

Abstract
Background and Purpose
As an osteoclast differentiation factor, receptor activator of NF‐κB ligand (RANKL) is produced by various immune cells and may be involved in the pathogenesis of osteoporosis and inflammation. Although RANKL is expressed in most immune cells and tissues, it is not clear how this might affect allergic inflammation.
Experimental Approach
The roles of RANKL in allergic rhinitis (AR) were analysed in an ovalbumin (OVA)‐induced animal model, human subjects, and a human mast cell line (HMC‐1). Small interfering RNA experiments were performed in an OVA‐induced AR model.
Key Results
RANKL and RANKL receptor (RANK) were up‐regulated in serum or nasal mucosal tissues of AR patients and AR mice. RANKL and RANK were colocalised in mast cells of nasal mucosa tissue. Depletion of RANKL by RANKL siRNA ameliorated AR symptoms and reduced AR‐related biomarkers, including thymic stromal lymphopoietin (TSLP), IgE, histamine, and inflammatory cell infiltration, whereas recombinant RANKL increased AR responses and TSLP levels. In addition, functional deficiency of TSLP decreased AR responses induced by RANKL. In human mast cells, interaction of RANKL with RANK increased production of TSLP and inflammatory cytokines. Production of TSLP by RANKL stimulation was mediated through activation of the PI3K, MAPK, caspase‐1, and NF‐κB pathways. Furthermore, dexamethasone alleviated RANKL‐induced inflammatory reactions in AR models.
Conclusion and Implications
Collectively, these data suggest that RANKL may induce development of AR through up‐regulation of TSLP.
What is already known
RANKL is an osteoclast differentiation factor.
RANKL‐RANK system plays important roles in bone homeostasis, organogenesis, immune regulation, and cancer.
What this study adds
RANKL is increased in patients with allergic rhinitis and in corresponding mouse models.
RANKL‐RANK system induces development of allergic rhinitis through up‐regulating the cytokine thymic stromal lymphopoietin.
What is the clinical significance
RANKL/RANK signalling pathway might be a target in the treatment of allergic rhinitis.
Abbreviations
- AR
allergic rhinitis
- DC
dendritic cells
- Foxp3
forkhead box P3
- ICAM‐1
intercellular adhesion molecule 1
- IMDM
Isocove's modified Dulbecco's medium
- MIP‐2
macrophage inflammatory protein 2
- OVA
ovalbumin
- PDTC
pyrrolidinedithiocarbonate
- TSLP
thymic stromal lymphopoietin
1. INTRODUCTION
Allergic rhinitis (AR) is a Type I hypersensitive inflammatory immune responses in the nasal mucosa caused by inhaled allergens, such as mould, animal dander, and pollen (Rondón et al., 2012). AR is induced by an allergen cross‐linking with IgE bound to a mast cell. Mast cells, which lead to acute clinical symptoms with early nasal responses such as itching, sneezing, congestion, and rhinorrhea by IgE‐dependent release of histamine, leukotrienes, and prostaglandins (May & Dolen, 2017), have a central role in inflammatory allergic reactions through the infiltration of inflammatory cells by the production of thymic stromal lymphopoietin (TSLP) and Th2 cytokines (Amin et al., 2012; Jeong et al., 2002; Moon et al., 2011; Oh, Ryu, Cha, Kim, & Jeong, 2012).
Receptor activator of NF‐κB ligand (RANKL, also known as osteoclast differentiation factor; osteoprotegerin ligand; and TNF‐related activation‐induced cytokine) is a member of the TNF ligand family that exists as a Type II membrane‐bound protein, a soluble fragment, and a secreted protein (Anderson et al., 1997). RANKL is expressed in a wide variety of cell lineages and tissues, including osteoblasts, activated T cells, keratinocytes, mammary gland epithelial cells, skeletal muscles, the lung, the stomach, and the brain (Anderson et al., 1997; Kartsoginannis et al., 1999; Loser et al., 2006; Schett, Hayer, Zerina, Redlich, & Smolen, 2005; Wada, Nakashima, Hiroshi, & Penninger, 2006). Its signalling receptor RANK, a member of the TNF receptor family, is expressed in dendritic cells (DCs), osteoclast progenitors, macrophages, and peripheral blood monocytes, and RANKL‐RANK signalling has been found to have roles beyond bone homeostasis in organogenesis, immune regulation, and cancer (Walsh et al., 2014; Wong et al., 1997; Theill, Boyle, & Penninger, 2002). Recently, mast cells were reported to produce RANKL in atherosclerotic plaques to induce the calcification of vascular smooth muscle cells (Ali et al., 2006). However, the core function of RANKL in AR is still not well understood.
TSLP is an IL‐7‐like cytokine that triggers DCs and mast cells to induce Th2 inflammatory responses (Allakhverdi et al., 2007) and has been linked to the pathogenesis of asthma, atopic dermatitis, and eosinophilic oesophagitis (Jariwala, Abrams, Benson, Fodeman, & Zheng, 2011; Rothenberg et al., 2010; Ying et al., 2005). Furthermore, TSLP is an important factor involved in the development of AR (Moon et al., 2011; Nam, Kim, & Jeong, 2017).
In the present study, we elucidated a novel role of RANKL/RANK in AR. Furthermore, the signal transduction pathways of RANKL/RANK were investigated in human mast cells. Our findings show that RANKL is a novel stimulator for the development of inflammatory responses in AR.
2. METHODS
2.1. Human tissue samples
The local ethics committee approved the study (KMC IRB 0915‐02), and informed consent was obtained from the donors of the tissue samples. Serum and nasal tissue was obtained from patients with AR and control subjects at the Ear, Nose, and Throat Department of the Kyung Hee University Hospital in Seoul, Korea, between o2010 and 2012. The baseline characteristics of normal healthy controls and AR patients are provided in Supporting Information Table S1. The normal nasal mucosa was obtained from the inferior turbinates of healthy control subjects who had normal nasal mucosa and who were admitted for augmentation rhinoplasty. Allergic nasal mucosa was obtained from patients with severe persistent AR during septoturbinate surgery; their turbinate mucosa showed an oedematous and congested appearance. These patients had been previously treated with medical management (antihistamine and intranasal steroid spray) without satisfactory improvement for at least 3 months. They had one or more of the following symptoms: sleep disturbance; impairment of daily activities, leisure and/or sports; and impairment of school or work. All tissue samples were divided into two portions. One portion was frozen in liquid nitrogen and stored at −70°C for protein isolation. For immunohistochemistry, another sample was also frozen in liquid nitrogen and stored at −70°C for a subsequent procedure.
2.2. Animals
All animal care and experimental procedures were conducted with approval from the animal care committee of Kyung Hee University (Approval no. KHUASP (SE)‐15‐118). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath & Lilley, 2015) and with the recommendations made by the British Journal of Pharmacology.
Six‐week‐old female BALB/c (20–22 g, Dae‐Han Experimental Animal Center, Eumsung, Republic of Korea) mice were maintained under pathogen‐free conditions (Cat# TAC:balb, RRID:IMSR_TAC:balb). Five animals were housed per cage (294 × 190 × 125 mm) in a laminar air‐flow room maintained at 22 ± 1°C and a relative humidity of 55 ± 10% under a 12:12 light/dark cycle with the light switched on at 7 a.m. throughout the study. The mice had access to food and water ad libitum.
2.3. Ovalbumin‐induced model of AR
Mice were sensitized (i.p.) with 100 μg of ovalbumin (OVA), emulsified with 20 mg of aluminium hydroxide (Sigma‐Aldrich) in 100 μl of PBS on Days 1, 5, and 14. Then, local immunization was performed once a day from Day 15 to Day 24 by instilling the OVA or recombinant mouse RANKL. For the siRNA treatment, mice were transfected with RANKL‐specific siRNA (2 nM), TSLP siRNA (2 nM), or negative control siRNA (2 nM) using lipofectamine 2000 (Invitrogen, Carlsbad, CA) as previously described (Hosoya et al., 2011) 20 min before intranasal OVA challenge for the entire 10‐day experimental period. Nasal symptoms were evaluated by counting the number of nasal rubs that occurred in the 10 min after OVA intranasal provocation at the 10‐day mark after the challenge. After scoring the nasal rubs, the animals were killed. We measured OVA‐specific IgE as the relevant endpoint for AR.
2.4. ELISA
Cytokine antibodies were purchased from BD Biosciences Pharmingen. Cytokines of serum, mucosa tissue, and cell supernatant were measured using an elisa. Nasal mucosa tissues were homogenized with protease and phosphatase inhibitors in homogenization buffer (Sigma, St. Louis, MO). The homogenates were transferred to 1.5‐ml tubes, centrifuged at 13,000× g for 10 min at 4°C, and the supernatant was stored at −80°C until analysed. elisa was performed by coating 96‐well plates with capture antibody (1 μg per well). Before the subsequent steps in the assay, the coated plates were washed twice with PBS containing 0.05% Tween 20. All reagents and coated wells used in this assay were incubated for 2 hr at room temperature. A standard curve was generated from the known concentrations of cytokine, as provided by the manufacturer. Detection antibodies were incubated at room temperature for 2 hr. After washing, avidin‐peroxidase was added and the plates were incubated at 37°C for 30 min. After additional washes, substrate (Pharmingen) was added. The plates were read at 405 nm. Appropriate specificity controls were included, and all samples were run in duplicate. Cytokine levels in the nasal mucosa were expressed relative to the total protein in the sample. The protein was estimated using the bicinchoninic acid method.
2.5. Histamine assay
Histamine was measured in serum and rat peritoneal mast cells by the OPA spectrofluorometric procedure (Han et al., 2010). The fluorescent intensity was measured at 460 nm (excitation at 355 nm) using a spectrofluorometer.
2.6. Cell culture and stimulation
HMC‐1 cells (Cat# SCC067, RRID:CVCL_H206) were generously provided by EichiMorri (Osaka University, Osaka, Japan). HMC‐1 cells were grown in an IMDM medium supplemented with 100 U·ml−1 of penicillin, 100 μg·ml−1 of streptomycin, 10 μM of monothioglycerol, and 10% heat‐inactivated FBS at 37°C, in 5% CO2 and 95% humidity. Human primary cord blood‐derived CD34+ progenitor cells were purchased from ATCC (Manassa, VA) and were cultured and grown in IMDM medium supplemented with 100 U·ml−1 of penicillin, 100 μg·ml−1 of streptomycin, 10% FBS, 50 μM of mercaptoethanol, recombinant human stem cell factor (100 ng·ml−1), IL‐6 (50 ng·ml−1), and IL‐3 (10 ng·ml−1) at 37°C in 5% CO2 with 95% humidity. After 6–7 weeks of culturing, more than 90% of the cells were identified as mast cells as determined by toluidine blue staining. Cells were treated with recombinant human RANKL 10 ng·ml−1 for 8 hr at 37°C.
2.7. RNA isolation and quantitative real‐time PCR
HMC‐1 cells were treated with RANKL (10 ng·ml−1) for 24 hr. The total RNA was isolated from the cells according to the manufacturer's specification using an easy‐BLUE RNA extraction kit (iNtRON Biotechnology, Kyunggi‐do, Korea). The concentrations of total RNA in the final elutes were determined by spectrophotometry. Total RNA (2.5 μg) was heated at 72°C for 5 min and then chilled on ice. Each sample was reverse‐transcribed to cDNA for 90 min at 42°C using a cDNA synthesis kit (iNtRON Biotechnology, Sungnam, Republic of Korea). For human TSLP, quantitative real‐time PCR was performed using an SYBR Green master mix with primers (forward 5′‐TATGAGTGGGACCAAAAGTACCG‐3′; reverse 5′‐GGGATTGAAGGTTAGGCTCTGG‐3′), and mRNA was analysed using an ABI StepOne real‐time PCR System (Applied Biosystems, Foster City, CA). All data were analysed using the ΔΔCT method. For human RANK, PCR was performed using the following primers for the human RANK (forward 5′‐AGGTGTCTTACTGACT CTGG‐3′; reverse 5′‐GCTGTCTTCCTCTATCTCGGTCT‐3′); GAPDH (forward 5′‐CAAAAGGGTCATCATCTCTG‐3′; reverse 5′‐CCTGCTTCACCACCTTCTTG‐3′). The annealing temperature was 57°C for RANK and 60°C for GAPDH respectively. Products were analysed by electrophoresis on a 1.5% agarose gel and visualized by staining with ethidium bromide.
2.8. Preparation of nuclear extracts and cytoplasmic extracts
Briefly, after cell activation, the cells were washed with ice‐cold PBS and resuspended in 60 μl of buffer A (10 mM of HEPES/KOH, 2 mM of MgCl2, 0.1 mM of EDTA, 10 mM of KCl, 1 mM of dithiothreitol, and 0.5 mM of phenylmethylsulfonyl fluoride, pH 7.9). The cells were left on ice for 15 min, lysed gently with 2.5 μl of 10% Nonidet P‐40, and centrifuged at 2,000× g for 10 min at 4°C. The supernatant was collected and used as the cytoplasmic extract. The nuclei pellets were resuspended in 40 μl of buffer B (50 mM of HEPES/KOH, 50 mM of KCl, 300 mM of NaCl, 0.1 mM of EDTA, 10% glycerol, 1 mM of dithiothreitol, and 0.5 mM of phenylmethylsulfonyl fluoride, pH 7.9), left on ice for 20 min and inverted. The nuclear debris was then centrifuged at 15,000× g for 15 min. The supernatant (nuclear extract) was collected and stored at −70°C until the analysis was performed.
2.9. Western blot analysis
All antibody‐based procedures used in this study complied with the recommendations made by the British Journal of Pharmacology. Cell extracts were prepared using a detergent lysis procedure. Samples were heated at 95°C for 5 min and briefly cooled on ice. After centrifugation, 50 μg aliquots were resolved by 10% SDS‐PAGE. After electrophoresis, the protein was transferred to nitrocellulose membranes, and then the membranes were blocked for 2 hr with 5% skim milk. The primary antibodies were diluted with PBS containing Tween‐20 (PBST) and incubated overnight at 4°C. Afterwards, the nitrocellulose membrane was washed five times for 15 min with PBST. For protein detection, the blot was incubated with secondary antibodies (1:3,000 in PBST) conjugated with peroxidase for 40 min. Finally, the protein bands were visualized using an enhanced chemiluminesence assay purchased from Amersham Co. (Newark, NJ) following the manufacturer's instructions.
2.10. Histological examination
Tissue samples were immediately fixed with 10% formaldehyde and embedded in paraffin. Next, nasal mucosa samples were cut into 4‐μm‐thick sections that were stained with haematoxylin and eosin (for eosinophils), Alcian blue and Safranin O (for mast cells), and Foxp3 (for Treg cells; Abbiotec, San Diego, CA) after dewaxing and dehydration. The sections were coded and randomly analysed by two observers, blinded to the treatment groups, by counting the numbers of eosinophils, mast cells, and Treg cells on both sides of the septal mucosa.
2.11. Confocal laser scanning microscopy
Nasal tissues were immediately fixed with 4% formaldehyde and embedded in paraffin. After dewaxing and dehydration, sections were blocked with BSA followed by 60‐min incubation with anti‐RANKL (Cat# sc‐9073, RRID:AB_2303585), anti‐RANK (Cat# sc‐374360, RRID:AB_10990136), anti‐C‐kit (Cat# sc‐168, RRID:AB_631033), and anti‐CD4 (Cat# sc‐19641, RRID:AB_627055) antibodies (Santa Cruz, CA) at a concentration of 1 μg·ml−1. The secondary antibodies, fluorescein isothiocyanate (green)‐conjugated anti‐goat IgG (Cat# ab6737, RRID:AB_955274), DyLight 649 (purple)‐conjugated anti‐mouse IgG (Cat# 015–490‐003, RRID:AB_2337220), or tetramethylrhodamine (red)‐conjugated anti‐rabbit IgG (Cat# ab6718, RRID:AB_955551; Abcam, Cambridge, MA), were added to the incubation for 30 min. Mounting medium containing 4′,6‐diamidino‐2‐phenylindole (Vector Laboratories, Burlingame, CA) was used to counterstain the DNA. All specimens were examined with a confocal laser‐scanning microscope.
2.12. Data and statistical analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018). All results are expressed as the mean ± SEM. In vitro studies were conducted independently at least five times (n = 5). A power analysis was performed to determine a suitable sample size of in vivo studies. Using two independent sample t‐tests, we calculated the sample size and G power (RRID:SCR_013726). Sample size (n = 5 mice per group: Type I error 0.05; power 99.86% and n = 10 mice per group: Type I error 0.05; power 98.97%) was determined in a pilot study. Statistical analyses were performed using SPSS statistical software (RRID:SCR_002865, SPSS 11.5, Armonk, NY). Independent t‐test or one‐way ANOVA followed by Tukey's post hoc test was used when there were two or multiple groups to compare respectively. For all one‐way ANOVAs, post hoc tests were run only if F achieved P < 0.05 and there was no significant variance inhomogeneity. In all cases, P values <0.05 were considered significant.
2.12. Materials
We purchased ovalbumin (OVA), phorbol 12‐myristate 13‐acetate, calcium ionophore (A23187), 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide, DMSO, PBS, BSA, bicinchoninic acid, dexamethasone, and avidin–peroxidase from Sigma Chemical Co. (St. Louis, MO); Isocove's modified Dulbecco's medium (IMDM), penicillin, streptomycin, and FBS from Gibco BRL (Grand Island, NY). ERK inhibitors N‐[(2R)‐2,3‐dihydroxypropoxy‐3,4‐difluoro‐2‐[(2‐fluoro‐4‐iodophenyl)amino]‐benzamide (PD03259010), c‐jun amino JNK inhibitors Anthra (1,9‐cd)pyrazol‐6(2H)‐one (SP600125), p38 inhibitors 4‐(4‐flurophenyl)‐2‐(4‐methylsulfinylphenyl)‐5‐(4‐pyridyl)imidazole (SB203580), NF‐κB inhibitors pyrrolidinedithiocarbonate (PDTC), PI3K inhibitors (wortmannin), and caspase‐1 inhibitor were purchased from Sigma‐Aldrich (St. Louis, MO). RANKL‐specific siRNA, TSLP‐specific siRNA, and a negative control siRNA were purchased form Dharmacon (Lafayette, LA). Antibody for RANKL (Cat# sc‐9073, RRID:AB_2303585), RANK (Cat# sc‐374360, RRID:AB_10990136), forkhead box P3 (Foxp3; Cat# sc‐130666, RRID:AB_2104931), ERK (Cat# sc‐94, RRID:AB_2140110), phosphorylated ERK (Cat# sc‐7383, RRID:AB_627545), JNK (Cat# sc‐571, RRID:AB_632385), pJNK (Cat# sc‐6254, RRID:AB_628232), p38 (Cat# sc‐535, RRID:AB_632138), pp38 (Cat# sc‐7973, RRID:AB_670359), pIκB‐α (Cat# sc‐8404, RRID:AB_627773), NF‐κB (Cat# sc‐8008, RRID:AB_628017), histone (Cat# sc‐10806, RRID:AB_2117992), caspase‐1 (Cat# sc‐56036, RRID:AB_781816), PI3K (Cat# sc‐1637, RRID:AB_628126), actin (Cat# sc‐8432, RRID:AB_626630), and α‐tubulin (Cat# B‐7, RRID:AB_628411) were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Recombinant human RANKL (Cat# CABT‐P1009H, RRID:AB_11466115) and anti‐human RANKL (neutralization, Cat# AF626, RRID:AB_355484) antibodies were purchased from R&D Systems Inc. (Minneapolis, MN). IgG Ab was purchased from BD Biosciences Pharmingen (San Diego, CA).
2.13. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander, Fabbro et al., 2017a; Alexander, Fabbro et al., 2017b).
3. RESULTS
3.1. RANKL is up‐regulated in AR patients and localized on mast cells
High levels of RANKL protein was detected in the serum and nasal mucosal tissue of AR patients, significantly above levels in samples from normal subjects. Most of the cells positive for RANKL were c‐Kit+ mast cells and CD4+ T cells (Figure 1a–d and Supporting Information Figure S1a,b). Additionally, quantitative scores for RANKL in the serum of the AR patients were significantly correlated with those for inflammation during eosinophil counting (Supporting Information Table S2). In the nasal mucosa tissues of mice with OVA‐induced AR, the levels of RANKL were higher than those in the tissues of the normal mice, and most of the cells staining positive for RANKL in the nasal mucosa tissues of the AR mice were mast cells (Figure 1e,f and Supporting Information Figure S1c). Moreover, the numbers of CD4+ T cells in the nasal mucosa of AR mice were significantly higher than those in normal mice (Supporting Information Figure S1d,e). Finally, colocalization of RANKL and RANK was observed in the mast cells of the nasal mucosa tissues (Figure 1g).
Figure 1.
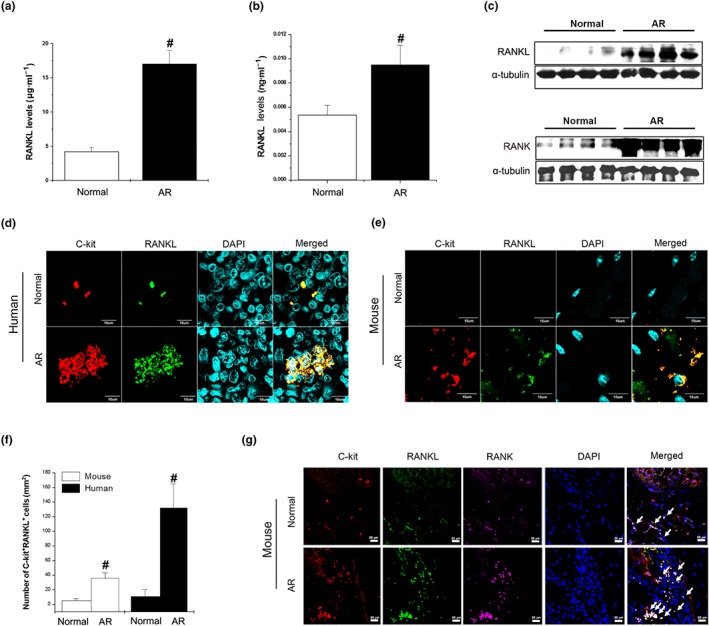
RANKL is up‐regulated in AR patients and localized on mast cells. (a) RANKL in serum (n = 40) and (b) homogenized nasal mucosal tissues (n = 20) of AR patients was analysed by elisa. # P < 0.05; significantly different from normal. (c) RANKL (upper panel) and RANK (lower panel) expression in the nasal mucosa of AR patients was determined by western blot analysis. (d) Immunostaining for RANKL (green) and staining for C‐kit (red) is shown in the AR patients (magnification, ×294). Mice were sensitized on Days 1, 5, and 14 by i.p. injections of 100 μg of OVA emulsified with 20 mg of aluminium hydroxide and then challenged with 1.5 mg of OVA. (e) Immunostaining for RANKL (green) and staining for C‐kit (red) is shown in nasal mucosal tissues from normal mice and AR mice (magnification, ×294). (f) C‐kit+RANKL+ cells were counted in AR patients and animal tissues by three individuals (n = 5 per group). Data shown are the means ± SEM. # P < 0.05; significantly different from normal. (g) Co‐localization of C‐kit (red), RANKL (green), and RANK (purple) in the nasal mucosa of AR animal (magnification, ×100). Arrows indicate C‐kit+RANKL+RANK+ cells
3.2. Knockdown of RANKL alleviates AR symptoms and responses in AR mice
We sought to confirm the potential role of RANKL in the AR mice model, using RANKL siRNA. Sensitized mice received local injections of RANKL siRNA or control siRNA prepared in lipofectamine and were challenged with intranasal OVA for 10 days. The knock‐down efficiencies of these siRNAs were then examined by elisa and Western blotting for the RANKL protein in the nasal mucosa tissues. RANKL‐specific siRNA transfection effectively reduced the protein levels of RANKL relative to the control siRNA in the nasal mucosal tissues (Figure 2a,b).
Figure 2.
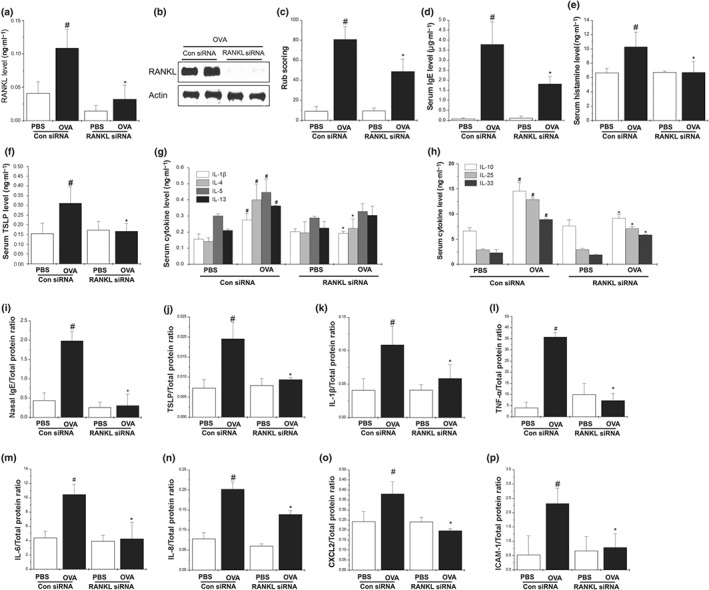
Knockdown of RANKL alleviates AR symptoms in AR mice. Mice were sensitized on Days 1, 5, and 14 by i.p. injections of 100 μg of OVA emulsified in 20 mg of aluminium hydroxide, after which they received local injections of RANKL siRNA and control siRNA, or PBS alone into the nasal cavity, and were challenged with intranasal OVA for 10 days. (a) The production of RANKL in the nasal mucosal tissues of mice was determined by elisa and (b) western blot analysis. (c) The number of nose rubs that occurred 10 min after OVA intranasal provocation. (d) OVA‐specific IgE levels in serum were measured using the elisa method. (e) The levels of histamine in serum were measured by a histamine assay method. (f–h) The levels of cytokines in serum were measured using the elisa method. (i) OVA‐specific IgE levels in the nasal mucosa were measured by elisa. (j–n) The levels of cytokine in the nasal mucosa were measured by elisa. (o,p) The levels of CXCL2 and ICAM in nasal mucosa tissue were measured using elisa method. Data shown are the means ± SEM from n = 10 mice per group. # P < 0.05; significantly different from the OVA‐unsensitized mice. *P < 0.05; significantly different from the control siRNA OVA‐sensitized mice
Animals with AR usually develop sneezing and nose rubbing symptoms as well as increased nasal secretions (Nam, Kim, & Jeong, 2017; Oh, Ryu, Cha, Kim, & Jeong, 2012). Assessment of nasal rubbing immediately after the last nasal challenge showed that it increased for 10 min after OVA challenge, then decreased (data not shown). Additionally, the frequency of nasal rubbing in the RANKL siRNA mice was significantly lower than in the mice that received the control siRNA (Figure 2c). These results clearly indicate that down‐regulation of RANKL in the nasal mucosa tissues ameliorated the AR‐related clinical symptoms in the OVA‐sensitized mice. Serum IgE and histamine levels were significantly attenuated in the RANKL siRNA mice relative to that in the control siRNA mice (Figure 2d,e). Serum TSLP, IL‐1β, IL‐4, IL‐25, and IL‐33 levels were significantly reduced in the RANKL siRNA mice compared with the control siRNA mice (Figure 2f–h). Serum IL‐5 and IL‐13 in the RANKL siRNA also tended to decrease when compared with the control siRNA mice, although this decrease was not significant (Figure 2g). IL‐10, which is also known as Th2 cytokine, is an anti‐inflammatory cytokine that is up‐regulated in OVA‐sensitized mice (Kleinjan et al., 2013). In the present study, the serum IL‐10 levels were significantly reduced in the RANKL siRNA mice compared with the control siRNA mice (Figure 2h). Taken together, these data suggest that RANKL induced an elevated Th2 immune response.
We next examined the effects of RANKL silencing on allergic inflammation in the nasal mucosal tissues. Increased IgE, TSLP, and pro‐inflammatory cytokines (IL‐1β, TNF‐α, IL‐6, and IL‐8) were significantly reduced in the RANKL siRNA mice compared with the control siRNA mice (Figure 2i–n;). Moreover, the increased levels of the chemokine CXCL2 and the adhesion molecule ICAM‐1 in the control siRNA mice were significantly reduced by the RANKL siRNA (Figure 2o,p).
To determine if RANKL silencing reduced infiltration of immune cells including mast cells, eosinophils, T cells, and Foxp3+ Treg cells into the nasal mucosal tissues, we used immunohistochemistry. The counts of these cells in the nasal mucosa tissues are shown in Figure 3 and Supporting Information Figure S1d,e. The respective numbers of inflammatory cells including mast cells in the nasal mucosal tissues of the AR mice were significantly greater than those in the OVA‐unsensitized mice. Histologically, mice treated with the RANKL siRNA exhibited a decreased infiltration of inflammatory cells compared with the control siRNA mice (Figure 3a,b and Supporting Information Figure S1d,e). In addition, the number of Treg cells was significantly reduced by the RANKL siRNA treatment (Figure 3a,b).
Figure 3.
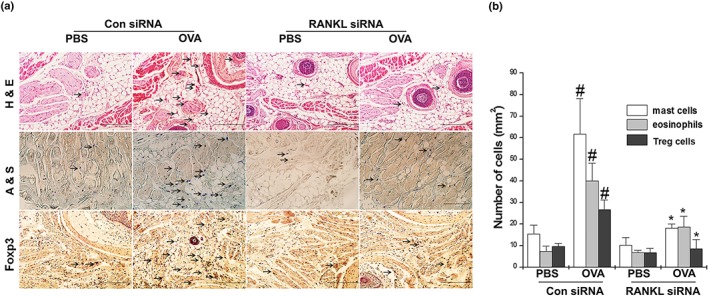
Knockdown of RANKL reduces infiltration of eosinophils, mast cells, and Treg cells into the AR nasal mucosa tissues. Mice were sensitized on Days 1, 5, and 14 by i.p. injections of 100 μg of OVA emulsified in 20 mg of aluminium hydroxide, after which they received local injections of RANKL siRNA and control siRNA, or PBS alone into the nasal cavity, and were challenged with intranasal OVA for 10 days. (a) Nasal mucosa were stained with haematoxylin and eosin (h,e) for eosinophils, Alcian blue and Safranin O (a,s) for mast cells, and immunohistochemical diaminobenzidine stain (for Foxp3) for Treg cells. (b) Mast cells, eosinophils, and Foxp3+ Treg cells were counted by two individuals, after which five randomly selected tissue sections per mouse were counted. The absolute number of cells is shown as the mean ± SEM. # P < 0.05; significantly different from OVA‐unsensitized mice. *P < 0.05; significantly different from control siRNA OVA‐sensitized mice. Scale bar = 100 μm
3.3. Exogenous RANKL exaggerates Th2 immune responses in AR mice
We next determined whether RANKL alone could modulate AR responses. Mice were challenged with RANKL (3 μg), which significantly increased the rub scoring compared with the PBS‐treated mice (Figure 4a). Moreover, exogenous RANKL also increased the serum concentration of IgE and histamine compared with the PBS‐treated mice (Figure 4b,c). To determine the effects of RANKL on the Th2 cytokine levels in the serum, the levels of TSLP, IL‐1β, IL‐4, IL‐5, IL‐10, IL‐13, IL‐25, and IL‐33 were analysed by elisa. The levels of TSLP, IL‐1β, IL‐5, IL‐10, IL‐13, IL‐25, and IL‐33 were significantly increased in the RANKL‐treated mice, but those of IL‐4 were not (Figure 4d–k). However, RANKL did reduce serum IFN‐γ, compared with the PBS‐treated mice (Figure 4l). Co‐challenge with RANKL and OVA synergistically increased the AR responses compared with the OVA‐sensitized mice (Supporting Information Figure S2).
Figure 4.
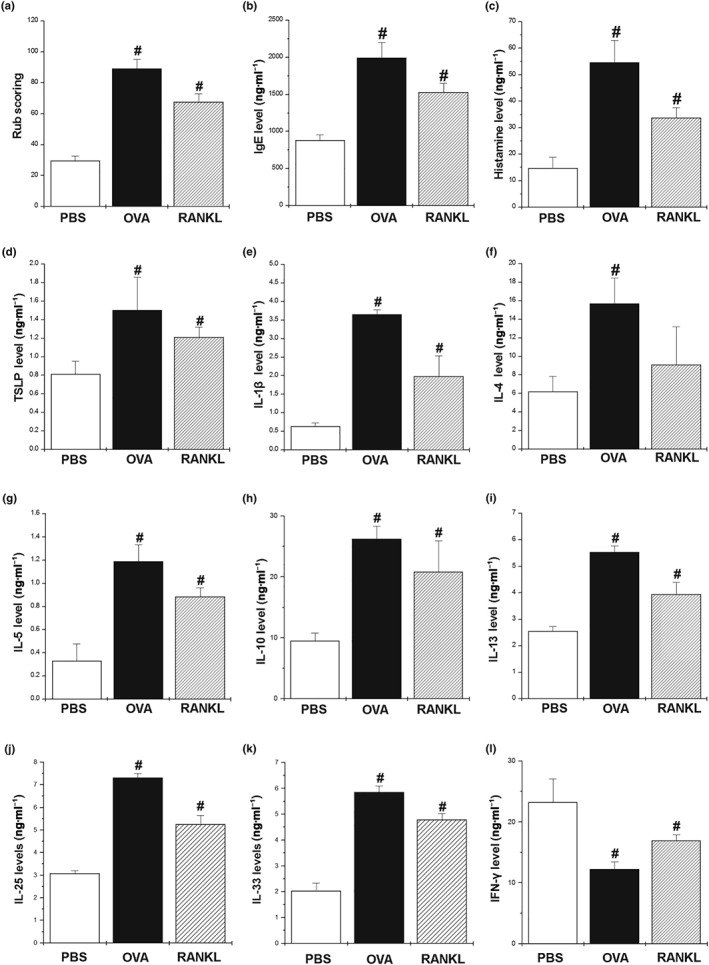
Exogenous RANKL exaggerates Th2 immune responses in AR mice. We sensitized mice on Days 1, 5, and 14 by i.p. injections of 100 μg of OVA emulsified in 20 mg of aluminium hydroxide and challenged mice with 1.5 mg of OVA or recombinant mouse RANKL for 10 days. (a) The number of nose rubs that occurred 10 min after OVA or RANKL intranasal provocation. (b) IgE levels in the serum were measured by elisa. (c) The levels of histamine in serum were measured by a histamine assay method. (d–l) The levels of cytokines in the serum were measured by elisa. Data shown are the means ± SEM from n = 5 mice per group. # P < 0.05; significantly different from OVA‐unsensitized mice (PBS)
3.4. RANKL induces AR by increasing TSLP levels
TSLP is a key factor of AR. To evaluate whether RANKL induced AR responses by increasing the TSLP levels, TSLP was knocked down using TSLP siRNA in RANKL‐induced AR mice. Down‐regulation of TSLP (Figure 5a) in the nasal mucosal tissues ameliorated the rub scoring in the RANKL‐treated mice (Figure 5b), and TSLP‐deficient mice produced markedly less IgE and histamine than that of the control siRNA mice (Figure 5c,d). In addition, increases in the Th2 cytokine levels by RANKL were significantly reduced by the TSLP siRNA treatment (Figure 5e–h).
Figure 5.
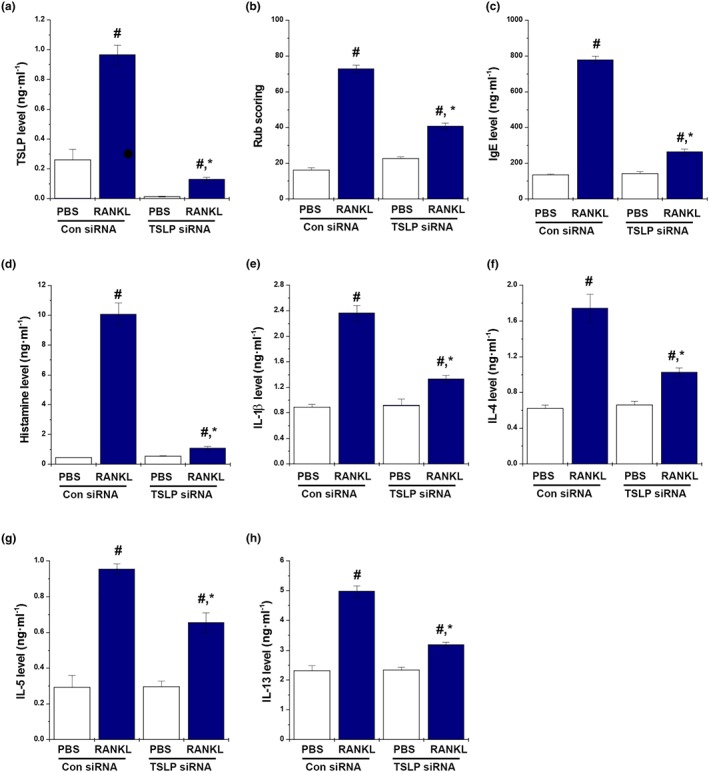
RANKL induces AR via increased TSLP levels. Mice were sensitized on Days 1, 5, and 14 by i.p. injections of 100 μg of OVA emulsified in 20 mg of aluminium hydroxide, after which they received local injections of TSLP siRNA and control siRNA, or PBS alone into the nasal cavity, and were challenged with intranasal RANKL for 10 days. (a) The number of the nose rubs that occurred 10 min after OVA intranasal provocation. (b) IgE levels in the serum were measured by elisa. (c) The levels of histamine in serum were measured as described. (d–h) The levels of cytokine in the serum were measured by elisa. Data shown are the means ± SEM from n = 5 mice per group. # P < 0.05; significantly different from OVA‐unsensitized mice. *P < 0.05; significantly different from control siRNA RANKL‐sensitized mice
Production of TSLP by human mast cells is induced by RANKL. We observed that RANKL was up‐regulated in mast cells and CD4+ T cells. As shown above, the TSLP levels were reduced by RANKL siRNA, and the RANKL‐induced AR responses were inhibited by TSLP siRNA. It is known that RANKL is expressed in activated T cells (Wong et al., 1997), and mast cells are major effectors in AR that induce TSLP production (Moon et al., 2011; Oh, Ryu, Cha, Kim, & Jeong, 2012). Therefore, we investigated whether RANKL can increase inflammatory immune responses by paracrine signalling in mast cells. To determine if RANKL can regulate expression of RANK, human cord blood‐derived mast cells and HMC‐1 cells were treated with RANKL (10 ng·ml−1). Upon stimulation with RANKL, RANK protein and mRNA expressions were clearly detected (Figure 6a and Supporting Information Figure S3). Co‐localization of RANKL and RANK was observed on the surface of the mast cells (Figure 6a).
Figure 6.
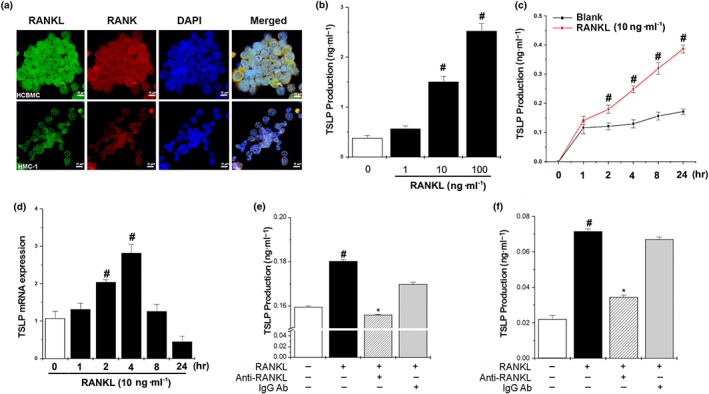
RANKL induces production of TSLP from human mast cells. (a) RANK was expressed on the surface of human cord blood‐derived mast cells (Original magnification ×800) and HMC‐1 cells (Original magnification ×800). (b) HMC‐1 cells were stimulated with various concentrations of RANKL (1, 10, and 100 ng·ml−1) for 24 hr, after which the production of TSLP in the supernatant was measured by elisa. (c) HMC‐1 cells (3 × 105) were stimulated with RANKL (10 ng·ml−1) for various times. The production of TSLP in the supernatant was measured by elisa. (d) HMC‐1 cells (3 × 106) were treated with RANKL (10 ng·ml−1) for various times, after which TSLP mRNA expression was analysed by RT‐PCR. (e) HMC‐1 cells were treated with RANKL (10 ng·ml−1), RANKL neutralizing antibodies (0.5 μg·ml−1), and/or IgG isotype for 24 hr, after which the production of TSLP in the supernatant was measured by elisa. (f) Human cord blood‐derived mast cells (HCBMC ) were treated with RANKL (10 ng·ml−1), RANKL neutralizing antibodies (0.5 μg·ml−1), and/or IgG isotype for 24 hr, after which the production of TSLP in the supernatant was measured by elisa. Data shown are the means ± SEM from n = 5 per group. # P < 0.05; significantly different from unstimulated cells' value. *P < 0.05; significantly different from RANKL value
To test whether interaction of RANKL and RANK could functionally activate human mast cells, we stimulated HMC‐1 cells for various times with RANKL and analysed the mRNA expression of TSLP, which leads to pathogenic inflammatory Th2 cell responses in an allergic reaction (Jariwala et al., 2011). As shown in Figure 6b, RANKL (10 and 100 ng·ml−1) significantly increased the production of TSLP in aconcentration‐dependent manner. Therefore, RANKL (10 ng·ml−1) was selected to stimulate mast cells. Additionally, RANKL increased the TSLP production in a time‐dependent manner (Figure 6c). The mRNA expression of TSLP increased when the cells were exposed to RANKL, reaching a plateau at 4 hr (Figure 6d). In addition, RANKL increased the TSLP production in human cord blood‐derived mast cells, while the increased TSLP levels were significantly reduced by anti‐RANKL neutralizing antibody (Figure 6e,f).
To identify the effects of RANKL on the production of other proinflammatory cytokines in HMC‐1 cells, HMC‐1 cells were treated with RANKL (1–100 ng·ml−1) for 24 hr. The production of TNF‐α, IL‐6, and IL‐8 was significantly increased by RANKL in a dose‐dependent manner (Supporting Information Figure S4a–c), while neutralization of RANKL significantly reduced the production of TNF‐α, IL‐6, and IL‐8 compared with the RANKL group (Supporting Information Figure S4d–f). In the presence of the anti‐IgG antibody , the RANKL‐induced TSLP production was not significantly reduced, compared to the RANKL control (Figure 6e,f and Supporting Information Figure S4d–f). Finally, RANKL or compound 48/80 (mast cell degranulator) induced the release of histamine, PGD2, and LTC4 from ex vivo primary mast cells (Supporting Information Figure S5).
3.5. Involvement of MAPKs, NF‐κB, PI3K, and caspase‐1 in RANKL‐induced TSLP production
Interaction of RANKL and RANK activates many intracellular signalling pathways. To identify the signalling pathways involved in RANKL‐induced TSLP production, we used specific pharmacological inhibitors (PD98059, SP600125, SB203580, wortmannin, PDTC, and caspase‐1 inhibitor) targeting ERK, JNK, p38, PI3K, NF‐κB, and caspase‐1. Notably, the increase in TSLP production induced by RANKL was significantly blocked by PD98059, SP600125, SB203580, wortmannin, PDTC, and caspase‐1 inhibitor (Supporting Information Figure 7a). Additionally, SB203580, SP600125, or wortmannin almost completely inhibited the RANKL‐induced TSLP production (Figure 7a). To gain insight into the role of RANKL on PI3K, MAPKs, NF‐κB, and caspase‐1 signalling, western blotting was performed. The results revealed that RANKL markedly increased the activation of PI3K, p38, JNK, and ERK within 30 min (Figure 7b–e). Moreover, RANKL markedly increased the activation of NF‐κB and caspase‐1 within 24 hr (Figure 7f–h), while neutralization of RANKL markedly suppressed the activation of PI3K, p38, JNK, ERK, NF‐κB, and caspase‐1 (Figure 7i,j). However, these effects were not changed by anti‐IgG antibody (Figure 7i,j).
Figure 7.
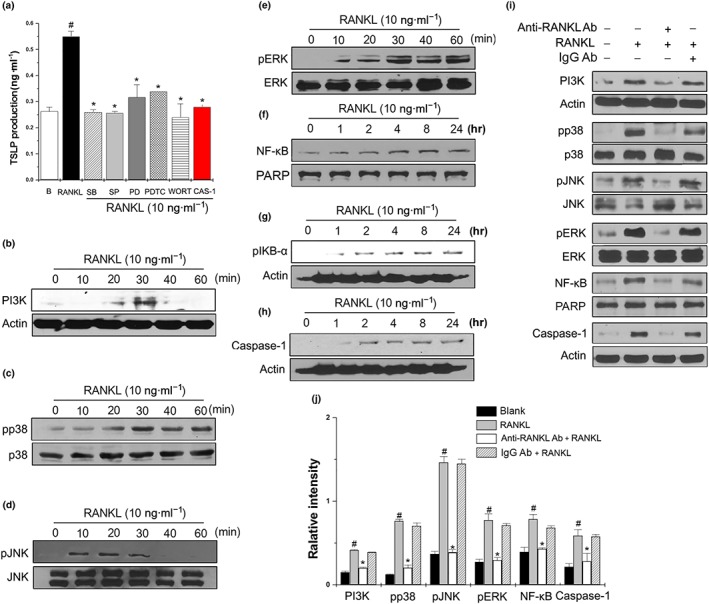
Involvement of MAPKs, NF‐κB, PI3K, and caspase‐1 in RANKL‐induced TSLP production. (a) HMC‐1 cells (3× 105) were pretreated with 0.2 μM SB203580 (SB), 10 μM SP600125 (SP), 1 μM PD98059 (PD), 10 μM PDTC, 1 μM wortmannin (WORT), or 0.05 μM caspase‐1 inhibitor (CAS‐1) for 1 hr, then incubated with 10 ng·ml−1 RANKL for 24 hr. The production of TSLP was measured by elisa. Data shown are the means ± SEM from n = 5 per group. # P < 0.05; significantly different from unstimulated cells' value. *P < 0.05; significantly different from RANKL' value. (b–h) HMC‐1 cells (3 × 106) were stimulated with 10 ng·ml−1 of RANKL for the indicated times. The activation of PI3K, MAPKs, NF‐κB, and caspase‐1 were determined by Western blot analysis. (i) HMC‐1 cells were treated with RANKL (10 ng·ml−1), RANKL neutralizing antibodies (0.5 μg·ml−1), and/or IgG isotype for the indicated times, after which the activation of PI3K, MAPKs, NF‐κB, and caspase‐1 were determined by Western blot analysis. (j) Relative intensities of protein levels were quantified by densitometry. Data shown are the means ± SEM from n = 5 per group. # P < 0.05; significantly different from unstimulated cells' value. *P < 0.05; significantly different from RANKL value
3.6. Regulatory effect of dexamethasone in RANKL‐induced inflammatory reactions in in vivo and in vitro models
We showed that the RANKL/RANK system might be one of the key signals accelerating mast cell‐mediated allergic inflammatory responses. To further demonstrate our hypothesis, we evaluated the effect of dexamethasone (corticosteroid) on the RANKL/RANK systems both in vivo and in vitro. The increase in rub scoring induced by RNAKL was significantly decreased by administration of dexamethasone (Figure 8a), while the up‐regulation of serum levels of histamine and IgE by RANKL were significantly down‐regulated by administration of dexamethasone (Figure 8b,c). In addition, dexamethasone significantly down‐regulated the levels of serum TSLP and Th2 cytokines in RANKL‐treated mice (Figure 8d–h). Next, we investigated the effects of dexamethasone in RANKL‐stimulated HMC‐1 cells and found that it decreased TSLP production through inhibition of PI3K, phosphorylated ERK, and caspase‐1 activation (Figure 8i,j).
Figure 8.
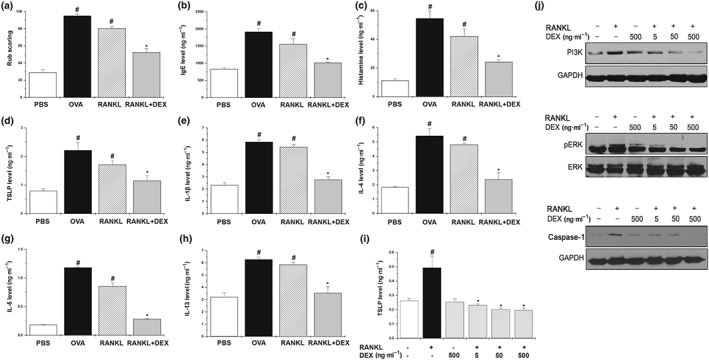
Regulatory effect of dexamethasone in RANKL‐induced inflammatory reactions in vivo and in vitro. Mice were treated orally with dexamethasone (DEX; 5 mg·kg−1) for 10 days before the intranasal RANKL challenge. (a) The number of the nasal rubs that occurred in the 10 min after the RANKL intranasal provocation. Serum was isolated from blood and then assayed for (b) IgE, (c) histamine, (d) TSLP, (e) IL‐1β, (f) IL‐4, (g) IL‐5, and (h) IL‐13. Data shown are the means ± SEM from n = 5 mice per group. (i) HMC‐1 cells (3 × 105) were pretreated with dexamethasone for 1 hr, then incubated with 10 ng·ml−1 of RANKL for 24 hr. The production of TSLP was measured by elisa. Data are represented as the mean ± SEM with n = 5 per group. (j) HMC‐1 cells (3 × 106) were stimulated with 10 ng·ml−1 of RANKL for the indicated times. The activation of PI3K, phosphorylated ERK (pERK), and caspase‐1 was determined by Western blot analysis. # P < 0.05; significantly different from the untreated group. *P < 0.05; significantly different from the RANKL‐treated group
4. DISCUSSION
AR is a persistent inflammatory disease of the upper airways regulated by a number of cytokines (Galli et al., 2012; Isobe et al., 2012). In this study, we found that RANKL expression was up‐regulated in AR patients compared with the normal group and was strongly inducible in both CD4+ T cells and mast cells in the nasal mucosal tissues of the AR group. In addition, we found that RANK was expressed in mast cells and colocalized with RANKL in the mast cells of AR nasal mucosal tissues. Therefore, we suggest that RANKL has an important role in AR responses by binding to RANK on the mast cell surface.
RANKL has a critical part in many inflammatory disorders and its overexpression is related to the development of postmenopausal osteoporosis, rheumatoid arthritis, bone metastasis, and experimental autoimmune encephalomyelitis (Guerrini et al., 2015; Melagraki et al., 2018; Theill et al., 2002). In the present study, we showed that the RANKL levels in AR patients and animals were higher than normal. In addition, the levels of RANKL in the serum were correlated with the severity of the AR symptoms. From this, we suggest that RANKL is an important factor in AR.
In this study, the RANKL levels in the serum were obtained from patients with AR that had the same sex ratio and similar ages as the normal subjects. However, the RANKL levels in the nasal mucosa were obtained from a higher percentage of male patients with AR. Findlay et al. (2008) reported that the RANKL levels in female osteoarthritis patients were higher than in male patients. Doumouchtsis et al. (2007) reported that the levels of RANKL in male haemodialysis patients were significantly higher than in female patients. In this study, the difference in the RANKL levels between male and female AR groups was not statistically significant (data not shown). Further study is necessary in many more patients to clarify the effect of sex on levels of RANKL in AR. In addition, we suggest that it would have been more informative to obtain serum from the same patients who provided the nasal mucosal samples.
In our subsequent experiments, we investigated the role of RANKL/RANK signalling in an AR mouse model through the addition of exogenous RANKL into the nostrils of mice or deletion of RANKL by siRNA treatment. Intranasal treatment with exogenous RANKL increased the levels of clinical nasal symptoms, IgE, histamine, Th2 cytokines, TSLP, IL‐25, IL‐33, CXCL2, and ICAM‐1, whereas RANKL deficiency effectively inhibited the symptoms of clinical AR and the production of AR‐related biomarkers including TSLP. AR is characterized by an inflammatory infiltrate consisting of mast cells, eosinophils, T cells, Tregs, and DCs (May & Dolen, 2017). Infiltrations of eosinophils and mast cells increase in response to ICAM‐1 and CXCL2 (Gonzalo et al., 1996). Eosinophils have an important role in Th2‐driven immune responses (Isobe et al., 2012) and contribute to the pathogenesis of AR by secreting eosinophil‐derived neurotoxin, eosinophil peroxidase, and lipid mediators (Rondón et al., 2012; Isobe et al., 2012). In addition, eosinophils release cytokines such as IL‐3, IL‐5, IL‐32, granulocyte‐macrophage colony‐stimulating factor, proinflammatory cytokines and chemokines, which have crucial roles in late phase and on‐going allergic inflammation (Jeong et al., 2011). The CD4+CD25+Foxp3+Treg cells regulate allergic inflammation by inhibiting inappropriate effector Th2 responses in AR (Pawankar et al., 2011). Mast cells are mostly found in the epithelial compartment of the nasal mucosa and are considered to be key regulators of inflammatory and immediate allergic reactions including AR, asthma, and atopic dermatitis (Moon et al., 2011; Oh, Ryu, Cha, Kim, & Jeong, 2012; Galli et al., 2012). These cells release potent inflammatory mediators such as histamine, proteases, and cytokines and induce eosinophilic inflammation by IgE‐dependent and IgE‐independent pathways (Han et al., 2010; Galli et al., 2012). We previously reported that mast cells produced TSLP, which increased the mast cell proliferation and allergic reactions (Moon et al., 2011; Han et al., 2014). Our previous study also showed that the depletion of TSLP ameliorated clinical symptoms in mice with OVA‐induced AR (Nam et al., 2018). In the present study, the silencing of RANKL significantly decreased the number of mast cells, eosinophils, T cells, and Foxp3+Tregs in the nasal mucosa, and TSLP deficiency markedly reduced the RANKL‐induced AR reaction. Taken together, these results suggest that RANKL promotes allergic inflammatory reaction by inducing TSLP.
The epithelial‐derived alarmins, TSLP, IL‐25, and IL‐33 amplify allergic inflammation by up‐regulating Th2 type inflammation (Mitchell & O'Byrne, 2017; Wang et al., 2007). In this study, RANKL increased the levels of Th2 cytokines, TSLP, IL‐25, and IL‐33, whereas it reduced the levels of IFN‐γ, compared with PBS‐treated mice. Collectively, our findings support the notion that RANKL/RANK is involved in the inflammation of AR through the induction of Th2 type immune responses and suggest that RANKL may be a crucial mediator for the development of AR.
Jung et al. (2014) recently reported that allergic diseases are related to clinically meaningful reductions in bone mineral density. As an osteoclast differentiation factor RANKL is involved in the reduction of bone mineral density and induces osteoporosis (Anderson et al., 1997). In addition, it promotes an inflammatory reaction by an autocrine mechanism (Ock et al., 2012). In the present study, we have shown that RANKL and RANK were co‐localized on the surface of mast cells and we detected the mRNA expression of RANK in human mast cells. Accumulating evidence suggests that RANKL is frequently involved in inflammatory and immune responses by inducing proinflammatory cytokine production and is a chemotactic factor for monocytes and neutrophils (Riegel et al., 2012; Seshasayee et al., 2004). Blocking of RANKL by RANK‐Fc protected mice from death that was induced by sepsis and inflammation‐mediated arthritis (Seshasayee et al., 2004). Recently, RANKL was shown to induce myocardial inflammation by stimulating expression of proinflammatory cytokines, including TNF‐α, IL‐1, and IL‐1β (Ock et al., 2012). TSLP is a newly discovered cytokine that has an important role in the AR reaction and mast cell differentiation (Han et al., 2014). In addition, TSLP is a regulator of Th2 responses because it promotes Th2 cytokine production, which orchestrates allergic inflammation (Leonard, 2002). In the present study, we showed that RANKL induced the production of TSLP and that the neutralization of RANKL significantly decreased TSLP production in human mast cells. Therefore, our results suggest that RANKL accelerates allergic inflammatory reactions by increasing TSLP production in mast cells.
The results of our studies indicate that the biochemical pathways involved in RANKL signalling in human mast cells appear to be mediated through the MAPKs, NF‐κB, PI3K, and caspase‐1 pathways. Previous studies have shown that RANKL induces the secretion of proinflammatory cytokines through the activation of the p38 MAPK, JNK, ERK, and NF‐κB pathways (Kim et al., 2010; Riegel et al., 2012; Seshasayee et al., 2004). Moreover, PI3K activates intracellular calcium levels, and its blocking by Wortmannin inhibits antigen‐mediated mast cell degranulation and cytokine production in both rodent and human mast cells (Okayama et al., 2003). Furthermore, caspase‐1 is required for the release of IL‐1β and TSLP in HMC‐1 cells (Moon et al., 2011). In the present study, we showed that the inhibition of MAPKs, NF‐κB, PI3K, and caspase‐1 by specific inhibitors significantly suppressed the TSLP production by mast cells, while dexamethasone reduced the RANKL‐induced inflammatory reactions in the in vivo and in vitro models. Therefore, we suggest that RANKL signalling in mast cells occurs through the activation of the MAPKs, NF‐κB, PI3K, and caspase‐1 pathways.
In conclusion, we have described here, for the first time, that RANKL is increased in the mast cells of AR patients and mice. We also showed that the RANKL/RANK system is one of the key signals accelerating mast cell‐mediated allergic inflammatory responses (Figure 9). Therefore, RANKL/RANK signalling might be a therapeutic target for the treatment of AR.
Figure 9.
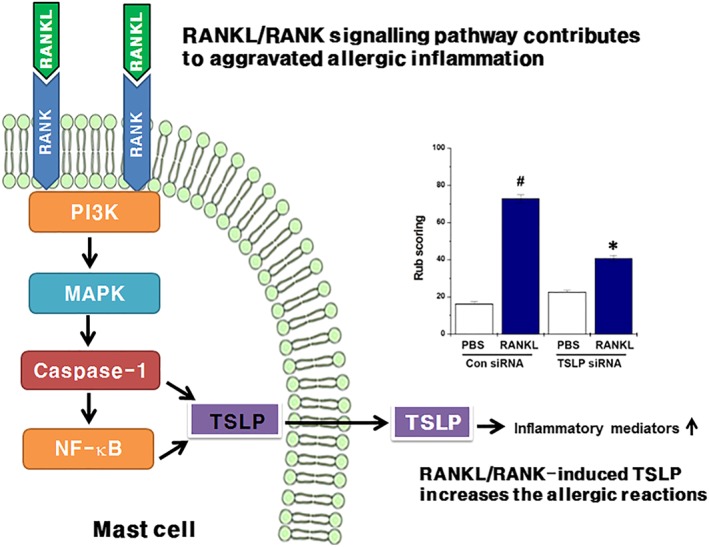
Schematic diagram of the mechanisms involved in allergic inflammatory signalling by the RANKL/RANK system. RANKL/RANK stimulation activates the PI3K, MAPK, caspase‐1, and NF‐κB signalling pathways in mast cells. Activated caspase‐1 and NF‐κB induces the production and transcription of TSLP. Finally, RANKL/RANK‐induced TSLP increases the allergic reactions through the up‐regulation of inflammatory mediators
AUTHOR CONTRIBUTIONS
H.‐J.J. and H.‐M.K. performed overall study design and supervision; S.‐Y.N. and H.‐Y.K. did the experiments and data analysis; J.‐Y.M. collected and analysed the samples; H.‐J.J., H.‐M.K., S.‐Y.N., and H.‐Y.K. wrote, revised, and edited the manuscript.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, Immunoblotting and Immunochemistry, and Animal Experimentation, and as recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1.
RANKL is up‐regulated in T cells. (A) Nasal mucosa tissues from AR patients were double stained by immunofluorescence with anti‐CD4 (green) and anti‐RANKL antibody (red) (magnification, × 60). (B) T cells were counted by three individuals in human tissues. Data are represented as the mean ± S.E.M. #P < 0.05; significantly different from the normal. Mice were sensitized on days 1, 5, and 14 by intraperitoneal injections of 100 μg of OVA emulsified in 20 mg of aluminium hydroxide and then received local injections of RANKL siRNA and control siRNA, or PBS alone into the nasal cavity, and challenged with intranasal OVA for 10 days. (C) Fluorescence intensities in nasal mucosa tissues of normal mice and AR mice were determined as threshold values. (D) Nasal mucosa tissues from normal mice and AR mice were double stained by immunofluorescence with anti‐CD4 (green) and anti‐RANKL antibody (red) (magnification, × 60). (E) T cells were counted by three individuals in animal tissues. Data are represented as the mean ± S.E.M. #P < 0.05; significantly different from the PBS. *P < 0.05; significantly different from the control siRNA OVA.
Figure S2.Co‐challenge of RANKL and OVA synergistically increased AR responses. We sensitized mice on days 1, 5, and 14 by intraperitoneal injections of 100 μg of OVA emulsified in 20 mg of aluminium hydroxide and we challenged mice with 1.5 mg of OVA. Mice received rmRANKL before the intranasal OVA challenge for 10 days. (A) The rub number of the nose that occurred 10 min after OVA intranasal provocation. Levels of (B) IgE, (C) histamine, and (D‐G) cytokine in the serum. Data are represented as the mean ± S.E.M. with n = 5 mice per group. #P < 0.05; significantly different from the OVA‐unsensitized mice. *P < 0.05; significantly different from the OVA‐sensitized mice.
Figure S3. RANK was expressed on human mast cell. RANK mRNA expressions in human cord blood‐derived mast cells (HCBMC) and HMC‐1 cells were analysed by the RT‐PCR.
Figure S4. RANKL induces production of pro‐inflammatory cytokines from HMC‐1 cells. (A‐C) HMC‐1 cells (1 × 105) were stimulated with RANKL (1, 10, and 100 ng/ml) for 24 hr. The production of pro‐inflammatory cytokines in the supernatant was measured by the elisa method. (D‐F) HMC‐1 cells were treated with RANKL (10 ng/ml), RANKL neutralizing antibodies (0.5 μg/ml), and/or IgG isotype for 24 hr. The production of pro‐inflammatory cytokines in the supernatant was measured by the elisa method. Each datum represents the mean ± SEM of five independent experiments. #P < 0.05; significantly different from unstimulated cells. *P < 0.05; significantly different from RANKL.
Figure S5. RANKL induces degranulation of rat peritoneal mast cells (RPMCs). RPMCs were stimulated with RANKL (10 ng/ml), compound 48/80 (6 μg/ml), RANKL + Anti‐RANKL Ab (Anti‐RANKL) for 20 and 60 min. The levels of (A) histamine, (B) PGD2, and (C) LTC4 were measured from cell supernatant. Data were expressed as mean ± SEM of five independent experiments with duplicate samples. #P < 0.05; significantly different from unstimulated cells. *P < 0.05; significantly different from RANKL’ value.
Table S1 Baseline characteristics for the normal and AR groups
ACKNOWLEDGEMENTS
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2015R1D1A1A01056607).
Nam S‐Y, Kim H‐Y, Min J‐Y, Kim H‐M, Jeong H‐J. An osteoclastogenesis system, the RANKL/RANK signalling pathway, contributes to aggravated allergic inflammation. Br J Pharmacol. 2019;176:1664–1679. 10.1111/bph.14615
Contributor Information
Hyung‐Min Kim, Email: hmkim@khu.ac.kr.
Hyun‐Ja Jeong, Email: hjjeong@hoseo.edu.
REFERENCES
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. British Journal of Pharmacology, 174, S225–S271. 10.1111/bph.13876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. British Journal of Pharmacology, 174, S272–S359. 10.1111/bph.13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali, A. , Lax, A. S. , Liljeström, M. H. , Paakkari, I. , Ashammakhi, N. , Kovanen, P. T. , & Konttinen, Y. T. (2006). Mast cells in atherosclerosis as a source of the cytokine RANKL. Clinical Chemistry and Laboratory Medicine, 44, 672–674. [DOI] [PubMed] [Google Scholar]
- Allakhverdi, Z. , Comeau, M. R. , Jessup, H. K. , Yoon, B. R. , Brewer, A. , Chartier, S. , … Delespesse, G. (2007). Thymic stromal lymphopoietin is released by human epithelial cells in response to microbes, trauma, or inflammation and potently activates mast cells. The Journal of Experimental Medicine, 204, 253–258. 10.1084/jem.20062211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin, K. (2012). The role of mast cells in allergic inflammation. Respiratory Medicine, 106, 9–14. 10.1016/j.rmed.2011.09.007 [DOI] [PubMed] [Google Scholar]
- Anderson, D. M. , Maraskovsky, E. , Billingsley, W. L. , Dougall, W. C. , Tometsko, M. E. , Roux, E. R. , … Galibert, L. (1997). A homologue of the TNF receptor and its ligand enhance T‐cell growth and dendritic‐cell function. Nature, 390, 175–179. 10.1038/36593 [DOI] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175, 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doumouchtsis, K. K. , Kostakis, A. I. , Doumouchtsis, S. K. , Tziamalis, M. P. , Tsigris, C. , Kostaki, M. A. , & Perrea, D. N. (2007). sRANKL/osteoprotegerin complex and biochemical markers in a cohort of male and female hemodialysis patients. Journal of Endocrinological Investigation, 30, 762–766. 10.1007/BF03350814 [DOI] [PubMed] [Google Scholar]
- Findlay, D. , Chehade, M. , Tsangari, H. , Neale, S. , Hay, S. , Hopwood, B. , … Fazzalari, N. (2008). Circulating RANKL is inversely related to RANKL mRNA levels in bone in osteoarthritic males. Arthritis Research & Therapy, 10, R2 10.1186/ar2348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli, S. J. , & Tsai, M. (2012). IgE and mast cells in allergic disease. Nature Medicine, 18, 693–704. 10.1038/nm.2755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalo, J. A. , Lloyd, C. M. , Kremer, L. , Finger, E. , Martinez, A. C. , Siegelman, M. H. , … Gutierrez‐Ramos, J. C. (1996). Eosinophil recruitment to the lung in a murine model of allergic inflammation. The role of T cells, chemokines, and adhesion receptors. The Journal of Clinical Investigation, 98, 2332–2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrini, M. M. , Okamoto, K. , Komatsu, N. , Sawa, S. , Danks, L. , Penninger, J. M. , et al. (2015). Inhibition of the TNF family cytokine RANKL prevents autoimmune inflammation in the central nervous system. Immunity, 43, 1174–1185. 10.1016/j.immuni.2015.10.017 [DOI] [PubMed] [Google Scholar]
- Han, D. , Wang, C. , Lou, W. , Gu, Y. , Wang, Y. , & Zhang, L. (2010). Allergen‐specific IL‐10‐secreting type I T regulatory cells, but not CD4(+)CD25(+)Foxp3(+) T cells, are decreased in peripheral blood of patients with persistent allergic rhinitis. Clinical Immunology, 136, 292–301. [DOI] [PubMed] [Google Scholar]
- Han, N. R. , Oh, H. A. , Nam, S. Y. , Moon, P. D. , Kim, D. W. , Kim, H. M. , & Jeong, H. J. (2014). TSLP induces mast cell development and aggravates allergic reactions through the activation of MDM2 and STAT6. The Journal of Investigative Dermatology, 134, 2521–2530. 10.1038/jid.2014.198 [DOI] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , et al. (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoya, K. , Satoh, T. , Yamamoto, Y. , Saeki, K. , Igawa, K. , Okano, M. , et al. (2011). Gene silencing of STAT6 with siRNA ameliorates contact hypersensitivity and allergic rhinitis. Allergy, 66, 124–131. 10.1111/j.1398-9995.2010.02440.x [DOI] [PubMed] [Google Scholar]
- Isobe, Y. , Kato, T. , & Arita, M. (2012). Emerging roles of eosinophils and eosinophil‐derived lipid mediators in the resolution of inflammation. Frontiers in Immunology, 3, 270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jariwala, S. P. , Abrams, E. , Benson, A. , Fodeman, J. , & Zheng, T. (2011). The role of thymic stromal lymphopoietin in the immunopathogenesis of atopic dermatitis. Clinical & Experimental Allergy, 41, 1515–1520. 10.1111/j.1365-2222.2011.03797.x [DOI] [PubMed] [Google Scholar]
- Jeong, H. J. , Koo, H. N. , Na, H. J. , Kim, M. S. , Hong, S. H. , Eom, J. W. , … Kim, H. M. (2002). Inhibition of TNF‐alpha and IL‐6 production by Aucubin through blockade of NF‐kappa B activation RBL‐2H3 mast cell. Cytokine, 18, 252–259. 10.1006/cyto.2002.0894 [DOI] [PubMed] [Google Scholar]
- Jeong, H. J. , Shin, S. Y. , Oh, H. A. , Kim, M. H. , Cho, J. S. , & Kim, H. M. (2011). IL‐32 up‐regulation is associated with inflammatory cytokine production in allergic rhinitis. The Journal of Pathology, 224, 553–563. 10.1002/path.2899 [DOI] [PubMed] [Google Scholar]
- Jung, J. W. , Kang, H. R. , Kim, J. Y. , Lee, S. H. , Kim, S. S. , & Cho, S. H. (2014). Are asthmatic patients prone to bone loss? Annals of Allergy, Asthma & Immunology, 112, 426–431. 10.1016/j.anai.2014.02.013 [DOI] [PubMed] [Google Scholar]
- Kartsoginannis, V. , Zhou, H. , Horwood, N. J. , Thomas, R. J. , Hards, D. K. , Quinn, J. M. , et al. (1999). Localization of RANKL (receptor activator of NF kappa B ligand) mRNA and protein in skeletal and extraskeletal tissues. Bone, 25, 525–534. 10.1016/S8756-3282(99)00214-8 [DOI] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). NC3Rs reporting guidelines working group. British Journal of Pharmacology, 160, 1577–1579. 10.1111/j.1476-5381.2010.00872.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J. , Min, J. K. , Park, J. A. , Doh, H. J. , Choi, Y. S. , Rho, J. , … Kwon, Y. G. (2010). Receptor activator of nuclear factor kappaB ligand is a novel inducer of tissue factor in macrophages. Circulation Research, 107, 871–876. 10.1161/CIRCRESAHA.110.221168 [DOI] [PubMed] [Google Scholar]
- Kleinjan, A. , van Nimwegen, M. , Leman, K. , Hoogsteden, H. C. , & Lambrecht, B. N. (2013). Topical treatment targeting sphingosine‐1‐phosphate and sphingosine lyase abrogates experimental allergic rhinitis in a murine model. Allergy, 68, 204–212. 10.1111/all.12082 [DOI] [PubMed] [Google Scholar]
- Leonard, W. J. (2002). TSLP: Finally in the limelight. Nature Immunology, 3, 605–607. 10.1038/ni0702-605 [DOI] [PubMed] [Google Scholar]
- Loser, K. , Mehling, A. , Loeser, S. , Apelt, J. , Kuhn, A. , Grabbe, S. , … Beissert, S. (2006). Epidermal RANKL controls regulatory T‐cell numbers via activation of dendritic cells. Nature Medicine, 12, 1372–1379. 10.1038/nm1518 [DOI] [PubMed] [Google Scholar]
- May, J. R. , & Dolen, W. K. (2017). Management of allergic rhinitis: A review for the community pharmacist. Clinical Therapeutics, 39, 2410–2449. 10.1016/j.clinthera.2017.10.006 [DOI] [PubMed] [Google Scholar]
- McGrath, J. C. , & Lilley, E. (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): New requirements for publication in BJ P. British Journal of Pharmacology, 172, 3189–3193. 10.1111/bph.12955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melagraki, G. , Leonis, G. , Ntougkos, E. , Rinotas, V. , Papaneophytou, C. , Mavromoustakos, T. , et al. (2018). Current status and future prospects of small‐molecule protein‐protein interaction (PPI) inhibitors of tumor necrosis factor (TNF) and receptor activator of NF‐κB ligand (RANKL). Current Topics in Medicinal Chemistry, 18, 661–673. 10.2174/1568026618666180607084430 [DOI] [PubMed] [Google Scholar]
- Mitchell, P. D. , & O'Byrne, P. M. (2017). Epithelial‐derived cytokines in asthma. Chest, 151, 1338–1344. [DOI] [PubMed] [Google Scholar]
- Moon, P. D. , & Kim, H. M. (2011). Thymic stromal lymphopoietin is expressed and produced by caspase‐1/NF‐κB pathway in mast cells. Cytokine, 54, 239–243. 10.1016/j.cyto.2011.03.007 [DOI] [PubMed] [Google Scholar]
- Nam, S. Y. , Kim, H. M. , & Jeong, H. J. (2017). The potential protective role of taurine against experimental allergic inflammation. Life Sciences, 184, 18–24. 10.1016/j.lfs.2017.07.007 [DOI] [PubMed] [Google Scholar]
- Nam, S. Y. , Kim, H. Y. , Han, N. R. , Moon, P. D. , Cho, J. S. , Kim, H. M. , & Jeong, H. J. (2018). Src‐type tyrosine kinase p56lck is critical for thymic stromal lymphopoietin‐induced allergic rhinitis. Clinical and Experimental Allergy, 48, 875–889. [DOI] [PubMed] [Google Scholar]
- Ock, S. , Ahn, J. , Lee, S. H. , Park, H. , Son, J. W. , Oh, J. G. , … Kim, J. (2012). Receptor activator of nuclear factor‐kB ligand is a novel inducer of myocardial inflammation. Cardiovascular Research, 94, 105–114. 10.1093/cvr/cvs078 [DOI] [PubMed] [Google Scholar]
- Oh, H. A. , Ryu, J. G. , Cha, W. S. , Kim, H. M. , & Jeong, H. J. (2012). Therapeutic effects of traditional Korean medicine, Jeechool‐Whan in allergic rhinitis model. Tang, 2, e9. [Google Scholar]
- Okayama, Y. , Tkaczyk, C. , Metcalfe, D. D. , & Gilfillan, A. M. (2003). Comparison of FcεRI‐ and FcγRI‐mediated degranulation and TNF‐α synthesis in human mast cells: Selective utilization of phosphatidylinositol‐3‐kinase for FcγRI‐induced degranulation. European Journal of Immunology, 33, 1450–1459. 10.1002/eji.200323563 [DOI] [PubMed] [Google Scholar]
- Pawankar, R. , Mori, S. , Ozu, C. , & Kimura, S. (2011). Overview on the pathomechanisms of allergic rhinitis. Asia Pacific Allergy, 1, 157–167. 10.5415/apallergy.2011.1.3.157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riegel, A. , Maurer, T. , Prior, B. , Stegmaier, S. , Heppert, V. , Wagner, C. , & Hänsch, G. M. (2012). Human polymorphonuclear neutrophils express RANK and are activated by its ligand, RANKL. European Journal of Immunology, 42, 975–981. 10.1002/eji.201141786 [DOI] [PubMed] [Google Scholar]
- Rondón, C. , Campo, P. , Togias, A. , Fokkens, W. J. , Durham, S. R. , Powe, D. G. , … Blanca, M. (2012). Local allergic rhinitis: Concept, pathophysiology, and management. The Journal of Allergy and Clinical Immunology, 129, 1460–1467. 10.1016/j.jaci.2012.02.032 [DOI] [PubMed] [Google Scholar]
- Rothenberg, M. E. , Spergel, J. M. , Sherrill, J. D. , Annaiah, K. , Martin, L. J. , Cianferoni, A. , … Hakonarson, H. (2010). Common variants at 5q22 associate with pediatric eosinophilic esophagitis. Nature Genetics, 42, 289–291. 10.1038/ng.547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schett, G. , Hayer, S. , Zerina, J. , Redlich, K. , & Smolen, J. S. (2005). Mechanisms of disease: The link between RANKL and arthritic bone disease. Nature clinical practice. Rheumatology, 1, 47–54. [DOI] [PubMed] [Google Scholar]
- Seshasayee, D. , Wang, H. , Lee, W. P. , Gribling, P. , Ross, J. , Van Bruggen, N. , et al. (2004). A novel in vivo role for osteoprotegerin ligand in activation of monocyte effector function and inflammatory response. Journal of Biological Chemistry, 279, 30202–30209. [DOI] [PubMed] [Google Scholar]
- Theill, L. E. , Boyle, W. J. , & Penninger, J. M. (2002). RANK‐L and RANK: T cells, bone loss, and mammalian evolution. Annual Review of Immunology, 20, 795–823. 10.1146/annurev.immunol.20.100301.064753 [DOI] [PubMed] [Google Scholar]
- Wada, T. , Nakashima, T. , Hiroshi, N. , & Penninger, J. M. (2006). RANKL‐RANK signaling in osteoclastogenesis and bone disease. Trends in Molecular Medicine, 12, 17–25. 10.1016/j.molmed.2005.11.007 [DOI] [PubMed] [Google Scholar]
- Walsh, M. C. , & Choi, Y. (2014). Biology of the RANKL‐RANK‐OPG system in immunity, bone and beyond. Frontiers in Immunology, 5, 511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. H. , Angkasekwinai, P. , Lu, N. , Voo, K. S. , Arima, K. , Hanabuchi, S. , et al. (2007). IL‐25 augments type 2 immune responses by enhancing the expansion and functions of TSLP‐DC‐activated Th2 memory cells. The Journal of Experimental Medicine, 204, 1837–1847. 10.1084/jem.20070406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, B. R. , Josien, R. , Lee, S. Y. , Sauter, B. , Li, H. L. , Steinman, R. M. , & Choi, Y. (1997). TRANCE (tumor necrosis factor‐ related activation‐induced cytokine), a new TNF family member predominantly expressed in T cells, is a dendritic cell‐specific survival factor. The Journal of Experimental Medicine, 186, 2075–2080. 10.1084/jem.186.12.2075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying, S. , O'Connor, B. , Ratoff, J. , Meng, Q. , Mallett, K. , Cousins, D. , … Corrigan, C. (2005). Thymic stromal lymphopoietin expression is increased in asthmatic airways and correlates with expression of Th2‐attracting chemokines and disease severity. Journal of Immunology, 174, 8183–8190. 10.4049/jimmunol.174.12.8183 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1.
RANKL is up‐regulated in T cells. (A) Nasal mucosa tissues from AR patients were double stained by immunofluorescence with anti‐CD4 (green) and anti‐RANKL antibody (red) (magnification, × 60). (B) T cells were counted by three individuals in human tissues. Data are represented as the mean ± S.E.M. #P < 0.05; significantly different from the normal. Mice were sensitized on days 1, 5, and 14 by intraperitoneal injections of 100 μg of OVA emulsified in 20 mg of aluminium hydroxide and then received local injections of RANKL siRNA and control siRNA, or PBS alone into the nasal cavity, and challenged with intranasal OVA for 10 days. (C) Fluorescence intensities in nasal mucosa tissues of normal mice and AR mice were determined as threshold values. (D) Nasal mucosa tissues from normal mice and AR mice were double stained by immunofluorescence with anti‐CD4 (green) and anti‐RANKL antibody (red) (magnification, × 60). (E) T cells were counted by three individuals in animal tissues. Data are represented as the mean ± S.E.M. #P < 0.05; significantly different from the PBS. *P < 0.05; significantly different from the control siRNA OVA.
Figure S2.Co‐challenge of RANKL and OVA synergistically increased AR responses. We sensitized mice on days 1, 5, and 14 by intraperitoneal injections of 100 μg of OVA emulsified in 20 mg of aluminium hydroxide and we challenged mice with 1.5 mg of OVA. Mice received rmRANKL before the intranasal OVA challenge for 10 days. (A) The rub number of the nose that occurred 10 min after OVA intranasal provocation. Levels of (B) IgE, (C) histamine, and (D‐G) cytokine in the serum. Data are represented as the mean ± S.E.M. with n = 5 mice per group. #P < 0.05; significantly different from the OVA‐unsensitized mice. *P < 0.05; significantly different from the OVA‐sensitized mice.
Figure S3. RANK was expressed on human mast cell. RANK mRNA expressions in human cord blood‐derived mast cells (HCBMC) and HMC‐1 cells were analysed by the RT‐PCR.
Figure S4. RANKL induces production of pro‐inflammatory cytokines from HMC‐1 cells. (A‐C) HMC‐1 cells (1 × 105) were stimulated with RANKL (1, 10, and 100 ng/ml) for 24 hr. The production of pro‐inflammatory cytokines in the supernatant was measured by the elisa method. (D‐F) HMC‐1 cells were treated with RANKL (10 ng/ml), RANKL neutralizing antibodies (0.5 μg/ml), and/or IgG isotype for 24 hr. The production of pro‐inflammatory cytokines in the supernatant was measured by the elisa method. Each datum represents the mean ± SEM of five independent experiments. #P < 0.05; significantly different from unstimulated cells. *P < 0.05; significantly different from RANKL.
Figure S5. RANKL induces degranulation of rat peritoneal mast cells (RPMCs). RPMCs were stimulated with RANKL (10 ng/ml), compound 48/80 (6 μg/ml), RANKL + Anti‐RANKL Ab (Anti‐RANKL) for 20 and 60 min. The levels of (A) histamine, (B) PGD2, and (C) LTC4 were measured from cell supernatant. Data were expressed as mean ± SEM of five independent experiments with duplicate samples. #P < 0.05; significantly different from unstimulated cells. *P < 0.05; significantly different from RANKL’ value.
Table S1 Baseline characteristics for the normal and AR groups