

Abstract
Background and Purpose
Although protein phosphatases regulate multiple cellular functions, their modulation under hypoxia remains unclear. We investigated expression of the protein phosphatase system under normoxic/hypoxic conditions and the mechanism by which hypoxia alters protein phosphatase 2A (PP2A) activity.
Experimental Approach
Human cardiovascular cells were cultured in cell type specific media under normoxic or hypoxic conditions (1% O2). Effects on mRNA expression, phosphatase activity, post‐translational modification, and involvement of hypoxia inducible factor 1α (HIF‐1α) were assessed using RT‐PCR, immunoblotting, an activity assay, and siRNA silencing.
Key Results
All components of the protein phosphatase system studied were expressed in each cell line. Hypoxia attenuated mRNA expression of the transcripts in a cell line‐ and time‐dependent manner. In human aortic smooth muscle cells (HASMC) and AC16 cells, hypoxia decreased PP2Ac activity and mRNA expression without altering PP2Ac abundance. Hypoxia increased demethylated PP2Ac (DPP2Ac) and phosphatase methylesterase 1 (PME‐1) abundance but decreased leucine carboxyl methyltransferase 1 (LCMT‐1) abundance. HIF‐1α siRNA prevented the hypoxia‐mediated decrease in phosphatase activity and expression of the catalytic subunit of protein phosphatase 2A (PPP2CA), independently of altering pPP2Ac, DPP2Ac, LCMT‐1, or PME‐1 abundance.
Conclusion and Implications
Cardiovascular cells express multiple components of the PP2A system. In HASMC and AC16 cells, hypoxia inhibits PP2A activity through HIF‐1α‐dependent and ‐independent mechanisms, with the latter being consistent with altered PP2A holoenzyme assembly. This indicates a complex inhibitory effect of hypoxia on the PP2A system, and highlights PP2A as a therapeutic target for diseases associated with dysregulated protein phosphorylation.
What is already known
Hypoxia modulates PP2Ac abundance and catalytic activity albeit with no consensus at a tissue level.
What this study adds
An overview of the PP2A system in cardiovascular cells and its modulation by hypoxia.
Demonstrates that hypoxia modulates the PP2A system through HIF‐1α‐dependent and ‐independent mechanisms.
What is the clinical significance
Identifies potential druggable targets to modulate dysregulation of phosphorylation associated with cardiovascular disease.
Abbreviations
- CIP2A
cancerous inhibitor of protein phosphatase 2A
- DMOG
dimethyloxalylglycine
- DPP2Ac
demethylated catalytic subunit of protein phosphatase 2A
- HAEC
human aortic endothelial cells
- HASMC
human aortic smooth muscle cells
- HCF‐av
human cardiac ventricular fibroblasts
- HIF‐1α
hypoxia inducible factor 1α
- LCMT‐1
leucine carboxyl methyltransferase 1
- PHD2
prolyl‐4‐hdroxylase enzyme 2
- PME‐1
protein phosphatase methylesterase 1
- PP2A
protein phosphatase 2A
- PP2Ac
catalytic subunit of PP2A
- pPP2Ac
phosphorylated catalytic subunit of PPA2
- PPP2CA
gene coding for PP2Ac
- PTEN
phosphatase and tensin homologue
1. INTRODUCTION
Protein phosphorylation is a dynamic and reversible process utilizing the activity of multiple kinases and phosphatases. In man, it is estimated that approximately half of all proteins are regulated through phosphorylation primarily at serine, threonine, and tyrosine residues (Cohen, 2002; Shi, 2009). Within the protein phosphatase family, the serine/threonine phosphatases, which include protein phosphatase (PP)1, PP2A, and PP2B, account for around 90% of phosphatase activity in the heart (Heijman, Dewenter, El‐Armouche, & Dobrev, 2013). The ability of such a small group of enzymes to dephosphorylate a wide range of substrates is dependent upon their capacity to interact with scaffolding and regulatory subunits to form numerous holoenzymes (Shi, 2009). For instance, the catalytic subunits of PP2A (PP2Acα and PP2Acβ) can bind with two different scaffolding subunits (PP2A‐Aα and PP2A‐Aβ) and four regulatory subunit families (PP2A‐B/B′/B″/B‴), each of which contain several isoforms and splice variants (Price & Mumby, 2000).
Regarding PP2A, it is regulated through transcription, post‐translational modification of its catalytic subunits, and via interaction with several endogenous inhibitors including the cancerous inhibitor of PP2A (CIP2A) and the SET protein (Sangodkar et al., 2016). Post‐transcriptional modification of PP2Ac entails phosphorylation of Thr304 and Tyr307 and methylation of Leu309 within a conserved motif in the C‐terminal. This is key in mediating not only catalytic activity but also association with the regulatory subunits (Longin et al., 2007). It is facilitated by several kinases including GSK‐3β (Yao et al., 2011) and the counter regulatory action of leucine carboxyl methyltransferase‐1 (LCMT‐1) and protein phosphatase methylesterase‐1 (PME‐1; Leulliot et al., 2004; Ogris et al., 1999).
Within the cardiovascular system, protein phosphatases modulate adrenoceptor signalling, excitation contraction coupling, Ca2+ handling, cardiac metabolism, and tissue remodelling (Heijman, Ghezelbash, Wehrens, & Dobrev, 2017). While most catalytic subunits of PP1, PP2A, and PP2B (calcineurin) are found in cardiac tissue, there are differences in their expression between ventricles and atria, and from the left to right chambers (DeGrande et al., 2013; Luss et al., 2000). Both scaffolding subunits (DeGrande et al., 2013; Wijnker et al., 2011) and several regulatory subunits including PP2R2A, PP2R2B, PP2R5A, PP2R5B, PP2R5D, and PP2R3A are also found in cardiac tissue (DeGrande et al., 2013; Herzig & Neumann, 2000; Kirchhefer et al., 2014; Varadkar et al., 2014; Wijnker et al., 2011). However, few groups have looked at the distribution pattern of the protein phosphatases between the different cell types within the heart and cardiovascular system. Importantly, information on the expression of the endogenous inhibitors and post‐translational modifiers of PP2A are lacking. These are important omissions as they affect enzyme activity, substrate specificity, and subcellular localization of PP2A (Wlodarchak & Xing, 2016).
While it is clear that phosphoprotein phosphatases have a role in the pathophysiology of heart failure, cardiac arrhythmias, and hypoxic disease (cardiac and peripheral; Lubbers & Mohler, 2016), the mechanisms by which they are modulated remain poorly understood. Although several groups have investigated the effect of hypoxia on the PP2A system, there is little consensus in the results obtained, as hypoxia was found to increase (Larsen et al., 2008; Zhu et al., 2015), decrease (Truttmann, Ashraf, Mishra, & Delivoria‐Papadopoulos, 2004), or have no effect (Kučera et al., 2017) on PP2A abundance or activity. Similarly, the hypoxia inducible factor 1α (HIF‐1α), a key mediator of the hypoxic response, activates PP2A in ovarian clear cell carcinoma but, in RMG‐1 cells, has no effect on PP2A abundance or activity (Takai et al., 2015). To complicate things further, PP2A can modulate the HIF‐1α signalling cascade through dephosphorylation of prolyl‐hydroxylase enzyme 2 (PHD2), to fine‐tune HIF‐1α abundance in colorectal cancer cells (Di Conza, Trusso Cafarello, Loroch, et al., 2017). Given the importance of HIF‐1α signalling in cardiac hypoxia, and the universal role played by PP2A in counter‐regulation of kinase signalling in the heart, it is surprising that the hypoxia‐HIF‐1α‐PP2A axis has not been more widely investigated. A fundamental knowledge of this axis will have a significant effect on our understanding of cardiovascular (patho)physiology and highlight new opportunities to identify druggable targets to modulate dysregulation of phosphorylation associated with cardiovascular disease.
Therefore, the aims of the present study were threefold: (a) to establish the expression profile of key protein phosphatases, associated subunits and other regulatory components in human aortic endothelial, smooth muscle, ventricular fibroblast cells, and ventricular cardiomyocytes; (b) investigate the effects of hypoxia on expression of the protein phosphatase system in these cell lines; and (c) specifically delineate the mechanism(s) by which hypoxia regulates PP2A in human primary aortic smooth muscle cells and ventricular cardiomyocytes.
2. METHODS
2.1. Cell culture and hypoxic insult
Human aortic endothelial cells (HAEC), human aortic smooth muscle cells (HASMC), human cardiac ventricular fibroblasts (HCF‐av), and immortalized adult ventricular cardiomyocytes (AC16 cells) were cultured in cell type specific media (EBM2; SMCM; FM2 and DMEM/F12, respectively) supplemented with FBS (1–12.5%), 100‐U·ml−1 penicillin, 100‐μg·ml−1 streptomycin, and appropriate growth supplements as required. Cells were cultured at 37°C under 5% CO2 in a humidified atmosphere and used up to passage 10. AC16 cells are a proliferating cell line derived from the fusion of primary cells from adult human ventricular heart tissues with SV40 transformed human fibroblasts, devoid of mitochondrial DNA (Davidson et al., 2005), and have been used as a cardiomyocyte model to study the effect of mechanical stretch and hypoxia (Lee et al., 2018; Wong et al., 2018).
For the hypoxia experiments, cells were plated at the density of 7 × 103 cells per well in 96‐well plates, 2 × 105 cells per well in 6‐well plates, or 2 × 106 in T75 flasks and allowed to attach for 24 hr under normoxic conditions before placement in a hypoxia chamber (Billups‐Rothenberg Inc.). Two hours prior to placement in the hypoxia chamber, the medium was changed to one without growth factors. For induction of hypoxia, the chamber was flushed with premixed gas (BOC) consisting of 1% O2, 5% CO2, and balanced N2 at the rate of 20 L·min−1 for 7–10 min, sealed, and the chamber re‐flushed 1 hr later. Normoxic controls were cultured in 5% CO2 and 95% air at 37°C. In HASMC, the effect of hypoxia and role of HIF‐1α were mimicked using the prolyl hydroxylase inhibitor dimethyloxalylglycine (100‐μM DMOG; Sigma‐Aldrich) to assess HIF‐1α in modulation of the PP2A system.
2.2. Semi‐quantitative real time PCR
Total cellular RNA was isolated using Tri‐reagent and treated with DNase I (Sigma‐Aldrich Ireland), prior to being reverse transcribed using random hexamers and MMLV reverse transcriptase (RevertAid, ThermoFisher Scientific). mRNA expression was determined using target specific primers (Table S1) and normalized to the geometric mean of the housekeeping genes, glucose‐6‐phosphate isomerase, and GAPDH. The housekeeping genes were validated using geNorm (RRID:SCR_006763; validation data are presented in Figure S1). For each primer set, a no template and no reverse transcriptase control were included. All samples were analysed as a technical duplicate to insure reliability of single values from six independent experiments. The presence and size of the amplified products were checked following agarose gel electrophoresis and visualization under UV transillumination.
2.3. Protein phosphatase activity
Total protein phosphatase and PP2A activities were determined using a PP2A immunoprecipitation phosphatase assay according to the manufacturer's protocol (Merck Millipore). In brief, cells were pelleted (400 x g, 5 min) and lysed in a buffer composed of 50‐mM Tris–HCl pH 7.4, 150‐mM NaCl, 1‐mM EDTA, and 1% (v/v) NP40 supplemented with protease inhibitors (SigmaFAST, Sigma‐Aldrich Ireland). For determination of PP2Ac activity, whole cell lysates (100 μg protein) were incubated with an anti‐PP2Ac antibody (4 μg; clone 1D6) along with protein A‐agarose. The mix was incubated for 2 hr at 4°C. The immunoprecipitate was collected, washed, and phosphatase activity determined according to the manufacturer's instructions. For determination of total protein phosphatase activity, 1 μg of protein was utilized. Absorbance was determined at 650 nm, and phosphatase activity calculated from the standard curve and expressed as a percentage of the free phosphate of the test samples to that of the control samples. Each sample was assayed in triplicate (technical replicate) to ensure reliability of single values.
2.4. Immunoblotting
The antibody‐based procedures used in this study comply with the recommendations made by the British Journal of Pharmacology. Protein was extracted in modified RIPA buffer (Trizma Base 50 mM, NaCl 150 mM, EDTA 2 mM, and NP40 0.5% v/v) supplemented with a protease inhibitor cocktail (Sigma‐Aldrich Ireland). For analysis of protein phosphorylation, the RIPA buffer was modified to contain Na3VO4 2 mM, NaF 5 mM, and sodium deoxycholate 0.25%. Samples (20 μg protein) were boiled in lithium dodecyl sulphate sample loading buffer (lithium dodecyl sulphate 5%, glycerol 50%, Tris–HCl [1 M, pH 6.8] 25%, bromophenol blue 2.5 mg, phenol red 2.5 mg, and Ficoll 400 5%) containing 10% β‐mercaptoethanol (Sigma Aldrich) and subjected to SDS‐PAGE (8% gel). Following separation, the protein samples were transferred (transfer buffer: Tris‐base 50 mM, glycine 40 mM, methanol 20% v/v, SDS 0.037% w/v, dH2O) to PVDF membranes and blocked in TBS‐T (Tris‐base 10 mM, NaCl 100 mM containing 5% marvel, and 0.1% Tween 20, pH 7.6). Membranes were probed overnight (4°C) with 1° antibodies (see Materials for details) directed against PP2Ac, phosphorylated PP2Ac (pPP2Ac), demethylated catalytic subunit of protein phosphatase 2A (DPP2Ac), PME‐1, LCMT‐1, CIP2A, PP2R1A, or PP2R5A (diluted 1:1000 in TBS‐T containing 5% non‐fat dry milk), washed and incubated with an appropriate 2° antibody conjugated to HRP. All antibody solutions were use once. Immunoreactive bands were visualized using chemiluminescence detection (3.2 μl of 30% hydrogen peroxide/6 ml of 250‐mM Luminol, 90‐mM 4‐iodophenylboronic acid, and 100‐mM Tris–HCl) and recorded on a Fusion FX imaging system (Vilber Lourmat); images were captured within the dynamic range of the cooled CCD camera. Densitometric analysis was undertaken using Bio1D software with the image analyst blinded with respect to group designation. Membranes were stripped (62.5 mM, pH 6.8 Tris–HCl, 2% SDS, and 0.83 μl β‐mercaptoethanol/100 ml) and re‐probed with an anti‐β‐actin antibody conjugated to HRP. An EZ‐RUN™ pre‐stained molecular weight ladder (Fisher Scientific, Dublin, Ireland) was included on all gels.
2.5. Cell viability and mitochondrial membrane potential
Cell viability was assessed using a 3‐(4,5‐dimethylthiazol‐2yl)‐2,5‐diphenyltetrazolium bromide (MTT, Sigma‐Aldrich) dye reduction assay. Cells were allowed to attach overnight in 96‐well plates in complete medium. The medium was then changing to one without growth factors, and the cells exposed to hypoxia, normoxia, dimethyloxalylglycine (DMOG; 100 μM), or doxorubicin (50 μM, positive control) for 24 hr. Three hours prior to the end of the incubation period, MTT (5 mg·ml−1) was added. The culture media was removed 3 hr later and the purple formazan crystals dissolved in DMSO. Colour development was quantified by measuring the absorbance at λ 540 nm on a spectrophotometer (BIO‐TEK EL808). Cell viability was expressed as a percentage of the absorbance in cells under normoxic conditions at the 24‐hr time point.
In HASMC, the effect of hypoxia on the mitochondrial membrane potential was assessed using MitoTracker Red CMXros (1:2000 dilution; Invitrogen, Oregon, USA). Cells were stained with MitoTracker Red CMXros and Hoechst 33342 (HQ; 1:1000 dilution; ThermoFisher Scientific, Dublin, Ireland), incubated for 1 hr at 37°C, washed with PBS, and fixed in 3% paraformaldehyde at 37°C for 30 min. To quantify cell viability and mitochondrial membrane permeability, the plates were scanned using a Cytell ™ imaging system and the data analysed using IN Cell investigator software (version 1.6). Doxorubicin (50 μM) was included as a positive control.
2.6. Quantification of HIF‐1α expression by ELISA
HASMCs and AC16 cells were lysed in RIPA buffer (Trizma Base 50 mM, sodium chloride 150 mM, EDTA 2 mM, and NP40 0.5% v/v) supplemented with a protease inhibitor cocktail (Sigma‐Aldrich Ireland) and incubated on ice for 15 min. Following disruption of the cells by three freeze thaw cycles at −80°C, supernatants were collected following centrifugation (400 x g for 10 min, 4°C) and protein concentration determined using a Bradford assay. Following standardization, HIF‐1α protein expression was determined by ELISA (HIF‐1A ELISA kit, ThermoFisher Scientific, Dublin, Ireland) according to the manufacturer. Absorbance was measured using a spectrophotometer (BIO‐TEK EL808) at 450 nm with a reference wavelength of 550 nm. The intra and inter assay coefficients of variation were <10%, while the assay sensitivity was ≤30 pg·ml−1. All samples and standards were analysed in duplicate (technical replicate) to ensure reliability of single values, and HIF‐1α protein expression determined from the standard curve.
2.7. Cell transfection and siRNA knockdown
HASMCs and AC16 cells were seeded in 6‐well plates and grown to 80% confluence before transfection with siRNA (25 nM) directed against HIF1A (TriFECTa Kit DsiRNA Duplex; IDT) using 7.5 μl of TransIT‐X2 transfection reagent (Mirus, MSC Medical Supply Co. Ltd, Dublin, Ireland). Mock transfected and non‐targeting siRNA were included as controls. Post transfection (24 hr), the medium was replaced by one without growth factors and the cells subjected to hypoxia for 24 hr. mRNA and cell lysates were collected for downstream analysis of PP2A, LCMT‐1, PME‐1, and CIP2A.
2.8. Materials
HASMC and HAEC were purchased from ScienCell Research Laboratories (Buckingham, UK), while ventricular fibroblasts (HCF‐av) and ventricular cardiomyocytes (AC16, Cat# SCC10; RRID:CVCL_4U18) were obtained from TCS Cellworks (Buckingham, UK) and Merck Millipore (Temecula, USA) respectively. Cell line specific culture media were procured from Lonza (EBM2; Walkersville, MD USA) and ScienCell (SMCM). Mouse anti‐PP2AC (Cat# 05‐421; RRID:AB_30972) was bought from Merck Millipore (Temecula, CA92590, USA), whereas mouse anti‐phospho‐PP2AC (Cat# sc‐271903; RRID:AB_10611810), anti‐demethylated PP2AC (Cat# sc‐13601; RRID:AB_675841), anti‐LCMT‐1 (Cat# sc‐365064; RRID:AB_10846026), anti‐PME‐1 (Cat# sc‐137145; RRID:AB_2170375), CIP2A (Cat# sc‐80662, RRID:AB_2130800), and anti‐β‐actin antibody conjugated to HRP (Cat# sc‐47778‐HRP; RRID:AB_2714189) were obtained from Santa Cruz Biotechnology (CA 95060, USA). Rabbit anti‐PP2R1A (polyclonal; IgG; residues 308‐589; Cat# ab154551; lot# GR125965‐18) and anti‐PP2R5A (Cat# ab72028, RRID:AB_1269943) were purchased from Abcam (Cambridge, UK). Goat anti‐mouse antibody (Cat# P0447; RRID:AB_261713) and swine anti‐rabbit immunoglobulin (Cat# P0217; RRID:AB_272871) conjugated to HRP were from DAKO (Agilent, Cork, Ireland). DNA primers were purchased from Integrated DNA Technologies (Leuven, Belgium), while RevertAid (reverse transcription kit) and GoTaq (DNA polymerase) were obtained from ThermoFisher (Dublin, Ireland) and Promega (Madison, WI53711‐5399 USA) respectively. The protein phosphatase assay kit was procured from Merck Millipore (Billerica, MA01821, USA), while the HIF‐1A ELISA Kit was obtained from ThermoFisher Scientific (Dublin, Ireland). Doxorubicin (Cat. No. sc‐280681 )was supplied by Santa Cruz Biotechnology (Santa Cruz, CA). All other reagents were acquired from Sigma‐Aldrich (Arklow, Ireland) unless otherwise specified.
2.9. Data and statistical analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology. All samples were randomly assigned in the normoxia or hypoxia groups, using an online randomizer (GraphPad QuickCalcs: https://www.graphpad.com/quickcalcs/randomize1.cfm). PCR data were normalized to the geometric mean of the house‐keeping genes, while data from immunoblots were normalized to β‐actin from the same blot. Both are expressed relative to values obtained under normoxic conditions as either a fold or percentage in order to set the Y‐axis to 1 or 100%. Similarly, cell viability and protein phosphatases activity (total and PP2Ac mediated) were normalized to the mean of the control group (normoxia) and expressed as a percentage in order to set the Y‐axis to 100%. All treatments were randomized to avoid systematic bias. Data are presented as the mean ± SEM of at least five independent experiments (“n”) and were analysed using an independent Student t‐test or by one‐way ANOVA with post hoc analysis (Bonferroni or Dunnett as appropriate; GraphPad Prism version 7: RRID:SCR_002798 and Statistical Package for the Social Sciences version 25: RRID:SCR_002865). Post hoc analysis was only performed when ANOVA showed a statistical difference between group means (F achieved P < 0.05). Statistical analysis of the western blots was only carried out between bands from the same blot. The data analyst was blinded (unaware of group designation) when undertaking the statistical analysis. A value of P < 0.05 was taken to indicate statistical significance.
2.10. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander, Christopoulos et al., 2017; Alexander, Fabbro et al., 2017).
3. RESULTS
3.1. Basal expression of the protein phosphatase system
All 14 components of the protein phosphatase system investigated under basal conditions were detectable at the mRNA level in HAEC, HASMC, HCF‐av, and AC16 cells. Expression of the genes PPP1CA, PPP2CA, and PPP4C were comparable in HAEC and HCF‐av cells, but lower by 27%, 39%, and 22% respectively in HASMC (Figure 1a,b). In AC16 cells, expression of PPP1CA and PPP4C was higher than in the HAEC and HASMC cells, while expression of pPP2Ac was similar to that in HASMC and HCF‐av cells. Regarding phosphatase and tensin homologue (PTEN), its rank order of expression was HCF‐av > HAEC > HASMC > AC16 (P < 0.05; Figure 1b). Although the relative expression of the PPP2R1A scaffolding subunit was highest in HCF‐av and lowest in HASMC and AC16 cells (Figure 1c), expression of the PPP2R1B subunit was similar across all four cell lines.
Figure 1.
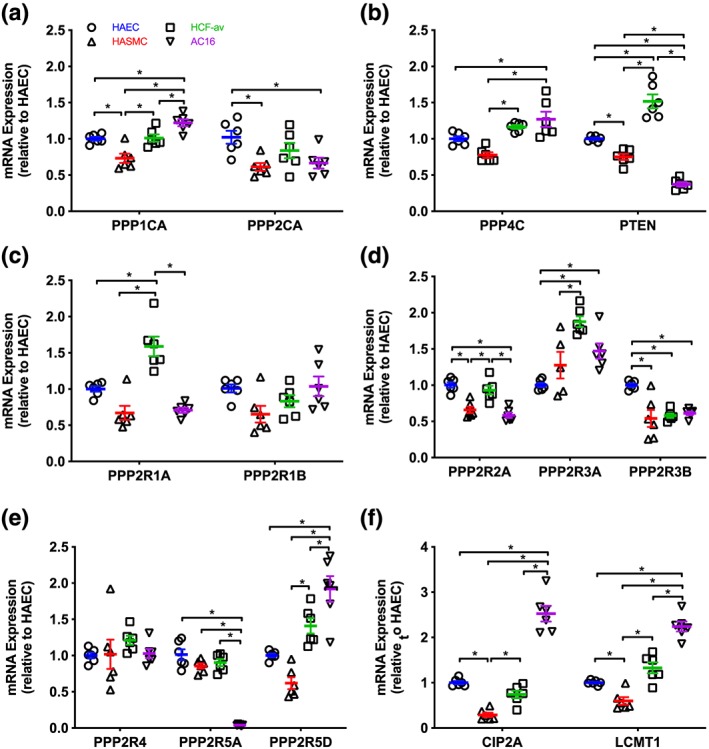
Expression pattern of key components of the protein phosphatase system in primary human aortic endothelial (HAEC), aortic smooth muscle (HASMC), cardiac fibroblast (HCF‐av) cell lines, and AC16 ventricular cardiomyocytes: catalytic subunit expression (a, b), scaffolding subunit expression (c), regulatory subunit expression (d, e), and endogenous regulators (f). The mRNA expression profile was determined using semi‐quantitative RT‐PCR‐based SYBR green chemistry. Data are presented relative to the expression level in HAEC (mean ± SEM) and were analysed using one‐way ANOVA with post hoc analysis (Bonferroni). *P < 0.05 (n = 6). CIP2A, gene coding cancerous inhibitor of PP2A; LCMT‐1, gene coding leucine carboxyl methyltransferase 1; PPP1CA, gene coding protein phosphatase 1 catalytic subunit A; PPP2CA, gene coding protein phosphatase 2A catalytic subunit A; PPP4C, gene coding protein phosphatase 4 catalytic subunit; PPP2R1A/1B, genes coding PP2A scaffolding subunit Aα/β; PP2R2A/3A/3B/4/5A/5D, genes coding PP2A regulatory subunits; PTEN, gene coding phosphatase and tensin homologue
Expression of PPP2R2A was lower in HASMC and AC16 cells than HAEC (P < 0.05; Figure 1d), while PPP2R3B was higher in HAEC than HASMC, HCF‐av, and AC16 cells (Figure 1d). Conversely, PPP2R3A expression was lower in HAEC than either HCF‐av or AC16 cells (Figure 1d). The expression of PPP2R4 was similar in all four cell lines (Figure 1e). Expression of PPP2R5A was lower in AC16 cells than in HAEC, HASMC, and HCF‐av cells (Figure 1e). PPP2R5D was higher in AC16 cells than in HAEC, HASMC, and HCF‐av cells (Figure 1e).
The rank order for the expression of CIP2A in the cell lines was AC16 > HAEC = HCF‐av > HASMC (Figure 1d), while that of LCMT‐1 was AC16 > HCF‐av = HAEC > HASMC (Figure 1f).
3.2. Effect of hypoxia on protein phosphatase activity and temporal expression of the phosphoprotein phosphatase system
Hypoxia (1% O2, 24 hr) did not alter total phosphatase activity in HAEC compared to those cultured under normoxic conditions (Figure 2a). However, hypoxia decreased protein phosphatase activity by ~20% to ~30% in lysates from HASMC, HCF‐av, and AC16 cells (P < 0.05; Figure 2b–d). Notably, cell viability as assessed by MTT assay, cell counting and change in mitochondrial membrane potential were unaffected by hypoxia over the time studied (Figure S2a–i). To further investigate the effect of hypoxia on total protein phosphatase activity, we explored if the temporal expression profile of the protein phosphatase system varied in a cell line‐dependent manner.
Figure 2.
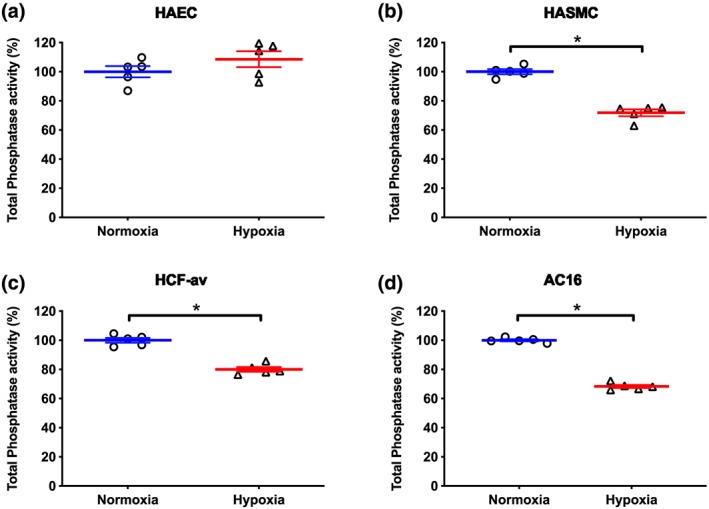
Effect of hypoxia (1% O2, 24 hr) on total protein phosphatase activity in (a) HAEC, (b) HASMC, (c) HCF‐av, and (d) AC16 cells. Data are presented as a percentage of total phosphatase activity (means ± SEM; n = 5) under normoxic conditions and were analysed using an unpaired Student t‐test. *P < 0.05, significantly different as indicated
3.2.1. HAEC mRNA expression profile
In HAEC, hypoxia did not alter mRNA expression of the catalytic subunits studied at 24 hr. However, by 48 hr, the expression of PPP1CA and PPP2CA were decreased by 25% and 66% respectively. By 72 hr, the expression of PPP1CA, PPP2CA, and PPP4C were decreased by ~35–45%. In contrast, hypoxia did not affect expression of PTEN mRNA at any time point (Table 1).
Table 1.
Temporal effect of hypoxia on the change in mRNA expression of the protein phosphatase system in HAEC, HASMC, HCF‐av, and AC16 cells
| Transcript | HAEC | HASMC | HCF‐av | AC16 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 24 hr | 48 hr | 72 hr | 24 hr | 48 hr | 72 hr | 24 hr | 48 hr | 72 hr | 24 hr | 48 hr | 72 hr | ||
| Catalytic subunits | PPP1CA | NS | −25.47 ± 6.3 | −44.46 ± 4.75 | −65.54 ± 9.4 | −24.0 ± 4.01 | NS | −56.09 ± 10.38 | NS | −30.61 ± 3.39 | −49.10 ± 3.79 | −52.51 ± 3.95 | −78.44 ± 2.28 |
| PPP2CA | NS | −66.1 ± 15.2 | −46.64 ± 2.43 | −88.7 ± 2.24 | NS | −46.7 ± 9.9 | −47.38 ± 8.24 | NS | NS | −52.00 ± 5.85 | −55.69 ± 1.55 | −88.30 ± 1.24 | |
| PPP4C | NS | NS | −36.69 ± 4.87 | NS | NS | −38.45 ± 4.34 | NS | NS | −23.96 ± 5.61 | NS | −59.00 ± 4.00 | −66.20 ± 2.17 | |
| PTEN | NS | NS | NS | −65.07 ± 9.35 | NS | −24.75 ± 6.56 | NS | NS | NS | −58.93 ± 5.18 | −43.91 ± 3.02 | −55.77 ± 3.91 | |
| Scaffold subunits | PPP2R1A | −16.28 ± 2.17 | −23.1 ± 4.25 | −40.82 ± 2.92 | −49.43 ± 5.04 | −20.26 ± 3.8 | −36.79 ± 3.46 | −64.67 ± 1.7 | NS | −34.94 ± 1.73 | −50.62 ± 3.37 | −46.48 ± 1.45 | −65.90 ± 2.84 |
| PPP2R1B | NS | −39.7 ± 10.24 | NS | −60.56 ± 4.18 | NS | −47.85 ± 1.94 | −72.04 ± 1.48 | NS | −28.45 ± 10.34 | −41.92 ± 1.84 | −57.44 ± 5.91 | −76.84 ± 3.33 | |
| Regulatory subunits | PPP2R2A | NS | −37.9 ± 6.53 | NS | −51.44 ± 5.36 | −27.57 ± 0.74 | −34.16 ± 4.8 | −74.15 ± 9.8 | NS | −17.14 ± 4.32 | −51.05 ± 2.27 | −56.49 ± 1.35 | −71.51 ± 4.52 |
| PPP2R3A | NS | NS | NS | −49.12 ± 3.84 | NS | −33.16 ± 6.38 | −53.49 ± 3.24 | NS | −16.8 ± 3.32 | −44.54 ± 4.11 | −42.12 ± 3.23 | −86.63 ± 5.52 | |
| PPP2R3B | −31.61 ± 2.66 | −36.54 ± 4.41 | −43.64 ± 6.64 | −56.28 ± 1.87 | −26.58 ± 3.42 | −49.67 ± 2.56 | −68.87 ± 1.91 | NS | −35.44 ± 3.18 | −53.12 ± 2.33 | −53.21 ± 5.43 | −31.30 ± 9.08 | |
| PPP2R4 | −39.8 ± 2.1 | −30.61 ± 4.86 | −51.07 ± 1.89 | −63.47 ± 3.4 | −31.51 ± 4.26 | −49.83 ± 2.38 | −74.17 ± 0.63 | NS | −37.1 ± 2.47 | −56.09 ± 2.34 | −43.91 ± 3.02 | −75.90 ± 2.62 | |
| PPP2R5A | NS | −48.8 ± 5.24 | NS | −72.58 ± 4.8 | NS | −41.58 ± 4.15 | −71.31 ± 4.04 | NS | NS | −77.40 ± 4.32 | −52.52 ± 3.85 | −56.03 ± 3.81 | |
| PPP2R5D | NS | −25.67 ± 4.06 | −41.32 ± 5.31 | −65.84 ± 4.48 | NS | −36.47 ± 2.7 | −56.34 ± 5.36 | NS | −23.29 ± 2.75 | −54.33 ± 1.79 | −46.07 ± 1.96 | −25.34 ± 3.61 | |
| Other | CIP2A | −54.9 ± 8.46 | −70.94 ± 2.22 | −79.50 ± 1.82 | −50.5 ± 6.06 | NS | −42.57 ± 5.55 | −73.77 ± 3.69 | −60.98 ± 5.2 | −25.62 ± 3.06 | −63.20 ± 3.45 | −51.68 ± 4.46 | −53.18 ± 5.36 |
| LCMT1 | −31.43 ± 8.89 | −28.23 ± 4.06 | −42.31 ± 6.3 | −59.03 ± 7.26 | NS | −31.52 ± 3.15 | −50.64 ± 5.5 | −54.31 ± 2.2 | −21.96 ± 4.15 | −59.52 ± 1.90 | −47.51 ± 6.26 | −52.21 ± 3.92 | |
Data are presented as percentage change (means ± SEM; n = 6) and were analysed using a Student t‐test. All numerical values shown were significantly different (P<0.05) from the corresponding values under normoxia; CIP2A, cancerous inhibitor of PP2A; LCMT‐1, leucine carboxyl methyltransferase 1; NS, no significant effect of hypoxia; PPP1CA, protein phosphatase 1 catalytic subunit A; PPP2Ac, protein phosphatase 2A catalytic subunit A; PPP2R1A/1B, PP2A scaffolding subunit Aα/β; PPP4C, protein phosphatase 4 catalytic subunit; PP2R2A/3A/3B/4/5A/5D, PP2A regulatory subunits; PTEN, phosphatase and tensin homologue.
Regarding the scaffolding subunits, hypoxia attenuated expression of PPP2R1A by ~16%, 23%, and 41% at 24, 48, and 72 hr respectively, whereas expression of PPP2R1B only decreased at 48 hr. Likewise, hypoxia decreased the expression of the regulatory subunits PPP2R2A and PPP2R5A at 48 hr only. Expression of PPP2R5D was reduced by ~25% and ~41% at 48 and 72 hr respectively following hypoxia, while expression of PPP2R3B and PPP2R4 were attenuated at all time points. Hypoxia did not affect expression of PPP2R3A but decreased expression of CIP2A and LCMT‐1 by ~55–75% and ~30–42% respectively at all time points (Table 1).
3.2.2. HASMC mRNA expression profile
In HASMC, hypoxia reduced expression of PPP1CA by ~65% and 25% at 24 and 48 hr respectively but had no effect at 72 hr. Expression of PP2CA and PTEN were attenuated by hypoxia at 24 and 72 hr, whereas expression of PPP4C was reduced at 72 hr. Hypoxia diminished expression of PPP2R1A at all time points, while that of PPP2R1B was decreased at 24 and 72 hr (Table 1).
Regarding the regulatory subunits, hypoxia decreased expression of PPP2R2A, PPP2R3B, and PPP2R4 all time points and that of PPP3R3A, PPP2R5A, and PPP2R5D at 24 and 72 hr. Expression of CIP2A and LCMT1 were diminished at 24 and 72 hr (Table 1).
3.2.3. HCF‐av mRNA expression profile
In HCF‐av cells, hypoxia attenuated expression of PPP1CA at 24 and 72 hr, whereas expression of PPP2CA and PPP4C were decreased at 24 and 72 hr respectively. In contrast, hypoxia did not affect mRNA expression of PTEN. Expression of the scaffolding subunits, PPP2R1A and PPP2R1B, were reduced at 24 and 72 hr by hypoxia (Table 1).
There was a consistent pattern in the response of PPP2R2A, PPP2R3A, PPP2R3B, PPP2R4, and PPP2R5D to hypoxia, in that their expression was attenuated at 24 and 72 hr, but not at 48 hr. Interestingly, this effect was greatest at the 24‐hr time point. Expression of PPP2R5A was lower in the hypoxia exposed cells compared to those cultured under normoxic conditions at 24 hr only. CIP2A and LCMT‐1 expression was decreased at all time points compared to the normoxic controls (Table 1).
3.2.4. AC16 mRNA expression profile
In AC16, cells hypoxia decreased the expression of PPP1CA, PPP2CA, PPP4C, and PTEN at all time points studied, with the exception of PPP4C which was unaltered at 24 hr. Expression of scaffolding subunits, PPP2R1A and PPP2R1B, and the regulatory subunits (PPP2R2A, PPP2R3A, PPP2R3B, PPP2R4, PPP2R5A, and PPP2R5D) were attenuated following exposure to hypoxia for 24, 48, and 72 hr. Similarly, hypoxia reduced CIP2A and LCMT1 mRNA expression of over the epoch studied (Table 1).
3.3. Temporal effect of hypoxia on PP2Ac, PP2R1A, PP2R5A, and CIP2A abundance
To establish if changes in the mRNA expression profiling were reflected at the protein level, selected targets were investigated using immunoblotting. In HAEC, hypoxia did not alter the abundance of PP2Ac, PP2R5A, and CIP2A at 24 and 48 hr but decreased them all by ~20–50% at the 72‐hr time point. Abundance of PPP2R1A was unaffected by hypoxia at all time points studied (Figure 3a–d).
Figure 3.
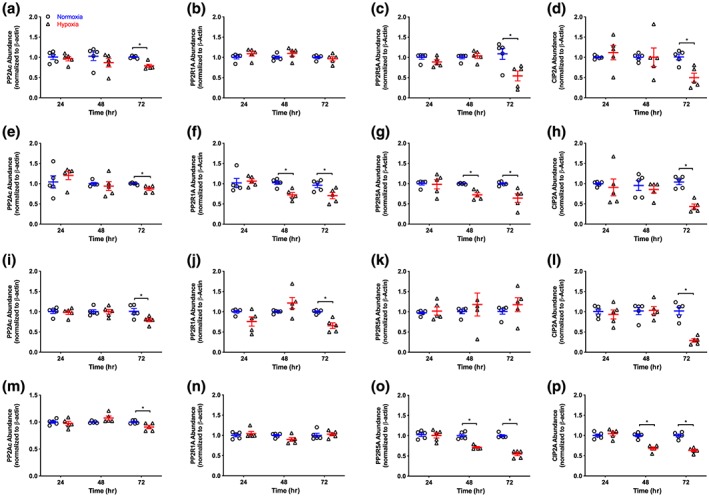
Temporal effect of hypoxia on PP2Ac, PP2R1A, PP2R5A, and CIP2A protein abundance in HAEC (a–d), HASMC (e–h), HCF‐av (i–l), and AC16 (m–p) cells. Data are normalized to the corresponding normoxia time point and presented as means ± SEM; n = 5. Data were analysed using an unpaired Student t‐test. *P < 0.05, significantly different as indicated. CIP2A, cancerous inhibitor of protein phosphatase 2A; PP2Ac: protein phosphatase 2A catalytic subunit C; PP2R1A: PP2A scaffolding subunit Aα; PP2R5A: PP2A regulatory subunit B'α (B56α)
In HASMC, hypoxia attenuated PP2R1A and PP2R5A protein expression by ~28% at the 48‐hr time point and remained depressed thereafter. The abundance of PP2Ac and CIP2A were unaffected by hypoxia (24 and 48 hr exposure) but decreased by ~13% and ~57% respectively following exposure to hypoxia for 72 hr (Figure 3e–h).
When the temporal effect of hypoxia was investigated in HCF‐av cells, the abundance of PP2Ac, CIP2A, and PP2R1A were attenuated only at the 72‐hr time point. Hypoxia did not alter the abundance of PP2R5A at any time point studied (Figure 3i–l).
In AC16 ventricular cardiomyocytes, hypoxia did not affect the abundance of PP2R1A, PP2Ac, PP2R5A, or CIP2A at the 24‐hr time point. Following more prolonged exposure to hypoxia (48 hr), the abundance of PP2R5A and CIP2A decreased by ~30% At the 72‐hr time point, PP2Ac, PP2R5A, and CIP2A abundance was attenuated while no effect was observed on the level of PP2R1A (Figure 3m–p).
3.4. Effect of DMOG on cell viability, phosphatase activity, and PP2Ac abundance
Under normoxic conditions, exposure of HASMC to DMOG (100 μM), a prolyl‐4‐hydoxylase inhibitor, for 24 hr did not alter cell viability compared to control (medium). Doxorubicin (50 μM, positive control) decreased cell viability by ~30% (Figure S3a). The total protein phosphatase activity was decreased by ~27% on incubation with DMOG (Figure S3b), without altering PP2Ac abundance in HASMC compared to control (Figure S3c). However, DMOG also decreased PP2CA mRNA expression by ~80% compared with control (Figure S3d). Together, these data suggested a role for HIF‐1α in hypoxia‐mediated modulation of PP2A.
To investigate the underlying mechanism(s) by which hypoxia could alter protein phosphatase activity, subsequent studies focused on the regulation of PP2A in only two of the cell lines used, the HASMC and AC16 adult ventricular cardiomyocytes.
3.5. Effect of hypoxia on PP2Ac (activity and post‐translational modification) in HASMC and AC16 cells
In HASMC and AC16 cells, hypoxia (1% O2, 24 hr) decreased PP2Ac activity by ~20% compared with that in the cells cultured under normoxic conditions (Figure 4a,b). Despite this, abundance of the catalytic subunit of PP2A (PP2Ac) was unaltered in either cell line (Figure 4c,d). However, hypoxia reduced PPP2CA mRNA expression by ~90% and 50% in HASMC and AC16 cells respectively (Figure 4e,f) compared with normoxia. Based on this, we postulated that hypoxia may modulate PP2A activity through post‐transcriptional modification of PP2Ac. Hypoxia increased the abundance of pPP2Ac by 0.8‐fold in HASMC (Figure 5a) but had no effect in AC16 cells (Figure 5b) compared to their respective normoxia control group. Interestingly, hypoxia increased the abundance of demethylated PP2Ac by ~55% in both cell lines (Figure 5c,d) compared with that in cells cultured under normoxic conditions.
Figure 4.
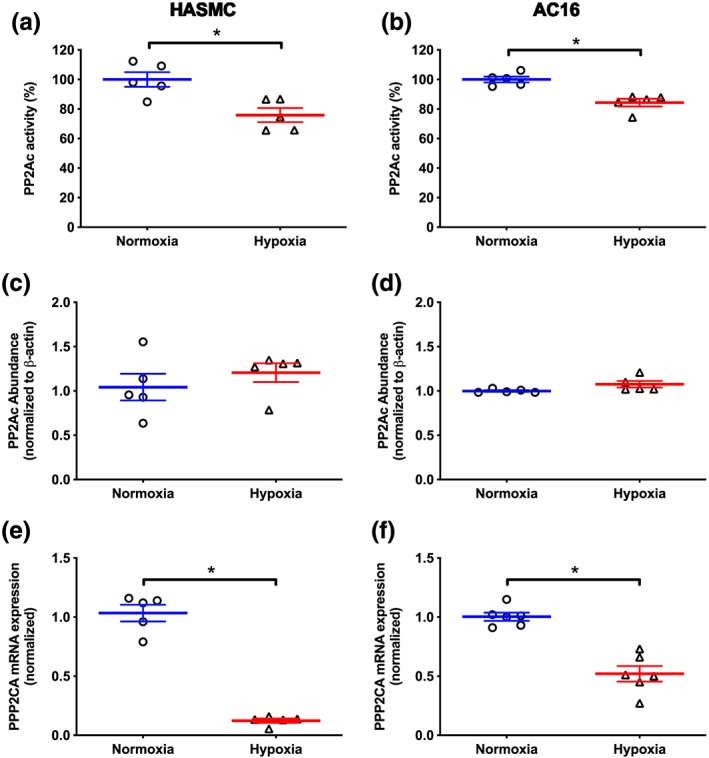
Effect of hypoxia (1% O2, 24 hr) on PP2Ac activity (a, b), PP2Ac abundance (c, d), and PPP2CA mRNA expression (e, f) in HASMC and AC16 ventricular cardiomyocytes. Protein phosphatase activity is presented as a percentage of the phosphatase activity under normoxia. PP2Ac abundance and mRNA expression are normalized (β‐actin and geometric mean of GPI and GAPDH as appropriate) and expressed relative to values under normoxic conditions. Data are presented as the mean ± SEM (n = 5) and were analysed using an unpaired Student t‐test. *P < 0.05, significantly different as indicated. PP2Ac: catalytic subunit of protein phosphatase 2A; PPP2CA, gene coding PP2Ac
Figure 5.
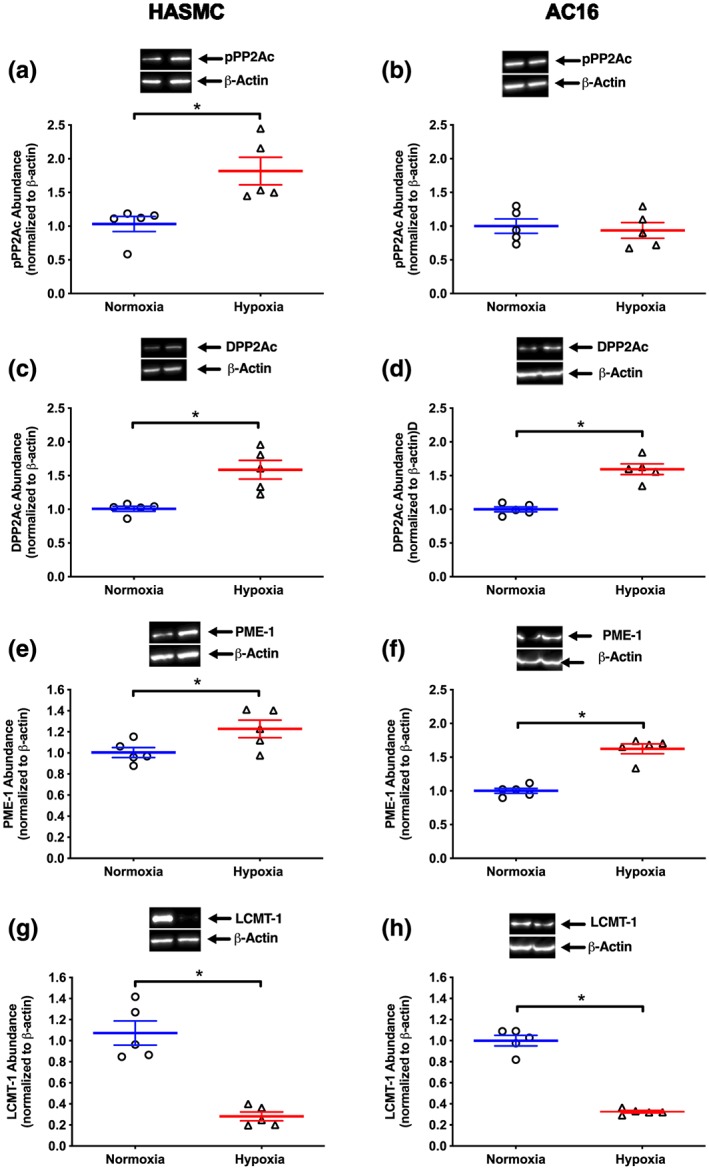
Effect of hypoxia (1% O2, 24 hr) on abundance of pPP2Ac (a, b), DPP2Ac (c, d), PME‐1 (e, f), and LCMT‐1 (g, h) in HASMC and AC16 ventricular cardiomyocytes. Representative immunoblots are inset. Normalized data are presented relative to abundance at 24 hr (mean ± SEM, n = 5) under normoxic conditions and were analysed using an unpaired Student t‐test. *P < 0.05, significantly different as indicated. DPP2Ac, demethylated protein phosphatase 2A; LCMT‐1, leucine carboxyl methyltransferase‐1; PME‐1, protein phosphatase methylesterase 1; pPP2Ac, phosphorylated catalytic subunit of protein phosphatase 2A
To extend the latter, we investigated if the change in the demethylated state of PP2Ac was reflected in alterations to the abundance of leucine carboxyl methyltransferase 1 (LCMT‐1) and protein phosphatase‐methylesterase (PME‐1). In HASMC and AC16 cells, PME‐1 and LCMT‐1 were detected under normoxic conditions (Figure 5e–h). Following exposure to hypoxia for 24 hr, the abundance of PME‐1 increased by ~20% (Figure 5e) in HASMC and 62% (Figure 5f) in AC16 cells. Moreover, the abundance of LCMT‐1 decreased by ~70% in both cell lines (Figure 5g,h).
3.6. Effect of HIF‐1α knockdown on phosphatase activity and PP2Ac expression
In HASMC and AC16 cells, hypoxia increased HIF‐1α abundance compared with values in cells cultured under normoxic conditions (Figure 6a,b). Transfection of HASMC and AC16 cells with three predesigned siRNAs directed against HIF‐1α, decreased HIF‐1α abundance by ~90% following 24 hr hypoxia, compared to untransfected cells subjected to hypoxia (Figure 6a,b). Mock transfection and transfection with a non‐target siRNA did not affect HIF‐1α abundance in either cell line compared to hypoxia alone (Figure 6a,b).
Figure 6.
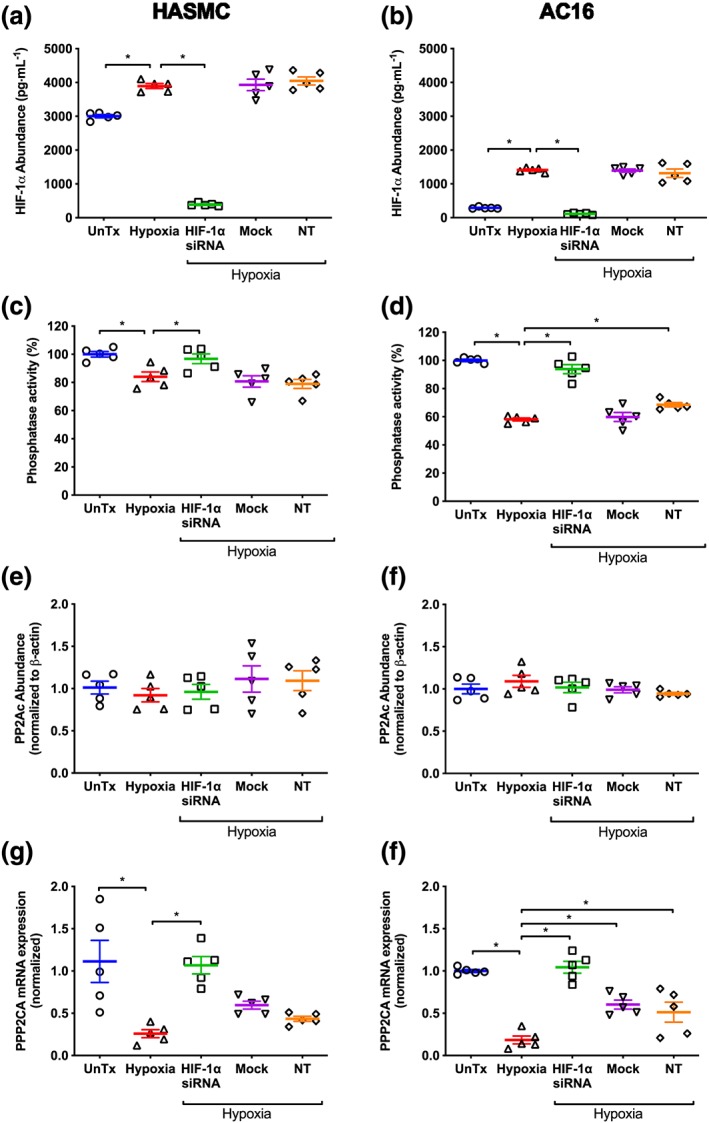
Effect of hypoxia (1% O2, 24 hr) on HIF‐1α abundance (a, b), total protein phosphatase activity (c, d), PP2Ac abundance (e, f), and PPP2CA mRNA expression (g, h) following HIF‐1α silencing in HASMC and AC16 ventricular cardiomyocytes. Normoxia, non‐target siRNA, and mock transfection were included as controls. HIF‐1α abundance was determined by ELISA, while total phosphatase activity, PP2Ac abundance, and PPP2CA mRNA expression were determined using a phosphatase activity assay, immunoblotting, and semi‐quantitative PCR respectively. HIF‐1α abundance is expressed in pg·ml−1. All other data are expressed relative to normoxia and where appropriate were normalized to β‐actin or geometric mean of the housekeeping genes (GPI and GAPDH). Data are presented as mean ± SEM (n = 5) and were analysed using one‐way ANOVA with Dunnett's post hoc test. *P < 0.05, significantly different as indicated. HIF‐1α, hypoxia inducible factor‐1 α; NT, non‐target siRNA; PP2Ac, catalytic subunit of protein phosphatase 2A; PPP2CA, gene coding PP2Ac; UnTx, untreated cells
Hypoxia (1% O2, 24 hr) decreased total phosphatase activity by ~16%, compared to cells cultured under normoxic conditions. Knockdown of HIF‐1α restored total phosphatase activity in HASMC and AC16 cells exposed to hypoxia to levels comparable to those cultured under normoxic conditions (Figure 6c,d). Transfection with non‐target siRNA and mock transfection did not alter the effect of hypoxia on total phosphatase activity in HASMC compared to untransfected cells exposed to hypoxia (Figure 6c). Likewise, mock transfection had no effect in AC16 cells, although there was a small increase in PP2A activity following transfection with non‐target siRNA (Figure 6d).
PP2Ac abundance was unaffected by hypoxia (Figure 6e,f) and remained unaltered following knockdown of HIF‐1α in HASMC and AC16 cells. Mock transfection and transfection with non‐target siRNA had no effect on PP2Ac abundance in either cell line compared to those cultured under hypoxic conditions (Figure 6e,f).
In HASMC and AC16 cells exposed to hypoxia, PPP2CA mRNA expression was decreased by 75% and 80% (Figure 6g,h) respectively, compared to their normoxic counterparts. This was abolished following silencing of HIF‐1α (P < 0.05; Figure 6g,h). Although mock transfection and transfection with non‐target siRNA did not influence the depressive effect of hypoxia on PPP2CA mRNA expression in HASMC (Figure 6g), they had a small effect in AC16 cells, compared to untransfected cells cultured under hypoxia (Figure 6h).
3.7. Effect of silencing HIF‐1α on post‐translational modification of PP2Ac and associated enzymes
In HASMC, hypoxia (24 hr) increased abundance of phosphorylated PP2Ac by 52% compared to cells cultured under normoxic conditions (Figure 7a). This effect was unaffected by knockdown of HIF‐1α. In contrast, hypoxia in the absence or presence HIF‐1α knockdown had no effect on pPP2Ac abundance in AC16 cells, (Figure 7b). In both cell lines, mock transfection or transfection with non‐target siRNA did not alter the effect of hypoxia on the level of phosphorylated PP2Ac (Figure 7a,b).
Figure 7.
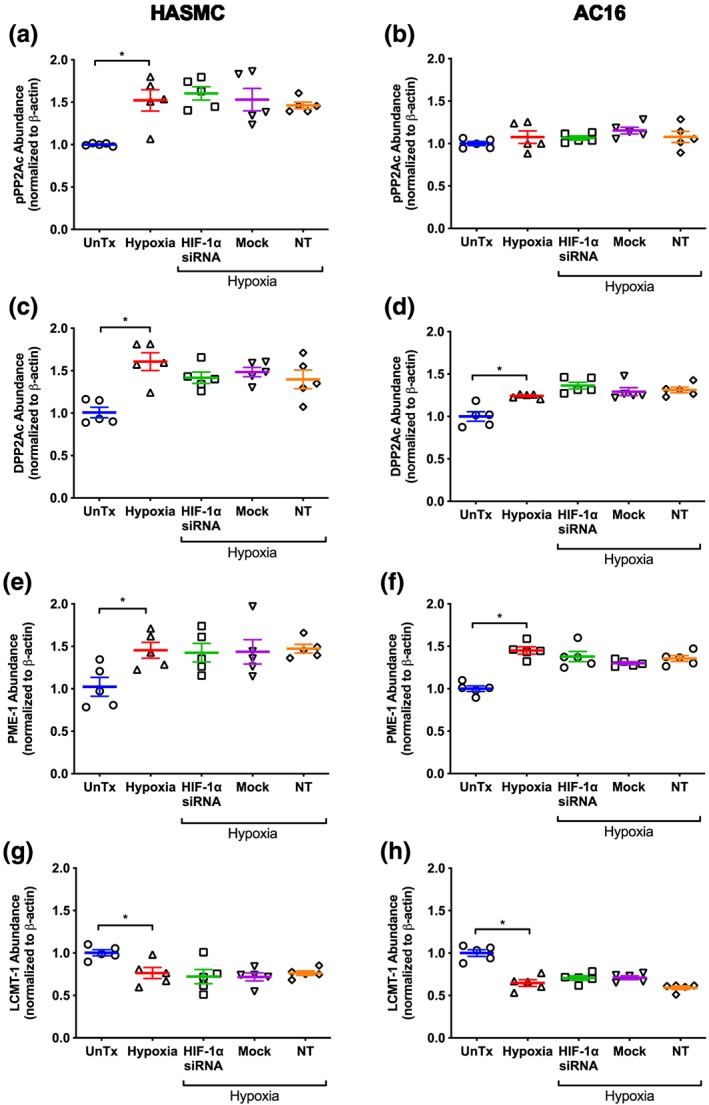
Effect of hypoxia (1% O2, 24 hr) on total phospho‐PP2Ac (a, b), demethylated PP2Ac (c, d), PME‐1 (E, F), and LCMT‐1 (g, h) abundance in HASMC and AC16 ventricular cardiomyocytes following silencing of HIF‐1α. Protein abundance was determined using immunoblotting, and all values are expressed relative to normoxia‐treated cells following normalization to β‐actin. Data are presented as mean ± SEM (n = 5) and were analysed using one‐way ANOVA with Dunnett's post hoc test. *P < 0.05, significantly different as indicated. DPP2Ac, demethylated catalytic subunit of protein phosphatase 2A; HIF‐1α, hypoxia inducible factor‐1 α; LCMT‐1, leucine carboxyl methyltransferase 1; NT, non‐target siRNA; PME‐1, protein phosphatase‐methylesterase 1; UnTx, untreated cells
Hypoxia increased the abundance of demethylated PP2Ac by 60% and 24% respectively in HASMC and AC16 cells compared to normoxia (Figure 7c,d). Knockdown of HIF‐1α did not affect the hypoxia‐mediated increase in the abundance of demethylated PP2Ac compared to the corresponding hypoxia controls (Figure 7c,d). Mock transfection and transfection with non‐target siRNA did not alter demethylated PP2Ac abundance in either cell line relative to untransfected cells cultured under hypoxia.
In HASMC and AC16 cells, hypoxia increased abundance of PME‐1 by 45% (Figure 7e,f) and decreased abundance of LCMT‐1 by 24% and 35% respectively (Figure 7g,h). Neither of these effects were altered by hypoxia following knockdown of HIF‐1α compared to untransfected cells subjected to hypoxia (Figure 7e–h). Mock transfection and transfection with non‐target siRNA had no effect on LCMT‐1 or PME‐1 abundance in either cell line under hypoxic conditions compared to untransfected cells exposed to hypoxia (Figure 7e–h).
3.8. Effect of HIF‐1α knockdown on CIP2A
In HASMC and AC16 cells, hypoxia decreased CIP2A mRNA expression by 90% and 65% respectively compared cells cultured under normoxic conditions (Figure 8a,b). Knockdown of HIF‐1α increased CIP2A mRNA expression compared to untransfected cells exposed to hypoxia (Figure 8a). However, this effect was very minor and not different from the response observed following mock transfection and transfection with non‐target siRNA (Figure 8a). In AC16 cells, knockdown of HIF‐1α, mock transfection, and transfection with non‐target siRNA had no effect on the hypoxia‐mediated attenuation of CIP2A mRNA expression (Figure 8b).
Figure 8.
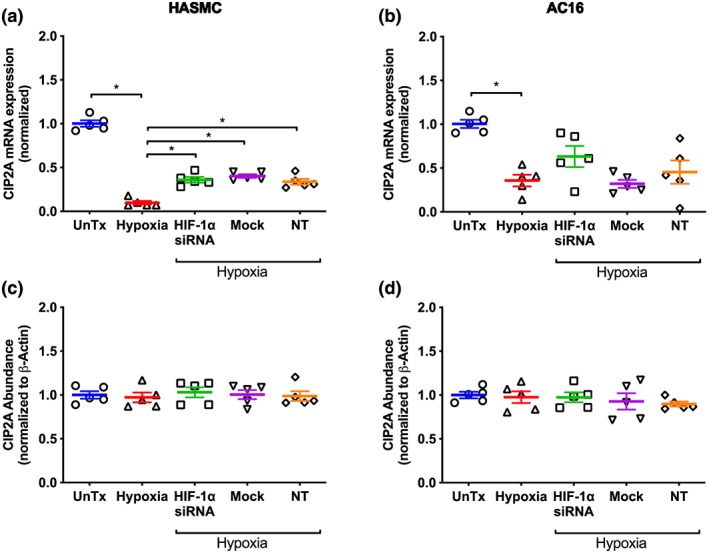
Effect of hypoxia (1% O2, 24 hr) on CIP2A mRNA expression (a, b) and protein abundance (c, d) in HASMC and AC16 ventricular cardiomyocytes following silencing of HIF‐1α. Protein abundance was determined using immunoblotting, and all values are expressed relative to normoxia‐treated cells following normalization to β‐actin. Data are presented as mean ± SEM (n = 5) and were analysed using one‐way ANOVA with Dunnett's post hoc test. *P < 0.05, significantly different as indicated. CIP2A, cancerous inhibitor of protein phosphatase 2A; HIF‐1α, hypoxia inducible factor‐1 α; NT, non‐target siRNA; UnTx, untreated cells
Hypoxia (24 hr) did not alter CIP2A abundance in HASMC or AC16 cells compared to their counterparts cultured under normoxic conditions or following knockdown of HIF‐1α (Figure 8c,d). Mock transfection and transfection with non‐target siRNA had no effect on CIP2A abundance in either cell line compared to untransfected cells exposed to hypoxia (Figure 8c,d).
4. DISCUSSION
To date, many of the studies investigating the effect of hypoxia on the expression profile of the protein phosphatase system have focused on the brain, with few looking at the heart and cardiovascular system. Those that do have focused primarily on whole tissue homogenates (DeGrande et al., 2013; Herzig & Neumann, 2000; Luss et al., 2000) and show that the distribution of PP1, PP2A, and associated regulatory and scaffolding subunits are differentially expressed in atria and ventricle (DeGrande et al., 2013; Luss et al., 2000). For example, PP1 (α, δ, γ) and PP2Aα expression is higher in right ventricles than in right atria, while there is no difference in the expression of PP2Aβ (Luss et al., 2000). Although informative, these studies do not localize the differences to a specific cell type and are confounded by the underlying species differences and medication history of patients; both of which influence expression of the protein phosphatase system (DeGrande et al., 2013; Wijnker et al., 2011). We have circumvented these confounding issues by using cultures of HAEC, aortic smooth muscle cells, ventricular fibroblasts, and immortalized adult human ventricular cardiomyocyte cells, as a cellular snapshot of the cardiovascular system.
Under normoxic conditions, all catalytic, scaffold, and regulatory subunits of the protein phosphatase system studied (PPP1CA, PPP2CA, PPP4C, PTEN, PPP2R1A, PPP2R1B, PP2R2A, PPP2R3A, PPP2R3B, PPP2R4A, PPP2R5A, and PPP2R5D) were expressed in HASMC, HAEC, HCF‐av cells, and AC16 cells, albeit differentially. This expands the observation that there are regional variations in protein phosphatase expression in heart (DeGrande et al., 2013; Luss et al., 2000) and kidney (Everett, Xue, & Stoops, 1999). We also showed that CIP2A and LCMT‐1 were expressed in HAEC, HASMC, HCF‐av, and ventricular cardiomyocytes (AC16 cells), which adds to earlier studies showing them to be expressed in murine fibroblasts (Gomes, Menck, & Cuervo, 2017; Hwang, Lee, & Pallas, 2016) and synoviocytes from patients with rheumatoid arthritis (Lee et al., 2013). Although the present study did not investigate why the protein serine/threonine phosphatase system is differentially expressed among HAEC, HASMC, HCF‐av, and AC16 cells, such differences may arise because of cell line‐dependent differences in cleavage and polyadenylation processes that influence mRNA stability and localization, or differences in trans‐acting factors and mRNA processing arising from variance in their 3′‐untranslated regions, which also affect mRNA stability (Matoulkova, Michalova, Vojtesek, & Hrstka, 2012). Differential expression of the PP2A subunits between cell lines may have significant functional implications, as the composition of the PP2A holoenzyme complex modulates its cellular localization and substrate specificity (Shi, 2009), and ultimately cell/tissue phenotype.
In the present study, hypoxia (24 hr) decreased total protein phosphatase activity in HASMC, HCF‐av cells, and AC16 cells, but not in HAEC. This extends previous reports demonstrating hypoxia to decrease total phosphatase activity, and PP2A/PP1 activity in animal, and human colorectal cancer cell lines (Cristobal et al., 2014; Krtolica, Krucher, & Ludlow, 1998; Truttmann et al., 2004). However, this is not an universal observation as hypoxia increased PP1 and PP2A activity in glioblastoma multiform‐derived tumour stem‐like cells (Hofstetter et al., 2012), and in left ventricular tissue from mice exposed to alveolar hypoxia (Larsen et al., 2008). The lack of consensus mirrors data obtained following ischaemia and ischaemia/reperfusion injury showing phosphoprotein phosphatase activity to be increased (Tian, Qiu, Zhao, Li, & Guo, 2009), decreased (Martin de la Vega, Burda, Toledo Lobo, & Salinas, 2002), or unaffected (Morioka et al., 1999). Regarding the other catalytic subunits investigated in the present study, we showed that PPP4 expression was depressed in all cell lines, while PTEN mRNA expression was only attenuated in HASMC and AC16 cells following exposure to hypoxia. The latter complements work of others showing ischaemia/ischaemic preconditioning to decrease PTEN activity and phospho‐PTEN abundance in the perfused isolated rat heart (Zhou et al., 2014). However, hypoxia increases PTEN expression in astrocytes (Jackson et al., 2013).
It was interesting to note that we did not find total phosphatase activity to be altered in endothelial cells. There may be several reasons for this. Firstly, if PP2A activity is mediated through HIF‐1α, as occurs in HASMC and AC16 cells, it might reflect lower expression of HIF‐1α in endothelial cells than in smooth muscle cells (Chi et al., 2006). Alternatively, it might simply reflect differences in the experimental models used (Martin de la Vega et al., 2002; Santos et al., 2016) or species (human, rat, and mouse). Nevertheless, at this time point (24‐hr hypoxia), it is not due to a change in PP2Ac or CIP2A abundance. The lack of effect of hypoxia on total protein phosphatase activity could also reflect the up‐regulation of other protein phosphatases. This is unlikely as hypoxia did not increase expression of the mRNA for PPP1, PPP4, or PTEN. Importantly, there is a clear difference between “short‐term” (24 hr) and “long‐term” exposure to hypoxia (72 hr) on PP2Ac abundance, as the latter deceased PP2Ac abundance in HAEC, HASMC, HCF‐av, and AC16 cells. This is consistent with hypoxia‐mediated attenuation of PPP2CA mRNA expression, which would not manifest at the protein level until much later, as PP2Ac protein has a t ½ of ~17 hr (Baharians & Schonthal, 1998). Intriguingly, the lack of effect of “short” term exposure to hypoxia on PPP2CA mRNA expression in HAEC could also be linked to low levels of HIF‐1α in endothelial cells (Chi et al., 2006), as HIF‐1α mediates the acute (<24 hr) hypoxic response, while HIF‐2 or HIF‐3 mediate the effect of chronic hypoxia (>24 hr; Holmquist‐Mengelbier et al., 2006; Koh, Lemos, Liu, & Powis, 2011).
When the effect of hypoxia (24 hr) was investigated on PP2Ac activity in HASMC and AC16 cells, this activity decreased independently of a change in PP2Ac protein abundance. For PP2A to be fully active, it must form a holoenzyme through interaction with scaffolding and regulatory subunits. We chose to investigate the effects on both scaffolding subunits and selected regulatory subunits (PP2R2A, PP2R3A, PP2R3B, PP2R4, PP2R5A, and PP2R5D) representative of members from all four regulatory subunit families that are known to be expressed in cardiac tissue (DeGrande et al., 2013). We showed that hypoxia decreased mRNA expression of the scaffolding subunits (PP2R1A and PPP2R1B) in all four cell lines, consistent with data from patients with ischaemic heart failure, but not with data from a canine model of coronary occlusion (DeGrande et al., 2013). Likewise, hypoxia‐mediated alteration of the regulatory subunits could account for the decrease in total protein phosphatase activity in HASMC, HCF‐av, and AC16 cells, and PP2Ac‐mediated phosphatase activity in HASMC and AC16 cells. While there is no report of the effect of hypoxia, per se, on the regulatory transcripts we studied, hypoxia increases PPP1R3C (PP1 regulatory subunit) expression in MCF7 cells (Shen, Zhang, Liu, & Zhang, 2010) and in liver, muscle, and spleen, but not brain of fish (Mohindra, Tripathi, Singh, & Lal, 2013). However, ischaemia attenuated expression and abundance of PP2R2A (B55), PPP2R5A, PPP2R5B, PPP2R5E, PPP2R3A, and PPP2R4 following focal cerebral occlusion in the rat (Koh, 2013) and in cardiac tissue from patients with both ischaemic or non‐ischaemic heart failure (Nicolaou, Hajjar, & Kranias, 2009; Wijnker et al., 2011). Although work by De Grande shows that expression of PPP2R3A and PPP2R5B was increased in ischaemic heart failure, while PPP2R5D expression was increased in cardiac tissue from patients with non‐ischaemic heart failure (DeGrande et al., 2013).
While the role of the regulatory subunits in ischaemia is unclear, expression of B55 (PP2R2A) but not B56 enhances ischaemia‐induced cell death (Tsao et al., 2007), which would indicate that down‐regulation of certain regulatory subunits may have a protective role in ischaemia. As we primarily investigated the effect of hypoxia on scaffolding and regulatory subunit expression, it is not possible to ascertain if this translates through to altered protein abundance, and hence inhibition of protein phosphatase activity. While an obvious limitation of our work, along with the fact that we did not investigate the effect of hypoxia on other regulatory subunits, it nevertheless generates a base for further work, which should include determining the effect of hypoxia on PP2A holoenzyme assembly. Regarding PP2R1A and PP2R5A, the effect of hypoxia on mRNA expression was not consistently reflected at the protein level across all cell lines studied and hence is unlikely to account for hypoxia‐mediated inhibition of PP2Ac activity.
PP2A activity is also modulated through interaction with several endogenous inhibitors including CIP2A, and via post‐translational modification of a conserved 304TPDYFL309 motif in the C‐terminal tail of PP2Ac (Cho & Xu, 2007; Xing et al., 2006). Importantly, methylation of Leu309 by LCMT‐1 enhances PP2A activity and promotes holoenzyme assembly (Bryant, Westphal, & Wadzinski, 1999). In the present study, the decrease in PP2Ac activity following exposure to hypoxia for 24 hr was associated with decreased CIP2A and LCMT‐1 mRNA expression in all cell lines. While LCMT‐1 abundance was decreased after 24 hr in HASMC and AC16 cells, CIP2A abundance was only attenuated following prolonged exposure (72 hr). In addition, PME‐1 and demethylated PP2Ac abundance were increased, consistent with post‐translational modification of PP2Ac and not CIP2A contributing to hypoxia‐mediated inhibition of PP2Ac activity at this time point. This complements data from a recent study showing hypoperfusion to increase PME‐1 and decreased LCMT‐1 abundance in rat brain (Liu et al., 2016). Furthermore, in ventricular tissue from patients with heart failure, LCMT‐1 abundance is decreased while that of PME‐1 is unaffected (DeGrande et al., 2013).
Phosphorylation at Thr304 and Tyr307 in the C‐terminal tail of PP2Ac is another critical post‐translational modification modulating enzyme activity. While we found hypoxia (24 hr) increased phosphorylation of PP2Ac in HASMC, this was not observed in AC16 cells, suggesting that phosphorylation may not be a unifying mechanism contributing to loss of PP2Ac activity following “short” term hypoxia. Nonetheless, phosphorylation at Tyr307 is associated with reduced PP2A activity in hypo‐perfused rat brain (Liu et al., 2016) and in cardiac tissue from patients with heart failure (DeGrande et al., 2013). Although we and others (DeGrande et al., 2013; Liu et al., 2016) have not investigated which kinase(s) catalyse the phosphorylation of PP2Ac, a variety of kinases (GSK‐3β, PTP1B, and insulin receptor kinase) have been implicated in earlier studies (Chen, Martin, & Brautigan, 1992; Yao et al., 2011).
Studies are now emerging to indicate crosstalk between HIF‐1α and the protein phosphatase system. For example, during hypoxia, PP2R2A can inactivate PHD2 through dephosphorylation of Ser125 in colorectal cancer cells (Di Conza, Trusso Cafarello, Loroch, et al., 2017). Conversely, in breast cancer, PHD2 hydroxylates Pro319 in PP2R2A increasing its degradation (Di Conza, Trusso Cafarello, Zheng, Zhang, & Mazzone, 2017). More specifically, hypoxia‐induced HIF‐1α expression is associated with increased PP2A activity and proliferation of glioblastoma multiform‐derived cells (Hofstetter et al., 2012). Our data complement these studies by demonstrating that the prolyl hydroxylase inhibitor DMOG (100 μM) mimicked the inhibitory effect of hypoxia on total phosphatase activity, PPP2CA mRNA, and HIF‐1α mRNA expression without affecting PP2Ac abundance or cell viability. Importantly, silencing of HIF‐1α reversed the effect of hypoxia on total protein phosphatase activity and PPP2CA mRNA expression, without affecting PP2Ac abundance in HASMC and AC16 cells. This is the first data establishing that hypoxia directly inhibits PPP2CA mRNA expression and phosphoprotein phosphatase activity through a HIF‐1α‐dependent mechanism in a non‐cancerous cell line.
To gain further insight into the role of HIF‐1α in attenuating PP2A activity, the effect of HIF‐1α knockdown on the phosphorylation and methylation state of PP2Ac and the associated enzymes were investigated. Interestingly, knockdown of HIF‐1α did not alter hypoxia‐induced phosphorylation or demethylation of PP2Ac, nor its effects on PME‐1 or LCMT‐1 abundance, consistent with a HIF‐1α‐independent mechanism. Similarly, HIF‐1α did not mediate the “short” term effect of hypoxia (24 hr) on CIP2A mRNA expression in either HASMC or AC16 ventricular cardiomyocytes; a finding not reported before for hypoxia or ischaemia. It is difficult to ascribe a functional consequence to this but it would increase PP2A activity and development of heart failure and cardiac arrhythmias (Little et al., 2015; Lubbers & Mohler, 2016). However, PP2Ac activity is clearly attenuated in the present study. This dichotomy may reflect the complexity of the PP2A holoenzyme and its multifaceted regulation. While these findings have not been reported before, the paradigm exists for HIF‐1α‐independent regulation of the PP2A system as hypoxia modulates autophagy and orexin A‐mediated glucose metabolism via a HIF‐1α‐independent mechanism in nucleus pulposus and hepatocellular carcinoma cell lines (Choi et al., 2016; Wan et al., 2017). Clearly, the mechanism by which hypoxia, both short term and long term, modulates protein phosphatase activity requires further investigation.
In conclusion, all components of the protein phosphatase system studied are expressed in cultures of HAEC, HASMC, HCF‐av, and ventricular cardiomyocytes. Functionally, hypoxia decreased global phosphoprotein phosphatase activity in human primary cardiovascular cell lines in a manner that is cell line, time, and subunit dependent. In HASMC and ventricular cardiomyocytes, hypoxia (1% O2; 24 hr) inhibits PP2A activity through a HIF‐1α‐dependent mechanism without altering PP2Ac abundance. However, PP2Ac undergoes HIF‐1α‐independent phosphorylation and demethylation during hypoxia consistent with changes in the abundance of PME‐1 and LMCT‐1. The post‐translational modification of PP2Ac may also contribute to decreased PP2A activity by altering assembly of the PP2A holoenzyme (Figure 9). It is unlikely that CIP2A is involved in diminishing global phosphatase activity as hypoxia does not alter its abundance, although its expression is decreased. Together, our study demonstrates that many components of the protein phosphatase system are expressed in cardiovascular cell lines and that there is a complex interplay between hypoxia, HIF‐1α, and the PP2A system. This has major implications for not only our understanding of cardiovascular disease but also its treatment. For example, understanding and targeting of the protein phosphatase system presents an alternative strategy to modulate adrenergic signalling and more generally kinase signalling pathways involved in cardiovascular disease. Indeed, the PP2A system is of particular interest in oncology and may explain why targeted kinase therapies fail (Westermarck, 2018).
Figure 9.
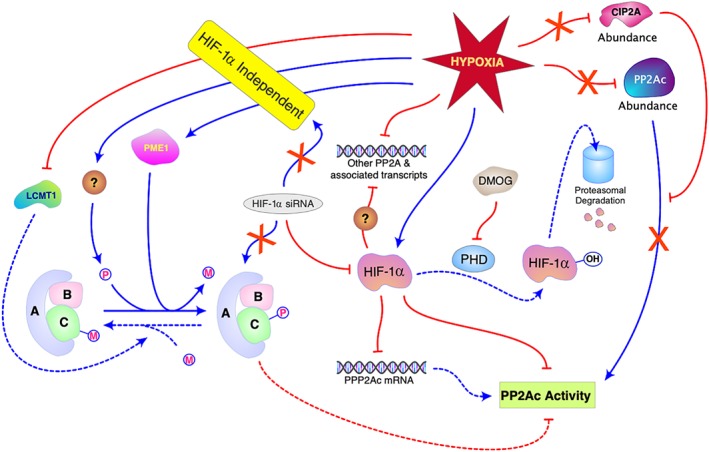
A schematic representation of HIF‐1α‐dependent and ‐independent regulation of the post‐translation modulation of PP2A and PP2A activity in human aortic smooth muscle cells and ventricular cardiomyocytes exposed to hypoxia (24 hr). Solid lines show experimental findings while dashed lines are inferred consequences; red and blue signify inhibition and activation respectively. CIP2A, cancerous inhibitor of PP2A; DMOG, dimethyloxalylglycine; HIF‐1α hypoxia inducible factor 1α LCMT‐1, leucine carboxyl methyltransferase 1; PDH, prolyl‐4‐hydroxylase enzyme; PP2Ac, Protein phosphatase 2A catalytic subunit A; PME‐1, protein phosphatase methylesterase‐1
ACKNOWLEDGEMENTS
This work was supported by the Ministry of Higher Education and Scientific Research/Libya, Grant 469/2009.
AUTHOR CONTRIBUTIONS
Both authors conceived and designed the study. I.S.E. carried out the experiments, while J.P.S. undertook the statistical analysis. Both authors discussed the results, their interpretation, and contributed to the writing of the final manuscript.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, and Immunoblotting and Immunochemistry, and as recommended by funding agencies, publishers and other organizations engaged with supporting research.
Supporting information
Table S1 Reference ID and primer sequences used in mRNA expression profiling along with their annealing temperature (Tm).
Figure S1 Validation of β‐actin, GAPDH and GPI as housekeeping genes in: (A) HAEC, (B) HASMC and (C) HCF‐av exposed to hypoxia for 24 h (1% O2, 5% CO2, and balanced N2 for 24 h) as assessed using geNORM software. Abbreviations: GAPDH, glyceraldehyde‐3‐phosphated dehydrogenase; GPI, glucose‐6‐phosphate isomerase
Figure S2 Temporal effect of hypoxia (1% O2; 24–72 h) on HAEC (A, D, G), HASMC (B, E, H) and HCF‐av (C, F, I) cell viability over 72 h was assessed using an MTT assay (A‐C), cell counting (D‐F) and mitochondrial membrane permeability (G‐I). Doxorubicin (50 μM) was included as a positive control. Data are presented normalised (mean ± S.E.M.) to the corresponding normoxia control and were analysed using one‐way ANOVA with post hoc analysis (Dunnett). * P < 0.05 (n = 5). Abbreviations: Dox, doxorubicin
Figure S3 Effect of DMOG (100 μM, 24 h) on: (A) cell viability, (B) total phosphatase activity, (C) PP2Ac abundance and (D) PPP2CA mRNA expression in HASMC. PP2Ac activity is presented as a percentage of the phosphatase activity under normoxia. In the cell viability assay, doxorubicin (50 μM) was includes as a positive control. PP2Ac abundance and mRNA expression were normalised (β‐actin and geometric mean of GPI and GAPDH, as appropriate), and expressed relative to normoxia. Data are presented as the mean ± S.E.M. (n = 5) and were analysed using an unpaired Student t‐test. * P < 0.05. Abbreviations: UnTx, Untreated cells; DMOG, dimethyloxalylglycine; Dox, doxorubicin.
Elgenaidi IS, Spiers JP. Hypoxia modulates protein phosphatase 2A through HIF‐1α dependent and independent mechanisms in human aortic smooth muscle cells and ventricular cardiomyocytes. Br J Pharmacol. 2019;176:1745–1763. 10.1111/bph.14648
REFERENCES
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators (2017). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. British Journal of Pharmacology, 174, S272–S359. 10.1111/bph.13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Christopoulos, A. , Davenport, A. P. , Kelly, E. , Marrion, N. V. , Peters, J. A. , … CGTP Collaborators (2017). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. British Journal of Pharmacology, 174, S17–S129. 10.1111/bph.13878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baharians, Z. , & Schonthal, A. H. (1998). Autoregulation of protein phosphatase type 2A expression. The Journal of Biological Chemistry, 273(30), 19019–19024. 10.1074/jbc.273.30.19019 [DOI] [PubMed] [Google Scholar]
- Bryant, J. C. , Westphal, R. S. , & Wadzinski, B. E. (1999). Methylated C‐terminal leucine residue of PP2A catalytic subunit is important for binding of regulatory Bα subunit. The Biochemical Journal, 339(Pt 2), 241–246. 10.1042/bj3390241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J. , Martin, B. L. , & Brautigan, D. L. (1992). Regulation of protein serine‐threonine phosphatase type‐2A by tyrosine phosphorylation. Science, 257(5074), 1261–1264. 10.1126/science.1325671 [DOI] [PubMed] [Google Scholar]
- Chi, J. T. , Wang, Z. , Nuyten, D. S. , Rodriguez, E. H. , Schaner, M. E. , Salim, A. , … Brown, P. O. (2006). Gene expression programs in response to hypoxia: Cell type specificity and prognostic significance in human cancers. PLoS Medicine, 3(3), e47 10.1371/journal.pmed.0030047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho, U. S. , & Xu, W. Q. (2007). Crystal structure of a protein phosphatase 2A heterotrimeric holoenzyme. Nature, 445(7123), 53–57. 10.1038/nature05351 [DOI] [PubMed] [Google Scholar]
- Choi, H. , Merceron, C. , Mangiavini, L. , Seifert, E. L. , Schipani, E. , Shapiro, I. M. , & Risbud, M. V. (2016). Hypoxia promotes noncanonical autophagy in nucleus pulposus cells independent of MTOR and HIF1A signaling. Autophagy, 12(9), 1631–1646. 10.1080/15548627.2016.1192753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen, P. T. W. (2002). Protein phosphatase 1—Targeted in many directions. Journal of Cell Science, 115(2), 241–256. [DOI] [PubMed] [Google Scholar]
- Cristobal, I. , Manso, R. , Rincon, R. , Carames, C. , Senin, C. , Borrero, A. , … Garcia‐Foncillas, J. (2014). PP2A inhibition is a common event in colorectal cancer and its restoration using FTY720 shows promising therapeutic potential. Molecular Cancer Therapeutics, 13(4), 938–947. 10.1158/1535-7163.MCT-13-0150 [DOI] [PubMed] [Google Scholar]
- Davidson, M. M. , Nesti, C. , Palenzuela, L. , Walker, W. F. , Hernandez, E. , Protas, L. , … Isaac, N. D. (2005). Novel cell lines derived from adult human ventricular cardiomyocytes. Journal of Molecular and Cellular Cardiology, 39(1), 133–147. 10.1016/j.yjmcc.2005.03.003 [DOI] [PubMed] [Google Scholar]
- DeGrande, S. T. , Little, S. C. , Nixon, D. J. , Wright, P. , Snyder, J. , Dun, W. , … Mohler, P. J. (2013). Molecular mechanisms underlying cardiac protein phosphatase 2A regulation in heart. The Journal of Biological Chemistry, 288(2), 1032–1046. 10.1074/jbc.M112.426957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Conza, G. , Trusso Cafarello, S. , Loroch, S. , Mennerich, D. , Deschoemaeker, S. , Di Matteo, M. , … Mazzone, M. (2017). The mTOR and PP2A pathways regulate PHD2 phosphorylation to fine‐tune HIF1α levels and colorectal cancer cell survival under hypoxia. Cell Reports, 18(7), 1699–1712. 10.1016/j.celrep.2017.01.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Conza, G. , Trusso Cafarello, S. , Zheng, X. , Zhang, Q. , & Mazzone, M. (2017). PHD2 targeting overcomes breast cancer cell death upon glucose starvation in a PP2A/B55α‐mediated manner. Cell Reports, 18(12), 2836–2844. 10.1016/j.celrep.2017.02.081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett, A. D. , Xue, C. , & Stoops, T. (1999). Developmental expression of protein phosphatase 2A in the kidney. J. Am. Soc. Nephrol., 10(8), 1737–1745. [DOI] [PubMed] [Google Scholar]
- Gomes, L. R. , Menck, C. F. M. , & Cuervo, A. M. (2017). Chaperone‐mediated autophagy prevents cellular transformation by regulating MYC proteasomal degradation. Autophagy, 13(5), 928–940. 10.1080/15548627.2017.1293767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR . (2018). The IUPHAR/BPS guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46(D1), D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heijman, J. , Dewenter, M. , El‐Armouche, A. , & Dobrev, D. (2013). Function and regulation of serine/threonine phosphatases in the healthy and diseased heart. Journal of Molecular and Cellular Cardiology, 64, 90–98. 10.1016/j.yjmcc.2013.09.006 [DOI] [PubMed] [Google Scholar]
- Heijman, J. , Ghezelbash, S. , Wehrens, X. H. , & Dobrev, D. (2017). Serine/Threonine phosphatases in atrial fibrillation. Journal of Molecular and Cellular Cardiology, 103, 110–120. 10.1016/j.yjmcc.2016.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig, S. , & Neumann, J. (2000). Effects of serine/threonine protein phosphatases on ion channels in excitable membranes. Physiological Reviews, 80(1), 173–210. 10.1152/physrev.2000.80.1.173 [DOI] [PubMed] [Google Scholar]
- Hofstetter, C. P. , Burkhardt, J. K. , Shin, B. J. , Gursel, D. B. , Mubita, L. , Gorrepati, R. , … Boockvar, J. A. (2012). Protein phosphatase 2A mediates dormancy of glioblastoma multiforme‐derived tumor stem‐like cells during hypoxia. PLoS ONE, 7(1), e30059 10.1371/journal.pone.0030059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmquist‐Mengelbier, L. , Fredlund, E. , Löfstedt, T. , Noguera, R. , Navarro, S. , Nilsson, H. , … Påhlman, S. (2006). Recruitment of HIF‐1α and HIF‐2α to common target genes is differentially regulated in neuroblastoma: HIF‐2α promotes an aggressive phenotype. Cancer Cell, 10, 413–423. 10.1016/j.ccr.2006.08.026 [DOI] [PubMed] [Google Scholar]
- Hwang, J. , Lee, J. A. , & Pallas, D. C. (2016). Leucine carboxyl methyltransferase 1 (LCMT‐1) methylates protein phosphatase 4 (PP4) and protein phosphatase 6 (PP6) and differentially regulates the stable formation of different PP4 holoenzymes. The Journal of Biological Chemistry, 291(40), 21008–21019. 10.1074/jbc.M116.739920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, T. C. , Verrier, J. D. , Drabek, T. , Janesko‐Feldman, K. , Gillespie, D. G. , Uray, T. , … Kochanek, P. M. (2013). Pharmacological inhibition of pleckstrin homology domain leucine‐rich repeat protein phosphatase is neuroprotective: Differential effects on astrocytes. The Journal of Pharmacology and Experimental Therapeutics, 347(2), 516–528. 10.1124/jpet.113.206888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhefer, U. , Brekle, C. , Eskandar, J. , Isensee, G. , Kucerova, D. , Muller, F. U. , … Boknik, P. (2014). Cardiac function is regulated by B56α‐mediated targeting of protein phosphatase 2A (PP2A) to contractile relevant substrates. The Journal of Biological Chemistry, 289(49), 33862–33873. 10.1074/jbc.M114.598938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh, M. Y. , Lemos, R. Jr. , Liu, X. , & Powis, G. (2011). The hypoxia‐associated factor switches cells from HIF‐1α‐ to HIF‐2α‐dependent signaling promoting stem cell characteristics, aggressive tumor growth and invasion. Cancer Research, 71(11), 4015–4027. 10.1158/0008-5472.CAN-10-4142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh, P. O. (2013). Ferulic acid attenuates the injury‐induced decrease of protein phosphatase 2A subunit B in ischemic brain injury. PLoS ONE, 8(1), e54217 10.1371/journal.pone.0054217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krtolica, A. , Krucher, N. A. , & Ludlow, J. W. (1998). Hypoxia‐induced pRB hypophosphorylation results from downregulation of CDK and upregulation of PP1 activities. Oncogene, 17(18), 2295–2304. 10.1038/sj.onc.1202159 [DOI] [PubMed] [Google Scholar]
- Kučera, J. , Netušilová, J. , Sladeček, S. , Lánová, M. , Vašíček, O. , Štefková, K. , … Pacherník, J. (2017). Hypoxia downregulates MAPK/ERK but not STAT3 signaling in ROS‐dependent and HIF‐1‐independent manners in mouse embryonic stem cells. Oxidative Medicine and Cellular Longevity, 2017, 4386947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen, K. O. , Lygren, B. , Sjaastad, I. , Krobert, K. A. , Arnkvaern, K. , Florholmen, G. , … Christensen, G. (2008). Diastolic dysfunction in alveolar hypoxia: A role for interleukin‐18‐mediated increase in protein phosphatase 2A. Cardiovascular Research, 80(1), 47–54. 10.1093/cvr/cvn180 [DOI] [PubMed] [Google Scholar]
- Lee, J. , Jeong, H. , Park, E. J. , Hwang, J. W. , Huang, B. , Bae, E. K. , … Koh, E. M. (2013). CIP2A facilitates apoptotic resistance of fibroblast‐like synoviocytes in rheumatoid arthritis independent of c‐Myc expression. Rheumatology International, 33(9), 2241–2248. 10.1007/s00296-013-2711-6 [DOI] [PubMed] [Google Scholar]
- Lee, W. H. , Tsai, M. J. , Chang, W. A. , Wu, L. Y. , Wang, H. Y. , Chang, K. F. , … Kuo, P. L. (2018). Deduction of novel genes potentially involved in hypoxic AC16 human cardiomyocytes using next‐generation sequencing and bioinformatics approaches. International Journal of Molecular Medicine, 42(5), 2489–2502. 10.3892/ijmm.2018.3851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leulliot, N. , Quevillon‐Cheruel, S. , Sorel, I. , Li de La Sierra‐Gallay, I. , Collinet, B. , Graille, M. , … van Tilbeurgh, H. (2004). Structure of protein phosphatase methyltransferase 1 (PPM1), a leucine carboxyl methyltransferase involved in the regulation of protein phosphatase 2A activity. The Journal of Biological Chemistry, 279(9), 8351–8358. 10.1074/jbc.M311484200 [DOI] [PubMed] [Google Scholar]
- Little, S. C. , Curran, J. , Makara, M. A. , Kline, C. F. , Ho, H. T. , Xu, Z. , … Mohler, P. J. (2015). Protein phosphatase 2A regulatory subunit B56α limits phosphatase activity in the heart. Science Signaling, 8(386), ra72 10.1126/scisignal.aaa5876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, C. D. , Wang, Q. , Zong, D. K. , Pei, S. C. , Yan, Y. , Yan, M. L. , … Ai, J. (2016). Knockdown of microRNA‐195 contributes to protein phosphatase‐2A inactivation in rats with chronic brain hypoperfusion. Neurobiology of Aging, 45, 76–87. 10.1016/j.neurobiolaging.2016.05.010 [DOI] [PubMed] [Google Scholar]
- Longin, S. , Zwaenepoel, K. , Louis, J. V. , Dilworth, S. , Goris, J. , & Janssens, V. (2007). Selection of protein phosphatase 2A regulatory subunits is mediated by the C terminus of the catalytic Subunit. The Journal of Biological Chemistry, 282(37), 26971–26980. 10.1074/jbc.M704059200 [DOI] [PubMed] [Google Scholar]
- Lubbers, E. R. , & Mohler, P. J. (2016). Roles and regulation of protein phosphatase 2A (PP2A) in the heart. Journal of Molecular and Cellular Cardiology, 101, 127–133. 10.1016/j.yjmcc.2016.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luss, H. , Klein‐Wiele, O. , Boknik, P. , Herzig, S. , Knapp, J. , Linck, B. , … Neumann, J. (2000). Regional expression of protein phosphatase type 1 and 2A catalytic subunit isoforms in the human heart. Journal of Molecular and Cellular Cardiology, 32(12), 2349–2359. 10.1006/jmcc.2000.1265 [DOI] [PubMed] [Google Scholar]
- Martin de la Vega, C. , Burda, J. , Toledo Lobo, M. V. , & Salinas, M. (2002). Cerebral postischemic reperfusion‐induced demethylation of the protein phosphatase 2A catalytic subunit. Journal of Neuroscience Research, 69(4), 540–549. 10.1002/jnr.10306 [DOI] [PubMed] [Google Scholar]
- Matoulkova, E. , Michalova, E. , Vojtesek, B. , & Hrstka, R. (2012). The role of the 3′ untranslated region in post‐transcriptional regulation of protein expression in mammalian cells. RNA Biology, 9(5), 563–576. 10.4161/rna.20231 [DOI] [PubMed] [Google Scholar]
- Mohindra, V. , Tripathi, R. K. , Singh, R. K. , & Lal, K. K. (2013). Molecular characterization and expression analysis of PPP1R3C in hypoxia‐tolerant Indian catfish, Clarias batrachus (Linnaeus, 1758) under hypoxia. Gene, 530(1), 127–133. 10.1016/j.gene.2013.07.042 [DOI] [PubMed] [Google Scholar]
- Morioka, M. , Fukunaga, K. , Hasegawa, S. , Okamura, A. , Korematsu, K. , Kai, Y. , … Ushio, Y. (1999). Activities of calcineurin and phosphatase 2A in the hippocampus after transient forebrain ischemia. Brain Research, 828(1–2), 135–144. 10.1016/S0006-8993(99)01349-9 [DOI] [PubMed] [Google Scholar]
- Nicolaou, P. , Hajjar, R. J. , & Kranias, E. G. (2009). Role of protein phosphatase‐1 inhibitor‐1 in cardiac physiology and pathophysiology. Journal of Molecular and Cellular Cardiology, 47(3), 365–371. 10.1016/j.yjmcc.2009.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogris, E. , Du, X. , Nelson, K. C. , Mak, E. K. , Yu, X. X. , Lane, W. S. , & Pallas, D. C. (1999). A protein phosphatase methylesterase (PME‐1) is one of several novel proteins stably associating with two inactive mutants of protein phosphatase 2A. The Journal of Biological Chemistry, 274(20), 14382–14391. 10.1074/jbc.274.20.14382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price, N. E. , & Mumby, M. C. (2000). Effects of regulatory subunits on the kinetics of protein phosphatase 2A. Biochemistry (Mosc), 39(37), 11312–11318. 10.1021/bi0008478 [DOI] [PubMed] [Google Scholar]
- Sangodkar, J. , Farrington, C. C. , McClinch, K. , Galsky, M. D. , Kastrinsky, D. B. , & Narla, G. (2016). All roads lead to PP2A: exploiting the therapeutic potential of this phosphatase. The FEBS Journal, 283(6), 1004–1024. 10.1111/febs.13573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos, C. X. , Hafstad, A. D. , Beretta, M. , Zhang, M. , Molenaar, C. , Kopec, J. , … Shah, A. M. (2016). Targeted redox inhibition of protein phosphatase 1 by Nox4 regulates eIF2α‐mediated stress signaling. The EMBO Journal, 35(3), 319–334. 10.15252/embj.201592394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, G. M. , Zhang, F. L. , Liu, X. L. , & Zhang, J. W. (2010). Hypoxia‐inducible factor 1‐mediated regulation of PPP1R3C promotes glycogen accumulation in human MCF‐7 cells under hypoxia. FEBS Letters, 584(20), 4366–4372. 10.1016/j.febslet.2010.09.040 [DOI] [PubMed] [Google Scholar]
- Shi, Y. (2009). Serine/threonine phosphatases: Mechanism through structure. Cell, 139(3), 468–484. 10.1016/j.cell.2009.10.006 [DOI] [PubMed] [Google Scholar]
- Takai, M. , Nakagawa, T. , Tanabe, A. , Terai, Y. , Ohmichi, M. , & Asahi, M. (2015). Crosstalk between PI3K and Ras pathways via protein phosphatase 2A in human ovarian clear cell carcinoma. Cancer Biology & Therapy, 16(2), 325–335. 10.1080/15384047.2014.1002362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian, H. P. , Qiu, T. Z. , Zhao, J. , Li, L. X. , & Guo, J. (2009). Sphingomyelinase‐induced ceramide production stimulate calcium‐independent JNK and PP2A activation following cerebral ischemia. Brain Injury, 23(13–14), 1073–1080. 10.3109/02699050903379388 [DOI] [PubMed] [Google Scholar]
- Truttmann, A. C. , Ashraf, Q. , Mishra, O. P. , & Delivoria‐Papadopoulos, M. (2004). Effect of hypoxia on protein phosphatase 2A activity, subcellular distribution and expression in cerebral cortex of newborn piglets. Neuroscience, 127(2), 355–363. 10.1016/j.neuroscience.2004.05.033 [DOI] [PubMed] [Google Scholar]
- Tsao, C. C. , Nica, A. F. , Kurinna, S. M. , Jiffar, T. , Mumby, M. , & Ruvolo, P. P. (2007). Mitochondrial protein phosphatase 2A regulates cell death induced by simulated ischemia in kidney NRK‐52E cells. Cell Cycle, 6(19), 2377–2385. 10.4161/cc.6.19.4737 [DOI] [PubMed] [Google Scholar]
- Varadkar, P. , Despres, D. , Kraman, M. , Lozier, J. , Phadke, A. , Nagaraju, K. , & Mccright, B. (2014). The protein phosphatase 2A B56γ regulatory subunit is required for heart development. Developmental Dynamics, 243(6), 778–790. 10.1002/dvdy.24111 [DOI] [PubMed] [Google Scholar]
- Wan, X. , Liu, Y. , Zhao, Y. , Sun, X. , Fan, D. , & Guo, L. (2017). Orexin A affects HepG2 human hepatocellular carcinoma cells glucose metabolism via HIF‐1α‐dependent and ‐independent mechanism. PLoS ONE, 12(9), e0184213 10.1371/journal.pone.0184213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermarck, J. (2018). Targeted therapies don't work for a reason; the neglected tumor suppressor phosphatase PP2A strikes back. The FEBS Journal, 285, 4139–4145. 10.1111/febs.14617 [DOI] [PubMed] [Google Scholar]
- Wijnker, P. J. , Boknik, P. , Gergs, U. , Muller, F. U. , Neumann, J. , dos Remedios, C. , … Kirchhefer, U. (2011). Protein phosphatase 2A affects myofilament contractility in non‐failing but not in failing human myocardium. Journal of Muscle Research and Cell Motility, 32(3), 221–233. 10.1007/s10974-011-9261-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlodarchak, N. , & Xing, Y. (2016). PP2A as a master regulator of the cell cycle. Critical Reviews in Biochemistry and Molecular Biology, 51(3), 162–184. 10.3109/10409238.2016.1143913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, T. Y. , Juang, W. C. , Tsai, C. T. , Tseng, C. J. , Lee, W. H. , Chang, S. N. , & Cheng, P. W. (2018). Mechanical stretching simulates cardiac physiology and pathology through mechanosensor Piezo1. J Clin Med, 7(11). 10.3390/jcm7110410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing, Y. , Xu, Y. H. , Chen, Y. , Jeffrey, P. D. , Chao, Y. , Lin, Z. , … Shi, Y. (2006). Structure of protein phosphatase 2A core enzyme bound to tumor‐inducing toxins. Cell, 127(2), 341–353. 10.1016/j.cell.2006.09.025 [DOI] [PubMed] [Google Scholar]
- Yao, X. Q. , Zhang, X. X. , Yin, Y. Y. , Liu, B. , Luo, D. J. , Liu, D. , … Liu, G. P. (2011). Glycogen synthase kinase‐3β regulates Tyr307 phosphorylation of protein phosphatase‐2A via protein tyrosine phosphatase 1B but not Src. The Biochemical Journal, 437(2), 335–344. 10.1042/BJ20110347 [DOI] [PubMed] [Google Scholar]
- Zhou, Y. , Wang, D. , Gao, X. , Lew, K. , Richards, A. M. , & Wang, P. (2014). mTORC2 phosphorylation of Akt1: A possible mechanism for hydrogen sulfide‐induced cardioprotection. PLoS ONE, 9(6), e99665 10.1371/journal.pone.0099665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, M. , Ding, J. , Jiang, H. , Kong, L. , Sun, Z. , Chen, J. , & Miao, C. (2015). Propofol ameliorates endothelial inflammation induced by hypoxia/reoxygenation in human umbilical vein endothelial cells: Role of phosphatase A2. Vascular Pharmacology, 73, 149–157. 10.1016/j.vph.2015.06.002 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Reference ID and primer sequences used in mRNA expression profiling along with their annealing temperature (Tm).
Figure S1 Validation of β‐actin, GAPDH and GPI as housekeeping genes in: (A) HAEC, (B) HASMC and (C) HCF‐av exposed to hypoxia for 24 h (1% O2, 5% CO2, and balanced N2 for 24 h) as assessed using geNORM software. Abbreviations: GAPDH, glyceraldehyde‐3‐phosphated dehydrogenase; GPI, glucose‐6‐phosphate isomerase
Figure S2 Temporal effect of hypoxia (1% O2; 24–72 h) on HAEC (A, D, G), HASMC (B, E, H) and HCF‐av (C, F, I) cell viability over 72 h was assessed using an MTT assay (A‐C), cell counting (D‐F) and mitochondrial membrane permeability (G‐I). Doxorubicin (50 μM) was included as a positive control. Data are presented normalised (mean ± S.E.M.) to the corresponding normoxia control and were analysed using one‐way ANOVA with post hoc analysis (Dunnett). * P < 0.05 (n = 5). Abbreviations: Dox, doxorubicin
Figure S3 Effect of DMOG (100 μM, 24 h) on: (A) cell viability, (B) total phosphatase activity, (C) PP2Ac abundance and (D) PPP2CA mRNA expression in HASMC. PP2Ac activity is presented as a percentage of the phosphatase activity under normoxia. In the cell viability assay, doxorubicin (50 μM) was includes as a positive control. PP2Ac abundance and mRNA expression were normalised (β‐actin and geometric mean of GPI and GAPDH, as appropriate), and expressed relative to normoxia. Data are presented as the mean ± S.E.M. (n = 5) and were analysed using an unpaired Student t‐test. * P < 0.05. Abbreviations: UnTx, Untreated cells; DMOG, dimethyloxalylglycine; Dox, doxorubicin.