein concentration was determined using the bicinchoninic acid quantification method (BCA assay) with BSA serving as the standard.
For brain homogenates, mice (8 to 12 weeks of age) were killed by cervical dislocation. Whole‐brain tissue, from the optic chiasmus forward, was removed immediately and chilled in ice‐cold buffer (50 mM Tris base, pH 7.4). Membrane homogenates were prepared as previously described (Lester & Traynor, 2006). Final membrane pellets were resuspended in 50 mM Tris base, pH 7.4, aliquoted, and stored at −80°C. Protein content was determined using bicinchoninic acid assay with BSA as the standard.
2.4. Stimulation of GTPγ35S binding
Agonist stimulation of GTPγ35S binding was measured as described previously (Clark et al., 2003). Homogenates of HEK cells expressing FLAG‐tagged‐human δ‐receptors, CHO cells expressing wild‐type human δ‐receptors, or mouse brain (15–20 μg per well) were incubated in “GTPγS buffer” (50 mM Tris–HCl, 100 mM NaCl, and 5 mM MgCl2, pH 7.4) containing 0.1 nM GTPγ35S and 30 μM GDP and varying concentrations of BMS 986187 or δ‐receptor agonists for 1 hr in a shaking water bath at 25°C. The reaction was terminated by vacuum filtration through GF/C filters using a Brandel harvester and washed five times with ice‐cold GTPγS buffer. Filters were dried and, following the addition of EcoLume scintillation cocktail, counted in a Wallac 1450 MicroBeta Liquid Scintillation and Luminescence Counter (PerkinElmer). The level of GTPγ35S binding was expressed as fmol bound·mg−1 protein or by comparison with the full δ‐receptor agonist SNC80 at 10 μM to account for variability between membrane preparations.
2.5. Internalization of δ‐receptors
As described previously (Bradbury, Zelnik, & Traynor, 2009), FLAG‐tagged HEK‐hDOPr cells were plated at a density of 0.5 × 106 cells per well in poly‐d‐lysine‐coated, 24‐well plates. When cells reached 80% confluency, they were treated with vehicle (1% DMSO) or indicated drugs and rocked at room temperature for the indicated times. Cells were then washed three times with ice‐cold tris‐buffered saline (TBS) and fixed with 3.7% paraformaldehyde in TBS at room temperature for 15 min. After fixing, cells were washed three times with cold TBS and blocked at room temperature with 1% BSA in TBS for 60 min. Following block, cells were washed two times with TBS and incubated with FLAG M2‐Alkaline Phosphatase Antibody at a 1:625 dilution for 60 min. Cells were then washed five times with TBS and treated with p‐nitrophenylphosphate for 8 min. The reaction was stopped with 3 N NaOH, and 200 μl from each well was transferred to a 96‐well plate for reading at 405 nm on VERSAmax tunable microplate reader (Molecular Devices, Sunnyvale, CA, USA). The percentage of internalized receptors was determined as loss of surface receptors using the following equation: [1 − (Drug OD − Background OD∕Control OD − Background OD) × 100]. Background was determined as the absorbance of non‐transfected HEK cells, and control was the absorbance of cells incubated in the absence of drug.
2.6. β‐Arrestin 2 recruitment
2.6.1. Confocal microscopy
Recruitment of β‐arrestin 2 in FLAG‐tagged HEK‐DOPr cells was performed as described previously (Bradbury et al., 2009). Briefly, cells were seeded into 24‐well plates containing poly‐d‐lysine‐coated glass coverslips. Cells were transfected using Lipofectamine 2000 with 0.4 μg of β‐arrestin 2‐GFP cDNA and incubated for 48 hr, then treated with vehicle, 10 μM SNC80, or 10 μM BMS 986187 for 5 min. Following fixation with 3.7% paraformaldehyde, cells were incubated with M2 mouse anti‐FLAG primary antibody followed by AlexaFluor 594 goat anti‐mouse secondary antibody. Images were obtained using a NikonA1R confocal microscope and quantified using ImageJ software (National Institutes of Health; ImageJ, RRID:SCR_003070).
2.6.2. PRESTO‐TANGO arrestin recruitment
For the PRESTO‐TANGO assay, HTLA cells at 15,000 cells per well were transfected with plasmids (20 ng) encoding FLAG‐tagged hDOPr‐TANGO (OPRD1‐TANGO; Thermo Fisher Scientific) using Lipofectamine 2000 and plated in Greiner Bio‐One cell culture microplates. After 24 hr, cells were treated with the indicated drug at the indicated concentrations. After 48 hr, One‐Glo solution was added to each well, and luminescence was measured using a Pherastar plate reader (BMG Labtech, Germany). Data were normalized to per cent of standard full agonist (10 μM SNC80) to account for variability between assays in plating and transfection efficiency.
2.7. ERK1/ERK2 phosphorylation
HEK293 cells stably expressing FLAG‐tagged human δ‐receptors were transiently transfected with the ERK activity reporter cEKAR (Fritz et al., 2013). ERK activity in response to SNC80 and BMS 986187 was assessed as previously described (Weinberg, Zajac, Phan, Shiwarski, & Puthenveedu, 2017). Briefly, cells were plated at low density, allowed to grow for 2 days, and then serum starved for 4 hr. Cells were labelled with Alexa Fluor 647 antimouse M1 antibody for 10 min. Single‐cell fluorescence for cyan fluorescent protein (405‐nm excitation, 470/50 emission filter), FRET (405‐nm excitation, 530lp emission filter), and M1 (647‐nm excitation, 700/75 emission) was collected every 30 s for 22.5 min, with addition of drug (1 μM SNC80 or 10 μM BMS 986187) occurring after 2.5 min of no‐treatment baseline. The ratio of FRET to cyan fluorescence protein fluorescence was calculated for each cell on a frame‐by‐frame basis and normalized to the average ratio during baseline. For calculating total response, the mean AUC was taken for the vehicle condition, and that mean was subtracted from the individual AUC for each cell in the treatment conditions. Each experiment was conducted using the same batch of transiently transfected cells from the same stable cell line and passage number and carried out on the same day under all conditions (vehicle, SNC80, and BMS 986187) to ensure that any nonresponding cells were represented equally across treatment conditions.
2.8. Western blot for phospho‐Ser363
As described previously (Bradbury et al., 2009), HEK cells stably expressing FLAG‐tagged human δ‐receptors were plated at a density of 0.5 × 106 cells per well in poly‐d‐lysine‐coated, 24‐well plates, and experiments were performed when cells were at 80% confluency. Cells were treated with vehicle (1% DMSO), TAN‐67, DPDPE, SNC80, or BMS 986187 for 1 hr. Cells were then rinsed with PBS, and lysates were collected with RIPA (50 mM Tris, pH 7.4, 150 mM NaCl, 1% Triton X‐100, 1% sodium deoxycholic acid, and 0.1% sodium dodecyl sulfate [SDS]) plus protease inhibitor cocktail, 2 mM EDTA, 100 mM NaF, 100 mM phenylmethanesulfonyl fluoride, and 10 mM sodium orthovanadate. Lysates were then sonicated briefly and centrifuged at 10,000x g for 10 min. Equal amounts of protein samples were diluted in SDS sample buffer (62.5 mM Tris–HCl, pH 6.8, 2% SDS, 10% glycerol, and 0.0008% bromophenol blue) and β‐mercaptoethanol, loaded onto 10% polyacrylamide gels. Following transfer to nitrocellulose, membranes were blocked for 1 hr with 5% nonfat dried milk in PBS then incubated with 1:1,000 dilution of rabbit anti‐phosphorylated δ‐receptor antibody overnight at 4°C. Membranes were washed and incubated with 1:10,000 HRP–goat antirabbit IgG for 1 hr. To probe total FLAG‐tagged δ‐receptors, the membranes were stripped using mild stripping buffer (distilled water, pH 2.2, 1.5% glycine, 0.1% SDS, and 1% Tween 20), washed, then blocked with 5% nonfat dried milk for 1 hr. Following block, membranes were incubated with 1:1,000 mouse anti‐FLAG for 1 hr in 5% nonfat dried milk, TBS Tween containing 1 mM CaCl2. Membranes were washed and treated with 1:10,000 HRP–goat anti‐mouse IgG for 1 hr. Following washing, membranes were treated with 1:1 SuperSignal chemiluminescent substrate, and bands were detected using the EpiChemi3 darkroom (UVP, Upland, CA, USA). Band intensity was quantitated using ImageJ (National Institutes of Health) and normalized to total human δ‐receptors, as determined by FLAG staining, to account for any differences in total protein.
2.9. Receptor desensitization
Desensitization was determined by incubating CHO‐hDOPr cells with either vehicle or drug for indicated time periods at 37°C. Following incubation, cells were washed five times with PBS, and membranes were prepared as described above. For the time course of desensitization, maximum GTPγ35S binding was measured using 10 μM SNC80 in vehicle‐treated cells; drug treated conditions were expressed as per cent of this maximal binding. For concentration response, GTPγ35S binding elicited by SNC80 was measured in membranes pretreated with 500 nM SNC80, 10 μM BMS 986187, or vehicle for 30 min and expressed as fmol bound·mg−1 of protein.
2.10. Data and statistical analyses
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology. All in vitro assays were a mean of at least five separate preparations, except where stated, and each was run in duplicate or triplicate as shown in the relevant figure legend, to ensure the reliability of the single values. None of the in vitro biochemical experiments were performed or analysed in a blinded manner.
Data were graphed as individual experiments for analyses unless otherwise stated, and statistical analysis was performed using Graphpad Prism 6.5. Concentration–effect curves were analysed using a three‐parameter curve fit with Hill slopes set to 1.0. Maximal values were not constrained; minimum values were constrained to zero if contained in the 95% confidence intervals. For internalization, the GTPγ35S assay, and confocal microscopy, one‐way ANOVA was performed, and Tukey post hoc test for multiple comparisons was applied if F was significant. The desensitization time course was analysed by two‐way ANOVA. Bias calculations were performed as described by Kenakin (2017) as follows: For each ligand and respective response, individual experimental curves were used to calculate log(max/EC50). The difference in log(max/EC50) between arrestin recruitment and GTPγ35S, Δlog(max/EC50), was then calculated. Individual results were combined to give means ± SEM values shown in Table 1. Finally, the differences between the Δlog(max/EC50) values for the reference ligand (SNC 80) and test ligand were calculated to give a ΔΔlog(max/EC50) values, the antilog of which is the bias factor. For all analyses, significance was set at 5% (0.05 P value).
Table 1.
Summary of bias calculations
| Ligand | β‐Arrestin 2 | Bias (towards G‐protein) | GTPγ35S | ||||
|---|---|---|---|---|---|---|---|
| Max | EC50 (nM) | Arrestin/G‐protein fold EC50 shift | ΔΔlog(Max/EC50) | Bias factor | EC50 (nM) | Max | |
| SNC80 | 1 | 353 ± 141 | 18.6 | 0 | 1 | 19.0 ± 6 | 1 |
| BMS 986187 | 1a | 578,500 ± 419,100 | 1787 | 1.91 ± 0.7 | 81 | 323 ± 96 | 0.92 ± 0.03 |
| 0.5a | 238,179 ± 188,400 | 737 | 1.53 ± 0.82 | 34 | |||
| DPDPE | 1 | 16,100 ± 805 | 85 | 1.03 ± 0.67 | 11 | 189 ± 25 | 0.85 ± 0.09 |
Calculations were performed from the data generated in Figure 5, as described in Section 2 to determine ΔΔlog(max/EC50) (Kenakin, 2017).
In order to extrapolate an accurate EC50 value for BMS 986187‐mediated arrestin recruitment, the E max was constrained to 1, equivalent to the level of recruitment by SNC80 or 0.5, assuming the maximum value is that observed at the point of solubility (Figure 5b).
2.11. Materials
Guanosine‐5′‐O‐(3‐[35S]thio)triphosphate (GTPγ35S) and [3H]diprenorphine were from PerkinElmer Life and Analytical Sciences (Boston, MA, USA). BMS 986187 was synthesized and characterized as previously described (Burford, Livingston, et al., 2015; Burford, Traynor, & Alt, 2015). Naltrindole (NTI), naloxone, TAN‐67, DPDPE, SNC80, protease inhibitor cocktail, GDP, p‐nitrophenyl phosphate, M1 mouse anti‐FLAG antibody (Cat# F3040, RRID:AB_439712), and M2 mouse anti‐FLAG antibody conjugated to alkaline phosphatase (Cat# A9469, RRID:AB_439699) were from MilliporeSigma (St Louis, MO, USA). Goat anti‐rabbit (Cat# sc‐2004, RRID:AB_631746) or mouse (Cat# sc‐2005, RRID:AB_631736) antibodies conjugated to HRP were from Santa Cruz Biotechnology (Santa Cruz, CA, USA) and rabbit anti‐phospho‐Ser363‐ δ‐receptor antibody (Cat# 3461, RRID:AB_2768155) from Cell Signaling (Danvers, MA, USA). β‐Arrestin 2‐GFP cDNA was a gift from Marc Caron (Duke University, Durham, NC, USA). Poly‐D‐lysine‐coated 24‐well plates and poly‐D‐lysine‐coated 12‐mm, no. 1 coverslips were from BD Biosciences (San Jose, CA, USA). EcoLume scintillation cocktail and ultrapure formaldehyde were obtained from MP Biomedicals (Aurora, OH, USA) and Polysciences Inc. (Warrington, PA, USA) respectively. ProLong Gold antifade reagent, Alexa 594 goat anti‐mouse IgG (Thermo Fisher Scientific, Cat# A‐11032, RRID AB_2534091), and Lipofectamine 2000 were from Invitrogen (Carlsbad, CA, USA). One‐Glo solution was purchased from Promega (Madison, WI, USA). SuperSignal chemiluminescent substrate was purchased from Thermo Fisher (Waltham, MA, USA).
2.12. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017).
3. RESULTS
3.1. BMS 986187 stimulates binding of GTPγ35S by δ‐receptors
BMS 986187 has been shown to produce inhibition of forskolin‐stimulated cAMP production in the absence of orthosteric ligand, demonstrating that it has direct agonist action via an allosteric site (Burford, Livingston, et al., 2015; Burford, Traynor, & Alt, 2015). However, inhibition of AC is a highly amplified signalling output and requires low efficacy in a compound, although it does require prior stimulation of heterotrimeric Gαi/o proteins. To demonstrate that BMS 986187 can directly stimulate δ‐receptors to activate Gαi/o, we performed GTPγ35S‐binding assays as previously described (Traynor & Nahorski, 1995) in HEK293 cells expressing human‐δ‐receptors (Figure 1a). BMS 986187 stimulated GTPγ35S binding in a concentration‐dependent manner, giving a potency value (EC50) of 301 ± 85 nM. significantly less potent than SNC80 (19 ± 11nM). In brain homogenates from C57/BL6 mice, the δ‐receptor full agonist SNC80 produced GTPγ35S binding with an EC50 of 203 ± 31 nM (Figure 1b). BMS 986187 also stimulated GTPγ35S binding with a significantly lower potency (EC50 of 1681 ± 244 nM), but the maximal GTPγ35S response to BMS 986187 was 38% greater than that produced by SNC80, although this difference did not reach significance. To confirm the response to BMS 986187 was due to activation of δ‐receptors, we repeated the experiments in brain tissue from δ‐receptor knockout mice. In brain homogenates from these mice, a small degree of BMS 986187‐stimulated GTPγ35S binding remained, representing 20% of the BMS 986187 response observed in tissue from wild‐type mice, with an EC50 of 600 ± 397 nM. In contrast, SNC80 produced no appreciable binding over baseline in δ‐receptor knockout mice. These findings suggest that BMS 986187 activates G‐protein through the δ‐receptors at physiological receptor expression levels while also eliciting a very low level of G‐protein activation through a non‐δ‐receptor‐mediated pathway.
Figure 1.
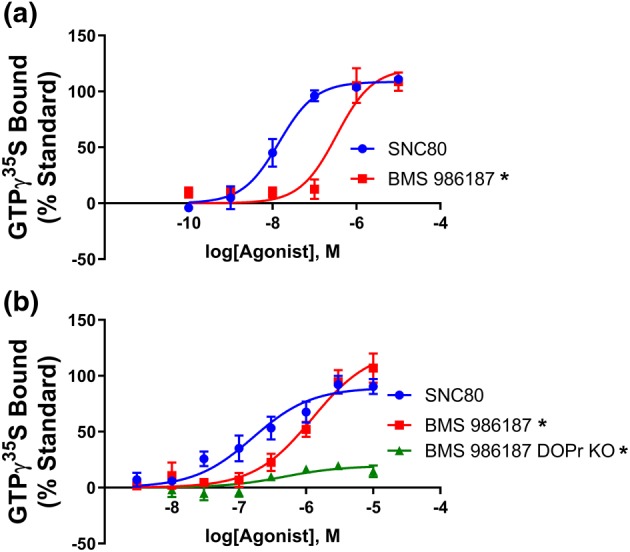
BMS 986187 elicits G‐protein activation. The capacity for increasing concentrations of BMS 9861897 and SNC80 to increase GTPγ35S binding was measured in (a) membranes from FLAG‐tagged HEK‐hDOPr cells or in (b) brain homogenates from wild‐type or δ‐receptor knockout (KO) mice. Data are presented as percentage of the response to a maximal concentration (10 μM) of SNC80. All plotted points are the means ± SEM of five independent experiments, each run in duplicate. *P<0.05, BMS 986187 concentration‐response curves significantly different from SNC80 concentration‐response curves, as determined by one‐ (A) or two‐ (B) way ANOVA with Dunnett's post hoc test
3.2. BMS 986187 stimulates GTPγ35S binding through an allosteric site on δ‐receptors
Previous work has shown that BMS 986187 does not displace the antagonist 3H‐diprenorphine bound to the orthosteric site (Burford, Livingston, et al., 2015; Burford, Traynor, & Alt, 2015). To confirm that the agonist action of BMS 986187 was not due to interaction at the orthosteric site, GTPγ35S binding was performed in membranes from HEK‐hDOPr cells in the presence or absence of various concentrations of orthosteric antagonists. The δ‐receptor antagonist NTI (100 nM) reduced the maximal GTPγ35S binding evoked by 10 μM BMS 986187 from 99 ± 6% to 51 ± 17% (Figure 2a). Increasing the concentration of NTI by 100‐fold (to 10 μM) caused no additional inhibition of BMS 986187‐stimulated GTPγ35S binding (50 ± 4%; Figure 2a). NTI (10 μM) alone failed to produce any appreciable stimulation of GTPγ35S binding, consistent with its classification as a neutral antagonist (Tryoen‐Toth et al., 2005). The partial loss of BMS 986187‐stimulated GTPγ35S binding was also observed in the presence of 10 μM of the non‐specific opioid antagonist naloxone. Using CHO‐hDOPr cells as an alternative cell line, NTI showed a concentration‐dependent, but saturable, inhibition of BMS 986187 stimulation of GTPγ35S binding with the lack of parallel shifts confirming the agonist action of the modulator is not due to competition at the orthosteric site but rather due to negative cooperativity between the orthosteric and allosteric sites. To further confirm this, we evaluated the effect of increasing concentrations of NTI on the maximal stimulation elicited by BMS 986187 and SNC80 using membranes from CHO‐hDOPr cells (Figure 2c). NTI showed a saturable inhibition of BMS 986187 stimulation of GTPγ35S binding. In contrast, SNC80‐mediated stimulation of GTPγ35S binding was fully inhibited by NTI, consistent with a competitive mechanism.
Figure 2.
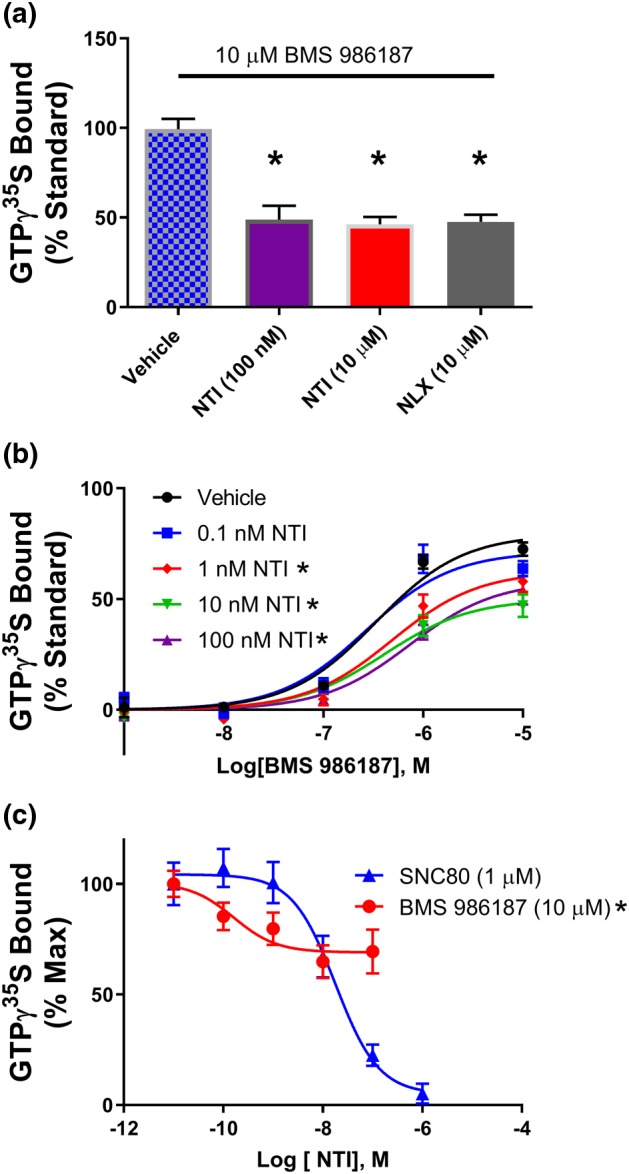
Antagonists show non‐competitive interaction with BMS 986187. (a) Antagonists naltrindole (NTI) and naloxone (NLX) reduce maximal GTPγ35S binding caused by BMS 986187 in FLAG‐tagged HEK‐hDOPr cells, normalized to per cent of effect of 10 μM SNC80 to control for variation between the different preparations. *P<0.05, significantly different from BMS 986187 with vehicle, as determined by one‐way ANOVA with Tukey's post hoc test. (b) GTPγ35S concentration response curves for BMS 986187 with increasing NTI concentrations. *P<0.05, significantly different from BMS 986187 + vehicle as determined by two‐way ANOVA with Tukey's post hoc test. (c) GTPγ35S binding concentration response of NTI with fixed concentration BMS 986187 (10 μM) or SNC80 (1 μM), in CHO cells expressing human δ‐receptors. *P<0.05, significantly different from SNC80 as determined by two‐way ANOVA with Tukey's post hoc test. All plotted points are the mean ± SEM of (b, c) 5 or (a) 10 individual experiments, each performed in duplicate
3.3. BMS 986187 causes a low level of δ‐receptor internalization
Previous studies of the δ‐receptors suggest that ligands with high efficacy at activating G‐protein while maintaining low efficacy at promoting receptor internalization show reduced tolerance in animal models (Pradhan et al., 2009). To this end, we next sought to determine whether BMS 986187 would cause internalization of δ‐receptors relative to the orthosteric partial agonists TAN‐67 and DPDPE and the full agonist SNC80. Preliminary studies indicated that 10 μM would be a maximal effect for all ligands, and from initial time‐course studies (Figure 3a), we chose 1 hr to evaluate and compare the ligands. Due to the small effect window, data were pooled for analysis to provide maximum and EC50 values with 95% confidence intervals. BMS 986187 treatment resulted in low levels of internalization (7 [3.9–10.0] %) relative to TAN‐67 (11 [5.2–16.2] %) < DPDPE (31 [17.7–44.1] %) and SNC80 (33 [24.7–40.9] %) with a potency order TAN‐67 (1.3 [0.15–11.0] nM) = SNC80 (3.7 [1.5–9.2] nM) > BMS968187 (94 [59–1007] nM) = DPDPE (212 [72–623] nM).
Figure 3.
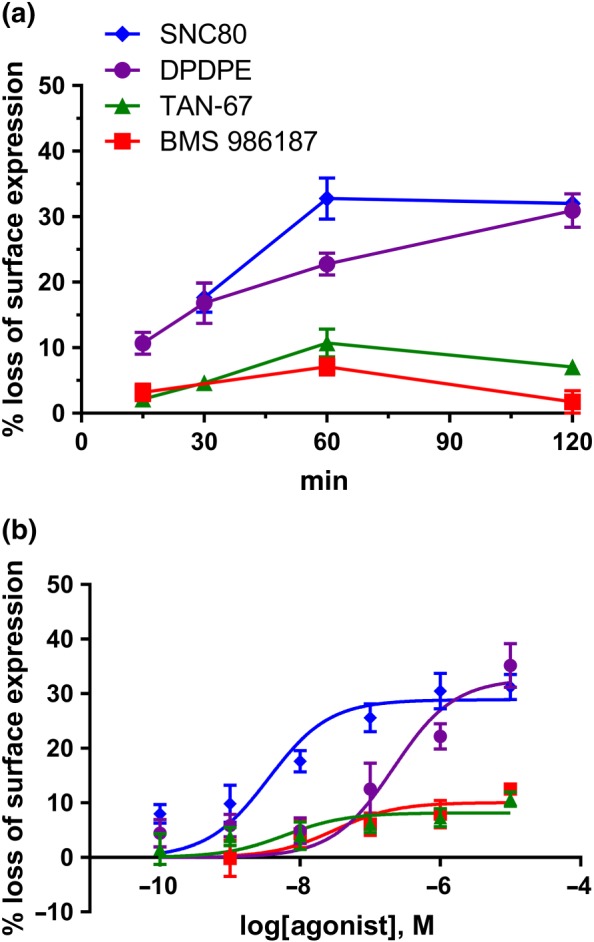
Internalization of δ‐receptors. Receptor internalization by δ‐receptor ligands in FLAG‐tagged HEK‐hDOPr cells. (a) Preliminary time‐course studies (means ± SEM, n = 3 experiments in triplicate) with 10 μM concentrations of ligands were to identify an appropriate time (1 hr) to determine (b) concentration–response studies for the different δ‐receptor ligands (means ± SEM, n = 5 experiments in triplicate). The symbols in (a) also refer to (b)
To contrast the propensity of BMS 986187 to cause internalization with its ability to activate G‐protein at δ‐receptors, we measured the maximal GTPγ35S binding by BMS 986187 and compared this with saturating concentrations (10 μM) of the partial agonist peptide DPDPE, the partial agonist TAN‐67, and the full agonist SNC80, which was used as the standard. As shown in Figure 4b, BMS 986187 elicited 99 ± 6% of GTPγ35S binding relative to SNC80 versus 63 ± 7% and 75 ± 6% for TAN‐67 and DPDPE respectively. Thus, BMS 986187 gives a greater level of G‐protein activation than DPDPE but a reduced level of internalization. Additionally, BMS 986187 affords a level of internalization similar to that after TAN‐67, but stimulates a higher level of GTPγ35S binding.
Figure 4.
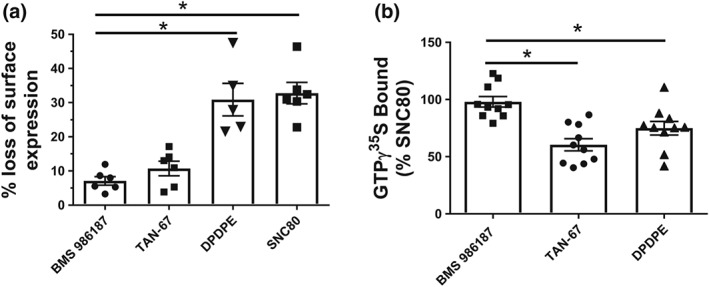
BMS 986187 shows biased activation of GTPγ35S relative to receptor internalization. δ‐receptor ligands were evaluated in FLAG‐tagged HEK‐hDOPr cells by measuring (a) receptor internalization or (b) stimulation of GTPγ35S bound. BMS 986187 showed significantly lower internalization relative to DPDPE and SNC80, despite showing significantly greater GTPγ35S binding relative to TAN‐67 and DPDPE. GTPγ35S binding is normalized as per cent of 10 μM SNC80 to control for variation between the different preparations. All experiments were performed using saturating concentrations of compounds (10 μM). Data are expressed as mean ± SEM of 5 (internalization) or 10 (GTPγ35S) individual experiments, each performed in duplicate. *P<0.05, significantly different from BMS 986187 as determined by one‐way ANOVA with Tukey's post hoc test
3.4. BMS 986187 is G‐protein‐biased, relative to β‐arrestin 2 recruitment
The fact that BMS 986187 affords greater GTPγ35S stimulation that DPDPE and TAN‐67 while causing a low level of δ‐receptor internalization is indicative of ligand bias. Internalization of receptors is largely β‐arrestin dependent. In order to explore this further, we directly compared concentration responses for BMS 986187 and SNC80 to recruit β‐arrestin 2 to δ‐receptors and stimulate GTPγ35S binding. BMS 986187 and SNC80 stimulated a similar level of GTPγ35S binding in HEK‐hDOPr cells (Figure 5b), in agreement with our previous result, (Figure 1a) although BMS 986187 was less potent (Table 1). In contrast, using the PRESTO‐TANGO assay, BMS 986187 recruited β‐arrestin 2 very weakly up to 100 μM, the limit of solubility (Figure 5a). Extrapolation of the BMS 986187 concentration–response curve assuming a similar maximum to SNC80 afforded an EC50 for BMS 986187 of 579 μM. In comparison, TAN‐67 and DPDPE recruited similar levels of β‐arrestin 2 as SNC80, although DPDPE (16.1 ± 8.0 μM) was much less potent than TAN‐67 (327 ± 176 nM) or SNC80 (353 ± 141 nM). From these data, the relative bias of BMS 986187 for GTPγ35S stimulation over β‐arrestin 2 recruitment was evaluated with SNC80 serving as a reference agonist using the log(max/EC50) function as described by Kenakin (2017; Table 1). This shows BMS 986187 is G‐protein biased, relative to β‐arrestin 2, when compared with SNC80, with a bias factor of 82 (Table 1). It should be noted that the PRESTO‐TANGO assay employs a chimeric δ‐receptor with a vasopressin receptor tail, although the effect of this modification should be eliminated by using SNC80 as a reference ligand (Kenakin, 2017). Utilizing this assay, we found DPDPE to be biased towards G‐protein compared with SNC80 with a calculated bias factor of 11 (Table 1). This is similar to the bias of DPDPE compared with SNC80 of 6, calculated using the same equation from data in Chiang, Sansuk, and van Rijn (2016), who employed a complementation assay for β‐arrestin recruitment in CHO cells. Moreover, the bias of BMS 986187 fits with the change in ligand order when comparing internalization with GTPγ35S binding (Figure 4), and the PRESTO‐TANGO assay has been previously used in studies of receptor bias (Che et al., 2018).
Figure 5.
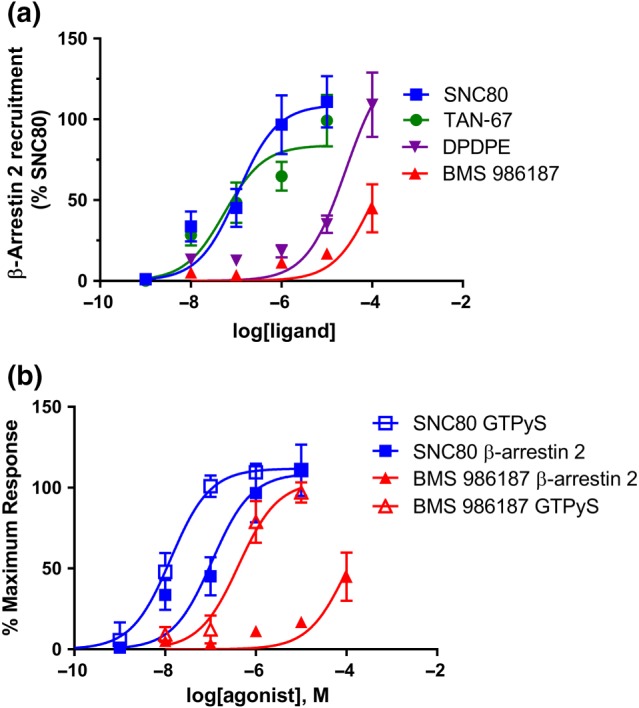
BMS 986187 is G‐protein biased over β‐arrestin 2. BMS 986187 bias was evaluated between (a) β‐arrestin 2 recruitment and (b) GTPγ35S binding relative to the standard orthosteric agonist SNC80. Normalization was performed to control for sources of variation across preparation and to allow for comparison across both assays. β‐Arrestin assays were performed in HTLA cells transiently transfected with hDOR‐TANGO and GTPγ35S assays in membranes from FLAG‐tagged HEK‐hDOPr cells, as described in Methods . Data are presented as the mean of five (GTPγ35S) or seven (β‐arrestin 2) independent experiments, each performed in duplicate and expressed as mean ± SEM
To confirm the low degree of β‐arrestin 2 recruitment to δ‐receptors by BMS 986187, we performed confocal microscopy in FLAG‐tagged HEK‐DOPr cells transfected with 0.4 μg β‐arrestin 2 GFP as shown in Figure 6. Cells were incubated for 5 min with 10 μM of either SNC80 or BMS 986187, a saturating concentration for G‐protein activation. β‐Arrestin 2 localization was then quantified as a ratio of fluorescent intensity at the cell membrane divided by the cytoplasmic intensity using line scan analysis. Consistent with the findings in Figure 5b and published literature (Chiang et al., 2016), SNC80 afforded significant translocation of β‐arrestin 2 to the plasma membrane, whereas localization in cells treated with BMS 986187 was not significantly different from baseline. This suggests the maximal β‐arrestin 2 recruitment in response to BMS 986187 is much less than recruited by SNC80, so we recalculated the bias factor assuming the 100 μM data point in Figure 5a (the point of solubility) was the maximal effect; this calculation yielded a bias factor of 34 (Table 1).
Figure 6.
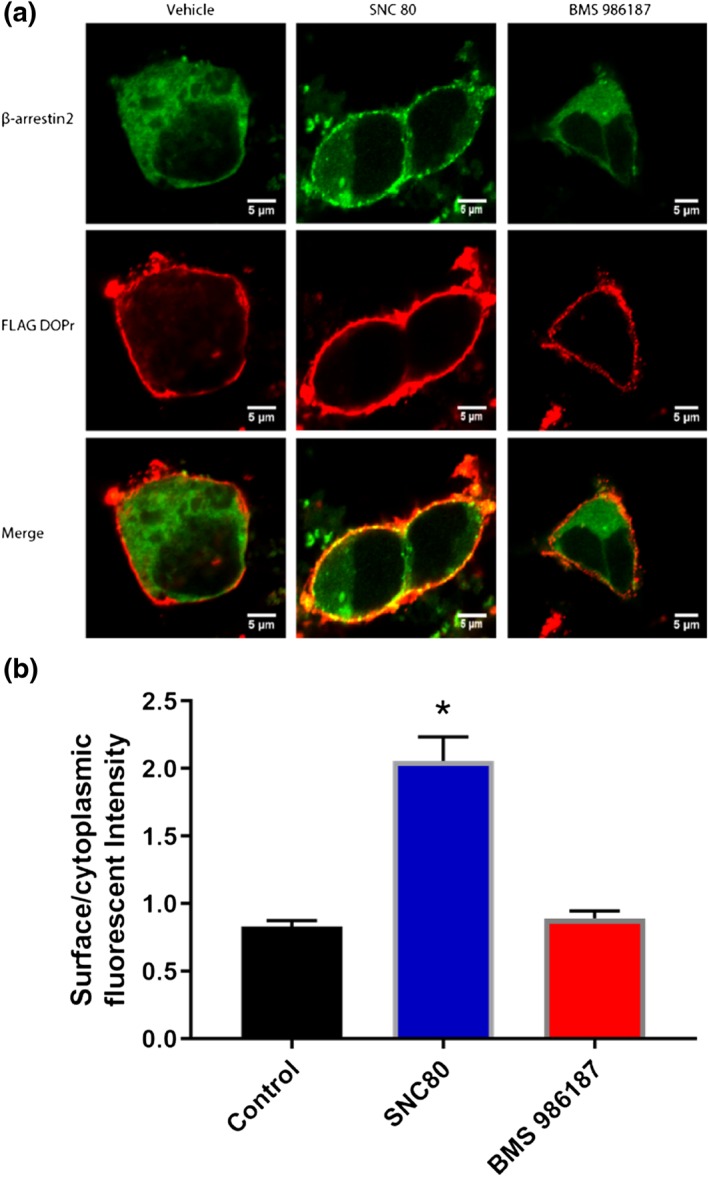
Effect of SNC80 and BMS 986187 on β‐arrestin 2 recruitment. FLAG‐tagged HEK‐hDOPr cells were transfected with 0.4 μg β‐arrestin 2 GFP and treated with 10 μM of either SNC80 or BMS 986187 for 5 min and imaged using confocal microscopy. FLAG‐tagged δ‐receptors are represented in the red channel with the green channel representing β‐arrestin 2. (a) Representative images and (b) β‐arrestin 2 recruitment expressed as surface/cytoplasmic GFP intensity. Data represent the means ± SEM of 34 cells per condition from five independent drug treatments. *P<0.05, significantly different from control and BMS 986187 as determined by one‐way ANOVA with Tukey's post hoc test
3.5. BMS 986187 shows low levels of ERK1/ERK2 activation
Agonists at opioid receptors have been shown to signal through ERK1/ERK2 via both G‐protein‐ and β‐arrestin‐mediated pathways, and previous work has shown that BMS 986187 acting as a PAM can increase the potency of orthosteric δ‐receptor agonists in promoting ERK1/ERK2 phosphorylation (Burford, Livingston, et al., 2015; Burford, Traynor, & Alt, 2015). However, BMS 986187 (10 μM) alone failed to elicit significant ERK1/ERK2 phosphorylation relative to vehicle in HEK‐hDOPr cells, whereas SCN80 (1 μM) afforded robust phosphorylation of ERK1/ERK2 (Figure 7). As expected, we observed SNC80‐promoted δ‐receptor internalization (Figure 7a) and we also saw internalization at later time points in approximately 50% of cells treated with BMS 986187 (Figure 7a). A similar percentage of vehicle‐treated cells showed δ‐receptor internalization, although to a lesser degree than BMS 986187, suggesting that the modulator is promoting constitutive internalization of δ‐receptors in these cells (Ong, Xue, Olmstead, & Cahill, 2015; Trapaidze, Gomes, Bansinath, & Devi, 2000). This agrees with the BMS 986187‐induced enhancement of δ‐receptor internalization shown in Figure 3.
Figure 7.
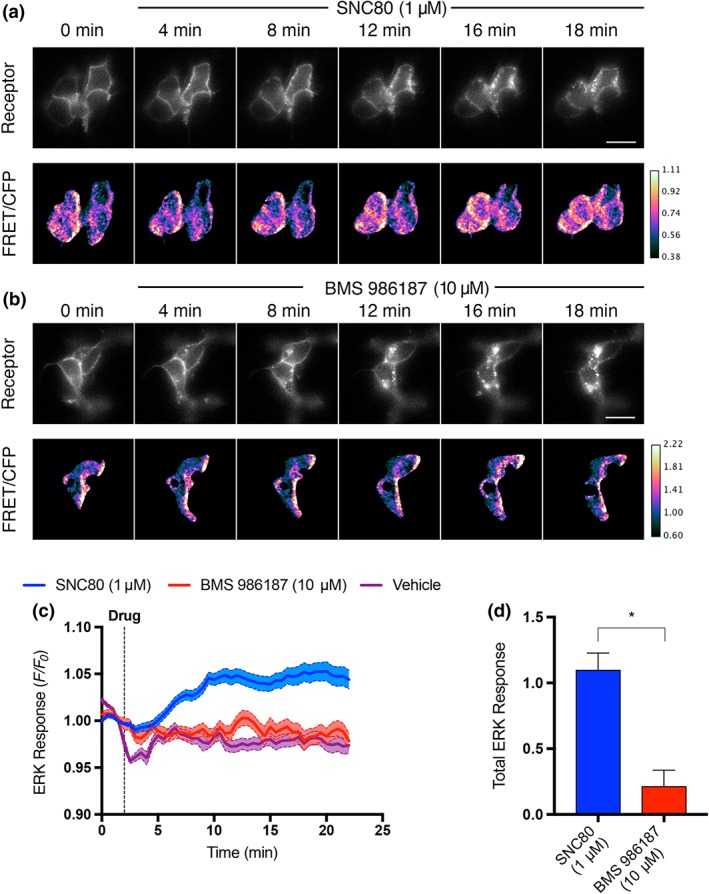
BMS 986187 elicits low ERK1/ERK2 phosphorylation. (a) Example montage of cytoplasmic ERK response in FLAG‐tagged HEK‐hDOPr cells in response to 1 μM SNC80. Top row: FLAG‐tagged δ‐receptors labelled with Alexa647‐conjugated M1 antibody. Bottom row: ratio of FRET/cyan fluorescence protein (CFP) fluorescence of expressed cEKAR sensor. Agonist added at 2.5 min. Scale bar is 20 μm, frames every 4 min. (b) Representative montage of cytoplasmic ERK response measured in HEK‐hDOPr cells, in response to 10 μM BMS 986187. (c) Average ERK response over time of HEK‐hDOPr cells treated with 1 μM SNC80 (n = 47 cells), 10 μM BMS 986187 (n = 34 cells), or vehicle (n = 39 cells). Responses are represented as fractional change over baseline. Solid line is the mean response, and shaded region inside dotted lines represents ± SEM. (d) SNC80 produced a significantly higher total ERK response compared with BMS 986187. Total response is measured as AUC of treatment condition minus AUC of vehicle. Values shown are means ± SEM. *P<0.05, significantly different as determined by unpaired two‐tailed t‐test
3.6. BMS 986187 induces low levels of phosphorylation and desensitization in δ‐receptors
Phosphorylation, arrestin recruitment, and receptor internalization of δ‐receptors are initiated by a phosphorylation event at Ser363 in the C‐tail of these receptors (El Kouhen et al., 2000; Qiu, Loh, & Law, 2007). As BMS 986187 promotes only a low level of receptor internalization or β‐arrestin 2 recruitment, we hypothesized this was due to inefficient phosphorylation of this residue. To assess this, we performed Western blot analysis for Ser363 phosphorylation of FLAG‐tagged δ‐receptors expressed in HEK293 cells treated with various δ‐receptor agonists for 1 hr as shown in Figure 8. Consistent with the internalization data, BMS 986187 did not induce significant phosphorylation of this residue compared with the vehicle control. Similar findings were seen with TAN‐67, whereas the higher internalizing agonists DPDPE and SNC80 caused a marked degree of phosphorylation.
Figure 8.
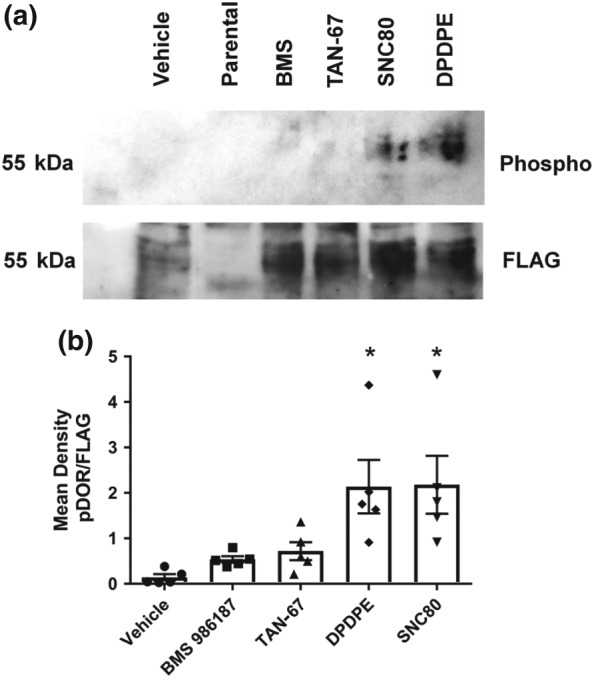
BMS 986187 poorly phosphorylates Ser363 on δ‐receptors. (a) Western blot of membranes from FLAG‐tagged HEK‐hDOPr cells, incubated with 10 μM concentrations of various δ‐receptor ligands probed for phosphorylated Ser363. (b) Data are expressed as a ratio of phosphorylated receptor to total receptor (FLAG). Each column is the mean of five independent experiments ± SEM. *P<0.05, significantly different from vehicle as determined by one‐way ANOVA with Tukey's post hoc test
Because BMS 986187 induced only a low level of receptor phosphorylation, arrestin recruitment, and internalization, we predicted there would be a reduced desensitization of δ‐receptors, measured as a loss of receptor signalling, when cells were treated with BMS 986187 compared with SNC80. CHO‐hDOPr cells were incubated for varying times with equipotent concentrations (as determined by GTPγ35S binding, Figure 1) of 10 μM BMS 986187 or 500 nM SNC80. Membranes homogenates were then prepared, and GTPγ35S binding was determined following a challenge with a maximal concentration (10 μM) of SNC80. Membranes from cells pretreated with SNC80 or BMS 986187 showed a reduction in the maximal GTPγ35S response, but the loss was more rapid for SNC80 (Figure 9), such that a 1 hr pretreatment with SNC80 resulted in a 86% loss compared with only a 39% loss with BMS 986187. Likewise, preincubation of cells for 30 min with BMS 986187 caused a lesser effect on the concentration response curve for SNC80 (maximum response = 68 ± 7%; EC50 = 7.4 ± 1.6 nM) than preincubation with SNC80 (maximal response = 31 ± 7%; EC50 = 25 ± 3.5 nM).
Figure 9.
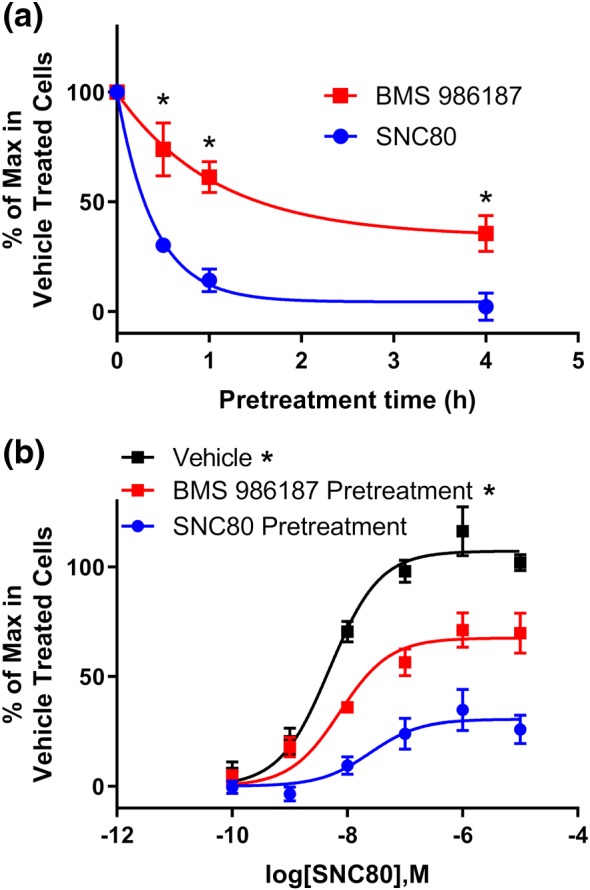
BMS 986187 treatment produces significantly less loss of agonist activity at δ‐receptors than SNC80. (a) CHO‐hDOPr cells were pretreated with either 500 nM SNC80 or 10 μM BMS 986187 for the indicated times, and then membrane homogenates were prepared as described in Section 2. The level of GTPγ35S binding induced by a challenge with 10 μM SNC80 in the membranes was determined and plotted against the time of cell pretreatment with SNC80 or BMS 986187. (b) CHO‐hDOPr cells were pretreated as above with SNC80 or BMS 986187 for 30 min, membranes homogenates were prepared, and SNC80 concentration response curves for stimulation of GTPγ35S binding were constructed. Data are expressed as % of maximum binding in untreated cells ± SEM from five independent experiments, each performed in duplicate. *P<0.05, significantly different from SNC80 as determined by two‐way ANOVA with Tukey's post hoc test
4. DISCUSSION
The data presented indicate that BMS 986187 is a biased allosteric agonist at the δ‐receptor, giving a maximal response in the GTPγ35S assay of equivalent magnitude to that seen with the orthosteric full agonist SNC80, albeit BMS 986187 is considerably less potent. In contrast to this strong response observed in G‐protein activation, BMS 986187 did not significantly recruit β‐arrestin 2. This is a consequence of low levels of receptor phosphorylation and leads to a low level of receptor internalization and desensitization. When compared with SNC80, BMS 986187 is significantly biased towards G‐protein activation, relative to the recruitment of β‐arrestin 2. This is the first evidence of an allosteric agonist displaying bias at an opioid receptor.
The direct agonist activity of BMS 986187 in the GTPγ35S assay in HEK‐DOPr cells agrees with the results from the original report of PAMs at δ‐receptors, using AC inhibition in CHO cells as a readout (Burford, Livingston, et al., 2015; Burford, Traynor, & Alt, 2015). The allosteric nature of the observed agonism was suggested by the inability of BMS 986187 to compete with the orthosteric ligand 3H‐diprenophine (Burford, Livingston, et al., 2015; Burford, Traynor, & Alt, 2015). In the present study, we confirmed that BMS 986187 agonist activity occurred via binding to an allosteric site because the orthosteric antagonists NTI and naloxone only partly inhibited the ability of BMS 986187 to stimulate GTPγ35S binding, and increasing concentrations of the NTI did not give parallel shifts in the BMS 986187 concentration–response curve. This confirms that agonism can be mediated by sites on the receptor other than the orthosteric site and demonstrates an indirect interaction between the allosteric and the orthosteric sites. Although these results in transfected HEK cells are encouraging, δ‐receptors in these cells are expressed at supraphysiological levels, which may not translate to relevant in vivo agonism (Kelly, 2013; Langmead & Christopoulos, 2006). Using mouse brain homogenates, we confirmed that the level of G‐protein activation elicited by BMS 986187 was similar to that of the full agonist, SNC80. However, these data also indicated that BMS 986187 was not completely selective for δ‐receptors, because the same assay performed using mouse brain homogenates from δ‐receptor knockout mice still afforded a low level of GTPγ35S stimulation. Previous work has indicated that BMS 986187 can act as a PAM for the μ‐ and κ‐opioid receptors, although no significant direct ago‐PAM activity has so far been detected at either of these receptors (Livingston et al., 2018). Alternatively, BMS 986187 could be acting at another, so far unidentified, GPCR.
Despite stimulating a higher level of GTPγ35S binding than the peptidic δ‐receptor agonist DPDPE, BMS 986187 produced significantly lower receptor internalization. Thus, maximal G‐protein stimulation was in the order BMS 986187 = SNC80 > DPDPE > TAN‐67, whereas maximal internalization was in the order SNC80 = DPDPE > TAN‐67 = BMS 986187. This striking change of order of maximal effect across the two different signalling outputs using the same cell line indicates the bias resulting from BMS 986187 occupancy of its allosteric binding site. We confirmed this finding by calculating the degree of bias for the signalling preference of BMS 986187‐occupied δ‐receptors for G‐protein activation over β‐arrestin 2 recruitment, compared with SNC80 as a reference ligand. The very low level of β‐arrestin 2 recruitment by BMS 986187 was confirmed by the lack of observable recruitment of GFP‐labelled β‐arrestin 2 to the plasma membrane in δ‐receptor‐expressing HEK cells.
Comparing β‐arrestin recruitment using the PRESTO‐TANGO assay with GTPγ35S binding, we find DPDPE to be G‐protein biased at δ‐receptors expressed in HEK cells, relative to SNC80. This agrees with studies that indicate SNC80 is a “super recruiter” of β‐arrestin 2, compared with DPDPE, at δ‐receptors in CHO cells, whereas both have similar activity as inhibitors of AC (Chiang et al., 2016) and with predictions from studies in vivo that SNC80 is a “high‐internalizing agonist” when compared with the low‐internalizing δ‐receptor agonist ARM390 and partial agonists such as TAN‐67 (Pradhan et al., 2009, 2010; Pradhsan et al., 2012). In contrast, bias calculations (Charfi, Audet, Bagheri Tudashki, & Pineyro, 2015) based on data obtained at the δ‐receptors expressed in HEK cells (Charfi et al., 2013) suggest SNC80 is highly biased towards AC inhibition, relative to internalization, when compared with DPDPE, although these data also show that SNC80 recruits much more β‐arrestin than DPDPE. Overall, these findings highlight the importance of understanding the relativity of bias, where the chosen reference ligand can have a significant influence on the direction, magnitude, and interpretation of observed results. Here, we chose to use SNC80 as a reference ligand as it is a “standard” δ‐receptor ligand characterized in many in vitro and behavioural assays (Chung et al., 2015; Danielsson et al., 2006; Dripps et al., 2017; Jutkiewicz et al., 2005; Pradhan et al., 2010). On the other hand, we have shown DPDPE to be 11‐fold biased towards G‐protein, relative to β‐arrestin recruitment, using SNC80 as a reference. Consequently, if DPDPE is used as a reference ligand, the bias of BMS 986187 towards G‐protein activation is reduced to ninefold.
BMS 986187 also failed to produce δ‐receptor‐mediated phosphorylation of ERK1/ERK2 in the MAPK pathway, although it showed significant G‐protein activation. This implies phosphorylation of ERK1/ERK2 via the allosteric site on δ‐receptors may be a β‐arrestin 2‐mediated process, a ligand‐dependent effect observed at other GPCRs (Shukla et al., 2008). However, this contrasts with the finding that BMS 986187 when acting as a PAM for δ‐receptors promotes ERK1/ERK2 phosphorylation (Burford, Livingston, et al., 2015; Burford, Traynor, & Alt, 2015). These apparently conflicting data imply that the BMS 986187‐occupied δ‐receptors may signal differently depending on whether or not the orthosteric site is occupied. We are currently investigating the effect of BMS 986187 on ERK1/ERK2 phosphorylation using a variety of δ‐receptor agonists. PAM activity arises at opioid receptors by a negative, indirect action with the Na+ ion‐binding site. Na+ holds the receptor in inactive conformations (R), and loss of Na+ ion binding allows the receptor to adopt ensembles of active receptor (R*) states (Liu et al., 2012; Livingston & Traynor, 2014; Pert, Pasternak, & Snyder, 1973). PAMs with greater efficacy to displace Na+ ions would then be predicted to show allosteric agonism by the same process. However, driving receptor activation through an allosteric site, there is no a priori reason why an allosteric agonist could not show functional selectivity at δ‐receptors by promoting a different ensemble of R* conformations from that promoted by an orthosteric ligand. Indeed, such an effect has been observed at the muscarinic ACh receptor where allosteric agonists promoted bias, comparing G‐protein activation and ERK1/ERK2 phosphorylation (Gregory, Hall, Tobin, Sexton, & Christopoulos, 2010).
Recruitment of β‐arrestin and δ‐receptor internalization requires sequential phosphorylation of several Ser and Thr residues in the C‐terminal tail of the receptor by G‐protein receptor kinases (Ferguson, 2001; Qiu et al., 2007; Stoffel, Pitcher, & Lefkowitz, 1997). Mutagenesis studies have indicated that an important initial phosphorylation site in the δ‐receptor phosphorylation cascade is Ser363 (El Kouhen et al., 2000). BMS 986187 produced a low level of Ser363 phosphorylation, compared with DPDPE and SNC80, and similar to the low‐internalizing agonist TAN‐67. This explains why the allosteric agonist induces significantly less desensitization than SNC80. However, it is perhaps surprising given the low level of phosphorylation and β‐arrestin recruitment that BMS 986187 caused δ‐receptor desensitization. It is possible that over extended periods, even with limited phosphorylation and β‐arrestin recruitment, BMS 986187 is able to drive significant desensitization. Alternatively, the BMS 986187‐occupied δ‐receptor is in different conformational states from those in orthosteric agonist‐occupied δ‐receptors and so may employ different desensitization mechanisms. In this regard, it should be noted that mutants of δ‐receptors expressed in HEK cells showing no detectable phosphorylation still desensitize over time (El Kouhen et al., 2000). Nonetheless, the low level of phosphorylation observed establishes that the bias driven by BMS 986187 results from reduced phosphorylation of the receptor, even though BMS 986187 does recruit G‐protein and presumably G‐protein receptor kinases. This signifies that the δ‐receptor conformations induced in the presence of BMS 986187 differ from those adopted in the presence of SNC80. A greater understanding of these conformations would provide insight into the driving force behind G‐protein versus β‐arrestin‐mediated signalling for both orthosteric and allosteric ligands.
In conclusion, BMS 986187 is a biased allosteric agonist of the δ‐receptor. Biased allosteric agonism at δ‐receptors could represent a novel strategy for treating chronic pain and depression, while potentially avoiding limiting factors such as rapid tolerance development and induction of convulsions.
AUTHOR CONTRIBUTIONS
M.A.S. and J.R.T. designed the experiments and analysed the data. M.A.S., K.E.L., and L.C. performed the experiments and analysed the data. Z.W. performed and analysed the ERK1/ERK2 experiments with supervision from M.P. M.A.S. and J.R.T. wrote the manuscript. J.R.T. provided the funding and supervision for the overall project.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, Immunoblotting and Immunochemistry, and Animal Experimentation, and as recommended by funding agencies, publishers, and other organizations engaged with supporting research.
ACKNOWLEDGEMENTS
This work was supported by a grant from the National Institutes of Health, USA (National Institute on Drug Abuse, R37 DA39997 to J.R.T.; GM117425 to M.A.P.). M.A.S. was also supported by NIH training grant (National Institute of General Medical Sciences, T32 GM007767).
Stanczyk MA, Livingston KE, Chang L, Weinberg ZY, Puthenveedu MA, Traynor JR. The δ‐opioid receptor positive allosteric modulator BMS 986187 is a G‐protein‐biased allosteric agonist. Br J Pharmacol. 2019;176:1649–1663. 10.1111/bph.14602
REFERENCES
- Alexander, S. P. H. , Christopoulos, A. , Davenport, A. P. , Kelly, E. , Marrion, N. V. , Peters, J. A. , … CGTP Collaborators (2017). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. British Journal of Pharmacology, 174(Suppl 1), S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audet, N. , Galés, C. , Archer‐Lahlou, É. , Vallières, M. , Schiller, P. W. , Bouvier, M. , & Pineyro, G. (2008). Bioluminescence resonance energy transfer assays reveal ligand‐specific conformational changes within preformed signaling complexes containing δ‐opioid receptors and heterotrimeric G proteins. The Journal of Biological Chemistry, 283, 15078–15088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bie, B. , & Pan, Z. Z. (2007). Trafficking of central opioid receptors and descending pain inhibition. Molecular Pain, 3, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury, F. A. , Zelnik, J. C. , & Traynor, J. R. (2009). G protein independent phosphorylation and internalization of the δ‐opioid receptor. Journal of Neurochemistry, 109, 1526–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burford, N. T. , Livingston, K. E. , Canals, M. , Ryan, M. R. , Budenholzer, L. M. L. , Han, Y. , … Alt, A. (2015). Discovery, synthesis, and molecular pharmacology of selective positive allosteric modulators of the δ‐opioid receptor. Journal of Medicinal Chemistry, 58, 4220–4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burford, N. T. , Traynor, J. R. , & Alt, A. (2015). Positive allosteric modulators of the μ‐opioid receptor: A novel approach for future pain medications. British Journal of Pharmacology, 172, 277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill, C. M. , Holdridge, S. V. , & Morinville, A. (2007). Trafficking of δ‐opioid receptors and other G‐protein‐coupled receptors: Implications for pain and analgesia. Trends in Pharmacological Sciences, 28, 23–31. [DOI] [PubMed] [Google Scholar]
- Charfi, I. , Audet, N. , Bagheri Tudashki, H. , & Pineyro, G. (2015). Distinguishing biased responses from ligand bias. British Journal of Pharmacology, 172, 435–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charfi, I. , Nagi, K. , Mnie‐Filali, O. , Thibault, D. , Balboni, G. , Schiller, P. W. , … Pineyro, G. (2013). Ligand‐ and cell‐dependent determinants of internalization and cAMP modulation by delta opioid receptor (DOR) agonists. Cellular and Molecular Life Sciences, 71, 1529–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Che, T. , Majumdar, S. , Zaidi, S. A. , Ondachi, P. , McCorvy, J. D. , Wang, S. , … Roth, B. L. (2018). Structure of the nanobody‐stabilized active state of the kappa opioid receptor. Cell, 172, 55–67.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang, T. , Sansuk, K. , & van Rijn, R. M. (2016). β‐Arrestin 2 dependence of δ opioid receptor agonists is correlated with alcohol intake. British Journal of Pharmacology, 173, 332–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung, P. C. , Boehrer, A. , Stephan, A. , Matifas, A. , Scherrer, G. , Darcq, E. , … Kieffer, B. L. (2015). Delta opioid receptors expressed in forebrain GABAergic neurons are responsible for SNC80‐induced seizures. Behavioural Brain Research, 278, 429–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark, M. J. , Harrison, C. , Zhong, H. , Neubig, R. R. , & Traynor, J. R. (2003). Endogenous RGS protein action modulates δ‐opioid signaling through Gio: Effects on adenylyl cyclase, extracellular signal‐regulated kinases, and intracellular calcium pathways. The Journal of Biological Chemistry, 278, 9418–9425. [DOI] [PubMed] [Google Scholar]
- Conn, P. J. , Christopoulos, A. , & Lindsley, C. W. (2010). Allosteric modulators of GPCRs: A novel approach for the treatment of CNS disorders. Nature Reviews. Drug Discovery, 8, 41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielsson, I. , Gasior, M. , Stevenson, G. W. , Folk, J. E. , Rice, K. C. , & Negus, S. S. (2006). Electroencephalographic and convulsant effects of the delta opioid agonist SNC80 in rhesus monkeys. Pharmacology, Biochemistry, and Behavior, 85, 428–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dripps, I. J. , Boyer, B. T. , Neubig, R. R. , Rice, K. C. , Traynor, J. R. , & Jutkiewicz, E. M. (2017). Role of signaling molecules in behaviors mediated by the δ‐receptor agonist SNC80. British Journal of Pharmacology, 175, 891–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Kouhen, O. M. , Wang, G. , Solberg, J. , Erickson, L. J. , Law, P. Y. , & Loh, H. H. (2000). Hierarchical phosphorylation of δ‐opioid receptor regulates agonist‐induced receptor desensitization and internalization. The Journal of Biological Chemistry, 275, 36659–36664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson, S. S. (2001). Evolving concepts in G protein‐coupled receptor endocytosis: The role in receptor desensitization and signaling. Pharmacological Reviews, 53, 1–24. [PubMed] [Google Scholar]
- Filliol, D. , Ghozland, S. , Chluba, J. , Martin, M. , Matthes, H. W. D. , Simonin, F. , … Kieffer, B. L. (2000). Mice deficient for δ‐ and μ‐opioid receptors exhibit opposing alterations of emotional responses. Nature Genetics, 25, 195–200. [DOI] [PubMed] [Google Scholar]
- Fritz, R. D. , Letzelter, M. , Reimann, A. , Martin, K. , Fusco, L. , Ritsma, L. , … Pertz, O. (2013). A versatile toolkit to produce sensitive FRET biosensors to visualize signaling in time and space. Science Signaling, 6, 1–14. [DOI] [PubMed] [Google Scholar]
- Gregory, K. J. , Hall, N. E. , Tobin, A. B. , Sexton, P. M. , & Christopoulos, A. (2010). Identification of orthosteric and allosteric site mutations in M2 muscarinic acetylcholine receptors that contribute to ligand‐selective signaling bias. The Journal of Biological Chemistry, 285, 7459–7474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46, D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jutkiewicz, E. M. , Kaminsky, S. T. , Rice, K. C. , Traynor, J. R. , & Woods, J. H. (2005). Differential behavioral tolerance to the δ ‐opioid agonist SNC80 ([(+)‐4‐[(alphaR)‐alpha‐[(2S,5R)‐2,5‐dimethyl‐4‐(2‐propenyl)‐1‐piperazinyl]‐(3‐methoxyphenyl)methyl]‐N,N‐diethylbenzamide) in Sprague‐Dawley rats. The Journal of Pharmacology and Experimental Therapeutics, 315, 414–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabli, N. , & Cahill, C. M. (2007). Anti‐allodynic effects of peripheral delta opioid receptors in neuropathic pain. Pain, 127, 84–93. [DOI] [PubMed] [Google Scholar]
- Kelly, E. (2013). Efficacy and ligand bias at the δ‐opioid receptor. British Journal of Pharmacology, 169, 1430–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin, T. (2007). Allosteric agonist modulators. Journal of Receptors and Signal Transduction, 27, 247–259. [DOI] [PubMed] [Google Scholar]
- Kenakin, T. (2017). A system‐independent scale of agonism and allosteric modulation for assessment of selectivity, bias and receptor mutation. Molecular Pharmacology, 92, 414–424. [DOI] [PubMed] [Google Scholar]
- Kenakin, T. , & Christopoulos, A. (2013). Signalling bias in new drug discovery: Detection, quantification and therapeutic impact. Nature Reviews Drug Discovery, 12, 205–216. [DOI] [PubMed] [Google Scholar]
- Kenakin, T. , Watson, C. , Muniz‐Medina, V. , Christopoulos, A. , & Novick, S. (2012). A simple method for quantifying functional selectivity and agonist bias. ACS Chemical Neuroscience, 3, 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keov, P. , Sexton, P. M. , & Christopoulos, A. (2011). Allosteric modulation of G protein‐coupled receptors: A pharmacological perspective. Neuropharmacology, 60, 24–35. [DOI] [PubMed] [Google Scholar]
- Kessler, R. C. , & Bromet, E. J. (2013). The epidemiology of depression across cultures. Annual Review of Public Health, 34, 119–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead, C. J. , & Christopoulos, A. (2006). Allosteric agonists of 7TM receptors: Expanding the pharmacological toolbox. Trends in Pharmacological Sciences, 27, 475–481. [DOI] [PubMed] [Google Scholar]
- Lester, P. A. , & Traynor, J. R. (2006). Comparison of the in vitro efficacy of μ, δ, κ and ORL1 receptor agonists and non‐selective opioid agonists in dog brain membranes. Brain Research, 1073–1074, 290–296. [DOI] [PubMed] [Google Scholar]
- Liu, W. , Chun, E. , Thompson, A. A. , Chubukov, P. , Xu, F. , Katritch, V. , … Stevens, R. C. (2012). Structural basis for allosteric regulation of GPCRs by sodium ions. Science, 337, 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livingston, K. E. , Stanczyk, M. A. , Burford, N. T. , Alt, A. , Canals, M. , & Traynor, J. R. (2018). Pharmacologic evidence for a putative conserved allosteric site on opioid receptors. Molecular Pharmacology, 93, 157–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livingston, K. E. , & Traynor, J. R. (2014). Disruption of the Na+ ion binding site as a mechanism for positive allosteric modulation of the mu‐opioid receptor. Proceedings of the National Academy of Sciences, 111, 18369–18374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz, P.‐E. , & Brigitte, K. (2014). Opioid receptors: Distinct roles in mood disorders. Trends in Neurosciences, 36, 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNicol, E. , Horowicz‐Mehler, N. , Fisk, R. A. , Bennett, K. , Gialeli‐Goudas, M. , Chew, P. W. , … Americal Pain Society (2017). Management of opioid side effects in cancer‐related and chronic noncancer pain: A systematic review. The Journal of Pain, 4, 231–256. [DOI] [PubMed] [Google Scholar]
- National Research Council . (1996). Guide for the care and use of laboratory animals. Washington, D.C.: National Academy Press. [Google Scholar]
- Ong, E. W. , Xue, L. , Olmstead, M. C. , & Cahill, C. M. (2015). Prolonged morphine treatment alters δ opioid receptor post‐internalization trafficking. British Journal of Pharmacology, 172, 615–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pert, C. B. , Pasternak, G. , & Snyder, S. H. (1973). Opiate agonists and antagonists discriminated by receptor binding in brain. Science, 182, 1359–1361. [DOI] [PubMed] [Google Scholar]
- Pradhan, A. A. , Befort, K. , Nozaki, C. , Gavériaux‐Ruff, C. , & Kieffer, B. L. (2011). The delta opioid receptor: An evolving target for the treatment of brain disorders. Trends in Pharmacological Sciences, 32, 581–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan, A. A. , Smith, M. L. , Kieffer, B. L. , & Evans, C. J. (2012). Ligand‐directed signalling within the opioid receptor family. British Journal of Pharmacology, 167, 960–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan, A. A. A. , Becker, J. A. J. , Scherrer, G. , Tryoen‐Toth, P. , Filliol, D. , Matifas, A. , … Kieffer, B. L. (2009). In vivo delta opioid receptor internalization controls behavioral effects of agonists. PLoS ONE, 4, e5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan, A. A. A. , Walwyn, W. , Nozaki, C. , Filliol, D. , Erbs, E. , Matifas, A. , … Kieffer, B. L. (2010). Ligand‐directed trafficking of the δ‐opioid receptor in vivo: Two paths toward analgesic tolerance. The Journal of Neuroscience, 30, 16459–16468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przewłocki, R. , & Przewłocka, B. (2001). Opioids in chronic pain. European Journal of Pharmacology, 429, 79–91. [DOI] [PubMed] [Google Scholar]
- Qiu, Y. , Loh, H. H. , & Law, P.‐Y. (2007). Phosphorylation of the δ‐opioid receptor regulates its β‐arrestins selectivity and subsequent receptor internalization and adenylyl cyclase desensitization. The Journal of Biological Chemistry, 282, 22315–22323. [DOI] [PubMed] [Google Scholar]
- Reinke, T. (2014). Providers need to boost efforts to prevent abuse of narcotics. Managed Care, 23, 11–12. [PubMed] [Google Scholar]
- Rush, A. J. , Trivedi, M. H. , Wisniewski, S. R. , Nierenberg, A. A. , Stewart, J. W. , Warden, D. , … Fava, M. (2006). Acute and longer‐term outcomes in depressed outpatients requiring one or several treatment steps: A STAR*D report. The American Journal of Psychiatry, 163, 1905–1917. [DOI] [PubMed] [Google Scholar]
- Saitoh, A. , & Yamada, M. (2012). Antidepressant‐like effects of δ opioid receptor agonists in animal models. Current Neuropharmacology, 10, 231–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid, C. L. , Kennedy, N. M. , Ross, N. C. , Lovell, K. M. , Yue, Z. , Morgenweck, J. , … Bohn, L. M. (2017). Bias factor and therapeutic window correlate to predict safer opioid analgesics. Cell, 171(1165), e13–e1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiwarski, D. J. , Darr, M. , Telmer, C. A. , Bruchez, M. P. , & Puthenveedu, M. A. (2017). PI3K class II α regulates δ‐opioid receptor export from the trans‐Golgi network. Molecular Biology of the Cell, 28, 2202–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla, A. K. , Violin, J. D. , Whalen, E. J. , Gesty‐palmer, D. , Shenoy, S. K. , & Lefkowitz, R. J. (2008). Distinct conformational changes in β‐arrestin report biased agonism at seven‐transmembrane receptors. Proceedings of the National Academy of Sciences 105, 9988–9993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoffel, R. H. 3rd , Pitcher, J. A. , & Lefkowitz, R. J. (1997). Targeting G protein‐coupled receptor kinases to their receptor substrates. The Journal of Membrane Biology, 157, 1–8. [DOI] [PubMed] [Google Scholar]
- Trapaidze, N. , Gomes, I. , Bansinath, M. , & Devi, L. A. (2000). Recycling and resensitization of delta opioid receptors. DNA and Cell Biology, 19, 195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynor, J. R. , & Nahorski, S. R. (1995). Modulation by mu‐opioid agonists of guanosine‐5′‐O‐(3‐[35S]thio)triphosphate binding to membranes from human neuroblastoma SH‐SY5Y cells. Molecular Pharmacology, 47, 848–854. [PubMed] [Google Scholar]
- Tryoen‐Toth, P. , Decaillot, F. M. , Filliol, D. , Befort, K. , Lazarus, L. H. , Schiller, P. W. , … Kieffer, B. L. (2005). Inverse agonism and neutral antagonism at wild‐type and constitutively active mutant delta opioid receptors. The Journal of Pharmacology and Experimental Therapeutics, 313, 410–421. [DOI] [PubMed] [Google Scholar]
- Weinberg, Z. Y. , Zajac, A. S. , Phan, T. , Shiwarski, D. J. , & Puthenveedu, M. A. (2017). Sequence‐specific regulation of endocytic lifetimes modulates arrestin‐mediated signaling at the μ opioid receptors. Molecular Pharmacology, 91, 416–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whalen, E. J. , Rajagopal, S. , & Lefkowitz, R. J. (2011). Therapeutic potential of β‐arrestin‐ and G protein‐biased agonists. Trends in Molecular Medicine, 17, 126–139. [DOI] [PMC free article] [PubMed] [Google Scholar]